

Abstract
Background
Obesity is an established risk factor for hypertension. Although obesity‐induced gut barrier breach leads to the leakage of various microbiota‐derived products into host circulation and distal organs, the roles of microbiota in mediating the development of obesity‐associated adrenomedullary disorders and hypertension have not been elucidated. We seek to explore the impacts of microbial DNA enrichment on inducing obesity‐related adrenomedullary abnormalities and hypertension.
Methods and Results
Obesity was accompanied by remarkable bacterial DNA accumulation and elevated inflammation in the adrenal glands. Gut microbial DNA containing extracellular vesicles (mEVs) were readily leaked into the bloodstream and infiltrated into the adrenal glands in obese mice, causing microbial DNA enrichment. In lean wild‐type mice, adrenal macrophages expressed CRIg (complement receptor of the immunoglobulin superfamily) that efficiently blocks the infiltration of gut mEVs. In contrast, the adrenal CRIg+ cell population was greatly decreased in obese mice. In lean CRIg−/− or C3−/− (complement component 3) mice intravenously injected with gut mEVs, adrenal microbial DNA accumulation elevated adrenal inflammation and norepinephrine secretion, concomitant with hypertension. In addition, microbial DNA promoted inflammatory responses and norepinephrine production in rat pheochromocytoma PC12 cells treated with gut mEVs. Depletion of microbial DNA cargo markedly blunted the effects of gut mEVs. We also validated that activation of cGAS (cyclic GMP‐AMP synthase)/STING (cyclic GMP–AMP receptor stimulator of interferon genes) signaling is required for the ability of microbial DNA to trigger adrenomedullary dysfunctions in both in vivo and in vitro experiments. Restoring CRIg+ cells in obese mice decreased microbial DNA abundance, inflammation, and hypertension.
Conclusions
The leakage of gut mEVs leads to adrenal enrichment of microbial DNA that are pathogenic to induce obesity‐associated adrenomedullary abnormalities and hypertension. Recovering the CRIg+ macrophage population attenuates obesity‐induced adrenomedullary disorders.
Keywords: adrenomedullary inflammation, catecholamine, CRIg+ macrophage, microbial DNA, obesity‐induced hypertension
Subject Categories: Hypertension, High Blood Pressure
Nonstandard Abbreviations and Acronyms
- CRIg
complement receptor of the immunoglobulin superfamily
- HFD
high‐fat diet
- KC
Kupffer cells
- mEVs
microbial DNA containing intestinal extracellular vesicles
Clinical Perspective
What Is New?
Gut extracellular vesicles connect host adrenomedullary cells and microbiota in obesity.
Microbial DNA containing intestinal extracellular vesicles promote obesity‐associated adrenal gland inflammation and dysfunction.
What Are the Clinical Implications?
Restoring CRIg+ (complement receptor of the immunoglobulin superfamily) macrophage population could blunt obesity‐induced adrenomedullary disorders.
Obesity is recognized as an independent risk factor for arterial hypertension. It has been reported by the Framingham Heart Study that 60% to 70% of essential hypertension results from obesity, and patients who are obese are 3.5 times more susceptible to develop high blood pressure. 1 , 2 Although various mechanisms underlying obesity‐associated hypertension have been suggested, including increased sympathetic tone, 3 , 4 , 5 overactivation of the renin–angiotensin system, 6 elevation of aldosterone levels, endothelial dysfunction, 7 and mechanical compression of the kidney, 8 it is yet to be established how obesity triggers the above changes to cause hypertension. Because dysregulated secretion of norepinephrine and epinephrine is a feature in chronic obesity in both animal and human models, 9 we sought to explore whether microbial DNA enrichment because of obesity‐induced increase in gut permeability causes inflammation of the adrenal medulla, 10 resulting in increased secretion of catecholamines and the subsequent development of hypertension. Although chronic and low‐degree tissue inflammation is a hallmark of obesity, the role of obesity‐induced adrenal inflammation in mediating the adrenomedullary function is much less clearly defined. 10 , 11 , 12
Gut barrier breach is one of the characteristics of obesity, resulting in elevated levels of translocation of microbiota‐derived products into host circulation and distal tissues. 13 , 14 , 15 , 16 , 17 , 18 Compelling evidence indicates that obesity is concomitant with microbial DNA enrichment in host circulation and tissues. 19 , 20 , 21 , 22 , 23 In addition, several studies have suggested that circulating microbial DNA species could be biomarkers that precisely mirror the stages of metabolic disease development. 19 , 22 , 24 Extracellular vesicles (EVs) are important carriers harboring various cargo, such as RNA, DNA, lipids, and proteins, transporting through circulation between the neighboring or distant cells. 25 It has been known that microbiota can secrete EVs. 26 , 27 More importantly, our previous study has shown that microbiota‐derived bacterial DNA‐containing EVs (mEVs) are readily translocated from the gut lumen into the host bloodstream and distal metabolic tissues in obesity, resulting in obesity‐related tissue inflammation and insulin resistance. 28 Therefore, these lead us to hypothesize that, in the context of obesity, gut mEVs could infiltrate into adrenal glands and cause microbial DNA accumulation that triggers adrenomedullary inflammation and dysfunction.
Although liver immunoglobulin superfamily (CRIg [complement receptor of the immunoglobulin superfamily])+ macrophages play important roles in filtering bacteria and their byproducts in the blood flowing from the intestine through the hepatic portal vein, there is a remarkable reduction in CRIg+ macrophages in the context of obesity. 28 , 29 , 30 We have shown that CRIg+ macrophages can capture gut mEVs from the bloodstream through a complement protein C3 (complement component 3)‐mediated mechanism, whereas depletion of either CRIg or C3 leads to the spread of gut mEVs into distal tissues. 28 , 30 , 31 , 32 , 33 In addition to the liver, CRIg+ macrophages also reside in other tissues such as pancreatic islets. 34 , 35 An early study also reports that human adrenal glands harbor a high level of CRIg+ macrophages. 30 However, whether adrenal gland resident CRIg+ macrophages exert protection from the infiltration of gut mEVs and bacterial DNA accumulation is still unknown.
Here, we report that microbial DNA enrichment causes adrenomedullary dysfunction in the context of obesity, resulting in elevated circulating norepinephrine levels and the subsequent development of hypertension. Adrenal gland CRIg+ macrophages exert sufficient protection from the penetration of gut mEVs, whereas the CRIg+ cell population is greatly reduced in obese adrenal glands. We further confirm that microbial DNA cargo contributes to the effects of gut mEVs by activating the cGAS (cyclic GMP‐AMP synthase)/STING (cyclic GMP–AMP receptor stimulator of interferon genes) pathway in adrenal chromaffin cells.
Methods
The data that support the findings of this study are available from the corresponding author on reasonable request.
Animal Care and Use
cGAS−/− (stock number 026554), C3−/− (stock number 029661), Cre‐inducible DTR mice (ROSA26iDTR) (stock number 007900), and C‐type lectin domain family 4, member f promoter‐driven Cre recombinase (Clec4f‐Cre) (stock number 033296) mice were received from the Jackson Laboratory. CRIg−/− mice (C57BL/6J background) were a generous gift from Dr Wenxian Fu (University of California, San Diego, CA). CRIg wild‐type (WT) mice were produced by crossing CRIg heterozygous mice together. All mice were maintained on a 12:12‐hour light–dark cycle. At 8 weeks of age, male mice were used as recipients for EV injection or fed ad libitum a high‐fat diet (HFD; 60% fat calories, 20% protein calories, and 20% carbohydrate calories; Research Diets) or a normal chow diet (Lab Diet) for various durations. Germ‐free C57BL/6 mice were maintained in the University of California, San Diego Gnotobiotic Mouse Facility. Germ‐free mice were fed an irradiated sterilized 60% HFD (06414; Teklad). To deplete Kupffer cells (KC), diphtheria toxin was intraperitoneally (200 ng/mouse) injected into Clec4fCre+DTR+ lean mice (KC‐KO) for 3 days, and these mice were treated with diphtheria toxin (200 ng/mouse) every 2 days to prevent KC recovery. For tissue collection, mice were anesthetized with an intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg), followed by euthanizing by cervical dislocation. All animal procedures were done in accordance with University of California, San Diego Research Guidelines for the Care and Use of Laboratory Animals, and all animals were randomly assigned to cohorts when used.
Mouse Blood Pressure Detection
In regard to noninvasive tail‐cuff measurement of blood pressure, systolic blood pressure was measured indirectly using tail‐cuff plethysmography in a mouse tail‐cuff blood pressure system (IITC Life Sciences, Woodland Hills, CA) as described previously. 36 Before any measurements, the plexiglass restrainers were warmed in 34 °C warming chambers for 20 minutes. The mice were then loaded into their restrainers and allowed to incubate in the same warming chambers for 10 to 15 minutes. The tails were placed inside inflatable cuffs with a photoelectric sensor that measured tail pulses. Systolic blood pressure was measured over 5 separate days with an average of at least 2 well‐defined blood pressure cones detected by the tail‐cuff plethysmograph per day.
