

Abstract
Background
Changes in arterial wall viscosity, which dissipates the energy stored within the arterial wall, may contribute to the beneficial effect of heart rate (HR) reduction on arterial stiffness and cardiovascular coupling. However, it has never been assessed in humans and could be altered by aging. We evaluated the effect of a selective HR‐lowering agent on carotid arterial wall viscosity and the impact of aging on this effect.
Methods and Results
This randomized, placebo‐controlled, double‐blind, crossover study performed in 19 healthy volunteers evaluated the effects of ivabradine (5 mg BID, 1‐week) on carotid arterial wall viscosity, mechanics, hemodynamics, and cardiovascular coupling. Arterial wall viscosity was evaluated by the area of the hysteresis loop of the pressure‐lumen cross‐sectional area relationship, representing the energy dissipated (WV), and by the relative viscosity (WV/WE), with WE representing the elastic energy stored.
HR reduction by ivabradine increased WV and WE whereas WV/WE remained stable. In middle‐aged subjects (n=11), baseline arterial stiffness and cardiovascular coupling were less favorable, and WE was similar but WV and therefore WV/WE were lower than in youth (n=8). HR reduction increased WV/WE in middle‐aged but not in young subjects, owing to a larger increase in WV than WE. These results were supported by the age‐related linear increase in WV/WE after HR reduction (P=0.009), explained by a linear increase in WV.
Conclusion
HR reduction increases arterial wall energy dissipation proportionally to the increase in WE, suggesting an adaptive process to bradycardia. This mechanism is altered during aging resulting in a larger than expected energy dissipation, the impact of which should be assessed.
Registration
URL: https://www.clinicaltrials.gov; Unique identifier: 2015/077/HP. URL: https://www. eudract.ema.europa.eu; Unique identifier: 2015‐002060‐17.
Keywords: aging, artery, heart rate, stiffness, wall viscosity
Subject Categories: Clinical Studies, Hemodynamics, Pathophysiology, Physiology
Nonstandard Abbreviations and Acronyms
- AIx
augmentation index
- AP
augmentation pressure
- AWV
arterial wall viscosity
- cfPWV
carotidofemoral pulse wave velocity
- Einc
incremental Young’s elastic modulus
- P‐LCSA
pressure‐lumen cross‐sectional area
- SBP
systolic blood pressure
- WE
elastic energy stored
- WV
dissipated energy
- WV/WE
relative viscosity
Clinical Perspective
What Is New?
Heart rate reduction increases energy dissipation in the carotid artery wall proportionally to the increase in elastic energy stored.
Aging affects this adaptative viscous response by increasing energy dissipation.
What Are the Clinical Implications?
Arterial wall viscosity should be considered when assessing the benefit of heart rate lowering strategies because the increased energy dissipation with heart rate reduction during aging could reduce the energy transmitted to the periphery but also could alter cardiovascular coupling.
High resting heart rate (HR) is increasingly recognized as a cardiovascular risk factor. 1 This may be related to the parallel increase in HR and arterial stiffness, a major independent predictor of cardiovascular events. 2 , 3 , 4 , 5 , 6 However, benefit of HR reduction on arterial stiffness and cardiovascular prognosis is more controversial in particular during aging. 7 , 8 , 9 , 10 , 11 , 12 , 13 Although it has not been not evaluated, some authors suggested that this lack of benefit is due to unfavorable change in arterial wall viscosity (AWV) with HR reduction. 3 , 4 , 6 , 14 , 15 , 16
In fact, the mechanical behavior of arteries is viscoelastic, but the viscous component, which is time dependent, has been less investigated than elasticity. At present, the impact of HR on AWV assessed in vitro and in animal models remains unclear, putatively because of differences in the methods used to assess AWV. 15 , 17 , 18 , 19 , 20 In a thermodynamic point of view, AWV is responsible for the dissipation as heat of a part of the mechanical energy stored within the arterial wall during each cardiac cycle. Thus, the hysteresis loop resulting from the shift between the loading and unloading phases of the pressure‐lumen cross‐sectional area curves (P‐LCSA) corresponds to the dissipated energy (WV), whereas the area under the loading curve reflects the elastic energy stored (WE). 21 , 22 Consequently, AWV can be expressed as the absolute value of the loop area or as a ratio (WV/WE), considering thus one of its major determinants. 21 , 22 Some in vitro and animal in vivo pacing studies suggested that an increase in HR is associated with a decrease in AWV but only 1 animal study explored the effect of HR reduction on AWV and showed its increase. 15 , 18 , 23 The mechanism by which HR modulates AWV is unknown but could be related to a direct effect on intrinsic viscous properties by modifying the time and the magnitude of stretch 4 , 14 , 24 or an indirect effect of HR on the vascular endothelium through changes in shear stress. 25 , 26 Moreover, in aging or in different pathological conditions, such as hypertension and stable coronary artery disease, the positive effect of HR reduction on arterial stiffness could be overridden by the increase in energy dissipation and thus lead to cardiovascular uncoupling. 10 , 12 , 27 This may have reduced the expected benefit of HR‐lowering agents such as betablockers or selective If current inhibitor without negative inotropic effect, ivabradine, on arterial wall mechanics and on cardiovascular outcomes. 7 , 8 , 9 , 10 , 11 , 12 , 13 , 28 Whether HR lowering in healthy volunteers is beneficial on arterial wall mechanics, including AWV, is unknown. Moreover, how aging could have an impact on this effect needs to be evaluated. Our main unexplored hypothesis is that HR reduction by ivabradine could increase AWV to damp the increase in elastic energy stored, but that this response is altered by aging.
In this context, the objectives of the study were to assess the effects of bradycardia induced by repeated administration of ivabradine on carotid AWV considering associated changes in arterial mechanics and hemodynamics in healthy volunteers and to evaluate the impact of aging on these effects.
Methods
Volunteers
Twenty volunteers were included between 2016 and 2019 with an age between 25 and 65 years old, a resting HR >70 bpm after 15 minutes of rest, and deemed healthy based on interview, clinical examination, and electrocardiographic and routine biological evaluation. Detailed exclusion criteria are available in Data S1. The study was approved by the local ethics committee (CPP Nord‐Ouest I, n°CPP01/004/2014), and all participants gave written informed consent. The study was conducted according to the Principles of Good Clinical Practice and the Declaration of Helsinki. The study was registered at ClinicalTrials.gov Identifier: 2015/077/HP and EudraCT Number: 2015‐002060‐17. The data that support the findings of this study are available from the corresponding author upon reasonable request.
Study Design
This monocentric, randomized, placebo‐controlled, double‐blind (volunteers and investigators), crossover study included 1 inclusion visit (V0), 4 investigation visits (V1 to V4), and an end‐of‐study visit (V5). Inclusion criteria were checked at V0 visit and then randomization was performed. Volunteers were randomly allocated to ivabradine 5 mg BID and a placebo in a crossover design during a period of 8 days: 1 pill at day 1 at the end of the V1 or V3 and then 1 pill BID for 6 days and a last pill at day 8, before V2 or V4 investigations. V2 and V3 were separated by a 14‐day wash‐out period (Figure S1).
Ivabradine is the only If current inhibitor approved in humans. Ivabradine prolongs diastolic depolarization resulting in a slowing of the sinoatrial node. In fact, contrary to digoxin or beta blockers, this treatment has mainly a chronotropic effect and no direct effect on systemic arterial pressure or cardiac inotropism. 29 Thus, as performed in previous studies, we chose ivabradine to pharmacologically manipulate HR and to assess the effect of bradycardia on cardiac and vascular systems. 10 , 30 This dosage and this duration of treatment have been chosen expecting a 10 bpm decrease of HR and/or an HR lower than 60 bpm and to reach the steady‐state concentration of ivabradine according to the half‐life of the treatment (12 hours). 29
General Procedure (V1 to V4)
Each investigation visit was performed according to the same design. Measurements were performed in the morning while volunteers were in a supine position in a quiet air‐conditioned room maintained at a stable temperature (22–24 °C). Volunteers were allowed to take a light breakfast, without tea, coffee, sugar, or fat. They were not allowed to smoke for 12 hours. After 15 minutes, resting HR, brachial systolic blood pressure (SBP), and brachial diastolic blood pressure were measured 3 times on the right arm using an oscillometric device (Omron® 750IT) and an ECG (Phillips®) was performed.
