

Abstract
The Minisci reaction, which encompasses the radical C–H alkylation of heteroarenes, has undergone revolutionary development in recent years. The application of photoredox catalysis for alkyl radical generation has given rise to a multitude of methods that feature enhanced functional group tolerance, generality, and operational simplicity. The intent of this short review is to bring readers up to date on this rapidly expanding field. Specifically, we will highlight key examples of visible light-driven Minisci alkylation strategies that represent key advancements in this area of research. The scope and limitation of these transformations will be discussed, with a focus on examining the underlying pathways for alkyl radical generation. Our goal is to make this short review a stepping stone for further synthetic research development.
Keywords: Minisci reaction, photoredox catalysis, visible light, radical alkylation, late-stage functionalization, heteroarenes
Graphical Abstract
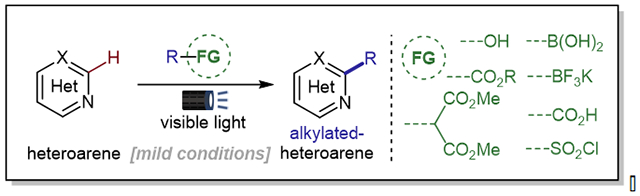
1. Introduction
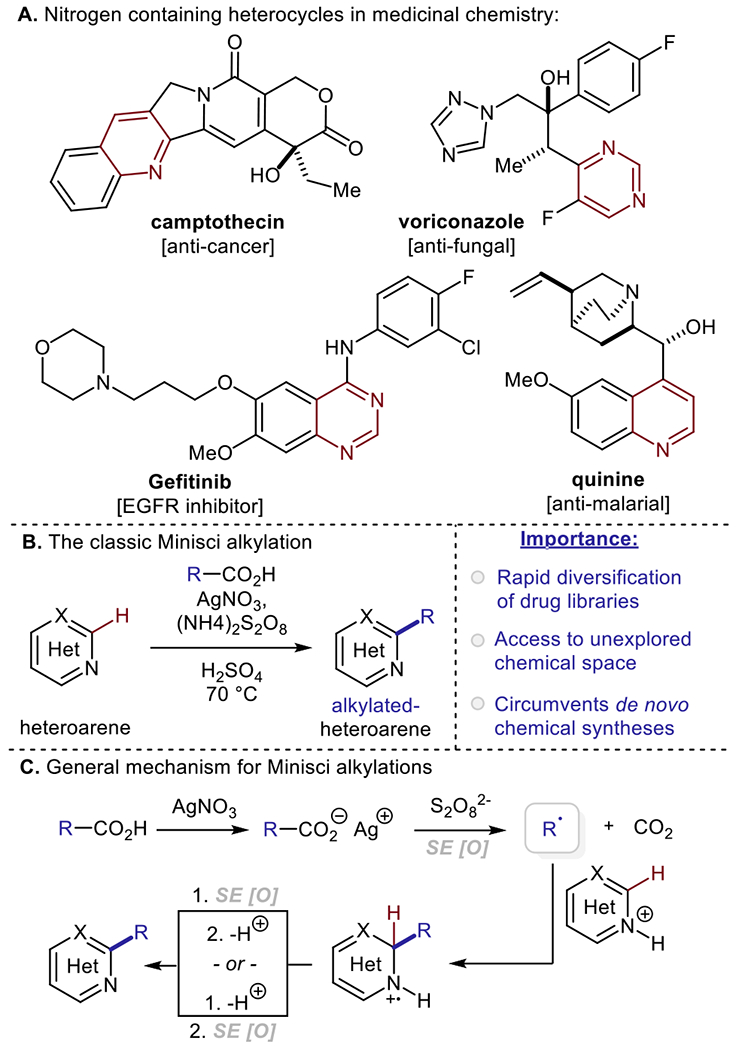
Nitrogen-containing heterocycles constitute the backbone of natural products, medicinally valuable small molecules and agrochemicals (Scheme 1A).1–2 Methodologies for the direct C–H alkylation and perfluoroalkylation of N-heteroarenes enable both the late-stage modification of clinical leads and rapid diversification of drug-like libraries.3–4 These strategies allow for expedient access to unexplored chemical space and circumvent conventional de novo chemical syntheses.5 Notably, the medicinal chemistry community has placed growing interest on late-stage functionalization technologies, as they allow for rapid modulation of drug metabolism and pharmacokinetic profiles of lead compounds.3–5 Thus, synthetic approaches which are not dependent on strong oxidants/reductants, high reaction temperatures, or pre-functionalized substrates are of high-value to both academic and industrial sectors.
Scheme 1.
The Minisci alkylation of N-heteroarenes
The addition of open-shell alkyl and perfluoroalkyl radical intermediates to heteroarenes is referred to as the Minisci reaction (Scheme 1B).6–9 Minisci’s original protocol relied on free radical formation from carboxylic acids via formation of their corresponding silver salts, followed by oxidative decarboxylation upon treatment with a persulfate oxidizing agent. Addition of an alkyl radical intermediate onto a protonated heteroarene, followed by rearomatization, yields the desired alkylated heterocyclic product (Scheme 1C). Based on Studer and Curran’s mechanistic studies, rearomatization is proposed to occur via deprotonation and sequential single electron oxidation of the functionalized heteroarene upon radical addition.10 Since Minisci’s seminal contributions, this reactive paradigm for the alkylation of (hetero)arenes has been a stalwart foundation for modern drug discovery and development.11 Furthermore, renewed interest in the mild and operationally simple generation of radical intermediates has spurred rapid evolution in the area of (hetero)arene alkylation.12–14 In part, the driving inertia for this interest has been the emergence of visible light-mediated photoredox catalysis, which facilitates exceptionally mild single electron transfer (SET) events with organic substrates.15–17 Importantly, the pharmaceutical industry has recognized the transformative impact of photoredox catalysis,18–19 as it has far reaching implications in harnessing sustainable energy sources, reducing waste streams, and avoiding hazardous and/or toxic reagents classically employed for carbon-centered radical formation (e.g. Bu3SnH, BEt3/O2). Given that state-of-the-art visible light-based methods are already employed in drug development (e.g. elbasvir20 and artemisinin21) drug discovery efforts, we anticipate that the photoredox radical (perfluoro)alkylation of (hetero)arenes will be an invaluable synthetic technology for years to come.
In light of the importance of such transformations, we have decided to summarize recently reported methods for visible light-enabled radical C(sp2)–H alkylations and perfluoroalkylations of medicinally relevant (hetero)arenes. The sections to follow are organized based on radical precursor reagents. When appropriate, the discussions will aim to highlight the unique selectivity outcomes dictated by the electronic properties of alkyl and perfluoroalkyl radicals. This Short Review is not intended to be comprehensive and is aimed at emphasizing novel photoredox catalysis technologies for the radical alkylation of (hetero)arenes, which we anticipate will have an enduring impact in academic and industrial settings.
2. Carboxylic Acids and Carboxylic Acid Derivatives
Alkyl carboxylic acids are versatile feedstock chemicals that are ubiquitous throughout nature and have been widely used as chemical building blocks.22–23 Owing to their low cost, stability, minimal toxicity, and commercial availability, alkyl carboxylic acids have been widely utilized across a variety of synthetic transformations and represent preeminent building blocks for combinatorial chemistry (e.g. amide bond formation). In recent years, the radical decarboxylation of aliphatic carboxylic acids and their activated derivatives has emerged as a powerful strategy for the Minisci functionalization of bioactive organic molecules.
