

Summary
Planar cell polarity (PCP) signaling plays a critical role in coordinating cell polarity during various organogenesis processes in mammals, and its disruption is causal to numerous congenital disorders in humans. To elucidate its actions in mammals, mouse genetics is an indispensable approach. Given that both gain- and loss-of-function of many PCP genes often cause similar defects, the standard mouse transgenic approach may not always be ideal for studying PCP genes in their wild-type and mutant forms. Here we describe using BAC (bacterial artificial chromosomes) transgenes as a versatile and effective alternative. Transgenes made from BACs, which are genomic clones 100-200 kb in size, can more faithful recapitulate endogenous gene expression levels and patterns. Bacterial based recombination system can be used to efficiently introduce mutations, fluorescent protein tags, and LoxP sites for conditional expressions. Cre can also be inserted into BACs to map the contribution of cells expressing any PCP gene of interest, and study PCP mediated tissue morphogenesis.
Keywords: bacterial artificial chromosome, transgenesis, planar cell polarity, morphogenesis
1. Introduction
Planar cell polarity (PCP) refers to the cellular polarity on the plane of the epithelium, perpendicular to the apicobasal (A-B) polarity of the cell [1–4]. The pathway that coordinates PCP was initially discovered in Drosophila as a branch of the non-canonical Wnt pathway, as it shares receptor Frizzled (Fz) and cytoplasmic protein Dishevelled (Dsh/Dvl) with the canonical Wnt pathway but functions in a β-catenin independent fashion [5,6]. Subsequent genetic studies identified four distinct proteins that function with Fz and Dvl to regulate PCP, including the atypical cadherin Flamingo (Fmi), the four-pass transmembrane protein Van gogh (Vang), as well as cytoplasmic proteins Prickle (Pk) and Diego (Dgo) [7]. The six proteins are collectively referred to as the core PCP members. Through complex intra- and inter-cellular feedback interactions, they partition into asymmetric clusters located on the opposing cell cortex to coordinate cell polarity across the entire field of epithelium [8–10].
Studies of vertebrate orthologs of the fly core PCP genes revealed that they also play a critical role in a morphogenetic process known as convergent extension (CE) [11,12]. Originally discovered in Xenopus, CE has now been recognized as a universal morphogenetic engine where polarized cell behaviors, including oriented cell intercalation, migration or division, shape the overall dimension of tissues by promoting their simultaneous elongation and narrowing [13]. A characteristic defect associated with disruption of core PCP genes is perturbation of neural plate morphogenesis, leading to a severe neural tube closure defect known as craniorachischisis where the entire neural tube from the midbrain to tail fails to close [14–18]. In humans, missense mutations in core PCP genes have been found to cause a spectrum of neural tube closure defects [19].
Besides the core PCP genes, regulation of CE in vertebrates also requires non-canonical Wnt ligands Wnt5a, 5b and 11, as well as a unique set of co-receptors with tyrosine kinase domains including Ryk (receptor like tyrosine kinase), Ptk7 (protein tyrosine kinase 7), Ror1/2 (RTK-like orphan receptor) and MuSK (muscle-specific kinase) [20]. Mutations in WNT5A and ROR2 are known to cause skeletal dysplasia such as Brachydactyly type B and Robinow syndrome [21–24].
To elucidate the action of PCP in mammalian development and human diseases, the mouse is an indispensable model organism. Some of the unique properties of PCP signaling, however, pose challenges to the conventional mouse transgenic techniques. For instance, PCP genes tend to be dosage sensitive, where their significantly decreased and increased expression can both abolished the pathway and lead to similar morphogenetic defects [25,11]. Therefore, we have adapted BAC (bacterial artificial chromosome) transgenesis as the genetic tool to study PCP gene functions in the mouse.
BACs are low-copy number plasmids that contain ~100-200 kb genomic DNA. Their large size makes BAC-derived transgenes more likely to contain the necessary enhancer elements to mimic the endogenous spatial and temporal pattern of gene expression, while at the same time provides more insulation to minimize position effect variegation when they are inserted randomly into the host genome [26,27]. Furthermore, because BAC transgenes tend to integrate as low copy concatamers [28], they can also mimic endogenous gene expression levels better than conventional transgenes. To this end, we have created a set of BAC transgenes to express wild-type and mutant Dvl2 (Dishevelled2) with various domain deletions and point mutation to genetically dissect its function in canonical Wnt vs. PCP signaling . These Dvl2 BAC transgenes could also be tagged with EGFP fused in-frame to the C-terminal end of Dvl2 to study the subcellular localization of wild-type and mutant Dvl2 in different tissues [16,29]. Moreover, by inserting LoxP sites to flank the Dvl2 locus in the BAC, these transgenes can also function as conditional alleles [30]. On the other hand, we have also used BACs to establish tamoxifen-inducible Wnt11-CreER and Wnt5a-CreER transgenes to map the spatial/ temporal expression pattern of these non-canonical Wnts in mice, and performed lineage tracing to study PCP-mediated morphogenetic in the cardiopulmonary progenitor field [31–33].
The versatility of BAC transgenes rely on the highly efficient homologous recombination systems developed in E. coli to make various modifications including insertions, deletions and single nucleotide substitutions. Collectively termed BAC recombineering, these techniques utilize λ prophage Red system or Rac phage RecET system to modify BACs using linear DNA templates with homology arms as short as 30-50 bases [34–36]. Therefore, in many cases one can use oligos or PCR products as the homologous DNA template to quickly and precisely engineer any genetic changes in any locations in the BAC [37].
In this chapter, we will describe BAC recombineering protocol using three modified DH10B bacterial strains (Table 1), which have stably integrated the defective prophage and can express the λ Red system in a temperature control fashion [34]. The integrated λ Red system consists of three recombination proteins Gam, Beta and Exo. Gam protects linear DNA template from degradation by E. coli nuclease RecBCD. Beta binds to single strand (ss) DNA and promotes annealing to its complementary DNA sequence. Exo is a 5’ -> 3’ exonuclease that can degrade double strand (ds) DNA template to expose ssDNA ends for recombination. Working together, they can promote highly efficient homologous recombination to induce any desired genetic changes in the BAC using either ssDNA or dsDNA as templates [34,35]. Finally, a temperature sensitive λ repressor (allele CI857) was incorporated to control Gam, Beta and Exo transcription to avoid their toxic effect. The repressor is active at low temperature (~32 °C), but can be rapidly inactivate with a 15 minutes incubation at 42 °C to provide a pulse of Gam, Beta and Exo expression. We will provide a step-by-step protocol on how to 1) identify the appropriate BAC clone, 2) introduce the BAC into the recombination competent strains, 3) design the strategy to create the desired changes, 4) carry out the BAC recombineering, and 5) purify the engineered BAC for pronuclear injection to acquire transgenes.
Table 1.
