

Abstract
Background
Heart failure with preserved ejection fraction (HFpEF) remains an increasing public health problem with substantial morbidity and mortality but with few effective treatments. A novel inflammatory mechanism has been proposed, but the inflammatory signals promoting the development of HFpEF remain greatly unknown.
Methods and Results
Serum of patients with HFpEF was collected for measurement of circulating neutrophils and markers of neutrophil extracellular traps (NETs). To induce HFpEF phenotype, male C57BL/6 mice underwent uninephrectomy, received a continuous infusion of d‐aldosterone for 4 weeks, and maintained on 1.0% sodium chloride drinking water. Heart tissues were harvested, immune cell types determined by flow cytometry, NETs formation by immunofluorescence, and western blotting. Differentiated neutrophils were cultured to investigate the effect of HMGB1 (high mobility group protein B1) and SGLT2 (sodium‐glucose cotransporter‐2) inhibitor on NETs formation in vitro. Circulating neutrophils and NETs markers are elevated in patients with HFpEF, as are cardiac neutrophils and NETs formation in HFpEF mice. NETs inhibition with deoxyribonuclease 1 in experimental HFpEF mice reduces heart macrophages infiltration and inflammation and ameliorates cardiac fibrosis and diastolic function. Damage‐associated molecular pattern HMGB1 expression is elevated in cardiac tissue of HFpEF mice, and HMGB1 inhibition reduces heart neutrophil infiltration and NETs formation and ameliorates diastolic function. Lastly, SGLT2 inhibitor empagliflozin down‐regulates heart HMGB1 expression, attenuates NETs formation and cardiac fibrosis, and improves diastolic function in HFpEF mice.
Conclusions
NETs contribute to the pathogenesis of HFpEF, which can be ameliorated by HMGB1 inhibition and SGLT2 inhibitors. Thus, HMGB1 and NETs may represent novel therapeutic targets for the treatment of HFpEF.
Keywords: empagliflozin, heart failure with preserved ejection fraction, HMGB1, neutrophil extracellular traps
Subject Categories: Heart Failure, Inflammation, Pathophysiology
Nonstandard Abbreviations and Acronyms
- DAMP
damage‐associated molecular pattern
- HFpEF
heart failure with preserved ejection fraction
- HFrEF
heart failure with reduced ejection fraction
- HMGB1
high mobility group protein B1
- NETs
neutrophil extracellular traps
- SGLT2
sodium‐glucose cotransporter‐2
Clinical Perspective
What Is New?
Neutrophil extracellular traps (NETs) formation increases in heart failure with preserved ejection fraction (HFpEF) and inhibition of NETs ameliorates cardiac diastolic function in HFpEF mice.
Damage‐associated molecular pattern HMGB1 (high mobility group protein B1) contributes to NETs formation and HMGB1 inhibition ameliorates diastolic function in HFpEF.
SGLT2 (sodium‐glucose cotransporter‐2) inhibitor empagliflozin improves diastolic function in HFpEF mice, which might be partially dependent on down‐regulating HMGB1 and NETs.
What Are the Clinical Implications?
Our study expands knowledge of the pathophysiology of HFpEF, emphasizing the contribution of NETs and HMBG1.
NETs and HMBG1may represent novel therapeutic targets for the treatment of HFpEF.
Our study makes important links to clinical HFpEF and mechanisms through which SGLT2 inhibitors are beneficial in HFpEF.
The prevalence of heart failure is ≈1%–2% and up to 50% of patients with heart failure have a preserved ejection fraction (HFpEF). 1 HFpEF has long been a condition with no convincing drugs, including neurohormonal antagonists that are effective in patients with heart failure and a reduced ejection fraction (HFrEF), shown to significantly improve clinical outcomes. The publication of the EMPERORPreserved (The Empagliflozin Outcome Trial in Patients with Chronic Heart Failure with Preserved Ejection Fraction) phase III trial documented empagliflozin, a SGLT2 (sodium‐glucose cotransporter‐2) inhibitor, as the first drug therapy to produce broad‐based benefits for patients with HFpEF, reducing both inpatient and outpatient heart failure events. 2 , 3 The remarkable response differences to therapies between HFrEF and HFpEF suggest potentially fundamental differences in the underlying pathophysiology.
A systemic proinflammatory state induced by comorbidities (such as overweight/obesity, diabetes, and salt‐sensitive hypertension) has been proposed as the cause of myocardial structural and functional alterations in HFpEF. 4 Clinical biomarker analyses reveal that profiles specific for HFpEF are related to inflammation and extracellular matrix reorganization, 5 different from those for HFrEF. Notably, in heart tissues from both humans and mice with HFpEF, many more neutrophils recruitments are observed than in control hearts. 6 However, the role of neutrophils in HFpEF pathogenesis is unknown.
Neutrophils are major effectors of acute inflammation and also contribute to chronic inflammatory conditions. 7 In response to danger signals, neutrophils can eject their DNA decorated with antimicrobial proteins, thus forming large web‐like structures that are capable of trapping and killing pathogens, a process termed neutrophil extracellular traps (NETs) formation. 8 NETs are networks of decondensed chromatins containing modified histones (CitH3) and granule‐derived enzymes, such as myeloperoxidase (MPO) and neutrophil elastase, which can be used to identify NETs. Of interest, sterile inflammation can also mediate the formation of NETs. NETs can enhance inflammation by different mechanisms but may also lead to tissue injury. As a consequence, NETs have emerged as an active player in a host of noninfectious conditions, including diabetes and rheumatoid arthritis. 9
HMGB1 (high mobility group protein B1) acts as a damage‐associated molecular pattern (DAMP) with proposed functions in the regulation of inflammation. Previous data have suggested that HMGB1 mediates neutrophils recruitment toward necrosis 10 and might trigger the formation of NETs. 11 However, the potential impact of HMGB1 and NETs in HFpEF has not been investigated. In this study, we revealed the functional importance of NETs for HFpEF pathogenesis and delineate the possible mechanistic link between HMGB1 and NETs. We reported a critical role of HMGB1 in neutrophils recruitment to the heart and formation of NETs and showed that targeting NETs or HMGB1 improves cardiac function in a mouse model of HFpEF. We also showed that SGLT2 inhibitor empagliflozin ameliorated cardiac diastolic function at least partially by inhibiting the HMGB1‐NETs axis.
Methods
The data that support the findings of this study are available from the corresponding author on reasonable request. This study was conducted in accordance with the ethical guidelines of the Declaration of Helsinki. The institutional review boards at the affiliated Drum Tower Hospital, Nanjing University School of Medicine approved the study, and written informed consent was obtained from all participants.
Animals and Experiment Protocol
Eight‐week‐old male C57BL/6 mice weighing about 20–22 g were obtained (Qinglongshan, Nanjing, China) and maintained in a specific‐pathogen‐freeenvironment at a temperature of 20±2 °C, with a relative humidity of 50%±1% and a light/dark cycle of 12/12 hours. Mouse care and in vivo experimental procedures were approved by the Institutional Animal Care and Use Committee of Nanjing University according to institutional animal ethics guidelines with an ethical clearance number of 2018‐035210‐225A. Free access to food and water was provided.
Mice were anesthetized with 80 to 100 mg/kg ketamine and 5 to 10 mg/kg xylazine intraperitoneally, underwent uninephrectomy, and then received either a continuous infusion of saline (control, n=10) or d‐aldosterone (0.3 μg/h, Sigma‐Aldrich, USA) (HFpEF, n=26) for 4 weeks via osmotic minipumps (Alzet, USA). Heat support was provided throughout the procedure and recovery period. All mice were maintained on 1.0% sodium chloride drinking water. 6 One day post surgery, mice were randomized to receive saline, deoxyribonuclease (DNase) 1 (50 µg intraperitoneal injection per mouse twice daily, Roche), glycyrrhizic acid (10 mg/kg per day, Sigma), or empagliflozin (10 mg/kg per day, MedChemExpress). All mice were weighed once every other day, and euthanized after 4 weeks.
Exercise Exhaustion Test
All groups of mice were trained for the treadmill endurance test the day before (SA101, SANS, China) according to the protocol previously described. 12 On the experimental day, the treadmill was set at the same conditions; running time was measured and running distance calculated.