A broad selection of methods have been developed to promote the decarboxylation of alkyl carboxylic acid derivatives through a reductive pathway. In the context of photoredox catalysis, the formation of alkyl radicals via a reductive pathway would enable a net redox neutral catalytic cycle, thereby eliminating the need for a terminal oxidant. At the same time, a reductive alkylation strategy has the potential to expand upon the scope of alkylation reagents, allowing access to compounds with significantly higher oxidation potentials.24 Pioneering studies on the reductive decarboxylative generation of alkyl radicals were conducted by Barton and co-workers in the 1960s.25–26 Barton et al. utilized N-hydroxypryidine-2-thione in the reductive activation of carboxylic acids for applications such as carbonyl reduction and reductive halogenation. In 1991, Oda and Okada disclosed the use of N-(acyloxy)phthalimides (NAP) as redox auxiliaries to enable the decarboxylative generation of alkyl radicals upon single electron reductive fragmentation (E1/2 = −1.26 to −1.39 V vs. SCE (saturated calomel electrode)), using visible light-mediated photoredox catalysis.27
Since 2017, NAP esters have been employed in several visible light-driven Minisci alkylation protocols to promote reductive alkyl radical generation.28–32 Notably, Phipps and co-workers have reported an enantioselective variant of the Minisci reaction (Scheme 2) which utilizes a combination of asymmetric Brønsted acid catalysis and photoredox catalysis.30 The use of a chiral phosphoric acid catalyst provides both stereo- and regiocontrol in the direct addition of prochiral α-amino alkyl radicals to the 2-position of a variety of pyridine and quinoline-based substrates. This strategy elegantly facilitates the synthesis of enantioenriched α-heterocyclic amines through an efficient late-stage functionalization approach. Jiang and co-workers have also designed an alternative, organocatalytic approach for constructing α-isoquinoline-substituted secondary amines in an enantioselective manner.31 Nonetheless, the use of NAP esters for photoredox Minisci alkylations typically necessitates a separate isolation step following ester formation, resulting in an overall two-step procedure. In 2018, Sherwood and co-workers at Bristol-Meyers Squibb developed an operationally simple, one-pot protocol for the in situ generation of NAP esters, which obviates the need for isolating the pre-functionalized alkyl partner and facilitates the rapid generation of analog libraries.32
Scheme 2.
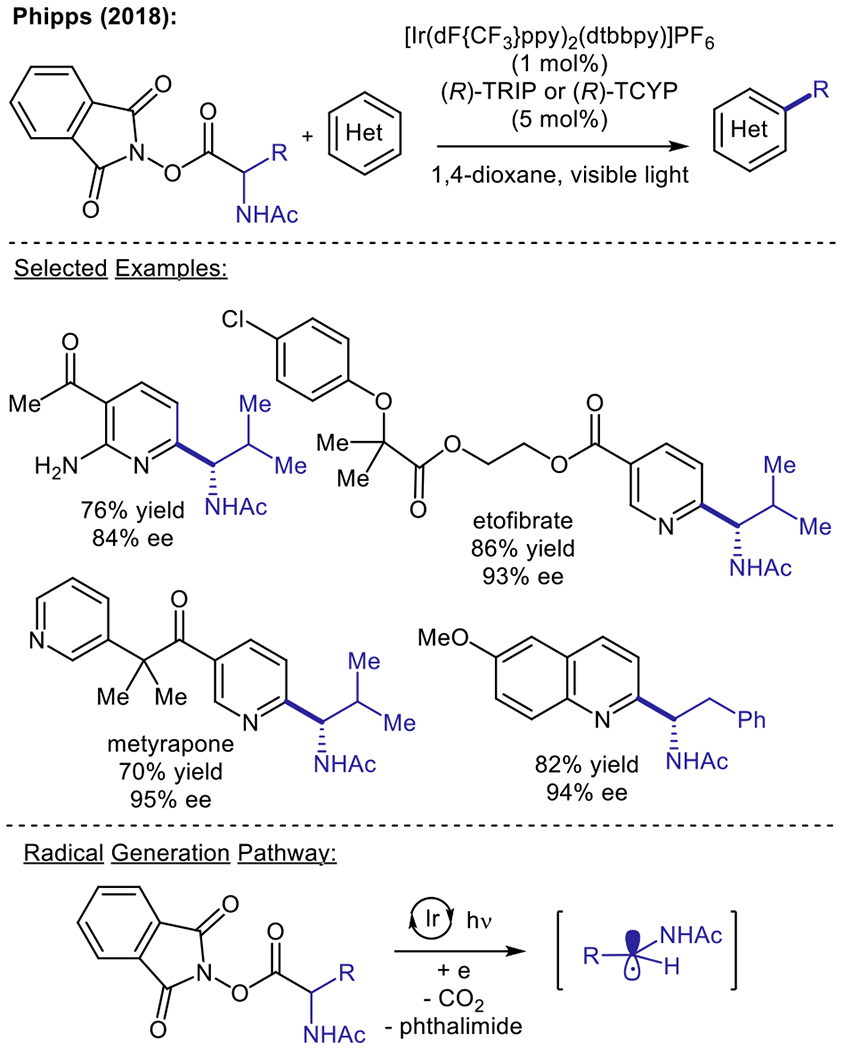
Enantioselective synthesis of α-heterocyclic amines using a Brønsted acid/photoredox catalytic platform
In 2015, the Stephenson group developed a novel strategy for the visible light-driven trifluoromethylation of electron-rich (hetero)arenes (Scheme 3), by using pyridine N-oxide to induce reductive radical generation from trifluoroacetic anhydride (TFAA).24 With respect to considerations including safety, material availability, and reagent price (TFAA $35 per kg at 1,000 kg), trifluoroacetic acid (TFA) and its derivatives represent highly attractive sources of CF3. Given the prohibitively high oxidation potential of the TFA anion (F3CCO2Na Ep/2ox > +2.4 V vs. SCE), the authors were able to promote a mild reductive decarboxylation of a TFAA/pyridine N-oxide adduct (Ep/2red = −1.10 V vs. SCE) to access the trifluoromethyl radical within the electrochemical window of [Ru(bpy)3]Cl2. Notably, following reductive cleavage of the weak N–O bond and CO2 extrusion, the generation of pyridine as a byproduct resolves the need for an exogenous base. Furthermore, TFAA and pyridine N-oxide are used in equal stoichiometry with respect to the substrate, and this reagent combination is sufficiently inexpensive for large-scale operations (pyridine N-oxide $40-$70 per kg at 1,000 kg). The authors have demonstrated the efficacy of this design in the C–H trifluoromethylation of a number of electron-rich heterocyclic and aromatic substrates, including medicinally important MIDA boronates. Through a collaboration with Eli Lilly, the trifluoromethylation of a tert-butoxycarbonyl (Boc)-protected pyrrole substrate was carried out on 1.2 kilogram scale in a continuous flow system, which produced the trifluoromethylated product in 50% yield at production rates of 87.2 mmol per hour (approx. 0.5 kg per day).33
Scheme 3.