Summary of three commonly used recombineering bacterial strains.
| List of recombineering competent strains | ||
|---|---|---|
| Name | Genotype | Note |
| DY380 | DH10B [λcl857 (cro-bioA < > tet] | DH10B cell with integrated λ prophage that expresses Gam, Beta and Exo under the control of temperature sensitive cl857 repressor. Tetracycline (12.5 ug/ml) resistant. |
| EL250 | DH10B [λcl857 (cro-bioA < > araC-PBADflpe] | Same as DY380 except also containing an arabinose-inducible flippase that can excise sequence flanked by FRT sites, such as FRT-Neo-FRT introduced through construct PL451 (see Note 8). Not tetracycline resistant. |
| EL350 | DH10B [λcl857 (cro-bioA < > araC-PBADcre] | Same as DY380 except also containing an arabinose-inducible Cre recombinase that can excise sequence flanked by LoxP sites, such as LoxP-Neo-LoxP introduced through construct PL452 (see Note 8). Can also be used to test any BACs that are designed to function as conditional transgenes with floxed sequence. Not tetracycline resistant. |
These strains can be requested from NCI through the following link: https://frederick.cancer.gov/Science/BrbRepository/#/productDataSheets/Bacteria
PL451 and 452 plasmids can be requested from NCI through the following link: https://frederick.cancer.gov/Science/BrbRepository/#/productDataSheets/Plasmid
2. Materials
2.1. Media and antibiotics for BAC culture
Chloramphenicol stock: make 25 mg/ml stock in ethanol; aliquot and store at −20 °C.
LB (Luria-Bertani) media and agar;
LB/ chloramphenicol plate: LB plate with 12.5 ug/ ml chloramphenicol.
LB/ chloramphenicol media: LB media with 12.5 ug/ ml chloramphenicol.
2.2. Solutions, materials and equipment for BAC DNA mini prep
P1 solution: 50 mM Tris-HCl pH 8.0; 10 mM EDTA; 100 μg/ml RNaseA.
P2 solution:200 mM NaOH; 1% SDS.
P3 solution: 3.0 M potassium acetate, pH 5.5.
Ethanol.
TE: 10 mM Tris, 1 mM EDTA, pH 8.0.
15 and 50 ml conical tubes.
Eppendorf tubes.
Centrifuge.
2.3. Restriction digest and pulse field gel electrophoresis (PFGE)
Restriction enzyme and buffer.
PFGE system: for instance, the CHEF-DR II system from BIO-RAD.
PFGE marker (such as MidRange PFG Marker from NEB cat.# N0342S)
Agarose.
0.5x TBE (Tris/Boric Acid/EDTA) running buffer: 45 mM Tris; 45 mM boric acid; 1 mM EDTA, pH8.3.
Nucleic acid dye such as ethidium bromide for DNA staining
2.4. Electroporation of BAC DNA into recombineering competent strains
Autoclaved water, chilled to 4 °C.
1 mm electroporation cuvettes.
An electroporator such as the BIO-RAD Gene Pulser Xcell system.
Recombineering competent strains (table 1). The strains can be requested from NCI through the following link: https://frederick.cancer.gov/Science/BrbRepository/#/productDataSheets/Bacteria
2.5. BAC recombineering with linear template
2.6. Purification of BAC DNA for pronuclear injection
Qiagen Large Construct Kit; cat.# 12462.
Ethanol.
Phenol: chloroform: isoamyl alcohol (25:24:1).
Chloroform.
Isoamyl alcohol.
TE: 10 mM Tris, 1 mM EDTA, pH 8.0.
Sodium acetate, pH 5.2.
Low TE (10 mM Tris, 0.1 mM EDTA, pH7.5).
Injection buffer: 10 mM Tris-HCl, pH7.5, 0.5 mM EDTA, 30 nM spermine, 70 nM spermidine, 100 mM NaCl.
Fluorometer and/or spectrometer.
Centrifuges.
2.7. Use BAC transgenic mice to study polarized tissue morphogenesis
4% PFA: 4% paraformaldehyde in PBS, pH7.5.
PBS with 15% sucrose.
PBS with 15% sucrose.
OCT: Optimal cutting temperature compound.
Ethanol or butanol and dry ice for snap freezing the specimen.
1x PBS, 0.5% Tween 20.
FITC-conjugated phalloidin
PBST: 1x PBS, 0.1% Tween 20
RIMS (Refractive index matching solution): 88% (weight by volume) Histodenz (Sigma); 0.02 M phosphate buffer; 0.01% sodium azide; 0.1% Tween 20 (Sigma); 2.5% 1,4-Diazabicyclo[2.2.2]octane (Sigma).
Superglue for making imaging chamber
Cryostat and blades for trimming the embryo
Petri dish (with cover slip bottom for inverted microscope; or regular to make imaging chamber for upright microscope).
Confocal microscope.
3. Methods
3.1. Identify and acquire the desired BAC clones
Go to the Ensembl genome browser Archive 88 (http://mar2017.archive.ensembl.org/index.html). See Note 1.
Under “Search”, select the desired species and enter the gene name (for instance, select “mouse” and enter “Rosa26”), then click “Go”.
From the search results displayed, click the link of the desired gene name (for instance for Rosa26, click the link “Gt(ROSA)26Sor (Mouse Gene, Strain: reference (CL57BL6))”).
On the gene summary page, scroll down and click “Region in Detail”; then click the “Configure this page” pane on the left hand side.
An “Active tracks” window will appear. Click the “Clones & misc. regions” pane on the left hand side.
Check the square boxes for the clones to be displayed. For mouse BAC clones, click on “RP23” and “RP24 mouse clones”; then click the check mark on the upper right hand corner to confirm the selection and close the window. See Note 2.
Scroll down to “Contigs” to view the BAC clones. To better view the gene in the context of the BAC clone, it might be necessary to zoom out by clicking the arrows in the “Location” pane, or by dragging the zoom bar to the right (towards “-“).
Choose the clone(s) in which the gene of interest is located in the middle. Click on the clone to display the information on its genome location and size. For instance, when clicking on the BAC clone RP24-85L15 that contain the Rosa26 gene, it will display its genome coordinates as “Range: 112952746-113158583” and size as “Length: 205838 bps”.
Copy the clone name. Go to the website “bacpacresources.org” (a distributor of BAC clones). Paste or enter the clone name under the “Clone Information Search” pane, click “Search”.
Click “LINK” under “ORDERING & PRICING” and follow the instruction displayed to order the BAC clone(S).
3.2. DNA purification from BAC clones
BAC clones are shipped as bacterial stab cultures (usually DH10B cells) instead of DNA. This is because due to its large size, purified BAC DNA cannot be stored as frozen stock but only temporarily at 4 °C. For long term storage, bacteria stock of BAC clones are kept at −80 °C. Therefore, upon receiving the BAC clone culture from BACPAC, it is necessary to amply the bacteria for restriction digest to confirm that they contain the correct BAC DNA. The bacterial culture containing the confirmed BAC DNA should then be frozen down and store at −80 °C. The purified DNA will also need to be electroplated into the recombinant competent strains (table 1) for BAC recombineering.
