

SUMMARY
The presence and abundance of viral proteins within host cells are part of the essential signatures of the cellular stages of viral infections. However, methods that can comprehensively detect and quantify these proteins are still limited, particularly for viruses with large protein coding capacity. Here, we design and experimentally validate a mass spectrometry-based Targeted herpesviRUS proTEin Detection (TRUSTED) assay for monitoring human viruses representing the three Herpesviridae subfamilies—herpes simplex virus type 1, human cytomegalovirus (HCMV), and Kaposi sarcoma-associated herpesvirus. We demonstrate assay applicability for (1) capturing the temporal cascades of viral replication, (2) detecting proteins throughout a range of virus concentrations and in in vivo models of infection, (3) assessing the effects of clinical therapeutic agents and sirtuin-modulating compounds, (4) studies using different laboratory and clinical viral strains, and (5) discovering a role for carbamoyl phosphate synthetase 1 in supporting HCMV replication.
In brief
Herpesviruses encode many proteins, making it difficult to comprehensively monitor viral protein levels by traditional approaches. Kennedy et al. develop a set of targeted mass spectrometry-based assays for measuring herpesvirus protein levels spanning all virus subfamilies (α, β, and γ) and demonstrate their usefulness for a wide range of applications.
Graphical Abstract
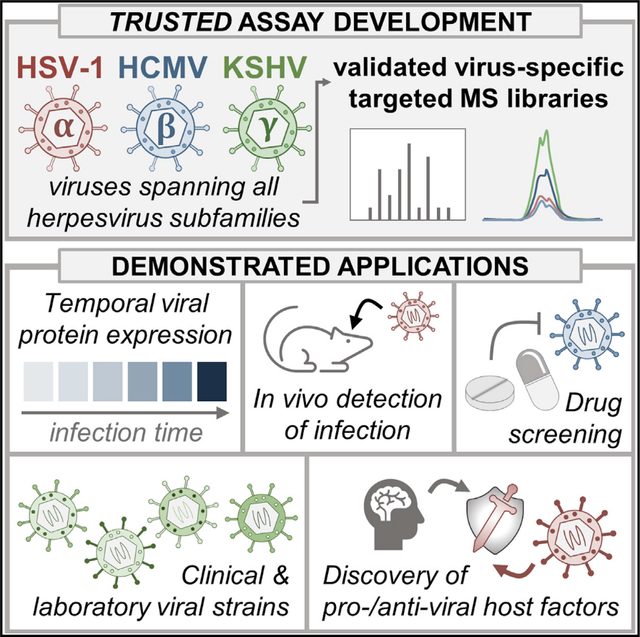
INTRODUCTION
As evidenced by the global burden of viral infectious disease, there is a need for methods that can quickly and accurately detect and monitor viral infections in both laboratory and clinical settings. An indicator of the presence of a viral infection and the stage of a replication cycle is the expression and abundance of viral proteins (Greco et al., 2014; Gruffat et al., 2016). Numerous human viruses proceed through their replication cycles by initiating a temporal cascade of viral gene expression, and these different viral proteins can provide signatures of infection progression. However, the genome size and subsequent number of proteins expressed by different viruses varies widely. For example, viruses range from those expressing a single polyprotein that is cleaved into 10–20 individual proteins (e.g., hepatitis C virus, coronaviruses, poliovirus) to those with hundreds (e.g., human cytomegalovirus [HCMV]) or thousands (e.g., pandoravirus) of predicted open reading frames (Philippe et al., 2013; Spall et al., 1997; Stern-Ginossar et al., 2012). Consequently, it can be challenging to comprehensively monitor viral protein levels for viruses with large protein coding capacity, given that the complexity of such a detection method would scale with the size of the viral proteome. Additionally, the study of viruses with large proteomes has historically suffered from the small percentage of viral proteins for which commercially produced antibodies are available.
Among these large viruses are herpesviruses, which emerged more than 200 million years ago and have coevolved with their hosts into modernity. This long history of virus-host co-evolution has allowed these viruses to acquire relatively large proteomes (70–250 putative proteins) that facilitate their diverse means for co-opting cellular processes and evading host defenses. The herpesvirus family consists of three subfamilies (α, β, and γ), each of which encompass prevalent human pathogens that establish latent, life-long infections that can sporadically reactivate to cause acute disease. For example, α-herpesviruses, like herpes simplex virus types 1 (HSV-1) and 2 (HSV-2), cause symptoms ranging from skin lesions to deadly encephalitis (Whitley and Roizman, 2001), and remains the leading cause of virally induced blindness. The β-herpesvirus HCMV is linked to cardiac disease (Courivaud et al., 2013) and is the leading viral cause of birth defects (Cheeran et al., 2009). Furthermore, some herpesviruses can exacerbate infections with other viral agents. HSV-2 infection increases the likelihood of contraction and spread of HIV (Zhu et al., 2009), and infection with the γ-herpesvirus Kaposi sarcoma-associated herpesvirus (KSHV) is the leading cause of cancer in individuals infected with HIV (Mesri et al., 2010). Despite their prevalence as human pathogens and the global health burden of herpesvirus-induced diseases, vaccines are lacking and antiviral treatments suffer from toxicity issues.
In addition to causing critical diseases, herpesviruses also share a common structure and replication cycle (Figure 1A). As enveloped, double-stranded DNA viruses, herpesviruses enter the cell, traffic to the nucleus where they replicate their viral genomes, and finally package newly synthesized viral DNA into progeny virions to continue the infection cycle. Although many of these stages are shared between these viruses, they replicate over different lengths of time. HSV-1 replication requires less than 24 h, while KSHV takes approximately 3 days, and HCMV takes 4–5 days. Despite these differences, a shared feature of these viruses is the tightly regulated temporal cascade of viral gene expression that ensues following entry into the cell, which can include the expression of immediate-early (IE), early (E), delayed early (DE), leaky late (LL), and late (L) classes of viral genes. Consequently, monitoring the levels of herpesvirus proteins not only reveals the presence of infection, but also the stage at which a particular sample is in the infection cycle. The monitoring of several marker proteins is standard for assessing replication progression using conventional approaches, such as antibody-based techniques like western blot (Sheng and Cristea, 2021) and ELISA (Inoue et al., 2000) or nucleic acid-based approaches such as microarrays (Bresnahan and Shenk, 2000) and RNA sequencing (Boldogkői et al., 2018} ). However, these methods suffer from drawbacks including that RNA-based approaches often do not accurately reflect the protein abundances that drive cellular phenotypes (Vogel and Marcotte, 2012) and antibodies against viral proteins either do not exist or are insufficiently characterized. Being able to accurately monitor the abundances of most viral proteins would provide the ability to better characterize specific stages of infection and to identify the temporal regulation of viral effectors that inhibit host defense factors and modulate cellular processes. Such a detection method would also allow for the screening of small molecules for their potential anti- or pro-viral activities and discovering their putative viral targets.
Figure 1. Developing and validating TRUSTED, a PRM-based method for monitoring HSV-1, HCMV, and KSHV proteins.

(A) Schematic representation of the herpesvirus replication cycle. Timeline below the schematic depicts the relative timescale of replication in HPI for HSV-1, HCMV, and KSHV.
(B) Overview of the PRM assay development process and its subsequent applications.
(C) Table of PRM assay specifications and protein targets.
(D) Traces of maximum concurrent precursors versus retention time (RT) for different RT windows. Dashed gray line denotes 30 concurrent precursors, which is the maximum number of precursors that can be monitored at a given RT in a single injection to achieve reliable quantitation with the instrument settings used in this study.
(E) Normalized abundances across infection time in MRC5 (HSV-1 and HCMV) or iSLK.219 (KSHV) cells for selected host proteins that can be used for data normalization.
(F) Average coefficient of variation between normalized abundance values across different peptides from a given protein within the same biological replicate (left), biological replicates for a given peptide (middle), or peptides and biological replicates for a given protein (right). Error bars represent a 95% confidence interval across biological replicates (HSV-1: n = 3; HCMV: n = 8; KSHV: n = 2).
To better understand the temporal regulation of herpesvirus protein abundances, several studies have monitored HSV-1, HCMV, or KSHV protein levels across infection time using shotgun proteomics approaches (Gabaev et al., 2020; Kulej et al., 2017; Weekes et al., 2014). Although shotgun methods allow both viral and host protein levels to be measured, they often lack viral proteome depth and can suffer from missing values and inconsistent detection due to changes in sample complexity during infection. Targeted mass spectrometry (MS) offers a robust method to directly detect and quantify specific proteins of interest with high sensitivity, reproducibility, and accuracy. Such methods, including parallel reaction monitoring (PRM), rely on the curation of libraries with signature peptides that fulfill a series of detection requirements (e.g., being unique to a given protein, well-ionized, amenable to chromatography separation and MS/MS fragmentation). With iterative development and validation, these methods can be scaled up for high-throughput monitoring of hundreds of proteins in a single run (Ebhardt et al., 2015; Lum et al., 2018). Once developed, targeted MS approaches are known to be applicable and transferrable across different instrumentation platforms and experimental workflows (Abbatiello et al., 2015; Zhang et al., 2019).
Here, we design and experimentally validate a PRM library for the broad detection of viral proteins from all three herpesvirus families: the α-herpesvirus HSV-1, the β-herpesvirus HCMV, and the γ-herpesvirus KSHV. We call this assay the Targeted herpesviRUS proTEin Detection (TRUSTED) assay. We demonstrate that our method captures the canonical cascades of viral protein expression and show that these proteins can be detected across different infection levels and models (e.g., cell culture, animal models). We next show our PRM assays accurately quantify the effects of clinically relevant antiviral agents and capture their temporal regulation of specific viral proteins. Further supporting the usefulness of this method for characterizing small molecules, we investigate the effects of a drug that modulates the antiviral activity of sirtuin proteins. We demonstrate the applicability of TRUSTED across different viral strains via computational analyses of peptide conservation and experimental validation using a low-passage virus strain. Finally, we leverage our HCMV assay to discover an unappreciated pro-viral role for the mitochondrial protein CPS1 early during infection. Overall, our method provides sensitive, reliable, and scalable assays for monitoring herpesvirus protein levels and has been deposited to the ProteomeXchange repository for use by other research groups. Our results support the broad applicability of these assays for probing viral protein abundances in various model systems and contexts, including antiviral drug screening, detecting infections in clinical settings, and genetic manipulations of virus or host factors.
RESULTS
A targeted MS assay for detecting and quantifying signature α-, β-, and γ-herpesvirus proteins
Considering the biological and clinical relevance of herpesviruses and the lack of methods to comprehensively monitor herpesvirus protein expression in laboratory and clinical settings, we aimed to develop a targeted PRM assay that offers the ability to systematically quantify viral protein abundances during HSV-1, HCMV, and KSHV infections. We performed infections in human fibroblast cells for HSV-1 and HCMV, and used a latently infected cell model (iSLK.219) that can be reactivated to study lytic KSHV infection (Myoung and Ganem, 2011). While these cell types represent standard model systems for studying these viruses, our assays are applicable to other cell culture models or tissues.
To capture the temporal replication stages of these viruses, we targeted proteins across all classes of herpesvirus gene expression and different virion components. We focused on the detection of both canonical markers of infection progression and viral proteins with diverse functions and localizations. The assays were designed to monitor peptides generated by trypsin digestion given the widespread use of this enzyme. Additionally, we found that the predicted lysine/arginine and tryptic peptide content of these viruses was well suited to MS analysis (Figures S1A and S1B). We manually curated a set of signature peptides for each virus by performing an iterative process of exploratory, data-dependent MS analyses of infected samples, and experimental validation of peptide detection and reliability by PRM (Figures 1B and S1C). We also performed unscheduled PRM injections to further advance our method to capture proteins and peptides not identified in our exploratory analyses. Most HSV-1, HCMV, and KSHV proteins were represented in these assays by 2–4 peptides ranging from 6–36 amino acids in length, with few viral proteins being captured by only one peptide (Figures S1D and S1E). This allowed us to monitor proteins belonging to all temporal classes of viral genes (e.g., IE, DE, E, LL, and L), as well as components of the virion (e.g., capsid, tegument, and envelope proteins) for all three viruses (Figures 1C and Table S1).
Overall, our assays measure the levels of proteins representing approximately 50%–80% of the reported proteomes for each virus. Of these three viruses, HSV-1 encodes the smallest number of proteins and replicates most quickly, and our PRM assay quantifies 59 viral proteins. Comparatively, HCMV and KSHV express substantially more proteins, and our assays monitor 90 and 62 viral proteins, respectively. Moreover, more than 75% of the proteins quantified by our assays represent targets without commercially available antibodies.