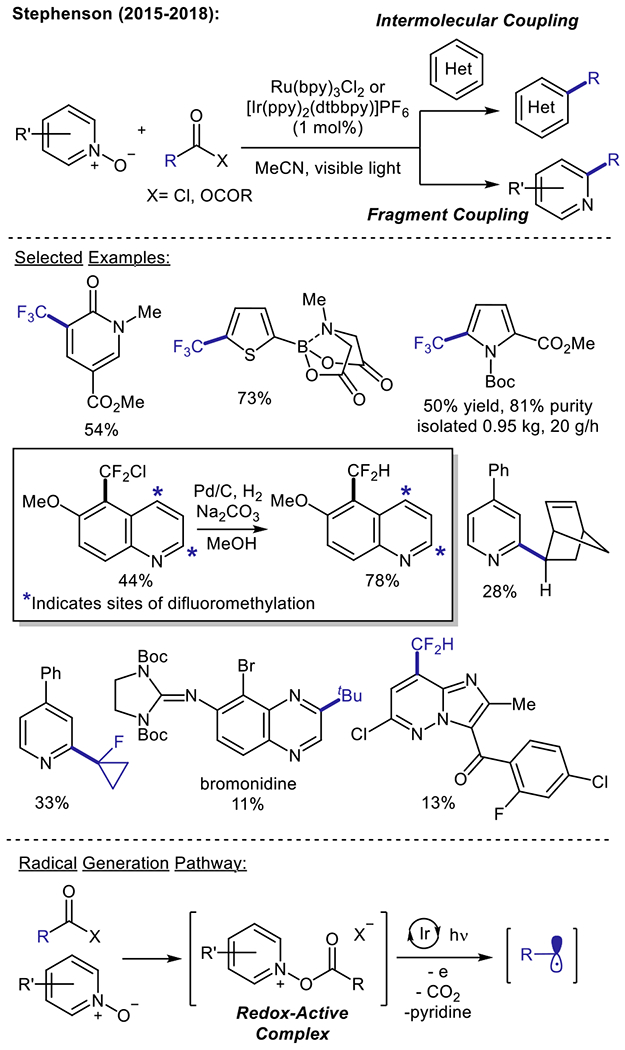
Reductive decarboxylative (perfluoro)alkylation of heteroarenes using pyridine N-oxides
Stephenson and co-workers have further expanded upon this methodology to achieve the radical perfluoroalkylation of a variety of electron-rich (hetero)arene substrates.33 In particular, they have designed a radical chlorodifluoromethylation strategy that provides a valuable synthetic entryway to accessing electron-rich difluoromethylated (hetero)arenes.34 Moreover, chlorodifluoromethylation, followed by hydrogenolysis, of 6-methoxyquinoline was demonstrated to furnish the 7-difluoromethylated product. This electronically mis-matched product is otherwise inaccessible via the nucleophilic difluoromethyl radical, which is selective for the electrophilic 2- and 4- positions of the quinoline core. An orthogonal, fragment coupling approach (Scheme 3) has recently been developed by Stephenson et al. for the addition of electron-rich (fluoro)alkyl radicals onto electron-deficient heteroarenes.35 Notably, this fragment coupling manifold minimizes chemical waste production, since the dual role of the heterocyclic N-oxide as both a redox auxiliary and a coupling partner avoids the use of stoichiometric additives. This methodology enables access to a wide range of alkyl coupling partners, including medicinally-relevant motifs such as tertiary azetidines, fluorinated cyclopropyl groups, and a norbornene bicyclic scaffold. A variety of pharmaceutically-important heterocyclic N-oxides derived from pyridine, quinoline, and azaindole cores were reported to undergo successful alkylation in modest to good yields. By using pyridine N-oxide as a sacrificial redox auxiliary, the authors were able to further expand upon their scope of heteroarene substrates to access electron-deficient heteroarenes, such as quinoxaline, as well as more complex pharmaceutical scaffolds.
With the goal of designing a Minisci alkylation strategy for the late-stage functionalization of advanced pharmaceutical intermediates, DiRocco and co-workers at Merck disclosed the innovative use of stable organic peroxides as alkylating reagents under photoredox conditions (Scheme 4).36 Reaction parameters were optimized using a high-throughput experimentation platform, and the use of cyclometallated Ir(III)+ photocatalysts [Ir(dF{CF3)ppy}2(dtbbpy)]PF6 and [Ir(ppy)2(dtbbpy)]PF6 provided access to methyl, ethyl and cyclopropyl radical intermediates from bench-stable and inexpensive alkyl peracetates. The methodology was shown to be amenable to the late-stage alkylation of an array of complex medicinal and agrochemical agents bearing both 6- and 5-membered heterocyclic scaffolds. Most importantly, the transformation proceeded smoothly in the presence of functionalities such as basic amines, alcohols, amides, and esters, without the need for protecting groups. With respect to methyl radical generation, the authors propose a mechanistic pathway involving the activation of tert-butylhydroperoxide through a reductive proton-coupled electron transfer (PCET) process. The resulting α-peroxy radical subsequently undergoes homolytic O–O bond cleavage to afford acetic acid and a tert-butoxy radical species. The authors hypothesize that methyl radical formation arises from β-scission of the tert-butoxy radical, thereby producing acetone as a byproduct.
Scheme 4.
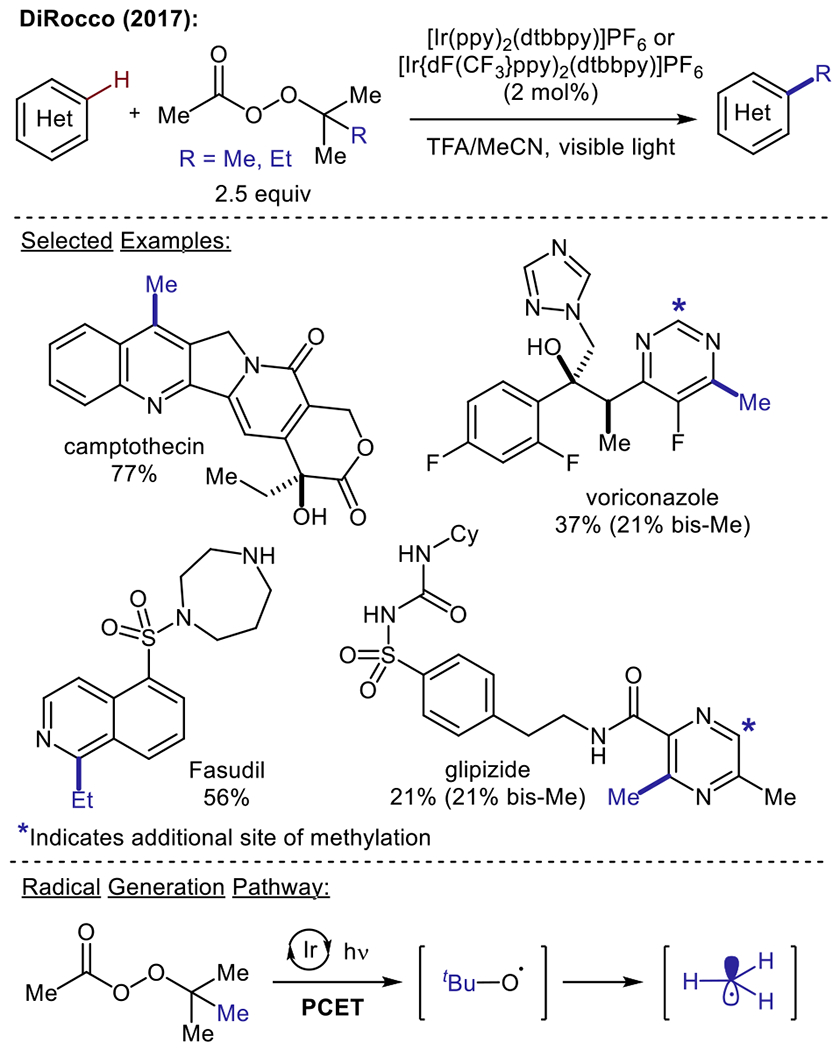
Late-stage functionalization of biologically active heterocycles using alkyl peracetates
In 2014, MacMillan et al. reported the first use of photoredox catalysis for the oxidative decarboxylation of alkyl carboxylic acids in the arylation of α-amino acids.37 In 2017, Glorius and co-workers disclosed a Minisci alkylation strategy that enables access to alkyl radical intermediates through the oxidative decarboxylation of carboxylic acids.38 Sodium persulfate is used as an external oxidant to mediate alkyl radical formation, as well as facilitate photocatalyst turnover. The authors propose that the generation of desired alkyl radicals occurs through a hydrogen-atom transfer (HAT) event between a reduced sulfate radical anion species and a carboxylic acid precursor, resulting in oxidative decarboxylation. This reaction manifold enables the expedient functionalization of heterocylic scaffolds, including pyridine, quinoline, and quinazoline cores. A range of primary, secondary, and tertiary alkyl radicals could be accessed from their corresponding alkyl carboxylic acid and amino acid precursors. The following year, Genovino and Frenette disclosed a separate visible light-driven Minisci alkylation protocol using hypervalent iodine reagents and organophotocatalysis to facilitate alkyl radical generation from carboxylic acids.39
3. Alkyl Boronic Acids
Over the past decade, aryl/alkyl-boron reagents have been identified to serve as radical precursors for C−C bond forming processes via oxidative C−B bond cleavage.40–47 In 2016, Chen and coworkers disclosed the Minisci C–H alkylation of N-heteroarenes with primary and secondary alkyl boronic acids using the photocatalyst Ru(bpy)3Cl2 and acetoxybenziodoxole as a sacrificial oxidant (Scheme 5). Diversely substituted primary and secondary boronic acids (e.g., alkyl bromide, aryl iodide, ester, amide, carbamate, terminal alkyne, and benzyl chloride) were well tolerated. Pyridines, pyrimidines, and a purine riboside substrate were all efficiently functionalized. It should be noted that more electron-rich heteroarenes, including benzothiazole and benzoimidazole, could also be successfully alkylated. The authors propose that the reaction is initiated by a single-electron reduction from the photoexcited Ru(II)* to acetoxybenziodoxole, providing an oxygen-centered radical intermediate. This radical species is then proposed to react with the alkyl boronic acid reagent to form the desired alkyl radical via a radical “ate” transition state. DFT calculations support that this is a facile and highly exothermic process at room temperature.