Common Carotid Geometry and AWV
Assessment of common carotid AWV was developed based on previous studies in animal models and radial artery in humans. 21 , 22 , 26 , 31 P‐LCSA relationship was obtained by the continuous and simultaneous measurement of local pressure by aplanation tonometry (Millar Instruments® SPT 301B) and local diameter by high‐resolution echotracking (WallTrack System®, Esaote Pie Medical) at the level of the right and left common carotid arteries respectively. 32 , 33 , 34 All measurements were performed by the same trained operator pair. Localization of the probes was carefully verified at each visit, to avoid misalignment related to change in probe localization. External diameter and intima‐media thickness were measured at the level of carotid posterior wall, 1 cm beneath the carotid bifurcation as previously described. 34 Despite the simultaneous recording of the pressure and diameter waveforms at the same level of each carotid, we systematically visually checked the quality of the acquisition. Our visual check confirmed the synchronization of the feet of the waves for each acquisition, so no additional postacquisition resynchronization was necessary (Figure S2A). 22 Thus, the P‐LCSA relationship was obtained and AWV was estimated from the hysteresis loop as previously described. 21 , 22 WE was assessed for each cardiac cycle by integrating the P‐LCSA area during the loading phase, that is, from diastolic to systolic pressure, and was thus graphically bounded by the area under the systolic P‐LCSA relationship, the pulse pressure, and the pulse diameter (Figure S2B). The area of the P‐LCSA loop obtained during the loading and unloading phases, which has a dimension of energy, corresponds to the energy dissipated in viscous work (WV) by the arterial wall during 1 cardiac cycle. The loop area was measured using image analysis software (ImageJ). 22 Values of AWV are the mean of at least 3 cardiac cycles on 3 different acquisitions. Energies are expressed in joules per meter during 1 cycle equivalent to the pressure*area units. AWV is expressed either in absolute value of WV or as a percentage of the energy stored during the loading phase (relative viscosity=WV/WE.100) (Figure S2B). 22 , 26 The practical precision of this method is given by the precision of pressure and diameter measurements (2 mm Hg and 21 μm, respectively) and can be estimated as 2.79×10−3 J.m−1. 22 , 35 Detailed methods are available in Data S1.
Pulse Wave Analysis
Right radial and carotid artery pressure waveforms were recorded noninvasively by applanation tonometry and processed with dedicated software (SphygmoCor® version 7, AtCor Medical). The pressure at the first shoulder of the wave, the augmentation pressure (AP), the augmentation index (AIx), and the AIx normalized to the HR at 75 bpm, the carotid‐to‐brachial amplification, the reflection time, the duration of ejection, and the period were calculated from the carotid artery pressure waveforms with the same dedicated software. The Buckberg index was calculated as the ratio of the central diastolic to systolic pressure time integral. 30 Carotidofemoral pulse wave velocity (cfPWV) was determined as previously described. 2 Detailed methods are available in Data S1.
Common Carotid Artery Elastic Properties, Blood Flow, and Shear Stress
Circumferential wall stress, arterial compliance, arterial distensibility, and the incremental Young’s elastic modulus (Einc) were estimated through the variations in arterial LCSA (ΔLCSA) and blood pressure (ΔP). 34 , 36 The systolic and mean wall shear stress were calculated based on the systolic and mean carotid blood velocity (v) evaluated by Doppler (ArtLab system, Esaote Pie Medical®), μ the total blood viscosity, and internal diameter. Detailed methods are available in Data S1.
Systemic Hemodynamics and Cardiac Parameters
Systemic hemodynamics and cardiac parameters were evaluated by impedance cardiography (PhysioFlow® PF‐05 Lab1TM, software version 2.7.0, Manatec Biomedical). 37 The following parameters were obtained: cardiac output, stroke volume, ejection fraction, and end‐diastolic volume. Moreover, total peripheral resistance was calculated from the ratio of mean blood pressure to cardiac output. Left ventricular end‐systolic elastance, a measure of cardiac contractility, was calculated as the ratio of end‐systolic pressure obtained with carotid tonometry and the end‐systolic volume calculated as end‐systolic volume=end‐diastolic volume−stroke volume. 13
Sample Size Calculation
The difference in the percent change from baseline of carotid relative viscosity between ivabradine and placebo was the main outcome. In the absence of previous results concerning AWV under ivabradine or beta blockers, the sample size was obtained according to the crossover design of the study and previous results obtained with inhibitors of endothelial factors release expecting an absolute difference of 13% between the change in the value of WV/WE under ivabradine and placebo with a common SD of 10%. 22 Thus, a sample of 18 subjects was needed with an alpha risk of 5% and a power of 80%.
Statistical Analysis
Results are expressed as mean±SD unless indicated otherwise. Statistical analysis was performed in the per‐protocol population using SAS® and R studio Version 1.4.1106 with lme4 package. Randomization was performed by the Department of Biostatistics using SAS®, accounting for period effect and in accordance with the crossover design of the study. Homogeneity of the groups was checked for each parameter investigated by the comparison between the 2 treatment sequences (ivabradine/placebo versus placebo/ivabradine) at baseline using Mann Whitney U test and chi‐square or Fisher exact test as appropriate and by the comparison of baseline parameters at V1 and V3 using paired t test. For all parameters, changes from baseline were compared between treatment and placebo using a repeated measure ANCOVA with subjects as the grouping variable. The changes in circumferential wall stress were added to the model (as a covariable) to evaluate the carotid wall mechanics (arterial compliance, arterial distensibility, and Einc). Moreover, AWV percent change from baseline was also compared between treatment and placebo using a linear mixed model with sex and mean baseline value of the parameter as cofactors and subject as random effect. Considering the crossover design of the study, all these analyses were performed with the evaluation of the interaction between the treatment and period (sequences of treatment). Period×treatment interactions were nonsignificant in all the analyses performed demonstrating the absence of significant carryover effect. The impact of aging on the treatment effect, a prespecified outcome of the study, was evaluated by comparing 2 age classes, the young (<45 years) and middle‐aged (>45 years) volunteers. Baseline parameters of these 2 groups were compared with a general linear model according to age class. Circumferential wall stress was added to the model for carotid wall mechanics. Moreover, changes from mean baselines were compared for all parameters between treatment and placebo using linear mixed model according to age class with sex and mean baseline value of the parameter as cofactors and subject as random effect. Finally, effect of treatment on AWV according to age was evaluated with 2 methods. First, percent changes of AWV from baseline were compared between treatment and placebo using linear mixed model according to age class with sex and the baseline value of the parameter (WV, WE, or WV/WE as appropriate) as cofactors and subject as random effect. Second, the same analysis was performed according to age (quantitative values) rather than age class. In addition to this analysis based on AWV percent change, the same analysis was performed considering absolute changes in AWV (available in Data S1). Thus, age class×treatment and age×treatment interaction were respectively evaluated. Period×treatment interaction was not included in the final models exploring the age effect because it was nonsignificant in the whole population. However, for exploratory purposes, we also performed a full linear mixed model with treatment×age×period interaction, sex and baseline parameter as cofactors, and subject as random effect that did not show any period×treatment interaction. P<0.05 was considered statistically significant.
Results
Baseline Characteristics
Twenty healthy volunteers (14 women, mean age 47 [26–62] years) were included in the study. Baseline characteristics of the volunteers at V0 are presented in Table S1. There was no difference for these parameters between the treatment sequences (Table S1).
One randomized volunteer was withdrawn from the study before treatment administration because of a low HR (55 bpm) at V1 despite a HR >70 bpm at V0 (Figure S3). Thus, 9 volunteers received ivabradine then placebo and 10 received placebo then ivabradine.
There was no difference between V1 and V3 for all parameters (Table S2). At baseline, resting HR was 71.4±7.4 bpm (Table 1). Mean baselines for all cardiovascular parameters are presented in Tables 1, 2 and 3.
Table 1.
Effect of Ivabradine Versus Placebo on Brachial and Central Pressure and Systemic and Cardiac Hemodynamics
| Baseline | Delta ivabradine | Delta placebo | P value | |
|---|---|---|---|---|
| Heart rate (ECG), bpm | 71±7 | −10±7 | −2±5 | <0.001* |
| Brachial SBP, mm Hg | 117±9 | 0±6 | −2±5 | 0.216 |
| Brachial DBP, mm Hg | 71±6 | −1±4 | −2±4 | 0.901 |
| Brachial MBP, mm Hg | 87±7 | −1±4 | −2±5 | 0.682 |
| Brachial pulse pressure, mm Hg | 46±7.7 | 1±4 | 0±3 | 0.118 |
| Cyclic stretch, mm Hg×bpm | 3408±774 | −423±504 | −150±344 | 0.001* |
| Central SBP, mm Hg | 106±9 | 1±7 | −2±5 | 0.127 |
| Central DBP, mm Hg | 72±7 | −2±4 | −2±4 | 0.899 |
| Central MBP, mm Hg | 87±7 | −1±4 | −2±4 | 0.549 |
| Central pulse pressure, mm Hg | 35±6 | 2±5 | 0±3 | 0.076 |
| Carotid‐to‐brachial amplification, unitless | 1.3±0.2 | −0.04±0.1 | −0.02±0.1 | 0.581 |
| Stroke volume, 10−3 L | 90±10 | 10±10 | 0±7 | <0.001* |
| End‐diastolic volume, 10−3 L | 139±22 | 12±15 | 1±13 | 0.001* |
| End‐systolic volume, 10−3 L | 49±19 | 2±8 | 2±11 | 0.843 |
| Cardiac output, L/min | 6±1.1 | −0.2±0.4 | −0.2±0.5 | 0.674 |
| Ejection fraction, % | 65±9 | 1±3 | −1±4 | 0.142 |
| Total peripheral resistance, mm Hg/L per min | 14.8±2.3 | 0.3±1.1 | 0.3±1.1 | 0.947 |
| Left ventricular end‐systolic elastance, mm Hg/10−3 L | 2.1±0.8 | −0.1±0.2 | −0.1±0.4 | 0.539 |
Data are mean±SD. DBP indicates diastolic blood pressure; MSP, mean blood pressure; and SBP, systolic blood pressure.
