

Abstract
The study of mitosis has always relied on bulk-preparation biochemistry techniques (Mazia and Dan, 1952), but very early on lent itself to living, single cell microscopic techniques (Inoue, 1953; Taylor, 1958). Here we describe several of the methods used by our lab to study cell division in living cultured cells, including cold-induced mitotic arrest, cold-induced chromosome missegregation, same-cell live and fixed cell imaging, and microinjection of inactivating antibodies. We detail our imaging system based on an upright fluorescent microscope and spinning disk confocal, as well as the customized “HEKS” metal support slide imaging chambers.
Keywords: Cell culture, cell cycle, chilling, chromosomes, cold-dependent, microinjection, microscopy, microtubule, spindle, spinning disk
I. Introduction
The study of cell division – mitosis and meiosis – is both exciting and challenging. Mitosis is a critical time during the cell cycle, where the genome is packaged in condensed form, and the pairs of sister chromatids must be segregated with absolute fidelity. This process depends on the assembly of a bipolar mitotic spindle, which entails a complete remodeling of the microtubule cytoskeleton, along with the de novo assembly of kinetochores on the chromatids. Similar mitotic remodeling events occurs in all compartments of the cell, membranous organelles, actin cytoskeleton, nuclear envelope, etc. Mistakes in any aspect of this coordinated series of processes can result in chromosome missegregation or damage. In either case, these mistakes have catastrophic consequences for the organism, and as such, highly sensitive regulatory and fail-safe mechanisms have evolved to ensure chromosome stability. Teasing apart these mechanisms continues to occupy the efforts of many research labs across the globe. However, these efforts face a major challenge: the timing and duration of M-phase, relative to the rest of the cell cycle. In mammalian cultured cells, the duration of mitosis is ~1 hr, and this occurs at a frequency of once every 24 hrs. In an asynchronous cell culture, the small percentage of mitotic cells can be difficult to study at the biochemical level. One solution is to use a synchronized population of cells, either using a naturally synchronized embryonic system (flies, urchins, frog egg extracts) or chemically treated cultured cells (thymidine block, nocodazole-treated, proteasome inhibitor-treated: Uetake and Sluder, 2007, 2010, 2018) to arrest the cells as they enter a particular stage, causing them to “pile-up” at a certain transition point, then releasing them where they can proceed in relative synchrony until the desired stage (prometaphase/metaphase, anaphase). This boosts the amount of mitotic material for investigation.
Another solution is to use an asynchronously dividing population and examine individual cells by live-cell microscopy. The advantage of the population study is that there is sufficient material to conduct biochemistry experiments on regulatory mechanisms, and post-translational modifications such as mitotic phosphorylation events. Single-cell experiments can study cytoskeletal organization, changes in protein distribution, and dynamics. These single cell experiments can provide a temporal control that may be difficult in a population of cells. Here we describe several of the techniques we use to study mitosis in individual cultured cells. Our focus is on examining transition periods, either from interphase in to prophase/prometaphase, from metaphase into anaphase, or the transition from mitosis into early interphase. These manipulations can be combined with molecular genetic techniques such as siRNA, CRIPSR insertion/deletion, or degron-mediated protein ablation (Fire et al.,1998; Elbashir et al., 2001; Komor et al., 2015; Natsume et al., 2016).
II. Cell culture
It is important to choose the right cells for live-imaging and mitotic manipulation studies. For cell division work, it is best to use cells that remain relatively flat during mitosis. Cells that grow in clumps or columns can complicate the live-cell imaging. The cells selected also have to be able to withstand fluorescence imaging conditions, because exposure to certain wavelengths of light can be cytotoxic – the toxicity may not be readily apparent. If a study is designed to test the response of cell proliferation after the manipulation of a mitotic component, then it is important to consider the cell cycle regulation of a chosen cell type as well. For instance, we have studied cell cycle arrest following loss of the centrosome, mistakes in spindle assembly, or missegregation of one or more chromosomes. We need to ensure that any arrest observed is due to the manipulation of the cellular component rather than in response to the imaging conditions. We do this by culturing cells at low density in our imaging chambers, and filming them by time-lapse for 72 hrs (Hinchcliffe, 2005). This ensures that the imaging methods do not interfere with cell proliferation. This is best determined empirically. If there is any question about a cell’s viability under fluorescence conditions, it can be imaged using a minimally destructive method, such as phase contrast, and obtain a baseline for cell cycle timing, mitotic duration, etc. These parameters can then be compared to those obtained from the same cell types using a fluorescence imaging system.
For rodent work normal rat kidney (NRK) cells have proven to be an excellent choice for mitosis research (Wheatley et al., 1997). For primate work, we use BSC1 (green monkey kidney cells), which are similar to the widely used COS7 primate cells, and are excellent for imaging (Hinchcliffe et al., 2001; Hornick et al., 2011; Hinchcliffe et al., 2016). Others have used RPE1 (hTERT-immortilzed human pigmented retinal cells) or BJ (hTERT-immortilzed human fibroblasts). However, RPE1 cells – thought to be “normal” human cells – harbor an activating KRAS mutation (Di Nicolantonio et al., 2009). This makes them a less ideal cell type for examining cell cycle regulation. We have also used various cancer cell lines, such as SF8628 pediatric glioma cells, which are excellent for imaging studies.
Regardless of the cell type used, it is important to consider the ability to introduce trans-genes (such as GFP-fusion proteins) into each cell type to visualize cellular components of interest (such as microtubules, microtubule plus-ends, cyclins, chromosomes, centrosomes, nuclear lamina, etc). We have adopted nucleofection using the Lonza nucleofector machine to introduce trans-genes, as this works well for many “hard-to-transfect” cell types. Other methods include chemical or viral transfection (Recillas-Targa, 2004). Cells can either be transfected transiently for each experiment or transfected and then selected to obtain a clone that constitutively expresses the transgene fusion (Recillas-Targa, 2004). Once we have generated a fusion protein-expressing clone, we grow large amounts of the cells and freeze them down as low-passage clones. This ensures we are working with cells that do not accumulate errors (as in the case of introducing proteins that trigger chromosomal instability).
Imaging media – we routinely use media supplemented with 12.5 mM Hepes buffer, because our imaging system does not use CO2 for bicarbonate buffer. We also use media that does not contain phenol red, which lowers the contrast of the fluorescence images.
III. Imaging chambers
Cell imaging occurs on one of two different types of microscopes: inverted or upright. The choice is largely dependent upon available instrumentation. Cultured cells grown on glass coverslips require some type of support to facilitate imaging. For those labs using inverted microscopes for imaging, there are two standard solutions for supports apparatus: the modified Rose chamber (Rose et al., 1958) or a “dish chamber”, which consists of a 35 mm culture dish with a center hole cut into it and glass coverslip affixed over the opening (Wadsworth, 1999). Details of the assembly of a modified Rose chambers are provided in Rieder and Cole, (1997) and Wadsworth (1999). To build a “dish chamber”, a standard 35mm culture dish has a hole cut in the center. We have found the easiest way to do this is to heat a metal cork-borer over a Bunsen burner and apply to the surface of the dish – this pushes through and punches out a small disk of plastic. The resulting hole is then rounded with a piece of sand paper to smooth the edges. To use for imaging cells, a number 1½ coverslip is fastened over the hole with silicon grease (Dow Corning high vacuum grease, Midland, MI), and then entire chamber sterilized by UV irradiation for 15 min in a culture hood. After sterilization the cells are seeded into the dish, and grown on the coverslip in a reservoir of culture medium. The dish is removed from the incubator, placed on the microscope and cells can be imaged. Media can be exchanged easily with a pipet. After the experiment, the coverslip is removed, the grease cleaned with 75% EtOH, then the chamber is washed, and re-assembled for the next experiment. Alternatively, commercial dishes may be purchased, which are both sterile and disposable (MatTek Corp. Ashland, MA, USA).
