

Abstract
Recent evidence suggests that alterations in oxidative metabolism induced by thiamine deficiency lead to neuronal cell death. However, the molecular mechanisms underlying this process are still under extensive investigation. Here, we report that rat pheochromocytoma PC-12 cells differentiated in the presence of NGF into neurons undergo apoptosis due to thiamine deficiency caused by antagonists of thiamine — amprolium, pyrithiamine and oxythiamine. Confocal laser scanning fluorescence microscopy revealed that annexin V binds to PC-12 cells in presence of thiamine antagonists after 72 h incubation. Results also show that thiamine antagonists trigger upregulation of gene expression of mitochondrial-derived apoptosis inducing factor, DNA fragmentation, cleavage of caspase 3 and translocation of active product to the nucleus. We therefore propose that apoptosis induced by amprolium, pyrithiamine or oxythiamine occurs via the mitochondria-dependent caspase 3-mediated signaling pathway. In addition, our data indicate that pyrithiamine and oxythiamine are more potent inducers of apoptosis than amprolium.
Keywords: thiamine deficiency, oxythiamine, pyrithiamine, amprolium, apoptosis
INTRODUCTION
The biologically active form of thiamine — thiamine diphosphate (ThDP) — is a cofactor for several key enzymes involved in energy metabolism, such as pyruvate dehydrogenase complex, α-ketoglutarate dehydrogenase complex, macromolecular complexes of enzymes that decarboxylate the α-keto acids derived from leucine, isoleucine and valine, and transketolase. Consequently, metabolic disturbances caused by thiamine deficiency (ThD) can stimulate neuronal cell loss through several mechanisms which include impairment in carbohydrate metabolism (Martin et al., 2003), energy failure and excess levels of free radicals (Pannunzio et al., 2000; Todd & Butterworth, 1999), lactic acidosis (Romanski & McMahon, 1999), stress of endoplasmic reticulum (Wang et al., 2007) as well as significant decrease in the voltage-dependent K+ membrane conductance (Oliveira et al., 2007). All the above-mentioned mechanisms can aggravate the neuronal damage resulting in ThD-related pathologies such as Wernicke–Korsakoff syndrome (Butterworth & Heroux, 1989; Pannunzio et al., 2000, Lonsdale, 2006) as well as some other neurodegenerative disorders (Heroux et al., 1996; Park et al., 2000; Kruse et al., 2004). Experimentally, ThD is commonly induced by depletion or decreasing of thiamine level in combination with thiamine antagonists (Rais et al., 1999; Park et al., 2000; Kruse et al., 2004). Thiamine antagonists such as pyrithiamine, oxythiamine or amprolium have been shown to inhibit thiamine transport and display complex and diverse action on thiamine metabolism and ThDP biosynthesis in rat nervous tissue in vivo (Rindi et al., 2003). Oxythiamine is recognized as a precursor of oxythiamine diphosphate — an anti-coenzyme form of vitamin B1 (Rais et al., 1999), while pyrithiamine is a known effective inhibitor of thiamine kinase (Rindi et al., 2003).
To date, many mechanisms have been proposed to explain the nature and molecular pathways associated with ThD and neuronal cell death. However, it remains contradictory whether neuronal cells undergo apoptosis (Hazell et al., 1998; Rais et al., 1999; Park et al., 2000; Wang et al., 2000; Chornyy & Parkhomenko, 2006; Oliveira et al., 2007) or necrosis (Bettendorff et al., 1995; Bakker et al., 1996), and what molecular mechanisms are involved in these processes. In the present investigation, we studied several key mechanisms involved in the regulation of apoptosis such as mitochondrial-derived apoptosis inducing factor, DNA fragmentation, cleavage of caspase 3 and translocation of the active product to the nucleus and demonstrated that neuronally differentiated PC-12 cells undergo apoptosis after induction of ThD.
MATERIALS AND METHODS
Materials.
Rat nerve growth factor (NGF), streptomycin, penicillin, amprolium, oxythiamine, pyrithiamine were obtained from Sigma-Aldrich (St Louis, MO, USA).
Cell culture.
Rat pheochromocytoma PC-12 cells were obtained from American Type Culture Collection (Manassas, VA, USA) and maintained in F-12K medium (ATCC) containing 15% (v/v) horse serum (ATCC), 2.5% (v/v) fetal clone III serum (FCIII) (Invitrogen Corp., Carlsbad, CA, USA), 1% streptomycin and 1% penicillin at 37°C in a humidified atmosphere of 5% CO2.
Detection of cell viability.
PC-12 cells were seeded on 96-well plates (BD Biosciences Clontech, Palo Alto, CA, USA) at a density of 1 × 103 cells/well in a final volume of 100 μl and incubated in complete media in the presence of 100 μM NGF for at least three days to achieve homogenous population of neurons, followed by incubation for 48, 72 and 96 h in F-12K containing ten times reduced serum supplementation and in the presence of 10, 100 or 1000 μM of oxythiamine, pyrithiamine or amprolium. Cell viability was measured using a WST-1 reagent (Roche Diagnostics Corp., Indianapolis, IN, USA) following the manufacturer’s instructions and Multiscan RC (Thermolabsystems, Helsinki, Finland).
Confocal immunofluorescence microscopy
Annexin V binding assay.
PC-12 cells were plated on Laboratory-Tek chamber slides (Nalge Nunc Int., Rochester, NY, USA) at a density of 1 × 105 cells/well and incubated in complete media in the presence of 100 μM NGF for three days to achieve homogenous population of neurons, followed by incubation for 72 h in F-12K containing ten times reduced serum supplementation and in the presence of 100 μM of thiamine antagonists — oxythiamine, pyrithiamine or amprolium. Treatment of cells with 4 μM staurosporine (Cell Signaling Technology, Beverly, MA, USA) for 24 h was used as a positive control to induce apoptosis (Schon & Manfredi, 2003). Next, cells were fixed for 5 min in PBS containing 3.7% formaldehyde, washed with PBS, permeabilized with 0.1% (v/v) Triton X-100 in PBS for 3 min, and washed three times with PBS, then incubated with annexin V conjugated to a fluorescent dye — alexa fluor 568 (Invitrogen Corp., Carlsbad, CA, USA) in annexin V binding buffer (10 mM Hepes/NaOH, pH 7.4, 140 mM NaCl, 5 mM CaCl2) for 20 min at room temp. followed by staining of nuclei with SYTOX Green (Invitrogen) for 20 min at room temp., washed with PBS and used for microscopy.
Detection of caspase 3 translocation to the nucleus.