Using an inoculation loop, steak the received BAC culture on an LB/ chloramphenicol plate (step 2 of 2.1), incubate at 37 °C overnight.
Pick 3 different colonies, and inoculate them each into 11 ml LB/ chloramphenicol media (step 3 of 2.1) in a 50 ml conical tube, shake at 250 rpm at 37 °C overnight.
Save 1 ml of the bacterial culture in an autoclaved Eppendorf tube to make frozen cell stock (see step 5 of 3.3). Spin down the rest of the bacterial culture. Re-suspend the pellet in 250 ul P1 solution and transfer to an 1.7 ml Eppendorf tube.
Add 250 ul P2 solution to each Eppendorf tube. Invert the tubes several times to mix P2 with the resuspended bacterial culture. Incubate 5 minutes at room temperature.
Add 350 ul P3 to each tube. Invert to mix. Spin 3 minutes at 13,000 rpm or above (~ 17,900 g or above).
Transfer supernatant to a new Eppendorf tube, spin again for 3 minutes to further clear the extract.
Transfer supernatant to a new Eppendorf tube (~800 ul), add 750 ul isopropanol. Invert the tubes to mix. Allow DNA to precipitate for 10 minutes at room temperature.
Centrifuge the tubes for 5 minutes to collect DNA pellets. Discard the solution and wash the DNA pellet with 1 ml 70% ethanol.
Spin again and discard ethanol. Do a quick spin and carefully suck up any remaining ethanol solution with a pipet. Let the pellets dry for 3 minutes at room temperature with the lid open. See Note 3.
Add 30 ul TE buffer to each tube. Let the solution soak the pellet for 10 minutes at room temperature to resuspend DNA. Gently tap the tube to help dissolving the DNA, or allow the DNA continuing to dissolve at 4 °C for a few hours or overnight.
Spin down the DNA solution at 13,000 rmp or above for 5 minute to clear the solution. Transfer the cleared supernatant to a new tube. See Note 4.
Store the DNA at 4 °C, or continue with step 3.3 for restriction digest.
3.3. Restriction digest and pulse field gene electrophoresis (PFGE) to characterize the purified BAC DNA
The standard method to determine the integrity of the BAC clone is to digest the purified DNA with rare-cutter enzymes (such as NotI). The resulting DNA fragments from the restriction digest are usually between ~20–100 kb and will require running PFGE for 20-24 hours to resolve them.
Use 10-15 ul of the purified DNA to set up overnight restriction digest with a rare-cutter such as NotI. See Note 5.
Set up PFGE with a system such as CHEF-DR II (Bio-Rad). Load the restriction digests and PFGE marker into each well of a 1% agarose gel made with 0.5x TBE, and run the gel in chilled 0.5x TBE with the following parameter: 5-25 second switch; 6 volts/ cm; 22 hours. Use a chiller to keep the buffer cool, or run the PFGE in a cold room.
After PFGE, stain the gel in 0.5 ug/ ml EtBr solution for 10 minutes and visualize under UV. Destain the gel in water if necessary.
Compare the observed restriction pattern to the predicated pattern (see Note 6), and calculate the size of the BAC clone based on the restriction fragments observed. See Note 7.
Make a frozen cell stock by adding 150 ul of glycerol to the 1 ml bacterial culture saved from step 3 of 3.2. Vortex to mix and store at −80 °C.
3.4. Electroporate BAC DNA into recombineering competent bacterial strains
Upon confirming the BAC DNA from 3.3, it will need to be electroporated into one of the recombineering competent strains (table 1). We will use EL350 as an example in the subsequent procedures, but the protocol will be identical for the other two strains.
Inoculate 1-2 ul of frozen EL350 cell stock in 1 ml LB media in a 15 ml conical tube. Shake at 250 rpm at 32 °C overnight. See Note 8.
The next morning, add 200 ul of the overnight culture in 10 ml LB media in a 50 ml conical tube, shake at 250 rpm at 32 °C until OD600 reaches 0.5-0.7 (~4 hours).
Chill the culture on ice for 20 minutes.
Spin down the cells at 5500 g for 5 minutes at 4°C. Remove the culture media and resuspend the cells in 1.5 ml of autoclaved water chilled to 4°C. Transfer the cells to an autoclaved Eppendorf tube.
Spin down the cells as in step 4, and repeat the wash with ice cold water for two more times.
After the last wash, resuspend the cells in 50 ul ice cold water. Add 1-3 ul of miniprepped BAC DNA from 3.2. Swirl the solution and tap the tube to help mixing BAC DNA with the cells.
Transfer the DNA-cell mixture to a 1 mm electroporation cuvette. Make sure the solution is dispensed into the slot between the two aluminum electrode plates. Incubate the cuvette on ice for 5-10 minutes.
Electroporate the cuvette with the following setting: 1.75 kV, 25 μF, 200 ohms.
Add 1 ml LB to the cuvette, and transfer the electroporated cells to a 15 ml conical tube, shake at 250 rmp at 32 °C for 1.5 hours.
Spin down the cells at 5500 g for 5 minutes. Resuspend the cells in 100 ul LB and plate them on an LB/ chloramphenicol plate. Incubate at 32 °C overnight.
Pick 3 colonies and culture each of them in 11 ml LB/ chloramphenicol media. Save 1 ml of the culture for frozen cell stock, and use the remaining 10 ml to purify BAC DNA as in 3.2. Characterize the DNA as in 3.3 to confirm that the BAC DNA in EL350 cells is the same as the original clone.
Make a frozen cell stock by adding 150 ul of glycerol to the 1 ml bacterial culture saved from step 11 above. Vortex to mix and store at −80 °C.
3.5. BAC recombineering with linear template
The design of the linear template for recombineering will depend on what type of modification one wants to engineer, and how precise the modification needs to be. See Note 9 for a general discussion of two main strategies that are commonly used to engineer different modifications ranging from insertions, deletions and point mutations. Once a strategy is chosen, either PCR or cloning can be used to add homology arms to flank the modification cassette (for instance, LoxP sites, Cre or fluorescent protein sequence that are to be inserted). If using the PCR method, primers can be designed to directly add homology arms up to 80 bp on each end to generate the template. Alternatively, for increased recombineering efficiency, longer homology arms (200-500 bp) can be added to each end of the modification cassette through PCR mediated cloning [38], and the template is then excised out of the cloning vector through restriction digest. We have found that 200 bp homology arms are sufficient for inserting a 9 kb cassette into the Rosa26 locus in a BAC through recombineering to [31]. Finally, if the modification to be made is a point mutation or deletion, the template can also be a single strand oligo with up to 50 base homology arms flanking the point mutation or deletion breakpoints.