Echocardiography
An experienced operator who was blinded to the study group performed the echocardiography test. Transthoracic echocardiography was performed using a VisualSonics Vevo 2100 system (Visual Sonics) after the induction of general anesthesia with isoflurane gas. Heart rate was monitored and adjusted to maintain a heart rate of 500±50 beats/minute through anesthesia delivery. Systolic function was evaluated by calculating ejection fraction from images obtained from short‐axis M‐mode scans at the midventricular level. Diastolic function was assessed from the apical 4‐chamber views using pulsed‐wave and tissue Doppler imaging at the level of the mitral valve. We collected diastolic parameters including isovolumic relaxation time, deceleration time, E/A, and E/e′. All parameters were measured at least 3 times, and means are presented.
Blood Pressure Measurements
Blood pressure was measured using a noninvasive blood pressure system (BP‐2000, Visitech), All mice were trained the day before the measurements, and blood pressure was measured 5 times; the means are presented.
Flow Cytometry
To obtain single‐cell suspensions, hearts were extracted and perfused with PBS, cut in small pieces, and incubated with 450 U/mL collagenase I, 125 U/mL collagenase XI, 60 U/mL DNase I, and 60 U/mL hyaluronidase (all Sigma‐Aldrich) for 1 hour at 37 °C. After incubation, the suspension was mixed gently, filtered through a 40‐µm cell strainer (Falcon), washed, and suspended in PBS. Cells were labeled with a fluorescein isothiocyanate‐conjugated rat anti‐mouse CD11b Ab (101205; Biolegend), APC‐conjugated rat anti‐mouse Ly6G Ab (127613; BioLegend), Percy5.5–conjugated rat anti‐mouse F4/80 Ab (123127; BioLegend) and incubated for 30 minutes at room temperature, according to standard protocols. Matching isotype Abs served as controls. Thereafter, the mixture was washed, resuspended, and immediately subjected to flow cytometry. Data were acquired with a FACS Aria flow cytometer (BD Bioscience) and analyzed with FlowJo software (TreeStar, Ashland, OR).
Histology, Heart, and Weight Ratio
Lungs were excised and weighed before and after drying at 65 °C for 48 hours to calculate the lung wet‐to‐dry weight ratio. Heart weight and tibia length were also recorded to calculate the heart weight/tibia length ratio. For histopathological evaluation of cardiac tissue, mouse hearts were rinsed with PBS and fixed in 4% PFA for at least 24 hours. Tissue were gradually dehydrated, embedded in paraffin, cut into transverse sections (5 μm thickness), and then stained with PicroSirius Red (Sigma‐Aldrich) according to the manufacturer’s instructions.
Circulating Markers of NETs
We measured serum concentrations of circulating markers of NETs from patients with HFpEF and controls. Cell‐free double‐stranded DNA (dsDNA) was measured after phenol extraction using the Qubit 2.0 Fluorometer (Life Technologies). Elastase 2 concentrations were measured using commercially available ELISA kits (Elabscience, China) according to the manufacturer’s instructions.
Western Blot
Proteins were extracted from tissues or cells using RIPA lysis buffer (Beyotime Biotechnology, Shanghai, China) containing 1 mmol/L Pierce™ phosphatase inhibitor (Bimake) and 0.1% protease inhibitor cocktail (Bimake). The homogenate was centrifuged (17 000g, 4 ℃) and supernatants were collected. Protein concentrations were determined by bicinchoninic acid assay (ThermoFisher Scientific). 20 μg proteins were separated by SDS‐PAGE gel and the protein bands were transferred to polyvinylidene difluoride membranes (Merck Millipore). After blocking with 5% bovine serum albumin in TBS‐Tween (0.1%, Beyotime Biotechnology) for 1.5 hours, target bands were incubated with corresponding primary antibodies against HMGB1 (ab18256, Abcam, UK), histone H3 (ab1791, Abcam), and CitH3 (ab5103, Abcam) at 4 ℃ overnight. GAPDH (Bioworld) was used as loading controls. Appropriate horseradish peroxidase‐labeled secondary antibodies (Bioworld) were added and incubated for 1 hour at room temperature. Chemiluminescent (ECL, ThermoFisher Scientific) detection was used to visualize the bands, and densitometric analysis was performed with Image J.
Immunofluorescence and Immunohistochemistry
For immunofluorescence staining, tissues were embedded in paraffin, slides then were blocked in 1% bovine serum (Beyotime) for 1 hour at room temperature followed by overnight incubation with primary antibodies against MPO (ab208670, Abcam) and CitH3 (ab5103, Abcam) at 4 ℃ overnight and subsequently with fluorochrome‐conjugated secondary antibodies (Bioworld). DAPI (Beyotime) was added for nuclear staining. For immunochemistry staining, primary antibody against HMGB1 (ab18256, Abcam) was first added and an enzyme‐conjugated secondary antibody (Beyotime) was used. Stained sections were examined with a Leica immunofluorescence microscope. Images were captured and processed with Adobe Photoshop.
RNA Isolation and Real‐Time Polymerase Chain Reaction
Total RNA was extracted from heart tissues using Trizol reagent (Vazyme, China) following the manufacturer’s instructions. The concentration and purity of the RNA were determined using a NanoDrop Spectrophotometer (ThermoFisher Scientific). Using a reverse transcription kit (Vazyme, China), 1 µg total RNA was transcribed into cDNA. Quantitative real‐time polymerase chain reaction analyses were performed in triplicate using the ChamQTM SYBR color qPCR Master Mix (Vazyme, China). The expression of relative gene was normalized to the housekeeping gene β‐actin. Relative expression to the reference gene was calculated with the ‐Ct method using the following equations: ΔCt(sample)=Ct(target)–Ct(reference); relative quantity=2–ΔCt.
Cell Culture and Treatment
We cultured the HL60 cells in Iscove's Modified Dulbecco's Medium medium (Gibco) supplemented with 20% FBS (Gibco), and stimulated their differentiation into neutrophils with dimethylfumarate (70 mmol/L, Aladdin, China) for 5 days. 13 After 5 days of differentiation, cells were divided into 6‐well culture plates and incubated with glycyrrhizic acid (500 μmol/L, Sigma) or empagliflozin (500 nmol/L, MedChemExpress) for 24 hours. Thereafter, cells were activated to release NETs with 100 nmol/L PMA (MedChemExpress) for 3 hours at 37 °C. Proteins were extracted and loaded for western blotting. Culture supernatants were collected to measure the concentration of cell‐free DNA using the PicoGreen dsDNA Assay Kits (Solarbio, Beijing, China) according to the manufacturer’s instruction.
Humans
HFpEF was defined according to the inclusion criteria recommended by the European Society of Cardiology. 14 We obtained blood samples from 20 healthy individuals and 20 subjects with HFpEF, who were age and sex matched. Subjects were recruited only if they were <80 years old, not presenting signs of active infection, not on steroid or other immunosuppressive treatments, and without diagnosis of tumor in the past 5 years. None were decompensated or admitted to the hospital for heart failure at the time of enrollment.
Statistical Analysis
Data are presented as mean±SD unless otherwise indicated. No statistical method was used to predetermine the sample size. For comparisons of variables between patients with HFpEF and controls, paired t test was used. Differences between multiple groups in mice were compared using 1‐way ANOVA followed by Tukey’s post hoc tests. Results were considered significant when P<0.05. The statistical tests were performed with the GraphPad Prism software (version 5.0).
Results
Neutrophils and NETs Markers are Elevated in Patients With HFpEF
We identified 20 outpatients with HFpEF who were not decompensated for heart failure and 20 age‐ and sex‐matched controls absent from heart diseases in the analysis. The demographic and clinical characteristics were shown in Table S1. As expected, patients with HFpEF had a higher rate of related comorbidities, including obesity, diabetes, hypertension, and atrial fibrillation; they also showed higher serum concentrations of B‐type natriuretic peptide and lower level of estimated glomerular filtration rate, whereas level of cardiac troponin T was not statistically different. Left ventricular wall thickness was higher in patients with HFpEF whereas left ventricular ejection fraction (LVEF) was comparable between 2 groups. We examined circulating neutrophils from healthy controls and patients with HFpEF and found a significantly higher number of neutrophils in patients with HFpEF (3.54±versus 6.76±×106/mL, P=2.6×10−11), as were monocytes but not lymphocytes; the total number of white blood cells was increased in patients with HFpEF (Figure 1A through 1D). We then determined the concentration of circulating NETs‐related biomarkers in the sera by measuring cell‐free dsDNA and elastase 2. We found higher levels of circulating NETs in the sera of patients with HFpEF compared with healthy controls (cell‐free dsDNA: 1.79 versus 5.43 ng/mL, P=3.7×10−13; elastase 2: 2.09 versus 3.71 μg/mL, P=4.5×10−12; Figure 1E and 1F).