P<0.05.
Table 2.
Effect of Ivabradine Versus Placebo on Pressure Wave, Cardiovascular Coupling, and Carotid Hemodynamics
| Baseline | Delta ivabradine | Delta placebo | P value | |
|---|---|---|---|---|
| Period, ms | 945±119 | 125±86 | 24±96 | <0.001* |
| Ejection duration, ms | 320±18 | 12±11 | 3±11 | <0.001* |
| Ejection duration/period, % | 34±4 | −3±2 | −1±3 | <0.001* |
| Reflexion time, ms | 158±32 | −6±29 | 7±20 | 0.054 |
| P1 height, mm Hg | 30±6 | 2±5 | 1±3 | 0.333 |
| Augmentation pressure, mm Hg | 3±7 | 1±3 | −1±3 | 0.011* |
| AIx, % | 6.9±18.8 | 1.8±6.5 | −1.8±7.3 | 0.045* |
| AIx75, % | 1.8±17.7 | −1.9±5.9 | −2.7±7 | 0.648 |
| Buckberg index, % | 161±23 | 20±17 | 4±18 | 0.002* |
| Central systolic pressure time integral, mm Hg×s/min | 2015±296 | −169±178 | −85±166 | 0.095 |
| Central diastolic pressure time integral, mm Hg×s/min | 3194±251 | 104±205 | −27±201 | 0.014* |
| Carotid‐to‐femoral pulse wave velocity, m/s | 7.3±0.9 | −0.2±0.6 | −0.1±0.5 | 0.614 |
| Mean blood velocity, cm/s | 27.6±4.3 | −1.0±3.8 | −0.4±3.3 | 0.426 |
| Systolic blood velocity, cm/s | 60.8±11.9 | 1.4±6.5 | −0.4±5.4 | 0.105 |
| Mean wall shear stress, Pa | 0.87±0.18 | −0.01±0.14 | −0.002±0.12 | 0.717 |
| Systolic wall shear stress, Pa | 1.93±0.50 | 0.09±0.24 | 0.01±0.20 | 0.071 |
Data are mean±SD. AIx indicates augmentation index; and AIx75, AIx indexed on heart rate (75bpm).
P<0.05.
Table 3.
Effect of Ivabradine Versus Placebo on Carotid Geometry, Mechanics, and Arterial Wall Viscosity
| Mean baseline | Delta ivabradine | Delta placebo | P value | |
|---|---|---|---|---|
| Diastolic diameter, mm | 5.548±0.520 | −0.086±0.262 | −0.07±0.195 | 0.742 |
| Carotid distension, mm | 0.583±0.142 | 0.076±0.041 | 0.007±0.048 | <0.001* |
| Intima‐media thickness, µm | 449±101 | 5±10 | 7±13 | 0.601 |
| Circumferential wall stress, kPa | 78±18 | −3±6 | −4±7 | 0.435 |
| Cross‐sectional compliance coefficient, 10−8×m2/kPa | 122±38 | 4±15 | −1±15 | 0.113† |
| Cross‐sectional distensibility coefficient, 10−3/kPa | 51±18 | 4±8 | 1±7 | 0.169‡ |
| Elastic modulus, kPa | 252±100 | −24±47 | −16±43 | 0.306§ |
| WV, J×m−1 | 1.74±1.28 | 0.35±1.07 | −0.20±0.48 | 0.035* |
| WE, J×m−1 | 11.07±4.89 | 2.65±2.23 | −0.02±1.25 | 0.013* |
| WV/WE, % | 15.1±6.5 | −0.4±7.9 | −1.5±4.3 | 0.52 |
Data are mean±SD. † P=0.07, ‡ P=0.04, and § P=0.05 after adjustment on circumferential wall stress. WE indicates elastic energy stored at each cycle; WV/WE, relative viscosity; and WV, viscous energy dissipation.
P<0.05.
Effect of Treatment on Cardiac and Systemic Hemodynamics
Ivabradine significantly decreased HR compared with placebo (P<0.001) and did not significantly change brachial SBP, brachial diastolic blood pressure, brachial mean blood pressure, and brachial pulse pressure. Thus, cyclic stretch, the product of HR and brachial pulse pressure, significantly decreased (P=0.001) under ivabradine (Table 1).
Ivabradine increased stroke volume and end‐diastolic volume (P<0.001 and P=0.001 versus placebo respectively) without change in end‐systolic volume, cardiac output, and ejection fraction, suggesting an increase in preload of the left ventricle without change in contractility, as evidenced by a stable left ventricular end‐systolic elastance. At the same time, total peripheral resistance did not change.
Effect of Treatment on Central Pressure and Cardiovascular Coupling
Compared with placebo, ivabradine nonsignificantly increased central pulse pressure, because of the opposite evolution of central SBP and central diastolic blood pressure. There was no change in the carotid‐to‐brachial amplification under treatment (Table 1). The HR reduction due to ivabradine resulted in an increase in the cardiac period associated with an increase in ejection duration. However, the ejection duration/period ratio decreased related to a more pronounced increase in diastolic than systolic time with the HR reduction (Table 2). Moreover, reflection time decreased but this effect was not significant. In this context, AP increased with ivabradine whereas pressure at the first shoulder of the wave remained stable. The marked increase in AP and the slight increase in central pulse pressure resulted in an increase in AIx under ivabradine and, as expected, this effect disappeared when adjusting on HR at 75 bpm (AIx75). Buckberg index increased with ivabradine compared with placebo owing to the increase in diastolic pressure time integral but not in systolic pressure time integral. In addition, treatment with ivabradine did not modify cfPWV (Table 2).
Effect of Treatment on Carotid Geometry, Mechanics, and Hemodynamics
Treatment with ivabradine did not change diastolic internal diameter but increased carotid distension compared with placebo (P<0.001). Ivabradine did not modify circumferential wall stress but arterial compliance and arterial distensibility increased and Einc decreased after adjusting on circumferential wall stress (P=0.07; P=0.04, and P=0.05 respectively; Table 3).
Mean carotid blood flow and mean wall shear stress were stable but carotid systolic blood flow and systolic wall shear stress nonsignificantly increased with ivabradine compared with placebo.
Effect of Treatment on AWV
Baseline WV and WE were 1.74±1.28 J.m−1 and 11.07±4.89 J.m−1. Thus, relative viscosity was 15.1%±6.5%. WV and WE increased under ivabradine compared with placebo (median[interquartile range 25–75]: 46[−22;122]% versus −10[−33;8]%, P=0.039, and 26[16;41]% versus −1[−11;9]%, P=0.007, respectively; Figure 1A and 1B, Table 3), leading to a stable relative viscosity WV/WE (Figure 1C). The same results were observed when considering absolute changes in AWV rather than percent changes (Figures S4A, S4B and S4C, Table 3).
Figure 1. Percent changes in viscous energy dissipation (A, WV), elastic energy stored at each cardiac cycle (B, WE), and relative viscosity (C, WV/WE) after 1‐week treatment with placebo (blue dots) or ivabradine (yellow dots) in the overall population of 19 healthy volunteers.