PC-12 cells were treated with thiamine antagonists as above. Then, cells were fixed for 30 min at 4°C with cold 70% ethanol, treated with 100 μg/ml RNase for 2 min at room temp. and blocked in 10% horse serum in PBS for 30 min followed by incubation with goat anti-rat cleaved caspase 3 antibody conjugated to fluorescein (Santa Cruz Biotech, Santa Cruz, CA, USA) at a dilution 1 : 1000, followed by 10 min incubation with 50 μg/ml propidium iodide (Invitrogen) for staining of nuclei, washed with PBS and used for microscopy.
TUNEL assay.
Detection of cell apoptosis was carried out with the DeadEnd fluorometric TUNEL system (Promega, Madison, WI, USA), following the manufacturer’s instructions. After performing the TUNEL assay, cells were co-stained with 50 μg/ml propidium iodide for determination of the total number of cells in each experiment, then washed with PBS and used for microscopy.
Images were acquired using a Zeiss LSM-5 Pascal scanning confocal microscope (Carl Zeiss Ltd) at magnifications 10× and 40×. Final image composites were created using Zeiss LSM-5 Pascal Image software, version 3.2.
RNA isolation.
Total RNA was isolated using the TRIZOL® (Invitrogen) reagent following manufacturer’s instructions. Briefly, the PC-12 cells were seeded initially at 0.5 × 106 cells/well in 6-well plates and differentiated with 100 μM NGF for three days followed by treatment with 100 μM of either antagonist for 72 h. After that cells were lysed with the TRIZOL® reagent by repeated pipetting. Cell homogenates were incubated for 5 min at room temp. to allow complete dissociation of the nucleoprotein complexes. After a chloroform extraction, the RNA-containing aqueous phase was separated and mixed with isopropyl alcohol. Precipitated RNA was washed once with 75% (v/v) ethanol, centrifuged at 8 200 × g for 5 min at 4°C and resuspended in RNase-free water. Removal of DNA was accomplished through treatment with DNase I (Roche) followed by RNA precipitation and assessment of its integrity by 1% (w/v) agarose gel electrophoresis.
RT-PCR.
RT-PCR was performed with total RNA isolated as described above. After DNase I treatment, 2 μg of RNA was used for cDNA synthesis with a Reverse Transcription System kit (Promega) following the manufacturer’s instructions. The reactions were performed with a Perkin-Elmer Gene-Amp® PCR System 2 400 at 42°C for 60 min and reactions were stopped by cooling at 4°C. PCR was performed using 2 μl of cDNA template from the reverse transcription reactions in a total volume of 25 μl of PCR Master Mix (Promega). The following cDNAs were amplified with the indicated sets of primers (Invitrogen) at 1 μM: rat aif (Gene Bank accession number: XM_342137; primers corresponding to 1112–1131 and 1371–1392 nt); rat gapdh (accession number: BC059110; primers corresponding to 497–516 and 785–804 nt). The latter was used as a positive control for the presence and integrity of the cDNA. The conditions for PCR amplification of cDNAs were as follows: an initial denaturation step at 95°C for 5 min, 30 cycles of denaturation at 95°C for 60 s, annealing at 52°C for 60 s and extension at 72°C for 60 s, and a final 7 min extension step at 72°C. Each PCR product (23 μl) was electro-phoresed in 1 × TAE buffer (40 mM Tris/acetate and 1 mM EDTA) on 1% (w/v) agarose gels containing 0.6 μg/ml ethidium bromide. A 100-bp DNA Ladder (Promega) was used as a marker for cDNA fragment size. The fluorescence of bands was quantified using BioRad (Philadelphia, PA, USA) Versa Doc® and QuantityOne® software.
Western blot analysis.
Cells were platted in six-well plates and incubated as above. After incubation for the time indicated in the figure legends, the cells were washed with ice-cold PBS and lysed with 50 μl of lysis buffer (25 mM Tris/HCl, pH 7.4, 25 mM NaCl, 1 mM Na3VO4, 10 mM NaF, 10 mM Na4P2O7, 25 mM β-glycerophosphate, 25 mM p-nitrophenylphosphate, 0.5 mM EGTA, 0.5% (v/v) Triton X-100, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, 10 μg/ml leupeptin and 10 nM okadaic acid). The cell extracts were centrifuged (13 000 × g for 10 min at 4°C) to remove insoluble material and then 15 μl of 3 × Laemmli sample buffer (187.5 mM Tris/HCl, pH 6.8, 6% (w/v) sodium dodecyl sulfate (SDS), 1.8% (v/v) β-mercaptoethanol and 0.003% (v/v) bromophenol blue) was added to 30 μl of supernatant. The mixture was heated for 5 min at 96°C, and 100 μg of cell lysate protein as determined by Bradford’s assay (Bradford, 1976), was subjected to 15% SDS/PAGE. Proteins were transferred to nitrocellulose membranes for Western blot analysis. Detection was performed by using rabbit anti-rat cleaved caspase-3 and rabbit anti-rat caspase-3 antibodies conjugated to horse radish peroxidase (HRP) (Cell Signaling, Beverly, MA, USA) at 1 : 1000 dilution that were visualized by chemiluminescence and quantitated using the Super-Signal West Dura Luminol/Enhancer Solution (Pierce, Woburn, MA, USA) and a GS-525 Molecular Imager using MultiAnalyst™ software (Bio-Rad).
Statistical analysis.
One-way multiple Tukey-Kramer comparison post-test analysis of variance (ANOVA) was used for evaluation of data obtained between a control and all other means (i.e. treatment of cells with thiamine antagonists) to test whether the mean of a variable differs among groups. P < 0.05 between control and experimental groups was considered to be statistically significant, P < 0.01 (**) — very significant and P < 0.001 (***) — extremely significant. If P obtained between variants was more than 0.05, then it was considered to be non-significant. All analyses were performed with InStat software, version 3.06 (GraphPad Software Inc., San Diego, CA, USA).
RESULTS
Effects of amprolium, oxythiamine and pyrithiamine on viability of neuronally differentiated PC-12 cells
Treatment of neuronally differentiated PC-12 cells with thiamine antagonists: amprolium, oxythiamine and pyrithiamine revealed dose- and time-dependent cell responses for each compound (Fig. 1). Cell viability was initially reduced in a dose-dependent manner starting after 48 h of treatment with all tested antagonists. Further reduction in cell viability was observed after incubation for 72 and 96 h (Fig. 1). ANOVA analysis revealed that the differences between the effects of the antagonists in reduction of viability of PC-12 cells treated for 48 h were not significant (10 μM — P = 0.2526; 100 μM — P = 0.539; 1000 μM — P = 0.4727). However, prolonged treatment with the antagonists for 72 h (10 μM — P = 0.0275; 100 μM — P = 0.0166; 1000 μM — P = 0.0020) or 96 h (10 μM — P = 0.0213; 100 μM — P = 0.0001; 1000 μM — P = 0.0083) demonstrated a significantly higher reduction of cell viability oxythiamine or pyrithiamine than by amprolium (Fig. 1).