On our instruments, these assays can monitor the peptides of interest using 6-min retention time windows across a series of one (KSHV) or two (HSV-1 and HCMV) 60-min injections (Figure 1D). Internal reference standard peptides are also provided to help account for variability in input material across samples due to natural variation in sample preparation. To serve this purpose we chose several ubiquitously expressed cytoskeletal factors, including tubulin (TUBA1A), myosin 5A (MYO5A), and a myosin II heavy chain (MYH9), which exhibit stable expression levels throughout infection (Figures 1E and S1F). Although these peptides are stably expressed in our model systems, differences may exist in different contexts (e.g., different cell lines, drug treatments, genetic manipulations). In these cases, MS1-based normalization provides the ability to account for differences in input sample amount without monitoring internal reference peptides (Federspiel et al., 2019). After normalization, we obtained reproducible measurements across the different infections, with coefficients of variation averaging less than 25% (Figures 1F and S2–S4). Altogether, this process culminated in the establishment of virus-specific peptide libraries that proved effective at detecting HSV-1, HCMV, and KSHV peptides during wild type infections. Given the robustness and accuracy of detection offered by targeted MS, we named this assay TRUSTED.
Herpesvirus PRM assay captures the signature temporal cascade of viral gene expression
An essential aspect of herpesvirus replication is the temporal cascade of gene expression. Having shown that our assays can accurately detect viral proteins, we next asked whether they can capture the temporality of these protein abundances during infection. We harvested infected fibroblasts at 2, 6, 12, and 18 h post-infection (HPI) for HSV-1, and at 24, 48, 72, 96, and 120 HPI for HCMV. For KSHV, we reactivated the latent virus in iSLK.219 cells and collected samples at 24, 48, and 72 h post-reactivation (HPR). For each virus, these time points represent specific stages of virus gene expression, virion assembly, and egress. In agreement with the known cascades of virus gene expression, these measurements captured the sequential nature of viral protein levels (depicted as log2 fold change relative to the first time point of detection in Figure 2 and as mean-normalized abundances in Figure S5, see also Table S2).
Figure 2. Herpesvirus PRM assay captures the signature temporal cascade of viral gene expression.

(A–D) Abundance plots of: HSV-1 proteins (A), antiviral host proteins of interest during HSV-1 infection (B), HCMV proteins (C), and KSHV proteins (D). Plots are stratified by temporal expression class. Protein abundance levels are represented as log 2-fold change relative to the first time point at which peptides were detected. Error bars represent a 95% confidence interval across biological replicates (HSV-1: n = 3; HCMV: n = 8; KSHV: n = 2) and peptides for a given protein.
For HSV-1, viral protein levels generally increased throughout the course of infection, with an approximately 12-fold median increase observed at 18 HPI relative to the first time point of detection for each protein (Figures 2A and S5A). An exception was US12 (ICP47) levels, which peaked at 6 HPI, then decreased throughout the rest of the infection cycle, in agreement with another study (Soh et al., 2020). As US12 inhibits MHC-I peptide presentation (Hill et al., 1995), this downregulation of US12 levels may be a host antiviral response. Alternatively, transcription of the US12 gene may decrease at later time points or have a high turnover rate. We also noted that, by 2 HPI, we already detected approximately 80% of the late temporal class HSV-1 proteins monitored by our assay, and all but two of these factors have previous evidence for being incorporated into the virion (Figure S5B). Given that de novo HSV-1 late gene synthesis does not begin until 2.5–3 HPI, this suggests that these viral proteins are associated with incoming virions, further underscoring the sensitivity of this method. We also designed the PRM assay to include peptides from host factors known to be inhibited by HSV-1. Indeed, in accordance with previous studies (Rodríguez et al., 2020), we observed virus-induced degradation of the host defense factors interferon-inducible protein 16, promyelocytic leukemia protein, and SP100 (Figure 2B).
During HCMV infection, viral protein levels increased up to 1,000-fold, with a median increase of approximately 15-fold by 120 HPI (Figures 2C and S5C). We also noted that some HCMV IE proteins (UL13, UL36, UL37 [vMIA], and UL123 [IE1]) exhibited less induction (<2-fold, on average), compared with other HCMV proteins. This agrees with reports that these IE proteins are highly induced early in infection, afterward maintaining their levels throughout the replication cycle (Jean Beltran et al., 2016; Weekes et al., 2014). Exceptions to this observation included UL122 (IE2), IRS1, and TRS1, which increased throughout the course of infection (also observed in Weekes et al., 2014).
The reactivation of KSHV led to milder temporal increases of approximately 5-fold by 72 HPR (Figures 2D and S5D). Like most HSV-1 and HCMV proteins, KSHV protein levels also generally increased after reactivation, except for DE protein K2, which was decreased by approximately 40% by 72 HPR. This agrees with a report that K2 is robustly expressed in latently infected iSLK.219 cells, but its levels decrease following reactivation (Park et al., 2019).
Application of TRUSTED to different levels of infection (multiplicity of infection) and in vivo infection
Having established that the TRUSTED assays reliably capture temporal herpesvirus gene expression, we went on to characterize their performance in recognizing different infection levels, i.e., the number of incoming viral particles per cell (multiplicity of infection [MOI]). To this end, we performed PRM on cells that were infected with increasing amounts of HCMV virus at MOIs of 0.05, 0.25, 1.25, and 6.25. Even at low MOIs (0.05 or 0.25), we found that nearly all targeted peptides (Figure 3A) and proteins (Figures 3B and 3C) reached detectable levels by 120 HPI, and at higher MOIs (1.25 or 6.25) were detectable by 24–72 HPI. Detected at low MOIs early in infection were IE proteins, such as UL122, UL123, UL13, UL36, and UL37, as well as the most abundant HCMV viral tegument protein, UL83 (pp65) (Figure 3C). As expected, MOI-dependent increased abundances were seen for most proteins (Figures 3D and Table S3A) and the increase was approximately linear with respect to the percent of cells infected at a given MOI (Figure S6A). However, we noticed that a subset of US12 family proteins did not conform to this pattern, including US12 and US15 (Figure 3E). Proteins within the US12 family play immunomodulatory roles and have been shown to be targeted for degradation (Fielding et al., 2017). As such, their decrease in abundance at high MOIs is perhaps unsurprising—yet, our results demonstrating that these proteins do not seem to be degraded at a low MOI suggests that there may be a threshold of US12 family protein expression that must be reached before these proteins are degraded. Overall, our results demonstrate the applicability of the PRM assays to a range of MOIs, with low MOIs being closer to physiological levels and high MOIs being often employed in research studies (e.g., for synchronous infections).
Figure 3. Application of TRUSTED to different levels of infection (MOI) and in vivo infection.

(A) Percent of HCMV peptides detected at different time points across increasing amounts of input virus (e.g., MOIs). A peptide was considered detected if it was observed in at least one biological replicate (n = 3).
(B) Number of viral proteins detected at increasing MOIs.
(C) Time point of first detection for HCMV proteins at increasing MOIs. Proteins are color coded by which part of the virion they are reported to associate with.
(D) Average HCMV protein abundance across infection time for different MOIs, stratified by temporal class. Error bars represent a 95% confidence interval across all proteins for a given temporal class after averaging across biological replicates (n = 3) and peptides.
(E) Protein abundance plots of HCMV proteins US12 and US15 at increasing MOIs. Error bars represent a 95% confidence interval across biological replicates(n = 3) and peptides.
(F) Schematic depiction of HSV-1 infection and PRM analysis of mouse frontal lobes.
(G) Distribution of HSV-1 protein detection in mouse frontal lobe tissues across different temporal classes and virion components. Proteins with at least one detected peptide were included in this analysis.
Given that physiological levels of infection tend to be low, we wanted to ask whether our PRM assays could be leveraged to quantify protein levels in infected tissues. We infected mice with HSV-1 (strain 17) and performed PRM on frontal lobe tissues collected from encephalitic mice at 8 days post-infection (Figure 3F). From this analysis, we detected approximately 80% (47) of the viral proteins monitored by our assay, and all temporal classes of gene expression and virion components were represented (Figures 3G and Table S3B). Notably, shotgun-based analysis of the same sample detected only three viral proteins with a single peptide each (UL12, UL13, and UL48; Table S3C). Hence, these assays are sensitive enough to detect viral infections in both cell culture and animal models of infection.
Investigations of clinically employed herpesvirus antiviral drugs
To demonstrate the usefulness of our PRM assays for screening antiviral compounds, we next monitored viral protein abundances upon treatment with canonical antiviral drugs. We treated fibroblasts with acyclovir (ACV) or cidofovir (CDV), two compounds used as treatments for HSV-1 and HCMV infections, respectively. Both ACV and CDV hinder viral replication by acting as nucleoside (ACV) or nucleotide (CDV) analogues that inhibit viral DNA polymerases (Biron, 2007). While both drugs target the same viral process, ACV is a more potent inhibitor of HSV-1 than HCMV and the converse is true for CDV. Although their mechanism of action and impact on virus production are well established, how these drugs broadly affect the landscape of viral protein abundances remains less understood, with the exception of one HSV-1 proteomics study performed after ACV treatment (Bell et al., 2013). Therefore, we asked whether our PRM assay can provide context to viral protein regulation upon drug treatment during HSV-1 and HCMV infection.
First, we confirmed that treatment with either ACV or CDV inhibited virus genome replication and production (Figures 4A and 4B), while not affecting cell viability (Figures S6B). We then treated cells with ACV or CDV and performed PRM after infecting with HSV-1 or HCMV, respectively (Figures 4C–4F, and Table S3D–S3F). We expected that both treatments would result in decreased viral protein levels after the block in DNA replication. Indeed, upon ACV treatment, we observed L HSV-1 proteins to decrease by approximately 40% by and after 12 HPI, with IE and E proteins showing fewer changes (Figure 4C). Similar results were observed for two ACV concentrations (1 and 3 mM) (Figures S6C–S6E). Among the E gene products affected by ACV treatment were proteins involved in viral DNA replication, such as DBP (ICP8), HELI (UL5), UL9, UL12, and UL42. Overall, our results are consistent with a prior transcriptome study (Dremel and DeLuca, 2019) demonstrating that ACV decreased HSV-1 gene transcription and has a differential impact on γ1 and γ2 late genes. Among these gene products, the subset that requires DNA replication for initiation of synthesis (e.g., γ2 genes) is more significantly affected by ACV treatment compared with the subset that is more robustly transcribed in the presence of DNA replication (e.g., γ1 genes) (Figure 4E). From our results at both concentrations of ACV, we observed that decreases in protein levels were correlated with substantial decreases in virus titers. Specifically, we observed that decreases in protein levels of approximately 25% and 40% (after treatments with 1 μM and 3 μM ACV, respectively) resulted in 85% and 99% decreases in virus production, respectively.
Figure 4. PRM application to investigations of clinically employed herpesvirus antiviral drugs.

(A and B) Viral genome (A) and virus titer (B) quantification after treatment with ACV or DMSO (control [CTRL]) during HSV-1 infection (left) or CDV or PBS (CTRL) during HCMV infection (right). Error bars represent a 95% confidence interval across biological (n = 3) and technical replicates (n = 2). Significance was determined by two-tailed Student’s t-test; * p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001.
(C and D) Average protein abundance plots of protein levels after treatment with ACV during HSV-1 infection (C) or CDV during HCMV infection (D). Error bars represent a 95% confidence interval across all proteins for a given temporal class after averaging across biological replicates (n = 3) and peptides.
(E) Heatmaps of viral protein levels after treatment with ACV during HSV-1 infection. γ1 and γ2 genes (Dremel and DeLuca, 2019) are denoted by their kinetic class determination. ND, not detected.
(F) Heatmaps of HCMV protein levels after treatment with CDV during HCMV infection.
For HCMV, we found that CDV treatment led to a 55% decrease in protein levels and a 99% decrease in titers. In contrast with the varied response to ACV, CDV treatment resulted in substantial decreases in HCMV protein levels across all temporal classes after 24 HPI, which marks the onset of robust viral genome replication (Figures 4D and 4F). By 72 HPI, more than 85% of the proteins monitored exhibited decreases of at least 35% compared with the control. Moreover, among all 90 proteins, only two (UL54 and UL102) were decreased by less than 20% across all time points, underscoring the global effect of CDV on HCMV protein levels. Nevertheless, a phenotype conserved from our observations for ACV was that most IE genes were less impacted by CDV compared with other gene classes. In both cases, this likely reflects the relative independence of IE gene expression as these proteins are characterized by their ability to be transcribed in the absence of de novo protein synthesis. An exception to this observation, however, was that the abundance of the IE protein UL122 was decreased by approximately 70% by 120 HPI. This observation may be explained by the fact that the UL122 locus produces at least two additional protein isoforms that are expressed from alternative downstream promoters with late kinetics and depend on successful viral genome replication (Puchtler and Stamminger, 1991). The peptides monitored by our PRM assay are within the C-terminal region of the UL122 protein, and thus common to both full-length and shorter isoforms.
