

Abstract
Autoimmune hypophysitis is classified as primary if its origin is idiopathic, and secondary if it develops as a consequence of treatment with immune checkpoint inhibitors (ICI). Expanding use of immunotherapy has been paralleled by the increasing hypophysitis prevalence. However, understanding of the immune responses driving the disease remains limited. Using a mouse model of primary hypophysitis we have identified CD4+ T lymphocytes to be the main pituitary-infiltrating immune cell population. Functional analysis showed that they display a Th17 and Th1/Th17 phenotype. In order to examine involvement of pro-inflammatory Th1, Th17 and Th1/17 subsets in hypophysitis, we have isolated RNA from the formalin-fixed paraffin-embedded (FFPE) pituitary specimens from 16 hypophysitis patients (3 of whom had hypophysitis secondary to ICI), 10 patients with adenoma and 23 normal pituitaries obtained at autopsy. Transcript levels of IFN-γ, IL-17A, IL-4, IL-10, TGF-β, CD4, CD8α and CIIT were analyzed by the reverse transcription- quantitative polymerase chain reaction (RT-qPCR). Pituitary glands of patients with hypophysitis showed significantly higher IL-17A, CD4 and CIIT mRNA levels compared to adenoma and normal pituitaries. All three secondary hypophysitis patients showed detectable IL-17A levels, but other cytokines were not detected in their pituitaries. Levels of IFN-γ, IL-4, IL-10 and TGF-β did not differ between the groups. TGF-β transcript was found in significantly fewer hypophysitis pituitaries (2/16) compared to adenoma (7/10) and normal pituitaries (11/23). Presence of TGF-β in two hypophysitis patients was associated with significantly lower IL-17A mRNA level compared to hypophysitis patients with no detectable TGF-β (p=0.03).
Introduction
Autoimmune hypophysitis is a disorder of the pituitary gland that in its primary (idiopathic) form is exceedingly rare (1–3). Awareness and knowledge about this condition has expanded rapidly after the outset of clinical trials testing immune checkpoint inhibitors (ICI) to treat melanoma and other cancers. Hypophysitis, in fact, emerged as one of the most common endocrine-related adverse events in patients treated with antibodies blocking CTLA-4 (ipilimumab) and/or PD-1 (nivolumab). Hypophysitis secondary to ICI has been reported in about 10% of cancer patients (3–6). Increase of the number of hypophysitis cases contributed to an advancement in the diagnosis and clinical management of the disease. However, understanding of the autoimmune events involved in the inflammatory process and, more importantly, specific autoantigen(s) driving it, remains limited (7). Recent study by Sharma et al (8) identified antibodies against integral membrane protein 2B (ITM2B) and guanine nucleotide-binding protein G(olf) subunit alpha (GNAL) as potential markers of ICI-induced hypophysitis. Immunotherapy increased their levels compared to baseline in patients who developed hypophysitis, but the anti-ITMB2 and anti-GNAL antibody expression did not change in patients without pituitary autoimmunity. Since only 8 patients with hypophysitis were included in the study, the validity of anti-ITMB2 and anti-GNAL antibodies as markers of (secondary) hypophysitis has to be confirmed in a larger independent cohort. Similarly, autoantibodies in patients with primary hypophysitis described by several reports, have not been established as clinically useful predictive markers. Using sections of human pituitary gland or baboon hypothalamus, it has been demonstrated that presence of serum anti-pituitary (APA) or anti-hypothalamus antibodies (AHA), respectively, did not help to discriminate between autoimmune hypophysitis and other pituitary diseases (9–13). APAs were also detected in ~5% of healthy subjects (9). Several studies evaluated specificity for primary hypophysitis of antibodies against pituitary autoantigens such as growth hormone (GH), pituitary gland specific factor 1a (PGSF1a) and PGSF2 (10, 11), chromodomain-helicase-DNA binding protein 8 (CHD8), presynaptic cytomatrix protein (Piccolo), calcium-dependent secretion activator (CADPS), neuron-specific enolase (NSE) (10) and alpha-enolase (12, 13). Those antibody specificities were not detected exclusively in hypophysitis patients but also in healthy subjects (10, 11) or patients with hypopituitarism, pituitary adenomas and other autoimmune diseases (10, 12, 13).
Available evidence strongly suggest that hypophysitis is a T-cell mediated, rather than autoantibody-mediated autoimmune disease. Report of two primary hypophysitis cases demonstrated T lymphocytes to be the largest fraction of the pituitary immune cell infiltrate (14). Unlike T cells, other immune cells, such as B cells, plasma cells, macrophages, neutrophils, eosinophils, or multinucleated giant cells are not detected in pituitary glands of all patients with a clinical diagnosis of hypophysitis (2, 14). T cells have been shown to be the main player in the pituitary extract-induced mouse model of hypophysitis (15). T cells from pituitaries of mice with GH-induced hypophysitis showed features of activation in situ (expression of PCNA and BrdU) and secreted IFN-γ and IL-17 in response to GH in vitro (16). However, a more detailed analysis of the phenotypic and functional profile of T cells involved in hypophysitis has not been performed yet.
In the present study, we have described a cytokine profile of T cells driving hypophysitis in a mouse model, and compared it to the cytokine profile of pituitary glands of patients with primary and secondary hypophysitis.
Material & Methods
Animals
SJL mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA), and bred in house. For all experiments, female 9–13 week-old SJL/J mice were used.
Experimental procedures were approved by the Johns Hopkins University Animal Care and Use Committee.
Pituitary extract preparation
Pituitary protein extract for induction of hypophysitis in mice was prepared as previously described (17). Briefly, pituitary glands isolated from mice of various strains were snap-frozen in liquid nitrogen and stored at −80°C. To prepare the protein extract, frozen pituitaries were homogenized in cold phosphate-buffered saline (PBS, pH 7.4, Ca/Mg/Mn-free) containing protease inhibitor cocktail (cOmplete™ Protease Inhibitor Cocktail, Sigma-Aldrich). Approximately 400 ul of the homogenization buffer was used for every 100 mg of pituitary tissue. The homogenate was centrifuged at 1000g, 10 min in the pre-cooled centrifuge, and the post-nuclear supernatant was collected and stored on ice. The remaining pellet was used for the re-extraction as described above, and the post-nuclear supernatant fractions were pooled. Protein concentration was measured by the bicinchoninic acid (BCA) assay, according to the manufacturer’s instructions (Thermo Fisher Scientific). Protein concentration was adjusted to 20 mg/ml, and the extract was stored at −80°C.
To induce hypophysitis, the extract was emulsified 1:1 in Complete Freund’s Adjuvant (CFA, Sigma-Aldrich) supplemented with 5 mg/ml of heat-killed Mycobacterium tuberculosis (strain H37Ra, BD).
Induction of hypophysitis
Autoimmune inflammation of the pituitary gland was induced as described previously (17). Mice were exposed to isoflurane to induce general anesthesia. Pituitary extract/CFA emulsion was injected subcutaneously in the dorsal region of a hind leg and then in the contralateral inguinal region, 50 μl/site (total 100 μl, corresponding to 1 mg pituitary proteins per mouse). Mice were injected twice (on days 0 and 7) and then sacrificed on day 28. The control group was injected with PBS/CFA (schematic outline of the experimental approach for disease induction is shown in Fig.1A).
Fig.1. Predominance of CD4+ T cells in pituitaries of mice with primary hypophysitis.