Figure 1. Thiamine antagonists amprolium, oxythiamine and pyrithiamine affect viability of PC-12 cells in time- and dose-dependent manner.
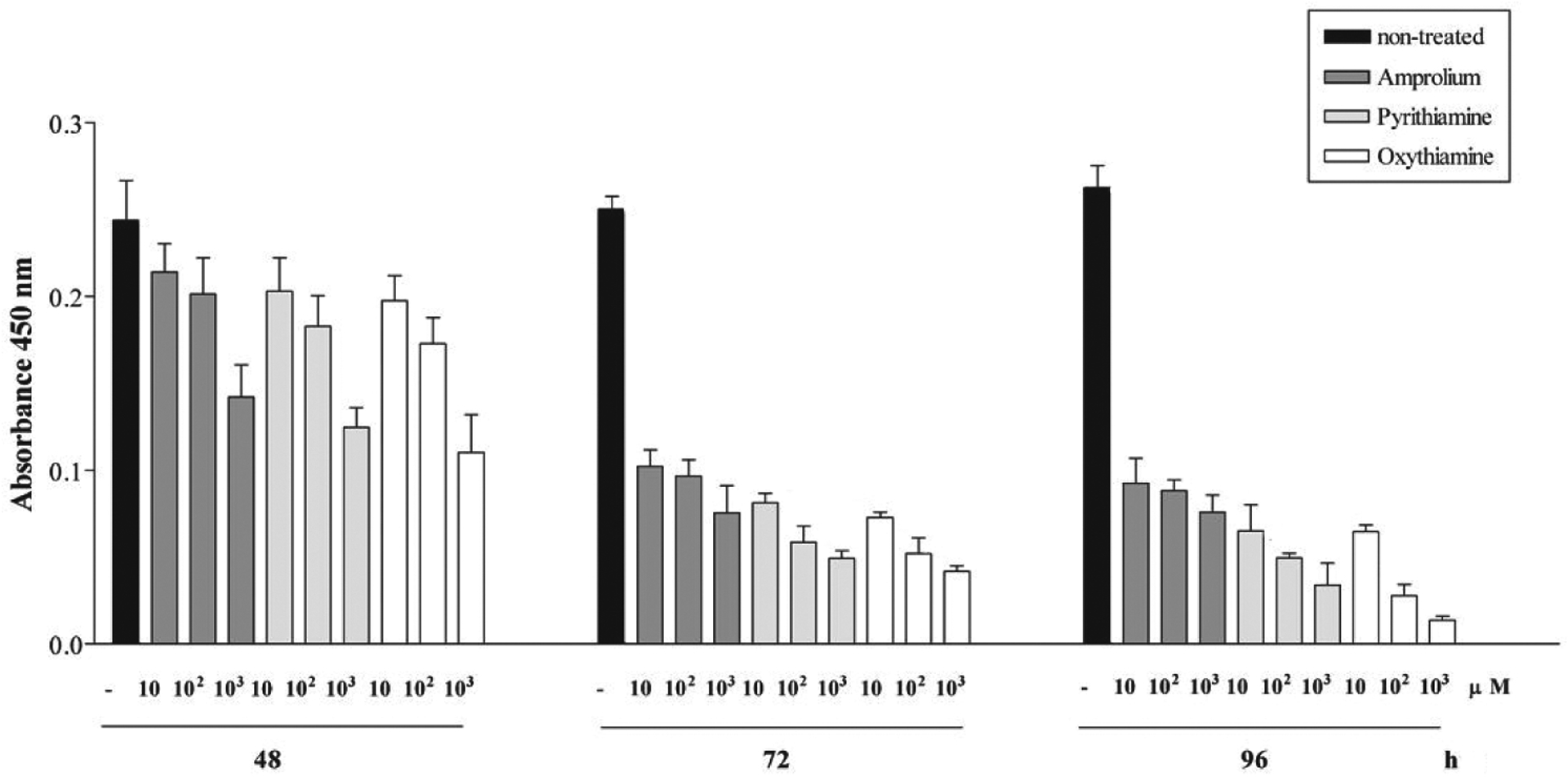
Viability of neuronally differentiated PC-12 cells was measured using WST-1 reagent and Multiscan RC (Thermolabsystems) upon treatment with 10, 100 or 1000 μM oxythiamine, pyrithiamine or amprolium in serum-reduced media for 48, 72 and 96 h. The viability rates represent the means ± S.E.M. for non-treated cells and cells treated with tested thiamine antagonists in which each value was determined from assays performed in quadruplicate. Further experimental details are given in Materials and Methods.
Amprolium, oxythiamine and pyrithiamine induce binding of annexin V to surface of cellular membrane
We have shown previously the diverse effects of amprolium, oxythiamine and pyrithiamine on stimulation of apoptosis in rat DITNC cells (Chornyy & Parkhomenko, 2006). Thus, 100 μM oxythiamine or pyrithiamine were shown to induce annexin V binding to cell surface in rat DITNC cells, while amprolium had no such effect. To further establish the role of thiamine antagonists on stimulation of apoptosis in neuronal cells, neuronally differentiated PC-12 cells were incubated for 72 h in the presence of 100 μM amprolium, oxythiamine or pyrithiamine. The duration of exposition and the dose were chosen after analysis of data obtained in cell viability studies as described above (Fig. 1). Untreated cells were examined as a control. Treatment of cells with 4 μM staurosporine for 24 h was used as a positive control in detection of morphological changes. We found that the percentage of apoptotic cells after treatment with oxythiamine or pyrithiamine was significantly higher than with amprolium (Fig. 2A). Thus, the percentage of cells stained by annexin V after exposure to amprolium was 65.57% ± 1.7 (total number of cells analyzed, (n) = 454), pyrithiamine — 87.2% ± 0.6 (n = 470) and oxythiamine — 87.8% ± 3 (n = 454) (Fig. 2A). In most cells the nucleus and cytoplasmic membrane were intact and apoptotic bodies were observed in very few cells (not shown). These experiments suggest that early signs of apoptosis induced by thiamine antagonists in neuronally differentiated PC-12 cells can be detected as early as after 72 h of incubation. In addition, our data indicate that pyrithiamine and oxythiamine are more potent inducers of apoptosis than amprolium and correlate with our previous report (Chornyy & Parkhomenko, 2006).