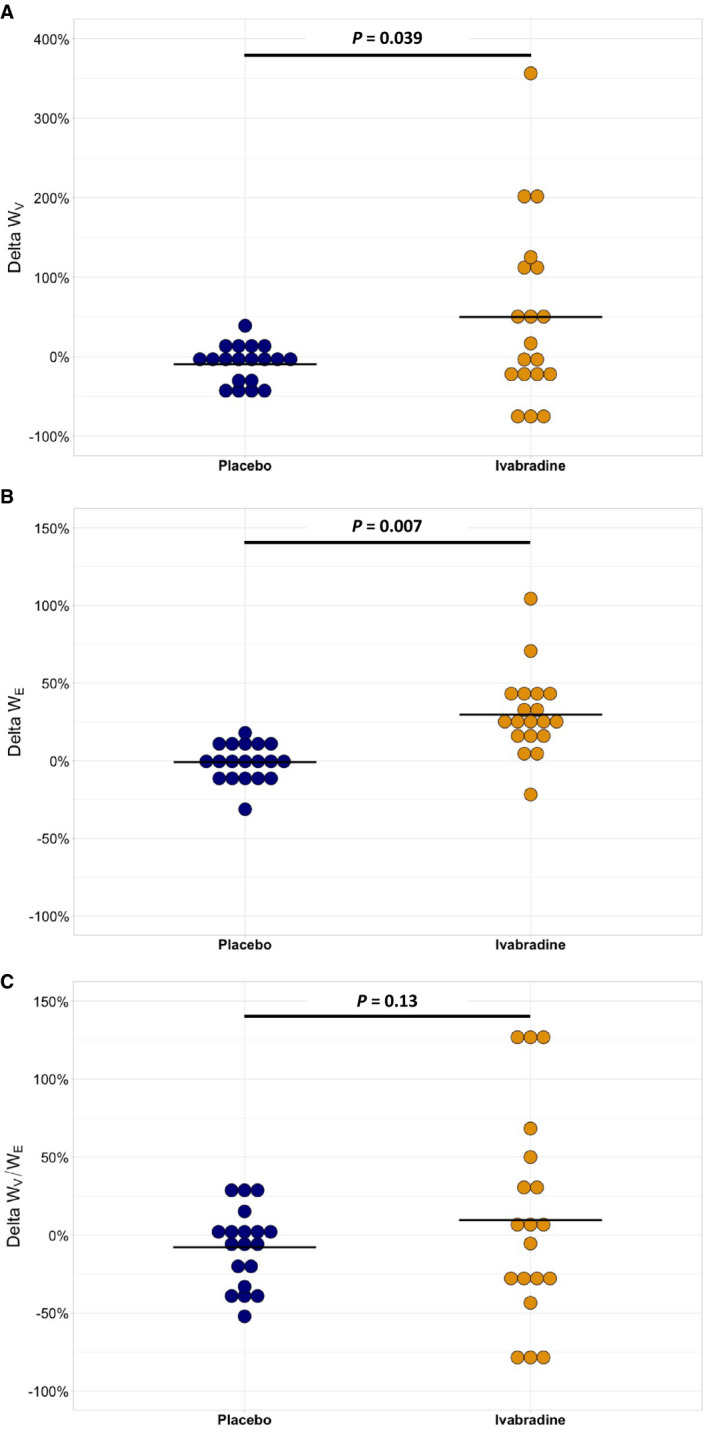
WV, WE, or WV/WE percent changes from baseline were compared between treatment and placebo using a linear mixed model with sex and mean baseline value of the parameter as cofactors and subject as random effect. Period*treatment interaction was evaluated in the model and was not significant.
Effect of Aging on Cardiovascular Parameters
Eight volunteers were in the young group (age=33[26–42] years, 6 women) and 11 volunteers were in the middle‐aged group (age=53[47–62] years, 8 women). At baseline, HR, peripheral and central blood pressure, and diastolic internal diameter were similar in the 2 groups. Conversely, carotid distension was lower (P=0.039) and intima‐media thickness was higher (P=0.001) in the middle‐aged group. As expected with aging and despite a lower circumferential wall stress (P=0.004), the middle‐aged group had a lower compliance and distensibility (P=0.03 and P=0.009, respectively) and a higher cfPWV (P=0.001) and elastic modulus (P=0.028 after adjusting on circumferential wall stress). Increase in arterial stiffness and total peripheral resistance were associated with a higher reflection of the pressure wave, explaining a higher AP and AIx and a lower carotid‐to‐brachial amplification (P=0.002, P=0.003, and P=0.12 respectively). Thus, cardiovascular coupling was less favorable in the middle‐aged group compared with the younger one (Table S3).
In this context, WE was similar in middle‐aged and young volunteers, especially after adjustment on circumferential wall stress (P=0.89). However, WV was lower (P=0.017) in the middle‐aged group, although this association disappeared after adjustment on circumferential wall stress (P=0.13). Thus, relative viscosity WV/WE was lower in the middle‐aged group even after adjustment on circumferential wall stress (P=0.004), suggesting that the lower circumferential wall stress in the middle‐aged group only partially explained the lower absolute and relative viscosity.
Effect of Aging on the Impact of HR Reduction on Cardiovascular Parameters
In this planned exploratory analysis, ivabradine markedly increased WV in middle‐aged but not in young volunteers (Figure 2A, P=0.009 for age class×treatment interaction) whereas it similarly increased WE in both groups (Figure 2B, P=0.29 for age class×treatment interaction). As a result, ivabradine induced an increase in WV/WE in the middle‐aged group but not in the young group (Figure 2C, P=0.004 for age class×treatment interaction). In parallel, the effect of ivabradine compared with placebo according to age class was similar on all other parameters (data not shown). In the same way, there was a linear relationship between ivabradine and the increase in WV according to age in the whole population explored (Figure 3A, P=0.004 for age×treatment interaction) whereas the effect of ivabradine on the increase in WE was similar regardless of age (Figure 3B, P=0.44 for age×treatment interaction). Consequently, there was a linear relationship between ivabradine and the increase in WV/WE according to age (Figure 3C, P=0.007 for age×treatment interaction). When considering absolute change in AWV rather than percent changes, results followed the same trend in both analyses performed according to age class or age (Figures S5A, S5B, and S5C and S6A, S6B, and S6C respectively).
Figure 2. Percent changes in viscous energy dissipation (A, WV), elastic energy stored at each cardiac cycle (B, WE), and relative viscosity (C, WV/WE) after 1‐week treatment with placebo (blue dots) or ivabradine (yellow dots) in 8 young and 11 middle‐aged healthy volunteers.
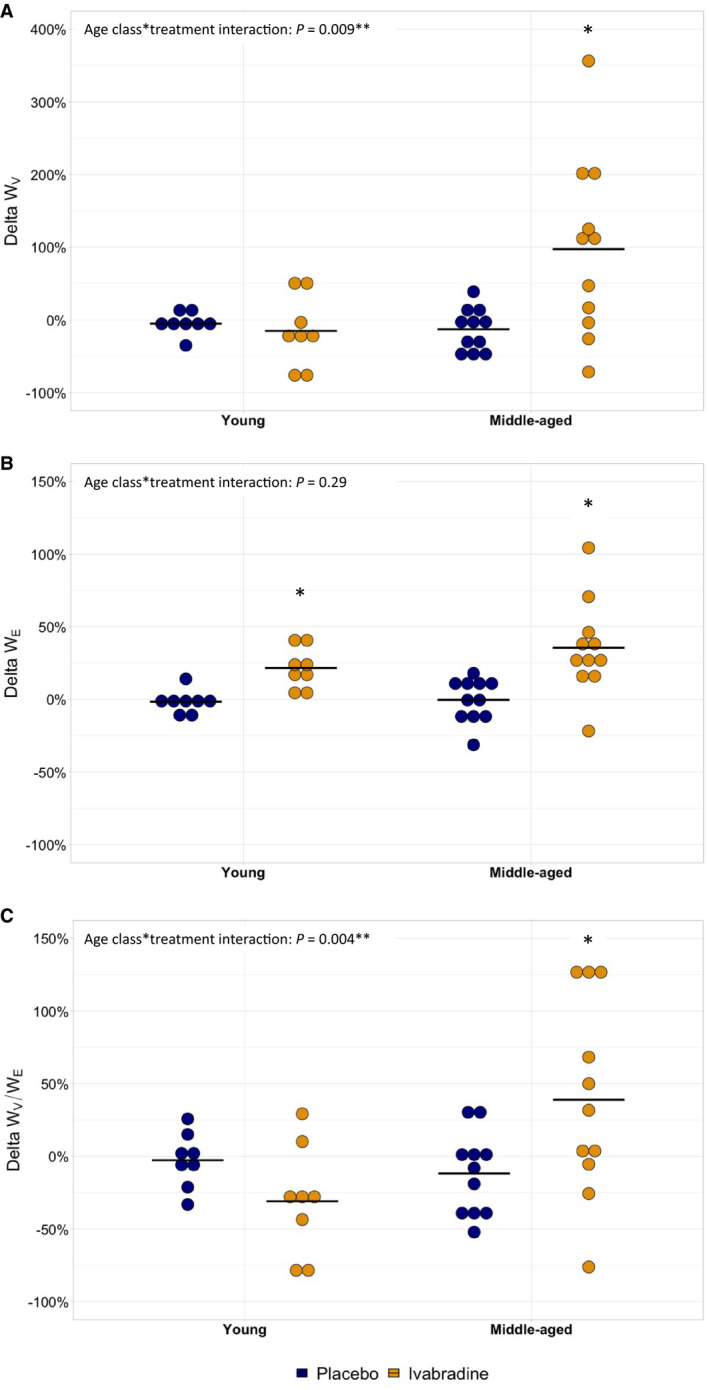
Percent changes of WV, WE, or WV/WE from baseline were compared between treatment and placebo using linear mixed model according to age class with sex and mean baseline value of the parameter as cofactors and subject as random effect. Interaction between age class and treatment is shown. *P<0.05 for the comparison of WV, WE, or WV/WE values under ivabradine or placebo vs baseline in each age subgroup.
Figure 3. Relationship between aging and the percent changes in viscous energy dissipation (A, WV), elastic energy stored at each cardiac cycle (B, WE), and relative viscosity (C, WV/WE) under ivabradine (yellow triangles/line) or placebo (blue dots/line).