Measurement of Catecholamines
Mice were anesthetized by inhalation of isoflurane, and blood was collected from the heart in potassium‐EDTA tubes. Plasma catecholamines were measured by an ACQUITY UPLC H‐Class System fitted with an Atlantis dC18 column (100A, 3 µm, 3×100 mm) and connected to an electrochemical detector (ECD model 2465; Waters, Milford, MA). The mobile phase (isocratic: 0.3 mL/min) consisted of phosphate‐citrate buffer and acetonitrile at 95:5 (vol/vol). The data were analyzed using Empower 3 software (Waters). Catecholamine levels were normalized with the recovery of the internal standard 3,4‐dihydroxybenzylamine. Catecholamines were expressed as nanomole per liter (plasma) or nanogram per milligram protein (rat pheochromocytoma PC12 cells) or picograms per milligram protein (kidney).
In Vivo and In Vitro EV Treatment
For in vitro assays, 1×107 EVs as determined by NanoSight analysis were added to 0.1×106 for 24 hours. In addition, for the in vitro rat pheochromocytoma PC12 catecholamine production assay, 5×108 EVs were added to 5×106 rat pheochromocytoma PC12 cells. For in vivo treatment, recipient mice were tail‐vein injected with 5×109 EVs twice per week. Our previous study has shown that the amount of bacterial DNA within 5×109 gut mEVs isolated from HFD WT mice is close to the amount of bacterial DNA in the bloodstream of 16 weeks HFD WT mice. 28
EV Purification and Characterization
The intestinal EVs were prepared from the small intestine lumen contents of 16 weeks HFD mice with sterile tools. Debris and dead cells in the lumen contents were removed by centrifugation at 1000g for 10 minutes and then filtered through a 0.2‐µm filter. The supernatant was then subjected to ultracentrifugation at 100 000g for 4 hours at 4 °C with a Type 70 Ti fixed‐angle rotor (Beckman Coulter). The EV‐containing pellet was resuspended in 1‐mL sterile PBS and passed through a 0.2‐µm filter to remove large particles. The particle size and concentration of intestinal EVs were measured by NanoSight analysis (Malvern Instruments, Westborough, MA). For electron microscopy, EVs were fixed with 2% paraformaldehyde and loaded on Formvar and carbon‐coated copper grids. Then, the grids were placed on 2% gelatin at 37 °C for 20 minutes, rinsed with 0.15 mol/L glycine/PBS, and the sections were blocked using 1% cold water fish‐skin gelatin. Grids were viewed using a JEOL 1200EX II (JEOL) transmission electron microscope and photographed using a Gatan digital camera (Gatan). To monitor EV trafficking, EVs were labeled with PKH26 fluorescent dye using the PKH26 fluorescent cell linker kit (catalog number PKH26GL‐1KT; Sigma). After PKH26 staining, the EVs were washed with sterile PBS and collected by ultracentrifugation (100 000g for 2 hours) at 4 °C. Finally, PKH26‐labeled EVs were resuspended in sterile PBS, and the particle concentration was calculated by NanoSight analysis.
In Vivo EV Trafficking Assays
PKH26‐labeled EVs (5×109 Evs per mouse) were delivered to either normal chow diet or HFD recipient mice through either injection into the tail vein or jejunum section. Sixteen hours after EV injection, adrenal glands were collected for detection of the appearance of PKH26 red fluorescence.
DNA Depletion in Intestinal EVs
The intestinal EV pellet was dissolved in 100‐µL PBS. As previously described, these EVs were loaded into a Gene Pulser/MicroPulser Cuvettes (Bio‐Rad) for electroporation (GenePulser Xcell electroporator; Bio‐Rad) and then treated with DNase (deoxyribonuclease) I (300 U) for 30 minutes at 37 °C. The depletion of bacterial DNA was confirmed by 16s ribosomal RNA (16s rRNA) quantitative polymerase chain reaction (qPCR) analysis.
Quantification of Bacterial DNA Using Real‐Time Polymerase Chain Reaction
Levels of bacterial DNA were assessed by qPCR using a Femto Bacterial DNA Quantification Kit (catalog number E2006; Zymo Research) by following the manufacturer’s instructions. Briefly, bacterial DNA were extracted from EVs or plasma using the ZymoBIOMICS DNA extraction kits (catalog number D4301; Zymo Research) according to the manufacturer’s instructions. The concentration of bacterial DNA in each sample was determined from the standard curve using a nonlinear regression 4‐parameter variable slope analysis.
siRNA Transfection
Small interfering RNA‐cGAS (catalog number J‐055608‐09‐0002; Horizon; 20 pmol small interfering RNA/0.1×106 rat pheochromocytoma PC12 cells) was transfected into recipient cells with the lipofectamine RNAiMAX reagent (catalog number 13778075; ThermoFisher). Small interfering RNA‐cGAS was mixed with RNAiMAX reagent and then incubated at room temperature for 15 minutes. This mixture was added into the medium of cells. After 24 hours, cells were collected and measured.
CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)‐Cas9 (CRISPR‐Associated Protein 9) System for Transcriptional Activation
Plasmids containing deactivated Cas9‐VPR (VP64, p65, and Rta) system (catalog number CAS11915) or guide RNA for V‐set and immunoglobulin domain containing 4 (Vsig4) (catalog number GSGM11893‐247477006) were obtained from Horizon. The lentivirus packing these plasmids were prepared by the University of California, San Diego vector core. Twelve weeks HFD WT mice were intravenously injected with these lentiviruses (1×108 particles per mouse). Control mice were treated with lentivirus containing deactivated Cas9‐VPR and control guide RNA (catalog cnumber VSGC10215; Horizon). After 4 weeks, CRIg expression in adrenal glands was evaluated by Western blot analysis.
Immunofluorescence Staining
Adrenal glands of normal chow diet– or HFD‐fed mice were snap frozen in optimum cutting temperature compound (Fisher Healthcare) with dry ice. Next, 6‐µm cryosections of tissue were cut and fixed with precold acetone for 20 minutes. Slides were blocked with 5% normal donkey serum for 60 minutes at room temperature. The samples were then incubated with anti‐CRIg (catalog number 17‐5752‐82; ThermoFisher) antibody diluted 1:100 in PBS at 4 °C overnight. After washing, nuclei were stained with 4′,6‐diamidino‐2‐phenylindole for 10 minutes at room temperature. Mounting media and cover slips were then added to slides for imaging. Images were acquired on a Keyence Fluorescent Microscope and processed with ImageJ (National Institutes of Health, Bethesda, MD).
RNAscope In Situ Hybridization Combined With Immunofluorescence
We performed RNAscope in situ hybridization to detect 16s rRNA. Mouse adrenal glands were frozen in optimum cutting temperature compound with dry ice. Next, 10‐µm cryosections of tissue were prepared and fixed with 4% Paraformaldehyde for 15 minutes at 4 °C and then dehydrated with 50%, 70%, and 100% ethyl alcohol gradients for 5 minutes each at room temperature. Tissue sections were then treated with hydrogen peroxide and protease IV at room temperature for 10 minutes each. 16s RNA probes (catalog number 464461; Advanced Cell Diagnostics) were then added for 2 hours at 40 °C. Signal amplification and detection reagents were applied sequentially and incubated in AMP 1, AMP 2, AMP 3, HRP‐C1 (RNAscope Multiplex fluorescent reagent kit v2, catalog number 323100; Advanced Cell Diagnostics), Opal 520 (catalog number PNFP1487001KT; Akoya Biosciences), or Opal 690 (catalog number FP1497001KT; Akoya Biosciences). Then, samples were immediately processed for immunofluorescence. Images were captured using Leica SP8 confocal microscope.
Isolation of Adrenal Chromaffin Cells
Mouse adrenal glands were digested in Hanks’ balanced salt solution containing papain (40 U/mL) as described in previous studies. 37 , 38
Flow Cytometry Analysis
Cells were dispersed and then stained with fluorescence‐tagged antibodies. These cells were analyzed by MA900 flow cytometer (Sony). Data were analyzed using Flowjo software.
Quantitative Reverse Transcriptase‐Polymerase Chain Reaction Analysis
Total RNA was extracted using the RNA extraction protocol according to the manufacturer’s instructions. Complementary DNA was synthesized using SuperScript III and random hexamers (high‐capacity complementary DNA reverse transcription kit, catalog number 4368813; ThermoFisher). qPCR was performed in 10‐μL reactions using the iTaq SYBR Green supermix (catalog number 172‐5125; Bio‐Rad) on a StepOnePlus Real‐Time PCR System (ThermoFisher). The data presented correspond to the mean of 2−ΔΔCt from at least 3 independent experiments after being normalized to β‐actin.
Western Blot Analysis
Cells or tissues were homogenized in RIPA buffer supplemented with protease and phosphatase inhibitors. Equal amounts of cell lysate proteins (30 μg of protein per lane for phosphorylated cyclic GMP–AMP receptor stimulator of interferon genes (pSTING), 10 μg of protein per lane for cGAS or CRIg detection) from each biological replicate were subjected to Western blotting. Using the ChemiDoc XRS imaging system (Bio‐Rad), the protein bands on blots were detected with the SuperSignal West Pico Chemiluminescent Substrate (catalog number 34077; ThermoFisher). Protein bands were analyzed using Image Lab software (Bio‐Rad). Western blot data in figures and supplemental figures are all representative of >3 independent experiments. pSTING (catalog number 72971), STING (catalog number 50494), and cGAS (catalog number 316595) antibodies were obtained from Cell Signaling Technology.