Figure 2. Thiamine antagonists induce binding of annexin V to cell surface membrane and translocation of active caspase 3 to the nucleus.
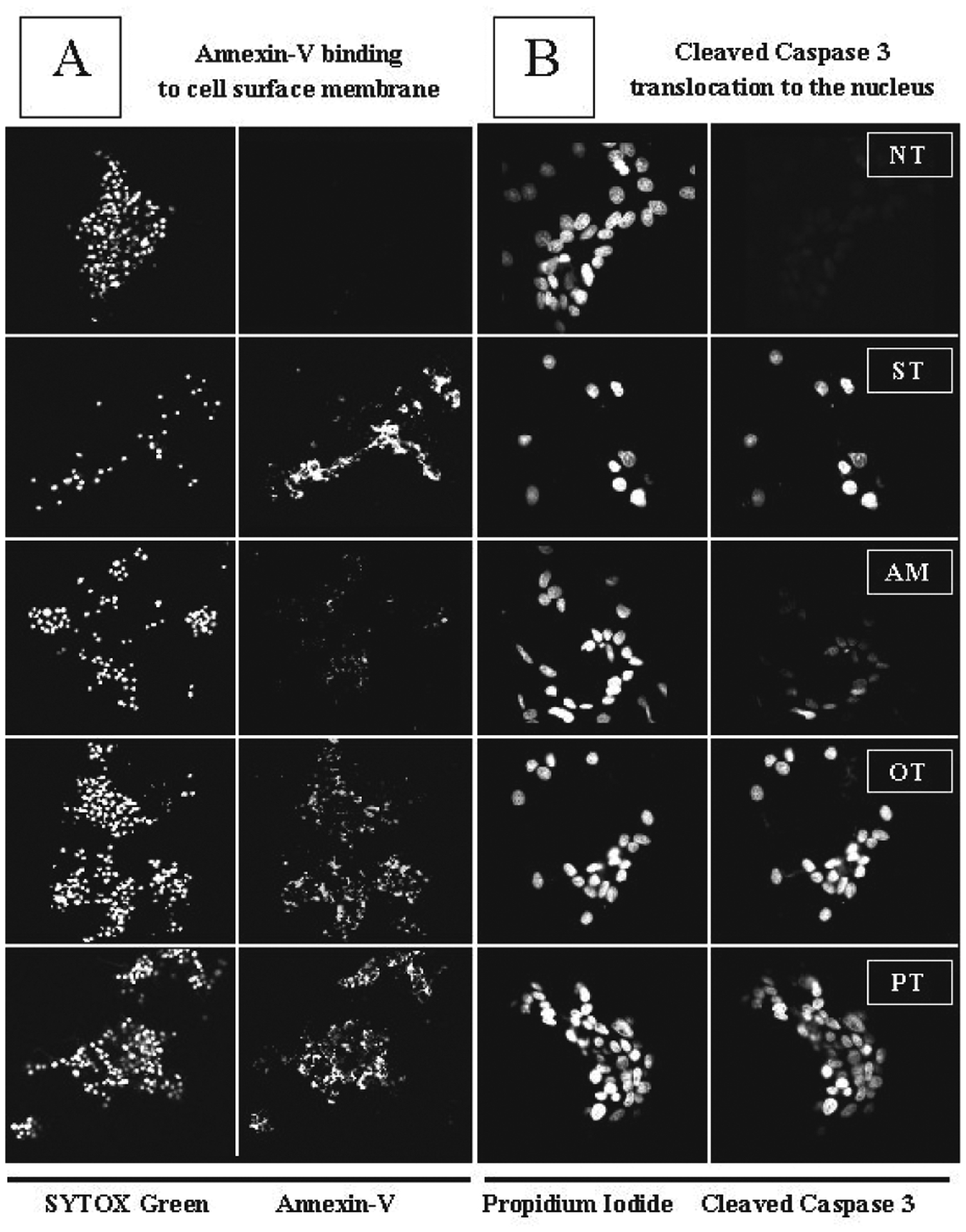
Neuronally differentiated PC-12 cells were treated for 72 h with 100 μM of thiamine antagonists — amprolium (AM), pyrithiamine (PT) or oxythiamine (OT). Non-treated cells were examined as control (NT). Cells treated with 4 μM staurosporine (ST) for 24 h were used as positive control. (A) Annexin V binding (magnification 10×) and (B) translocation of cleaved caspase 3 to the nucleus (magnification 40×) were analyzed by confocal laser scanning immunofluorescence microscopy. SYTOX Green — nucleic acid stain; annexin V (annexin V-alexa fluor 568) — fluorescent conjugate which detects apoptotic cells; propidium iodide — fluorescent DNA stain; cleaved caspase 3 was detected by goat anti-rat antibody conjugated to fluorescein. Staining of cell nuclei allowed counting of cells. Each experiment was performed in triplicate.
Effect of thiamine antagonists on cleavage of caspase 3
Caspases are aspartate-specific cysteine pro-teases that play critical roles in apoptosis, which can occur after treatment of cells with different chemical agents or under pathological conditions (Thornberry & Lazebnik, 1998). Our experiments suggest that treatment of neuronally differentiated PC-12 cells with 100 μM of either of the three thiamine antagonists investigated resulted in translocation of cleaved caspase 3 to the nucleus (Fig. 2B). Analysis of the effects caused by the antagonists on the activation of caspase 3 revealed a maximal response for oxythiamine (96.68% ± 1.12, n = 197) and pyrithiamine (95.03% ± 2.94, n = 124), and a weaker one for amprolium-treated cells (53.9% ± 2.11, n = 194), whereas a very low expression of active caspase 3 was detected in untreated cells (10.55% ± 0.82, n = 246) (Fig. 2B). Similar data were obtained by Western blot analysis as evidenced by the appearance of cleaved 19 kDa fragment of caspase 3 at 72 h (Fig. 3A). Furthermore, we observed a reduction of the level of caspase 3 in cells treated with the thiamine antagonists. Thus, the amount of caspase 3 was decreased by amprolium by 1.39-fold (± 0.22), by oxythiamine by 2.08-fold (± 0.06) and by pyrithiamine by 2.36-fold (± 0.33). At the same time, the levels of cleaved caspase 3 were correspondingly higher in cells treated with oxythiamine- or pyrithiamine-treated cells than in those treated with amprolium (Fig. 3B). As shown in Fig. 3A and 3B, the ratios between cleaved caspase 3 and caspase 3 were as follows: non-treated cells 0.34 (± 0.06), amprolium 1.16 (± 0.18), oxythiamine 2.05 (± 0.8) and pyrithiamine 1.99 (± 0.13). ANOVA analysis demonstrated that the difference in the ratios between oxythiamine- and pyrithiamine-treated cells are non-significant (P > 0.05), while the effect of amprolium was significantly lower (Figs. 3A and B). These observations indicate that, the tested thiamine antagonists promote apoptosis in neuronally differentiated PC-12 cells via activation of caspase 3 signalling, with amprolium being less effective than oxythiamine and pyrithiamine.