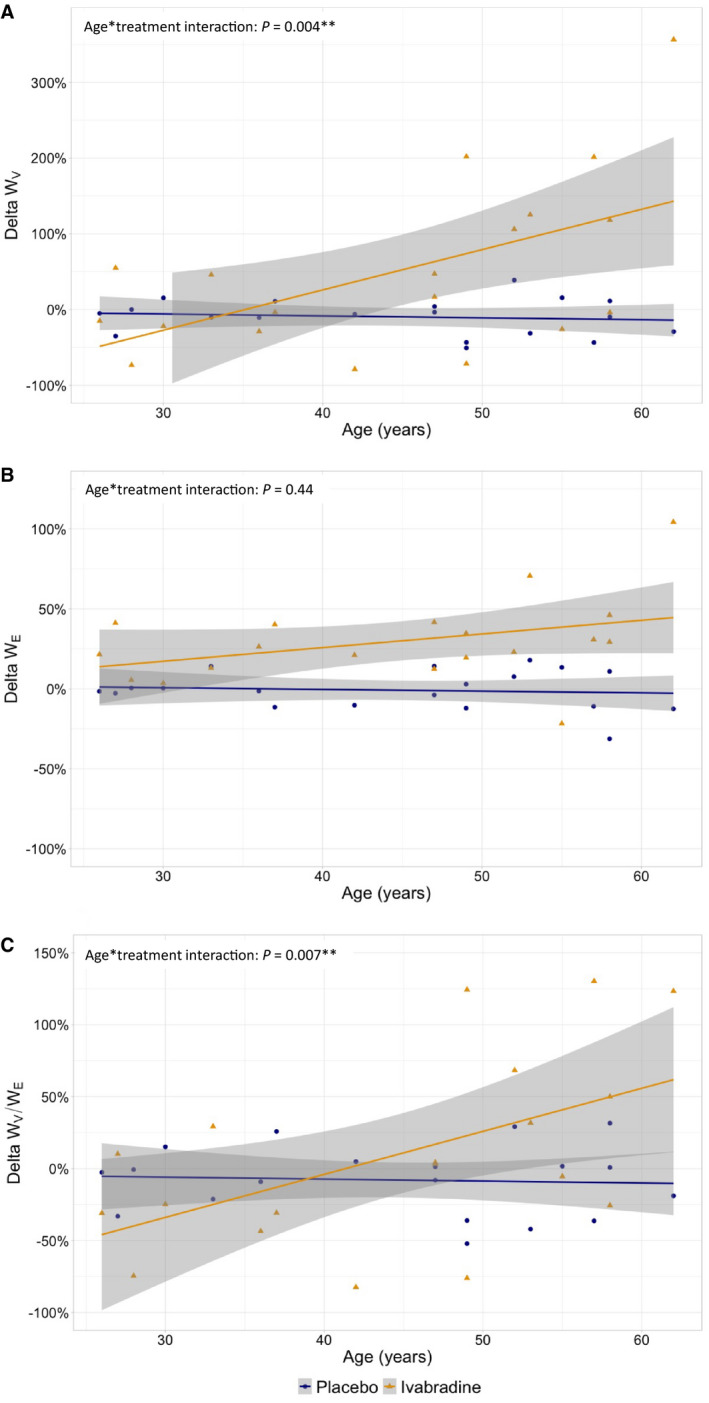
Percent changes of WV, WE, or WV/WE from baseline were compared between treatment and placebo using linear mixed model according to age (quantitative values) with sex and mean baseline value of the parameter as cofactors and subject as random effect. Interaction between age and treatment is shown. **P<0.05.
Discussion
This study demonstrates for the first time in healthy humans that HR reduction increases energy dissipation in the carotid artery wall. This increase is proportional to the increase in elastic energy stored within the arterial wall during cardiac cycle, resulting in a stable relative viscosity and suggesting an adaptive response. However, physiological aging appears to affect the viscous response to HR lowering, the energy dissipation being more pronounced than expected from the energy stored, resulting in an increase in relative viscosity.
We performed a study with a robust design, in healthy volunteers, to assess the effect of HR reduction on AWV and mechanics of the common carotid artery, a large conductance elastic artery close to the aorta and that is known to have reduced elastic properties with aging. 33 Viscous behavior was evaluated by measuring the hysteresis loop of the P‐LCSA relationship obtained using validated high‐resolution echotracking method and tonometry. 32 , 34 , 35 , 36 Continuous measurements of local pressure and diameter were performed simultaneously allowing for direct synchronization of waveforms without need for postprocedure treatment, unlike the few noninvasive studies exploring AWV in humans. 38 , 39 Thus, we evaluated the energy exchanged within the arterial wall and expressed the viscous behavior as the absolute value of energy dissipation and related to the elastic energy stored, one of its major determinants. 21 , 22
Moreover, repeated 8‐day ivabradine administration allowed the attainment of steady‐state concentration of ivabradine resulting in an expected 10‐bpm HR decrease, without long‐term structural changes, which was not expected from the design of the study.
In addition, as opposed to beta blockers, ivabradine has a highly specific chronotropic effect without inotropic effect, allowing a specific assessment of the effect of HR reduction. 29 , 40 , 41 In these conditions, we performed a comprehensive assessment of carotid, cardiac, and systemic hemodynamics and of the carotid mechanics to interpret the effect of HR reduction on carotid AWV and cardiovascular coupling.
In this context, HR reduction was responsible for an increase in stroke volume and in end‐diastolic volume directly related to the increase in cardiac preload. At the same time, the absence of change in end‐systolic volume, cardiac output, ejection fraction, and left ventricular end‐systolic elastance support the absence of negative inotropic effect of ivabradine. In parallel no change in peripheral blood pressure and total peripheral resistance was observed. These effects were in accordance with previous studies performed in healthy subjects and in coronary artery disease patients. 29 , 30 , 42
In parallel, AP and AIx increased, confirming the unfavorable effect of bradycardic agents on central hemodynamics. 8 , 9 , 10 Thus, the longer systolic ejection time increases the likelihood that the arterial wave reflection returns to the proximal aorta relatively earlier during systole. 9 , 43 This mechanism is supported by the stable AIx75, suggesting more a direct HR effect than a change in reflected wave amplitude. The absence of a large increase in central SBP whereas the AP increase could be explained by a nonsignificant decrease in the central diastolic blood pressure secondary to the lengthening of the diastolic period. Although this absence of effect on central blood pressure could be related to a small sample size, it has already been reported in coronary artery disease patients, for short time exposure to ivabradine. In contrast, an increase in AIx75 and in central SBP was observed after long‐time treatment, suggesting delayed effects from HR decrease related to structural changes. 10 , 30 , 44 However, these differences on the HR‐dependent effects of ivabradine on AP and AIx could also be explained by small differences between studies in baseline HR and percentage of patients already receiving beta blockers, the impact of bradycardia being more pronounced in patients with the higher HR at baseline. Anyway, this unfavorable cardiovascular coupling, that may be accompanied by a decreased myocardial perfusion during proto‐diastolic time, was compensated by an increase in the length of the coronary perfusion in diastole, as suggested from the higher Buckberg index with ivabradine. 30
Furthermore, cfPWV was not affected by ivabradine. However, modifications in cfPWV with short‐term HR changes are usually low, resulting in conflicting results. 3 , 4 Thus, a 10‐bpm increase in HR seems to be responsible for a 0.17 m/s increase in cfPWV, near to that we observed. 3 More significant changes in cfPWV were usually observed for more prolonged duration of treatment. 10 , 45
In contrast, carotid compliance and distensibility increased and Einc decreased confirming the decrease in arterial stiffness secondary to HR reduction. Even suggested previously, 5 , 6 , 10 , 27 this is the first demonstration that a selective HR reduction quickly decreases local arterial stiffness in human. This suggests a nonstructural effect and could be related to change in arterial tone noticeable at the carotid level. In fact, HR reduction nonsignificantly increase carotid systolic blood velocity and therefore systolic wall shear stress, which could have affected wall mechanics by potentiating the release of endothelium‐derived relaxing factors. 26 Moreover, change in cyclic stretch could explain the effect of HR reduction on vascular system by modulating the smooth muscle cells’ phenotype or changing the ability of endothelial cells to release endothelial factors, to remodel extracellular matrix, or to interact with smooth muscle cells. 46 , 47 , 48 , 49 , 50 More recently, experimental studies showed that ivabradine also has pleiotropic effects independent from the HR reduction, including antioxidant or anti‐inflammatory actions. 51 , 52 Although the treatment duration is probably too short, we cannot exclude that part of the effects observed in our study could be related to these pleiotropic mechanisms.