Figure 1. Neutrophils and NETs markers are elevated in patients and mice with HFpEF.
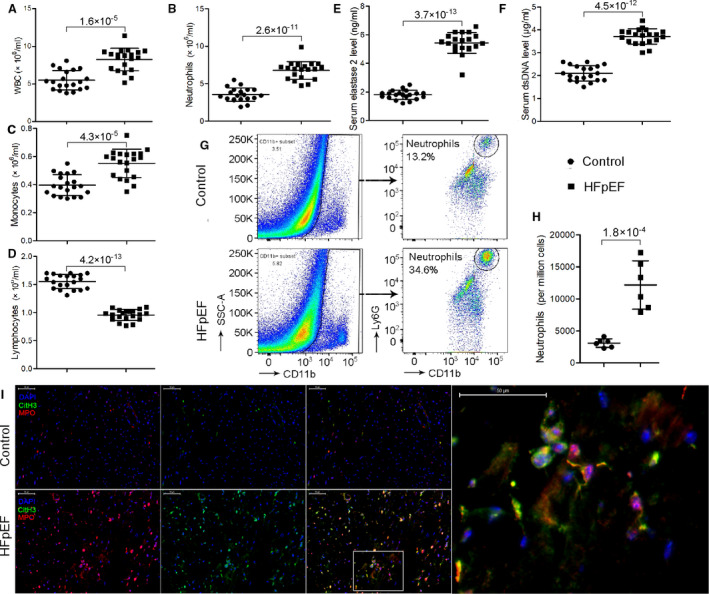
A through D, Patients with HFpEF showed significantly higher levels of circulating white blood cells (A), particularly neutrophils (B) and monocytes (C) but not lymphocytes (D) compared with matched healthy controls. E through F, Circulating NETs markers, that is, elastase 2 (E) and cell‐free dsDNA (F) were significantly higher in the sera of patients with HFpEF compared with healthy controls. G through H, Flow cytometric analysis showed that HFpEF mice had an over 3‐fold increase in neutrophil counts in the heart. I, In situ immunofluorescence identifying NETs by extracellular MPO (red), CitH3 (green), and DNA (blue) deposits in the heart, showing increased NETs formation in HFpEF mice than controls. Scale bar represents 50 µm. CitH3 indicates citrullinated histone 3; dsDNA, double‐stranded DNA; HFpEF, heart failure with preserved ejection fraction; MPO, myeloperoxidase; NETs, neutrophil extracellular traps; and WBC, white blood cell.
Neutrophils Infiltrate Murine Hearts and Undergo NETs Formation in HFpEF
To investigate the functional relevance of NETs in HFpEF, we constructed a mouse model of HFpEF. Mice received salty drinking water, unilateral nephrectomy, and chronic exposure to aldosterone (SAUNA) to mimic the phenotypes of HFpEF. Four weeks after SAUNA treatment, mice exhibited significant diastolic function, as evidenced by an increase in peak early diastolic transmitral velocity/spectral tissue Doppler‐derived peak early diastolic velocity (E/e′) (27.8±1.0 versus 39.1±5.1, P=1.3×10−6; Figure 2C and 2D), a reliable predictor of left ventricular filling pressure. An increase in E/A ratio (1.5±0.06 versus 2.3±0.37, P=9.1×10−7) and shortening of E wave deceleration time (19.6±1.04 versus 16.6±1.04, P=3.4×10−8) was also observed (Figure 2B, 2E and 2G), suggesting a restrictive pattern of left ventricular diastolic filling. Isovolumic relaxation time (17.4±0.50 versus 12.7±0.78, P=2.5×10−8) was elevated (Figure 2F) whereas LVEF was preserved after treatment (62.5±5.07 versus 64.4%±3.14%, P=0.93; Figure 2A and 2J). Consistently, SAUNA‐treated mice showed a significant reduction in running distance (indicative of exercise tolerance: 271.8±16.68 versus 147.3±10.34 m, P=2.8×10−8; Figure 2I) and increasement in wet/dry lung ratio (indicative of pulmonary congestion: 2.7±0.27 versus 4.1±0.29, P=7.2×10−6; Figure 2H) compared with the controls, all surrogate markers for congestive heart failure. Cardiac hypertrophy (Figure 2L and 2O) and increased heart fibrosis were also noted in SAUNA‐treated mice (Figure 2Q), as revealed by Sirius red staining. Body weight and heart rate were not statistically different between 2 groups (Figure 2N and 2P).
Figure 2. NETs inhibition reduces cardiac macrophage infiltration, attenuates cardiac fibrosis, and ameliorates diastolic function in HFpEF mice.
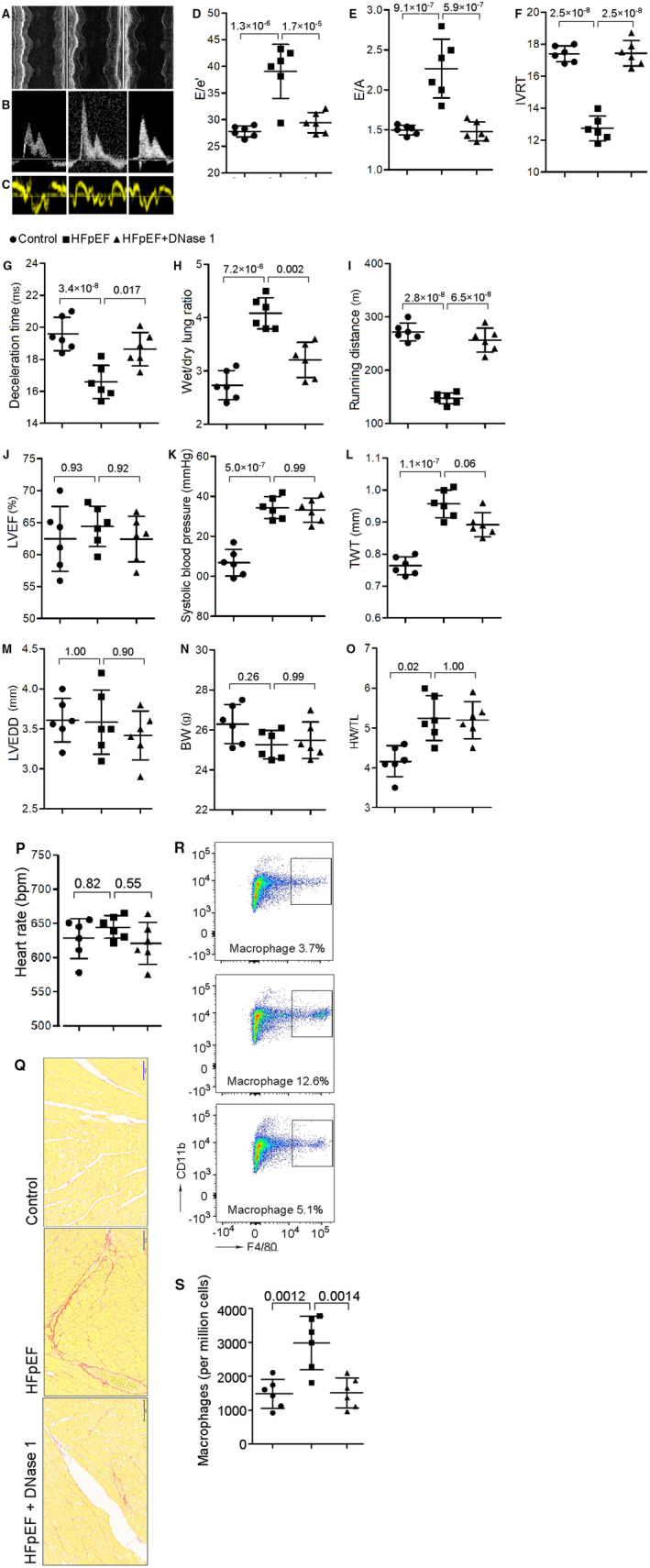
A through C, Representative echocardiography images of left ventricular M‐mode echocardiography (A), pulsed‐wave Doppler (B) and tissue Doppler (C) tracings. D, Ratio between mitral E wave and e′ wave (E/e′). E, Ratio between mitral E wave and A wave (E/A). F, Isovolumic relaxation time (IVRT). G, Deceleration time; H, Ratio between wet and dry lung weight. I, Running distance during exercise exhaustion test. J, Percentage of LVEF. K, Systolic blood pressure. L, Total wall thickness (TWT). M, Left ventricular end‐diastolic diameter (LVEDD). N, Body weight (BW). O, Ratio between heart weight and tibia length (HW/TL). P, Heart rate. These results showed that DNase 1 significantly attenuated diastolic dysfunction, improved exercise tolerance, reduced lung congestion, but had no significant effect on LVEF, blood pressure or cardiac hypertrophy. Q, Representative images of Sirius red staining of heart tissue, showing an increment of cardiac fibrosis in HFpEF and an attenuation after DNase 1 treatment. Scale bar represents 50 µm. R and S, Flow cytometric analysis showing that HFpEF mice had a significant increase in macrophage counts in the heart, and NETs inhibition with DNase 1 treatment significantly reduced the number of macrophages in the heart. BW indicates body weight; DNase 1, deoxyribonuclease; HFpEF, heart failure with preserved ejection fraction; LVEF, left ventricular ejection fraction; NETs, neutrophil extracellular traps; and TWT, total wall thickness.