If the linear recombineering template is made via PCR or restriction digest of a construct after cloning, it should be gel purified with Qiagen Gel Extraction Kit (or something similar) and eluted in sterile water before proceeding to the following step. It is important that the template is free of contamination from primers, nucleotides and other DNA, and ideally with a concentration above 50 ug/ul. Oligos can be directly diluted in water to ~100 ng/ul.
Inoculate 1-2 ul of frozen cell stock from step 12 of 3.4 in 1 ml LB/ chloramphenicol media in a 15 ml conical tube. Shake at 250 rpm at 32 °C overnight.
The next morning, add 200 ul of the overnight culture in 10 ml LB/ chloramphenicol media in a 50 ml conical tube, shake at 250 rpm at 32 °C until for ~4 hours until OD600 reaches 0.5-0.7.
Place the culture tube in a 42 °C shaking water bath for 15 minutes to induce the expression of Gam, Beta and Exo
Chill the culture on ice for 20 minutes.
pin down the cells at 5500 g for 5 minutes at 4°C. Remove the culture media and resuspend the cells in 1.5 ml of autoclaved water chilled to 4°C. Transfer the cells to an autoclaved Eppendorf tube.
Spin down the cells as in step 5, and repeat the wash with ice cold water for two more times.
After the last wash, resuspend the cells in 50 ul ice cold water. Add 100-300 ng linear DNA template. Mix the template DNA with the cells.
Transfer the template DNA-cell mixture to a 1 mm electroporation cuvette. Incubate the cuvette on ice for 5-10 minutes.
Electroporate the cuvette with the following setting: 1.75 kV, 25 μF, 200 ohms.
Add 1 ml LB/ chloramphenicol to the cuvette, and transfer the electroporated cells to a 15 ml conical tube, shake at 250 rmp at 32 °C for 1.5 hours.
Spin down the cells at 5500 g for 5 minutes. Resuspend the cells in 100 ul LB and plate them on an LB/ chloramphenicol plate. Incubate at 32 °C overnight.
Pick colonies and culture them in 100 ul LB/ chloramphenicol in a 96 well plate. Shake at 125 rpm at 32 °C overnight. Use 1-2 ul of the bacterial culture as PCR template to screen for recombined clones.
Once candidate clones are identified, streak the culture on LB/ chloramphenicol plates and incubate at 32 °C overnight. Pick single colonies and grow in 11 ml LB/ chloramphenicol media. Save ~1 ml to make frozen cell stock, and to further confirm that they contain the correct modification(s) by PCR and/or sequencing of the PCR reaction. Use the remaining 10 ml to purify BAC DNA as in 3.2. Characterize the DNA as in 3.3 to confirm that the modified BAC has the correct size and restriction digest pattern in comparison to the original clone.
3.6. Purification of BAC DNA for pronuclear injection
The easiest method to acquire high quality BAC DNA suitable for pronuclear injection in mice is to first perform purification with Qiagen Large Construct Kit (cat.# 12462), and then extract DNA with phenol/ chloroform [39]. It requires no special equipment such as ultracentrifuge, and can be carried out within three days in any labs with basic molecular biology set up.
Steak frozen cell stock from step 12 of 3.5 onto an LB/ chloramphenicol plate. Incubate at 32 °C overnight.
Inoculate a single colony in 2 ml LB/ chloramphenicol media. Shake at 250 rpm at 32 °C overnight.
Dilute the overnight culture 1:1,000 into 500 ml LB/ chloramphenicol media, Shake at 250 rpm at 32 °C for 20-24 hours. See Note 10.
Proceed with BAC DNA purification with Qiagen Large Construct Kit. After ethanol precipitation of the BAC DNA eluted from column, skip the 70% ethanol wash and instead dissolve the BAC DNA pellet in 500 ul TE. There is no need to dry the pellet before dissolving in TE either.
Determine the DNA concentration with a spectrometer or fluorometer. Typical yield of BAC DNA from the large construct kit is 10 to 50 ug/ 500 ml culture, so the expected concentration usually ranges from 20 to 100 ng/ul.
Transfer the DNA solution to a clean Eppendorf tube and mix DNA solution with equal volume (500 ul) of phenol: chloroform: isoamyl alcohol (25:24:1). Do not vortex but invert tube multiple time to gently mix DNA with phenol/chloroform. Centrifuge the tube at 10,000 rpm for 3 minutes. Transfer the upper DNA solution to a clean autoclaved Eppendorf tube.
Repeat step 3 once with phenol: chloroform: isoamyl alcohol (25:24:1), and once with chloroform: isoamyl alcohol (24:1).
Add 1/10 volume (~50 ul) of 3 M sodium acetate and two volume of ethanol (~1 ml). Invert the tube to mix. Centrifuge at 13,000 g for 15 minutes at room temperature to precipitate DNA.
Wash the pellet with 70% ethanol. Centrifuge at 13,000 g for 3 minutes and remove the liquid. Centrifuge again briefly and use a pipet and pipet tip to suck out any residual liquid.
Air dry for 3-5 minutes. Add 100 ul low TE to the tube. Gently tap the tube and let the pellet dissolve overnight at 4 °C.
Determine DNA concentration using a spectrometer or fluorometer. The concentration is usually between 50 and 300 ug/ul. Store the DNA at 4 °C
Characterize the DNA as in 3.3 to confirm that the purified BAC has the correct size and restriction digest pattern.
The day before the scheduled injection, spin the DNA solution at 13,000 g for 15 minutes to precipitate any particles that may interfere with pronuclear injection. Carefully transfer the upper half of the DNA solution into a clean autoclaved Eppendorf tube. Measure the DNA concentration in the upper fraction .
Depending on the preference of the transgenic facility, dilute the DNA in either injection buffer or low TE. 200-300 ul of the diluted DNA solution is usually enough. Invert the tube to mix.
Spin at 13,000 g for 15 minutes at room temperature to precipitate any residual particles/ aggregates. Carefully transfer the upper half of the DNA solution to a clean autoclaved Eppendorf tube.
Store the DNA at 4 °C before providing to the transgenic facility for pronuclear injection.
3.7. Use BAC transgenic mice to study polarized tissue morphogenesis
Core PCP genes and presumptive PCP ligands such as Wnt5a/ 11 regulates diverse morphogenetic events in the mouse. To elucidate their function at the tissue level, we created BAC transgenes in which the tamoxifen-inducible CreERT2 is inserted into BAC clones containing Wnt5a and Wnt11. We used these BAC transgenes in conjunction with the Cre reporter Rosa26tdTomato for genetic labeling to 1) map the expression of Wnt5a and Wnt11 with high spatial and temporal resolution, 2) lineage trace the contribution of Wnt5a- and Wnt11-expressing cells in different organs and tissues, and 3) study the origin and impact of morphogenetic defects in PCP mutants [31–33]. We present below a method to visualize these genetically labeled embryos using wholemount imaging with confocal microscopy and 3 dimensional reconstruction.