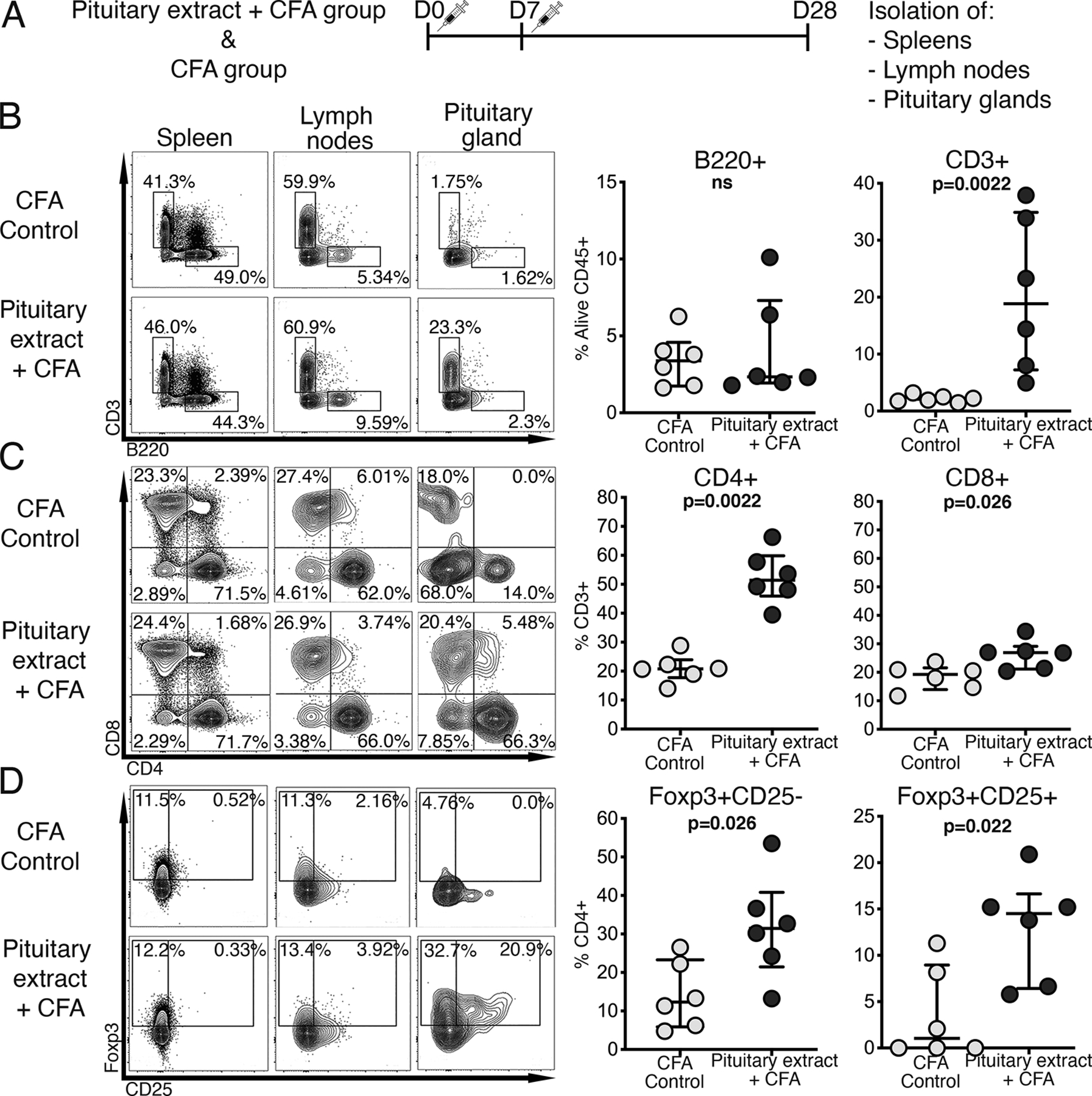
A) Outline of the protocol for inducing primary hypophysitis in mice. At day 28 after the pituitary extract injections, mice were sacrificed. Pituitary glands, as well as spleens and lymph nodes were used for flow cytometric analysis of the immune cell infiltrate (representative staining pattern of each organ from a mouse from control and experimental group is shown). Frequency of B) B220+ B cells and CD3+ T cells in pituitary glands; C) CD4+ and CD8+ cells within the CD3+ T cell gate and D) Foxp3+ regulatory T cells within the CD4+ T cell gate.
Mann-Whitney test was used.
In vivo blocking of IFN-γ and IL-17A
To assess the role of IFN-γ and IL-17A in the pathophysiology of hypophysitis, antibodies neutralizing IFN-γ (clone XMG1.2) or IL-17A (clone 17F3, both from Bio X Cell) were injected in the peritoneum three times at a dose of 500 μg/mouse (a total of 1.5 mg/mouse). The injections were performed on days −1, 6, 14 (to investigate the effect of blocking the cytokines before disease onset/early in the disease process) or on days 11, 15, 19 (after disease induction).
Cell isolation and flow cytometry
Upon sacrifice (day 28 after the first immunization) and perfusion with cold PBS, pituitary glands, spleens, and lymph nodes (superficial cervical, deep cervical) were harvested into RPMI1640 (MilliporeSigma) supplemented with L-glutamine and 10% fetal calf serum (FCS, Corning). Single cell suspensions were prepared by first mashing the organs between frosted ends of microscope slides, passed through a 70-μm cell strainer, and pelleted.
For a detailed characterization of the immune infiltrate of pituitary glands, cells from pituitaries, lymph nodes and spleens were washed with PBS, stained with Live/Dead Aqua viability dye (Thermo Fisher Scientific) according to the manufacturer’s instructions, and incubated with antibodies against surface markers: CD45 (BUV563), CD4 (BUV805), CD62L (BUV395), B220 (BUV395), CD64 (BV650), CD1d (BV786), Siglec-F (PE-CF594; all from BD), CD3 (APC/Fire 750), CD8 (PE/Cy7), TCRgd (BV 605), CD44 (BV785), CD49b (AF488), NKp46 (BV711), CD5 (APC/Fire 750), CCR2 (PE/Cy7), CD11b (PE), Ly6C (BV421), Ly6G (PerCP-Cy5.5), CD11c (APC), MHC II (AF700), F4/80 (BV605; all from BioLegend), CD25 (AF488; from eBioscience). After incubation for 30 min at 4°C, cells were fixed and permeabilized with the eBioscience Foxp3 / Transcription Factor Staining Buffer Set (Thermo Fisher Scientific), and stained with antibodies against intracellular markers: T-bet (BV711), Ki-67 (AF700; both from BioLegend), Foxp3 (PerCP-Cy5.5; Thermo Fisher Scientific).
To characterize their cytokine profile, T cells from pituitaries, lymph nodes and spleens were stimulated with phorbol myristate acetate (PMA) at a final concentration of 20 ng/ml and ionomycin at 1 μg/ml (both from Sigma-Aldrich) in RPMI1640 (with L-glutamine) and 10% FCS. To quantify T cell degranulation potency, anti- CD107a antibody was added to the cell suspensions (0.5 μg/100 μl). After an hour of stimulation at 37°C, brefeldin A (GolgiPlug) and monensin (GolgiStop, both from BD) were added and incubation was continued for another 4 hours.
Cells were washed in PBS, stained with Live/Dead Aqua viability dye (Thermo Fisher Scientific) according to the manufacturer’s instructions, and incubated with antibodies against surface markers: anti-CD3 (APC/Fire 750), CD8 (PE-Cy7), CD25 (Alexa Fluor 700, all from BioLegend), CD4 (BUV805), B220 (BUV395, both from BD) for 30 min at 4°C. Following fixation and permeabilization with eBioscience Foxp3 / Transcription Factor Staining Buffer Set, antibodies against intracellular markers were used: anti-TNF-α (PE), IL-17A (AF647), Granzyme B (Pacific Blue, all from BioLegend), IFN-γ (PE-CF594), IL-10 (BV711, both from BD), Foxp3 (PerCP-Cy5.5; Thermo Fisher Scientific). Cells were washed with FACS staining buffer (PBS with 1% bovine serum albumin [BSA] and 2 mM EDTA), acquired with the LSR Fortessa (BD), and data was analyzed with the FlowJo software (Tree Star, Inc.).