Modulation of antiviral sirtuin enzymatic activity differentially regulates viral protein levels during herpesvirus infections
In addition to those targeting DNA replication, a variety of other small molecules have been shown to impact herpesvirus infections. These include compounds that target sirtuin proteins, which we have previously shown to exhibit antiviral activity against several viruses, including HSV-1 and HCMV (Koyuncu et al., 2014). Sirtuins are a diverse family of seven (SIRT1–7) NAD+-dependent deacetylases and deacylases that regulate a range of cellular processes including metabolism, the cell cycle, and gene expression (Michan and Sinclair, 2007). Accumulating evidence suggests that sirtuins could serve as potential targets for antiviral therapies (Budayeva et al., 2016). For example, activation of SIRT1 activity using the compound CAY10602 results in decreased HCMV titers (Koyuncu et al., 2014). However, the effects of this compound on the HCMV viral proteome have not been investigated, nor has its impact on HSV-1 or KSHV replication and viral protein levels been tested.
To characterize the effects of SIRT1 activation on viral protein levels during HCMV infection, we treated cells with CAY10602 and performed our PRM assay (Figures 5A and 5B, and Table S3G). Of the small subset of proteins that we had previously quantified by western blot (UL123, UL26, and UL99) after CAY10602 treatment (Koyuncu et al., 2014), the PRM results are in agreement with our previous observations. UL123 levels are unchanged at 24 HPI, UL26 levels are decreased at 48 HPI, and UL99 (pp28) levels are robustly decreased by 72 HPI (Figure 5A). However, our results also revealed that treatment with CAY10602 globally decreased viral protein levels by approximately 65% at 120 HPI, and this was not due to drug cytotoxicity (Figure S6F). Moreover, several viral proteins (e.g., NEC1 [UL53], UL76, UL79, UL87, and IR10 [TRL10]) become undetectable at early HPIs, displaying delayed expression kinetics upon treatment. For most viral proteins, the sirtuin-modulatory effects became more pronounced as the infection progressed. Viral protein levels were globally decreased approximately 85% by the end of the infection cycle, which translated into an approximately 150-fold decrease in virus titers by 120 HPI (Figure 5C), in agreement with our previous measurements (Koyuncu et al., 2014). Our results demonstrate that the impact of SIRT1 activation on HCMV titers is driven by alterations in virus protein levels.
Figure 5. Modulation of sirtuin enzymatic activity differentially regulates HSV-1, HCMV, and KSHV protein levels.

(A) Heatmap of HCMV protein levels after treatment with CAY10602 or DMSO (control).
(B and C) Averaged HCMV protein levels across temporal classes (B) or virus titers (C) after CAY10602 treatment.
(D) Heatmap of HSV-1 protein levels after CAY10602 treatment.
(E and F) Averaged HSV-1 protein levels across temporal classes (E) or virus titers (F) after CAY10602 treatment.
(G) Heatmap of KSHV protein levels after CAY10602 treatment.
(H and I) Averaged KSHV protein levels across temporal classes (H) or virus titers (I) after CAY10602 treatment. For (B), (E), and (H), the error bars represent a 95% confidence interval across all proteins for a given temporal class after averaging across biological replicates (HSV-1 and HCMV: n = 3; KSHV: n = 2) and peptides. For (C), (F), and (I), the error bars represent a 95% confidence interval across biological (n = 3) and technical (HSV-1 and HCMV: n = 2; KSHV: n = 3) replicates. Significance was determined by two-tailed Student’s t-test; * p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001.
We next asked whether treatment with this compound, at the same concentration, would also impact viral protein levels in the context of HSV-1 infection or KSHV reactivation. In comparison with the robust downregulation of HCMV protein levels, CAY10602 treatment during HSV-1 infection did not globally impact viral protein levels (Figures 5D, 5E, and Table S3H). In fact, by 18 HPI, no proteins exhibited levels that were decreased by more than 20% compared with the control, and virus titers were not affected by CAY10602 treatment (Figure 5F). This is likely due to diminished NAD+ levels during HSV-1 infection, unlike the relatively unchanged NAD+ levels observed during HCMV infection (Vastag et al., 2011). Finally, for KSHV, CAY10602 treatment led to a decrease in protein levels by 72 HPR, albeit to a lesser extent than HCMV (Figures 5G and 5H, and Table S3I). Specifically, viral protein levels were decreased by approximately 20% at 72 HPR, which resulted in an approximately 80% decrease in virus titers (Figure 5I). These results support an antiviral role for SIRT1 during KSHV infection, and knockdown (KD) of SIRT1 has been shown to increase KSHV reactivation from latency (Li et al., 2014). Together, these results reveal how sirtuin activity-modulating treatments impact protein expression throughout the course of HCMV, HSV-1, and KSHV infections. Our results also demonstrate the ability of these assays to contextualize the effects of small molecule treatments, both at the individual and global viral protein levels.
Conservation of TRUSTED peptides indicates assay utility across diverse virus strains
An important consideration when developing a detection assay is its broad applicability—in our case, whether these assays are suitable for detecting viral proteins from a range of HSV-1, HCMV, and KSHV strains. Several laboratory and clinical strains are implemented for the study of each of these viruses, and many have readily accessible complete genome sequences available in online databases. To address the applicability of our assay to different strains, we performed a computational analysis of potential peptide sequences represented by the genomes of different HSV-1, HCMV, and KSHV strains in the NCBI nucleotide database to determine the extent of conservation for peptides targeted by our PRM assay (Figures 6A and 6B, and Table S4A). As expected, our analysis demonstrated that approximately 100% of the PRM peptides were conserved for HSV-1 strain 17 and HCMV strain AD169, the models upon which our PRM assays were developed. A direct comparison with the type of KSHV virus produced by iSLK.219 cells was not possible, given the lack of a fully sequenced genome in the NCBI database. However, we observed nearly full conservation when compared with BrK.219, a B cell line latently infected with the same type of KSHV that is also harbored by iSLK.219 cells (Kati et al., 2015).
Figure 6. Conservation of TRUSTED peptides indicates assay utility across laboratory and clinical virus strains.

(A and B) Percentage of peptides (A) or number of proteins (B) targeted by the PRM assays that are conserved across different herpesvirus strains. For (B), proteins with at least one conserved peptide were included in this analysis.
(C–E) Numbers of conserved peptides for all proteins targeted in the PRM assays for HSV-1 (C), HCMV (D), and KSHV (E). The number of conserved peptides is denoted within each box and its color corresponds with the percent of peptides that are conserved for a given protein.
(F) Percentage of HCMV PRM peptides computationally predicted to be detectable in the TB40/E strain compared to peptides that were experimentally validated and detected at 96 HPI in cells infected with HCMV TB40/E.
(G) Alignment between computational predictions and experimentally detected/not detected peptides for the TB40/E HCMV strain.
We next compared the peptide sequences targeted by the TRUSTED assay to those predicted to be present in other HSV-1 strains: F, H129, KOS, MacIntyre, McKrae, and SC16 (Figure 6C). Among these strains we observed approximately 100% conservation for most proteins targeted by our assay, supporting its broader use for studies with a range of HSV-1 strains. Considering the clinical importance of being able to discriminate between HSV-1 and HSV-2 infections, we also asked whether our peptides were conserved in HSV-2. We observed that approximately 25% of the peptides targeted by our HSV-1 PRM assay were also present in HSV-2, with approximately 50% of the proteins targeted by our assay having at least one peptide in common between the two viruses. It is, thus, possible that our HSV-1 PRM assay could be leveraged to discriminate between HSV-1 and HSV-2 infections, given the detection or lack thereof for the 75% of peptides in the assay that are specific to HSV-1.
For both HCMV and KSHV, we observed greater than 90% conservation among the different strains assessed in this analysis (Figures 6D and 6E). A comparison of laboratory/high-passage (AD169 and Towne) and clinical/low-passage (Toledo, TR, TB40/E, and Merlin) strains of HCMV demonstrated conservation across most proteins, with more than 85% of the proteins targeted by our PRM assay having at least one conserved peptide across all strains tested. Similar levels of conservation were observed for the different KSHV strains assessed, which included the laboratory strain BAC16, as well as two clinical strains (GK18 and DG-1). A limitation of this analysis, however, is that protein segments resulting from alternative splicing may not be captured by our computational predictions. For both HCMV and KSHV, we observed that there was one protein for each virus with peptides targeted by our PRM assays that were not predicted to be fully conserved across any of the strains. In both cases, the proteins in question (UL128 for HCMV and K8 for KSHV) are known to be produced as the result of alternative splicing, likely explaining why they were not detected by this analysis.
We next aimed to experimentally validate our computational predictions. Upon infection of cells with the low-passage HCMV strain TB40/E, we observed high agreement (approximately 95%) between our computational predictions and the experimentally detected peptides (Figures 6F, 6G, and Table S4B). Of the 223 viral peptides monitored by our assay, 189 peptides were computationally predicted to be present, and 176 peptides were experimentally validated by PRM. Additionally, 34 peptides were computationally predicted to be absent from this strain, and we did not experimentally detect any of these peptides. Altogether, these results highlight the validity of this computational approach for predicting how these assays will perform and generalize across other viral strains.
The mitochondrial protein CPS1 supports HCMV genome replication early in infection
We next tested the ability of our assays to aid the characterization of host factors during infection. As HCMV is known to alter the metabolic state of infected cells (Shenk and Alwine, 2014), of interest has been our previous observation that the metabolic enzyme carbamoyl phosphate synthetase 1 (CPS1) increases in abundance throughout infection relative to uninfected cells (Sheng and Cristea, 2021) (Figures S6G). CPS1 catalyzes the first committed step of the urea cycle, a pathway commonly dysregulated in cancer cells (Keshet et al., 2018). Considering that the metabolic phenotype during HCMV infection resembles that of cancer cells (Chambers et al., 2010; Rodríguez-Sánchez and Munger, 2019), we hypothesized that CPS1 may also play a role during HCMV replication.
To investigate this, we performed a small interfering RNA (siRNA)-mediated KD of CPS1 and used our PRM assay to assess HCMV protein levels. We confirmed the efficiency of CPS1 KD, observing a 20-fold decrease in CPS1 transcript abundance and limited cytotoxicity (Figures S6H and S6I). Given that HCMV globally rewires host cell metabolism starting at approximately 48 HPI, we predicted that we would observe effects of CPS1 KD on HCMV protein levels between 24 and 72 HPI. Indeed, when we leveraged our PRM assay to measure a representative subset of proteins from each temporal class, we observed that CPS1 KD resulted in a nearly global decrease in viral protein abundances by 48–72 HPI (Figures 7A–7C). Moreover, CPS1 KD led to a more than 300-fold decrease in virus production (Figure 7D). Together, these results demonstrate that CPS1 supports HCMV replication. We next sought to understand which stage of the viral replication cycle is impacted by CPS1. We noted that IE protein levels were not globally decreased at 24 HPI, indicating that CPS1 KD does not inhibit viral entry or trafficking to the nucleus. CPS1 KD also resulted in a nearly global decrease in virus protein abundances by 72 HPI, suggesting that CPS1 plays a role before viral assembly. Hence, we hypothesized that CPS1 might impact viral genome replication. To test this, we quantified the abundance of viral genomes at 24, 48, and 72 HPI (Figure 7E). We found that CPS1 KD results in a progressive defect in viral genome production, beginning as early as 24 HPI and peaking at 72 HPI. Altogether, the application of our assay uncovered a pro-viral role for CPS1, pinpointing its importance for viral genome replication.
Figure 7. The mitochondrial protein CPS1 supports HCMV genome replication early in infection.

(A and B) Heatmaps of mean (A) or log 2-fold change (B) normalized HCMV protein abundances after treatment with siCPS1 or siCTRL (control) siRNAs.
(C) Average HCMV protein levels after treatment with siCPS1 or siCTRL. Error bars represent a 95% confidence interval across all proteins for a given temporal class after averaging across biological replicates (n = 2) and peptides.
(D) HCMV virus titers after treatment with siCPS1 or siCTRL. Error bars represent a 95% confidence interval across biological replicates (n = 3). Significance was determined by two-tailed Student’s t-test; * p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001.
(E) HCMV viral genome quantification following siCPS1 or siCTRL treatment. Error bars represent a 95% confidence interval across biological replicates (n = 2).
(F) Possible processes that may contribute to the ability of CPS1 to support HCMV replication.
DISCUSSION
Here, we present TRUSTED, a targeted MS assay for detecting and quantifying proteins from three model viruses across herpesvirus subfamilies. Our assays for HSV-1, HCMV, and KSHV allow for a comprehensive overview of replication cycle progression, while simultaneously quantifying locus-specific changes covering much of the proteomes of these viruses. By applying these assays, we were able to (1) capture the temporal characteristics of the herpesvirus gene expression cascade, (2) detect proteins throughout a range of virus concentrations and in in vivo models of infection, (3) further investigate the proteinlevel effects of canonical herpesvirus treatments, (4) screen putative antiviral compounds, as we show for the modulation of SIRT1 antiviral function, (5) propose and validate their utility across different laboratory and clinical viral strains, and (6) leverage our assays to discover a role for CPS1 in supporting HCMV replication. Ultimately, our approach is broadly applicable to investigating the progression of herpesvirus replication in diverse model systems and in the context of a wide variety of perturbations, including small molecule treatment, antiviral screening, and genetic perturbations.