Scheme 5.
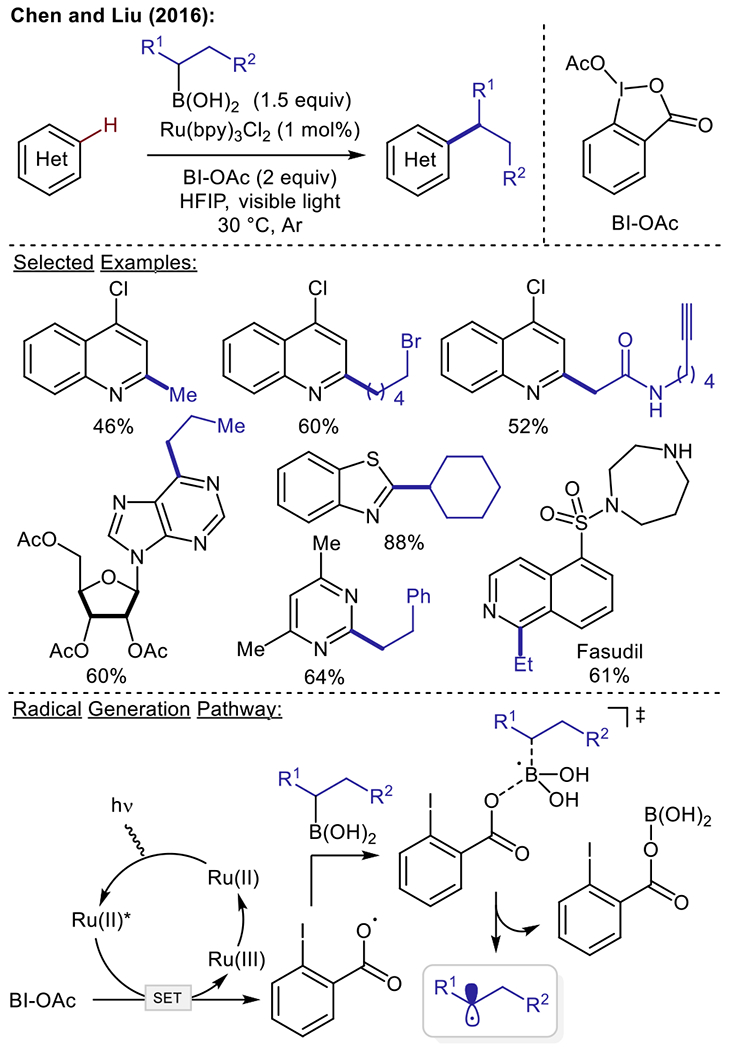
Photoredox Minisci alkylation using boronic acid alkylating reagents
4. Potassium Alkyl- and Alkoxymethyltrifluoroborates
Potassium organotrifluoroborates are considerably more attractive radical precursors than their corresponding boronic acids, given their lack of an empty p-orbital, which increases their overall stability and robustness toward harsh reaction conditions.48 In 2011, Molander and coworkers reported the first use of potassium alkyl- and alkoxymethyltrifluoroborates as radical precursors in the direct C−H alkylation of (hetero)arenes employing manganese(III) acetate as an oxidant in the presence of trifluoroacetic acid.50–51 Under the optimized reaction conditions, the authors were able to functionalize several nitrogen-containing heterocycles all in good to excellent yields.
In 2017, the Molander group reported an impressive advance from their earlier manganese(III) acetate-mediated Minisci chemistry by showcasing that alkyltrifluoroborates (many of which are commercially available) can be activated by an inexpensive, sustainable organophotocatalyst (Scheme 6).52 Following reaction optimization, the authors found the utility of a mesityl acridinium photocatalyst, potassium persulfate (as a sacrificial oxidant), and trifluoroacetic acid to be the optimal reagent combination for the C–H functionalization of heteroarenes. Under the title reaction conditions, medicinally important cores including quinolines, isoquinolines, indazoles, pyridines, and quinazolinones, could all be functionalized with an impressive scope of primary, secondary, and tertiary alkyltrifluoroborates in good to excellent yields. As expected, electron-rich cores such as benzimidazole, were unreactive toward these Minisci alkylation conditions. These conditions proved tolerant of a diverse array of functional groups including aryl halides, unprotected amines, thioethers, and amides. Notably, quinine, which features a free alcohol, terminal alkene, and a tertiary amine (which has a known propensity for competitive photocatalytic oxidation) was efficiently (54% yield) and selectively (C2-) functionalized. To showcase the late-stage functionalization utility of their developed protocol, the authors successfully functionalized camptothecin, an anti-cancer drug candidate, at the C7-position. Mechanistically, the authors propose single electron oxidation of the alkyltrifluoroborate reagent, which leads to generation of the desired alkyl radical intermediate and BF3.
Scheme 6.

Organophotocatalytic Minisci alkylation using alkyltrifluoroborate radical precursors
5. Alkyl Halides
Alkyl halides are among the most widely used materials in organic chemistry. However, their application as radical precursors has been hindered because of the harsh conditions required for radical generation, such as the use of highly toxic trialkyltin hydrides.53 The advent of modern photoredox catalysis provided a solution to this problem, as photoredox catalysts can be readily employed for the reductive dehalogenation of alkyl halides, resulting in the formation of free alkyl radicals. In 2010, the Stephenson group reported the seminal application of photoredox catalysis for the intramolecular alkylation of heteroarenes through reductive dehalogenation of activated alkyl bromides (Scheme 7A).54 This report represented a significant milestone, as it was the first Minisci alkylation that was promoted by photoredox catalysis. The authors’ proposed mechanism involved generation of a Ru(I) species through reductive quenching of the excited state photocatalyst. This Ru(I) species could then reduce malonyl bromides to produce a carbon-centered radical; subsequent trapping of the radical intermediate by electron rich indoles and pyrroles afforded the functionalized products. Following this initial report, the Stephenson group extended this methodology to access intermolecular C–H alkylations,55 as well as the intermolecular construction of quaternary centers (Scheme 7B).56
Scheme 7.