In this context, WE dramatically increased under ivabradine mainly because of the increased distension, which reflects the increased elastic energy stored within the arterial wall during systole. In parallel, WV increased, reflecting the increase in energy dissipation. Hence, relative viscosity remained stable suggesting that the increase in WV is proportional to the increase in elastic energy stored and not to a direct effect of the HR reduction on the viscous component of the carotid mechanics. This could be a passive mechanism but also related to a “smart damping” of the energy stored by the system as previously proposed by some authors. 38
Furthermore, a change in AWV is frequently evoked as a major determinant of change in arterial stiffness during HR variation, 4 , 14 , 15 , 16 but no study has evaluated the magnitude and the direction of this relationship. In this context, our results strongly support that the decrease in arterial stiffness with HR reduction is not explained by a parallel decrease in AWV evaluated by a thermodynamic approach. Only 1 previous animal study evaluated the effect of ivabradine on carotid AWV using this approach. 15 The authors found in accordance with our results an increase in arterial distension, WE, and WV but an increase in relative viscosity. 15 This discrepancy could be related to the experimental model used with an acute administration of ivabradine in rats that was associated with a large increase in pulse pressure and that could result in insufficient time for the arterial wall to adapt to these new hemodynamic conditions. 15 In addition, some in vitro and animal studies assessed the effect of HR modulation on AWV through the evaluation of the viscous coefficient η, representing the intrinsic viscosity of the material and obtained with frequency 19 , 20 or time‐domain analysis. 24 , 53 In fact, η is not equivalent to WV, which corresponds to the dissipated energy related to both intrinsic viscosity and arterial working conditions and can be approximated by the product of η, HR, and distension. 17 , 38 Thus, the decrease in η observed when HR increases is concordant with our result showing an increase in WV when HR decreases. 17 , 18 , 19 , 20 , 53
Furthermore, we performed a planned exploratory analysis to evaluate the impact of aging on the adaptation of AWV to HR reduction. At baseline, despite similar peripheral blood pressure and HR, middle‐aged volunteers had a less favorable cardiovascular coupling, as shown by the increased AP and AIx and the decrease in the carotid‐to‐brachialb amplification. This increase in pressure wave reflection is related to both an increase in cfPWV secondary to the aortic stiffening and an increase in the magnitude of the reflected pulse wave suggested from the increase in peripheral reflectivity with aging. 54 Moreover, carotid intima‐media thickness increased with aging without significant increase in diameter, resulting in a decrease in circumferential wall stress. This result has already been observed in healthy middle‐aged population, 36 , 39 although advanced aging is classically associated with an increase in carotid diameter, particularly in men. In this context, arterial distensibility was lower and Einc was higher in the middle‐aged group confirming the stiffening of large arteries with aging. 33 Aging was associated with a decrease in systolic carotid flow. Thus, in addition to the age‐related increase in flow fluctuations previously described, these impairments in carotid hemodynamics could decrease the release of endothelium‐derived relaxing factors. 55
In this context, WV, but not WE, was lower in the middle‐aged group, resulting in a lower relative viscosity. The stable WE could be explained by the decrease in pulse diameter and the nonsignificant increase in central pulse pressure. Few studies evaluated the impact of aging on AWV. In a typical result figure, Giannattasio et al showed a smaller hysteresis loop in aging people but did not measure the area. 56 However, this was in accordance with a previous in vitro study. 57 Using time‐domain assessment, Kawano et al found an higher η in unfit middle‐aged people. 39 However, in this study, brachial mean blood pressure increased with aging suggesting a higher circumferential wall stress, a major determinant of the viscous properties that we considered in our study. 22 Thus independently from HR, aging appears to be associated with an increase in arterial stiffness and a decrease in AWV that may be due, despite an increase in intima‐media thickness, to structural changes, such as a reduced vascular muscular cell content and replacement by less viscous elements such as extracellular matrix. 57
Finally, for a similar change in HR and arterial wall mechanics, AWV response to HR reduction was different between the 2 age groups, even after adjustment on sex and baseline AWV values. With similar increase in WE, the middle‐aged volunteers exhibited an increase in WV, and thus in their relative viscosity, compared with the younger subjects. These results were confirmed by the linear increase of relative viscosity under ivabradine observed during aging in the whole population. Conversely, younger volunteers tended to nonsignificantly decrease their relative viscosity under ivabradine. This could explain the neutral effect observed in the overall population and suggests that HR reduction is associated with an increase in WE but that the change in viscous work depends on age. One hypothesis is that HR reduction induces an overcompensation of the adaptive process maintaining AWV in younger subjects and that the age‐related progressive alteration of endothelial and arterial wall integrity and lesser release of endothelium‐derived relaxing factors could lead to a progressive loss of this compensatory mechanism and thus to a progressive increase in WV/WE under ivabradine during aging. 26 In addition, the relative heterogeneity in the response to ivabradine in the middle‐aged group could be related to heterogeneity in vascular aging secondary to lifestyle differences. For example, some studies showed that physical activities modified AWV. 39 In our study, even if we did not exhaustively assess physical activity, we excluded patients with sports activity >1 hour per day. All these results suggest that the change in AWV observed after HR reduction may be rather dependent on vascular than chronological aging. This alteration of AWV adaptation may minimize the beneficial effect of HR reduction on arterial stiffness. Whether this could have contributed to reduce the benefit of HR‐lowering strategies performed in a middle‐aged and older population in different pathological conditions remains to be investigated. 7 , 11 , 28 , 58
Limitations
Our study has some limitations. First, our sample size was calculated to explore the effect of ivabradine on AWV taking into consideration aging and may have been too small to detect other effects in particular when comparing young and middle‐aged groups. Second, our age‐related results are limited to the range 25 to 65 years, and we cannot extrapolate the linear relationship observed between HR reduction and AWV to extreme ages. Third, our study did not explore the mechanisms involved in the effect of HR reduction on AWV and thus, as discussed previously, we cannot exclude that part of our results are related to pleiotropic HR‐independent effects of ivabradine on vascular wall. However, these effects may be probably more effective after long‐term administration and the magnitude of them remain to be discussed in humans. 52
Finally, the measurement of AWV with 2 trained operators is complex, which likely limits the assessment of AWV in a very large cohort. Moreover, the temporal resolution of the LCSA acquisition was low in our study. This could limit a detailed analysis of the hysteresis loop and contribute to intersubject variability, explaining why some comparisons raise significance when considering percent but not absolute changes. However, our crossover design with a careful verification of the location of the probe limited the within‐subject variability.
Perspectives
Our study extends our knowledge about the physiology and the physiopathology of AWV, a less investigated aspect of the arterial viscoelastic properties, with a particular focus on the effects of bradycardia and aging. This may be of importance because several bradycardic agents are widely used in cardiovascular medicine and in older patients. Whether the low arterial damping at baseline observed in middle‐aged subjects is deleterious or not remains to be evaluated. In fact, less energy dissipation may be beneficial for cardiovascular coupling but could also be associated with an increase in the energy transmitted through the arterial wall to the downstream arterial bed and therefore lead to damage peripheral organs, such as brain or kidney. Conversely, the increase in energy dissipation with HR reduction during aging could reduce the energy transmitted to the periphery but also could alter cardiovascular coupling thus participating to the inconstant benefit of HR lowering strategies.
Conclusions
In conclusion, this study demonstrates for the first time that an adaptive increase in energy dissipation occurs in healthy humans during HR reduction with ivabradine and compensates for the increase in large artery elasticity and in the energy stored within the arterial wall. Aging is associated with a lower energy dissipation at baseline but a larger increase in energy dissipation after HR reduction. The long‐term impact of this age‐related increased energy dissipation secondary to HR reduction needs to be investigated.
Sources of Funding
This work was supported by institutional grants: “Appel a Projets Emergents en recherche Clinique‐Jeunes Chercheurs” from the Rouen University Hospital and the “Conseil Régional de Haute‐Normandie” and the “Appel d’Offres Annuel 2015” from the Charles Nicolle Foundation.
Disclosures
None.
Supporting information
Acknowledgments
The authors thank the Clinical Trial Unit of Rouen University Hospital Pharmacy for treatment packaging and delivery, Mrs Carine Daniel from the CIC‐CRB 1404 for helping in the recruitment of the volunteers and Mrs Mylène Hervet, Mrs Cécile Pourcher, and Mr Maxime Depierre from the Clinical Research and Innovation Department for administrative support and data management.
Supplemental Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.121.023409
For Sources of Funding and Disclosures, see page 13.