An important driver for the development of our assay was the lack of commercially available antibodies for most of the proteins expressed by these large viruses. By using targeted MS, we were able to directly measure viral peptide levels in an antibody-independent manner. An equally important driver was the need for methods that provide high throughput detection of viral proteins. In comparison with standard antibody-based methods (e.g., western blot, ELISA), this assay also has the advantage of being able to simultaneously measure a vast number of viral proteins. Although mRNA measurements also offer throughput, it is known that transcript levels do not always reflect the levels of functional protein products (Vogel and Marcotte, 2012). Our HSV-1, HCMV, and KSHV detection assays include peptides from viral proteins belonging to all temporal classes of viral genes, providing an informed snapshot of the virus replication state in one or two injections onto the instrument.
After the development of our assays, we aimed to validate their performance in the context of canonical herpesvirus treatments, ACV and CDV, which act as inhibitors of virally encoded DNA polymerases. For both established drugs, we observed a decrease in protein levels by late time points during HSV-1 and HCMV infections, concomitant with decreased viral titers and viral genome levels. However, for HCMV, we observed a much more global and severe decrease in viral protein levels across all temporal classes of genes, while for HSV-1 these effects were largely restricted to late proteins or early proteins involved in viral genome replication. Given the extended replication cycle for HCMV compared with HSV-1, these results may reflect a greater degree of feedback mechanisms at play during HCMV infection. Alternatively, it is also possible that this longer replication time allows for the effects of CDV treatment to become compounded over the 5-day replication span.
We next aimed to test the applicability of our assays for characterizing less well-established antiviral compounds. As we have previously shown that the sirtuin family of deacetylases can restrict herpesvirus replication (Koyuncu et al., 2014), we applied our assays to determine the effects of modulating sirtuin activity on viral protein levels. Although siRNA KD or small molecular modulation of SIRT1 has been shown to decrease HCMV titers (Koyuncu et al., 2014), it is not known how these effects are mediated or whether these changes in viral titer are also evident at the HCMV protein level. Here, we found that this was indeed the case; the treatment of HCMV-infected cells with the SIRT1 activator CAY10602 resulted in a global decrease in viral protein production by 48 HPI. Altogether, our results establish that SIRT1 enzymatic activity modulates HCMV protein expression—yet, whether these effects are mediated directly or indirectly remains to be investigated. Considering that one of the main targets of SIRT1 is histones, it is possible that SIRT1 directly regulates viral protein expression by deacetylating histones on viral genomes (Cliffe and Knipe, 2008). Alternatively, it remains to be seen whether SIRT1 can regulate the acetylation status of HCMV proteins, thereby impacting their levels and functions. It is also possible, however, that these effects are indirectly mediated by SIRT1. For example, it is well established that SIRT1 deacetylates and inhibits the transcription factor nuclear factor-κB (Kauppinen et al., 2013), which is essential for driving HCMV protein expression from the major IE promoter (MIEP) (Hancock and Nelson, 2017). Consistent with this notion, we observed decreases in UL122 and UL123 levels upon CAY10602 treatment, perhaps owing to differential MIEP activity. Moreover, considering the robust and global decrease in HCMV protein levels that we observed after SIRT1 activation by CAY10602, it follows that these effects could be driven by altering the levels of essential transcription factors like UL122 and UL123.
In comparison with HCMV, we did not observe a CAY10602-dependent effect on HSV-1 protein levels or titers. This is likely due to the already decreased NAD+ levels during HSV-1 infection (Vastag et al., 2011), which could constrict SIRT1 activity even in the presence of an agonist. For KSHV, SIRT1 activation led to a decrease in viral protein levels and virus production, albeit to a lesser extent than during HCMV infection. One potential explanation for these differences is that reactivation of KSHV in the model used in this study was achieved, in part, by the addition of sodium butyrate (NaB). NaB is a broad inhibitor of class I and II histone deacetylases, thereby promoting KSHV reactivation by preventing histone deacetylase-mediated silencing of the major lytic transactivator RTA (Lu et al., 2003). Considering that SIRT1 has been shown to inhibit the reactivation of KSHV by binding to this same region (Li et al., 2014), it is possible that the antiviral effects of SIRT1 on the RTA locus are muted in the context of NaB treatment.
The final goal of our study was to apply our PRM assay to aid the characterization of host factors during infection, and we focused on the urea cycle enzyme CPS1. We used our PRM assay to demonstrate that CPS1 functions in a pro-viral manner, further revealing that it impacts viral genome replication. Although very few studies have focused on investigating the urea cycle during HCMV infection, it is known that depletion of the urea cycle enzyme argininosuccinate synthetase 1 decreases HCMV virus production by 10-fold (Grady et al., 2013). Outside the context of infection, in cancer cells, recent evidence suggests that CPS1 facilitates the use of ammonia-derived nitrogen for pyrimidine biosynthesis (Kim et al., 2017). Given the similar metabolic phenotypes induced by HCMV infection and cancer, it is possible that CPS1 serves a similar function during HCMV replication. Furthermore, it has been shown that carbamoyl phosphate produced in the mitochondria can be used for cytoplasmic pyrimidine biosynthesis via an unknown mechanism (Wendler et al., 1983). Alternatively, CPS1-driven urea cycle functions may repurpose waste products from catabolic processes for building biosynthetic precursors necessary for HCMV replication, including amino acids. The urea cycle also generates fumarate, which is required for de novo arginine biosynthesis. Interestingly, it has been known for some time that arginine is required for HCMV replication (Garnett, 1975), and a recent study demonstrated an increased consumption of arginine during HCMV infection (Rodríguez-Sánchez et al., 2019). Given these pieces of evidence, it is tempting to speculate that the pro-viral function of CPS1 early during infection may derive from its ability to promote pyrimidine biosynthesis and/or the production of biomolecules (Figure 7F). Future investigations would be needed to delineate the precise mechanisms underlying the pro-viral functions of CPS1.
In summary, we have demonstrated the value of these TRUSTED assays for globally detecting and quantifying viral proteins from the three main Herpesviridae subfamilies with high accuracy and throughput. These targeted detection methods can offer information about virus biology, as well as provide the means to monitor the effects of small molecules or genetic perturbations in the context of infections. Given the promise for their broad applicability to a range of biological contexts and viral strains, we expect these assays to be of widespread utility. We further hope that our study will inspire the future development of additional targeted MS assays for the detection of diverse viral pathogens, leading to the development of highly needed repositories of signature peptides for virus detection.
Limitations of the study
The detection parameters that we established for these herpesvirus proteins can be leveraged by other groups in a wide variety of model systems (e.g., different cell types, tissues, animal models). Yet, in each of these contexts, it may be necessary to optimize the sample preparation procedure, for example by altering lysis conditions, or to reschedule retention times for the signature peptides defined in our assays on a new liquid chromatography (LC)-MS platform. From the results obtained in this study, our experiments using low MOIs and infected tissue samples suggest the promise of these assays for detecting viral proteins in clinical samples. However, future experiments would be needed to support their use in this context.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Ileana M. Cristea, Princeton University (icristea@princeton.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Skyline data analysis files and raw MS data have been deposited to PanoramaWeb at https://panoramaweb.org/HerpesvirusPRM.url (associated with the identifier ProteomeXchange: PXD025879) and are publicly available as of the date of publication. Accession numbers are listed in the Key resources table. Processed data are available in Tables S2, S3, S4, and S5. Code for data processing, normalization, and plotting has been deposited on GitHub and is publicly available as of the date of publication. The DOI is listed in the Key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| anti-IE1, Mouse polyclonal | Gift from Dr. Thomas Shenk | N/A |
| Goat anti-Mouse Alexa Fluor 488 conjugate | Invitrogen | Cat# A-11001; RRID:AB_2534069 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| HCMV strain AD169 | Gift from Dr. Thomas Shenk | N/A |
| HCMV strain TB40/E | Gift from Dr. Thomas Shenk | N/A |
| HSV-1 strain 17+ | Gift from Dr. Beate Sodeik | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Acyclovir | Cayman Chemical | Cat# 14160 |
| Cidofovir | Cayman Chemical | Cat# 13113 |
| CAY10602 | Cayman Chemical | Cat# 10009796 |
| DAPI | ThermoFisher Scientific | Cat# 62248 |
| HALT protease and phosphatase inhibitor | ThermoFisher Scientific | Cat# 78438 |
| TCEP | ThermoFisher Scientific | Cat# 77720 |
| Hygromycin | ThermoFisher Scientific | Cat# 10687010 |
| Sodium butyrate | Sigma-Aldrich | Cat# B5887 |
| Doxycycline | Sigma-Aldrich | Cat# D9891 |
| Polybrene | Millipore Sigma | Cat# TR-1003-G |
| SYBR green PCR master mix | ThermoFisher Scientific | Cat# 4368706 |
| Lipofectamine RNAiMAX | ThermoFisher Scientific | Cat# 13778150 |
|
| ||
| Critical commercial assays | ||
|
| ||
| BCA Assay | ThermoFisher Scientific | Cat# 23225 |
| in situ Cell Death Detection Kit - Fluorescein | Roche | Cat# 1168479 |
| in situ Cell Death Detection Kit - TMR red | Roche | Cat# 12156792910 |
| SuperScript IV First-Strand cDNA Synthesis Reaction Kit | ThermoFisher Scientific | Cat# 18091050 |
|
| ||
| Deposited data | ||
|
| ||
| PRM MS/MS data (Panorama) | This study | https://panoramaweb.org/HerpesvirusPRM.url |
| PRM MS/MS data (RAW files) | This study | ProteomeXchange (PXD025879) |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| MRC5 | ATCC | Cat# CCL-171; RRID:CVCL_0440 |
| HEK293T | ATCC | Cat# CRL-3216; RRID:CVCL_0063 |
| U2OS | ATCC | Cat# HTB-96; RRID:CVCL_0042 |
| iSLK.219 | Gift from Dr. Britt Glaunsinger | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: BALB/cAnNTac | Taconic Biosciences | Cat# BALB-F |
|
| ||
| Oligonucleotides | ||
|
| ||
| CPS1 siRNA | ThermoFisher Scientific | Cat# EHU002511 |
| For qPCR primers, see Table S6 | N/A | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Proteome Discoverer v2.4 | ThermoFisher Scientific | Cat# OPTON-31040 |
| Skyline | (MacLean et al., 2010) | https://skyline.ms/ |
| RawMeat | Vast Scientific | https://vastscientific.com/rawmeat |
| Custom Python Code | This study | https://doi.org/10.5281/zenodo.6468269 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and primary cultures
MRC5 primary human fibroblasts (HFs) (ATCC CCL-171; derived from normal lung tissue of a 14-week-old male fetus) were used as the model system for HSV-1 and HCMV infections and were cultured in complete growth medium (DMEM supplemented with 10% fetal bovine serum [FBS] and 1% penicillin-streptomycin antibiotics) at 37°C and 5% CO2. The iSLK.219 cells harboring latent KSHV (a gift from Dr Britt Glaunsinger, University of California, Berkeley) were grown in complete growth medium supplemented with 500 μg/mL hygromycin (ThermoFisher Scientific, 10687010) at 37°C and 5% CO2. For titering of virus supernatants, reporter plates consisting of U2OS (HSV-1), MRC5 (HCMV), or HEK293T (KSHV) cells were maintained in complete growth medium at 37°C and 5% CO2.
Virus strains and infections
Wild-type HSV-1 strain 17+ (a gift from Dr. Beate Sodeik, Hannover Medical School) was propagated as previously described (Diner et al., 2015). Briefly, P0 stocks were generated by electroporating pBAC-HSV-1 into U2OS cells. Working stocks were then generated from the P0 stock by infecting U2OS cells at a low level (approximately 0.001 plaque-forming units per milliliter [PFU]/cell) and virus was collected approximately 3 days later when cells exhibited 100% cytopathic effect. In a similar manner, wild-type HCMV strains AD169 and TB40/E (gifts from Dr. Thomas Shenk) were produced from BAC electroporation into HFs and working stocks were propagated by infecting fibroblasts at a low level. In both cases, cell-associated virus was released by sonication, combined with supernatant virus, then concentrated by ultracentrifugation (20,000 rpm, 2 h, 4°C with SW28 swinging bucket rotor [Beckman Coulter]) over a 10% Ficoll (HSV-1) or 20% sorbitol (HCMV) cushion. Virus stock titers were determined by plaque assay for HSV-1 or tissue culture infectious dose for HCMV and infections were performed at an MOI of 3. For HSV-1 infections, stock virus was diluted into DMEM media containing 2% FBS and added to MRC5 cells. After incubation at 37°C for 1 h, the inoculum was replaced with complete growth medium. HCMV infections were conducted similarly, except stock virus was diluted into complete growth medium. KSHV infections were performed by reactivating iSLK.219 cells with 1 mM NaB (Sigma-Aldrich, B5887) and 1 μg/mL doxycycline (Sigma-Aldrich, D9891), which resulted in 100% reactivation after 72 h.