Human pituitary specimens
Formalin-fixed paraffin-embedded (FFPE) pituitary specimens were obtained from 16 patients with hypophysitis (13 from surgical pathology and 3 from autopsy), 10 patients with surgically resected pituitary adenoma and 23 subjects with normal pituitary gland collected at autopsy (Table 1). Specimens containing inflamed pituitary tissue were obtained from the FFPE block archive of the Department of Pathology, Johns Hopkins Hospital (Table 2). From the total of 34 hypophysitis blocks banked between 1959 and 2018, 20 were collected after calendar year 2000 and selected for RNA isolation. Of them, 12 yielded sufficient RNA amount to enable detection of the reference gene glyceraldehyde 3-phosphate dehydrogenase (gapdh). Additional hypophysitis blocks used for RT-qPCR included 3 autopsies of cancer patients who had been treated with immune checkpoint inhibitors, and 1 surgical biopsy of a patient with primary hypophysitis who had their pituitary FFPE specimen sent for consult. Of the cancer immunotherapy patients, one had a clinical diagnosis of hypophysitis and featured in a previous, detailed study (18), while the other two were incidental findings at autopsy of a mononuclear cell infiltration in the pituitary gland.
Table.1.
Demographical and experimental data of all subjects whose FFPE pituitary glands were used for RT-qPCR.
| Autopsy pituitaries | Pituitary adenomas | Hypophysitis | P value* | |
|---|---|---|---|---|
| N | 23 | 10 | 16 | |
| Age; median (range) | 62 (17–98) | 53 (19–69) | 38 (14–79) | |
| Females | 7 (30%) | 4 (40%) | 10 (63%) | ns |
| Detectable IFN-γ and IL-17A | 16 (70%) | 7 (70%) | 7 (44%) | ns |
| Detectable IFN-γ only | 2 (9%) | 0 | 0 | ns |
| Detectable IL-17A only | 4 (17%) | 1 (10%) | 5 (31%) | ns |
| Detectable IL-10 | 1 (4%) | 3 (30%) | 2 (13%) | ns |
| Detectable IL-4 | 0 | 0 | 0 | ns |
| Detectable TGF-β | 11 (48%) | 7 (70%) | 2 (13%) | 0.01 |
Chi-square test was used; ns = not significant.
Table 2.
Demographical and clinical information of hypophysitis patients whose FFPE pituitaries were used for RT-qPCR.
| Patient no | Age | Sex | Race | Type of hypophysitis | Glucocorticoid use at the time of specimen collection |
|---|---|---|---|---|---|
| 1 | 36 | F | White | Lymphocytic | yes |
| 2 | 37 | F | Black | Lymphocytic | yes |
| 3 | 20 | F | Black | Granulomatous | yes |
| 4 | 50 | M | White | Xanthogranulomatous | yes |
| 5 | 41 | F | Other | Lymphocytic | yes |
| 6 | 23 | F | Black | Lymphocytic | no |
| 7 | 33 | F | White | Necrotizing | no |
| 8 | 36 | M | White | Lymphocytic and granulomatous | no |
| 9 | 33 | M | White | Granulomatous | yes |
| 10 | 38 | M | White | Lymphocytic | yes |
| 11 | 14 | F | White | Lymphocytic | no |
| 12 | 42 | F | Black | Granulomatous | yes |
| 13 | 39 | F | White | Lymphocytic | no data |
| 14 | 79 | F | White | Secondary to immune checkpoint blockade. Diagnosis based on clinical presentation and MRI imaging. |
yes |
| 15 | 65 | M | White | Secondary to immune checkpoint blockade. Diagnosis based on histological analysis of the pituitary gland at autopsy. |
no data |
| 16 | 59 | M | White | Secondary to immune checkpoint blockade. Diagnosis based on histological analysis of the pituitary gland at autopsy. |
no data |
The pituitary adenomas included 8 prolactin (PRL)- secreting, 1 adrenocorticotropic hormone (ACTH)- secreting, and 1 non-secreting adenoma. Normal pituitary glands were obtained through the Legacy Gift Rapid Autopsy program.
RT-PCR to detect cytokine expression in human pituitary glands
To isolate total RNA, 20-μm tissue scrolls were cut from the FFPE blocks and processed using RecoverAll Total Nucleic Acid Isolation kit, according to the manufacturer’s instructions. At least 200 ng of total RNA was used for reverse transcription performed with 250 U of the SuperScript III reverse transcriptase, 0.5 μg of oligo(dT)12–18 primer, 0.5 mM dNTP mix, 40 units of RNaseOUT™ Ribonuclease Inhibitor, 5 mM DTT in 1x first-strand buffer (all reagents from Thermo Fisher Scientific). After digestion of the RNA strand with RNase H (Thermo Fisher Scientific), cDNA was amplified using primers specific for human IFN-γ, IL-17A, IL-4, IL-10, TGF-β, CD4, CD8 alpha chain (CD8α) and class II transactivator (CIIT) transcripts. Primers were designed with Primer-BLAST (Suppl. Table 1) and purchased from Integrated DNA Technologies.
Each reaction was run in duplicate. For 10 μl PCR reaction, 1 μl of cDNA in Fast SYBR Green Master Mix (Thermo Fisher Scientific) and 0.2 μM of primers were used, and reactions were analyzed on the QuantStudio 6 Flex Real-Time PCR System (Thermo Fisher Scientific). Temperature cycling was performed as follows: an initial polymerase activation at 95°C for 20 s, followed by 40 cycles of DNA denaturation at 95°C for 1 s, and Annealing/Extension at 60°C for 20 s. Values were normalized to housekeeping expression of gapdh using the 2–dCt method. Amplicon sizes were assessed on 2% agarose gels.
Statistical analysis
Percent of lymphoid cells in the various organs were summarized by median and interquartile range, and compared by Mann Whitney U test or one-way ANOVA test with Tukey’s correction for multiple comparisons. Categorical variables were compared by Fisher’s exact test. Analyses were done in GraphPad Prism version 8 (GraphPad Software, San Diego, CA, USA). P<0.05 was considered statistically significant.
Results
Pituitary glands of mice with hypophysitis were infiltrated predominantly by CD4+ T cells.
CD3+ T cells significantly increased in the pituitary glands of mice immunized with pituitary extract and CFA, as compared to those injected with CFA only: from a median of 2.1% to 18.9% (p=0.002). No significant changes were seen in the B cell compartment (Fig.1B). Both pituitary CD4+ and CD8+ T cells increased upon hypophysitis induction, but the increase was more pronounced for CD4+ T cells (median of 51.4% vs. 20.8% in pituitary extract/CFA and CFA group, respectively; p=0.002) than for CD8+ T cells (median of 26.9% vs. 19.3% in pituitary extract/CFA and CFA group, respectively; p=0.026; Fig.1C). A large fraction (median of 46%) of pituitary-infiltrating CD4+ T cells showed regulatory phenotype defined by Foxp3 expression. Mouse Foxp3+ Treg population includes CD25+ and CD25- T cells, both showing suppressive activity (19, 20). Those two Treg subsets were increased in pituitaries of mice with hypophysitis (Fig.1D). The increase in CD4+ and CD8+ T cells was pituitary-specific, as it was not seen in the draining lymph nodes or spleens (data not shown).