For upright microscopes we use a modified “HEKS chamber”, named for the designers and users of this device: Hiramoto/Ellis/Kiehart/Sluder. These were originally designed for the micromanipulation and/or microinjection of echinoderm zygotes (Hiramoto, 1962; Begg and Ellis, 1979; Kiehart, 1982; Sluder and Begg, 1983; Hinchcliffe et al., 1998) and adapted by our lab for imaging cultured cells (Hinchcliffe et al., 2001; Sluder et al., 1999; Sluder et al., 2007). This “HEKS” chamber consists of an aluminum support slide, with the overall dimensions of a microscope slide (1” × 3”) that has been milled to form a “U-shaped” cutout in the center that is open to one side (Fig. 1). There are 1 mm standoffs on either end, so the surface of the chamber remains above the surface of the microscope stage. Over this center cutout on either side of the chamber is affixed a glass coverslip, creating a well that is filled with culture medium.
Fig. 1. Modified HEKS imaging chamber.
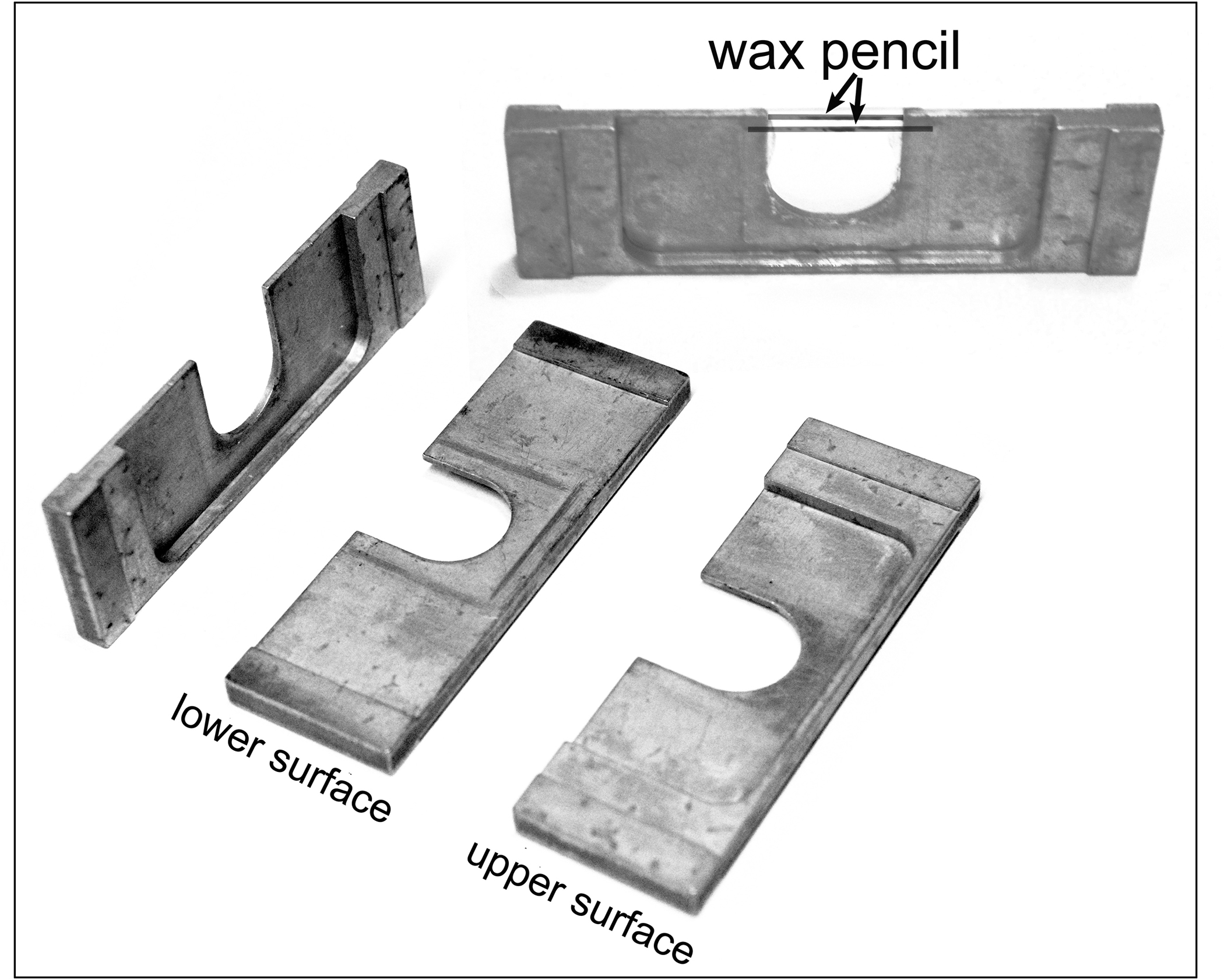
The chamber support slide is machined out of aluminum stock. This can also be made using a 3-D printer. The stand-off at either edge maintain the slide just above the surface of the microscope stage. The lower surface is flat, while the upper surface has a cutout to allow access for the microscope objective. When the chamber is assembled, the two coverslips – each with a wax pencil line – enclose the u-shaped cutout. The well below the pencil lines is filled with medium. The gap from the pencil lines to the edge of the coverslip is filled with mineral oil.
While we have had these slides milled in a traditional machine shop, they are also amenable to 3-D printing (Alessandri et al. 2017; Lerman et al., 2018). The only concern is that the medium used to print the support slide is stiff enough to provide a flat surface for the coverglass to adhere to. Otherwise, the plane of focus is uneven as the slide is moved in the XY.
The coverslip is held in place by a bead of silicon grease (Dow Corning high vacuum grease, Midland, MI). The cells are cultured on one of the coverslips, where they can be manipulated and observed. The open surface at the edge of the cutout remains open to the air, but retains the culture medium due to the presence of wax pencil lines (Sharpie Peel-Off Red China Marker, Atlanta, GA) applied to the inner surfaces of the coverslips. These lines face the interior of the preparation and act as a hydrophobic barrier that is still open. The gap between the pencil lines and the coverslip edge is filled with mineral oil (heavy white oil; sigma, St. Louis, MO). The advantage is that needles for microinjection or micromanipulation can be inserted into the chamber. Also, drugs can be introduced into the chamber through this opening using a standard narrow bore gel loading pipet tip. The wax is applied on the inner surface of the coverslips, in the same plane as the cells. This wax is auto-fluorescent and serves as a fiduciary mark for rapid focusing of the cells (Fig. 1).
IV. Microscope Setup for Live-Cell Imaging
Live-cell imaging requires a microscope that can support cells at biological conditions, including temperature, pH, humidity and light. For mammalian cells this means 37 °C, a pH of ~7, minimal exposure to blue and green wavelengths of light and blocking of longer (infrared) wavelengths of light. The choice of microscope to image the mitotic cell depends on the nature of the experiment and the available equipment. We have used a variety of imaging modes in the past, including phase contrast, differential interference contrast, polarization, wide-field fluorescence, and spinning disk confocal (Hinchcliffe et al., 1998, 1999; Hinchcliffe et al., 2001; Hinchcliffe, 2005; Hornick et al., 2011; Hinchcliffe et al., 2016).
For mammalian cultured cells, temperature regulation is very important. There are a variety of commercially available microscope environmental control incubator systems, and these can be obtained from the microscope manufacturing companies (Zeiss, Olympus, Leica or Nikon) or WPI (World Precision Instruments, Sarasota, FL). For most of these systems the microscope itself is enclosed in a plexiglas box and this is attached to a heating element and a system to regulate CO2 levels, just as in a cell incubator.
While complete turn-key cell incubator systems can be obtained commercially, we have found it far more economical to build our own with the help of a local machine shop. First, we mock-up an enclosure using pieces of cardboard cut with a box cutter and straight edge, and assembled using duct tape. This is placed on the microscope, and adjustments are made for protrusions, camera ports, etc. Make sure that the lamps and switches on the external surface of the scope are accessible. This mock-up provides a guide or a model to use in drawing plans for manufacture of a plexiglas version at a local machine shop. Measure twice.