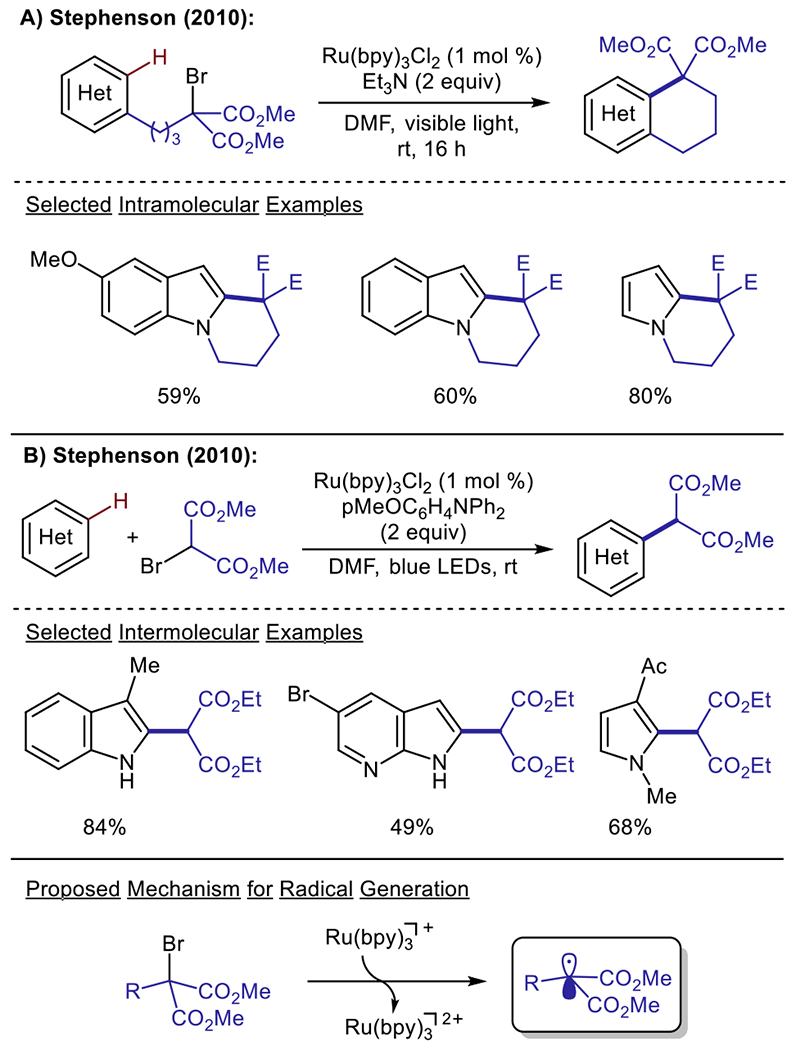
Visible light-driven dehalogenative alkylation of heteroarenes
Two recent reports have highlighted the continued expansion of Minisci protocols featuring dehalogenative radical generation. First, a group at Vertex Pharmaceuticals demonstrated the ability to predictably access C3- and C5-functionalized products by performing the Minisci reaction under basic conditions.57 This report featured the reductive dehalogenation of unactivated alkyl iodides and demonstrated the ability to predict the site of alkylation based upon the electronics of a heteroaryl substrate. Additionally, the Wang group has reported a separate Minisci alkylation protocol which utilizes a halogen atom abstraction event to promote radical generation.58 This work was enabled through the adaptation of conditions concurrently reported by the Stephenson59 and MacMillan60 groups for visible light-mediated bromide atom abstraction from alkyl and aryl bromides, facilitated by a tris(trimethylsilyl)silane radical [(Me3Si)3Si•] species generated in situ. The use of a halogen atom abstraction approach allowed Wang and co-workers to access a diverse scope of alkyl halides and heteroarenes.
The incorporation of trifluoromethyl groups onto (hetero)arenes represents an important transformation in medicinal chemistry applications. As such, dehalogenative Minisci alkylations have also been expanded upon to include the trifluoromethylation of heteroarenes. In 2011, the MacMillan group developed the first reported method for the visible light-driven radical trifluoromethylation of (hetero)arenes (Scheme 8).61 In this report, reduction of trifluoromethanesulfonyl chloride by a ruthenium photocatalyst induced the loss of sulfur dioxide, affording the reactive trifluoromethyl radical species. This species could be effectively trapped by a number of (hetero)arenes, resulting in C–H trifluoromethylation. This method demonstrated the applicability of photoredox catalysis in medicinal chemistry, as a number of trifluoromethylated pharmacophores could be easily accessed. Following this report, a collaborative effort by the Fukuzumi, Cho, and You groups described the use of a platinum(II)acetylacetonate (acac) photosensitizer for the reduction of trifluoromethyl iodide. The resultant trifluoromethyl radical was utilized in the subsequent C–H trifluoromethylation of heteroarenes.62
Scheme 8.
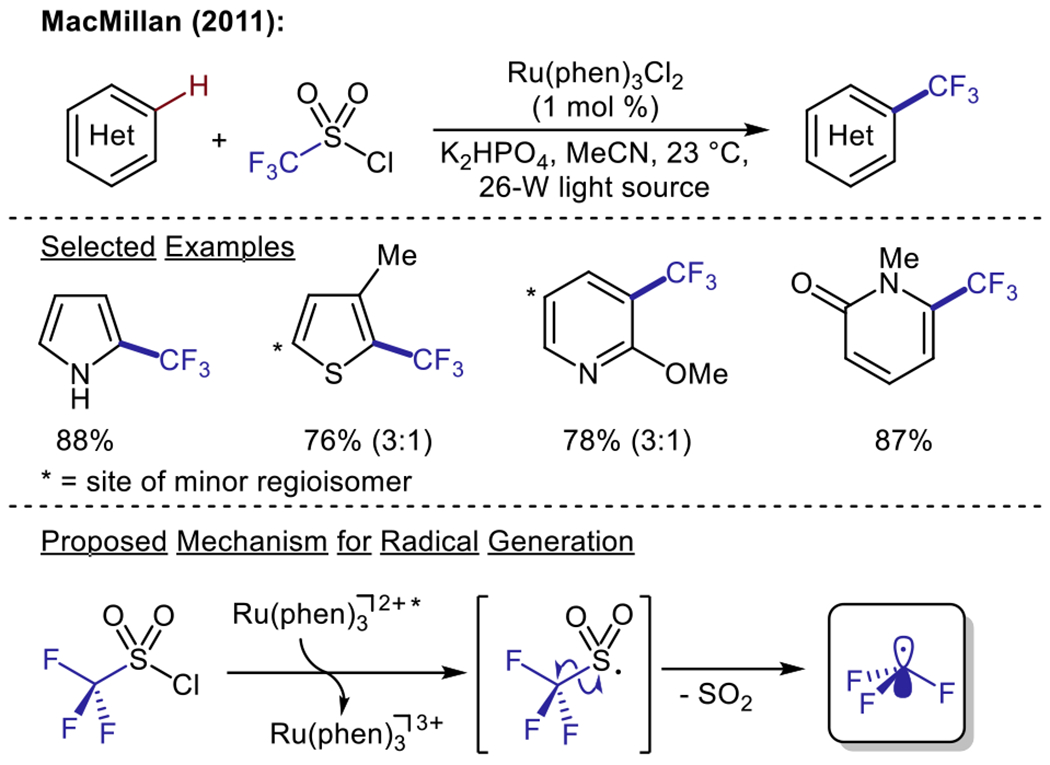
Photoredox trifluoromethylation of unactivated (hetero)arenes
In the aforementioned examples, catalysis is promoted by engaging the photosensitizer in outer sphere electron transfer events. At the same time, dehalogenative radical generation has also been demonstrated to be driven by non-canonical photocatalysts that engage the halide substrate through inner sphere electron transfer or direct halogen atom abstraction events. In 2015, the Barriault group described the use of gold photoredox catalysis for the application of unactivated alkyl bromides to the alkylation of N-heteroarenes through an intramolecular cyclization (Scheme 9).63 This methodology was extended to intermolecular radical additions in 2016. In this more recent study, the Barriault group proposed a mechanistic pathway involving an excited state exciplex which could undergo an inner-sphere electron transfer to furnish the alkyl radical species (Scheme 9).64 The development of these methods has provided mild conditions for accessing primary alkyl radical fragments. Recently, a group from Pfizer reported the use of manganese decacarbonyl (Mn2CO10) for the alkylation of heteroarenes utilizing simple alkyl iodides as substrates.65 The authors proposed that the Mn2CO10 catalyst undergoes Mn–Mn bond homolysis upon irradiated with blue light. The resultant (CO)5Mn• radical species can then abstract an iodine atom from the alkyl iodide reagent to enable alkyl radical generation.