Other immune cell populations, including NK cells (B220-CD49b+NKp46+), macrophages (CD11b+CD11c+Siglec-F-Ly6G-CD64+F4/80+), monocyte subsets (CD11b+CD11c+ Siglec-F-Ly6G-Ly6C+CCR2+/-), neutrophils (CD11b+CD11c+Siglec-F- Ly6G+) and eosinophils (CD11b+CD11c+Siglec-F+Ly6G-) did not differ in pituitary glands isolated from cases or CFA controls (data not shown; gating strategy used to identify outlined leukocyte populations is shown in Suppl. Fig.1).
CD4+ T cells infiltrating mouse pituitary showed predominantly a Th1/Th17 and Th17 cytokine profile.
In pituitary glands of mice with experimentally induced hypophysitis, the most notable finding was the appearance of the IFN-γ/IL-17A- double producing cells in both CD4+ (Fig.2A) and CD8+ (Fig. 2B) T cell subsets. The increase was more marked for CD4+ than CD8+ T cells. The median percent of pituitary IFN-γ/IL-17A- double positive cells increased from about 2.6% in controls to 18.4% (IQR from 13.3% to 28.0%) within CD4+, and from 1.1% to 11.4% (IQR from 4.2% to 20.7%) within CD8+ T cells. Pituitary-infiltrating CD4+ T cells were also enriched with cells of theTh17 phenotype (IL-17A- single producers p=0.002) and, to a lesser degree, with Th1 cells (IFN-g- single producers, p=0.041) compared to CD4+ T cells from control mice (Fig.2A). T cells infiltrating pituitary glands of pituitary extract/CFA- injected mice also showed significantly higher TNF-α production compared to CFA only controls. The increase was more pronounced in CD4+ (p=0.002, Fig.2C) than CD8+ T cells (p=0.015, Fig.2D). Induction of hypophysitis was also associated with increased expression of cytotoxicity markers CD107a and granzyme B by both CD4+ and CD8+ T cells (data not shown). No significant changes in the frequency of pro-inflammatory cytokines/ cytotoxicity markers-expressing cells were observed between draining lymph node cells in mice from control and experimental groups (data not shown).
Fig.2. CD4+ T cells infiltrating pituitary glands express both IFN-γ+ and IL-17A+ (Th1/Th17), IFN-γ+ alone (Th1) and IL-17A alone (Th17).
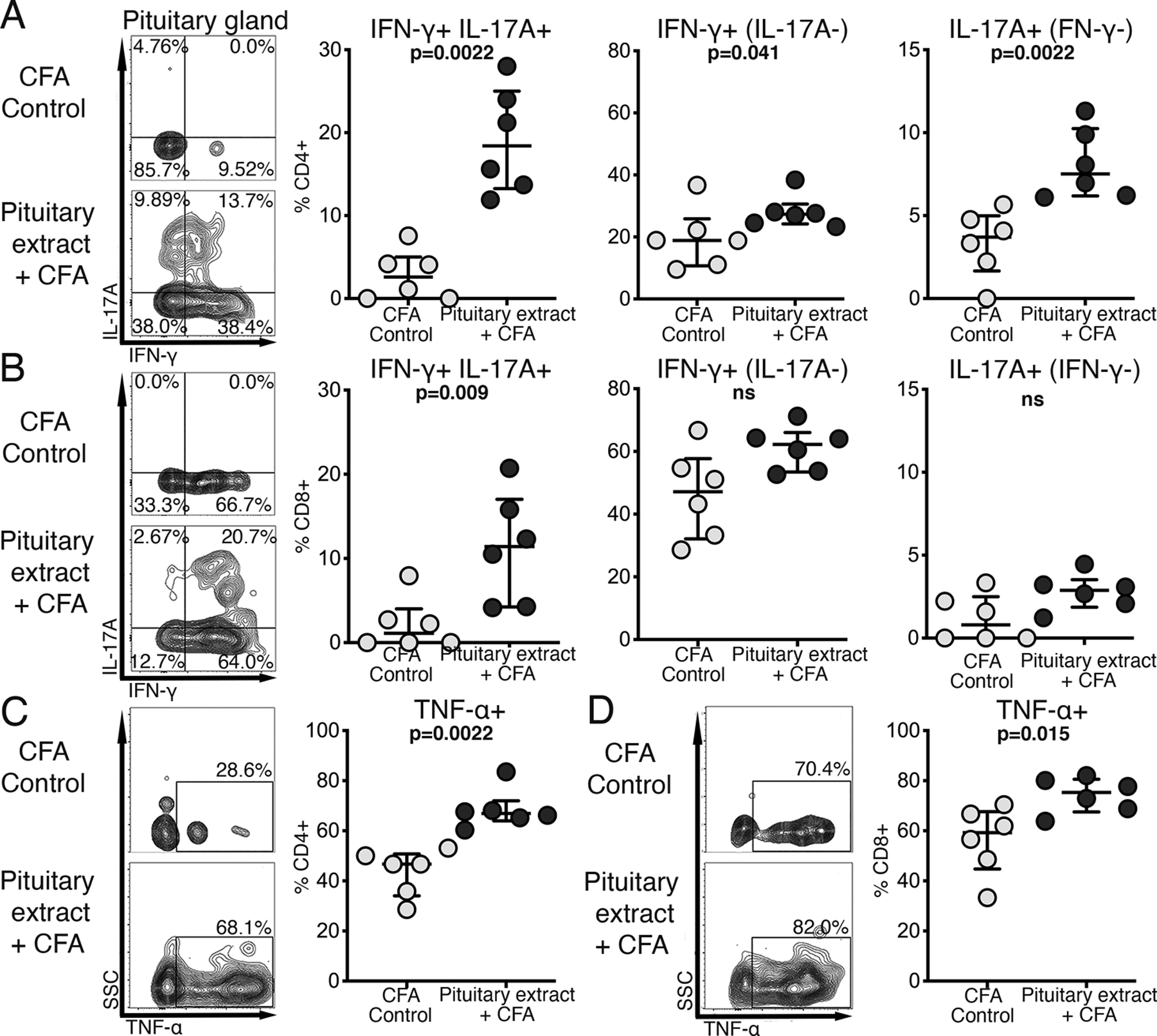
After induction of primary hypophysitis, as shown in Fig.1A, cells from pituitary glands as well as cells from spleens and lymph nodes were stimulated with PMA and Ionomycin, in the presence of brefeldin A and monensin. Cells were then stained with fluorochrome- conjugated antibodies and analyzed by flow cytometry. Representative staining pattern of a pituitary of a mouse from control and experimental group is shown. Frequency of IFN-γ+IL-17+ (Th1/Th17), IFN-γ+(IL-17A-) and IL-17A+(IFN-γ-) within A) CD4+ and B) CD8+ T cell populations; frequency of C) CD4+TNF-α+ and D) CD8+ TNF-α+ T cells in pituitary glands.
Mann-Whitney test was used.
Blocking IFN-γ before hypophysitis induction reduced subsequent infiltration of the pituitary by T cells.
Analysis of the cytokine expression by the ex vivo stimulated murine pituitary cells suggested that IFN-γ, IL-17A, or both drive hypophysitis. To assess the contribution of these cytokines to the disease process, mice were injected with antibodies neutralizing IFN-γ or IL-17A at an early (days −1, 6, 14) or late stage (days 11, 15, 19) of the hypophysitis inducing protocol (Fig.3A).
Fig.3. Blocking IFN-γ, but not IL-17A, at an early stage of the disease induction, leads to a reduction of CD3+ T cell infiltrate in a mouse model of hypophysitis.