For our home-built enclosure we use a heating system incorporating a proportional temperature controller and thermocouple element (Omega Engineering, Norwalk, CT USA), wired to an industrial heat gun (Master Mite heat gun; Master Tools, Racine, WI USA). The heating element of the heatgun is wired directly to the temperature controller using a standard electrical plug, whereas the fan on the heat gun is always on (controlled by master switch). This way, the heater cycles based on feedback from the thermocouple, with a constant stream of air through the heater element, because the master switch is always on. This is critical, because the heating element can produce very high temperatures. The constant flow of air through the heating element ensures that the heater does not cause a melt-down of the plastic housing. Make sure to shut off the temperature controller first, and check that the heating element has cooled to room temperature BEFORE turning off the master switch on the heatgun.
Regardless of the imaging modality, care must be taken to ensure the imaging conditions do not overly influence the cell behavior (Hinchcliffe, 2005; Magidson and Khodjakov, 2013; Douthwright and Sluder, 2017). While it is beyond the scope of this chapter to discuss the concerns involved with live-imaging of cells, the rule of thumb is to minimize total light exposure. If the experiment is relatively fast (on the order of minutes per cell), and you will not be examining the downstream behavior of that cell (cell cycle progression, etc.), then you can use greater light levels, high frequency image capture, and greater Z-plane accuracy. However, if you are interested in the longer-term behavior, then light must be decreased, the frequency of image capture must be decreased, and the size of the Z-steps used to generate a focal series must be increased. For our lowest light-level imaging of cells transitioning from metaphase to anaphase and then into G1 (using a spinning disk confocal), we use a 2 μm step size to generate the Z-series. The frequency of imaging varies, depending on the events we need to examine. For example, in a typical experiment we would follow an individual cell expressing histone H2B-GFP as it undergoes anaphase with a misaligned chromosome, then follow the two daughter cells (one containing a micronucleus) for 72 hrs post-anaphase, to assess its cell cycle response. For this experiment we would collect at intervals ranging from 90 sec (prior to anaphase onset, while the chromosomes are congressing), then switch to 10 min intervals after the cells have exited mitosis and enter into G1 (Hinchcliffe et al., 2016).
Recent advances in light-sheet microscopy have the advantage of providing high-resolution fluorescence imaging with dramatically lower phototoxicity, and going forward, these will undoubtedly become the primary fluorescence imaging modes for live cell microscopy (Wu et al., 2013; Chen et al., 2014; Kumar et al., 2014). However, these instruments require an investment of either money or time/expertise, and are not currently ubiquitous.
Currently we use a spinning disk confocal microscope for our live-cell fluorescence imaging experiments. This consists of a Yokogawa CSU-10 spinning disk confocal head, solid-state laser stack with beam switcher (Intelligent Imaging Innovations, Denver, CO), a back-thinned EMCCD camera (C9100–02: Hamamatsu, E. Bridgewater, NJ) and Slidebook software (Intelligent Imaging Innovations, Denver, CO). The confocal is mounted on a Leica DM RXA2 upright microscope with motorized stage and focus (Leica Microsystems, Buffalo Grove, IL). We have mounted two objectives on this microscope: a 63× 1.3 NA glycerol-immersion lens for high magnification imaging and a 20× 0.8 NA multi-immersion objective for low magnification imaging and for visual scanning of the field. We also have a diamond scribe mounted in the nosepiece. This allows us to mark the position of an individual cell with a circular scribe mark for relocating later after fixation and immuno-labelling. The microscope itself in placed on an anti-vibration isolation table (TMC, Peabody, MA).
V. Methods
a. Cell chilling to disassemble the mitotic spindle:
one method of manipulating mitotic cells that we have found to be particularly useful is the disassembly of the mitotic spindle by lowering the temperature of the culture media to 4 °C for 20 min. This causes the microtubule cytoskeleton to completely disassemble (Fig 2). When the temperature is returned to 37 °C, the spindle rapidly reforms, and the cell is capable of proceeding through the metaphase-anaphase transition. The major advantage of chilling over treatment of cells with chemical microtubule poisons is the complete and rapid reversal of the process. In a drug washout experiment, the cells have to undergo several washes with fresh media, and even then, not every cell exhibits a complete washout at the same time. In chilling/re-warming, each cell begins to reform the mitotic spindle as soon as the temperature rises to the level sufficient to promote microtubule growth. As all the cells lie at the surface of the coverslip, this happens with great synchrony (Fig. 2).
Fig. 2. Cell Chilling.
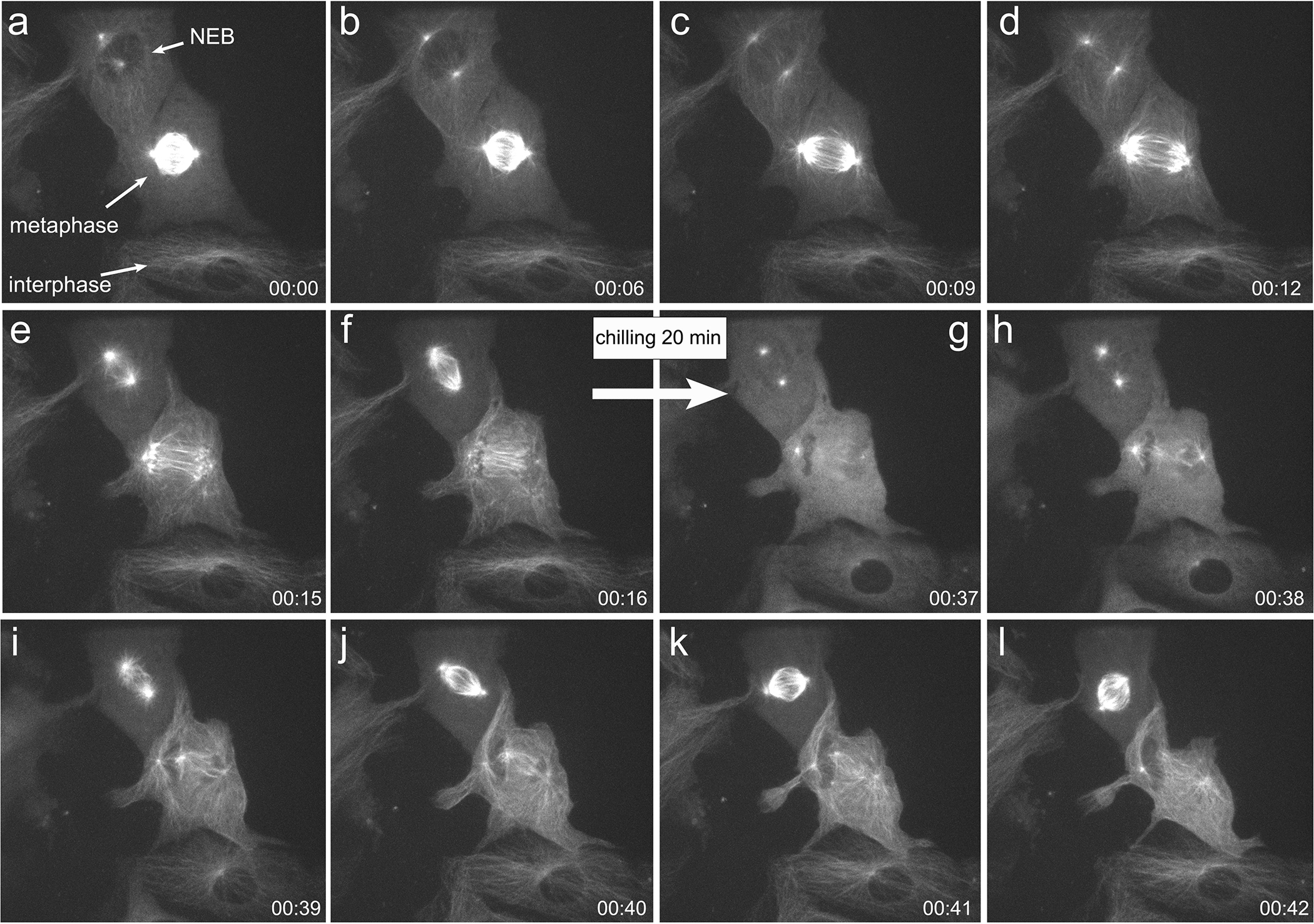
Frames from a time-lapse video, showing before and after chilling of BSC1 cells expressing α tubulin-GFP. a. In the field are three cells in different stages of the cell cycle: the top cell is just entering mitosis with a pair of duplicated, separating centrosomes (bright dots), the middle cell is in metaphase with a bipolar spindle, and the bottom cell is in interphase. c. The middle cell undergoes anaphase. f. At t=16 min, the image sequence was paused, the chamber was removed from the microscope and placed in the cold. g. After 20 min, the chamber was returned to the warm microscope, and imaging resumed. Not the microtubule networks are disassembled, but the position of the centrosomes can be visualized as a bright dot as the microtubules begin to nucleate from them. i. In less than two minutes the microtubules have re-grown, and the spindle in the top cell reforms. T = Hr:Min.