Scheme 9.
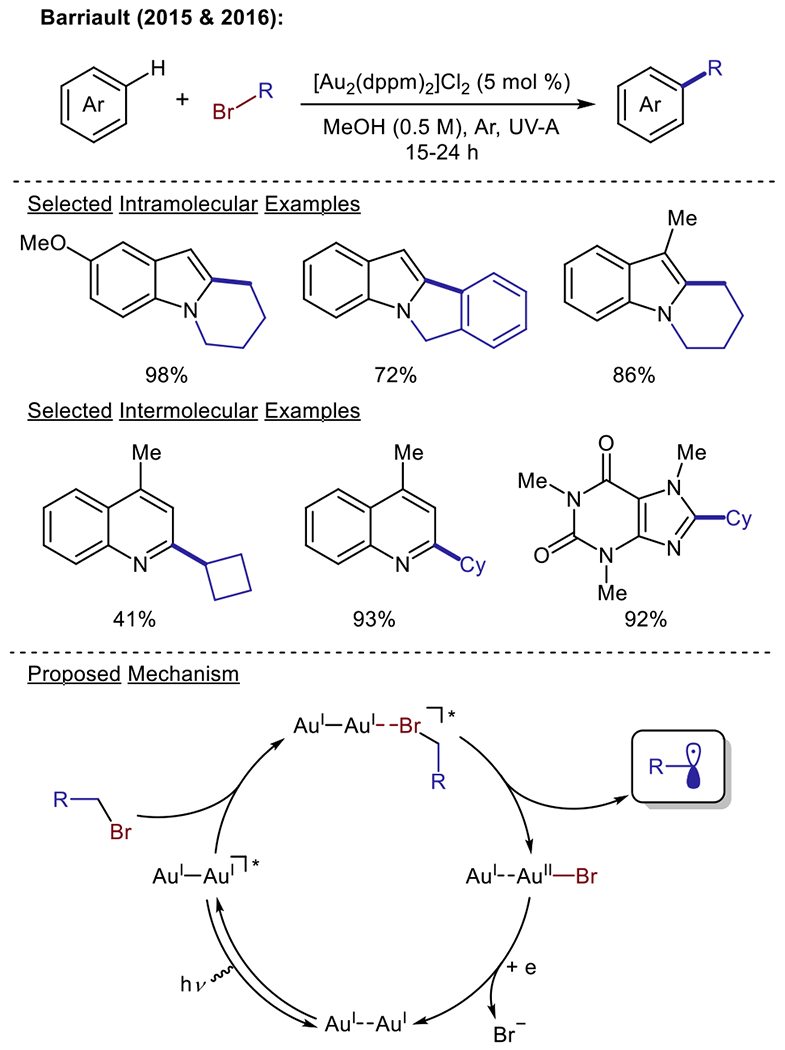
Dehalogenative alkylation using gold photoredox catalysis
6. Alcohols and Ethers
The late-stage incorporation of oxygenated functionality into complex molecules can have a significant impact on the physical properties (e.g. solubility) of a compound. For drug discovery, the optimization of these properties for a lead compound is vital to the development of clinical candidates.66 Thus, the development of methods for the installation of simple oxygenated fragments, such as those derived from alcohols and ethers, is an important point of development for the Minisci reaction.
The application of alcohols in the visible light-driven Minisci alkylation of heteroarenes was first reported in 2015 by the MacMillan group (Scheme 10).67 The authors proposed that the methylation of heteroarenes could be achieved through the initial addition of a carbon-centered hydroxymethyl radical onto a heteroarene substrate. The hydroxymethyl group could then be converted to the desired methyl fragment through a spin-center shift induced by the concomitant loss of water. The subsequent benzylic radical species was proposed to be reduced and protonated to furnish the final methylated product. Importantly, the proposed hydroxymethyl radical intermediate in this report was generated through C–H abstraction of methanol with a thiol co-catalyst. This method provided a general manifold for accessing Minisci reactivity, as a variety of alcohols, pyridines, quinolines, and isoquinolines were amendable to these alkylation conditions. Following this report, in 2016, DiRocco and co-workers utilized a radical relay reaction to promote the visible light-mediated hydroxymethylation of heteroarenes with methanol.68 This reaction was proposed to proceed through the generation of a phenyl radical species from the Ir(III)-catalyzed reductive decomposition of benzoyl. The phenyl radical intermediate then undergoes hydrogen atom abstraction from methanol, thereby generating the active hydroxymethyl radical species, which could be trapped by a variety of heteroarenes. This hydroxymethylation protocol allows for the late-stage functionalization of an array of pharmacophores. While the above two examples utilize iridium photocatalysts to promote reactivity, Minisci reactions featuring alkyl alcohol reagents have also been reported in the absence of photocatalysts. In 2017, the groups of Li69 and Barriault70 independently reported the application of near UV irradiation to promote the methylation of heteroarenes.
Scheme 10.