Enhanced circulating neutrophils and NETs formation in patients with HFpEF prompted us to assess neutrophilia and NETs formation in heart tissue from HFpEF mice. We explored the phenotype of heart infiltrating innate immune cells with flow cytometric analysis and found that SAUNA‐treated HFpEF mice had an over 3‐fold increase in neutrophil counts in the heart (3112±667 versus 12 168±3779 per million cells, P=1.8×10−4; Figure 1G and 1H). We then aimed to determine whether these infiltrated neutrophils would undergo NETs formation in the heart. Western blot analysis revealed that CitH3, a specific marker of NETs formation, was increased after SAUNA treatment (fold change: 1.0±0.21 versus 2.01±0.57, P=6.1×10−4; Figure 3B and 3D). We also performed immunofluorescence staining on heart tissue from HFpEF mice with anti‐CitH3 and anti‐MPO antibodies, together with DAPI, the most widely used and recommended method for NETs detection. NETs were identified by extracellular colocalization of DNA, CitH3, and neutrophil granule marker MPO, as per previous reports. 15 We found that NETs were not observed in the heart tissues obtained from control mice but revealed the presence of characteristic NETs in HFpEF mice after SAUNA treatment (Figure 1I).
Figure 3. HMGB1 inhibition and empagliflozin reduces heart neutrophil infiltration, NETs formation and ameliorates diastolic function in HFpEF mice.
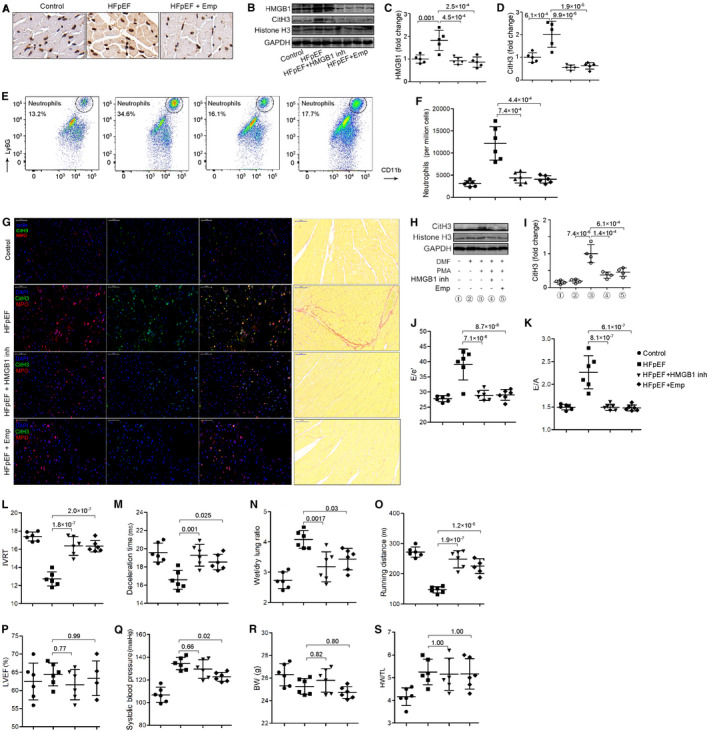
A, Heart sections of indicated mice immunostained for HMGB1, showing HMGB1 expression was increased in HFpEF heart but attenuated after empagliflozin treatment. B through D, Representative western blot of HMGB1, CitH3, histone H3, and GAPDH from left ventricles of indicated mice (B). Quantification of levels of HMGB1 (C) and CitH3 (D). These results showed that HMGB1 and CitH3 expressions were increased in HFpEF heart but attenuated after HMBG1 inhibition with glycyrrhizin or empagliflozin treatment. E and F, Flow cytometric analysis showed that neutrophil counts were increased in heart from HFpEF mice but attenuated after glycyrrhizin or empagliflozin treatment. G, In situ immunofluorescence identifying NETs by extracellular MPO (red), CitH3 (green), and DNA (blue) deposits in the heart (left), showing increased NETs formation in HFpEF mice than controls but attenuation after glycyrrhizin or empagliflozin treatment. Representative images of Sirius red staining of heart tissue (right), showing an increment of cardiac fibrosis in HFpEF and an attenuation after glycyrrhizin or empagliflozin treatment. Scale bar represents 50 µm. H and I, Representative western blot of CitH3, histone H3, and GAPDH from cultured differentiated neutrophils of indicated treatment (H). Quantification of levels of CitH3 (I) showed that HMBG1 inhibition with glycyrrhizin or empagliflozin decreased CitH3 expression in vitro. J, Ratio between mitral E wave and e′ wave (E/e′). K, Ratio between mitral E wave and A wave (E/A). L, Isovolumic relaxation time (IVRT). M, Deceleration time; (N) Ratio between wet and dry lung weight. O, Running distance during exercise exhaustion test. P, Percentage of LVEF. Q, Systolic blood pressure. R, Body weight (BW). S, Ratio between heart weight and tibia length (HW/TL). HMGB1 inhibition with glycyrrhizin or empagliflozin improved diastolic function and exercise tolerance and reduced lung congestion but had no significant effect on LVEF, blood pressure, or cardiac hypertrophy. BW indicates body weight; CitH3, citrullinated histone 3; HFpEF, heart failure with preserved ejection fraction; HMGB1, high mobility group protein B1; NETs, neutrophil extracellular traps; LVEF, left ventricular ejection fraction; and MPO, myeloperoxidase.
NETs Inhibition Reduces Cardiac Macrophage Infiltration
Previous study showed a significantly higher density of macrophages in the heart after SAUNA treatment, and cardiac macrophages activate fibroblasts and contribute to impaired myocardial relaxation. 6 NETs may support an important crosstalk between immune cells in chronic inflammation, such as neutrophils and macrophages. Therefore, we aimed to investigate whether NETs inhibition would affect macrophage infiltration. We confirmed a higher density of macrophages infiltration in the heart tissue after SAUNA treatment (1488±428 versus 2980±790 per million cells, P=0.0012; Figure 2R and 2S), and NETs breakdown by DNase 1 given through intraperitoneal injection, a common method to validate the function of NETs, significantly reduced the number of macrophages in the heart (2980±790 versus 1514±440 per million cells, P=0.0014; Figure 2R and 2S). Also, DNase 1 decreased heart expression of IL‐10 (Figure S1), which was found to be increased in macrophages during diastolic dysfunction. 6
NETs Inhibition Ameliorates Diastolic Function in HFpEF
To examine whether NETs contribute to the pathogenesis of HFpEF, we tested whether suppression of NETs would ameliorate cardiac relaxation and exercise intolerance in HFpEF mice. Compared with controls, DNase 1 significantly attenuated cardiac diastolic dysfunction after SAUNA treatment, as evident by normalization of E/e′ (39.1±5.1 versus 29.5±1.9, P=1.7×10−5), E/A (2.3±0.37 versus 1.5±0.12, P=5.9×10−7), isovolumic relaxation time (12.7±0.78 versus 17.4±0.79, P=2.5×10−8), and deceleration time (16.6±1.04 versus 18.6±1.04, P=0.017) (Figure 2D to 2G). In addition, DNase 1 treatment improved exercise tolerance (running distance: 147.3±10.34 versus 256.7±22.51 m, P=6.5×10−8; Figure 2I) and reduced lung congestion (wet/dry lung ratio: 4.1±0.29 versus 3.2±0.33, P=0.002; Figure 2H) but had no significant effect on left ventricular systolic function (LVEF: 64.4±3.14 versus 62.4%±3.57%, P=0.92; Figure 2J), blood pressure (Figure 2K), or cardiac hypertrophy (Figure 2O). Diastolic dysfunction is often attributed to cardiac fibrosis. We found that NETs blockade with DNase 1 significantly reduced cardiac fibrosis compared with controls (Figure 2Q). Together, these findings suggest that NETs inhibition improves cardiac function in this model.