3.7.1. Specimen preparation
For most conventional confocal microscopes, although objectives can have working distance up to 2 mm and fluorescent signal can be observed as deep as 1 mm into the specimen after clearing, the image resolution often drops significantly after 400~500 μm. Therefore, it is often necessary to first trim the specimens close to the plane where the imaging will start.
After tamoxifen induction, collect embryos at the desired times points and fix in 4% PFA at 4 °C for 30 minutes to overnight.
Place the fixed embryo in a 10 cm Petri Dish on its lateral side, and slowly pour melted 2% agarose to fill the entire dish and fully the overed the embryo (Figure 2a). See Note 11.
After the agarose is completely solidified, use a razor or microtome blade to roughly trim the embryo close to the scanning plane. For the remainder of the protocol, we will use imaging the aortic arch as an example so the cuts are around the lower jaw and behind the forelimb (Figure 2a).
Take the specimen out of the agarose and remove the forelimb. Wash it with PBS for 5 min.
Transfer the specimen into PBS with 15% sucrose and shake at room temperature until it sinks to the bottom.
Transfer the specimen into PBS with 30% sucrose and shake at room temperature until it sinks to the bottom (see Note 12).
Transfer the specimen into a mold and cover it completely with OCT (see Note 13). Leave the specimen submerged in OCT at room temperature for 15 ~ 30 minutes, then snap freeze the specimen by immersing the mold into dry ice chilled ethanol or butanol.
Mount the specimen into a cryostat to continue trimming until the surface is close to the desired scanning plane. Check the sections under the microscope frequently during sectioning.
Melt the OCT embedded specimen in PBS at room temperature then wash with PBS 5 times.
Figure 2. Whole-mount imaging of genetically labeled mouse embryo and 3-D reconstruction.
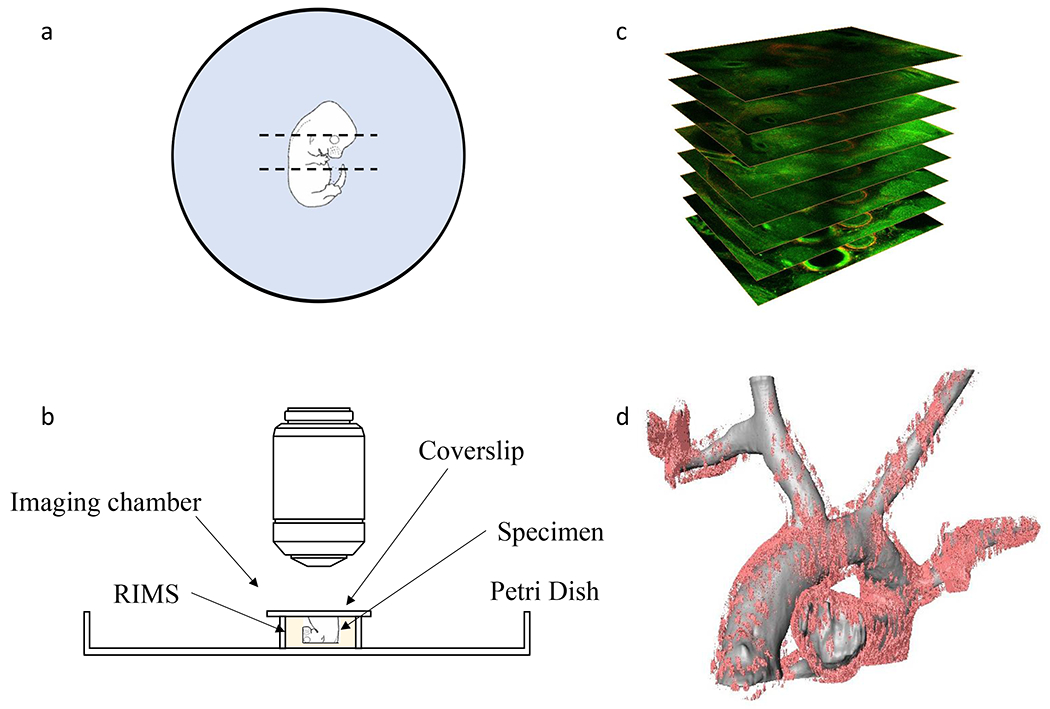
a. 4% PFA fixed embryo is embedded in 2% agarose in a 10 cm Petri Dish. The dashed line indicates the cutting position to image Wnt5a-CreER labeled cells in the aortic arch region. b. Schematic diagram of mounting the specimen in the RIMS in the imaging chamber for confocal microscopy. c. A set of aortic arch images taken by z-stack scanning. d. 3-D reconstructed aortic arch (grey) from an E14.5 Wnt5a-CreER; Rosa26td-Tomato embryo. Wnt5a-CreER labeled cells are marked in light pink.
3.7.2. Whole mount phalloidin staining and RIMS clearing
Permeabilize the specimen in 1x PBS, 0.5% Tween 20 at 4 °C overnight.
Dilute FITC-conjugated phalloidin 1 : 2000 in PBST. Stain the specimen with diluted phalloidin at 4 °C for 24 hours.
Wash the specimen with PBST at room temperature 4 times, 1-2 hours each time with sharking. Then transfer the specimen into fresh PBST and wash on a shaker at 4 °C overnight.
To clear the specimen, transfer it into a 2 ml Eppendorf tube with RIMS. Wrap the tube with foil and rotate it at room temperature for 24 hours or until the specimen becomes transparent.
3.7.3. Confocal imaging and 3D reconstruction
To image with an upright microscope, it will be necessary to make an imaging chamber to mount the cleared specimen with the scanning surface facing up (Figure 2b). To make the chamber, cut a 5 mm height ring from a 15 mL tube and glue it to a Petri dish. Check that the seal is tight and there is no leakage.
Gently transfer the specimen to the imaging chamber filled with RIMS, with the cryostat trimmed surface facing up. Carefully place a coverslip on the top (Figure 2b) without introducing any bubbles.
Place the mounted specimen on the stage of a confocal microscope. Determine the start and end position for the z-stack, set the step size to 2 ~ 4 μm (for 5-10x objectives). Adjust the laser power and PMT settings (see Note 14).
After scanning, the z-stack images (Figure 2c) can be import into Amira or other imaging software for 3D reconstruction.
If using Amira, split the channels if the sample is imaged for two or more fluorophores.
Perform “z-drop correction” to normalize the fluorescent intensity on all the images within the z-stack.
Run automatic alignment with the least-squares alignment mode.