Animal models of infection
Female 6- to 8-week-old BALB/cAnNTac (Murine Pathogen Free, Taconic Biosciences) were infected with 1 × 105 PFU HSV-1 (strain 17) via the ocular route and the frontal lobes from brains of encephalitic mice were isolated at 8 days post-infection. Mice were group housed in microisolator cages in an MPF facility. All animal care and handling were done in accordance with the U.S. National Institutes of Health Animal Care and Use Guidelines and as approved by the NIAID Animal Care and Use Committee (Protocol LVD40E, T.M.K.).
METHOD DETAILS
HSV-1 titering by plaque assay
Supernatant virus was 10-fold serially diluted into DMEM media +2% FBS and added to 80% confluent monolayers of U2OS cells in a 12-well plate. After 1 h, viral media were replaced with a DMEM solution containing 1% methocel and cells were incubated at 37°C. After 20 h, cells were rinsed with PBS and fixed for 15 min in crystal violet stain (50% methanol [v/v], 1% crystal violet [w/v]). Plaques were counted manually, and virus titers were calculated as plaque-forming units per milliliter based on the dilution factor.
HCMV titering by IE1 staining
After collection of virus supernatant, cellular debris was pelleted by centrifugation at 10,000×g. Collected supernatants were diluted in fresh complete growth media and added directly to a reporter plate of confluent wild-type fibroblasts (MRC5). After 24 h, the reporter cells were washed three times with cold PBS and then fixed with −20°C methanol at −20°C for 15 min. Cells were then washed three times with PBS, blocked in 3% BSA and incubated with IE1 primary antibody (a gift from Dr. Thomas Shenk; 1:100 in 0.3% BSA). After primary antibody incubation, cells were washed three times in PBS +0.2% Tween 20 (PBST) and incubated with secondary antibody Alexa 488 goat anti-mouse IgG (Invitrogen, A-11001; 1:1000) and DAPI (ThermoFisher Scientific, 62248). Finally, cells were washed three times in PBST and imaged on the Operetta imaging system (PerkinElmer) to count IE1-positive cells. The total number of IE1-positive cells across all imaged fields was used to calculate infectious units per milliliter in the original sample.
KSHV titering by supernatant transfer assay
To determine KSHV titers, supernatant transfer assays were performed as previously described (Davis et al., 2015). Briefly, viral supernatants were collected at 72 HPR, passed through an 0.45-μm filter and added to a confluent monolayer of HEK293T cells in a 96-well plate by spinfection (1,000×g for 1 h at room temperature) with 8 μg/mL polybrene (Millipore Sigma, TR-1003-G). After 48 h, cells were resuspended in the growth medium and infected (GFP-positive) cells were measured on a Countess II automated cell counter equipped with a GFP light cube (ThermoFisher Scientific). To ensure reliable measurements, each sample was measured in technical triplicate.
Viral genome isolation and quantification by qPCR
To quantify HSV-1 and HCMV intracellular viral genomes, frozen cell pellets were thawed slowly on ice, resuspended in 500 μL DMEM, and sonicated to release DNA. Cell lysates were added to a DNA resuspension buffer (400 mM NaCl, 10 mM Tris pH 8.0, 10 mM EDTA) and cellular protein was digested by treating with 20 μg proteinase K and 0.2% SDS overnight at 37°C. After overnight digestion, DNA was extracted from the samples using phenol-chloroform extraction followed by ethanol precipitation at −20°C. Precipitated DNA was recovered by centrifugation and resuspended in ultrapure water for a qPCR analysis. HSV-1 and HCMV genome abundances were quantified via qPCR (ViiA7 Real-Time PCR System) using the SYBR green PCR master mix (ThermoFisher Scientific, 4368706) with primers targeting either the HCMV IE1 gene (forward primer: 5′-TCGTTGCAATCCTCGGTCA-3′; reverse primer: 5′-ACAGTCAGCTGAGTCTGGGA-3′) or the HSV-1 UL30 gene (forward primer: 5′-GCGAAAAGACGTTCACCAAG-3′; reverse primer: 5′-GGAGACGGTATCGTCGTAA −3′) and the nuclear β2-microglobulin (B2M) gene (forward primer: 5′-TGCTGTCTCCATGTTTGATGTATCT-3′; reverse primer: 5′-TCTCTGCTCCCCACCTCTAAGT-3′). Viral genome abundances were quantified using the Comparative Ct method using B2M as an internal control (Schmittgen and Livak, 2008).
Cell death assays
Apoptosis-induced cell death after drug or siRNA treatments in MRC5 cells was monitored by terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay using the in situ Cell Death Detection Kit (Roche, 1168479 - fluorescein) as per manufacturer recommendations. Owing to the constitutive expression of GFP in iSLK.219 cells, apoptosis-induced cell death after drug treatment was monitored using the in situ Cell Death Detection Kit (Roche, 12156792910 – TMR red). In both cases, cell death was quantified by fluorescence microscopy on the Operetta imaging system (PerkinElmer), and TUNEL positive cells were identified by fluorescein or TMR red signal in the nucleus.
Small molecule treatments and sample collection
ACV (Cayman Chemical, 14160), CDV (Cayman Chemical, 13113), and CAY10602 (Cayman Chemical, 10009796) were resuspended in DMSO (ACV, CAY10602) or PBS (CDV) to generate 2,000× stocks that were stored at −80°C. At 12 h before virus infection or reactivation, cells were treated with either the small molecule drug or DMSO/PBS control at an equivalent volume. Cell culture concentrations of each drug were as follows: ACV (1 or 3 μM; median inhibition concentration [IC50] = 2–3 μM (Bacon et al., 1996; Brandi et al., 2001; Leary et al., 2002), CDV (1 μM; IC50 = 0.5–1 μM) (Beadle et al., 2002; Scott et al., 2007), and CAY10602 (12.5 μM). Drug cytotoxicity was evaluated by TUNEL assay 48 h after treatment. For infection cycles lasting longer than 24 h, small molecule drugs were re-added to the cell culture medium every 24 h. Upon collection, cells were rinsed with PBS, scraped into a microcentrifuge tube, pelleted by centrifugation, and rinsed again with PBS. After the addition of 2 μL of protease inhibitor cocktail (Sigma, P8340) sample pellets were snap frozen in liquid nitrogen and stored at −80°C until ready for MS analysis.
siRNA-mediated KD of CPS1
KD of CPS1 was carried out by transfection of MRC5 cells with siRNA against CPS1 (Sigma Aldrich, EHU002511). MRC5 cells plated in 6-well dishes were transfected at 60% confluency with 5.5 pM CPS1 siRNA or control siRNA and 9 μL lipofectamine RNAiMAX (ThermoFisher Scientific, 13778150) in 150 μL Opti-MEM Medium. Cell media were replaced with 1.8 mL complete growth media and the siRNA mixture was added dropwise to each well. KD of CPS1 was confirmed by qPCR 48 h after transfection and siRNA cytotoxicity was evaluated by TUNEL assay. To assess the effect of CPS1 KD during HCMV infection, transfected cells were infected with HCMV at an MOI of 3 at 48 h post-transfection. After the infection, virus-containing media were aspirated and replaced with 1.8 mL complete growth media and siRNA mixture was added dropwise, as for the initial transfection. Cells were collected at 24, 48, or 72 HPI for PRM and qPCR analysis. For virus titers, viral supernatants were collected at 120 HPI and titers were determined by IE1 staining.
RNA isolation and quantitative RT-qPCR
To verify siRNA-mediated KD of CPS1, RNA was extracted from transfected cells 48 h post transfection using the Qiagen RNeasy Mini kit (Qiagen, 74106). The Qiagen RNeasy Mini Kit protocol was followed as written. RNA was reverse-transcribed into cDNA using the SuperScript IV First-Strand cDNA Synthesis Reaction Kit (ThermoFisher Scientific, 18091050), following the manufacturer protocol. CPS1 cDNA abundance was quantified by qPCR (ViiA7 Real-Time PCR System) using the SYBR green PCR master mix (ThermoFisher Scientific, 4368706) and the following primers: CPS1 primer pair 1 (forward: 5′-AATGAGGTGGGCTTAAAGCAAG-3′; reverse: 5′-AGTTCCACTCCACAGTTCAGA-3′), CPS1 primer pair 2 (forward: 5′-ACTTCAGTTGAGTCCATTATGGC-3′; reverse: 5′-GGAACGGATCATCACTACTGGGTAG-3′), GAPDH primer pair (forward: 5′-TTCGACAGTCAGCCGCATCTTCTT-3′; reverse: 5′-CAGGCGCCCAATACGACCAAATC-3′). CPS1 transcript abundance in KD relative to control samples was quantified using the comparative Ct method using GAPDH as an internal control (Schmittgen and Livak, 2008).
Selection of target proteins and peptides for PRM
Initial whole proteome data-dependent analysis (DDA) runs were acquired for wild-type infected samples at the latest time point of infection (18 HPI for HSV-1, 120 HPI for HCMV, and 72 HPI for KSHV) to identify as many viral proteins and peptides as possible. Peptides and proteins were identified by searching the DDA data in Proteome Discoverer 2.4 and Sequest HT, then imported into Skyline (MacLean et al., 2010) for manual assessment. These preliminary analyses used the same chromatography conditions as the targeted analyses and provided a foundation of viral peptides that were abundant enough to be detected by shotgun-based methods. However, DDA analyses alone did not allow us to achieve the proteome coverage that we desired due to the low abundance nature of many viral proteins. Therefore, to discover more peptides corresponding with a greater number of proteins, we also performed unscheduled, targeted PRM analyses (monitoring ≤30 peptides/injection) on late infection time point samples. Potential peptides of interest were prioritized for unscheduled runs using the PREGO plugin for Skyline (Searle et al., 2015). Peptides that were reproducibly detected by these unscheduled analyses were then combined with those detected by DDA. Up to four proteotypic peptides for each viral protein detected were selected. In cases where more than four unique peptides were available, peptides were prioritized for selection based first on originating from different regions of the protein and second based on eluting at different points in the chromatogram. Finally, this list of peptides was validated by scheduled PRM across the entire time course of infection to ensure that peptides for a given protein followed the same trend and their abundances and elution times were relatively consistent across multiple injections. While not every viral protein was detected for each virus, proteins representing all of the temporal classes of viral protein expression and all virion components are present in the final targeted method. Our general approach to curating a set of peptides for monitoring infection progression are applicable to the study of other viruses. As such, we have also provided a tutorial on how to complete this process for a virus of interest (see TUTORIAL: Developing a targeted MS assay for a virus of interest section).
Preparation of cell culture samples for PRM analysis
For practical considerations and to demonstrate assay applicability to other peptide preparation methods, samples were prepared for MS analysis by one of two different methods (methanol-chloroform precipitation followed by C18 desalting or via S-Trap on-column digestion). The specific protocol used to prepare a given sample for the analyses performed in this paper can be found in Table S6A.
Methanol-chloroform precipitation followed by C18 desalting
Frozen cell pellets were resuspended in lysis buffer (4% SDS, 50 mM Tris pH 7.5, 100 mM NaCl, 0.5 mM EDTA) and lysed by repeated steps of incubation at 95°C for 3 min followed by sonication in a cup-horn sonicator for 20 pulses. Protein concentration was determined by BCA assay and 50–100 μg protein were then reduced and alkylated at 70°C for 20 min using 25 mM TCEP (ThermoFisher #77720) and 50 mM 2-chloroacetamide (MP Biomedicals #ICN15495580). Protein was then extracted by methanol-chloroform precipitation, resuspended in 25 mM HEPES buffer (pH 8.2), and digested for 16 h at 37°C using a 1:50 ratio of trypsin to protein (w/w). The resulting peptides were then adjusted to 1% trifluoroacetic acid (TFA) and desalted using the StageTip method (Rappsilber et al., 2007) with C18 material (3M #2215). Finally, bound peptides were washed with 0.5% TFA, eluted with 70% acetonitrile (ACN) and 0.5% formic acid (FA), dried via SpeedVac (ThermoFisher Scientific), and resuspended in 1% FA and 1% ACN to a concentration of 0.75 μg/mL for peptide LC-MS/MS analysis.
S-trap on-column digestion
Samples were prepared using S-Trap (Protifi, C02-micro-80) following the manufacturer’s protocol. Briefly, samples were resuspended in lysis buffer (9% SDS, 50 mM Tris pH 7.5, 100 mM NaCl, 0.5 mM EDTA) and lysed by repeated steps of incubation at 95°C for 3 min followed by sonication in a cup-horn sonicator for 20 pulses. Protein concentration was determined by BCA assay and 30 mg of protein was adjusted to a volume of 40 μL and reduced and alkylated at 70°C for 20 min using 25 mM TCEP and 50 mM 2-chloroacetamide. Samples were then acidified to a final concentration of 1.2% aqueous phosphoric acid, mixed with 165 μL of wash buffer solution (90% methanol, 100 mM triethanolamine bicarbonate [TEAB], pH 7.1), and loaded onto the S-trap column. Next, samples were washed five times with 150 μL wash buffer, and a 1-h on-column digestion was performed at 47°C using a 1:25 ratio of trypsin to protein (w/w) in 25 μL of 25 mM TEAB (pH 8). Digested peptides were then eluted with sequential addition of 40 μL of 25 mM TEAB (pH 8), 40 μL of 0.2% FA, and 70 μL of 50% ACN in 0.2% FA. Finally, pooled elutions were dried via SpeedVac and resuspended in 1% FA and 1% ACN to a concentration of 0.75 μg/μL for peptide LC-MS/MS analysis.