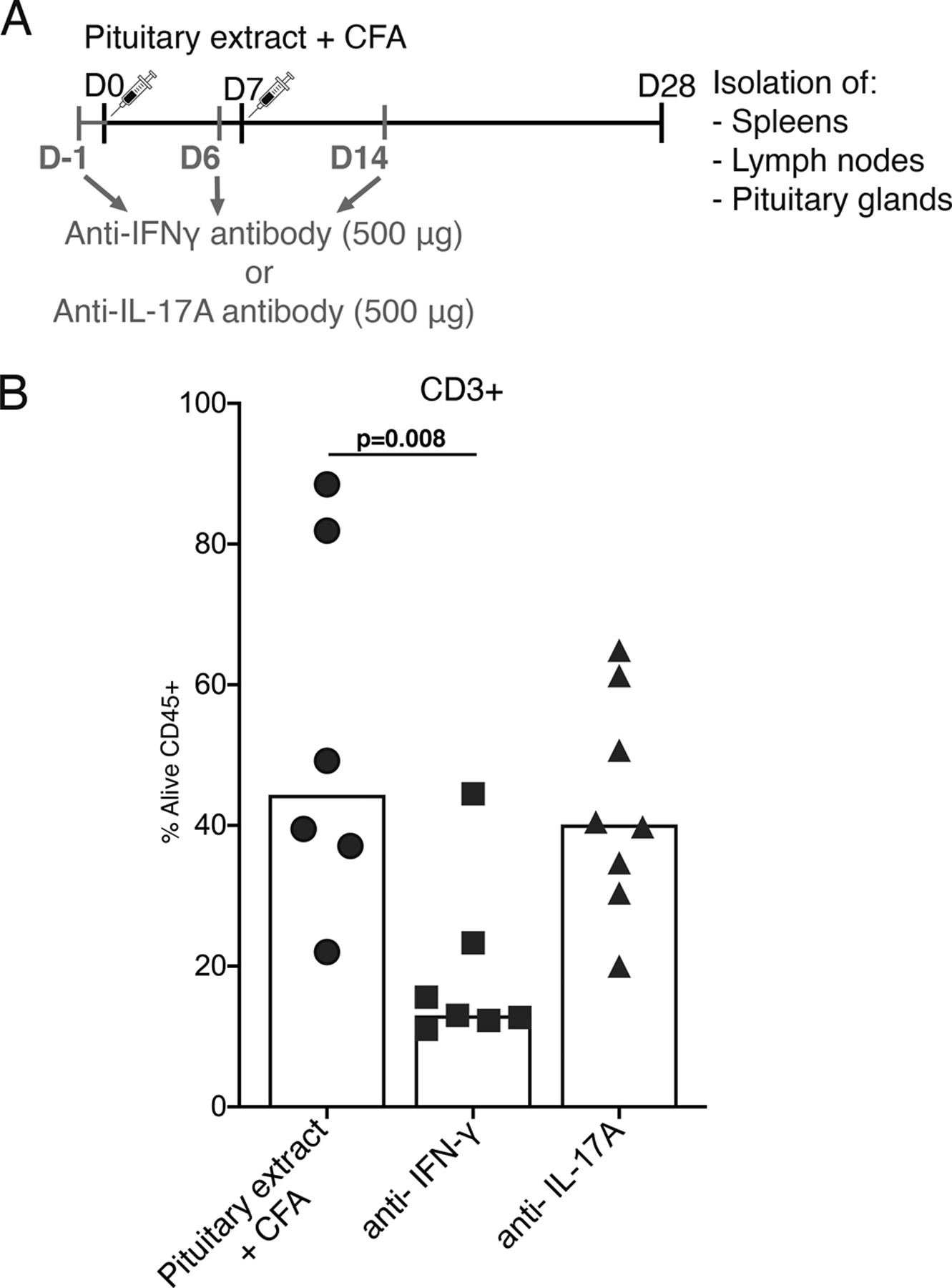
A) Outline of the experiment to assess a role of IFN-γ and IL-17A in the pathogenesis of primary hypophysitis. Mice were injected with the pituitary extract (+ CFA) as described in Fig.1A, or with the pituitary extract (+ CFA) as well as antibodies neutralizing IFN-γ or IL-17A. B) Flow cytometry analysis of the frequency of CD3+ T cells in pituitary glands.
One-way ANOVA and Tukey’s multiple comparisons test were used.
Early blocking of IFN-γ significantly decreased the percentage of pituitary-specific CD3+ T cells from a median of 44.4% in immunized mice not treated with neutralizing antibodies (n=6) to a median of 13.0% in those injected with the anti-IFN-γ antibody (n=7, p=0.008). On the contrary, early blockade of IL-17A had no effect and resulted in a percentage of pituitary CD3+ T cells similar to that found in mice injected with pituitary extract/CFA only (p= 0.85, Fig. 3B).
Blocking IFN-γ or IL-17A at a later stage of the disease induction (days 11, 15, 19) had no effect on the frequency of CD3+ T cells or B220+ B cells infiltrating mouse pituitary glands (data not shown). The composition of the CD3+ pool, including the frequency of CD4+, CD8+ or Treg cells did not differ between the groups (data not shown).
There were no differences in the immune cell composition at the level of lymph nodes between mice injected with the pituitary extract/CFA and anti- IFN-γ/IL-17A- antibodies and mice injected with the pituitary extract/CFA alone (data not shown).
Pituitaries of patients with hypophysitis showed increased levels of IL-17A mRNA expression.
Following analysis of the Th1/Th17 cell involvement in a mouse model of hypophysitis, we characterized cytokine profile of the inflammatory infiltrate in pituitary tissue from hypophysitis patients. Expression of IFN-γ, IL-17A, IL-10, IL-4 and TGF-β transcripts was measured by RT-qPCR and compared to that found in normal pituitary glands and pituitary adenomas. When scored dichotomously as present or absent, mRNA for both IFN-γ and IL-17A, IFN-γ only, IL-17A only, and IL-10 was found in a similar percentage of hypophysitis, adenoma, and normal pituitary gland specimens (Table 1). IL-4 was not expressed in any of the specimens. TGF-β mRNA was less prevalent in hypophysitis (2/16) than autopsy pituitaries (11/23) or adenomas (7/10).
When scored quantitatively, the levels of IL-17A mRNA were significantly higher in pituitaries from hypophysitis patients than autopsy or adenoma pituitaries (Fig. 4A). Patients with hypophysitis secondary to immune checkpoint blockade (open triangles in Fig.4A and delineated in Table 2) showed similarly high IL-17A transcript levels as patients with primary autoimmune hypophysitis (closed triangles in Fig.4A). None of the other cytokines was detected in pituitaries from the three secondary hypophysitis cases.
Fig.4. Expression of IL-17A mRNA is increased in pituitary glands of patients with hypophysitis.