For chilling in a dish chamber, the method is simple: the dish is removed from the warm microscope, and the culture media is removed with a pipet, and then pre-chilled culture media at 4 °C is added to the dish. After 20 min, the cold media is removed, and replaced with warm media. The dish is then returned to the microscope.
For out upright microscope system and HEKS chamber, cells are cultured on biocleaned coverslips (detailed in Sluder et al., 2007) with a wax pencil line and a 3-sided perimeter of silicon grease (Dow Corning high vacuum grease, Midland, MI) are assembled onto the HEKS metal support slide. A matching coverslip with wax line is assembled onto the opposing surface using silicon grease applied with a fine-tip metal spatula. The reservoir (up to the wax line) is filled with fresh media using a syringe and fine gauge needle. The opening between the coverslips is then capped with mineral oil (added with a syringe and fine gauge needle). This prevents the media from drying out. To depolymerize the microtubule cytoskeleton the chamber itself is rapidly chilled 4 °C. To do this, we first identify the cell of interest, and take a series of image stacks (Z-series) that serve as the pre-chill images (Fig. 3). Then the imaging is paused, the chamber is carefully removed from the microscope stage, the immersion media quickly aspirated from the coverslip, and the chamber is placed in a −20 °C freezer for 5 min (exactly). It is then quickly moved to the 4 °C refrigerator for 15 min. At the end of the chilling period, the chamber is returned to the warm 37 °C microscope, the cell quickly centered and focused, and the imaging series is allowed to proceed (Fig. 3). Note that in the time it takes to focus the cell and switch from widefield epifluorescent imaging (through the eyepieces) over to confocal imaging through the camera, the chamber warms up such that the microtubules have begun to re-assemble from the spindle poles (Fig. 3 arrows). The spindle then rapidly reforms and the cell undergoes anaphase.
Fig. 3. Anaphase offset following chilling/re-warming.
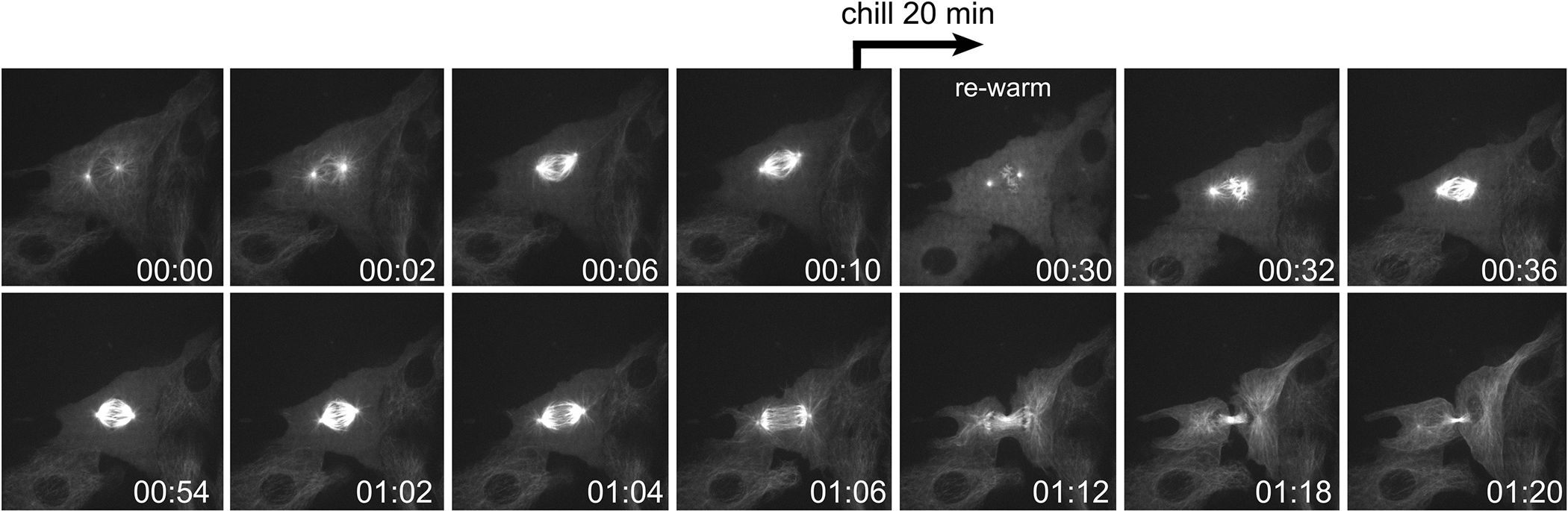
Frames from a time-lapse sequence of a mitotic BSC1 cell expressing α tubulin-GFP. The cell forms a bipolar spindle. At T = 10 min, the sequence was paused and the chamber placed in the cold. After 20 min, the chamber is returned to the warm microscope and imaging resumed. Following cold treatment the spindle microtubules have depolymerized. Within several minutes of rewarming, the bipolar spindle reforms, and at T = 64 min the cell undergoes anaphase and cleaves into two daughter cells. T = Hrs:Min.
These chilling experiments can also be coupled with the addition of a chemical inhibitor (drug) to measure the effects of inactivating a particular cellular component (such as a kinase or phosphatase) with loss of the microtubule network. Chilling can also be done, and then the cells rewarmed in the presence of a microtubule drug: nocodazole prevents the reformation of the spindle. While taxol causes a spindle to re-assemble with stabilized microtubules.
The timing of cell chilling can be set to a prescribed time after a marked mitotic event such as nuclear envelope breakdown or anaphase onset (Fig. 4). To do so, we identify a cell in prometaphase by its nuclear morphology (condensing chromosomes within a nuclear boundary). The cell is then imaged for a period of time, then chilled as it reaches the decided endpoint (i.e. anaphase onset). This requires that the cell be constantly observed until it reaches the point where it is to be chilled. As with chilling in prometaphase, this experiment can be coupled with drug treatments to separate out the activity of a particular regulatory component (kinase/phosphatase, etc.) from the signalling activity of the mitotic spindle.
Fig. 4. Chilling/re-warming an anaphase cell.