Figure 3. Differential effect of pyrithiamine, oxythiamine and amprolium on cleavage of caspase 3.
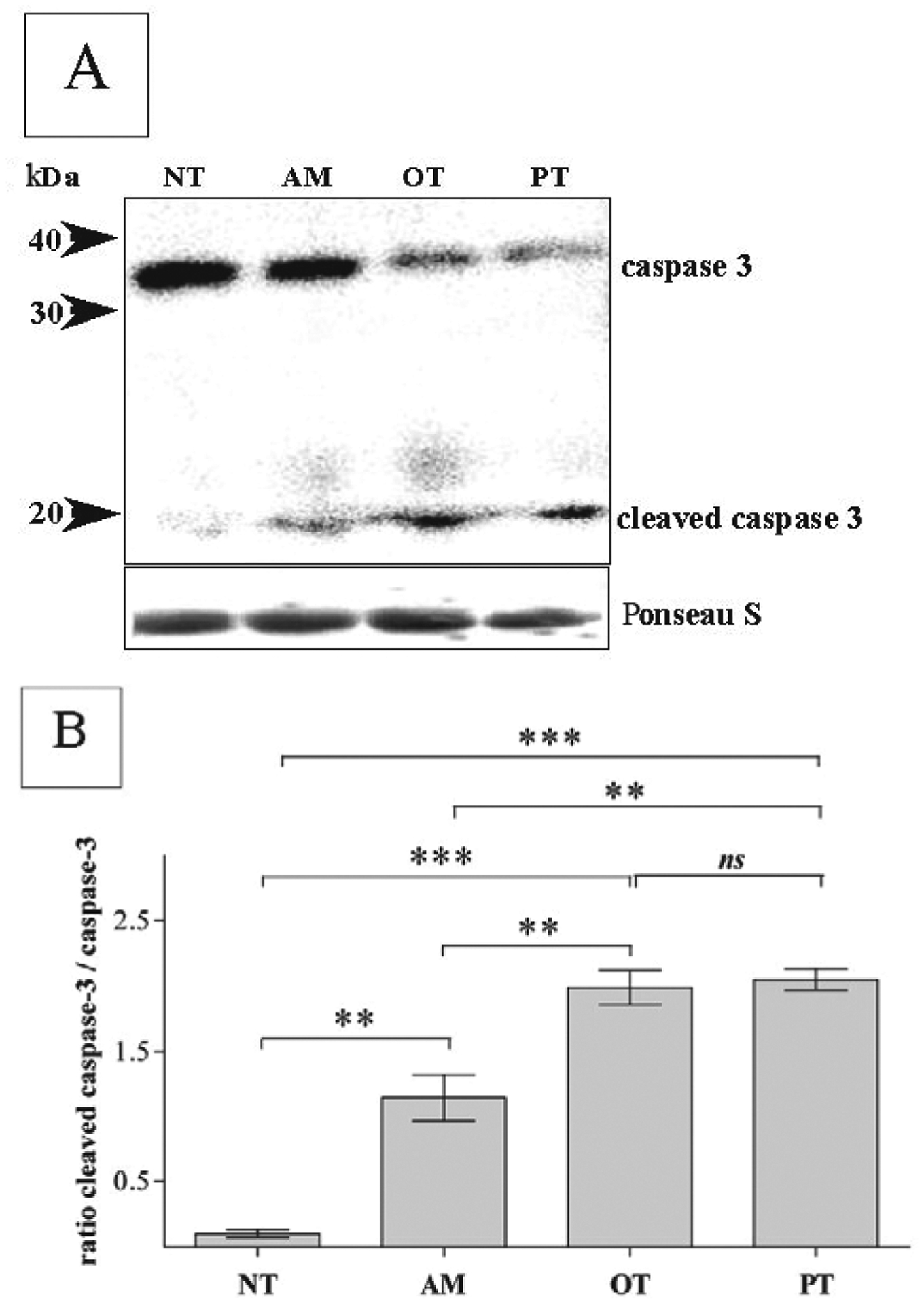
PC-12 cells differentiated with 100 μM NGF were treated for 72 h with 100 μM amprolium (AM), oxythiamine (OT) or pyrithiamine (PT). Non-treated cells were examined as control (NT). Western blots (A) of caspase 3 and cleaved caspase 3 after 72 h treatment were stained by appropriate antibodies as described in Materials and Methods. Equal protein loading (A) shown by Ponceau S staining of nitrocellulose membrane before application of antibodies. Data on the graph (in B) indicate ratios between cleaved caspase 3 and caspase 3 ± S.E.M. (n = 3) in response to treatment with antagonists.
Amprolium, pyrithiamine and oxythiamine cause upregulation of apoptosis inducing factor gene expression and DNA fragmentation
Apoptosis inducing factor (AIF) is a caspase-independent death effector that is released from mitochondria early in the apoptotic process (Susin et al., 1999). Our data indicate that ThD that occurs via incubation of PC-12 cells in the presence of 100 μM of amprolium, pyrithiamine or oxythiamine for 72 h causes upregulaton of aif gene expression (Figs. 4A). After induction of apoptosis, AIF translocates to the nucleus and induces chromatin condensation and large-scale (50 kb) DNA fragmentation (Daugas et al., 2000). Therefore, we decided to measure if the percentage of TUNEL-positive cells correlates with the levels of aif gene expressions. As expected, we obtained a similar percentage of TUNEL-positive cells (± S.E.M.) upon treatment of cells for 72 h with 100 μM pyrithiamine (65% ± 5, n = 67) or oxythiamine (64% ± 4, n = 243) and a significantly lover one upon treatment with amprolium (38 ± 5, n = 277) (Figs. 4B). In untreated cells only 5% ± 1 (n = 277) were TUNEL-positive.
Figure 4. Activation of aif gene expression and DNA fragmentation by thiamine antagonists.