Preparation of mouse frontal lobe tissues for PRM
Using a 7-mL Dounce homogenizer, pooled tissues from 3 mice were homogenized in 2 mL of lysis buffer without SDS (1× HALT protease inhibitor cocktail (ThermoFisher Scientific, 78,438) (50 mM Tris pH 7.5, 100 mM NaCl, 0.5 mM EDTA). After homogenization, samples were adjusted to 4.5% SDS by adding 2 mL lysis buffer with 9% SDS and lysed by repeated steps of incubation at 95°C for 3 min followed by sonication in a cup-horn sonicator for 20 pulses. Lysate was then subjected to Polytron homogenization for two cycles of 20 s at a speed of 20,000 rpm, and samples were centrifuged at 100×g to pellet any remaining insoluble particles. Proteins were reduced and alkylated using 25 mM TCEP (ThermoFisher Scientific #77720) and 50 mM 2-chloroacetamide (MP Biomedicals #ICN15495580), extracted by methanol-chloroform precipitation, resuspended in 25 mM HEPES buffer (pH 8.2), and prepared for MS analysis (via an on-column digest with trypsin) using an S-Trap column (Protifi, C02-micro-80) following the manufacturer’s protocol as described above.
Peptide LC-MS/MS analysis of cell culture samples
Samples prepared for PRM analysis were analyzed on a Q Exactive HF mass spectrometer (ThermoFisher Scientific) coupled to an EASYSpray ion source (ThermoFisher Scientific). Peptides were resolved for nLC-MS/MS analysis using a Dionex Ultimate 3000 nanoRSLC (ThermoFisher Scientific) equipped with a 25-cm EASYSpray C18 column (ThermoFisher Scientific, ES902). Peptides (1.5 μg) were separated by reverse phase chromatography with solvents A (0.1% FA) and B (90% ACN, 0.1% FA) at a flow rate of 250 nL/min using a two-phase linear gradient of 2%–22% solvent B for 45 min and 22%–38% solvent B for 15 min and were ionized at 1.7 kV. A single duty cycle consisted of an MS-SIM scan (400–2000 m/z range, 15,000 resolution, 15 ms max injection time [MIT], 3 × 106 automatic gain control [AGC] target) followed by 30 PRM scans (30,000 resolution, 60 ms MIT, 1 × 105 AGC target, 0.8 m/z isolation window, normalized collision energy (NCE) of 27, 125 m/z fixed first mass) and spectrum data were recorded in profile. Acquisition was controlled by scheduled inclusion lists containing no more than 30 concurrent precursors. For KSHV, all peptides were acquired in a single run. For HSV-1 and HCMV, the peptide inclusion list was split in half and two injections per sample were made to obtain sufficient scans across the peak.
Peptide LC-MS/MS analysis of mouse frontal lobe tissues
Samples prepared for PRM analysis were analyzed as above, except peptides were resolved on a 50 cm EASYSpray C18 column (ThermoFisher Scientific, ES903), and a single instrument duty cycle consisted of 8 PRM scans (30,000 resolution, 240 ms MIT, 1 × 105 AGC target, 1.0 m/z isolation window, NCE of 27, 125 m/z fixed first mass). Acquisition was controlled by scheduled inclusion lists containing no more than eight concurrent precursors.
PRM data processing and analysis
Raw files containing PRM spectra were imported into Skyline and peak quality for all peptides monitored was assessed manually and compared to a reference spectral library generated by searching the raw files using Sequest HT and Proteome Discoverer 2.4. Peptides without convincing spectra or spectra with excessive interference were manually discarded. Following quality control, peptide abundance was calculated from the summed area under the curve (total peak area) for the top three most abundant fragment ions per peptide and peptide quantification was exported as a csv file for programmatic analysis in Python. To normalize for differences in input sample, we performed MS1-based normalization as previously described (Federspiel et al., 2019). Briefly, the peptide peak areas were normalized by the average MS1 intensity of the corresponding raw file as reported by RawMeat (Vast Scientific) relative to other files within the same replicate (see Table S6B for normalization factors for all files generated by this study). Following MS1-level normalization, peptides were then mean normalized to their average abundance across all time points and conditions on a per replicate basis. This brings all peptide abundances to a similar scale such that multiple peptides from a single protein can now be compared, and accounts for natural variation in instrument performance when different replicates are run at different times. To further highlight the change in abundance of a given protein across infection time, we also calculated the log 2-fold change of all proteins during wild-type infection (relative to the first time point at which any of its associated peptides become detectable; see Table S6C for an example of how all normalization steps were performed). Our process of log-transforming the peptide abundances ultimately maintains the variability of the original peptide abundance values but centers them around zero at the first time point of detection. Values at subsequent time points then represent the relative induction of that protein compared with the earliest detection time point.
Analysis of PRM peptide conservation across herpesvirus species and strains
Peptide conservation analysis was performed by downloading all herpesvirus-associated complete genomes from the NCBI nucleotide database. Potential peptide sequences were then generated for both strands in all reading frames and compared to each peptide targeted by the PRM assay to determine if a given peptide could be produced from a given genome. For virus strains with more than one reported, complete genome deposited in the database, peptides were considered to be conserved as long as they were computationally detected in at least one of these genomes.
TUTORIAL: Developing a targeted MS assay for a virus of interest
Targeted MS is a proven method for the specific and sensitive detection of proteins in various biological contexts, and can be a powerful tool for monitoring the replication cycles of various viruses—particularly for those with large protein coding capacity that are tedious and/or expensive to monitor by other means (e.g., Western blot, ELISA). Not only is targeted MS a sensitive and reliable method for quantifying the abundance of viral proteins, but once these assays are developed and validated, they can be adapted to address a wide range of biological questions and can be distributed to other researchers. Developing a targeted MS assay for a virus of interest can be a straightforward but sometimes time-consuming task (Figure S1C). The following tutorial details how to create and validate a targeted proteomics assay for a given virus of interest using PRM.
Note
This tutorial represents one of many conceptual approaches to developing a targeted MS assay and does not attempt to exhaustively explain how to optimize all aspects of the sample collection, preparation, and MS quantification procedures.
Software, instrumentation, and materials
(1) Skyline (MacLean et al., 2010), (2) Software for peptide/protein assignment from LC-MS/MS data (Proteome Discoverer, MSFragger, etc.), (3) LC mass spectrometer with targeted MS capabilities, such as a triple quadrupole instrument for multiple reaction monitoring or a quadrupole-high resolution and accurate mass analyzer (i.e., Orbitrap or time-of-flight [ToF] mass analyzer) for PRM (a ThermoFisher QE-HF mass spectrometer was used in our study of herpesviruses), (4) Virus-infected samples (at time points covering the stages of entry, genome replication, assembly, and egress), and (5) Mock-infected (i.e., uninfected) samples (optional).
Setting up a skyline document for the virus of interest
Skyline is a versatile and open-source tool for targeted proteomics analyses. A more in-depth tutorial on how to generate a Skyline document for a targeted MS method can be found online (https://skyline.ms/wiki/home/software/Skyline/page.view?name=tutorial_method_edit). For the purposes of this tutorial, we have briefly outlined this process as it relates to creating targeted assays for viral proteins. An important but easily overlooked step in the initial setup of a Skyline document is the proper configuration of the document for the specific instrumentation that will be used. Depending on the type of instrumentation used (e.g., triple quadrupole, Orbitrap, or ToF), Skyline must be updated to reflect the type of analyzer used under the Full-Scan tab in the Transition settings. Downstream data analysis in Skyline depends on selecting the proper instrumentation settings. Next, Skyline must be configured to generate potential peptides of interest. The foundation of any targeted MS assay is based upon the identification and quantification of proteotypic peptides of your protein of interest, i.e., peptides that are unique and specific to a single protein of interest. The first step in this process is to develop a background proteome, which Skyline will compare against to determine if the peptides of interest are proteotypic of the selected protein for quantitation. As it relates to this tutorial, the background proteome will be a combination of the entire host proteome (e.g., the human proteome for our herpesviruses study) and the viral proteome(s) for the virus(es) of interest. Reference proteome FASTA files can be downloaded from UniProt. After importing a background proteome, the user can select a protease (usually trypsin) and perform an in silico digestion within Skyline to populate the document with all potential peptides that could be produced in the experimental samples, which will be leveraged in the following steps. Importantly, Skyline provides tools to filter the available peptides based on those that are unique to a given protein of interest (e.g., occur in just a single protein in the background proteome) to ensure faithful and accurate quantification. Additionally, the tryptic peptides within the document can be filtered to include only those with lengths that are likely to be detected by MS (e.g., approximately 6–30 amino acids).
Developing an initial peptide library via DDA
An important step in developing a PRM assay is to verify that the proteins of interest can be detected in the specific LC-MS/MS instrumentation that will be used for later PRM analysis. DDA (e.g., shotgun proteomics) methods detect the most abundant peptides across the retention time gradient, which is in contrast to scheduled targeted proteomics approaches that aim to detect a predefined list of peptide precursor ions with a defined m/z value within a given retention time window. As a result, DDA scans are often sufficient to detect high abundance viral proteins but fail to detect those that are of lower abundance. Nonetheless, DDA analyses are a good starting point for identifying viral proteins and peptides that are amenable to MS analysis and, when performed with the same gradient settings as for targeted MS runs, the observed retention times for peptides detected by DDA can be leveraged in subsequent scheduled PRM runs. These analyses should be performed on infected samples collected when viral protein levels are at their highest (usually late in infection). To increase viral proteome coverage, samples can be fractionated to decrease matrix complexity and increase the number of proteins identified in the DDA runs. Following these DDA analyses, the data can be searched in a proteomics software of the user’s choice and the resulting detected peptides and proteins can be imported into Skyline for manual inspection of peptide peaks. Peptide degradation and adsorption on MS vials is a common cause for failed peptide detection, so it can also be helpful to run these DDA analyses on samples as they become older to determine whether the viral peptides that are identified in the DDA runs are stable and reliably detected over an extended period of time. Similar exploratory analyses could be achieved using data-independent acquisitions, but is not discussed in this tutorial.
Predicting and validating MS-amenable peptides using skyline and PREGO
For viral proteins that are not sufficiently detected by DDA (e.g., those not having at least three or four detectable peptides), coverage can be increased by performing unscheduled targeted analyses. Unscheduled targeted runs aim to detect a predefined list of peptide precursor ions with a defined m/z value across the entire LC gradient, rather than within a specific retention time window (as is the case for scheduled PRM). However, depending on the instrumentation platform and PRM scan settings used, only approximately 30 peptides of interest can be monitored in a single unscheduled PRM injection. Moreover, larger proteins may contain upwards of 100 potential tryptic peptides. Thus, considering a hypothetical virus that expresses 40 proteins each with 30 potential peptides, would require 40 injections onto the instrument and significant instrument runtime. For viruses with large proteomes, it therefore becomes increasingly important to be able to prioritize which of these peptides are most likely to be detected by MS analysis. Fortunately, the Skyline plugin PREGO (Searle et al., 2015) can facilitate this process. Briefly, PREGO predicts peptides that are likely to be high responding peptides for targeted MS analyses. By leveraging this plugin, users can simply run the PREGO algorithm on a given set of peptides within a Skyline document, which will return the top-n scoring peptides on a per-protein basis (e.g., the top five peptides for every protein in the document). This set of peptides can then be used to populate inclusion lists for unscheduled PRM injections. Similar predictions can be achieved using another tool called Prosit (Gessulat et al., 2019), which predicts the in silico peptides that will generate fragment ions with high relative intensities. Following unscheduled PRM acquisition, the data can then be searched in proteomics software, imported into Skyline, and further rounds of exploratory unscheduled injections can be performed until the desired coverage is reached or all potential peptides for a given protein have been exhausted.