Targeting HMGB1 Reduces Neutrophil Infiltration and NETs Formation and Ameliorates Diastolic Function in HFpEF
Neutrophils respond not only to microbes but also to endogenous danger signals. We previously showed that HMGB1, a critical DAMP, was involved in diabetic‐induced inflammation and oxidative stress, 16 , 17 which might cause NETs formation. In other studies, HMGB1 has been shown to promote the recruitment and activation of neutrophils, stimulate NETs information, and drive organ fibrosis. 10 , 18 , 19 To test whether HMGB1 would also stimulate NETs formation in the context of HFpEF, 10 we first determined the expression of HMGB1 in heart tissues after SAUNA treatment in mice. We showed a remarkably increased expression of HMGB1 in the heart after SAUNA treatment, as revealed by immunohistochemistry analysis (Figure 3A) and western blotting (fold change: 1.0±0.18 versus 1.83±0.44, P=0.001; Figure 3B and 3C). We then investigated the effect of HMGB1 on NETs formation in vitro. We stimulated differentiated neutrophils with PMA to increase NETs formation 13 and revealed considerable expression of CitH3, as indicated by western blotting (Figure 3H). Adding glycyrrhizin, a widely used functional HMGB1 inhibitor, to the cultured cells significantly reduced CitH3 expression (fold change: 1.0±0.27 versus 0.37±0.09, P=1.4×10−4; Figure 3H and 3I) and cell‐free dsDNA concentrations (Figure S2), confirming the inhibitory effect of HMGB1 on NETs formation in vitro. To validate these effects in vivo, SAUNA‐treated HFpEF mice were treated with glycyrrhizin. Treatment of mice with HMGB1 inhibitor largely abrogated neutrophilic inflammation in the heart after SAUNA treatment (12 168±3779 versus 4411±1220 per million cells, P=7.4×10−4; Figure 3E and 3F). Also, HMGB1 inhibition reduced NETs formation in the heart, as confirmed by both immunofluorescent and western blot assays (fold change: 2.01±0.57 versus 0.55±0.11, P=9.9×10−6; Figure 3B, 3D and 3G). Similar to NETs inhibition, HMGB1 inhibition significantly improved diastolic function (E/e′: 39.1±5.1 versus 28.9±1.7, P=7.1×10−6; E/A: 2.3±0.37 versus 1.5±0.07, P=8.1×10−7), attenuated exercise intolerance (running distance: 147.3±10.34 versus 248.3±28.35 m, P=1.9×10−7), and reduced lung congestion (wet/dry lung ratio: 4.1±0.29 versus 3.2±0.50, P=0.0017) and cardiac fibrosis (Figure 3J through 3O and 3G). HMGB1 inhibition decreased SAUNA‐induced mRNA expression of Anp, Bnp, and collagen 1 but not collagen 3 (Figure S1). These results indicate that HMGB1 inhibition ameliorates diastolic function in HFpEF and provides a HMGB1‐dependent mechanistic link between SAUNA treatment and NETs formation in the heart.
Empagliflozin Down‐Regulates Heart HMGB1 Expression and Improves Diastolic Function in HFpEF
Large clinical trials have shown that SGLT2 inhibitors reduce the risk of important heart failure events in patients with type 2 diabetes and in those without diabetes. Animal studies and the recent EMPEROR‐Preserved trial have shown that SGLT2 inhibitors improve cardiac diastolic function and clinical outcomes. These beneficial effects could not be wholly explained by the antihyperglycemic effect or diuretic effects of this class of drugs. We aimed to investigate whether SGLT2 inhibitor empagliflozin achieves its cardiac benefits through the HMGB1‐NETs axis. In vitro experiments showed that empagliflozin inhibited the CitH3 expression in differentiated neutrophils stimulated with PMA (fold change: 1.0±0.27 versus 0.45±0.13, P=6.1×10−4; Figure 3H and 3I), suggesting a NETs‐inhibitory effect. In vivo, SAUNA induced HFpEF mice treated with empagliflozin showed significantly reduced expression of HMGB1 in the heart than controls (fold change: 1.83±0.44 versus 0.87±0.22, P=2.5×10−4; Figure 3B and 3C). The evident recruitment of neutrophils (12 168±3779 versus 4051±830 per million cells, P=4.4×10−4) and NETs formation (fold change: 2.01±0.57 versus 0.63±0.15, P=1.9×10−5) seen in HFpEF mice were also attenuated after empagliflozin treatment (Figure 3B, 3D and 3G). Consistent with human clinical findings, empagliflozin significantly improved diastolic function (E/e′: 39.1±5.1 versus 29.0±1.7, P=8.7×10−6; E/A: 2.3±0.37 versus 1.5±0.07, P=6.1×10−7) and exercise tolerance (running distance: 147.3±10.34 versus 225.7±22.49 m, P=1.2×10−5) and reduced lung congestion (wet/dry lung ratio: 4.1±0.29 versus 3.4±0.36, P=0.03) and cardiac fibrosis (Figure 3G, 3J to 3S).
Discussion
To our knowledge, this is the first study to show an excess of circulating NETs in patients with HFpEF, and a higher density of neutrophils and NETs in the heart tissues from HFpEF mice. Moreover, we found that inhibition of NETs significantly reduced the infiltration of macrophages, reduced cardiac fibrosis, and attenuated the development of HFpEF phenotypes in a clinically relevant mouse model of HFpEF. Our data also showed that DAMP molecular HMGB1 contributed to promote NETs formation and inhibition of HMGB1 improved diastolic function in HFpEF. These findings suggest that HMGB1 and NETs suppression might be a potential therapeutic strategy for patients with HFpEF.
HFpEF remains an increasing public health problem with substantial morbidity and mortality 20 and few effective treatments. HFpEF represents a unique pathophysiological phenotype distinct from HFrEF. Recent experimental and clinical evidence suggests an inflammatory and profibrotic pathophysiological mechanism underlying HFpEF. Biomarker studies have shown strong association between proinflammatory biomarker concentrations and the presence and severity of HFpEF, including growth differentiation factor‐15, soluble interleukin 1 receptor‐like 1, C‐reactive protein, and interleukin 6. 21 Two large biomarker studies showed that unique pathways in patients with HFpEF were related to inflammation, cytokine response, and extracellular matrix organization, remarkably different from those in HFrEF. 5 , 22 Moreover, longitudinal observational studies demonstrated a predictive role of circulating endothelial adhesion molecules for HFpEF occurrence in community‐based populations 23 , 24 . These molecules, which include E‐selectin, intercellular adhesion molecule 1, and vascular cell adhesion molecule, are all all critical for inflammatory cells adhesion.