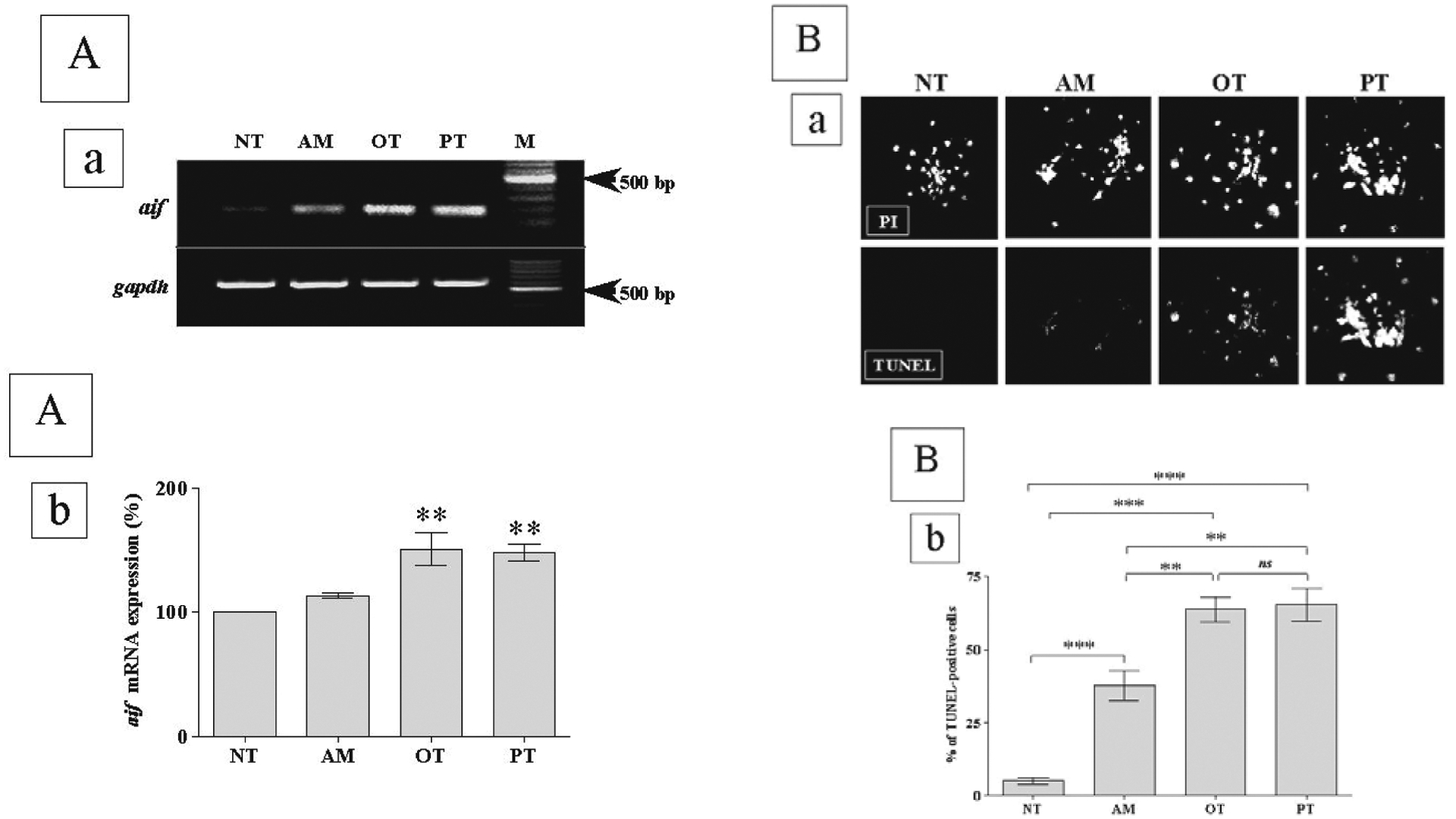
(A) Neuronally differentiated PC-12 cells were treated for 72 h with 100 μM amprolium (AM), oxythiamine (OT) or pyrithiamine (PT). Non-treated cells were examined as control (NT). Total RNA extracted, reverse transcribed and PCR was performed to quantitate the expression of aif mRNA, presented on the gel (a). Values (in b) are mean ± S.E.M. (n = 3) for aif gene expression were quantified using Versa Doc® and QuantityOne® software, normalized to gapdh mRNA levels and expressed as the percentage of increase over NT. (B) TUNEL fluorescence microscopy for evaluation of the apoptotic events was conducted on neuronally differentiated PC-12 cells (a) after treatment with amprolium (AM), oxythiamine (OT), or pyrithiamine (PT). Non-treated cells were examined as control (NT). Propidium iodide (PI) was used to stain all cells. Graph (in b) represents percentage of TUNEL-positive cells relative to total PI nuclei as evaluated by confocal laser scanning fluorescence microscopy at magnification 40×. Data on the graph indicate percentage of TUNEL-positive cells ± S.E.M. (n = 3) in response to treatment with antagonists.
DISCUSSION
It has been demonstrated that chronic impairment of oxidative metabolism caused by ThD accelerates microglial activation and results in neuronal cell loss (Todd & Butterworth, 1999; Calingasan et al., 1999). The possible metabolic consequences of ThD that trigger death signalling mechanisms involve cellular energy failure, oxidative stress, focal lactic acidosis, N-methyl-d-aspartate receptor-mediated excitotoxicity, increased expression of endothelial nitric oxide synthase, nitrotyrosine deposition, microglial activation, decreased expression of astrocytic glutamate transporters, mitochondrial dysfunction, decrease in the voltage-dependent K+ membrane conductance, stress of endoplasmic reticulum, lipid peroxidation and reactive oxygen species production (Todd & Butterworth, 1999; Calingasan et al., 1999; Park et al., 2000; Desjardins & Butterworth 2005; Oliveira et al., 2007; Wang et al., 2007). Among the diverse mechanisms proposed to explain the nature of neuronal loss due to ThD is programmed cell death — apoptosis (Hazell et al., 1998).