Statistical Analysis
Tests used for statistical analyses are described in the figure legends. To assess whether the means of 2 groups were statistically different from each other, an unpaired 2‐tailed Student t test was used for statistical analyses using Prism 8 software (version 8.0; GraphPad, La Jolla, CA). P values of 0.05 or less were considered to be statistically significant. Degrees of significance were indicated in the figure legends.
Results
Bacterial DNA Is Enriched in Adrenal Glands in the Context of Obesity
Previous studies have shown that obesity is accompanied with the accumulation of bacterial DNA in host circulation and tissues. 22 , 24 , 28 In line with these findings, we detected high levels of 16s rRNA in the adrenal glands of 16 weeks high‐fat diet (16 weeks HFD) fedWT mice, whereas no bacterial DNA was detected in lean adrenal glands (Figure 1A). In addition, qPCR analysis with 16s rRNA primers indicates that obese adrenal glands harbored much more 16s rRNA, compared with barely detectable 16s rRNA abundance in lean adrenal glands (Figure 1B). Concomitant with bacterial DNA enrichment in obesity, there were greater levels of proinflammatory cytokines in the adrenal glands of obese WT mice than that in lean WT mice (Figure 1C). To examine the impact of external bacterial DNA contamination during these experiments, we also measured 16s rRNA abundance in the tissues of HFD‐fed germ‐free mice using the same protocol. As shown in Figure S1A, there was no detectable 16s rRNA in these germ‐free tissues, thus validating a minimal level of exogenous bacterial DNA contamination in our assays. In addition, after 2 weeks of antibiotics treatment, bacterial DNA abundance was greatly reduced in obese adrenal glands (Figure S1B).
Figure 1. Obesity is associated with bacterial DNA enrichment and inflammation in the adrenal glands.
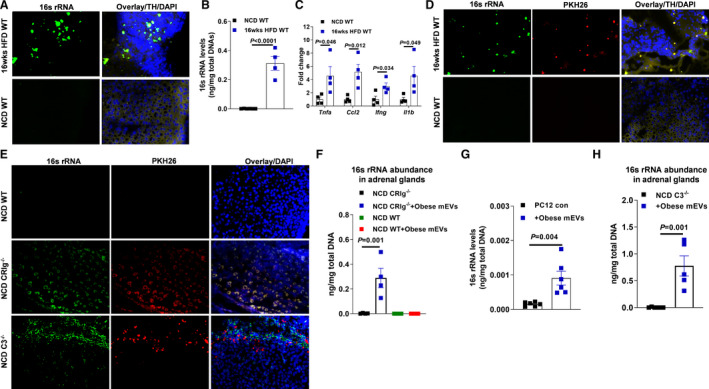
A, 16s rRNA abundance in the adrenal glands of both normal chow diet (NCD) or high‐fat diet (16 weeks HFD)‐fed wild‐type (WT) mice. B, Quantitative polymerase chain reaction (qPCR) analysis of 16s rRNA abundance in mouse adrenal glands. C, The expression levels of proinflammatory genes in the adrenal glands of NCD and 16 weeks HFD WT mice. D, The levels of PKH26 red fluorescence and 16s rRNA in the adrenal glands after injection of PKH26‐labeled gut microbial DNA‐containing extracellular vesicles (mEVs) into the jejunum sections of NCD or 16 weeks HFD WT mice. E, The abundance of PKH26 red fluorescence and 16s rRNA in the adrenal glands after intravenous injection of PKH26 gut mEVs into lean WT, CRIg−/− (complement receptor of the immunoglobulin superfamily), or C3−/− (complement component 3) mice. F, qPCR analysis of 16s rRNA levels in the adrenal glands of NCD WT or CRIg−/− mice after 4 weeks of treatment with obese mEVs. G, The abundance of 16s rRNA in rat pheochromocytoma PC12 cells after 24 hours of treatment with obese mEVs. H, 16s rRNA enrichment in the adrenal glands of lean C3−/− mice after 4 weeks of treatment with obese mEVs. Data are representative of 3 experiments (A, D, and E). Data are presented as mean±SEM. P values are determined by unpaired 2‐sided Student t test (B, C, G, and H) or 1‐way ANOVA analysis (F). DAPI indicates 4',6‐Diamidino‐2‐28 phenylindole dihydrochloride; TH tyrosine hydroxylase; and 16s rRNA, 16s ribosomal RNA.
Gut mEVs can pass through the impaired gut barrier into host circulation and distal tissues in the context of obesity. 17 Thus, to assess whether intestinal mEVs were translocated from gut lumen into host adrenal glands, gut mEVs were collected from the small intestinal lumen of 16 weeks HFD‐fed WT mice and labeled with PKH26 red fluorescent dye (Figure S1C through S1F). These PKH26 mEVs (5×109 EVs per mouse) were injected into the jejunum section of either lean or 12 weeks HFD/obese WT recipient mice. After 24 hours, we observed robust red fluorescent signals in the adrenal glands of obese recipients, demonstrating the leakage of PKH26 mEVs into adrenal glands in the context of obesity (Figure 1D). By contrast, PKH26 signals were barely detected in the adrenal glands of lean WT recipient mice (Figure 1D). Taken together, these data indicate that the obesity‐induced impaired gut barrier allows for the translocation of gut mEVs into adrenal glands.
Loss of CRIg+ Macrophages Lead to the Infiltration of Gut mEVs Into Adrenal Glands
Our previous study has shown that CRIg+ macrophages play a critical role in blocking the translocation of gut mEVs into host tissues, whereas the population of CRIg+ macrophages is remarkably diminished in both obese humans and mice. 28 Consistently, after an intravenous injection with PKH26 mEVs (5×109 EVs per mouse), we observed that red fluorescent signals were spread in the adrenal glands of lean CRIg−/− mice (Figure 1E). Given that gut EVs harbored bacterial DNA, the spread of gut EVs led to the enrichment of bacterial DNA in these tissues (Figure 1E and Figure S1E). In addition, qPCR analysis confirmed that 16s rRNA abundance was significantly increased in the adrenal glands of lean CRIg−/− mice after 4 weeks of treatment with gut mEVs (5×109 EVs per mouse, 2 injections per week; Figure 1F). We also observed a remarkable increase in 16s rRNAs within adrenal gland cell rat pheochromocytoma PC12 after 24 hours of treatment with obese mEVs (Figure 1G).
Complement protein C3 plays important roles in modulating the ability of CRIg+ macrophages to interact with gut mEVs. 28 , 30 Consistent with our previous observation, loss of C3 blunted the interaction between CRIg+ macrophages and gut mEVs, as evidenced by robust red fluorescent signals in the adrenal glands of lean C3−/− mice intravenously injected with PKH26 mEVs (5×109 EVs per mouse; Figure 1E). Concomitant with the infiltration of gut mEVs, bacterial DNA was enriched in the adrenal glands of lean C3−/− mice injected with gut mEVs (Figure 1H). Thus, these results indicate the importance of the CRIg–C3 regulatory axis on blocking the infiltration of gut mEVs.
Gut mEV Penetration Elevates Inflammatory Responses and Norepinephrine Secretion From Adrenal Medulla
Given that the infiltration of gut mEVs can result in host cell dysfunction, we next evaluated the impact of gut mEVs on the responses of adrenal glands in either lean CRIg−/− or C3−/− mice. After 4 weeks of intravenous injection with gut mEVs (5×109 EVs per mouse, 2 injections per week), lean CRIg−/− or C3−/− mice displayed elevated levels of inflammatory responses in adrenal glands compared with control mice treated with empty liposomes, as shown by greater abundance of proinflammatory cytokines in adrenal glands (Figure 2A and 2B). In addition, there were greater levels of circulating norepinephrine, but not epinephrine, in lean CRIg−/− or C3−/− mice after 4 weeks of treatment with gut mEVs (Figure 2C and 2D). However, gut mEV treatment did not affect the concentration of norepinephrine in the kidneys and the expression of monoamine oxidase in adrenal glands of lean CRIg−/− mice (Figure S2A and S2B). Consistent with gut mEV‐induced increase in circulating norepinephrine levels, we also observed that gut mEVs‐treated lean CRIg−/− or C3−/− mice exhibited augmented blood pressure than control mice (Figure 2E and 2F). By contrast, gut mEV treatment had minimal effects on the adrenal gland responses in lean WT mice, as evidenced by comparable levels of adrenal gland inflammation and blood pressure among lean WT recipient mice (Figures 1F and 2G and Figure S2C). In addition, without gut mEV treatment, lean CRIg−/− mice had comparable blood pressure with lean WT mice (Figure S2D). Given that marked in vivo effects of gut mEVs, we also evaluated the direct impacts of gut mEVs on cellular responses in rat pheochromocytoma PC12 cells. After 24 hours of treatment with gut mEVs (1×107 EVs/0.1×106 cells), rat pheochromocytoma PC12 cells expressed greater levels of proinflammatory cytokines than the control cells treated with empty liposomes (Figure 2H). In addition, gut mEV treatment significantly induced synthesis of norepinephrine in rat pheochromocytoma PC12 cells (Figure 2I and Figure S2E). We also observed that adrenal chromaffin cells isolated from lean WT mice resulted in increased expression of proinflammatory genes after gut mEV treatment (Figure S2F). Taken together, in the context of loss of CRIg+ macrophages, infiltration of gut mEVs causes functional abnormalities in adrenal glands, contributing to elevated blood pressure.