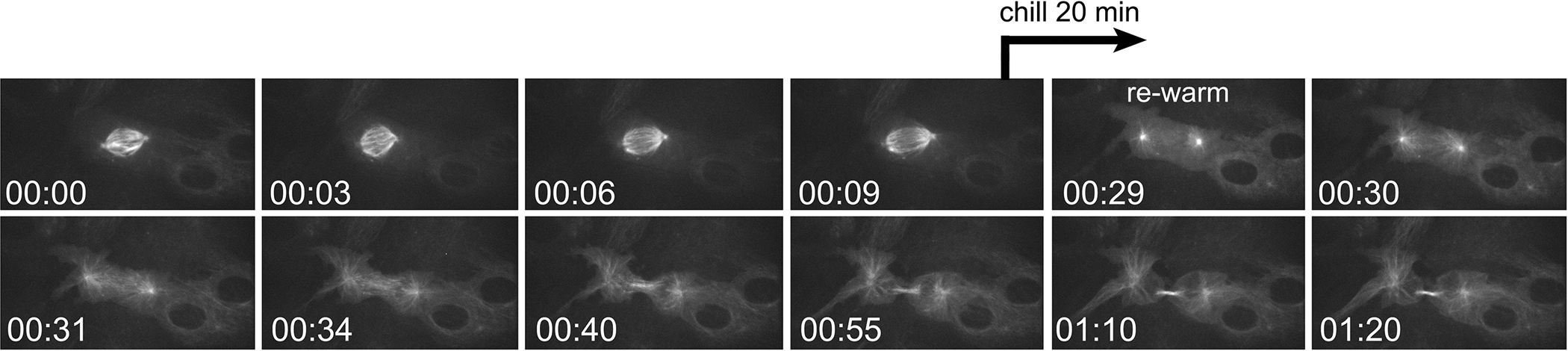
Frames from a time-lapse sequence of a mitotic BSC1 cell expressing α tubulin-GFP. At T = 10 min the cell enter anaphase. At this time, the imaging sequence was paused and the chamber placed in the cold. After 20 min, the chamber is returned to the warm microscope and imaging resumed. Following cold treatment the spindle microtubules have depolymerized, and there is no ingression of the cleavage furrow. The microtubules re-polymerize and the furrow begins to ingress. At T = 80 min, the furrow has completed ingression, and the midbody begins to form. T = Hrs:Min.
b. Chilling to induce chromosome missegregation:
during our chilling studies, we observed an interesting phenomenon – cells that had undergone transient spindle disassembly/re-assembly showed an increase in chromosome segregation anomalies, including chromosomes that fail to congress before anaphase onset, and chromosomes that lag in the central spindle region during anaphase. We have used this phenomenon to induce chromosome missegregation in normal, diploid cells (Hinchcliffe et al., 2016).
To induce missegregations, cells expression H2B-GFP (to fluorescently label chromosomes) are cultured at moderate density, then allowed to grow for ~36 hrs so they are at 75% density. We typically build six chambers at a time, because this experiment has a relatively low success rate (success is achieved by getting a normal diploid cell to undergo anaphase with a misaligned chromosome), and once chilled, either we achieve success, or have to start with a fresh chamber.
Once the cells are at the correct density, the coverslips are assembled into a HEKS chamber, and the chamber is chilled for 20 min (−20 °C for 5 min, then 4 °C for 15 min). After the chilling, the chamber is placed on the microscope, the wax pencil line used as a focus tool, and the coverslip is scanned using a 20× 0.8 NA objective; we are looking for a mitotic cell with one or more misaligned chromosomes, which is easily detected by the trained eye (Fig. 5). The objective is exchanged for the 63× 1.3 NA lens, and this cell is followed until it either undergoes anaphase with a misaligned chromosome (which is considered a success) or it congresses the last chromosome to the metaphase plate (which will most likely segregate all chromosomes properly), in which case we switch to another cell on that chamber. If we do not find a success within the first two cells imaged, we begin again with a new, un-chilled HEKS chamber.
Fig. 5. Chromosome missegregation following chilling/re-warming.
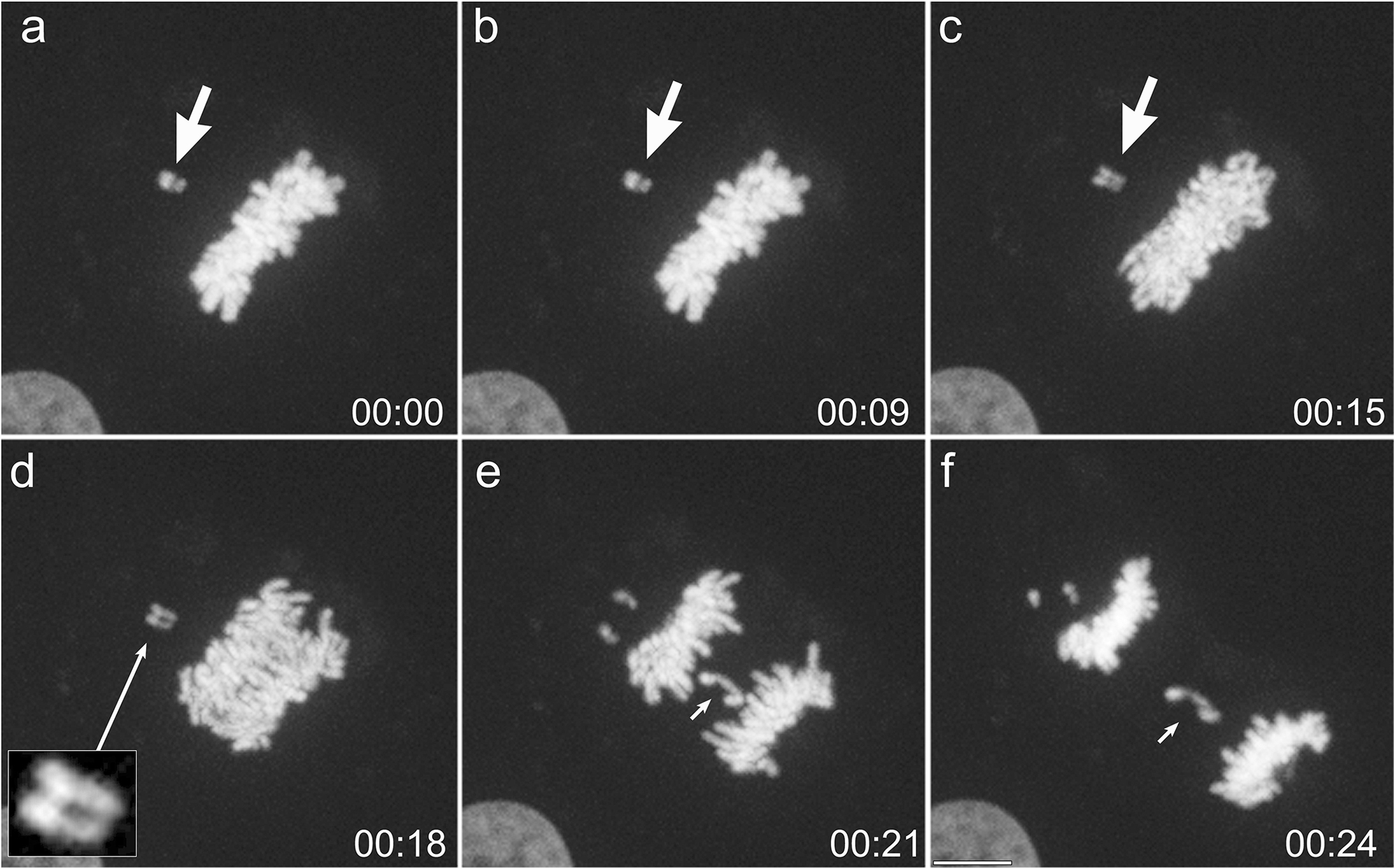
Frames from a time-lapse sequence of a mitotic BSC1 cell expressing H2B-GFP to visualize the mitotic chromosomes. the chamber was assembled then placed in the cold. After 20 min, the chamber is placed on the warm microscope, a metaphase cell with a misaligned chromosome (arrow) identified and the imaging sequence started (a). c. The cell begins to enter anaphase, and the sister chromatids are seen to begin to separate, including the misaligned chromosome (arrow). d. The separation of the misaligned pair of sister chromatids can be easily visualized (inset). e. As the sister chromatids segregate towards the spindle poles a pair of chromatids can been seen lagging in the central spindle region (small arrow). These lag as the cell begins to undergo cytokinesis (f). T = Hrs:Min.