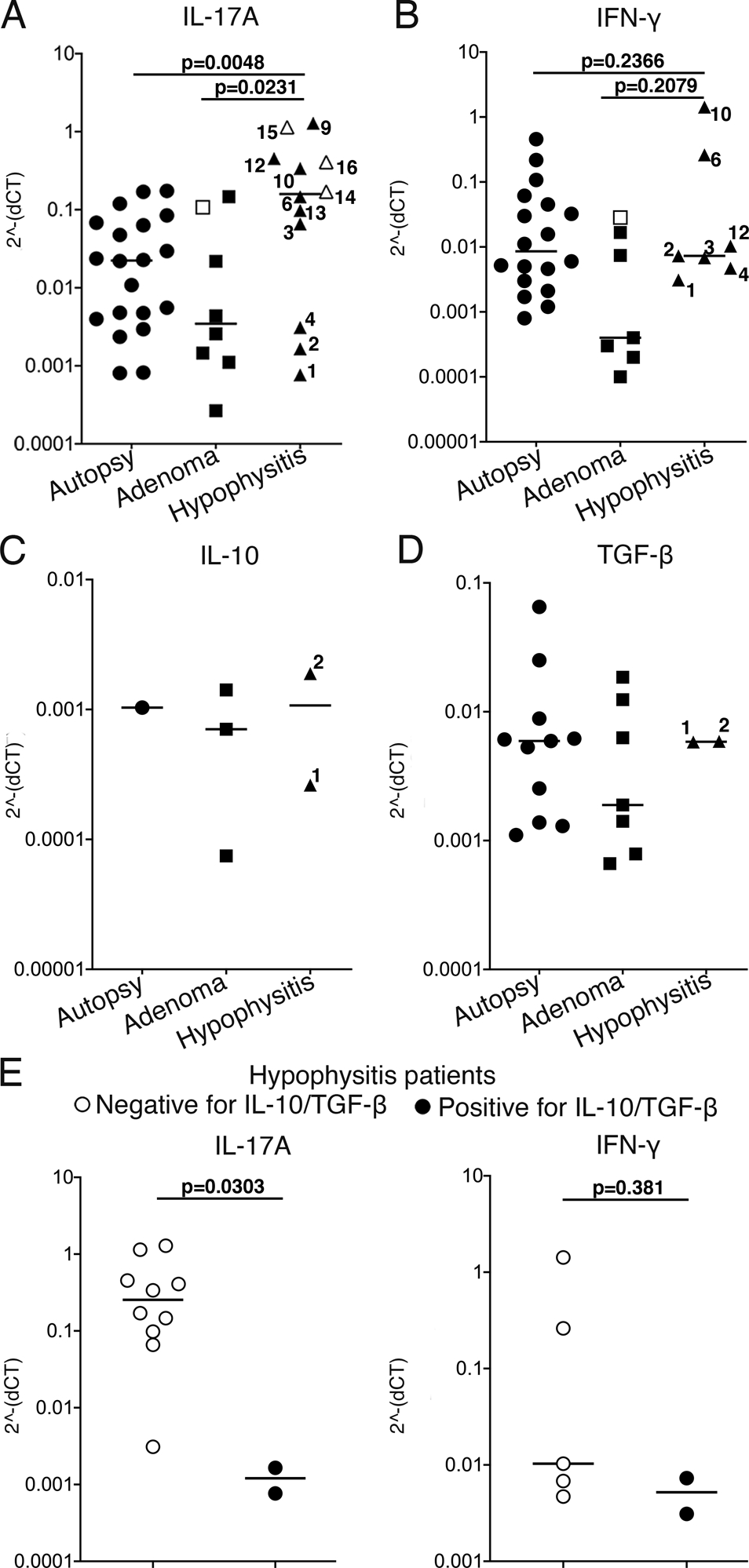
Levels of mRNA transcripts of human A) IL-17A, B) IFN-γ, C) IL-10 and D) TGF-β detected in FFPE pituitary glands derived from patients with hypophysitis, patients with pituitary adenomas and individuals with no pituitary gland diseases (autopsy cases). Cytokine transcript levels were normalized to the reference gene (gapdh) expression in each of the specimens. Numbers in the hypophysitis data set correspond to patients’ numbers shown in Table 2. Open triangles indicate patients with hypophysitis secondary to ICI; open rectangle indicates adenoma patient with reported accompanying pituitary lymphocytic infiltrate.
One-way ANOVA and Tukey’s multiple comparisons test were used.
E) Levels of IL-17A and IFN-γ mRNA in hypophysitis patients stratified according to the presence of detectable pituitary IL-10 and TGF-β mRNA.
Mann-Whitney test was used.
We stratified hypophysitis patients according to the glucocorticoid use at the time of the pituitary tissue collection (Table 2) and compared the number of cases with detectable IFN-γ or IL-17A mRNA in the “glucocorticoids” and “no glucocorticoids” subgroups. Only 1/4 of the glucocorticoids- free hypophysitis patients showed pituitary IL-17A expression, while this cytokine was detected in 8/9 of patients on glucocorticoids (p=0.052, Fisher’s exact test).
Two of the (primary) hypophysitis patients showed detectable levels of TGF-β and IL-10 in their pituitaries. These patients also showed pituitary IFN-γ and IL-17A expression, however presence of TGF-β/IL-10 at the inflammatory site was associated with a lower level of IL-17A (p=0.03), but not IFN-γ (p=0.38) as compared to patients with no detectable TGF-β/IL-10 mRNA (Fig.4E).
Of note, adenoma cases used in this study had no reported accompanying lymphocytic infiltrate, except for the one non-secreting adenoma where extensive multifocal lymphocytic infiltrates were described in the pathology report (this case is indicated by open rectangle in Fig.4A,B).
In order to support our claim that Th17 cells are the cellular source of IL-17A and are involved in the autoinflammation of the pituitary gland, we analyzed mRNA levels of CD4, CD8α and CIIT (class II transactivator, which was used as a marker of MHC II- expressing cells) in FFPE pituitary specimens (with the exception of 2 autopsy cases). CD4 mRNA was detected in almost all pituitary samples: 21/21 autopsy specimens, 10/10 adenomas and 14/16 hypophysitis specimens (of note, the 2 hypophysitis cases with no CD4 mRNA expression, had also no detectable mRNA of any of the cytokines). CD4 mRNA levels were significantly increased in hypophysistis compared to autopsy (p=0.017) and adenoma (p=0.043) cases (Suppl.Fig.2A). On the contrary, CD8α mRNA was detected in only 5/16 hypophysitis cases and its level was similar to that detected in autopsy and adenoma specimens (Suppl. Fig.2B). The notion that CD4+ IL-17A+ cells drive the inflammatory process in hypophysitis is further supported by the fact that CIIT mRNA was expressed at a significantly higher level in hypophysitis than in autopsy (p=0.011) and adenoma (p=0.019) cases (Suppl.Fig. 2C).
Discussion
This study reports that pituitary glands of mice with experimentally-induced primary hypophysitis are infiltrated predominantly by Th1/Th17 and Th17 CD4+ T cells, and pituitaries of patients with primary and secondary hypophysitis show increased IL-17A transcript level. These findings point to Th17 cells as the potential main driver of pituitary inflammation during hypophysitis. A role of Th1 cells, however, can also be implied based on the co-presence of IFN-γ and IL-17A (proteins expressed by the same pituitary-infiltrating CD4+ T cells in a mouse hypophysitis model and mRNA of both cytokines detected in 7/16 of hypophysitis patients), and a reduction of the T cell infiltration into pituitaries upon IFN-γ- blocking (but not IL-17A blocking).
Available data on the role of Th17 and Th1 subsets in other autoimmune diseases offer clues into the complexity of their interplay in the context of (auto)inflammatory environment and their plasticity route. It also undermines the notion that autoimmune diseases can be simply classified as Th1- or Th17- driven. Experiments performed with mouse models of autoimmune disorders-experimental autoimmune encephalomyelitis (EAE) (21, 22), experimental autoimmune uveitis (EAU) (23) and type 1 diabetes (T1D) (24) showed comparable disease induction upon transfer of Th1- and Th17-polarized cells.
Ustekinumab, an antibody that blocks both Th1 and Th17 differentiation by binding to the p40 peptide shared by IL-12 and IL-23, has been shown to be an effective treatment for psoriasis, psoriatic arthritis (25, 26) and Crohn’s disease (27).