Figure 2. Gut microbial DNA‐containing extracellular vesicles (mEVs) induces adrenal inflammation and dysfunction.
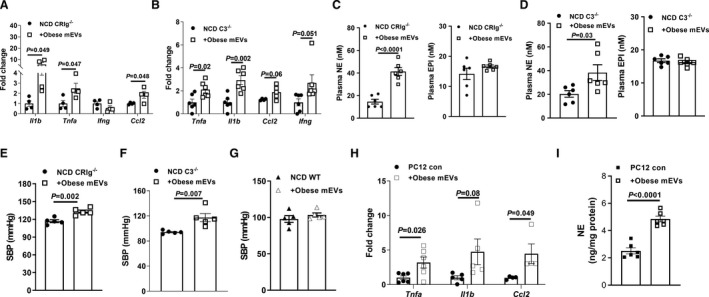
Quantitative polymerase chain reaction analysis of proinflammatory genes in the adrenal glands of either normal chow diet (NCD) CRIg−/− (complement receptor of the immunoglobulin superfamily) (A) or C3−/− (complement component 3) (B) mice after 4 weeks of treatment with obese mEVs. The effect of obese mEVs on circulating epinephrine (EPI) and norepinephrine (NE) levels in either lean CRIg−/− (C) or C3−/− (D) mice. Systolic blood pressure (SBP) levels in either lean CRIg−/− (E), C3−/− (F), or wild‐type (WT) (G) mice injected with obese mEVs. The abundance of proinflammatory gene abundance (H) and NE production levels (I) of rat pheochromocytoma PC12 cells after 24 hours treatment with obese mEVs. Data are presented as mean±SEM. P values are determined by unpaired 2‐sided Student t test (A through I).
Adrenal Gland Resident CRIg+ Macrophages Are Sufficient to Prevent the Effects of Gut mEVs
Although the liver contains a large amount of CRIg+ macrophages, earlier studies have suggested that adrenal gland is also one of the main sites harboring CRIg+ macrophages. 30 We can detect a group of CRIg+ cells in the adrenal glands of lean WT mice (Figure 3A). More importantly, in adrenal glands, almost all CRIg signals were colocalized with F4/80+ cells, suggesting a majority of adrenal gland resident macrophages express CRIg (Figure 3A). By contrast, the population of CRIg+F4/80+ cells were greatly reduced in obese adrenal glands (Figure 3A and Figure S3A and S3B). We next assessed whether adrenal gland CRIg+ macrophages, in the context of knockout of liver CRIg+ macrophages, protect adrenal glands from the infiltration of gut mEVs. Given that in the liver, CRIg is mainly expressed on KC, 28 , 29 , 30 Clec4fCre+DTR+ lean mice were treated with diphtheria toxin (200 ng/mouse, continuous 3 days of intraperitoneal injection) to deplete liver CRIg+ macrophages (KC‐KO mice; Figure S3C). In addition, diphtheria toxin treatment did not affect CRIg abundance in adrenal glands (Figure S3D and S3E). More importantly, there was no bacterial DNA detected in the adrenal glands of lean KC‐KO mice after 4 weeks of intravenous injection with gut mEVs (5×109 EVs per mouse, 2 injections per week; intraperitoneal injections of 200 ng diphtheria toxin per mouse every 2 days) (Figure 3B). In addition, after 2 hours of intravenous injection of PKH26‐labeled gut mEVs (5×109 EVs per mouse), adrenal macrophages in lean WT mice showed elevated PKH26 signals, compared with lean WT control mice without EV injection (Figure 3C). In contrast, in lean CRIg−/− mice, adrenal macrophages failed to capture PKH26 EVs, as evidenced by comparable low levels of PKH26 signals among lean CRIg−/− mice (Figure 3D). Consistent with the absence of bacterial DNA in the adrenal glands after 4 weeks of treatment with gut mEVs, lean KC‐KO mice had similar levels of adrenal gland inflammation and blood pressure with control KC‐KO mice (Figure 3E and 3F). Thus, these results demonstrate that CRIg+ macrophages residing in the adrenal glands exert profound protection from the penetration of gut mEVs.
Figure 3. Adrenal gland resident CRIg+ (complement receptor of the immunoglobulin superfamily) macrophages exert profound protection from the infiltration of gut microbial DNA‐containing extracellular vesicles (mEVs).
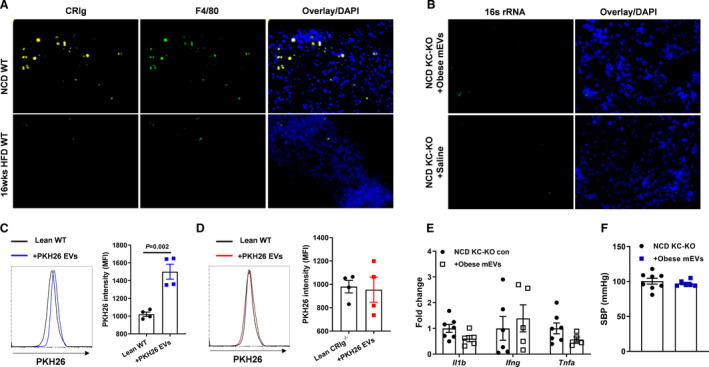
A, CRIg abundance within the adrenal glands of normal chow diet (NCD) or 16 weeks high‐fat diet (16 weeks HFD) wild‐type (WT) mice. B, The 16s rRNA levels in the adrenal glands of lean Kupffer cell‐depleted (KC‐KO) mice after 4 weeks of treatment with obese mEVs. After 2 hours of intravenous injection of PKH26‐labeled gut mEVs, the intensity of PKH26 signal was detected in adrenal macrophages of lean WT (C) or CRIg−/− (D) mice by flow cytometry analysis. Lean mice without EV injection were used as controls. After 4 weeks of injection with obese mEVs, proinflammatory gene abundance in the adrenal glands (E) and systolic blood pressure (SBP) (F) of lean KC‐KO mice. Data are the representative of 3 experiments (A and B). Data are presented as mean±SEM. P values are determined by unpaired 2‐sided Student t test (C through F). DAPI indicates 4',6‐Diamidino‐2‐28 phenylindole dihydrochloride; EV, extracellular vesicle; KC‐KO, depletion of Kupffer cells; MFI, median fluorescence intensity; and 16s rRNA, 16s ribosomal RNA.
Bacterial DNA Cargo Contribute to the Pathogenic Effects of Gut mEVs
Concomitant with the infiltration of gut mEVs, the abundance of bacterial DNA was remarkably elevated in the adrenal glands of lean CRIg−/− or C3−/− mice. In addition, our previous study has demonstrated the critical roles of bacterial DNA cargo within gut EVs. 28 We prepared DNA‐free gut EVs using our previously reported method (Figure S4A). 28 As expected, treatment with DNA‐free gut EVs did not cause bacterial DNA accumulation in rat pheochromocytoma PC12 cells (Figure S4B). Consistent with our previous findings, 28 depletion of bacterial DNA cargo blunted the effects of gut mEVs, as evidenced by nonsignificant effects of DNA‐free gut EVs on inflammatory responses and norepinephrine production in rat pheochromocytoma PC12 cells (Figure 4A and 4B and Figure S4C). In contrast, gut mEV treatment led to functional abnormalities in rat pheochromocytoma PC12 cells (Figure 4A and 4B and Figure S4C). In addition, we confirmed that DNA‐free EV treatment did not lead to bacterial DNA enrichment in the adrenal glands of lean CRIg−/− (Figure S4D). More importantly, these CRIg−/− mice had no significant changes in adrenal gland inflammation, circulating catecholamine levels, and blood pressure after treatment with DNA‐free gut EVs (Figure 4C through 4E). We also confirmed that microbiota‐derived EVs contributed to the pathogenic effects of gut EVs, as evidenced by minimal effects of gut EVs collected from 12 weeks HFD germ‐free WT mice on rat pheochromocytoma PC12 cells (Figures S1C, S1D, S1F, S4A, and S4E). To test the possibility that bacterial DNA could be bound outside of the EV membrane, we treated obese gut mEVs with DNase, and then used these EVs to treat rat pheochromocytoma PC12 cells. After 24 hours, these gut mEVs resulted in elevated inflammatory responses in rat pheochromocytoma PC12 cells (Figure S4F). Thus, these data demonstrate that microbial DNA is the key cargo contributing to the pathogenic effects of intestinal EVs.
Figure 4. Microbial DNA is pathogenic cargo for the effects of gut extracellular vesicles (EVs).