Validation of peptide targets by PRM
After DDA and unscheduled PRM analyses, detected peptides of interest should be combined into a single Skyline document for validation. Of note, users should ensure that their PRM method contains proteins that span different viral functions, kinetic classes of viral gene expression, and virion components. When possible, each protein should include more peptides than the final method will monitor, to account for peptides that do not pass the validation procedure. For example, if one wants to measure at least three to four peptides per protein, each protein should have five to six peptides at this point in the method development process. The validation of PRM peptides aims to ensure that (1) peptides for a given protein exhibit the same trends across infection time, and (2) a given peptide displays relatively stable retention times (±30-s retention time shifts) across multiple injections of the same sample and across multiple infection time points. To accomplish this task, one should collect samples of infected cells across time points that span all stages of the virus replication cycle (e.g., entry, genome replication, assembly, and egress). Optionally, one can also perform an analysis of uninfected (mock) samples to experimentally validate that the chosen viral peptides are not present in the host proteome. Given that peptides retention times are known from the DDA runs and unscheduled PRM analyses performed above, users can now perform scheduled PRM analyses monitoring up to 30 concurrent precursors (depending on the instrumentation used) across the LC gradient. The precise number of total precursors that can be monitored in a single injection will vary based on the instrumentation, PRM scan settings, and retention time window used. In our herpesvirus PRM assays, we were able to monitor approximately 150 peptides using a 6-min retention time window with the instrument and settings described in our STAR Methods section. After performing these scheduled PRM analyses for samples spanning the infectious cycle, peptides with high variability across the infection time course compared to other peptides from the same protein or peptides with variable retention times should be removed from the method. Further prioritization of peptides for the final method should then be performed based on fragment ion intensity (e.g., to select for higher abundance peptides that are detectable earlier during the infection cycle), reproducibility in detection (e.g., low detection variability across multiple technical replicate injections of the same sample), retention times (e.g., to distribute monitored peptides along the LC gradient, thus optimizing the number precursors that can be monitored in a single injection), and peptide positioning within the protein of interest (e.g., to select for peptides covering a range of domains). It is highly recommended to perform these validations in multiple technical and biological replicates. Additional validation assays can be performed, such as performing this validation process with different LC columns, gradients, and even different mass spectrometers. However, such validations are frequently beyond the scope of standard targeted MS analyses.
Assay application
Following the initial development of a PRM assay, it can then be quickly and easily applied to investigate diverse biological questions in different model systems. For example, consider a situation where one wants to determine the effects of knocking out a host gene on virus replication. The general workflow for applying the developed PRM assay to answer this question would include collecting knockout and control cells across the virus replication cycle. These samples would then be digested with a protease (e.g., trypsin), prepared for MS analysis, and virus peptide/protein levels would be determined by scheduled PRM acquisition. Despite being relatively straightforward, there are several considerations to keep in mind while acquiring PRM data over multiple days, months, or years to ensure the continued reliability of peptide measurements.
PRM method rescheduling
A normal reality of LC-MS/MS platforms is the drifting of peptide elution intervals (e.g., retention times) over time. For example, PRM analyses can take multiple days to perform, and this could result in a small drift in peptide retention times due to changes in column complexity or the use of a new LC column. Thus, the most frequent assay optimization that needs to performed after the initial development of the PRM assay is peptide retention time rescheduling. Although a given PRM assay can always be applied in an unscheduled manner (as mentioned above), scheduled PRM greatly improves the throughput of peptide detection. An efficient way to achieve rescheduling of the peptides of interest with minimal injections is to leverage Skyline’s retention time prediction calculator. Essentially, this calculator can compute the predicted retention time of a given peptide given known elution times for a subset of peptides in the document. In this way, a single optimization injection from a sample likely to contain most viral proteins can be used to reschedule all peptides in the Skyline document for subsequent injections. In our experience, this procedure works quite reliably for rescheduling peptides, and even works well to reschedule for different instruments, columns, and LC gradients.
Data quality assessment and quantification
Following data acquisition, it is essential to ensure that the peaks picked by the Skyline algorithm correspond with the correct peptide. To aid in this process, it is helpful to search the same PRM files imported into the Skyline document with other bioinformatic proteomics software (i.e., Proteome Discoverer, MSFragger). These searches can then be imported into Skyline to build a spectral library, allowing the user to see specific peptide spectrum matches overlaid on top of the chromatograms in Skyline. The generation of such peptide spectral libraries thus helps to demark which co-eluting transitions are associated with correct peptide assignments and which co-eluting peaks are spurious assignments.
QUANTIFICATION AND STATISTICAL ANALYSIS
Programs, software, and statistics
Data processing and analyses were performed using Python 3.7 in conjunction with Pandas, NumPy, SciPy, Biopython, Seaborn, and Matplotlib libraries. Significance was determined by two-tailed Student’s t-test using the Python SciPy library unless otherwise stated. Results of statistical tests are reported in the figure panels. Where applicable: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Specific statistical details (e.g., numbers of technical and biological replicates) are reported in the figure legends. Proteomic database searches were performed in Proteome Discoverer 2.4 using Sequest HT. Raw PRM spectra were analyzed in Skyline and MS1 intensities were extracted from PRM raw files using RawMeat. Figures were constructed in Microsoft PowerPoint.
Supplementary Material
Highlights.
TRUSTED is a set of targeted MS assays for monitoring herpesvirus protein levels
Assays for HSV-1, HCMV, and KSHV span all herpesvirus subfamilies (α, β, and γ)
Valid for screening responses to small molecule treatments and genetic perturbations
Applicable to a range of infection levels, to diverse viral strains, and in vivo
ACKNOWLEDGMENTS
We are grateful for funding provided by NIH NIGMS (GM114141), Stand Up To Cancer Convergence 3.1416, and the Edward Mallinckrodt, Jr. Foundation to I.M.C., NSF Graduate Research Fellowship (DGE-1656466) to M.A.K, NIH Ruth L. Kirschstein F31 fellowship to C.N.B. (1F31AI154796–01A1), the Intramural Research Program of NIAID to T.M.K, and the NIGMS training grant (T32GM007388). We also thank the Molecular Biology Confocal Imaging Facility at Princeton University, a Nikon Center of Excellence, for instrument use and technical advice.
Footnotes
DECLARATION OF INTERESTS
I.M.C. is a shareholder of Evrys Bio (previously Forge Life Science), which has licensed sirtuin-related technology from Princeton University. I.M.C., J.D.F., and M.A.K. have a provisional patent application on a “Method for quantitative monitoring of the progression of infections with herpesviruses.”
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110810.
REFERENCES
- Abbatiello SE, Schilling B, Mani DR, Zimmerman LJ, Hall SC, MacLean B, Albertolle M, Allen S, Burgess M, Cusack MP, et al. (2015). Large-scale interlaboratory study to develop, analytically validate and apply highly multiplexed, quantitative peptide assays to measure cancer-relevant proteins in plasma. Mol. Cell. Proteomics 14, 2357–2374. 10.1074/mcp.m114.047050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacon TH, Howard BA, Spender LC, and Boyd MR (1996). Activity of penciclovir in antiviral assays against herpes simplex virus. J. Antimicrob. Chemother. 37, 303–313. [DOI] [PubMed] [Google Scholar]
- Beadle JR, Hartline C, Aldern KA, Rodriguez N, Harden E, Kern ER, and Hostetler KY (2002). Alkoxyalkyl esters of cidofovir and cyclic cidofovir exhibit multiple-log enhancement of antiviral activity against cytomegalovirus and herpesvirus replication in vitro. Antimicrob. Agents Chemother. 46, 2381–2386. 10.1128/aac.46.8.2381-2386.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell C, Desjardins M, Thibault P, and Radtke K (2013). Proteomics analysis of Herpes Simplex Virus type 1-infected cells reveals dynamic changes of viral protein expression, ubiquitylation, and phosphorylation. J. Proteome Res. 12, 1820–1829. 10.1021/pr301157j. [DOI] [PubMed] [Google Scholar]
- Biron KK (2007). Candidate anti-herpesviral drugs; mechanisms of action and resistance. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis (Cambridge University Press; ), pp. 1219–1250. [PubMed] [Google Scholar]
- Boldogkői Z, Szűcs A, Balázs Z, Sharon D, Snyder M, and Tombaácz D (2018). Transcriptomic study of herpes simplex virus type-1 using full-length sequencing techniques. Sci. Data 5, 180266. 10.1038/sdata.2018.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandi G, Schiavano GF, Balestra E, Tavazzi B, Perno CF, and Magnani M (2001). The potency of acyclovir can be markedly different in different cell types. Life Sci 69, 1285–1290. [DOI] [PubMed] [Google Scholar]
- Bresnahan WA, and Shenk T (2000). A subset of viral transcripts packaged within human cytomegalovirus particles. Science 288, 2373–2376. 10.1126/science.288.5475.2373. [DOI] [PubMed] [Google Scholar]
- Budayeva HG, Rowland EA, and Cristea IM (2016). Intricate roles of mammalian sirtuins in defense against viral pathogens. J. Virol. 90, 5–8. 10.1128/jvi.03220-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers JW, Maguire TG, and Alwine JC (2010). Glutamine metabolism is essential for human cytomegalovirus infection. J. Virol. 84, 1867–1873. 10.1128/jvi.02123-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeran MCJ, Lokensgard JR, and Schleiss MR (2009). Neuropathogenesis of congenital cytomegalovirus infection: disease mechanisms and prospects for intervention. Clin. Microbiol. Rev. 22, 99–126. 10.1128/cmr.00023-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliffe AR, and Knipe DM (2008). Herpes simplex virus ICP0 promotes both histone removal and acetylation on viral DNA during lytic infection. J. Virol. 82, 12030–12038. 10.1128/jvi.01575-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courivaud C, Bamoulid J, Chalopin JM, Gaiffe E, Tiberghien P, Saas P, and Ducloux D (2013). Cytomegalovirus exposure and cardiovascular disease in kidney transplant recipients. J. Infect. Dis. 207, 1569–1575. 10.1093/infdis/jit064. [DOI] [PubMed] [Google Scholar]
- Davis ZH, Verschueren E, Jang GM, Kleffman K, Johnson JR, Park J, VonDollen J, Maher MC, Johnson T, Newton W, et al. (2015). Global mapping of herpesvirus-host protein complexes reveals a transcription strategy for late genes. Mol. Cell 57, 349–360. 10.1016/j.molcel.2014.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diner BA, Lum KK, Javitt A, and Cristea IM (2015). Interactions of the antiviral factor interferon gamma-inducible protein 16 (IFI16) mediate immune signaling and herpes simplex virus-1 immunosuppression. Mol. Cell. Proteomics 14, 2341–2356. 10.1074/mcp.m114.047068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dremel SE, and DeLuca NA (2019). Genome replication affects transcription factor binding mediating the cascade of herpes simplex virus transcription. Proc. Natl. Acad. Sci. U S A 116, 3734–3739. 10.1073/pnas.1818463116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebhardt HA, Root A, Sander C, and Aebersold R (2015). Applications of targeted proteomics in systems biology and translational medicine. Proteomics 15, 3193–3208. 10.1002/pmic.201500004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federspiel JD, Tandon P, Wilczewski CM, Wasson L, Herring LE, Venkatesh SS, Cristea IM, and Conlon FL (2019). Conservation and divergence of protein pathways in the vertebrate heart. PLoS Biol. 17, e3000437. 10.1371/journal.pbio.3000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielding CA, Weekes MP, Nobre LV, Ruckova E, Wilkie GS, Paulo JA, Chang C, Suárez NM, Davies JA, Antrobus R, et al. (2017). Control of immune ligands by members of a cytomegalovirus gene expansion suppresses natural killer cell activation. Elife 6, 22206. 10.7554/elife.22206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabaev I, Williamson JC, Crozier TWM, Schulz TF, and Lehner PJ (2020). Quantitative proteomics analysis of lytic KSHV infection in human endothelial cells reveals targets of viral immune modulation. Cell Rep. 33, 108249. 10.1016/j.celrep.2020.108249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett HM (1975). The effect of arginine deprivation on the cytopathogenic effect and replication of human cytomegalovirus. Arch. Virol. 48, 131–145. 10.1007/bf01318146. [DOI] [PubMed] [Google Scholar]