Use the “segmentation” tool to label and define the area of interest. For instance, in our images in the green channel (FITC-phalloidin), the blood vessel lumen can be labeled and defined as the aorta/ aortic arch and pulmonary trunk.
Create another material in the segmentation tool. Use the “grow” function to mark the vessel wall of the labelled aorta and pulmonary trunk.
Go to the red channel and use the “threshold tool” to identify Wnt5a-Cre lineage cells (labeled by td-Tomato) on the blood vessel wall.
Generate the surface from segmented blood vessel in the green channel and Cre lineage in the red channel to create the tetra grid and 3D view of the z-stack (Figure 2d).
Figure 1.
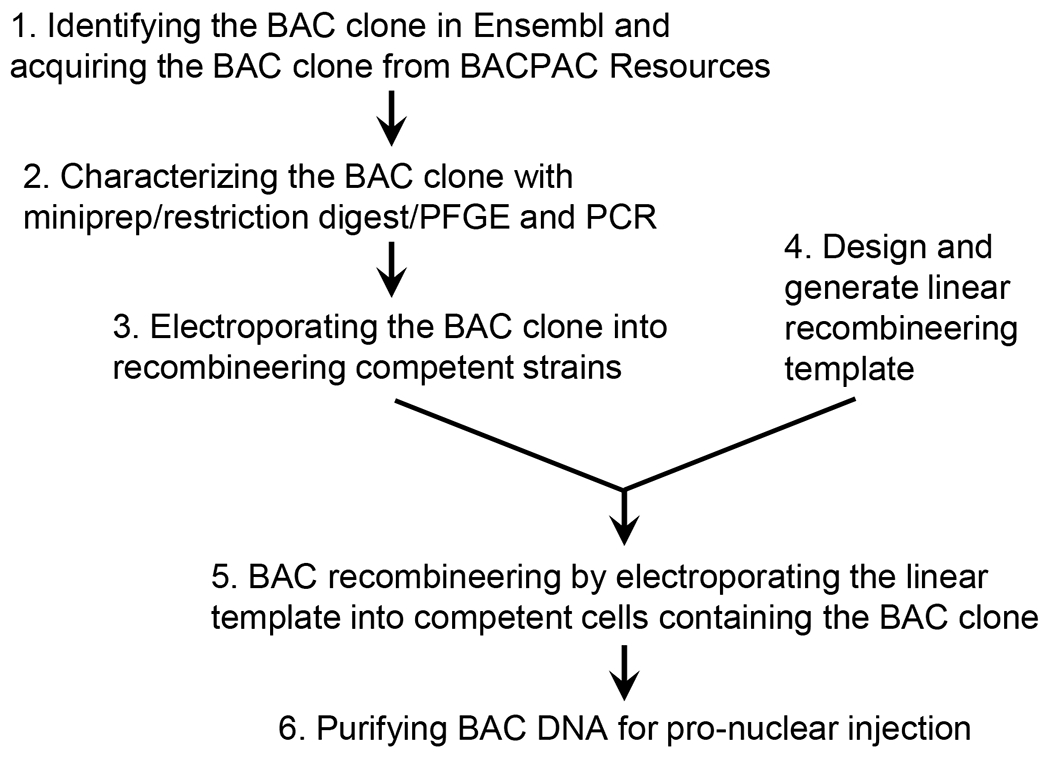
Workflow of BAC recombineering and transgenesis.
Acknowledgments
This work was supported by grants from the National Institutes of Health (HL109130 and HL138470) to J.W.
4. Notes
We have noticed that the more current versions of Ensembl browser display few choices of BAC clones, whereas the archives from 2017 have more BAC clone information.
RP23 (RPCI-23) BAC library was constructed from genomic DNA of female C57BL/6J mice using pBACe3.6 as cloning vector (https://bacpacresources.org/library.php?id=11); whereas RP24 (RPCI-24) BAC library was constructed from genomic DNA of male C57BL/6J mice using pTARBAC1 as cloning vector (https://bacpacresources.org/library.php?id=12).
Do not over dry the pellet, otherwise the DNA may be difficult to dissolve.
Because of their large size, BAC DNA will be more easy to break during pipetting. Therefore cautions should be taken throughout the protocol to avoid repeated pipetting of BAC DNA solution.
To be able to visualize all the bands, usually ~0.5-1 ug BAC DNA and 1 ul of enzyme will be needed for the restriction reaction.
The predicted restriction fragments can often be determined by analyzing the sequence of the BAC clones if their genome coordinates are given in Ensembl during the initial search in 3.1. For instance, BAC clone RP24-85L15 that contain the Rosa26 gene, has genome coordinates of 112952746-113158583. To download its sequence, go to http://useast.ensembl.org/index.html, click on “Mouse”, enter “6: 112952746-113158583” in the “Search all categories”, then click “Go” (6 stands for moue chromosome 6 on which Rosa26 is located). On the next page, click on “Export data” on the left hand to download the 205,838 bp sequence. If NotI is used for restriction digest, note that there is usually 1-2 NotI sites on the cloning vector besides the genomic sequence in the BAC DNA.
To further confirm that the BAC clone contains the gene(s) of interest, PCR can also be performed. The PCR reaction covering the region to be modified should also be sequenced and compare to the genome sequence in the database. This is critical for designing the primers that will serve as the homology arms for recombineering, as even minor difference between the designed homology arm and the actual sequence within the BAC will reduce the recombineering efficiency.
In the recombineering competent strains, transcription of Gam, Beta and Exo is controlled by a temperature sensitive λ repressor in order to minimize their toxic effect. The repressor is active at low temperature (~32 °C), but can be rapidly inactivate with a 15 minutes incubation at 42 °C to provide a pulse of Gam, Beta and Exo expression. Therefore, when growing these cells either in media or on plates, the temperature should always be at or lower than 32 °C.
There are two general strategies to make the desired modifications. The first is a two-step strategy using a dual-selectable marker. The marker is first selected for integration into the location to be modified because it provides resistance to an antibiotic, and is subsequently replaced (selected against) with the desired modification. This strategy is more difficult and time consuming, but can create precise, seamless modifications such as point mutations or in-frame insertions/deletions with minimal perturbation to the flanking genomic sequence. The most commonly used dual selectable markers include SacB-Neo [40,41] and rpsL-Neo [42,43], which confer resistance to kanamycin when inserted into the BAC, and sensitivity to sucrose (~7%) and streptomycin (60 ug/ml), respectively, in the subsequent step when selecting for their replacement by the desired modification.