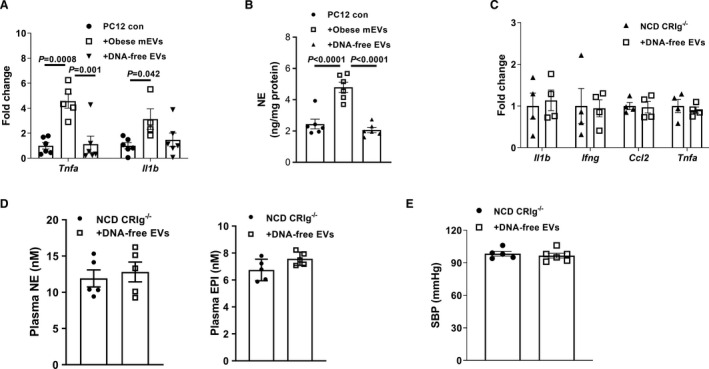
A, Quantitative polymerase chain reaction analysis of proinflammatory genes in rat pheochromocytoma PC12 cells after treatment with either obese microbial DNA containing intestinal extracellular vesicles (mEVs) or DNA‐free gut EVs. B, Effect of obese mEVs or DNA‐free EVs on norepinephrine (NE) production of rat pheochromocytoma PC12 cells. Proinflammatory gene abundance (C), circulating catecholamine levels (D), and systolic blood pressure (SPB) (E) of normal chow diet (NCD) CRIg−/− (complement receptor of the immunoglobulin superfamily). Data are presented as mean±SEM. P values are determined by 1‐way ANOVA analysis (A and B). EPI indicates epinephrine.
cGAS/STING Signaling Is Critical for the Ability of Microbial DNA to Induce Adrenal Gland Dysfunction
Previous studies, including ours, have demonstrated that the activation of the cGAS/STING pathway is required for bacterial DNA‐induced cellular responses. 28 , 39 , 40 Concomitant with bacterial DNA accumulation, we observed an enhancement on the abundance of cGAS and phosphorylated STING in the adrenal glands of obese WT mice, compared with lean WT mice (Figure 5A). After 4 weeks of treatment with gut mEVs, either lean CRIg−/− or C3−/− mice had greater levels of activation of cGAS/STING signaling in the adrenal glands (Figure 5B and Figure S5A). In addition, in vitro treatment with gut mEVs led to increased activation of the cGAS/STING pathway in rat pheochromocytoma PC12 cells (Figure 5C). By contrast, consistent with the absence of bacterial DNA accumulation, gut mEV treatment did not change the abundance of cGAS and phosphorylated STING in the adrenal glands of lean KC‐KO mice (Figure S5B).
Figure 5. cGAS/STING signaling activation is critical for the effects of microbial DNA.
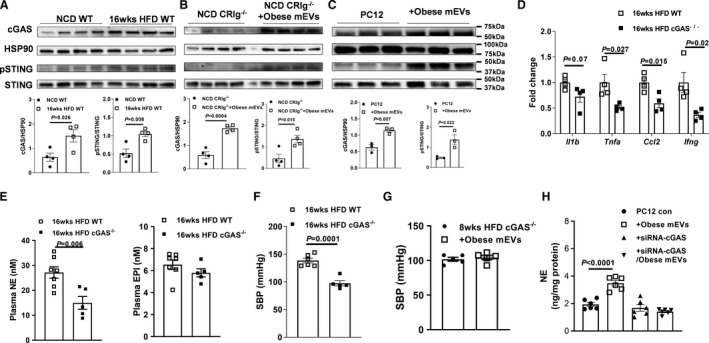
A, The abundance of cGAS, phospho‐STING, and total STING in the adrenal glands of both normal chow diet (NCD) and 16 weeks high‐fat diet (16 weeks HFD) wild‐type (WT) mice. B, Effects of obese gut microbial DNA‐containing extracellular vesicles (mEVs) on the activation of cGAS/STING signaling in NCD CRIg−/− (complement receptor of the immunoglobulin superfamily) adrenal glands (B) or rat pheochromocytoma PC12 cells (C). The levels of adrenal inflammation (D), circulating catecholamine (E), and systolic blood pressure (SBP) (F) of 16 weeks HFD WT vs cGAS−/− mice. Effects of obese mEVs on the levels of SBP (G) of 16 weeks HFD cGAS−/− mice. H, The norepinephrine (NE) production of rat pheochromocytoma PC12 cells treated with obese mEVs and/or siRNA‐cGAS. Data are presented as mean±SEM. P values are determined by unpaired 2‐sided Student t test (A through F) or 1‐way ANOVA analysis (H). cGAS indicates Cyclic GMP–AMP synthase ; EPI, epinephrine; HSP90, heat shock protein 90; pSTING, phosphorylated STING; siRNA, small interfering RNA; and STING, the cyclic GMP–AMP receptor stimulator of interferon genes.
To further assess the importance of the cGAS/STING pathway on the ability of microbial DNA to induce adrenal gland dysfunction, we compared the adrenal inflammation, circulating catecholamine levels, and blood pressure between 16 weeks HFD WT and cGAS−/− mice. As shown in Figure 5D through 5F, cGAS depletion significantly reduced the levels of obesity‐induced adrenal inflammation, plasma norepinephrine levels, and blood pressure, compared with obese WT mice. In addition, another cohort of 8 weeks HFD cGAS−/− mice were intravenously injected with gut mEVs (5×109 EVs/mouse, 2 injections per week), and control HFD cGAS−/− mice were treated with empty liposomes. After 4 weeks of treatment, there were comparable levels of adrenal gland inflammation and blood pressure among these cGAS−/− mice (Figure 5G and Figure S5C through S5E). In the in vitro experiments using rat pheochromocytoma PC12 cells pretreated with siRNA‐cGAS, we also observed that gut mEVs did not significantly affect cellular responses (Figure 5H and Figure S5F through S5H). Taken together, these results demonstrate the critical role of cGAS/STING signaling for the effects of microbial DNA on host adrenal cells.
Restoring CRIg+ Macrophage Population Attenuates Obesity‐Associated Adrenomedullary Dysfunction
Given the critical roles of CRIg+ macrophages in blocking the effects of gut mEVs, we next tested whether recovery of CRIg+ macrophage population could alleviate obesity‐induced adrenal disorders. Previous studies have demonstrated that deactivated Cas9 can fuse to the transcriptional activation domains by specifically designed guide RNA. 41 , 42 , 43 , 44 In addition, the VPR system (including 3 transcriptional activators VP64, p65, and Rta) linked to the C‐terminal end of deactivated Cas9 activates gene expression (Figure S6A). To reboot the expression of Vsig4 (a gene encoding CRIg), 12 weeks HFD WT mice were intravenously injected with lentivirus carrying both the deactivated Cas9‐VPR system (VP64, p65, and Rta) and guide RNA‐Vsig4 (1×108 particles per mouse; HFD CRIg overexpression). After 4 weeks of injection with these lentiviruses, we observed that the CRIg+ macrophage population was restored in the adrenal glands (Figure S6B). More importantly, compared with 16 weeks HFD WT controls, the levels of bacterial DNA and inflammatory responses were significantly reduced after recovery of the CRIg+ macrophage proportion (Figure 6A through 6C), concomitant with decreased levels of circulating norepinephrine and the consequent decrease in blood pressure (Figure 6D and 6E). Thus, these findings indicate that restoring the CRIg+ macrophage population mitigates obesity‐associated dysregulation of catecholamine secretion and the consequent development of hypertension.
Figure 6. Recovery of CRIg+ (complement receptor of the immunoglobulin superfamily) macrophage population attenuates obesity‐associated adrenal dysfunction.
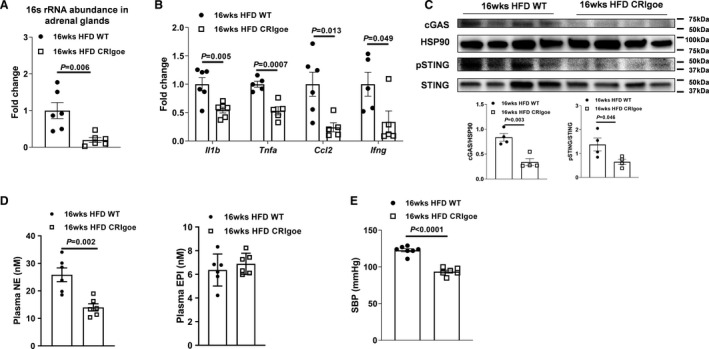
After 4 weeks of injection with lentivirus harboring deactivated Cas9 and guide RNA‐Vsig4, the abundance of bacterial DNA (A), proinflammatory genes (B), cGAS/STING activation (C) in the adrenal glands, circulating catecholamine (D), and systolic blood pressure (SBP) (E) of 16 weeks high‐fat diet (HFD) wild‐type (WT) mice were evaluated. Data are presented as mean±SEM. P values are determined by unpaired 2‐sided Student t test (A through E). Cas9 indicates CRISPR associated protein 9; cGAS, Cyclic GMP–AMP synthase; CRIgoe, overexpressing CRIg; EPI, epinephrine; HSP90, heat shock protein 90; NE, norepinephrine; pSTING, phosphorylated STING; STING, the cyclic GMP–AMP receptor stimulator of interferon genes; and Vsig4, V‐set and immunoglobulin domain containing 4.