In vitro ThD can be induced by “classic” thiamine structural analogs amprolium, oxythiamine and pyrithiamine, which metabolically interfere with thiamine metabolism and its functions (Singleton & Martin, 2001). Amprolium is known to inhibit thiamine transport but has no effect on thiamine diphosphorylation (Lumeng et al., 1979). Therefore, cells treated with amprolium are able to retain a low concentration of thiamine and undergo cell death slower than after treatment with oxythiamine or pyrithiamine. Pyrithiamine is one of the potent antagonists of vitamin B1, which completely imitate the neurological impairment seen in the paralytic form of beriberi disease or Wernicke–Korsakoff syndrome (Butterworth & Heroux, 1989). Pyrithiamine competes with thiamine transport as amprolium but, in addition, it interacts with thiamine kinase (TK) and inhibits the synthesis of ThDP by a currently unknown mechanism (Lumeng et al., 1979). Moreover, it has been reported that TK forms small amounts of the anti-coenzyme pyri-ThDP (Liu et al., 2006). However, the ability of pyri-ThDP to disrupt thiamine phosphoester homeostasis through interference with the production or function of thiamine monophosphate, diphosphate and triphosphate has been suggested to be a reason of severe neurological metabolic aberrations (Liu et al., 2006). Oxythiamine is another potent thiamine antagonist that penetrates the plasma membrane, competes with thiamine transport and interacts with TK (Chernikevich et al., 1995). Oxythiamine can also form the anti-coenzyme oxy-ThDP, which is totally inactive when bound in place of ThDP to the apoenzyme (Tylicki et al., 2003). Oxythiamine is known to induce such physiological aberrations as lethargy and anorexia seen in animal studies (Ali, 1985). As it was expected, our present studies demonstrated that ThD induced by pyrithiamine and oxythiamine had a more potent influence on the loss of neuronally differentiated PC-12 cells than amprolium (Figs. 1–4).
It is known that apoptosis occurs in several prominent neurodegenerative conditions associated with mitochondrial dysfunction (Linnik et al., 1993; Cotman & Anderson, 1995). To evaluate whether PC-12 cell loss in response to treatment with thiamine antagonists was related to programmed cell death — apoptosis, we investigated the expression of several key elements of apoptosis. First we analyzed the integrity of plasma membranes of cells subjected to ThD. It is known that in the early phases of apoptotic cell death, the asymmetry of phospholipids of the plasma membrane breaks up and exposes phosphatidylserine, which translocates to the outer layer of the membrane (Rucker-Martin et al., 1999). To identify cell surface changes, we used fluorescently labeled annexin V, which belongs to a recently discovered family of proteins with anticoagulant properties that preferentially bind to negatively charged phospholipids (Stuart et al., 1998). While the plasma membrane of PC-12 cells treated for 72 h with thiamine antagonists remained visually, intact more annexin V-positive cells were obtained after treatment with oxythiamine or pyrithiamine than with amprolium (Fig. 2A). This correlates with our previously reported data for DTNC cells (Chornyy & Parkhomenko, 2006). We next performed an analysis of activation of caspase 3, which included Western blot detection of the cleaved product in the cytoplasmic extracts (Fig. 3) and microscopic analysis of translocation of the cleaved product to the nucleus (Fig. 2B). Interestingly, active caspase 3 has been discussed to be a mediator in the process of DNA fragmentation (Schon & Manfredi, 2003).
Our data indicate that all thiamine antagonists tested trigger apoptosis via a caspase 3-dependent signaling pathway. However, pyrithiamine and oxythiamine displayed a higher potency to induce apoptotic cell death than amprolium.
We next suggested that mitochondrial dysfunction caused by ThD might also stimulate an alternative pathway of activation of mitochondria-dependent and caspase 3-independent apoptosis. This pathway may occur via activation and release of AIF followed by its translocation into the nucleus triggering chromatin condensation and DNA fragmentation (Porter & Urbano, 2006). We found that treatment of PC-12 cells with oxythiamine or pyrithiamine upregulates aif gene expression in a similar manner, and to a lesser extent after treatment with amprolium (Fig. 4A). Similar data were obtained by TUNEL analysis of DNA fragmentation (Fig. 4B).
Therefore, we propose that the observed neuronal PC-12 cell loss occurs due to apoptosis and presumably via mitochondria-dependent caspase 3-mediated signaling pathway. Furthermore, we suggest that thiamine antagonists trigger different progression of programmed cell death due to their diverse mechanisms of interference with thiamine metabolism. However, we do not exclude alternative mechanisms of non-coenzyme influence of thiamine and its antagonists on cell signalling in apoptosis.
Acknowledgements
We are grateful to Professor Fernando A. Gonzalez, from the Department of Chemistry at the University of Puerto Rico, Rio Piedras Campus, San Juan, Puerto Rico for supporting our investigation.
Abbreviations:
- AIF
apoptosis inducing factor protein
- aif
apoptosis inducing factor gene
- ANOVA
the one-way post test analyses of variance
- DITNC
rat immortalized primary cultures of type 1 astrocytes from brain diencephalon tissue
- NGF
nerve growth factor
- RT-PCR
reverse transcriptase-polymerase chain reaction
- PC-12 cells
rat pheochromocytoma PC-12 cells
- PBS
phosphate-buffered saline
- ThD
thiamine deficiency
- ThDP
thiamine diphosphate
REFERENCES
- Ali BH (1985) Effect of some monoamine oxidase inhibitors on the thiamin status of rabbits. Br J Pharmacol 86: 869–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker SJ, Yin M, Kootstra G (1996) Tissue thiamine and carnitine deficiency as a possible cause of acute tubular necrosis after renal transplantation. Transplant Proc 28: 314–315. [PubMed] [Google Scholar]
- Bettendorff L, Sluse F, Goessens G, Wins P, Grisar T (1995) Thiamine deficiency-induced partial necrosis and mitochondrial uncoupling in neuroblastoma cells are rapidly reversed by addition of thiamine. J Neurochem 65: 2178–2184. [DOI] [PubMed] [Google Scholar]
- Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248–254. [DOI] [PubMed] [Google Scholar]
- Butterworth RF, Heroux M (1989) Effects of thiamine deficiency on brain metabolism: implications for the pathogenesis of the Wernicke-Korsakoff syndrome. Alcohol Alcohol 24: 271–279. [DOI] [PubMed] [Google Scholar]