References
- 1. Custodis F, Schirmer SH, Baumhäkel M, Heusch G, Böhm M, Laufs U. Vascular pathophysiology in response to increased heart rate. J Am Coll Cardiol. 2010;56:1973–1983. doi: 10.1016/j.jacc.2010.09.014 [DOI] [PubMed] [Google Scholar]
- 2. The Reference Values for Arterial Stiffness’ TRV for AS . Determinants of pulse wave velocity in healthy people and in the presence of cardiovascular risk factors: ‘establishing normal and reference values’. Eur Heart J. 2010;31:2338–2350. doi: 10.1093/eurheartj/ehq165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tan I, Spronck B, Kiat H, Barin E, Reesink KD, Delhaas T, Avolio AP, Butlin M. Heart rate dependency of large artery stiffness. Hypertension. 2016;68:236–242. doi: 10.1161/HYPERTENSIONAHA.116.07462 [DOI] [PubMed] [Google Scholar]
- 4. Lantelme P, Mestre C, Lievre M, Gressard A, Milon H. Heart rate: an important confounder of pulse wave velocity assessment. Hypertension. 2002;39:1083–1087. doi: 10.1161/01.HYP.0000019132.41066.95 [DOI] [PubMed] [Google Scholar]
- 5. Giannattasio C, Vincenti A, Failla M, Capra A, Cirò A, De Ceglia S, Gentile G, Brambilla R, Mancia G. Effects of heart rate changes on arterial distensibility in humans. Hypertension. 2003;42:253–256. doi: 10.1161/01.HYP.0000085199.33254.15 [DOI] [PubMed] [Google Scholar]
- 6. Mircoli L, Mangoni AA, Giannattasio C, Mancia G, Ferrari AU. Heart rate‐dependent stiffening of large arteries in intact and sympathectomized rats. Hypertension. 1999;34:598–602. doi: 10.1161/01.HYP.34.4.598 [DOI] [PubMed] [Google Scholar]
- 7. Kang S, Li C‐J, Zhang X‐M. Ivabradine has a neutral effect on mortality in randomized controlled trials. Medicine. 2017;96:e8067. doi: 10.1097/MD.0000000000008067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Messerli FH, Rimoldi SF, Bangalore S, Bavishi C, Laurent S. When an increase in central systolic pressure overrides the benefits of heart rate lowering. J Am Coll Cardiol. 2016;68:754–762. doi: 10.1016/j.jacc.2016.03.610 [DOI] [PubMed] [Google Scholar]
- 9. Williams B, Lacy PS, Thom SM, Cruickshank K, Stanton A, Collier D, Hughes AD, Thurston H, O’Rourke M, CAFE Investigators , et al. Differential impact of blood pressure‐lowering drugs on central aortic pressure and clinical outcomes: principal results of the Conduit Artery Function Evaluation (CAFE) study. Circulation. 2006;113:1213–1225. doi: 10.1161/CIRCULATIONAHA.105.595496 [DOI] [PubMed] [Google Scholar]
- 10. Hohneck AL, Fries P, Ströder J, Schneider G, Wagenpfeil S, Schirmer SH, Böhm M, Laufs U, Custodis F. Effects of heart rate reduction with ivabradine on vascular stiffness and endothelial function in chronic stable coronary artery disease. J Hypertens. 2019;37:1023–1031. doi: 10.1097/HJH.0000000000001984 [DOI] [PubMed] [Google Scholar]
- 11. Khan N, McAlister FA. Re‐examining the efficacy of β‐blockers for the treatment of hypertension: a meta‐analysis. CMAJ: Can Med Assoc J. 2006;174:1737–1742. doi: 10.1503/cmaj.060110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bonadei I, Sciatti E, Vizzardi E, Fabbricatore D, Pagnoni M, Rossi L, Carubelli V, Lombardi CM, Metra M. Effects of ivabradine on endothelial function, aortic properties and ventricular‐arterial coupling in chronic systolic heart failure patients. Cardiovasc Ther. 2018;36:e12323. doi: 10.1111/1755-5922.12323 [DOI] [PubMed] [Google Scholar]
- 13. Reil J‐C, Tardif J‐C, Ford I, Lloyd SM, O’Meara E, Komajda M, Borer JS, Tavazzi L, Swedberg K, Böhm M. Selective heart rate reduction with ivabradine unloads the left ventricle in heart failure patients. J Am Coll Cardiol. 2013;62:1977–1985. doi: 10.1016/j.jacc.2013.07.027 [DOI] [PubMed] [Google Scholar]
- 14. Mangoni AA, Mircoli L, Giannattasio C, Ferrari AU, Mancia G. Heart rate‐dependence of arterial distensibility in vivo. J Hypertens. 1996;14:897–901. doi: 10.1097/00004872-199607000-00013 [DOI] [PubMed] [Google Scholar]
- 15. Albaladejo P, Challande P, Kakou A, Benetos A, Labat C, Louis H, Safar ME, Lacolley P. Selective reduction of heart rate by ivabradine: effect on the visco‐elastic arterial properties in rats. J Hypertens. 2004;22:1739–1745. doi: 10.1097/00004872-200409000-00018 [DOI] [PubMed] [Google Scholar]
- 16. Antonov P, Antonova M, Nikolova N, Antonova N, Vlaskovska M, Kasakov L. Age dependent changes of arterial wall viscoelasticity. Clin Hemorheol Micro. 2008;39:63–68. doi: 10.3233/CH-2008-1069 [DOI] [PubMed] [Google Scholar]
- 17. Salvucci FP, Schiavone J, Craiem D, Barra JG. Arterial wall mechanics as a function of heart rate: role of vascular smooth muscle. J Phys: Conf Ser. 2007;90:012010. doi: 10.1088/1742-6596/90/1/012010 [DOI] [Google Scholar]
- 18. Busse R, Bauer RD, Sattler T, Schabert A. Dependence of elastic and viscous properties of elastic arteries on circumferential wall stress at two different smooth muscle tones. Pflügers Archiv: Eur J Physiol. 1981;390:113–119. doi: 10.1007/BF00590192 [DOI] [PubMed] [Google Scholar]
- 19. Bergel DH. The dynamic elastic properties of the arterial wall. J Physiol. 1961;156:458–469. doi: 10.1113/jphysiol.1961.sp006687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gow BS, Schonfeld D, Patel DJ. The dynamic elastic properties of the canine left circumflex coronary artery. J Biomech. 1974;7:389–395. doi: 10.1016/0021-9290(74)90001-3 [DOI] [PubMed] [Google Scholar]
- 21. Boutouyrie P, Bézie Y, Lacolley P, Challande P, Chamiot‐Clerc P, Benetos A, de la Faverie JF, Safar M, Laurent S. In vivo/in vitro comparison of rat abdominal aorta wall viscosity. Influence of endothelial function. Arterioscler Thromb Vasc Biol. 1997;17:1346–1355. doi: 10.1161/01.ATV.17.7.1346 [DOI] [PubMed] [Google Scholar]
- 22. Roca F, Iacob M, Remy‐Jouet I, Bellien J, Joannides R. Evidence for a role of vascular endothelium in the control of arterial wall viscosity in humans. Hypertension. 2018;71:143–150. doi: 10.1161/HYPERTENSIONAHA.117.09870 [DOI] [PubMed] [Google Scholar]
- 23. Armentano R, Pessana F, Graf S, Romero L, Fischer EC. Frequency dependance of arterial wall young modulus after de‐endothelization. In: Memorias II Congreso Latinoamericano De Ingeneria Biomedica, La Habana. 2001.
- 24. Armentano RL, Barra JG, Levenson J, Simon A, Pichel RH. Arterial wall mechanics in conscious dogs. Assessment of viscous, inertial, and elastic moduli to characterize aortic wall behavior. Circ Res. 1995;76:468–478. doi: 10.1161/01.RES.76.3.468 [DOI] [PubMed] [Google Scholar]
- 25. Himburg HA, Dowd SE, Friedman MH. Frequency‐dependent response of the vascular endothelium to pulsatile shear stress. Am J Physiol Heart Circ Physiol. 2007;293:H645–H653. doi: 10.1152/ajpheart.01087.2006 [DOI] [PubMed] [Google Scholar]
- 26. Roca F, Bellien J, Iacob M, Joannides R. Endothelium‐dependent adaptation of arterial wall viscosity during blood flow increase is impaired in essential hypertension. Atherosclerosis. 2019;285:102–107. doi: 10.1016/j.atherosclerosis.2019.04.208 [DOI] [PubMed] [Google Scholar]
- 27. Boutouyrie P, Beaussier H, Achouba A, Laurent S, EXPLOR trialists . Destiffening effect of valsartan and atenolol: influence of heart rate and blood pressure. J Hypertens. 2014;32:108–114. doi: 10.1097/HJH.0000000000000014 [DOI] [PubMed] [Google Scholar]
- 28. Fox K, Ford I, Steg PG, Tardif J‐C, Tendera M, Ferrari R, SIGNIFY Investigators . Ivabradine in stable coronary artery disease without clinical heart failure. N Engl J Med. 2014;371:1091–1099. doi: 10.1056/NEJMoa1406430 [DOI] [PubMed] [Google Scholar]
- 29. Savelieva I, Camm AJ. Heart rate reduction by pharmacological if current inhibition. In: Camm AJ, Tendera M, eds. Heart Rate Slowing by if Current Inhibition. Basel: Karger; 2006. [Google Scholar]
- 30. Dillinger J‐G, Maher V, Vitale C, Henry P, Logeart D, Manzo Silberman S, Allée G, Levy BI. Impact of ivabradine on central aortic blood pressure and myocardial perfusion in patients with stable coronary artery disease. Hypertension. 2015;66:1138–1144. doi: 10.1161/HYPERTENSIONAHA.115.06091 [DOI] [PubMed] [Google Scholar]