Discussion
It is well established that obesity is associated with hypertension and is prevalent in both children and adults. 2 , 45 , 46 Despite intensive research, the mechanisms underlying obesity‐induced hypertension are incompletely understood. Existing literature reveals that high calorie loads in patients with obesity increase peripheral norepinephrine turnover, which increases sympathetic nervous system activity. 45 , 47 Diminished baroreflex and insulin sensitivity in patients with obesity have been implicated in further enhancing sympathetic nervous system activity. 48 Obesity‐induced hyperinsulinemia has also been reported to contribute to the development of hypertension through increased sodium retention, causing volume overload as well as endothelial dysfunction and favoring vasoconstriction. 45 , 49 , 50 Here we provide strong evidence that enrichment of microbial DNA in the adrenal medulla leads to inflammation of the adrenal medulla, resulting in increased catecholamine secretion and the consequent development of hypertension. We have recently reported that the lack of an anti‐inflammatory peptide catestatin results in infiltration of macrophages in the adrenal medulla, causing tissue inflammation, increased catecholamines secretion, and hypertension. 36 Inflammation is a key pathophysiologic factor driving hypertension, and obesity is characterized by chronic and low‐degree inflammation. The world‐wide obesity epidemic is paralleling a growth in hypertension prevalence. Therefore, understanding the mechanisms underlying obesity‐associated hypertension is necessary for future therapeutic development. In this study, we have assessed the impacts of microbial DNA enrichment on the incidence of obesity‐associated adrenomedullary inflammation and hypertension. We found that obesity is associated with a high abundance of bacterial DNA in the adrenal glands, concomitant with elevated level of adrenal inflammation. In the context of obesity, gut mEVs are readily translocated into distal adrenal glands and contribute to the enrichment of microbial DNA in these tissues. By contrast, there was no detectable bacterial DNA in the adrenal glands of lean and healthy WT mice. We found that adrenal gland resident macrophages are CRIg+ cells and exert profound protection from the infiltration of gut mEVs, whereas the population of these adrenal CRIg+ cells are largely diminished in obesity. In lean CRIg−/− or C3−/− mice treated with gut mEVs, adrenal microbial DNA enrichment results in elevated levels of adrenomedullary inflammation and catecholamine secretion, concomitant with hypertension. In addition, in vitro treatment with gut mEVs directly enhances inflammation and catecholamine secretion levels in rat pheochromocytoma PC12 cells. By contrast, depletion of microbial DNA blunts the effects of gut mEVs. However, these results cannot exclude the effects of other pathogenic factors leaked from gut lumen on obesity‐associated adrenal dysfunction. We also confirm that the activation of the cGAS/STING pathway is required for the ability of microbial DNA to trigger cellular dysfunctions. Finally, rebooting CRIg expression significantly attenuates obesity‐associated adrenal disorder.
The leakage of microbiota‐derived products is increased in obese human and animal models, because of the impaired gut barrier. 13 , 17 , 18 In line with previous findings, 22 , 24 , 28 a great amount of bacterial DNA was present in obese adrenal glands. We have demonstrated that microbiota‐derived EVs harbored microbial DNA and played a critical role in delivering this cargo into distal host tissues. There may be other ways that result in the enrichment of bacterial DNA in host tissues. For example, previous studies suggest that gut microbiota could penetrate through the obese gut barrier into host circulation and tissues. 19 , 20 , 51 However, Zulian et al reported that there is a rare existence of bacteria in obese host tissues, suggesting that bacteria hardly escape from gut lumen in obesity. 52 Previous studies suggest that a healthy gut barrier can selectively allow microbial product to secrete into host circulation. 53 , 54 However, in our studies, gut mEVs cannot pass through the healthy intact gut barrier, as evidenced by no detectable red fluorescent signals and bacterial DNA in the adrenal glands of lean WT mice after being injected with PKH26‐labeled gut mEVs into the jejunum section.
Our previous study has demonstrated the critical role of CRIg+ macrophages in blocking the spread of gut mEVs into host tissues. 28 In addition to the liver, consistent with previous findings, 30 we also observed that most of the adrenal tissue resident macrophages express CRIg in lean WT mice. In contrast, obese adrenal glands contained a much lower amount of CRIg+ macrophages. Using lean KC‐KO mice, we further validated that adrenal CRIg+ macrophages also exerted profound protection from the infiltration of gut mEVs.
It has been well known that obesity is accompanied by chronic and low‐degree inflammatory responses in various tissues. 11 Several studies have also reported that both human and animal adrenal cells can produce proinflammatory cytokines, including IL‐1β (interleukin‐1β), TNF⍺ (tumor necrosis factor‐⍺), IFN‐γ (interferon‐γ), and CCL2 (CC‐chemokine ligand 2), in response to various stress. 55 , 56 We also observed that obese adrenal chromaffin cells produced elevated levels of proinflammatory cytokines, suggesting an active inflammatory status in obese adrenal medulla. Although previous studies suggest that elevated circulating proinflammatory cytokines can trigger adrenomedullary abnormalities, obesity‐induced adrenomedullary production of proinflammatory cytokines may also act as autocrine factors eventuating in adrenal catecholamine secretion disorder. 10 , 57 , 58 More importantly, in both in vivo and in vitro experiments, we observed that gut mEVs‐induced accumulation of bacterial DNA promoted the production of adrenal proinflammatory mediators, whereas treatment with DNA‐free gut EVs had minimal effects on adrenal inflammation. Thus, these findings indicate that bacterial DNA plays an important role in inducing obesity‐associated adrenomedullary inflammation and hormonal dysfunction.
Previous studies, including ours, have demonstrated that cGAS/STING signaling is required for the ability of microbial DNA to modulate host cell responses. 28 , 40 Consistently, there was also improvement in the activation of cGAS/STING signaling in adrenal cells after gut mEV‐induced bacterial DNA accumulation in both in vivo and in vitro assays. More importantly, activation of cGAS/STING signaling plays a critical role in bacterial DNA‐mediated adrenomedullary inflammation and catecholamine secretion disorders, as shown by minimal functional changes in cGAS knockout adrenal cells in response to gut mEV treatment. Previous studies also indicate that canonical inflammatory pathways, such as NFκB (nuclear factor kappa B subunit)‐associated signaling, play critical roles in mediating adrenomedullary inflammatory responses. 59 , 60 , 61 In addition to the cGAS/STING pathway, other studies have reported that theTLR9 (toll‐like receptor 9) or the AIM2 (absent in melanoma 2) inflammasome exert profound function on sensing pathogenic DNA. 62 , 63 Endosomal membrane receptor TLR9 specifically detects CpG hypomethylated DNA to protect from virus and other pathogens. 64 AIM2 inflammasome activation is initiated by double stranded DNA and leads to proteolytic maturation of the pro‐inflammatory cytokines. 63 However, whether TLR9 or AIM2 participate in sensing microbial DNA in adrenal medulla is still unknown.
Conclusions
We have shown that microbial DNA accumulation leads to obesity‐associated adrenomedullary dysfunction leading to hypertension. We demonstrated the critical roles of adrenal resident CRIg+ macrophages and put forward a novel concept of how obesity induces hypertension through microbial DNA‐induced adrenomedullary abnormalities, thereby providing a potential therapeutic target for obesity‐related hypertension.
Sources of Funding
This study was funded by the University of California, San Diego School of Medicine Microscopy Core grant (P30 NS047101), and the U.S. National Institute of Diabetes and Digestive and Kidney Diseases R00 award (R00DK115998 to Dr Ying) and R01 award (R01DK125560 to Dr Ying), the UCSD/UCLA Diabetes Research Center Pilot and Feasibility grant (P30 DK063491 to Dr Ying), the Natural Science Foundation of China (81900786 to Dr Gao), and Veterans Affairs Merit Review grant (I01 BX003934 to Dr Mahata).
Disclosures
None.
Supporting information
Figures S1–S6
Acknowledgments
The authors thank Dr Fu for providing CRIg−/− mice; J. Santini for the confocal microscope, and University of California, San Diego vector core for lentivirus preparation.
Author contributions: Drs Ying and Mahata conceived the project and designed the experiments. Dr Gao, Z. Jin, and Dr Tang performed most of the experiments. Drs Ji, J. Suarez, J. A. Suarez, Cunha e Rocha, and Zhang assisted with tissue collection, cell culture, qPCR analysis, and flow cytometric analysis. Drs Ying, Dillmann, and Mahata supervised the project. Drs Gao, Ying, and Mahata analyzed and interpreted the data and cowrote the article.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.121.024561
For Sources of Funding and Disclosures, see page 13.
Contributor Information
Sushil K. Mahata, Email: smahata@health.ucsd.edu.
Wei Ying, Email: weying@health.ucsd.edu.