Once the cell missegregates a chromosome, we have the option of switching objectives back to the 20× 0.8 NA lens, and the resulting daughter cells imaged by time-lapse over several days to determine whether or not they re-divide.
c. Same cell live- + fixed imaging:
With all of our live-cell imaging experiments, we have the option of fixing the coverslip after the period of observations, marking the position of our cell of interest with the diamond scribe mounted in the nosepiece of the microscope, and then conducting same-cell immunolabelling experiments on them. At any time during the live-cell imaging, we can exchange the objective lens for the diamond scribe, which screws into an objective slot in the microscope nosepiece, and is roughly parfocal with all objectives. The scribe is brought in close apposition to the coverslip, and then slightly further down: because the scribe mount is spring-loaded, it will retract slightly when in contact with the coverslip surface. This is done extremely slowly, and is best viewed straight on by eye. Once the coverslip causes the scribe to slightly depress, the scribed is turned seven times to generate a circular mark on the coverslip surface. The specimen is then lowered, the scribe exchanged for the 63× 1.3 NA objective, and refocused. This is done to check that the cell of interest remains in the center of the field of view. Once the cell is confirmed, the chamber is removed, and the coverslip eased off the chamber mount with a pair of tweezers, and quickly fixed by the method of choice. After fixation, the coverslip is immuno-labeled and mounted on a slide.
To find the cell of interest, we use a separate microscope, equipped with wide-field fluorescence and phase contrast optics. The slide is placed on the scope, and the circular scribe mark found using a 10× 0.4 NA phase 1 objective. The surface of the coverslip should be well-cleaned, and free of salt crystals, dust, etc. in order to detect the scribe mark. If dirty, it should be cleaned first with diH2O, then 76% EtOH and a piece of lens tissue. The cell of interest should be visible within the center of the circular mark. We then image the immune-labeled cell with a 63 1.4 NA oil-immersion objective and wide-field fluorescence.
d. Microinjection:
The following procedure is for microinjecting antibodies into mitotic cells (Day et al., 2018). It is similar to protocols we have used for the injection of in vitro translated mRNAs (Wheatley et al, 1997) or purified proteins (Hinchcliffe et al., 1999). For those conducting microinjection experiments using Rose chambers, see Gorbsky et al., (1998); Campbell and Gorbsky, (1995); Wadsworth (1999), and Stout et al., (2009).
Microinjection microscope:
In order to conduct microinjections, it is critical to use a fixed stage microscope, where focus is achieved by moving the microscope objective relative to the specimen, rather than moving the stage relative to a fixed objective nosepiece. This allows the needle tip to be positioned at the same focal plane as the cells, and any subsequent focus changes do not disturb this relationship. Fixed stage microscopes can either be inverted or upright; inverted scopes tend to have more room above the stage, allowing easier access for the manipulator/needle.
For our microinjection studies we use a Leica DM IRE2 inverted microscope equipped for phase contrast microscopy and a rotating stage, which allows for precise positioning of the needle tip relative to the cell in the X-Y plane. Illumination is via a halogen lamp with a 546 nm green filter (Hinchcliffe, 2005). The stage is heated with a commercial hair dryer (Vidal Sasson, Santa Monica, CA). The dryer is switched off prior to the injection procedure, as it a source of vibration for the scope and microneedle.
Our scope is equipped with a 10× phase objective to allow rapid scanning of the cell culture field to select suitable cells for manipulation/injection. This also allows us to detect the circle scribe mark on the coverslip that denotes a cell of choice when we are injecting a cell after prolonged observation (Hinchcliffe et al., 2016). For the actual manipulation/injection procedure we use a 40× phase objective plus a 1.5× optivar to provide a high-magnification view of the cell.
Manipulator and Injector:
We use a Burleigh Mis-5000 piezo-driven micromanipulator, which is no longer manufactured. However, there are several commercially available manipulators from ThorLabs (Newton, NJ USA), Sutter (Novato, CA USA), Scientifica (Clarksburg, NJ USA), Eppendorf (Hauppauge, NY USA) and others. We have found piezo-driven manipulators provide superior control for working with relatively small cells, such as cultured mammalian cells, compared to manual manipulators, which are excellent for bigger cells like echinoderm zygotes.
For microinjection, we use an Eppendorf Femtojet (Eppendorf North America, Hauppauge, NY USA). The pulse duration and pressure settings are determined empirically for each cell type (described below in the microinjection method section). Our manipulator/injector and microscope are supported on a TMC vibration isolation table (TMC, Peabody, MA USA).
Microinjection Needles:
For microinjection, we start with Borosil capillary tubing (Frederick Haer Co, Bowdoinham, ME USA) using a 1.2mm OD × 0.75mm ID × 100mm length microcap tubes, with omega dot inner fiber for rapid filling (cat. #30–31-0). The inner fiber is essential, to allow the injectate liquid to move to the needle tip.
Microinjection needles are drawn using a Kopf vertical needle puller (Model 720, David Kopf Instruments, Tujunga, CA USA). The filament and solenoid settings are determined empirically for each needle type and experiment. The ideal needle shape incorporates a long, gradual taper, rather than an abrupt end with a slender tip.
Injection protocol:
Prior to microinjection, it is critical that the user be familiar with the use of the micro-injector and focus of the microscope. Practice which way the knob turns the focus up and down, and how the manipulator controls move up and down and side-to-side by looking through the eyepieces. It is recommended that unloaded needles and mock chambers be used, until the user can manipulate the system with minimal delay.
Dialyze the antibody solution into the appropriate microinjection buffer (10mM Na2PO4, pH 7.4, 100mM KCl, 1mM MgCl2), and concentrate with a centrifugation filter (Amicon Ultra filter with a 50 kDa NMWL - EMD/Millipore). Estimate the final protein concentration by spectrophotometry (A280). 20 μl aliquots are flash frozen and stored at −80 °C.
Pre-heat the stage of the injection microscope, turn on the FemtoJet, and place the Eppendorf tool in the arm of the manipulator.
A fresh injection needle is placed in the Eppendorf tip holder (with O-ring), and stored vertical by pressing it into a clump of modeling clay affixed to the side of a lab shelf (Fig. 6). This prevents the tip from being damaged and allows the injectate to flow into the fine needle tip.
For injection the concentrated antibody is thawed on ice and pre-cleared by centrifugation for 15 min at 14,000g in an Eppendorf tabletop centrifuge.
The pre-cleared antibody solution is drawn from the tube with a Hamilton syringe and appropriate gauge needle and back-loaded into the microinjection needle. The injectate will flow almost to the tip, but a tiny air bubble (just visible to the naked eye) will remain in the tip. This is removed by slowly and gently rolling the needle (in holder) between the thumb and forefinger. Gentle flicking and tapping of the needle helps to dislodge this bubble. If the needle tip is held up to the light, the bubble can be observed as it floats up the needle shaft to the surface of the injectate. Failure to remove this bubble will result in a blocked needle. The needle is returned to the modeling clay mount at room temperature.
Loaded needles should be used within 1 hr. Otherwise a new aliquot of antibody should be thawed, cleared and loaded into a fresh needle.
Cells cultured on biocleaned coverslips (detailed in Sluder et al., 2007) with a wax pencil line and a 3-sided perimeter of silicon grease (Dow Corning high vacuum grease, Midland, MI) are assembled onto the HEKS metal support slide. A matching coverslip with wax line is assembled onto the opposing surface using silicon grease applied with a fine-tip metal spatula. The reservoir (up to the wax line) is filled with fresh media using a syringe and fine gauge needle. The opening between the coverslips is then capped with mineral oil (added with a syringe and fine gauge needle). This prevents the media from drying out.
The blow dryer is turned off, and the loaded HEKS chamber is placed onto the stage. Using a 10× phase objective, focus on the wax line.
Assemble the needle onto the Eppendorf tool, attach the FemtoJet tube onto the pump (this will be at pressure. If not, wait for the pump to come to pressure before attaching the tubing).
Scan the coverslip for a suitable cell, selecting on near the front end of the chamber, near the wax line. If the cell to be injected was previously identified on the live-cell imaging scope, it will be marked by a circular scribe mark, which is readily visible by phase contrast.