Chronic neutrophil recruitment and inflammatory responses might cause detrimental consequences. In obesity, one of the most common comorbidities of HFpEF, there is persistent low‐grade cell death and sterile inflammation, a result of the release of DAMPs that activate the Nlrp3 inflammasome and alert circulating neutrophils adherent. 25 Neutrophils have been noted in the inflammatory cell infiltrates of HFpEF 6 , and an elevated neutrophil‐to‐lymphocyte ratio positively correlates with inflammatory markers and predicts worse outcomes in patients with HFpEF. 26 However, the role of neutrophils as potential drivers of HFpEF disease process has not been addressed yet. Upon activation by various stimuli such as cytokines, hypoxia, and activated platelets, neutrophils can release chromatin structures and form NETs, which in turn contribute to eliminate pathogens more efficiently, but can also cause tissue injury, autoimmunity, and other dysfunctional outcomes, such as metastasis, thrombosis, and inappropriate coagulation. 27 NETs have been described to contribute to the pathophysiology of common noninfectious diseases. 27
Human and animal studies suggest a possible role for NETs in diabetes, one of the most common comorbidities in HFpEF. Neutrophils isolated from patients with type 1 and type 2 diabetes were more susceptible to NETs release. 28 Skin wounds from diabetic mice showed higher NETs formation and a delayed wound healing, 28 whereas blockade of PAD4, the key enzyme in NETs formation, significantly accelerated wound healing in diabetic mice. Similar results were observed in foot ulcers of humans with diabetes. 29 This experimental data established the important role of NETs in promoting diabetes‐associated complications. Similarly, the role of NETs in atherosclerosis has also been documented in an increasing body of clinical and experimental studies. 30 , 31 , 32
It is notable that over 90% of patients with HFpEF showed evidence of epicardial coronary artery disease, coronary microvascular dysfunction, or both, 33 and many had diabetes. HFpEF might show similar etiological paradigm. Studies investigating the role of NETs in heart failure are rare. Martinod and colleagues 34 quantified circulating neutrophils and CitH3‐positive neutrophils from peripheral blood of aged c57Bl/6nIA mice and neutrophils and NETs from heart tissues of mice undergoing transverse aortic constriction. They found that neutrophils and NETs counts were higher from aged mice and mice after transverse aortic constriction surgery. Inhibition of NETs through PAD4 deficiency or DNase 1 prevented the decline in systolic and “diastolic” function, as indicated by preservation of LVEF and E/A. 34 However, both models used in the study of Martinod and colleagues showed considerable LVEF decline (which is statistically significant) but no convincing evidence of diastolic dysfunction. In fact, transverse aortic constriction‐induced pressure overload is one of the commonly used models of HFrEF but not HFpEF. 35 Likewise, isolated aged c57Bl/6nIA mice did not represent HFpEF phenotypes. In current literature, aging in senescence‐accelerated mice (but not c57Bl/6nIA) plus a western diet develops diastolic dysfunction but not substantial heart failure, reflected by less of an increase in diastolic pressure and pulmonary congestion; 36 and the combination of aging, long‐term high‐fat diet, and desoxycorticosterone pivalate could recapture the important features of HFpEF. 37 Martinod and colleagues used a single parameter—E/A—to evaluate diastolic function (to our knowledge, E/A is not a good surrogate for diastolic function); whereas other more specific parameters such as E/e′ and heart failure related symptoms (for example, exercise intolerance and fluid accumulation in the lung) were not tested. Therefore, the work of Martinod and colleagues reflected more of age‐related heart fibrosis but not HFpEF. In contrast, we first verified that NETs are highly formed in an experimental mouse model of HFpEF and then confirmed their contributory role in the pathogenesis. This evidence implicates for the first time that NETs inhibition might be a potential therapeutic target in HFpEF.
In response to cellular stress, endogenous molecules termed DAMPs are passively released into the extracellular milieu from dying cells or actively secreted by mononuclear and other cells, exerting an essential role in danger response. Among these DAMPs, HMGB1 has been suggested to amplify inflammation, which is of high relevance as excessive inflammation often results in tissue damage. 38 The function of HMGB1 is multidimensional and highly context dependent. HMGB1 induces cytokine, chemokine, and metalloproteinase synthesis; promotes the migration of transendothelial monocytes; and triggers neutrophils recruitment. 7 In our study, blockade of the HMGB1 substantially reduced neutrophils infiltration into cardiac tissues of HFpEF mice. In accordance with our work, diminished neutrophils migration was observed in HMGB1‐deficient mice after hepatic ischemic/reperfusion injury. 10 Similarly, in a model of ultraviolet irradiation‐induced inflammation, inhibition of HMGB1 largely abrogated ultraviolet‐dependent neutrophilic inflammation. 18 We also observed a reduction of NETs after HMGB1 inhibition in HFpEF mice. These findings indicate a dual function of HMGB1 by promoting the recruitment of neutrophils as well as the formation of NETs in the heart.
The exact mechanism by which HMGB1 promotes the recruitment of neutrophils and NETs formation during HFpEF remains unclear. It has been reported that HMGB1 binds to the diverse receptors, in which toll‐like receptor 4 and RAGE (receptor for advanced glycation end products) are most comprehensively investigated. 39 Toll‐like receptor4/RAGE engagement results in the expression of inflammatory cytokines, chemokines, and corresponding receptors. 7 HMGB1 was shown to participate in the pathogenesis of liver fibrosis by signaling via RAGE. 19 In a model of tropinin I‐induced experimental autoimmune myocarditis, HMGB1 and its main receptor RAGE are crucial factors in the pathogenesis; but other receptors such as toll‐like receptor 4 may also be involved. 40 β2‐intergrin is another important molecule that might contribute to HMGB1‐induced neutrophils recruitment and NETs formation. A large network analysis points to β2‐intergrin as one of the 2 central proteins in HFpEF. 5 In 2 transgenic models of Alzheimer’s disease, Aβ42 peptide was shown to promote rapid neutrophil adhesion to integrin ligands via the LFA‐1 (lymphocyte function‐associated antigen‐1), 41 one type of β2‐intergrin. In agreement, β2 integrin–mediated systemic NETs formation is a critical contributor to hantavirus‐associated disease such as kidney and lung damage. 42 The exact role of β2‐intergrin in HMBG1‐induced NETs formation remains to be determined. The exact source and localization of HMGB1 in HFpEF also need to be clarified in further studies.
SGLT2 inhibitors are among one of the few drugs showing prognosis‐improving effects in patients with heart failure since the past decade, which are not confined to HFrEF but also involved in HFpEF. 43 Human and animal studies have consistently shown that SGLT2 inhibitors significantly ameliorate diastolic function, 44 , 45 and a recent randomized trial documented the clinical benefits of SGLT2 inhibitors on HFpEF. 2 The mechanisms underlying the protective cardiovascular effects of SGLT2 inhibitors may not be primarily related to hemodynamic effects or glucose‐lowering or body‐weight‐lowering effects, indicating other undiscovered mechanisms. As SGLT2 is not expressed in the heart, an indirect mechanism is likely to be present, such as regulation of inflammation. 46 A recent study suggests that empagliflozin reduced cardiac inflammation via blunting activation of the NLRP3 inflammasome, 46 and previous studies have shown that SGLT2 inhibitor dapagliflozin slows the progression of renal complications through the suppression of renal inflammation, 47 and empagliflozin promotes fat use and browning and attenuates inflammation and insulin resistance. 48 Our study for the first time showed that empagliflozin inhibited neutrophils recruitment and NETs formation in HFpEF, which might be partially dependent on HMGB1.
Several limitations should be acknowledged in our study. First, rescue treatments were given 1 day after surgery, and therefore it remains unclear whether anti‐HMGB1 or DNase 1 therapy is able to reverse established HFpEF and future studies are needed. Second, our clinical studies of patients with HFpEF and their age‐ and sex‐matched controls were limited by baseline characteristics differences, including comorbidities such as obesity, hypertension, and diabetes, which might bias the NETs quantification attributed to HFpEF as NETs might also be related to other metabolic diseases. However, to our knowledge, metabolic comorbidities are key features of HFpEF and co‐contribute to the myocardial structural and functional abnormities in HFpEF; therefore, the collective changes of NETs related to the metabolic diseases might represent HFpEF. Meanwhile, our animal studies confirmed findings from human clinical studies, which reinforced the relationship between NETs and HFpEF. Third, although HMGB1 was able to induce NETs formation in vivo and in vitro, a contribution of other proinflammatory effectors to NETs formation in HFpEF is also possible and requires further investigation. Also, we showed that empagliflozin treatment decreased HMGB1 expression and NETs formation, but how much these effects contributed to the clinical benefits of SGLT2 inhibitors remains unclear, because it is quite clear that a host of mechanisms are involved.