References
- 1. Henry SL, Barzel B, Wood‐Bradley RJ, Burke SL, Head GA, Armitage JA. Developmental origins of obesity‐related hypertension. Clin Exp Pharmacol Physiol. 2012;39:799–806. doi: 10.1111/j.1440-1681.2011.05579.x [DOI] [PubMed] [Google Scholar]
- 2. Kotchen TA. Obesity‐related hypertension: epidemiology, pathophysiology, and clinical management. Am J Hypertens. 2010;23:1170–1178. doi: 10.1038/ajh.2010.172 [DOI] [PubMed] [Google Scholar]
- 3. Troisi RJ, Weiss ST, Parker DR, Sparrow D, Young JB, Landsberg L. Relation of obesity and diet to sympathetic nervous system activity. Hypertension. 1991;17:669–677. doi: 10.1161/01.HYP.17.5.669 [DOI] [PubMed] [Google Scholar]
- 4. Sowers JR, Nyby M, Stern N, Beck F, Baron S, Catania R, Vlachis N. Blood pressure and hormone changes associated with weight reduction in the obese. Hypertension. 1982;4:686–691. doi: 10.1161/01.HYP.4.5.686 [DOI] [PubMed] [Google Scholar]
- 5. Gudbjornsdottir S, Friberg P, Elam M, Attvall S, Lonnroth P, Wallin BG. The effect of metformin and insulin on sympathetic nerve activity, norepinephrine spillover and blood pressure in obese, insulin resistant, normoglycemic, hypertensive men. Blood Press. 1994;3:394–403. doi: 10.3109/08037059409102293 [DOI] [PubMed] [Google Scholar]
- 6. Reisin E, Weir MR, Falkner B, Hutchinson HG, Anzalone DA, Tuck ML. Lisinopril versus hydrochlorothiazide in obese hypertensive patients: a multicenter placebo‐controlled trial. Treatment in Obese Patients With Hypertension (TROPHY) Study Group. Hypertension. 1997;30:140–145. doi: 10.1161/01.HYP.30.1.140 [DOI] [PubMed] [Google Scholar]
- 7. Raitakari M, Ilvonen T, Ahotupa M, Lehtimaki T, Harmoinen A, Suominen P, Elo J, Hartiala J, Raitakari OT. Weight reduction with very‐low‐caloric diet and endothelial function in overweight adults: role of plasma glucose. Arterioscler Thromb Vasc Biol. 2004;24:124–128. doi: 10.1161/01.ATV.0000109749.11042.7c [DOI] [PubMed] [Google Scholar]
- 8. Sugerman H, Windsor A, Bessos M, Wolfe L. Intra‐abdominal pressure, sagittal abdominal diameter and obesity comorbidity. J Intern Med. 1997;241:71–79. doi: 10.1046/j.1365-2796.1997.89104000.x [DOI] [PubMed] [Google Scholar]
- 9. Reimann M, Qin N, Gruber M, Bornstein SR, Kirschbaum C, Ziemssen T, Eisenhofer G. Adrenal medullary dysfunction as a feature of obesity. Int J Obes (Lond). 2017;41:714–721. doi: 10.1038/ijo.2017.36 [DOI] [PubMed] [Google Scholar]
- 10. Byrne CJ, Khurana S, Kumar A, Tai TC. Inflammatory signaling in hypertension: regulation of adrenal catecholamine biosynthesis. Front Endocrinol (Lausanne). 2018;9:343. doi: 10.3389/fendo.2018.00343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee YS, Wollam J, Olefsky JM. An integrated view of immunometabolism. Cell. 2018;172:22–40. doi: 10.1016/j.cell.2017.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McMaster WG, Kirabo A, Madhur MS, Harrison DG. Inflammation, immunity, and hypertensive end‐organ damage. Circ Res. 2015;116:1022–1033. doi: 10.1161/CIRCRESAHA.116.303697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E. Dysbiosis and the immune system. Nat Rev Immunol. 2017;17:219–232. doi: 10.1038/nri.2017.7 [DOI] [PubMed] [Google Scholar]
- 14. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–1772. doi: 10.2337/db06-1491 [DOI] [PubMed] [Google Scholar]
- 15. Jin X, Yu CH, Lv GC, Li YM. Increased intestinal permeability in pathogenesis and progress of nonalcoholic steatohepatitis in rats. World J Gastroenterol. 2007;13:1732–1736. doi: 10.3748/wjg.v13.i11.1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Johnson AM, Costanzo A, Gareau MG, Armando AM, Quehenberger O, Jameson JM, Olefsky JM. High fat diet causes depletion of intestinal eosinophils associated with intestinal permeability. PLoS One. 2015;10:e0122195. doi: 10.1371/journal.pone.0122195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thaiss CA, Levy M, Grosheva I, Zheng D, Soffer E, Blacher E, Braverman S, Tengeler AC, Barak O, Elazar M, et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science. 2018;359:1376–1383. doi: 10.1126/science.aar3318 [DOI] [PubMed] [Google Scholar]
- 18. Tilg H, Zmora N, Adolph TE, Elinav E. The intestinal microbiota fuelling metabolic inflammation. Nat Rev Immunol. 2020;20:40–54. doi: 10.1038/s41577-019-0198-4 [DOI] [PubMed] [Google Scholar]
- 19. Puri P, Liangpunsakul S, Christensen JE, Shah VH, Kamath PS, Gores GJ, Walker S, Comerford M, Katz B, Borst A, et al. The circulating microbiome signature and inferred functional metagenomics in alcoholic hepatitis. Hepatology. 2018;67:1284–1302. doi: 10.1002/hep.29623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lelouvier B, Servant F, Païssé S, Brunet A‐C, Benyahya S, Serino M, Valle C, Ortiz MR, Puig J, Courtney M, et al. Changes in blood microbiota profiles associated with liver fibrosis in obese patients: a pilot analysis. Hepatology. 2016;64:2015–2027. doi: 10.1002/hep.28829 [DOI] [PubMed] [Google Scholar]
- 21. Amar J, Chabo C, Waget A, Klopp P, Vachoux C, Bermúdez‐Humarán LG, Smirnova N, Bergé M, Sulpice T, Lahtinen S, et al. Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: molecular mechanisms and probiotic treatment. EMBO Mol Med. 2011;3:559–572. doi: 10.1002/emmm.201100159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Anhê FF, Jensen BAH, Varin TV, Servant F, Van Blerk S, Richard D, Marceau S, Surette M, Biertho L, Lelouvier B, et al. Type 2 diabetes influences bacterial tissue compartmentalisation in human obesity. Nat Metab. 2020;2:233–242. doi: 10.1038/s42255-020-0178-9 [DOI] [PubMed] [Google Scholar]
- 23. Ortiz S, Zapater P, Estrada JL, Enriquez P, Rey M, Abad A, Such J, Lluis F, Frances R. Bacterial DNA translocation holds increased insulin resistance and systemic inflammatory levels in morbid obese patients. J Clin Endocrinol Metab. 2014;99:2575–2583. doi: 10.1210/jc.2013-4483 [DOI] [PubMed] [Google Scholar]
- 24. Oh TG, Kim SM, Caussy C, Fu T, Guo J, Bassirian S, Singh S, Madamba EV, Bettencourt R, Richards L, et al. A universal gut‐microbiome‐derived signature predicts cirrhosis. Cell Metab. 2020;32:878–888.e6. doi: 10.1016/j.cmet.2020.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mathieu M, Martin‐Jaular L, Lavieu G, Thery C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell‐to‐cell communication. Nat Cell Biol. 2019;21:9–17. doi: 10.1038/s41556-018-0250-9 [DOI] [PubMed] [Google Scholar]
- 26. Liu Y, Defourny KAY, Smid EJ, Abee T. Gram‐positive bacterial extracellular vesicles and their impact on health and disease. Front Microbiol. 2018;9:1502. doi: 10.3389/fmicb.2018.01502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chelakkot C, Choi Y, Kim D‐K, Park HT, Ghim J, Kwon Y, Jeon J, Kim M‐S, Jee Y‐K, Gho YS, et al. Akkermansia muciniphila‐derived extracellular vesicles influence gut permeability through the regulation of tight junctions. Exp Mol Med. 2018;50:e450. doi: 10.1038/emm.2017.282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Luo Z, Ji Y, Gao H, Gomes Dos Reis FC, Bandyopadhyay G, Jin Z, Ly C, Chang Y‐J, Zhang D, Kumar D, et al. CRIg(+) macrophages prevent gut microbial DNA‐containing extracellular vesicle‐induced tissue inflammation and insulin resistance. Gastroenterology. 2021;160:863–874. doi: 10.1053/j.gastro.2020.10.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zeng Z, Surewaard BG, Wong CH, Geoghegan JA, Jenne CN, Kubes P. CRIg functions as a macrophage pattern recognition receptor to directly bind and capture blood‐borne gram‐positive bacteria. Cell Host Microbe. 2016;20:99–106. doi: 10.1016/j.chom.2016.06.002 [DOI] [PubMed] [Google Scholar]
- 30. Helmy KY, Katschke KJ Jr, Gorgani NN, Kljavin NM, Elliott JM, Diehl L, Scales SJ, Ghilardi N, van Lookeren CM. CRIg: a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell. 2006;124:915–927. doi: 10.1016/j.cell.2005.12.039 [DOI] [PubMed] [Google Scholar]
- 31. He JQ, Katschke KJ Jr, Gribling P, Suto E, Lee WP, Diehl L, Eastham‐Anderson J, Ponakala A, Komuves L, Egen JG, et al. CRIg mediates early Kupffer cell responses to adenovirus. J Leukoc Biol. 2013;93:301–306. doi: 10.1189/jlb.0612311 [DOI] [PubMed] [Google Scholar]
- 32. Gorgani NN, He JQ, Katschke KJ Jr, Helmy KY, Xi H, Steffek M, Hass PE, van Lookeren CM. Complement receptor of the Ig superfamily enhances complement‐mediated phagocytosis in a subpopulation of tissue resident macrophages. J Immunol. 2008;181:7902–7908. doi: 10.4049/jimmunol.181.11.7902 [DOI] [PubMed] [Google Scholar]