Once the cell is identified, use the stage controls to move the chamber away from the needle in a straight line (so the cell can easily be re-acquired).
Bring the needle into view using the course controls and visualized by eye (not through the eye pieces). As the needle tip enters the light path, it will be visible. Once in view, switch to the eye pieces, and bring the tip into focus using the manipulator. At this point DO NOT CHANGE THE FOCUS of the microscope – it is set to the level of the cells. Make all adjustments using the manipulator to change the level of the microneedle.
Once the tip is in focus, slowly bring the chamber back into view, so the needle tip and the edge of the coverslips are visible but not touching.
Using the FINE controls (joystick), gently touch the tip of the needle to the coverslip edge. This will break the sealed tip, and allow injectate to flow. If this is done too forcefully, the tip diameter will become too large, and the cell cannot be injected (think stovepipe, rather than needle). In that case, start over.
Retract the needle, and using the fine Z-direction (top pot of the joystick) raise the needle a few turns, so it is above the plane of the coverslip. This allows the needle to be inserted into the chamber without dragging through the cells. It is still visible as an out-of-focus blurred line.
Use the stage controls to move the chamber until the out-of-focus needle is near (but not touching) the cell of interest.
Using the fine (joystick) controls, lower the needle until it is almost in focus and close to the cell. De-focus the microscope using the course adjustments, and swing in the higher magnification objective (change phase rings as needed). Re-focus to bring the cell and needle tip in focus.
Swing in the Optivar lens (1.5×) to provide narrow field of view. Readjust focus and illumination as needed.
Use the joystick to move needle tip to the cell, and gently puncture the membrane. Pause, then tap the foot pedal to deliver the injectate. Watch the cell as it is injected. It will swell briefly then return to its normal morphology. Withdraw the needle gently using the fine controls and raise it out of focus (so it does not injure the cells).
If the appearance of the cell in phase contrast dramatically changes (becomes darker), it is likely damaged or dead. Either choose a nearby cell to inject from the same needle, or start over with a fresh needle.
If the injection is successful, withdraw the needle from the chamber using the course controls.
De-focus and bring the low mag objective into place. Re-focus and confirm position of the injected cell. To mark its position, de-focus, and bring in the diamond scribe. By eye, bring the diamond up to the coverslip. Once the tip of the diamond touches the coverslip, the oil cap will become slightly deformed – this is easily observed by eye, and is a cue to twirl the tip, making the circular mark. Re-check with the 10× phase objective and confirm the scribe mark.
Transfer the chamber to the imaging microscope and precede with the experiment. The major complication with this microsurgery procedure is a clogged needle tip. Though microinjection pumps have a high pressure setting to purge a clogged needle, we have found that these rarely work. The only remedy is to begin again. The major cause of clogged needles is failure to pre-clear the injectate before loading the needle, or leaving it in the needle for longer than 1 hr prior to injection. It the injectate repeatedly clogs the needle, it can be diluted in injection buffer. Otherwise, begin again with a fresh sample.
Fig. 6. Microinjection setup.
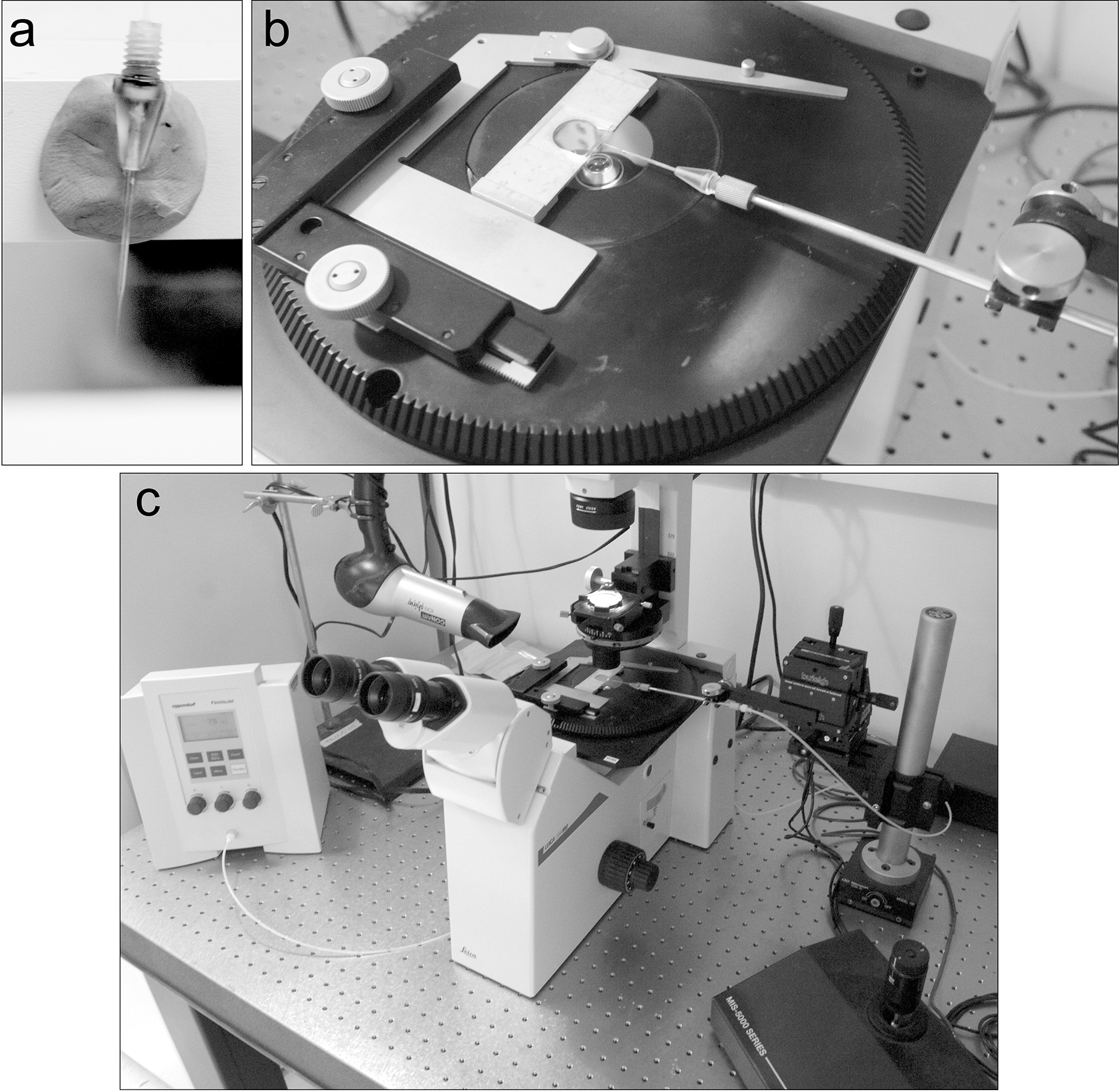
a. The needle holder is placed into a small piece of modeling clay that is affixed to a shelf above the lab bench to ensure it remains upright and the tip is not damaged. b. The assembled chamber on the injection microscope, with the needle assembled onto the injector and positioned in the chamber opening. c. Wide view of the injection microscope showing the injector pump, Vidal Sassoon heat gun, manipulator and joystick.
Acknowledgments
CA Day is supported by NIH T32 CA21783602. Neuro-Oncology Training Grant. Work in the author’s lab is supported by grants from NIH (HL125353) and the DoD CDMRP (CA171071 and CA130436), and support from the I.J. Holton Trust and the Hormel Foundation.