- Gessulat S, Schmidt T, Zolg DP, Samaras P, Schnatbaum K, Zerweck J, Knaute T, Rechenberger J, Delanghe B, Huhmer A, et al. (2019). Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat. Methods 16, 509–518. 10.1038/s41592-019-0426-7. [DOI] [PubMed] [Google Scholar]
- Grady SL, Purdy JG, Rabinowitz JD, and Shenk T (2013). Argininosuccinate synthetase 1 depletion produces a metabolic state conducive to herpes simplex virus 1 infection. Proc. Natl. Acad. Sci. U S A 110, E5006–E5015. 10.1073/pnas.1321305110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco TM, Diner BA, and Cristea IM (2014). The impact of mass spectrometry–based proteomics on fundamental discoveries in virology. Annu. Rev. Virol. 1, 581–604. 10.1146/annurev-virology031413-085527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruffat H, Marchione R, and Manet E (2016). Herpesvirus late gene expression: a viral-specific pre-initiation complex is key. Front. Microbiol. 7, 869. 10.3389/fmicb.2016.00869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock MH, and Nelson JA (2017). Modulation of the NFκb signalling pathway by human cytomegalovirus. Virology 1, 104. [PMC free article] [PubMed] [Google Scholar]
- Hill A, Jugovic P, York L, Russ G, Bennink J, Yewdell J, Ploegh H, and Johnson D (1995). Herpes simplex virus turns off the TAP to evade host immunity. Nature 375, 411–415. 10.1038/375411a0. [DOI] [PubMed] [Google Scholar]
- Inoue N, Mar EC, Dollard SC, Pau CP, Zheng Q, and Pellett PE (2000). New immunofluorescence assays for detection of Human herpesvirus 8- specific antibodies. Clin. Diagn. Lab. Immunol. 7, 427–435. 10.1128/cdli.7.3.427-435.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean Beltran PM, Mathias RA, and Cristea IM (2016). A portrait of the human organelle proteome in space and time during cytomegalovirus infection. Cell Syst. 3, 361–373.e6. 10.1016/j.cels.2016.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kati S, Hage E, Mynarek M, Ganzenmueller T, Indenbirken D, Grundhoff A, and Schulz TF (2015). Generation of high-titre virus stocks using BrK.219, a B-cell line infected stably with recombinant Kaposi’s sarcoma-associated herpesvirus. J. Virol. Methods 217, 79–86. 10.1016/j.jviromet.2015.02.022. [DOI] [PubMed] [Google Scholar]
- Kauppinen A, Suuronen T, Ojala J, Kaarniranta K, and Salminen A (2013). Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell. Signal. 25, 1939–1948. 10.1016/j.cellsig.2013.06.007. [DOI] [PubMed] [Google Scholar]
- Keshet R, Szlosarek P, Carracedo A, and Erez A (2018). Rewiring urea cycle metabolism in cancer to support anabolism. Nat. Rev. Cancer 18, 634–645. 10.1038/s41568-018-0054-z. [DOI] [PubMed] [Google Scholar]
- Kim J, Hu Z, Cai L, Li K, Choi E, Faubert B, Bezwada D, Rodriguez-Canales J, Villalobos P, Lin YF, et al. (2017). CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature 546, 168–172. 10.1038/nature22359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyuncu E, Budayeva HG, Miteva YV, Ricci DP, Silhavy TJ, Shenk T, and Cristea IM (2014). Sirtuins are evolutionarily conserved viral restriction factors. MBio 5, e02249–14. 10.1128/mbio.02249-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulej K, Avgousti DC, Sidoli S, Herrmann C, Della Fera AN, Kim ET, Garcia BA, and Weitzman MD (2017). Time-resolved global and chromatin proteomics during herpes simplex virus type 1 (HSV-1) infection. Mol. Cell. Proteomics 16, S92–S107. 10.1074/mcp.m116.065987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leary JJ, Wittrock R, Sarisky RT, Weinberg A, and Levin MJ (2002). Susceptibilities of herpes simplex viruses to penciclovir and acyclovir in eight cell lines. Antimicrob. Agents Chemother. 46, 762–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, He M, Zhou F, Ye F, and Gao S-J (2014). Activation of Kaposi’s sarcoma-associated herpesvirus (KSHV) by inhibitors of class III histone deacetylases: identification of sirtuin 1 as a regulator of the KSHV life cycle. J. Virol. 88, 6355–6367. 10.1128/jvi.00219-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Zhou J, Wiedmer A, Madden K, Yuan Y, and Lieberman PM (2003). Chromatin remodeling of the Kaposi’s sarcoma-associated herpesvirus ORF50 promoter correlates with reactivation from latency. J. Virol. 77, 11425–11435. 10.1128/jvi.77.21.11425-11435.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum KK, Song B, Federspiel JD, Diner BA, Howard T, and Cristea IM (2018). Interactome and proteome dynamics uncover immune modulatory associations of the pathogen sensing factor cGAS. Cell Syst. 7, 627–642.e6. 10.1016/j.cels.2018.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, and MacCoss MJ (2010). Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968. 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesri EA, Cesarman E, and Boshoff C (2010). Kaposi’s sarcoma and its associated herpesvirus. Nat. Rev. Cancer 10, 707–719. 10.1038/nrc2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michan S, and Sinclair D (2007). Sirtuins in mammals: insights into their biological function. Biochem. J. 404, 1–13. 10.1042/bj20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myoung J, and Ganem D (2011). Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: maintenance of tight latency with efficient reactivation upon induction. J. Virol. Methods 174, 12–21. 10.1016/j.jviromet.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MK, Cho H, Roh SW, Kim SJ, and Myoung J (2019). Cell type-specific interferon-γ-mediated antagonism of KSHV lytic replication. Sci. Rep. 9, 2372. 10.1038/s41598-019-38870-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe N, Legendre M, Doutre G, Couteé Y, Poirot O, Lescot M, Arslan D, Seltzer V, Bertaux L, Bruley C, et al. (2013). Pandoraviruses: amoeba viruses with genomes up to 2.5 Mb reaching that of parasitic eukaryotes. Science 341, 281–286. 10.1126/science.1239181. [DOI] [PubMed] [Google Scholar]
- Puchtler E, and Stamminger T (1991). An inducible promoter mediates abundant expression from the immediate-early 2 gene region of human cytomegalovirus at late times after infection. J. Virol. 65, 6301–6306. 10.1128/jvi.65.11.6301-6306.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, and Ishihama Y (2007). Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906. 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Sánchez I, and Munger J (2019). Meal for two: human cytomegalovirus-induced activation of cellular metabolism. Viruses 11, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Sánchez I, Schafer XL, Monaghan M, and Munger J (2019). The Human Cytomegalovirus U L 38 protein drives mTOR-independent metabolic flux reprogramming by inhibiting TSC2. PLoS Pathog. 15, e1007569. 10.1371/journal.ppat.1007569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez MC, Dybas JM, Hughes J, Weitzman MD, and Boutell C (2020). The HSV-1 ubiquitin ligase ICP0: modifying the cellular proteome to promote infection. Virus Res. 285, 198015. 10.1016/j.virusres.2020.198015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, and Livak KJ (2008). Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 3, 1101–1108. 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Scott GM, Weinberg A, Rawlinson WD, and Chou S (2007). Multidrug resistance conferred by novel DNA polymerase mutations in human cytomegalovirus isolates. Antimicrob. Agents Chemother. 51, 89–94. 10.1128/aac.00633-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searle BC, Egertson JD, Bollinger JG, Stergachis AB, and MacCoss MJ (2015). Using data independent acquisition (DIA) to model high-responding peptides for targeted proteomics experiments. Mol. Cell. Proteomics 14, 2331–2340. 10.1074/mcp.m115.051300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng X, and Cristea IM (2021). The antiviral sirtuin 3 bridges protein acetylation to mitochondrial integrity and metabolism during human cytomegalovirus infection. PLoS Pathog. 17, e1009506. 10.1371/journal.ppat.1009506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenk T, and Alwine JC (2014). Human cytomegalovirus: coordinating cellular stress, signaling, and metabolic pathways. Annu. Rev. Virol. 1, 355–374. 10.1146/annurev-virology-031413-085425. [DOI] [PubMed] [Google Scholar]
- Soh TK, Davies CTR, Muenzner J, Hunter LM, Barrow HG, Connor V, Bouton CR, Smith C, Emmott E, Antrobus R, et al. (2020). Temporal proteomic analysis of herpes simplex virus 1 infection reveals cell-surface remodeling via pUL56-mediated GOPC degradation. Cell Rep. 33, 108235. 10.1016/j.celrep.2020.108235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spall VE, Shanks M, and Lomonossoff GP (1997). Polyprotein processing as a strategy for gene expression in RNA viruses. Semin. Virol. 8, 15–23. 10.1006/smvy.1997.0102. [DOI] [Google Scholar]
- Stern-Ginossar N, Weisburd B, Michalski A, Le VTK, Hein MY, Huang SX, Ma M, Shen B, Qian SB, Hengel H, et al. (2012). Decoding human cytomegalovirus. Science 338, 1088–1093. 10.1126/science.1227919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vastag L, Koyuncu E, Grady SL, Shenk TE, and Rabinowitz JD (2011). Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog. 7, e1002124. 10.1371/journal.ppat.1002124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C, and Marcotte EM (2012). Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 13, 227–232. 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weekes MP, Tomasec P, Huttlin EL, Fielding CA, Nusinow D, Stanton RJ, Wang ECY, Aicheler R, Murrell I, Wilkinson GWG, et al. (2014). Quantitative temporal viromics: an approach to investigate host-pathogen interaction. Cell 157, 1460–1472. 10.1016/j.cell.2014.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendler PA, Blanding JH, and Tremblay GC (1983). Interaction between the urea cycle and the orotate pathway: studies with isolated hepatocytes. Arch. Biochem. Biophys. 224, 36–48. 10.1016/0003-9861(83)90188-1. [DOI] [PubMed] [Google Scholar]
- Whitley RJ, and Roizman B (2001). Herpes simplex virus infections. Lancet 357, 1513–1518. 10.1016/s0140-6736(00)04638-9. [DOI] [PubMed] [Google Scholar]
- Zhang B, Whiteaker JR, Hoofnagle AN, Baird GS, Rodland KD, and Paulovich AG (2019). Clinical potential of mass spectrometry-based proteogenomics. Nat. Rev. Clin. Oncol. 16, 256–268. 10.1038/s41571-018-0135-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Hladik F, Woodward A, Klock A, Peng T, Johnston C, Remington M, Magaret A, Koelle DM, Wald A, and Corey L (2009). Persistence of HIV-1 receptor-positive cells after HSV-2 reactivation is a potential mechanism for increased HIV-1 acquisition. Nat. Med. 15, 886–892. 10.1038/nm.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Skyline data analysis files and raw MS data have been deposited to PanoramaWeb at https://panoramaweb.org/HerpesvirusPRM.url (associated with the identifier ProteomeXchange: PXD025879) and are publicly available as of the date of publication. Accession numbers are listed in the Key resources table. Processed data are available in Tables S2, S3, S4, and S5. Code for data processing, normalization, and plotting has been deposited on GitHub and is publicly available as of the date of publication. The DOI is listed in the Key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| anti-IE1, Mouse polyclonal | Gift from Dr. Thomas Shenk | N/A |
| Goat anti-Mouse Alexa Fluor 488 conjugate | Invitrogen | Cat# A-11001; RRID:AB_2534069 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| HCMV strain AD169 | Gift from Dr. Thomas Shenk | N/A |
| HCMV strain TB40/E | Gift from Dr. Thomas Shenk | N/A |
| HSV-1 strain 17+ | Gift from Dr. Beate Sodeik | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Acyclovir | Cayman Chemical | Cat# 14160 |
| Cidofovir | Cayman Chemical | Cat# 13113 |
| CAY10602 | Cayman Chemical | Cat# 10009796 |
| DAPI | ThermoFisher Scientific | Cat# 62248 |
| HALT protease and phosphatase inhibitor | ThermoFisher Scientific | Cat# 78438 |
| TCEP | ThermoFisher Scientific | Cat# 77720 |
| Hygromycin | ThermoFisher Scientific | Cat# 10687010 |
| Sodium butyrate | Sigma-Aldrich | Cat# B5887 |
| Doxycycline | Sigma-Aldrich | Cat# D9891 |
| Polybrene | Millipore Sigma | Cat# TR-1003-G |
| SYBR green PCR master mix | ThermoFisher Scientific | Cat# 4368706 |
| Lipofectamine RNAiMAX | ThermoFisher Scientific | Cat# 13778150 |
|
| ||
| Critical commercial assays | ||
|
| ||
| BCA Assay | ThermoFisher Scientific | Cat# 23225 |
| in situ Cell Death Detection Kit - Fluorescein | Roche | Cat# 1168479 |
| in situ Cell Death Detection Kit - TMR red | Roche | Cat# 12156792910 |
| SuperScript IV First-Strand cDNA Synthesis Reaction Kit | ThermoFisher Scientific | Cat# 18091050 |
|
| ||
| Deposited data | ||
|
| ||
| PRM MS/MS data (Panorama) | This study | https://panoramaweb.org/HerpesvirusPRM.url |
| PRM MS/MS data (RAW files) | This study | ProteomeXchange (PXD025879) |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| MRC5 | ATCC | Cat# CCL-171; RRID:CVCL_0440 |
| HEK293T | ATCC | Cat# CRL-3216; RRID:CVCL_0063 |
| U2OS | ATCC | Cat# HTB-96; RRID:CVCL_0042 |
| iSLK.219 | Gift from Dr. Britt Glaunsinger | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: BALB/cAnNTac | Taconic Biosciences | Cat# BALB-F |
|
| ||
| Oligonucleotides | ||
|
| ||
| CPS1 siRNA | ThermoFisher Scientific | Cat# EHU002511 |
| For qPCR primers, see Table S6 | N/A | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Proteome Discoverer v2.4 | ThermoFisher Scientific | Cat# OPTON-31040 |
| Skyline | (MacLean et al., 2010) | https://skyline.ms/ |
| RawMeat | Vast Scientific | https://vastscientific.com/rawmeat |
| Custom Python Code | This study | https://doi.org/10.5281/zenodo.6468269 |