Conclusions
In conclusion, we showed that NETs substantially contribute to the pathogenesis of HFpEF, which can be ameliorated by HMGB1 inhibition and SGLT2 inhibitors. HMGB1 as well as NETs may represent novel therapeutic targets for the treatment of HFpEF.
Source of Funding
This study was supported by the National Natural Science Foundation of China (NO. 81800752) and Key Project supported by Medical Science and Technology Development Foundation, Nanjing Department of Health (CZLA1340‐2020).
Disclosures
None.
Supporting information
Table S1
Figures S1–S2
For Disclosures, see page 12.
Contributor Information
Yong Wang, Email: yongwang@nju.edu.cn.
Wei Xu, Email: yongwang@nju.edu.cn.
References
- 1. Roger VL. Epidemiology of heart failure: a contemporary perspective. Circ Res. 2021;128:1421–1434. doi: 10.1161/CIRCRESAHA.121.318172 [DOI] [PubMed] [Google Scholar]
- 2. Anker SD, Butler J, Filippatos G, Ferreira JP, Bocchi E, Böhm M, Brunner–La Rocca HP, Choi DJ, Chopra V, Chuquiure‐Valenzuela E, et al. Empagliflozin in heart failure with a preserved ejection fraction. N Engl J Med. 2021;385:1451–1461. doi: 10.1056/NEJMoa2107038 [DOI] [PubMed] [Google Scholar]
- 3. Packer M, Butler J, Zannad F, Filippatos G, Ferreira JP, Pocock SJ, Carson P, Anand I, Doehner W, Haass M, et al. Effect of empagliflozin on worsening heart failure events in patients with heart failure and a preserved ejection fraction: the EMPEROR‐Preserved trial. Circulation. 2021;144:1284–1294. doi: 10.1161/CIRCULATIONAHA.121.056824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62:263–271. doi: 10.1016/j.jacc.2013.02.092 [DOI] [PubMed] [Google Scholar]
- 5. Tromp J, Westenbrink BD, Ouwerkerk W, van Veldhuisen DJ, Samani NJ, Ponikowski P, Metra M, Anker SD, Cleland JG, Dickstein K, et al. Identifying pathophysiological mechanisms in heart failure with reduced versus preserved ejection fraction. J Am Coll Cardiol. 2018;72:1081–1090. doi: 10.1016/j.jacc.2018.06.050 [DOI] [PubMed] [Google Scholar]
- 6. Hulsmans M, Sager HB, Roh JD, Valero‐Muñoz M, Houstis NE, Iwamoto Y, Sun Y, Wilson RM, Wojtkiewicz G, Tricot B, et al. Cardiac macrophages promote diastolic dysfunction. J Exp Med. 2018;215:423–440. doi: 10.1084/jem.20171274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Harris HE, Andersson U, Pisetsky DS. HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat Rev Rheumatol. 2012;8:195–202. doi: 10.1038/nrrheum.2011.222 [DOI] [PubMed] [Google Scholar]
- 8. Thiam HR, Wong SL, Wagner DD, Waterman CM. Cellular mechanisms of NETosis. Annu Rev Cell Dev Biol. 2020;36:191–218. doi: 10.1146/annurev-cellbio-020520-111016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bonaventura A, Montecucco F, Dallegri F, Carbone F, Luscher TF, Camici GG, Liberale L. Novel findings in neutrophil biology and their impact on cardiovascular disease. Cardiovasc Res. 2019;115:1266–1285. doi: 10.1093/cvr/cvz084 [DOI] [PubMed] [Google Scholar]
- 10. Huebener P, Pradere J‐P, Hernandez C, Gwak G‐Y, Caviglia JM, Mu X, Loike JD, Jenkins RE, Antoine DJ, Schwabe RF. The HMGB1/RAGE axis triggers neutrophil‐mediated injury amplification following necrosis. J Clin Invest. 2015;125:539–550. doi: 10.1172/JCI76887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stark K, Philippi V, Stockhausen S, Busse J, Antonelli A, Miller M, Schubert I, Hoseinpour P, Chandraratne S, von Brühl M‐L, et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood. 2016;128:2435–2449. doi: 10.1182/blood-2016-04-710632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY, Luo X, Jiang N, May HI, Wang ZV, et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature. 2019;568:351–356. doi: 10.1038/s41586-019-1100-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Manda‐Handzlik A, Bystrzycka W, Wachowska M, Sieczkowska S, Stelmaszczyk‐Emmel A, Demkow U, Ciepiela O. The influence of agents differentiating HL‐60 cells toward granulocyte‐like cells on their ability to release neutrophil extracellular traps. Immunol Cell Biol. 2018;96:413–425. doi: 10.1111/imcb.12015 [DOI] [PubMed] [Google Scholar]
- 14. Pieske B, Tschöpe C, de Boer RA, Fraser AG, Anker SD, Donal E, Edelmann F, Fu M, Guazzi M, Lam CSP, et al. How to diagnose heart failure with preserved ejection fraction: the HFA‐PEFF diagnostic algorithm: a consensus recommendation from the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur Heart J. 2019;40:3297–3317. doi: 10.1093/eurheartj/ehz641 [DOI] [PubMed] [Google Scholar]
- 15. Kessenbrock K, Krumbholz M, Schonermarck U, Back W, Gross WL, Werb Z, Grone HJ, Brinkmann V, Jenne DE. Netting neutrophils in autoimmune small‐vessel vasculitis. Nat Med. 2009;15:623–625. doi: 10.1038/nm.1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wu H, Li R, Wei ZH, Zhang XL, Chen JZ, Dai Q, Xie J, Xu B. Diabetes‐induced oxidative stress in endothelial progenitor cells may be wustained by a positive feedback loop involving high mobility group box‐1. Oxid Med Cell Longev. 2016;2016:1943918. doi: 10.1155/2016/1943918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu H, Sheng ZQ, Xie J, Li R, Chen L, Li GN, Wang L, Xu B. Reduced HMGB 1‐mediated pathway and oxidative stress in resveratrol‐treated diabetic mice: a possible mechanism of cardioprotection of resveratrol in diabetes mellitus. Oxid Med Cell Longev. 2016;2016:9836860. doi: 10.1155/2016/9836860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bald T, Quast T, Landsberg J, Rogava M, Glodde N, Lopez‐Ramos D, Kohlmeyer J, Riesenberg S, van den Boorn‐Konijnenberg D, Hömig‐Hölzel C, et al. Ultraviolet‐radiation‐induced inflammation promotes angiotropism and metastasis in melanoma. Nature. 2014;507:109–113. doi: 10.1038/nature13111 [DOI] [PubMed] [Google Scholar]
- 19. Ge X, Arriazu E, Magdaleno F, Antoine DJ, Dela Cruz R, Theise N, Nieto N. High mobility group box‐1 drives fibrosis progression signaling via the receptor for advanced glycation end products in mice. Hepatology. 2018;68:2380–2404. doi: 10.1002/hep.30093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Paulus WJ, Zile MR. From systemic inflammation to myocardial fibrosis: the heart failure with preserved ejection fraction paradigm revisited. Circ Res. 2021;128:1451–1467. doi: 10.1161/CIRCRESAHA.121.318159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chia YC, Kieneker LM, van Hassel G, Binnenmars SH, Nolte IM, van Zanden JJ, van der Meer P, Navis G, Voors AA, Bakker SJL, et al. Interleukin 6 and development of heart failure with preserved ejection fraction in the general population. J Am Heart Assoc. 2021;10:e018549. doi: 10.1161/JAHA.120.018549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tromp J, Khan MA, Klip IT, Meyer S, de Boer RA, Jaarsma T, Hillege H, van Veldhuisen DJ, van der Meer P, Voors AA. Biomarker profiles in heart failure patients with preserved and reduced ejection fraction. J Am Heart Assoc. 2017;6:e003989. doi: 10.1161/JAHA.116.003989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Patel RB, Colangelo LA, Reiner AP, Gross MD, Jacobs DR Jr, Launer LJ, Lima JAC, Lloyd‐Jones DM, Shah SJ. Cellular adhesion molecules in young adulthood and cardiac function in later life. J Am Coll Cardiol. 2020;75:2156–2165. doi: 10.1016/j.jacc.2020.02.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Paulus WJ. Adhesion molecules in early adulthood predict heart failure with preserved ejection fraction at older age. J Am Coll Cardiol. 2020;75:2166–2168. doi: 10.1016/j.jacc.2020.03.016 [DOI] [PubMed] [Google Scholar]