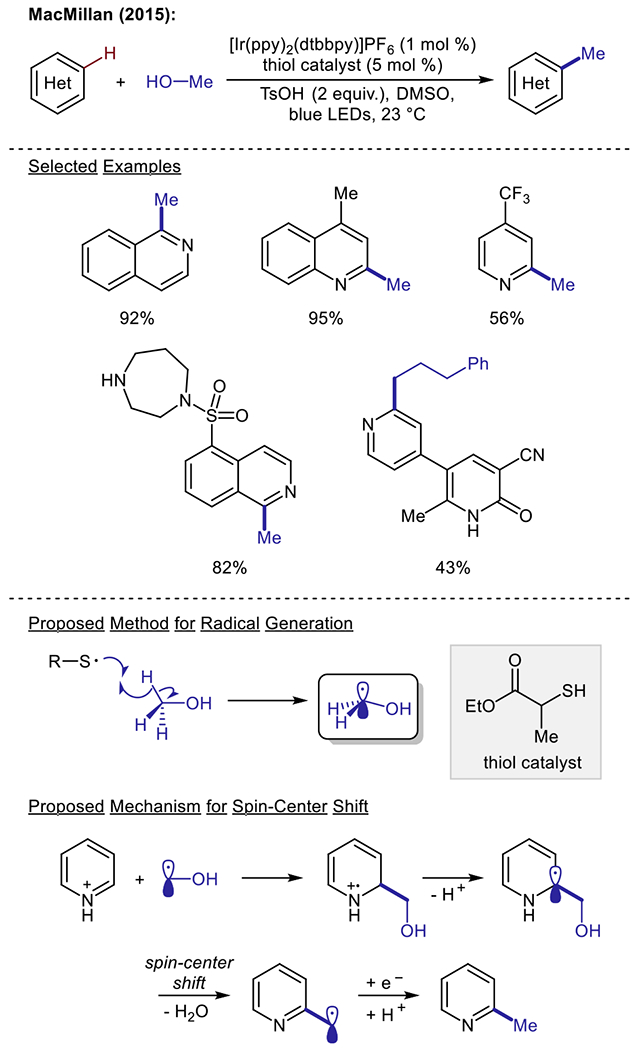
Visible light-driven Minisci alkylation reaction using alcohols as alkylating agents
In 2014, the MacMillan group reported the first application of ethers in conjunction with photoredox catalysis for Minisci reactivity.71 The developed method utilized persulfate salts as both an oxidant and C–H abstraction reagent. From a mechanistic standpoint, oxidative quenching of the photocatalyst by the persulfate salt generates an equivalent of sulfate radical anion, which readily abstracts a hydrogen atom from the ethereal substrate. This seminal report demonstrates the impact of photoredox catalysis on broadening the scope of Minisci reaction protocols, as both cyclic and acyclic ethers could be innovatively used as radical alkylating reagents under mild conditions. In 2017, the Ryu group described the use of a polyoxometalate potocatalyst tetrabutylammonium decatungstate (TBADT) for a visible light-driven Minisci alkylation reaction.72 In its excited state, the TBADT photcatalyst enabled the selective, oxidative generation of radical intermediates through the direct abstraction of electron-rich hydrogen atoms present across ether, alkane, and amide substrates. It is noteworthy that Minisci reactions enabled by the C–H abstraction of saturated molecules are not limited to oxygenated substrates, as this mechanistic paradigm has also been reported with the employment of protected amines73 and alkanes.72,74
6. Conclusion
As exemplified in this Short Review, the utility of photoredox catalysis for the Minisci alkylation reaction provides synthetic chemists with a myriad of opportunities to utilize inexpensive, commercially abundant alkylating reagents (e.g. carboxylic acids, alcohols, alkyltrifluoroborates, alkyl halides, etc.) for the direct, C–H alkylation of heteroarenes. Notably, visible light-driven Minisci alkylation reactions have been demonstrated to proceed under mild reaction conditions and are tolerant of a variety of complex functionalities. In particular, these strategies have been shown to hold significant value for late-stage functionalization efforts in drug discovery. The continued development of photoredox Minisci alkylation reactions that are amenable to a broader scope of complex heterocyclic compounds, while providing improved regioselectivity, is vital to enhancing the synthetic utility and impact of this transformation. Furthermore, demonstrating the scalability of photoredox Minisci alkylation protocols (e.g. using continuous flow systems) may offer valuable opportunities for bridging drug discovery efforts with process development needs.
Funding Information
The authors acknowledge the financial support for this work from the NIH NIGMS (R01-GM127774) and the University of Michigan. This work is supported by an NSF Graduate Research Fellowship for A.C.S. and R.C.M. (grant DGE 1256260).
Biographies
Biosketches
Corey R.J. Stephenson was born in Collingwood, Ontario, Canada and received his undergraduate degree from the University of Waterloo in 1998. He completed graduate studies under the direction of Professor Peter Wipf at the University of Pittsburgh before joining the lab of Professor Erick M. Carreira at ETH Zürich. In September 2007, he joined the Department of Chemistry at Boston University as an Assistant Professor and was granted tenure and promoted to Associate Professor in February, 2013. In July 2013, he joined the Department of Chemistry at the University of Michigan as Associate Professor of Chemistry. In September 2015, Corey was promoted to full Professor.

Alexandra C. Sun was born in Boston, Massachusetts, USA in 1993. She received her BS degree (Chemistry) in 2015 from Brandeis University, where she conducted undergraduate research with Professor Christine Thomas. Alexandra is now pursuing her PhD studies in the group of Professor Corey Stephenson at the University of Michigan, where she is currently working on the development of new photoredox catalysis methodology and continuous flow technology.
Rory C. McAtee was born in Phillipsburg, New Jersey, USA in 1992. He received his BS (Chemistry) from Lycoming College in 2015 performing undergraduate research with Professors Holly Bendorf and Charles Mahler. Rory then joined the group of Professor Corey Stephenson at the University of Michigan, where he is currently a PhD candidate studying photoredox catalysis in organic synthesis.
Edward J. McClain was born in Pittsburgh, Pennsylvania, USA in 1993. He attended West Virginia University for undergraduate studies, performing undergraduate research with Professors Xiaodong Shi and Brian V. Popp. Upon graduating from WVU in 2016, Edward joined the group of Professor Corey Stephenson at the University of Michigan, where he is currently a PhD candidate studying photoredox catalysis in organic synthesis.
References
- (1).Pozharskii AF; Soldatenkov AT; Katritzky AR Heterocycles in Life and Society: An Introduction to Heterocyclic Chemistry, Biochemistry, and Applications; John Wiley & Sons: 2011. [Google Scholar]
- (2).Vitaku E; Smith DT; Njardarson JT J. Med. Chem 2014, 57, 10257. [DOI] [PubMed] [Google Scholar]
- (3).Taylor AP; Robinson RP; Fobian YM; Blakemore DC; Jones LH; Fadeyi O Org. Biomol. Chem 2016, 14, 6611. [DOI] [PubMed] [Google Scholar]
- (4).Cernak T; Dykstra KD; Tyagarajan S; Vachal P; Krska SW Chem. Soc. Rev 2016, 45, 546. [DOI] [PubMed] [Google Scholar]
- (5).Schönherr H; Cernak T Angew. Chem., Int. Ed 2013, 52, 12256. [DOI] [PubMed] [Google Scholar]
- (6).Minisci F; Bernardi R; Bertini F; Galli R; Perchinummo M Tetrahedron 1971, 27, 3575. [Google Scholar]
- (7).Langlois BR; Laurent E; Roidot N Tetrahedron Lett 1991, 32, 7525. [Google Scholar]
- (8).Minisci F; Citterio A; Vismara E; Giordano C Tetrahedron 1985, 41, 4157. [Google Scholar]
- (9).Punta C; Minisci F Trends Heterocycl. Chem 2008, 13, 1. [Google Scholar]
- (10).Studer A; Curran DP Angew. Chem., Int. Ed 2011, 50, 5018. [DOI] [PubMed] [Google Scholar]
- (11).Duncton MA J. MedChemComm 2011, 2, 1135. [Google Scholar]
- (12).Fujiwara Y; Dixon JA; O’Hara F; Funder ED; Dixon DD; Rodriguez RA; Baxter RD; Herlé B; Sach N; Collins MR; Ishihara Y; Baran PS Nature 2012, 492, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ji Y; Brueckl T; Baxter RD; Fujiwara Y; Seiple IB; Su S; Blackmond DG; Baran PS Proc. Natl. Acad. Sci. USA 2011, 108, 14411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).O’Hara F; Blackmond DG; Baran PS J. Am. Chem. Soc 2013, 135, 12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Narayanam JMR; Stephenson CR J. Chem. Soc. Rev 2011, 40, 102. [DOI] [PubMed] [Google Scholar]