- 31. Boutouyrie P, Boumaza S, Challande P, Lacolley P, Laurent S. Smooth muscle tone and arterial wall viscosity: an in vivo/in vitro study. Hypertension. 1998;32:360–364. doi: 10.1161/01.HYP.32.2.360 [DOI] [PubMed] [Google Scholar]
- 32. Laurent S, Girerd X, Mourad JJ, Lacolley P, Beck L, Boutouyrie P, Mignot JP, Safar M. Elastic modulus of the radial artery wall material is not increased in patients with essential hypertension. Arterioscler Thromb Vasc Biol. 1994;14:1223–1231. doi: 10.1161/01.ATV.14.7.1223 [DOI] [PubMed] [Google Scholar]
- 33. Benetos A, Laurent S, Hoeks AP, Boutouyrie PH, Safar ME. Arterial alterations with aging and high blood pressure. A noninvasive study of carotid and femoral arteries. Arterioscler Thromb: A J Vasc Biol/Am Heart Assoc. 1993;13:90–97. doi: 10.1161/01.ATV.13.1.90 [DOI] [PubMed] [Google Scholar]
- 34. Maltete D, Bellien J, Cabrejo L, Iacob M, Proust F, Mihout B, Thuillez C, Guegan‐Massardier E, Joannides R. Hypertrophic remodeling and increased arterial stiffness in patients with intracranial aneurysms. Atherosclerosis. 2010;211:486–491. doi: 10.1016/j.atherosclerosis.2010.04.002 [DOI] [PubMed] [Google Scholar]
- 35. Meinders JM, Hoeks APG. Simultaneous assessment of diameter and pressure waveforms in the carotid artery. Ultrasound Med Biol. 2004;30:147–154. doi: 10.1016/j.ultrasmedbio.2003.10.014 [DOI] [PubMed] [Google Scholar]
- 36. Bussy C, Boutouyrie P, Lacolley P, Challande P, Laurent S. Intrinsic stiffness of the carotid arterial wall material in essential hypertensives. Hypertension. 2000;35:1049–1054. doi: 10.1161/01.HYP.35.5.1049 [DOI] [PubMed] [Google Scholar]
- 37. Bour J, Kellett J. Impedance cardiography: a rapid and cost‐effective screening tool for cardiac disease. Eur J Intern Med. 2008;19:399–405. doi: 10.1016/j.ejim.2007.07.007 [DOI] [PubMed] [Google Scholar]
- 38. Armentano RL, Barra JG, Santana DB, Pessana FM, Graf S, Craiem D, Brandani LM, Baglivo HP, Sanchez RA. Smart damping modulation of carotid wall energetics in human hypertension: effects of angiotensin‐converting enzyme inhibition. Hypertension. 2006;47:384–390. doi: 10.1161/01.HYP.0000205915.15940.15 [DOI] [PubMed] [Google Scholar]
- 39. Kawano H, Yamamoto K, Gando Y, Tanimoto M, Murakami H, Ohmori Y, Sanada K, Tabata I, Higuchi M, Miyachi M. Lack of age‐related increase in carotid artery wall viscosity in cardiorespiratory fit men. J Hypertens. 2013;31:2370–2379. doi: 10.1097/HJH.0b013e328364cbba [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ariff B, Zambanini A, Vamadeva S, Barratt D, Xu Y, Sever P, Stanton A, Hughes A, Thom S. Candesartan‐ and atenolol‐based treatments induce different patterns of carotid artery and left ventricular remodeling in hypertension. Stroke. 2006;37:2381–2384. doi: 10.1161/01.STR.0000236839.69658.c5 [DOI] [PubMed] [Google Scholar]
- 41. Mahmud A, Feely J. Beta‐blockers reduce aortic stiffness in hypertension but nebivolol, not atenolol, reduces wave reflection. Am J Hypertens. 2008;21:663–667. doi: 10.1038/ajh.2008.156 [DOI] [PubMed] [Google Scholar]
- 42. Joannides R, Moore N, Iacob M, Compagnon P, Lerebours G, Menard J‐F, Thuillez C. Comparative effects of ivabradine, a selective heart rate‐lowering agent, and propranolol on systemic and cardiac haemodynamics at rest and during exercise. Br J Clin Pharmacol. 2006;61:127–137. doi: 10.1111/j.1365-2125.2005.02544.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wilkinson IB, Mohammad NH, Tyrrell S, Hall IR, Webb DJ, Paul VE, Levy T, Cockcroft JR. Heart rate dependency of pulse pressure amplification and arterial stiffness. Am J Hypertens. 2002;15:24–30. doi: 10.1016/S0895-7061(01)02252-X [DOI] [PubMed] [Google Scholar]
- 44. Lopatin YM, Vitale C. Effect of ivabradine on central aortic blood pressure in patients with stable coronary artery disease: what do we know? Int J Cardiol. 2016;224:145–148. doi: 10.1016/j.ijcard.2016.09.054 [DOI] [PubMed] [Google Scholar]
- 45. Ait‐Oufella H, Collin C, Bozec E, Laloux B, Ong K‐T, Dufouil C, Boutouyrie P, Laurent S. Long‐term reduction in aortic stiffness: a 5.3‐year follow‐up in routine clinical practice. J Hypertens. 2010;28:2336–2341. doi: 10.1097/HJH.0b013e32833da2b2 [DOI] [PubMed] [Google Scholar]
- 46. Lamontagne D, Pohl U, Busse R. Mechanical deformation of vessel wall and shear stress determine the basal release of endothelium‐derived relaxing factor in the intact rabbit coronary vascular bed. Circ Res. 1992;70:123–130. doi: 10.1161/01.RES.70.1.123 [DOI] [PubMed] [Google Scholar]
- 47. Hutcheson IR, Griffith TM. Release of endothelium‐derived relaxing factor is modulated both by frequency and amplitude of pulsatile flow. Am J Physiol. 1991;261:H257–262. doi: 10.1152/ajpheart.1991.261.1.H257 [DOI] [PubMed] [Google Scholar]
- 48. Haga JH, Li Y‐SJ, Chien S. Molecular basis of the effects of mechanical stretch on vascular smooth muscle cells. J Biomech. 2007;40:947–960. doi: 10.1016/j.jbiomech.2006.04.011 [DOI] [PubMed] [Google Scholar]
- 49. Cummins PM, von Offenberg SN, Killeen MT, Birney YA, Redmond EM, Cahill PA. Cyclic strain‐mediated matrix metalloproteinase regulation within the vascular endothelium: a force to be reckoned with. AJP: Heart and Circulatory Physiology. 2006;292:H28–H42. doi: 10.1152/ajpheart.00304.2006 [DOI] [PubMed] [Google Scholar]
- 50. Califano JP, Reinhart‐King CA. Exogenous and endogenous force regulation of endothelial cell behavior. J Biomech. 2010;43:79–86. doi: 10.1016/j.jbiomech.2009.09.012 [DOI] [PubMed] [Google Scholar]
- 51. Kleinbongard P, Gedik N, Witting P, Freedman B, Klöcker N, Heusch G. Pleiotropic, heart rate‐independent cardioprotection by ivabradine. Br J Pharmacol. 2015;172:4380–4390. doi: 10.1111/bph.13220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Simko F, Baka T. Ivabradine and blood pressure reduction: underlying pleiotropic mechanisms and clinical implications. Front Cardiovasc Med. 2021;8:607998. doi: 10.3389/fcvm.2021.607998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bauer RD, Busse R, Schabert A, Summa Y, Wetterer E. Separate determination of the pulsatile elastic and viscous forces developed in the arterial wall in vivo. Pflügers Archiv: Eur J Physiol. 1979;380:221–226. doi: 10.1007/BF00582900 [DOI] [PubMed] [Google Scholar]
- 54. Avolio A. Ageing and wave reflection. J Hypertens. 1992;10:S83–S86. doi: 10.1097/00004872-199208001-00022 [DOI] [PubMed] [Google Scholar]
- 55. Hirata K, Yaginuma T, O’Rourke MF, Kawakami M. Age‐related changes in carotid artery flow and pressure pulses: possible implications for cerebral microvascular disease. Stroke. 2006;37:2552–2556. doi: 10.1161/01.STR.0000242289.20381.f4 [DOI] [PubMed] [Google Scholar]
- 56. Giannattasio C, Salvi P, Valbusa F, Kearney‐Schwartz A, Capra A, Amigoni M, Failla M, Boffi L, Madotto F, Benetos A, et al. Simultaneous measurement of beat‐to‐beat carotid diameter and pressure changes to assess arterial mechanical properties. Hypertension. 2008;52:896–902. doi: 10.1161/HYPERTENSIONAHA.108.116509 [DOI] [PubMed] [Google Scholar]
- 57. Dobrin PB. Mechanical properties of arteries. Physiol Rev. 1978;58:397–460. doi: 10.1152/physrev.1978.58.2.397 [DOI] [PubMed] [Google Scholar]
- 58. Fox K, Ford I, Steg PG, Tendera M, Ferrari R, BEAUTIFUL Investigators . Ivabradine for patients with stable coronary artery disease and left‐ventricular systolic dysfunction (BEAUTIFUL): a randomised, double‐blind, placebo‐controlled trial. Lancet. 2008;372:807–816. doi: 10.1016/S0140-6736(08)61170-8 [DOI] [PubMed] [Google Scholar]
- 59. Laurent S, Cockcroft J, Van Bortel L, Boutouyrie P, Giannattasio C, Hayoz D, Pannier B, Vlachopoulos C, Wilkinson I, Struijker‐Boudier H, et al. Expert consensus document on arterial stiffness: methodological issues and clinical applications. Eur Heart J. 2006;27:2588–2605. doi: 10.1093/eurheartj/ehl254 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.