- 33. Gorgani NN, Thathaisong U, Mukaro VR, Poungpair O, Tirimacco A, Hii CS, Ferrante A. Regulation of CRIg expression and phagocytosis in human macrophages by arachidonate, dexamethasone, and cytokines. Am J Pathol. 2011;179:1310–1318. doi: 10.1016/j.ajpath.2011.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fu W, Wojtkiewicz G, Weissleder R, Benoist C, Mathis D. Early window of diabetes determinism in NOD mice, dependent on the complement receptor CRIg, identified by noninvasive imaging. Nat Immunol. 2012;13:361–368. doi: 10.1038/ni.2233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yuan X, Yang BH, Dong Y, Yamamura A, Fu W. CRIg, a tissue‐resident macrophage specific immune checkpoint molecule, promotes immunological tolerance in NOD mice, via a dual role in effector and regulatory T cells. Elife. 2017;6:e29540. doi: 10.7554/eLife.29540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ying W, Tang K, Avolio E, Schilling JM, Pasqua T, Liu MA, Cheng H, Gao H, Zhang J, Mahata S, et al. Immunosuppression of macrophages underlies the cardioprotective effects of CST (Catestatin). Hypertension. 2021;77:1670–1682. doi: 10.1161/HYPERTENSIONAHA.120.16809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kolski‐Andreaco A, Cai H, Currle DS, Chandy KG, Chow RH. Mouse adrenal chromaffin cell isolation. J Vis Exp. 2007:129. doi: 10.3791/129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lukewich MK, Lomax AE. Endotoxemia enhances catecholamine secretion from male mouse adrenal chromaffin cells through an increase in Ca(2+) release from the endoplasmic reticulum. Endocrinology. 2014;155:180–192. doi: 10.1210/en.2013-1623 [DOI] [PubMed] [Google Scholar]
- 39. Ahn J, Barber GN. STING signaling and host defense against microbial infection. Exp Mol Med. 2019;51:1–10. doi: 10.1038/s12276-019-0333-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ablasser A, Chen ZJ. cGAS in action: expanding roles in immunity and inflammation. Science. 2019;363:eaat8657. doi: 10.1126/science.aat8657 [DOI] [PubMed] [Google Scholar]
- 41. Cheng AW, Wang H, Yang H, Shi L, Katz Y, Theunissen TW, Rangarajan S, Shivalila CS, Dadon DB, Jaenisch R. Multiplexed activation of endogenous genes by CRISPR‐on, an RNA‐guided transcriptional activator system. Cell Res. 2013;23:1163–1171. doi: 10.1038/cr.2013.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. Genome‐scale CRISPR‐mediated control of gene repression and activation. Cell. 2014;159:647–661. doi: 10.1016/j.cell.2014.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, et al. Genome‐scale transcriptional activation by an engineered CRISPR‐Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chavez A, Scheiman J, Vora S, Pruitt BW, Tuttle M, P R Iyer E, Lin S, Kiani S, Guzman CD, Wiegand DJ, et al. Highly efficient Cas9‐mediated transcriptional programming. Nat Methods. 2015;12:326–328. doi: 10.1038/nmeth.3312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kotsis V, Stabouli S, Papakatsika S, Rizos Z, Parati G. Mechanisms of obesity‐induced hypertension. Hypertens Res. 2010;33:386–393. doi: 10.1038/hr.2010.9 [DOI] [PubMed] [Google Scholar]
- 46. Wirix AJ, Kaspers PJ, Nauta J, Chinapaw MJ, Kist‐van Holthe JE. Pathophysiology of hypertension in obese children: a systematic review. Obes Rev. 2015;16:831–842. doi: 10.1111/obr.12305 [DOI] [PubMed] [Google Scholar]
- 47. Lambert EA, Straznicky NE, Dixon JB, Lambert GW. Should the sympathetic nervous system be a target to improve cardiometabolic risk in obesity? Am J Physiol Heart Circ Physiol. 2015;309:H244–H258. doi: 10.1152/ajpheart.00096.2015 [DOI] [PubMed] [Google Scholar]
- 48. Thorp AA, Schlaich MP. Relevance of sympathetic nervous system activation in obesity and metabolic syndrome. J Diabetes Res. 2015;2015:341583. doi: 10.1155/2015/341583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Reisin E, Jack AV. Obesity and hypertension: mechanisms, cardio‐renal consequences, and therapeutic approaches. Med Clin North Am. 2009;93:733–751. doi: 10.1016/j.mcna.2009.02.010 [DOI] [PubMed] [Google Scholar]
- 50. Hall JE, do Carmo JM, da Silva AA, Wang Z, Hall ME. Obesity‐induced hypertension: interaction of neurohumoral and renal mechanisms. Circ Res. 2015;116:991–1006. doi: 10.1161/CIRCRESAHA.116.305697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schierwagen R, Alvarez‐Silva C, Madsen MSA, Kolbe CC, Meyer C, Thomas D, Uschner FE, Magdaleno F, Jansen C, Pohlmann A, et al. Circulating microbiome in blood of different circulatory compartments. Gut. 2019;68:578–580. doi: 10.1136/gutjnl-2018-316227 [DOI] [PubMed] [Google Scholar]
- 52. Zulian A, Cancello R, Cesana E, Rizzi E, Consolandi C, Severgnini M, Panizzo V, Di Blasio AM, Micheletto G, Invitti C. Adipose tissue microbiota in humans: an open issue. Int J Obes (Lond). 2016;40:1643–1648. doi: 10.1038/ijo.2016.111 [DOI] [PubMed] [Google Scholar]
- 53. Kimura I, Miyamoto J, Ohue‐Kitano R, Watanabe K, Yamada T, Onuki M, Aoki R, Isobe Y, Kashihara D, Inoue D, et al. Maternal gut microbiota in pregnancy influences offspring metabolic phenotype in mice. Science. 2020;367:eaaw8429. doi: 10.1126/science.aaw8429 [DOI] [PubMed] [Google Scholar]
- 54. Vuong HE, Pronovost GN, Williams DW, Coley EJL, Siegler EL, Qiu A, Kazantsev M, Wilson CJ, Rendon T, Hsiao EY. The maternal microbiome modulates fetal neurodevelopment in mice. Nature. 2020;586:281–286. doi: 10.1038/s41586-020-2745-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Call GB, Husein OF, McIlmoil CJ, Adams A, Heckmann RA, Judd AM. Bovine adrenal cells secrete interleukin‐6 and tumor necrosis factor in vitro. Gen Comp Endocrinol. 2000;118:249–261. doi: 10.1006/gcen.2000.7458 [DOI] [PubMed] [Google Scholar]
- 56. Gonzalez‐Hernandez JA, Ehrhart‐Bornstein M, Spath‐Schwalbe E, Scherbaum WA, Bornstein SR. Human adrenal cells express tumor necrosis factor‐alpha messenger ribonucleic acid: evidence for paracrine control of adrenal function. J Clin Endocrinol Metab. 1996;81:807–813. doi: 10.1210/jcem.81.2.8636308 [DOI] [PubMed] [Google Scholar]
- 57. Willenberg HS, Path G, Vogeli TA, Scherbaum WA, Bornstein SR. Role of interleukin‐6 in stress response in normal and tumorous adrenal cells and during chronic inflammation. Ann N Y Acad Sci. 2002;966:304–314. doi: 10.1111/j.1749-6632.2002.tb04230.x [DOI] [PubMed] [Google Scholar]
- 58. Douglas SA, Sreenivasan D, Carman FH, Bunn SJ. Cytokine interactions with adrenal medullary chromaffin cells. Cell Mol Neurobiol. 2010;30:1467–1475. doi: 10.1007/s10571-010-9593-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Perez‐Rodriguez R, Roncero C, Olivan AM, Gonzalez MP, Oset‐Gasque MJ. Signaling mechanisms of interferon gamma induced apoptosis in chromaffin cells: involvement of nNOS, iNOS, and NFkappaB. J Neurochem. 2009;108:1083–1096. doi: 10.1111/j.1471-4159.2008.05862.x [DOI] [PubMed] [Google Scholar]
- 60. Samal B, Ait‐Ali D, Bunn S, Mustafa T, Eiden LE. Discrete signal transduction pathway utilization by a neuropeptide (PACAP) and a cytokine (TNF‐alpha) first messenger in chromaffin cells, inferred from coupled transcriptome‐promoter analysis of regulated gene cohorts. Peptides. 2013;45:48–60. doi: 10.1016/j.peptides.2013.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ait‐Ali D, Turquier V, Tanguy Y, Thouennon E, Ghzili H, Mounien L, Derambure C, Jegou S, Salier JP, Vaudry H, et al. Tumor necrosis factor (TNF)‐alpha persistently activates nuclear factor‐kappaB signaling through the type 2 TNF receptor in chromaffin cells: implications for long‐term regulation of neuropeptide gene expression in inflammation. Endocrinology. 2008;149:2840–2852. doi: 10.1210/en.2007-1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, et al. A Toll‐like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123 [DOI] [PubMed] [Google Scholar]
- 63. Hornung V, Ablasser A, Charrel‐Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase‐1‐activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hoelzer K, Shackelton LA, Parrish CR. Presence and role of cytosine methylation in DNA viruses of animals. Nucleic Acids Res. 2008;36:2825–2837. doi: 10.1093/nar/gkn121 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S6