- (16).Prier CK; Rankic DA; MacMillan DW C. Chem. Rev 2013, 113, 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Schultz DM; Yoon TP Science 2014, 343, 1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Blakemore DC; Castro L; Churcher I; Rees DC; Thomas AW; Wilson DM; Wood A Nat. Chem 2018, 10, 383. [DOI] [PubMed] [Google Scholar]
- (19).Douglas JJ; Sevrin MJ; Stephenson CR J. Org. Proc. Res. Dev 2016, 20, 1134. [Google Scholar]
- (20).Yayla HG; Peng F; Mangion IK; McLaughlin M; Campeau L-C; Davies IW; DiRocco DA; Knowles RR Chem. Sci 2016, 7, 2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Turconi J; Griolet F; Guevel R; Oddon G; Villa R; Geatti A; Hvala M; Rossen K; Göller R; Burgard A Org. Proc. Res. Dev 2014, 18, 417. [Google Scholar]
- (22).Li Y; Ge L; Muhammad MT; Bao H Synthesis 2017, 49, 5263. [Google Scholar]
- (23).Liu P; Zhang G; Sun P Org. Biomol. Chem 2016, 14, 10763. [DOI] [PubMed] [Google Scholar]
- (24).Beatty JW; Douglas JJ; Cole KP; Stephenson CR J. Nature Comm 2015, 6, 7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Barton DHR; Crich D; Motherwell WBJ Chem. Soc., Chem. Commun 1983, 939. [Google Scholar]
- (26).Barton DHR; Lacher BL; Zard SZ Tetrahedron 1986, 42, 2325. [Google Scholar]
- (27).Okada K; Okamoto K; Morita N; Okubo K; Oda M J. Am. Chem. Soc 1991, 113, 9401. [Google Scholar]
- (28).Cheng W-M; Shang R; Fu Y ACS Catal 2017, 7, 907. [Google Scholar]
- (29).Cheng W-M; Shang R; Fu M-C; Fu Y Chem. Eur. J 2017, 23, 2537. [DOI] [PubMed] [Google Scholar]
- (30).Proctor RSJ; Davis HJ; Phipps RJ Science 2018, 360, 419. [DOI] [PubMed] [Google Scholar]
- (31).Liu X; Liu Y; Chai G; Qiao B; Zhao X; Jiang Z Org. Lett 2018, 20, 6298. [DOI] [PubMed] [Google Scholar]
- (32).Sherwood TC; Li N; Yazdani AN; Dhar TGM J. Org. Chem 2018, 83, 3000. [DOI] [PubMed] [Google Scholar]
- (33).Beatty JW; Douglas JJ; Miller R; McAtee RC; Cole KP; Stephenson CR J. Chem 2016, 1, 456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).McAtee RC; Beatty JW; McAtee CC; Stephenson CR J. Org. Lett 2018, 20, 3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Sun AC; McClain EJ; Beatty JW, Stephenson CR J. Org. Lett 2018, 20, 3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).DiRocco DA; Dykstra K; Krska S; Vachal P; Conway DV; Tudge M Angew. Chem. Int. Ed 2014, 53, 4802. [DOI] [PubMed] [Google Scholar]
- (37).Zuo Z; MacMillan DWC J. Am. Chem. Soc 2014, 136, 5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Garza-Sanchez RA; Tlaheuxt-Aca A; Tavakoli G; Glorius F ACS Catal 2017, 7, 4057 [Google Scholar]
- (39).Genovino J; Lian Y; Zhang Y; Hope TO; Juneau A; Gagne Y; Ingle G; Frenette M Org. Lett 2018, 20, 3229. [DOI] [PubMed] [Google Scholar]
- (40).Seiple IB; Su S; Rodriguez RA; Gianatassio R; Fujiwara Y; Sobel AL; Baran PS J. Am. Chem. Soc 2010, 132, 13194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Ollivier C; Renaud P Chem. Rev 2001, 101, 3415. [DOI] [PubMed] [Google Scholar]
- (42).Darmency V; Renaud P In Radicals in Synthesis I, Springer: 2006. [Google Scholar]
- (43).Vogler T; Studer A Org. Lett 2008, 10, 129. [DOI] [PubMed] [Google Scholar]
- (44).Pouliot M; Renaud P; Schenk K; Studer A; Vogler T Angew. Chem., Int. Ed 2009, 48, 6037. [DOI] [PubMed] [Google Scholar]
- (45).Koike T; Akita M Org. Biomol. Chem 2016, 14, 6886. [DOI] [PubMed] [Google Scholar]
- (46).Yasu Y; Koike T; Akita M Adv. Synth. Catal 2012, 354, 3414. [Google Scholar]
- (47).Tellis JC; Primer DN; Molander GA Science 2014, 1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Li G-X; Morales-Rivera CA; Wang Y; Gao F; He G; Liu P; Chen G Chem. Sci 2016, 7, 6407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Sorin G; Martinez Mallorquin R; Contie Y; Baralle A; Malacria M; Goddard J-P; Fensterbank L Angew. Chem., Int. Ed 2010, 49, 8721. [DOI] [PubMed] [Google Scholar]
- (50).Molander GA; Colombel V; Braz VA Org. Lett 2011, 13, 1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Presset M; Fleury-Brégeot N; Oehlrich D; Rombouts F; Molander GA J. Org. Chem 2013, 78, 4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Matsui JK; Primer DN; Molander GA Chem. Sci 2017, 8, 3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Kuivila HG Synthesis 1970, 1970, 499. [Google Scholar]
- (54).Tucker JW; Narayanam JMR; Krabbe SW; Stephenson CR J. Org. Lett 2010, 12, 368. [DOI] [PubMed] [Google Scholar]
- (55).Furst L; Matsuura BS; Narayanam JMR; Tucker JW; Stephenson CR J. Org. Lett 2010, 12, 3104. [DOI] [PubMed] [Google Scholar]
- (56).Swift EC; Williams TM; Stephenson CR J. Synlett 2016, 27, 754. [Google Scholar]
- (57).Bissonnette NB; Boyd MJ; May GD; Giroux S; Nuhant P J. Org. Chem 2018, 83, 10933. [DOI] [PubMed] [Google Scholar]
- (58).Dong J; Lyu X; Wang Z; Wang X; Song H; Liu Y; Wang Q Chem. Sci 2018. DOI: 10.1039/c8sc04892d. [DOI] [Google Scholar]
- (59).Devery JJ; Nguyen JD; Dai C; Stephenson CR J. ACS Catal 2016, 6, 5962. [Google Scholar]
- (60).Zhang P; Le C; MacMillan DWC J. Am. Chem. Soc 2016, 138, 8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Nagib DA; MacMillan DWC Nature 2011, 480, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Choi WJ; Choi S; Ohkubo K; Fukuzumi S; Cho EJ; You Y Chem. Sci 2015, 6, 1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Kaldas SJ; Cannillo A; McCallum T; Barriault L Org. Lett 2015, 17, 2864. [DOI] [PubMed] [Google Scholar]
- (64).McCallum T; Barriault L Chem. Sci 2016, 7, 4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Nuhant P; Oderinde MS; Genovino J; Juneau A; Gagné Y; Allais C; Chinigo GM; Choi C; Sach NW; Bernier L; Fobian YM; Bundesmann MW; Khunte B; Frenette M; Fadeyi OO Angew. Chem., Int. Ed 2017, 56, 15309. [DOI] [PubMed] [Google Scholar]
- (66).Sloan KB; Bodor N Int. J. Pharm 1982, 12, 299. [Google Scholar]
- (67).Jin J; MacMillan DWC Nature 2015, 525, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Huff CA; Cohen RD; Dykstra KD; Streckfuss E; DiRocco DA; Krska SW J. Org. Chem 2016, 81, 6980. [DOI] [PubMed] [Google Scholar]
- (69).Liu W; Yang X; Zhou Z-Z; Li C-J Chem 2017, 2, 688. [Google Scholar]
- (70).McCallum T; Pitre SP; Morin M; Scaiano JC; Barriault L Chem. Sci 2017, 8, 7412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Jin J; MacMillan DWC Angew. Chem. Int. Ed 2014, 54, 1565. [Google Scholar]
- (72).Quattrini MC; Fujii S; Yamada K; Fukuyama T; Ravelli D; Fagnoni M; Ryu I Chem. Commun 2017, 53, 2335. [DOI] [PubMed] [Google Scholar]
- (73).Bosset C; Beucher H; Bretel G; Pasquier E; Queguiner L; Henry C; Vos A; Edwards JP; Meerpoel L; Berthelot D Org. Lett 2018, 20, 6003. [DOI] [PubMed] [Google Scholar]
- (74).Hu A; Guo J-J; Pan H; Zuo Z Science 2018, 361, 668. [DOI] [PubMed] [Google Scholar]