References
- Alessandri K, Andrique L, Feyeux M, Bikfalvi A, Nassoy P, and Recher G (2017). All-in-one 3D printed microscopy chamber for multidimensional imaging, the UniverSlide. Scientific Reports 7:42378. DOI: 10.1038/srep42378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begg DA and Ellis GW (1979). Micromanipulation studies of chromosome movement. I. Chromosome-spindle attachment and the mechanical properties of chromosomal spindle fibers. J Cell Biol. 82:528–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MS and Gorbsky GJ (1995). Microinjection of mitotic cells with the 3F3/2 anti-phosphoepitope antibody delays the onset of anaphase. J Cell Biol. 129:1195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BC, Legant WR, Wang K, Shao L, Milkie DE, Davidson MW, Janetopoulos C, Wu XS, Hammer JA 3rd, Liu Z, English BP, Mimori-Kiyosue Y, Romero DP, Ritter AT, Lippincott-Schwartz J, Fritz-Laylin L, Mullins RD, Mitchell DM, Bembenek JN, Reymann AC, Böhme R, Grill SW, Wang JT, Seydoux G, Tulu US, Kiehart DP, and Betzig E (2014). Lattice light-sheet microscopy: imaging molecules to embryos at high spatio-temporal resolution. Science. 346:439–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Nicolantonio F, Arena S, Gallicchio M, Zecchin D, Martini M, Flonta S, Stella G, Lamba S, Cancelliere C, Russo M, Geuna M, Appendino G, Fantozzi R, Medico E, and Bardelli A (2009). Replacement of normal with mutant alleles in the genome of normal human cells unveils mutation-specific drug responses. PNAS 105:20864–20869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douthwright S and Sluder G (2017). Live Cell Imaging: Assessing the Phototoxicity of 488 and 546 nm Light and Methods to Alleviate it. J Cell Physiol. 232:2461–2468. [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, and Tuschl T (2001). Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411:494–498. [DOI] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, and Mello CC (1998). Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391:806–811. [DOI] [PubMed] [Google Scholar]
- Gorbsky GJ, Chen RH, and Murray AW (1998). Microinjection of antibody to Mad2 protein into mammalian cells in mitosis induces premature anaphase. J Cell Biol. 141:1193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinchcliffe EH, Cassels GO, Rieder CL, and Sluder G, (1998). The coordination of centrosome reproduction with nuclear events during the cell cycle in the sea urchin zygote. J Cell Biol 140:1417–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinchcliffe EH, Thompson EA, Miller FJ, Yang J, and Sluder G (1999). Nucleo-cytoplasmic interactions in control of nuclear envelope breakdown and entry into mitosis in the sea urchin zygote. J Cell Sci 112:737–746. [DOI] [PubMed] [Google Scholar]
- Hinchcliffe EH, Miller FJ, Cham M, Khodjakov A, and Sluder G. (2001). Requirement of a centrosomal activity for cell cycle progression through G1 into S phase. Science 291:1547–1550. [DOI] [PubMed] [Google Scholar]
- Hinchcliffe EH (2005). Using long-term time-lapse imaging of mammalian cell cycle progression for laboratory instruction and analysis. Cell Biol Edu 4:284–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinchcliffe EH, Day CA, Karanjeet K, Fadness S, Langfald A, Vaughan KT, and Dong Z (2016). Chromosome missegregation during anaphase triggers p53 cell cycle arrest through histone H3.3 Ser31 phosphorylation. Nat Cell Biol 18:668–675. [DOI] [PubMed] [Google Scholar]
- Hiramoto Y (1962). Microinjection of the live spermatozoa into sea urchin eggs. Exp Cell Res. 27:416–26. [DOI] [PubMed] [Google Scholar]
- Hornick JE, Mader CC, Tribble EK, Bagne CC, Vaughan KT, Shaw SL, and Hinchcliffe EH (2011). Amphiastral mitotic spindle assembly in vertebrate cells lacking centrosomes. Curr. Biol. 21:598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue S (1953). Polarization optical studies of the mitotic spindle. I. The demonstration of spindle fibers in living cells. Chromosoma. 5:487–500. [DOI] [PubMed] [Google Scholar]
- Kiehart DP (1982). Microinjection of echinoderm eggs: apparatus and procedures. Methods Cell Biol 25 Pt B:13–31 [DOI] [PubMed] [Google Scholar]
- Komor AC, Badran AH, and Liu DR (2015). CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. 168:20–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Wu Y, Christensen R, P Chandris, Gandler W, McCreedy E, Bokinsky A, Colón-Ramos DA, Bao Z, McAuliffe M, Rondeau G, and Shroff H (2014). Dual-view plane illumination microscopy for rapid and spatially isotropic imaging. Nat Protoc. 9:2555–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerman MJ, Lembong J, Gillen G, and Fisher JP (2018). 3D printing in cell culture systems and medical applications. Applied Physics Reviews 5, 041109 10.1063/1.5046087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magidson V and Khodjakov A (2013). Circumventing photodamage in live-cell microscopy. Methods Cell Biol. 114:545–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazia D, and Dan K (1952). The Isolation and Biochemical Characterization of the Mitotic Apparatus of Dividing Cells. Proc Natl Acad Sci U S A. 38:826–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsume T, Kiyomitsu T, Saga Y and Kanemaki MT (2016). Rapid Protein Depletion in Human Cells by Auxin-Inducible Degron Tagging with Short Homology Donors. Cell Reports 15:210–218. [DOI] [PubMed] [Google Scholar]
- Recillas-Targa F (2004). Gene transfer and expression in mammalian cell lines and transgenic animals. Methods Mol Biol. 267:417–33. [DOI] [PubMed] [Google Scholar]
- Rieder CL and Cole RW (1998). Perfusion chambers for high-resolution video light microscopic studies of vertebrate cell monolayers: some considerations and a design. Methods Cell Biol. 56:253–75. [DOI] [PubMed] [Google Scholar]
- Rose GG, Pomerat CM, Shindler TO, and Trunnell JB (1958). A cellophane-strip technique for culturing tissue in multipurpose culture chambers. J Biophys Biochem Cytol. 4:761–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout JR, Rizk RS, and Walczak CE (2009). Protein inhibition by microinjection and RNA-mediated interference in tissue culture cells: complementary approaches to study protein function. Methods Mol Biol. 518:77–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluder G and Begg DA. (1983). Control mechanisms of the cell cycle: role of the spatial arrangement of spindle components in the timing of mitotic events. J Cell Biol. 97:877–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluder G, Miller FJ, and Hinchcliffe EH (1999). Using sea urchin gametes for the study of mitosis. Methods in Cell Biology 61:439–472. [DOI] [PubMed] [Google Scholar]
- Sluder G, Nordberg J, Miller F, and Hinchcliffe EH (2007). A sealed preparation for long–term observation of cultured cells. CSH Protoc :pdb.prot4660. doi: 10.1101/pdb.prot4660. [DOI] [PubMed] [Google Scholar]
- Taylor EW (1959). Dynamics of spindle formation and its inhibition by chemicals. J. Biophysic. Biochem. Cytol. 6:193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetake Y and Sluder G (2010). Prolonged prometaphase blocks daughter cell proliferation despite normal completion of mitosis. Curr Biol. 20:1666–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetake Y and Sluder G (2007). Cell-cycle progression without an intact microtubule cytoskeleton. Curr Biol. 17:2081–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetake Y and Sluder G (2018). Activation of the apoptotic pathway during prolonged prometaphase blocks daughter cell proliferation. Mol Biol Cell. 29:2632–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadsworth P (1999). Microinjection of mitotic cells. Methods Cell Biol. 61:219–31 [DOI] [PubMed] [Google Scholar]
- Wheatley SP, Hinchcliffe EH, Glotzer M, Hyman AA, Sluder G, and Wang YL (1997). Cdk1 inactivation regulates anaphase spindle dynamics and cytokinesis in vivo. Journal of Cell Biology 138:385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Wawrzusin P, Senseney J, Fischer RS, Christensen R, Santella A, York AG, Winter PW, Waterman CM, Bao Z, Colón-Ramos DA, McAuliffe M, and Shroff H (2013). Spatially isotropic four-dimensional imaging with dual-view plane illumination microscopy. Nat Biotechnol. 31:1032–8. [DOI] [PMC free article] [PubMed] [Google Scholar]