Data from independent teams and different autoimmune conditions point to the same possible mechanism of the autoimmune pathology- inducing Th17/Th1 interplay, namely inherent plasticity of Th17 subset resulting in it acquiring a Th1- like phenotype (up-regulating IFN-γ, T-bet, CXCR6, IL-12Rβ2 chain), while maintaining Th17-cell features (IL-17, RORγt, RORC, Aryl hydrocarbon receptor [AhR], IL-23R, CCR6, CD161) in the context of inflammatory environment (24, 28–32). Specifically, the switch from Th17 to Th1 (termed non-classic Th1 cells) has been shown to be induced by IL-12 (24, 28), IL-12 and IFN-γ (32) or TNF-α (33). This newly- emerged tissue- specific Th17/Th1 population displays more auto-aggressive function than the “parental” Th17 cells they differentiated from (24, 31, 33). Studies that demonstrated Th17 switching to Th1-like subset to be a disease-driving mechanism in T1D have also showed that IFN-γ was indispensable in that process. Treatment with anti- IFN-γ, but not anti-IL-17A neutralizing antibody at an early disease stage prevented diabetes onset in mice injected with either Th17- or Th1- polarized cells (24, 34).
Similar to experimental murine models of chronic inflammatory diseases and human autoimmune conditions outlined above, hypophysitis may be driven by the Th17/Th1 cells. Appearance of this subset, as well as Th17 cells (which the Th17/Th1 subset can derive from) was observed in the inflamed mouse pituitaries. Reduction of the lymphocytic infiltration in pituitary glands in the hypophysitis model upon early IFN-γ blocking is in line with the notion that the Th17-derived Th1 cells are responsible for the onset of tissue autoimmunity. However, our results of the cytokine expression in human pituitary specimens, namely significantly increased IL-17A, but not IFN-γ levels in hypophysitis cases, seem to contradict the hypothesis of Th17 conversion to Th1 as a prerequisite for the disease development. It is possible that the cytokine profile of the inflamed pituitary tissue reflects early stage of the disease- characterized by the initial accumulation of Th17 cells, rather than chronic inflammation that would result in the Th17 to Th1 conversion (31). In line with this hypothesis- cases of secondary hypophysitis included in this study represent a relatively recent disease onset, and showed high IL-17A mRNA expression, while IFN-γ was not detected in their pituitaries. Alternatively, as suggested by others (35), the discrepancy between our data derived from human specimens and animal model could be due to the use of CFA with Mycobacterium tuberculosis as an adjuvant in a mouse hypophysitis model. CFA may influence the plasticity/fate of the Th17 subset (i.e. by inducing IL-6 production), although no study has yet experimentally tested this hypothesis.
Our study also reveals the importance of TGF-β in controlling levels of IL-17A in pituitaries of hypophysitis patients, and possibly- the disease induction in general. Lack of TGF-β transcript at the level of pituitary gland discriminated between hypophysitis cases and normal pituitaries or adenomas. It was also associated with significantly lower IL-17A, but not IFN-γ, mRNA level in 2/16 hypophysitis patients that showed detectable TGF-β expression.
Available data do not provide a straightforward answer on the role of TGF-β in Th17 differentiation. TGF-β alone counterregulates murine Th17 development by skewing naïve T cells towards Treg phenotype (34), however TGF-β in synergy with pro-inflammatory cytokines, such as IL-6, IL-1β, IL-21, IL-23 promotes mouse and human Th17 differentiation (36–40). TGF-β- independent differentiation of murine Th17 has also been reported (41). Importantly, study by the F. Sallusto group demonstrated that in humans TGF-β inhibits Th17 differentiation. Moreover, cells producing only IL-17A were more resistant towards TGF-β- dependent inhibition than IL-17/IFN-γ- double producers (42). In view of these results, our observation on the presence of TGF-β being associated with lower IL-17A, but not IFN-γ, mRNA expression suggests that Th17, rather than Th17/Th1 cells are involved in the pituitary inflammation in hypophysitis patients.
In conclusion, the study adds to the understanding of the immune alterations underlying autoimmune response against the pituitary gland, and is particularly relevant in the current era of cancer immunotherapy as its expansion may be paralleled by the increasing number of hypophysitis cases.
Supplementary Material
Key points.
Th17 and Th1/Th17 cells infiltrate pituitaries in a mouse model of hypophysitis.
Hypophysitis patients show increased pituitary IL-17A, CD4 and CIIT mRNA levels.
Presence of TGF-β mRNA is associated with lower pituitary IL-17A in hypophysitis.
Acknowledgments
The authors would like to thank Dr. Jody E. Hooper (Associate Professor in the Department of Pathology, Johns Hopkins University School of Medicine, Director of the Legacy Gift Rapid Autopsy program) and her team.
This work was supported by the NIH grant R01 CA 194042.
References
- 1.Sen A, Kushnir VA, Barad DH, and Gleicher N. 2014. Endocrine autoimmune diseases and female infertility. Nat Rev Endocrinol 10: 37–50. [DOI] [PubMed] [Google Scholar]
- 2.Caturegli P, Newschaffer C, Olivi A, Pomper MG, Burger PC, and Rose NR. 2005. Autoimmune hypophysitis. Endocr Rev 26: 599–614. [DOI] [PubMed] [Google Scholar]
- 3.Prete A, and Salvatori R. 2000. Hypophysitis. In Endotext. Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dungan K, Grossman A, Hershman JM, Hofland HJ, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Purnell J, Singer F, Stratakis CA, Trence DL, and Wilson DP, eds, South Dartmouth (MA). [Google Scholar]
- 4.de Filette J, Andreescu CE, Cools F, Bravenboer B, and Velkeniers B. 2019. A Systematic Review and Meta-Analysis of Endocrine-Related Adverse Events Associated with Immune Checkpoint Inhibitors. Horm Metab Res 51: 145–156. [DOI] [PubMed] [Google Scholar]
- 5.Di Dalmazi G, Ippolito S, Lupi I, and Caturegli P. 2019. Hypophysitis induced by immune checkpoint inhibitors: a 10-year assessment. Expert Rev Endocrinol Metab 14: 381–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byun DJ, Wolchok JD, Rosenberg LM, and Girotra M. 2017. Cancer immunotherapy - immune checkpoint blockade and associated endocrinopathies. Nat Rev Endocrinol 13: 195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caturegli P 2007. Autoimmune hypophysitis: an underestimated disease in search of its autoantigen(s). J Clin Endocrinol Metab 92: 2038–2040. [DOI] [PubMed] [Google Scholar]
- 8.Tahir SA, Gao J, Miura Y, Blando J, Tidwell RSS, Zhao H, Subudhi SK, Tawbi H, Keung E, Wargo J, Allison JP, and Sharma P. 2019. Autoimmune antibodies correlate with immune checkpoint therapy-induced toxicities. Proc Natl Acad Sci U S A 116: 22246–22251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ricciuti A, De Remigis A, Landek-Salgado MA, De Vincentiis L, Guaraldi F, Lupi I, Iwama S, Wand GS, Salvatori R, and Caturegli P. 2014. Detection of pituitary antibodies by immunofluorescence: approach and results in patients with pituitary diseases. J Clin Endocrinol Metab 99: 1758–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanaka S, Tatsumi KI, Kimura M, Takano T, Murakami Y, Takao T, Hashimoto K, Kato Y, and Amino N. 2002. Detection of autoantibodies against the pituitary-specific proteins in patients with lymphocytic hypophysitis. Eur J Endocrinol 147: 767–775. [DOI] [PubMed] [Google Scholar]