The second strategy is to directly link the desired modification, such as Cre or EGFP, with a selection marker, such as Neo. PCR or cloning is then used to place homology arms around them. This strategy is considerably easier, but often leaves behind exogenous sequence around the desired modification. PL451 and PL452 [38] are constructs that contain Neo flanked by FRT and LoxP sites, respectively. They can be used to link a desired modification, such as Cre or EGFP, with Neo. Once the Cre-Frt-Neo-Frt or Cre-LoxP-Neo-LoxP cassette has been integrated into a desired location in the BAC through recombineering, Cre or Flippase expression can be induced by arabinose (0.1% for 1 hour) in EL350 or EL250 cells (table 1) to remove Neo [33,38]. This strategy can also be used to easily insert LoxP sites in introns to engineer conditional BAC transgenes [16,38]. Similar to the recombineering cells lines, PL451 and 452 constructs can be requested from NCI through the following link: https://frederick.cancer.gov/Science/BrbRepository/#/productDataSheets/Plasmid.
The handbook of Qiagen Large Construct Kit recommends culturing the bacterial for ~16 hours. The modified BAC in the recombineering strains, however, is culture at 32 °C instead of standard 37 °C, and therefore a longer culturing period is needed to achieve the same level of cell growth.
Regular agarose can be used but make sure the temperature is lower than 30 °C before pouring. Shaking the agarose during cooling to keep it from solidifying.
Can also leave the specimen in 30% sucrose at 4 °C overnight.
It is important to remove any residual PBS from the surface of the specimen with Kimwipes. Otherwise, the PBS on the surface will form a sheet of ice during the snap freezing processes, causing the specimen to detach from OCT during sectioning.
If the specimen to be scanned is too thick or specimen clearing is not optimal, it may be difficult to image the entire sample with one laser power setting. If this is the case, the imaging can be divided into several z-stacks with increasing laser power settings for deeper tissues. Images from different z-stacks can be stitched together during 3D reconstruction with various softwares.
References
- 1.Zallen JA (2007) Planar polarity and tissue morphogenesis. Cell 129 (6):1051–1063. doi: 10.1016/j.cell.2007.05.050 [DOI] [PubMed] [Google Scholar]
- 2.Humphries AC, Mlodzik M (2018) From instruction to output: Wnt/PCP signaling in development and cancer. Curr Opin Cell Biol 51:110–116. doi: 10.1016/j.ceb.2017.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goodrich LV, Strutt D (2011) Principles of planar polarity in animal development. Development 138 (10):1877–1892. doi: 10.1242/dev.054080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Butler MT, Wallingford JB (2017) Planar cell polarity in development and disease. Nat Rev Mol Cell Biol 18 (6):375–388. doi: 10.1038/nrm.2017.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Axelrod JD, Miller JR, Shulman JM, Moon RT, Perrimon N (1998) Differential recruitment of Dishevelled provides signaling specificity in the planar cell polarity and Wingless signaling pathways. Genes Dev 12 (16):2610–2622. doi: 10.1101/gad.12.16.2610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boutros M, Paricio N, Strutt DI, Mlodzik M (1998) Dishevelled activates JNK and discriminates between JNK pathways in planar polarity and wingless signaling. Cell 94 (1):109–118. doi: 10.1016/s0092-8674(00)81226-x [DOI] [PubMed] [Google Scholar]
- 7.Seifert JR, Mlodzik M (2007) Frizzled/PCP signalling: a conserved mechanism regulating cell polarity and directed motility. Nat Rev Genet 8 (2):126–138. doi: 10.1038/nrg2042 [DOI] [PubMed] [Google Scholar]
- 8.Axelrod JD, Tomlin CJ (2011) Modeling the control of planar cell polarity. Wiley Interdiscip Rev Syst Biol Med 3 (5):588–605. doi: 10.1002/wsbm.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strutt H, Gamage J, Strutt D (2019) Reciprocal action of Casein Kinase Iepsilon on core planar polarity proteins regulates clustering and asymmetric localisation. Elife 8. doi: 10.7554/eLife.45107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Y, Mlodzik M (2015) Wnt-Frizzled/planar cell polarity signaling: cellular orientation by facing the wind (Wnt). Annu Rev Cell Dev Biol 31:623–646. doi: 10.1146/annurev-cellbio-100814-125315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wallingford JB, Rowning BA, Vogeli KM, Rothbacher U, Fraser SE, Harland RM (2000) Dishevelled controls cell polarity during Xenopus gastrulation. Nature 405 (6782):81–85. doi: 10.1038/35011077 [DOI] [PubMed] [Google Scholar]
- 12.Heisenberg CP, Tada M, Rauch GJ, Saude L, Concha ML, Geisler R, Stemple DL, Smith JC, Wilson SW (2000) Silberblick/Wnt11 mediates convergent extension movements during zebrafish gastrulation. Nature 405 (6782):76–81 [DOI] [PubMed] [Google Scholar]
- 13.Keller R (2002) Shaping the vertebrate body plan by polarized embryonic cell movements. Science 298 (5600):1950–1954 [DOI] [PubMed] [Google Scholar]
- 14.Murdoch JN, Doudney K, Paternotte C, Copp AJ, Stanier P (2001) Severe neural tube defects in the loop-tail mouse result from mutation of Lpp1, a novel gene involved in floor plate specification. Human molecular genetics 10 (22):2593–2601 [DOI] [PubMed] [Google Scholar]
- 15.Kibar Z, Vogan KJ, Groulx N, Justice MJ, Underhill DA, Gros P (2001) Ltap, a mammalian homolog of Drosophila Strabismus/Van Gogh, is altered in the mouse neural tube mutant Loop-tail. Nature genetics 28 (3):251–255. [DOI] [PubMed] [Google Scholar]
- 16.Wang J, Hamblet NS, Mark S, Dickinson ME, Brinkman BC, Segil N, Fraser SE, Chen P, Wallingford JB, Wynshaw-Boris A (2006) Dishevelled genes mediate a conserved mammalian PCP pathway to regulate convergent extension during neurulation. Development 133 (9):1767–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y, Guo N, Nathans J (2006) The role of Frizzled3 and Frizzled6 in neural tube closure and in the planar polarity of inner-ear sensory hair cells. J Neurosci 26 (8):2147–2156. doi: 10.1523/JNEUROSCI.4698-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Curtin JA, Quint E, Tsipouri V, Arkell RM, Cattanach B, Copp AJ, Henderson DJ, Spurr N, Stanier P, Fisher EM, Nolan PM, Steel KP, Brown SD, Gray IC, Murdoch JN (2003) Mutation of Celsr1 disrupts planar polarity of inner ear hair cells and causes severe neural tube defects in the mouse. Current biology : CB 13 (13):1129–1133. doi: 10.1016/s0960-9822(03)00374-9 [DOI] [PubMed] [Google Scholar]