- 25. Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD. The NLRP3 inflammasome instigates obesity‐induced inflammation and insulin resistance. Nat Med. 2011;17:179–188. doi: 10.1038/nm.2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Curran FM, Bhalraam U, Mohan M, Singh JS, Anker SD, Dickstein K, Doney AS, Filippatos G, George J, Metra M, et al. Neutrophil‐to‐lymphocyte ratio and outcomes in patients with new‐onset or worsening heart failure with reduced and preserved ejection fraction. ESC Heart Fail. 2021;8:3168–3179. doi: 10.1002/ehf2.13424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med. 2017;23:279–287. doi: 10.1038/nm.4294 [DOI] [PubMed] [Google Scholar]
- 28. Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, Kahn CR, Wagner DD. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med. 2015;21:815–819. doi: 10.1038/nm.3887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fadini GP, Menegazzo L, Rigato M, Scattolini V, Poncina N, Bruttocao A, Ciciliot S, Mammano F, Ciubotaru CD, Brocco E, et al. NETosis delays diabetic wound healing in mice and humans. Diabetes. 2016;65:1061–1071. doi: 10.2337/db15-0863 [DOI] [PubMed] [Google Scholar]
- 30. Borissoff JI, Joosen IA, Versteylen MO, Brill A, Fuchs TA, Savchenko AS, Gallant M, Martinod K, ten Cate H, Hofstra L, et al. Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arterioscler Thromb Vasc Biol. 2013;33:2032–2040. doi: 10.1161/ATVBAHA.113.301627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stakos DA, Kambas K, Konstantinidis T, Mitroulis I, Apostolidou E, Arelaki S, Tsironidou V, Giatromanolaki A, Skendros P, Konstantinides S, et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur Heart J. 2015;36:1405–1414. doi: 10.1093/eurheartj/ehv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. 2015;349:316–320. doi: 10.1126/science.aaa8064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rush CJ, Berry C, Oldroyd KG, Rocchiccioli JP, Lindsay MM, Touyz RM, Murphy CL, Ford TJ, Sidik N, McEntegart MB, et al. Prevalence of coronary artery disease and coronary microvascular dysfunction in patients with heart failure with preserved ejection fraction. JAMA Cardiol. 2021;6:1130–1143. doi: 10.1001/jamacardio.2021.1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Martinod K, Witsch T, Erpenbeck L, Savchenko A, Hayashi H, Cherpokova D, Gallant M, Mauler M, Cifuni SM, Wagner DD. Peptidylarginine deiminase 4 promotes age‐related organ fibrosis. J Exp Med. 2017;214:439–458. doi: 10.1084/jem.20160530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Noll NA, Lal H, Merryman WD. Mouse models of heart failure with preserved or reduced ejection fraction. Am J Pathol. 2020;190:1596–1608. doi: 10.1016/j.ajpath.2020.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mishra S, Kass DA. Cellular and molecular pathobiology of heart failure with preserved ejection fraction. Nat Rev Cardiol. 2021;18:400–423. doi: 10.1038/s41569-020-00480-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Deng Y, Xie M, Li Q, Xu X, Ou W, Zhang Y, Xiao H, Yu H, Zheng Y, Liang Y, et al. Targeting mitochondria‐inflammation circuit by beta‐hydroxybutyrate mitigates HFpEF. Circ Res. 2021;128:232–245. doi: 10.1161/CIRCRESAHA.120.317933 [DOI] [PubMed] [Google Scholar]
- 38. Cai J, Wen J, Bauer E, Zhong H, Yuan H, Chen AF. The role of HMGB1 in cardiovascular biology: danger signals. Antioxid Redox Signal. 2015;23:1351–1369. doi: 10.1089/ars.2015.6408 [DOI] [PubMed] [Google Scholar]
- 39. Pellegrini L, Foglio E, Pontemezzo E, Germani A, Russo MA, Limana F. HMGB1 and repair: focus on the heart. Pharmacol Ther. 2019;196:160–182. doi: 10.1016/j.pharmthera.2018.12.005 [DOI] [PubMed] [Google Scholar]
- 40. Bangert A, Andrassy M, Müller A‐M, Bockstahler M, Fischer A, Volz CH, Leib C, Göser S, Korkmaz‐Icöz S, Zittrich S, et al. Critical role of RAGE and HMGB1 in inflammatory heart disease. Proc Natl Acad Sci USA. 2016;113:E155–E164. doi: 10.1073/pnas.1522288113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zenaro E, Pietronigro E, Bianca VD, Piacentino G, Marongiu L, Budui S, Turano E, Rossi B, Angiari S, Dusi S, et al. Neutrophils promote Alzheimer's disease‐like pathology and cognitive decline via LFA‐1 integrin. Nat Med. 2015;21:880–886. doi: 10.1038/nm.3913 [DOI] [PubMed] [Google Scholar]
- 42. Raftery MJ, Lalwani P, Krautkrmer E, Peters T, Scharffetter‐Kochanek K, Kruger R, Hofmann J, Seeger K, Kruger DH, Schonrich G. Beta2 integrin mediates hantavirus‐induced release of neutrophil extracellular traps. J Exp Med. 2014;211:1485–1497. doi: 10.1084/jem.20131092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Herrington WG, Savarese G, Haynes R, Marx N, Mellbin L, Lund LH, Dendale P, Seferovic P, Rosano G, Staplin N, et al. Cardiac, renal, and metabolic effects of sodium‐glucose co‐transporter‐2 inhibitors: a position paper from the European Society of Cardiology ad‐hoc task force on sodium‐glucose co‐transporter‐2 inhibitors. Eur J Heart Fail. 2021;23:1260–1275. doi: 10.1002/ejhf.2286 [DOI] [PubMed] [Google Scholar]
- 44. Habibi J, Aroor AR, Sowers JR, Jia G, Hayden MR, Garro M, Barron B, Mayoux E, Rector RS, Whaley‐Connell A, et al. Sodium glucose transporter 2 (SGLT2) inhibition with empagliflozin improves cardiac diastolic function in a female rodent model of diabetes. Cardiovasc Diabetol. 2017;16:9. doi: 10.1186/s12933-016-0489-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pabel S, Wagner S, Bollenberg H, Bengel P, Kovács Á, Schach C, Tirilomis P, Mustroph J, Renner A, Gummert J, et al. Empagliflozin directly improves diastolic function in human heart failure. Eur J Heart Fail. 2018;20:1690–1700. doi: 10.1002/ejhf.1328 [DOI] [PubMed] [Google Scholar]
- 46. Byrne NJ, Matsumura N, Maayah ZH, Ferdaoussi M, Takahara S, Darwesh AM, Levasseur JL, Jahng JWS, Vos D, Parajuli N, et al. Empagliflozin blunts worsening cardiac dysfunction associated with reduced NLRP3 (Nucleotide‐binding domain‐like receptor protein 3) inflammasome activation in heart failure. Circ Heart Fail. 2020;13:e006277. doi: 10.1161/CIRCHEARTFAILURE.119.006277 [DOI] [PubMed] [Google Scholar]
- 47. Jaikumkao K, Pongchaidecha A, Chueakula N, Thongnak LO, Wanchai K, Chatsudthipong V, Chattipakorn N, Lungkaphin A. Dapagliflozin, a sodium‐glucose co‐transporter‐2 inhibitor, slows the progression of renal complications through the suppression of renal inflammation, endoplasmic reticulum stress and apoptosis in prediabetic rats. Diabetes Obes Metab. 2018;20:2617–2626. doi: 10.1111/dom.13441 [DOI] [PubMed] [Google Scholar]
- 48. Xu L, Nagata N, Nagashimada M, Zhuge F, Ni Y, Chen G, Mayoux E, Kaneko S, Ota T. SGLT2 inhibition by empagliflozin promotes fat utilization and browning and attenuates inflammation and insulin resistance by polarizing M2 macrophages in diet‐induced obese mice. EBioMedicine. 2017;20:137–149. doi: 10.1016/j.ebiom.2017.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Figures S1–S2