- 11.Smith CJ, Bensing S, Burns C, Robinson PJ, Kasperlik-Zaluska AA, Scott RJ, Kampe O, and Crock PA. 2012. Identification of TPIT and other novel autoantigens in lymphocytic hypophysitis: immunoscreening of a pituitary cDNA library and development of immunoprecipitation assays. Eur J Endocrinol 166: 391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crock PA 1998. Cytosolic autoantigens in lymphocytic hypophysitis. J Clin Endocrinol Metab 83: 609–618. [DOI] [PubMed] [Google Scholar]
- 13.O’Dwyer DT, Smith AI, Matthew ML, Andronicos NM, Ranson M, Robinson PJ, and Crock PA. 2002. Identification of the 49-kDa autoantigen associated with lymphocytic hypophysitis as alpha-enolase. Journal of Clinical Endocrinology & Metabolism 87: 752–757. [DOI] [PubMed] [Google Scholar]
- 14.Mirocha S, Elagin RB, Salamat S, and Jaume JC. 2009. T regulatory cells distinguish two types of primary hypophysitis. Clinical and Experimental Immunology 155: 403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tzou SC, Lupi I, Landek M, Gutenberg A, Tzou YM, Kimura H, Pinna G, Rose NR, and Caturegli P. 2008. Autoimmune hypophysitis of SJL mice: clinical insights from a new animal model. Endocrinology 149: 3461–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin HH, Gutenberg A, Chen TY, Tsai NM, Lee CJ, Cheng YC, Cheng WH, Tzou YM, Caturegli P, and Tzou SC. 2017. In Situ Activation of Pituitary-Infiltrating T Lymphocytes in Autoimmune Hypophysitis. Scientific Reports 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tzou SC, Landek-Salgado MA, Kimura H, and Caturegli P. 2010. Preparation of mouse pituitary immunogen for the induction of experimental autoimmune hypophysitis. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caturegli P, Di Dalmazi G, Lombardi M, Grosso F, Larman HB, Larman T, Taverna G, Cosottini M, and Lupi I. 2016. Hypophysitis Secondary to Cytotoxic T-Lymphocyte-Associated Protein 4 Blockade Insights into Pathogenesis from an Autopsy Series. American Journal of Pathology 186: 3225–3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zelenay S, Lopes-Carvalho T, Caramalho I, Moraes-Fontes MF, Rebelo M, and Demengeot J. 2005. Foxp3+ CD25- CD4 T cells constitute a reservoir of committed regulatory cells that regain CD25 expression upon homeostatic expansion. Proc Natl Acad Sci U S A 102: 4091–4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coleman MM, Finlay CM, Moran B, Keane J, Dunne PJ, and Mills KH. 2012. The immunoregulatory role of CD4(+) FoxP3(+) CD25(-) regulatory T cells in lungs of mice infected with Bordetella pertussis. FEMS Immunol Med Microbiol 64: 413–424. [DOI] [PubMed] [Google Scholar]
- 21.Kroenke MA, Carlson TJ, Andjelkovic AV, and Segal BM. 2008. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med 205: 1535–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Domingues HS, Mues M, Lassmann H, Wekerle H, and Krishnamoorthy G. 2010. Functional and pathogenic differences of Th1 and Th17 cells in experimental autoimmune encephalomyelitis. PLoS One 5: e15531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luger D, Silver PB, Tang J, Cua D, Chen Z, Iwakura Y, Bowman EP, Sgambellone NM, Chan CC, and Caspi RR. 2008. Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J Exp Med 205: 799–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bending D, De La Pena H, Veldhoen M, Phillips JM, Uyttenhove C, Stockinger B, and Cooke A. 2009. Highly purified Th17 cells from BDC2.5NOD mice convert into Th1-like cells in NOD/SCID recipient mice. Journal of Clinical Investigation 119: 565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Papp KA, Langley RG, Lebwohl M, Krueger GG, Szapary P, Yeilding N, Guzzo C, Hsu MC, Wang YH, Li S, Dooley LT, Reich K, and Investigators P. 2008. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet 371: 1675–1684. [DOI] [PubMed] [Google Scholar]
- 26.Gottlieb AB, and Goldminz AM. 2012. Ustekinumab for Psoriasis and Psoriatic Arthritis. Journal of Rheumatology 39: 86–89. [DOI] [PubMed] [Google Scholar]
- 27.Feagan BG, Sandborn WJ, Gasink C, Jacobstein D, Lang Y, Friedman JR, Blank MA, Johanns J, Gao LL, Miao Y, Adedokun OJ, Sands BE, Hanauer SB, Vermeire S, Targan S, Ghosh S, de Villiers WJ, Colombel JF, Tulassay Z, Seidler U, Salzberg BA, Desreumaux P, Lee SD, Loftus EV, Dieleman LA, Katz S, Rutgeerts P, and Grp U-I-US. 2016. Ustekinumab as Induction and Maintenance Therapy for Crohn’s Disease. New England Journal of Medicine 375: 1946–1960. [DOI] [PubMed] [Google Scholar]
- 28.Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, Parente E, Fili L, Ferri S, Frosali F, Giudici F, Romagnani P, Parronchi P, Tonelli F, Maggi E, and Romagnani S. 2007. Phenotypic and functional features of human Th17 cells. Journal of Experimental Medicine 204: 1849–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Annunziato F, Cosmi L, and Romagnani S. 2010. Human and murine Th17. Current Opinion in Hiv and Aids 5: 114–119. [DOI] [PubMed] [Google Scholar]
- 30.Zhou L, Chong MMW, and Littman DR. 2009. Plasticity of CD4(+) T Cell Lineage Differentiation. Immunity 30: 646–655. [DOI] [PubMed] [Google Scholar]
- 31.Annunziato F, Santarlasci V, Maggi L, Cosmi L, Liotta F, and Romagnani S. 2013. Reasons for rarity of Th17 cells in inflammatory sites of human disorders. Seminars in Immunology 25: 299–304. [DOI] [PubMed] [Google Scholar]
- 32.Lexberg MH, Taubner A, Albrecht I, Lepenies I, Richter A, Kamradt T, Radbruch A, and Chang HD. 2010. IFN-gamma and IL-12 synergize to convert in vivo generated Th17 into Th1/Th17 cells. European Journal of Immunology 40: 3017–3027. [DOI] [PubMed] [Google Scholar]
- 33.Maggi L, Cimaz R, Capone M, Santarlasci V, Querci V, Simonini G, Nencini F, Liotta F, Romagnani S, Maggi E, Annunziato F, and Cosmi L. 2014. Etanercept Inhibits the Tumor Necrosis Factor alpha-Driven Shift of Th17 Lymphocytes Toward a Nonclassic Th1 Phenotype in Juvenile Idiopathic Arthritis. Arthritis & Rheumatology 66: 1372–1377. [DOI] [PubMed] [Google Scholar]
- 34.Martin-Orozco N, Chung Y, Chang SH, Wang YH, and Dong C. 2009. Th17 cells promote pancreatic inflammation but only induce diabetes efficiently in lymphopenic hosts after conversion into Th1 cells. European Journal of Immunology 39: 216–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steinman L 2008. A rush to judgment on Th17. Journal of Experimental Medicine 205: 1517–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, Ziegler SF, and Littman DR. 2008. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature 453: 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, and Weaver CT. 2006. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441: 231–234. [DOI] [PubMed] [Google Scholar]
- 38.Ivanov L. Zhou II, and Littman DR. 2007. Transcriptional regulation of Th17 cell differentiation. Seminars in Immunology 19: 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manel N, Unutmaz D, and Littman DR. 2008. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol 9: 641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, and Stockinger B. 2006. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24: 179–189. [DOI] [PubMed] [Google Scholar]
- 41.Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, Grainger JR, Chen Q, Kanno Y, Watford WT, Sun HW, Eberl G, Shevach EM, Belkaid Y, Cua DJ, Chen W, and O’Shea JJ. 2010. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature 467: 967–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, and Sallusto F. 2007. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol 8: 942–949. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.