- 19.Juriloff DM, Harris MJ (2012) A consideration of the evidence that genetic defects in planar cell polarity contribute to the etiology of human neural tube defects. Birth Defects Res A Clin Mol Teratol 94 (10):824–840. doi: 10.1002/bdra.23079 [DOI] [PubMed] [Google Scholar]
- 20.Roy JP, Halford MM, Stacker SA (2018) The biochemistry, signalling and disease relevance of RYK and other WNT-binding receptor tyrosine kinases. Growth Factors 36 (1–2):15–40. doi: 10.1080/08977194.2018.1472089 [DOI] [PubMed] [Google Scholar]
- 21.Wang B, Sinha T, Jiao K, Serra R, Wang J (2011) Disruption of PCP signaling causes limb morphogenesis and skeletal defects and may underlie Robinow syndrome and brachydactyly type B. Human molecular genetics 20 (2):271–285. doi: 10.1093/hmg/ddq462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeChiara TM, Kimble RB, Poueymirou WT, Rojas J, Masiakowski P, Valenzuela DM, Yancopoulos GD (2000) Ror2, encoding a receptor-like tyrosine kinase, is required for cartilage and growth plate development. Nature genetics 24 (3):271–274. doi: 10.1038/73488 [DOI] [PubMed] [Google Scholar]
- 23.Person AD, Beiraghi S, Sieben CM, Hermanson S, Neumann AN, Robu ME, Schleiffarth JR, Billington CJ Jr., van Bokhoven H, Hoogeboom JM, Mazzeu JF, Petryk A, Schimmenti LA, Brunner HG, Ekker SC, Lohr JL (2010) WNT5A mutations in patients with autosomal dominant Robinow syndrome. Developmental dynamics : an official publication of the American Association of Anatomists 239 (1):327–337. doi: 10.1002/dvdy.22156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Afzal AR, Rajab A, Fenske CD, Oldridge M, Elanko N, Ternes-Pereira E, Tuysuz B, Murday VA, Patton MA, Wilkie AO, Jeffery S (2000) Recessive Robinow syndrome, allelic to dominant brachydactyly type B, is caused by mutation of ROR2. Nature genetics 25 (4):419–422. doi: 10.1038/78107 [DOI] [PubMed] [Google Scholar]
- 25.Goto T, Keller R (2002) The planar cell polarity gene strabismus regulates convergence and extension and neural fold closure in Xenopus. Developmental biology 247 (1):165–181. doi: 10.1006/dbio.2002.0673 [DOI] [PubMed] [Google Scholar]
- 26.Ting JT, Feng G (2014) Recombineering strategies for developing next generation BAC transgenic tools for optogenetics and beyond. Front Behav Neurosci 8:111. doi: 10.3389/fnbeh.2014.00111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bian Q, Belmont AS (2010) BAC TG-EMBED: one-step method for high-level, copy-number-dependent, position-independent transgene expression. Nucleic Acids Res 38 (11):e127. doi: 10.1093/nar/gkq178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heaney JD, Bronson SK (2006) Artificial chromosome-based transgenes in the study of genome function. Mamm Genome 17 (8):791–807. doi: 10.1007/s00335-006-0023-9 [DOI] [PubMed] [Google Scholar]
- 29.Wang J, Mark S, Zhang X, Qian D, Yoo SJ, Radde-Gallwitz K, Zhang Y, Lin X, Collazo A, Wynshaw-Boris A, Chen P (2005) Regulation of polarized extension and planar cell polarity in the cochlea by the vertebrate PCP pathway. Nature genetics 37 (9):980–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sinha T, Wang B, Evans S, Wynshaw-Boris A, Wang J (2012) Disheveled mediated planar cell polarity signaling is required in the second heart field lineage for outflow tract morphogenesis. Developmental biology 370 (1):135–144. doi:S0012-1606(12)00407-1 [pii] 10.1016/j.ydbio.2012.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li D, Angermeier A, Wang J (2019) Planar cell polarity signaling regulates polarized second heart field morphogenesis to promote both arterial and venous pole septation. Development 146 (20). doi: 10.1242/dev.181719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sinha T, Li D, Theveniau-Ruissy M, Hutson MR, Kelly RG, Wang J (2015) Loss of Wnt5a disrupts second heart field cell deployment and may contribute to OFT malformations in DiGeorge syndrome. Human molecular genetics 24 (6):1704–1716. doi: 10.1093/hmg/ddu584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sinha T, Lin L, Li D, Davis J, Evans S, Wynshaw-Boris A, Wang J (2015) Mapping the dynamic expression of Wnt11 and the lineage contribution of Wnt11-expressing cells during early mouse development. Developmental biology 398 (2):177–192. doi: 10.1016/j.ydbio.2014.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee EC, Yu D, Martinez de Velasco J, Tessarollo L, Swing DA, Court DL, Jenkins NA, Copeland NG (2001) A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics 73 (1):56–65. doi: 10.1006/geno.2000.6451 [DOI] [PubMed] [Google Scholar]
- 35.Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL (2000) An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci U S A 97 (11):5978–5983. doi: 10.1073/pnas.100127597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, Buchholz F, Muyrers JP, Stewart AF (1998) A new logic for DNA engineering using recombination in Escherichia coli. Nature genetics 20 (2):123–128. doi: 10.1038/2417 [DOI] [PubMed] [Google Scholar]
- 37.Sharan SK, Thomason LC, Kuznetsov SG, Court DL (2009) Recombineering: a homologous recombination-based method of genetic engineering. Nat Protoc 4 (2):206–223. doi: 10.1038/nprot.2008.227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu P, Jenkins NA, Copeland NG (2003) A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res 13 (3):476–484. doi: 10.1101/gr.749203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gama SM, De Gasperi R, Wen PH, Gonzalez EA, Kelley K, Lazzarini RA, Elder GA (2002) BAC and PAC DNA for the generation of transgenic animals. Biotechniques 33 (1):51–53. doi: 10.2144/02331bm07 [DOI] [PubMed] [Google Scholar]
- 40.Gong S, Yang XW, Li C, Heintz N (2002) Highly efficient modification of bacterial artificial chromosomes (BACs) using novel shuttle vectors containing the R6Kgamma origin of replication. Genome Res 12 (12):1992–1998. doi: 10.1101/gr.476202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muyrers JP, Zhang Y, Benes V, Testa G, Ansorge W, Stewart AF (2000) Point mutation of bacterial artificial chromosomes by ET recombination. EMBO Rep 1 (3):239–243. doi: 10.1093/embo-reports/kvd049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y, Muyrers JP, Rientjes J, Stewart AF (2003) Phage annealing proteins promote oligonucleotide-directed mutagenesis in Escherichia coli and mouse ES cells. BMC Mol Biol 4 (1):1. doi: 10.1186/1471-2199-4-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bird AW, Erler A, Fu J, Heriche JK, Maresca M, Zhang Y, Hyman AA, Stewart AF (2011) High-efficiency counterselection recombineering for site-directed mutagenesis in bacterial artificial chromosomes. Nat Methods 9 (1):103–109. doi: 10.1038/nmeth.1803 [DOI] [PubMed] [Google Scholar]