- Calingasan NY, Chun WJ, Park LC, Uchida K, Gibson GE (1999) Oxidative stress is associated with region-specific neuronal death during thiamine deficiency. J Neuropathol Exp Neurol 58: 946–958. [DOI] [PubMed] [Google Scholar]
- Chornyy SA, Parkhomenko YM (2006) Activation of apoptosis in astroglial DTNC cells during metabolic disturbance caused by thiamine antagonists. Biopolymers and Cell 22: 290–298. [Google Scholar]
- Chernikevich IP, Gritsenko EA, Lisitskaia IM, Luchko TA (1995) On vitamin B1 metabolism in avitaminosis and its correction with thiamine and taurine. Vopr Med Khim 41: 36–42. [PubMed] [Google Scholar]
- Cotman CW, Anderson AJ (1995) A potential role for apoptosis in neurodegeneration and Alzheimer’s disease. Mol Neurobiol 10: 19–45. [DOI] [PubMed] [Google Scholar]
- Chinopoulos C, Adam-Vizi V (2006) Calcium, mitochondria and oxidative stress in neuronal pathology: Novel aspects of an enduring theme. FEBS J 273: 433–450. [DOI] [PubMed] [Google Scholar]
- Daugas E, Susin SA, Zamzami N, Ferri KF, Irinopoulou T, Larochette N, Prevost MC, Leber B, Andrews D, Penninger J, Kroemer G (2000) Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J 14: 729–739. [PubMed] [Google Scholar]
- Desjardins P, Butterworth R (2005) Role of mitochondrial dysfunction and oxidative stress in the pathogenesis of selective neuronal loss in Wernicke’s encephalopathy. Mol Neurobiol 31: 17–25. [DOI] [PubMed] [Google Scholar]
- Hazell AS, McGahan L, Tetzlaff W, Bedard AM, Robertson GS, Nakabeppu Y, Hakim AM (1998) Immediate-early gene expression in the brain of the thiamine-deficient rat. J Mol Neurosci 10: 1–15. [DOI] [PubMed] [Google Scholar]
- Heroux M, Raghavendra Rao VL, Lavoie J, Richardson JS, Butterworth RF (1996) Alterations of thiamine phosphorylation and of thiamine-dependent enzymes in Alzheimer’s disease. Metab Brain Dis 11: 81–88. [DOI] [PubMed] [Google Scholar]
- Kruse M, Navarro D, Desjardins P, Butterworth RF (2004) Increased brain endothelial nitric oxide synthase expression in thiamine deficiency: relationship to selective vulnerability. Neurochem Int 45: 49–56. [DOI] [PubMed] [Google Scholar]
- Linnik MD, Zobrist RH, Hatfield MD (1993) Evidence supporting a role for programmed cell death in focal cerebral ischemia in rats. Stroke 24: 2002–2009. [DOI] [PubMed] [Google Scholar]
- Liu JY, Timm DE, Hurley TD (2006) Pyrithiamine as a substrate for thiamine pyrophosphokinase. J Biol Chem 281: 6601–6607. [DOI] [PubMed] [Google Scholar]
- Lonsdale D (2006) A review of the biochemistry, metabolism and clinical benefits of thiamin(e) and its derivatives. Evid Based Complement Alternat Med 3: 49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng L, Edmondson JW, Schenker S, Li TK (1979) Transport and metabolism of thiamin in isolated rat hepatocytes. J Biol Chem 254: 7265–7268. [PubMed] [Google Scholar]
- Martin P, Singleton CK, Hiller-Sturmhöfel S (2003) The role of thiamine deficiency in alcoholic brain disease. Alcohol Res Health 27: 174–181. [PMC free article] [PubMed] [Google Scholar]
- Oliveira FA, Galan DT, Ribeiro AM, Santos Cruz J (2007) Thiamine deficiency during pregnancy leads to cerebellar neuronal death in rat offspring: Role of voltage-dependent K(+) channels. Brain Res 1134: 79–86. [DOI] [PubMed] [Google Scholar]
- Pannunzio P, Hazell AS, Pannunzio M, Rao KV, Butterworth RF (2000) Thiamine deficiency results in metabolic acidosis and energy failure in cerebellar granule cells: an in vitro model for the study of cell death mechanisms in Wernicke’s encephalopathy. J Neurosci Res 62: 286–292. [DOI] [PubMed] [Google Scholar]
- Park LC, Calingasan NY, Uchida K, Zhang H, Gibson GE (2000) Metabolic impairment elicits brain cell type-selective changes in oxidative stress and cell death in culture. J Neurochem 74: 114–124. [DOI] [PubMed] [Google Scholar]
- Porter AG, Urbano AG (2006) Does apoptosis-inducing factor (AIF) have both life and death functions in cells? Bioessays 28: 834–843. [DOI] [PubMed] [Google Scholar]
- Rais B, Comin B, Puigjaner J, Brandes JL, Creppy E, Saboureau D, Ennamany R, Lee WN, Boros LG, Cascante M (1999) Oxythiamine and dehydroepiandrosterone induce a G1 phase cycle arrest in Ehrlich’s tumor cells through inhibition of the pentose cycle. FEBS Lett 456: 113–118. [DOI] [PubMed] [Google Scholar]
- Rindi G, Patrini C, Nauti A, Bellazzi R, Magni P (2003) Three thiamine analogues differently alter thiamine transport and metabolism in nervous tissue: an in vivo kinetic study using rats. Metab Brain Dis 18: 245–263. [DOI] [PubMed] [Google Scholar]
- Romanski SA, McMahon MM (1999) Metabolic acidosis and thiamine deficiency. Mayo Clin Proc 74: 259–263. [DOI] [PubMed] [Google Scholar]
- Rucker-Martin C, Henaff M, Hatem SN, Delpy E, Mercadier JJ (1999) Early redistribution of plasma membrane phosphatidylserine during apoptosis of adult rat ventricular myocytes in vitro. Basic Res Cardiol 94: 171–179. [DOI] [PubMed] [Google Scholar]
- Schon EA, Manfredi G (2003) Neuronal degeneration and mitochondrial dysfunction. J Clin Invest 111: 303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton CK, Martin PR (2001) Molecular mechanisms of thiamine utilization. Curr Mol Med 1: 197–207. [DOI] [PubMed] [Google Scholar]
- Stuart MC, Reutelingsperger CP, Frederik PM (1998) Binding of annexin V to bilayers with various phospholipid compositions using glass beads in a flow cytometer. Cytometry 33: 414–419. [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G (1999) Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397: 441–446. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Lazebnik Y (1998) Caspases: enemies within. Science 281: 1312–1316. [DOI] [PubMed] [Google Scholar]
- Todd K, Butterworth RF (1999) Mechanisms of selective neuronal cell death due to thiamine deficiency. Ann N Y Acad Sci USA 893: 404–411. [DOI] [PubMed] [Google Scholar]
- Tylicki A, Lempicka A, Romaniuk-Demonchaux K, Czerniecki J, Dobrzyn P, Strumilo S (2003) Effect of oxythiamin on growth rate, survival ability and pyruvate decarboxylase activity in Saccharomyces cerevisiae. J Basic Microbiol 43: 522–529. [DOI] [PubMed] [Google Scholar]
- Wang JJ, Hua Z, Fentress HM, Singleton CK (2000) JNK1 is inactivated during thiamine deficiency-induced apoptosis in human neuroblastoma cells. J Nutr Biochem 11: 208–215. [DOI] [PubMed] [Google Scholar]
- Wang X, Wang B, Fan Z, Shi X, Ke ZJ, Luo J (2007) Thiamine deficiency induces endoplasmic reticulum stress in neurons. Neuroscience 144: 1045–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]