

Abstract
Ependymal neoplasms occur at all ages and encompass multiple tumor types and subtypes that develop in the supratentorial compartment, the posterior fossa, or the spinal cord. Clinically, ependymomas represent a very heterogeneous group of tumors from rather benign subependymomas to very aggressive and often deadly childhood ependymomas of the posterior fossa. Newly identified biological markers and classification schemes, e. g. based on global DNA methylation profiling, have led to the definition of 10 types of ependymal tumors and an improved prediction of patients’ outcome by applying the new classification system. While the exact genetic basis for several ependymoma types still remains unclear, the knowledge about ependymoma driving events has significantly increased within the last decade and contributed to a classification based on molecular characteristics and localization rather than histological features alone. Convincing evidence is now pointing towards gene fusions involving ZFTA or YAP1 causing the development of supratentorial ependymomas. Also, H3, EZHIP, or TERT mutations have been detected in a fraction of infratentorial ependymal tumors. Finally, MYCN amplifications have recently been identified in spinal ependymomas, in addition to the previously known mutations in NF2. This review summarizes how recent findings regarding biology, molecular tumor typing, and clinical outcome have impacted the classification of ependymomas as suggested by the updated 2021 WHO CNS tumor classification system. We focus on changes compared to the previous classification of 2016 and discuss how a formal grading could evolve in the future and guide clinicians to treat ependymoma patients.
Keywords: classification, DNA methylation, ependymoma, grading, histology, molecular pathology
Recent findings regarding biology, molecular tumor typing, and clinical outcome have significantly impacted the classification of ependymomas, which is now reflected by the updated 2021 WHO CNS tumor classification system. This review focuses on changes compared to the previous classification of 2016 and discusses how a formal grading could evolve in the future and guide clinicians to treat ependymoma patients.

1. INTRODUCTION
The current classification of ependymoma has significantly changed since the release of the past WHO classification of CNS tumors in 2016. [1, 2] First and foremost, these changes include the definition of ependymal tumors. Some tumors that, based on their histological features, may have previously been categorized into other brain tumor entities (e. g. glioblastoma, astroblastoma, or embryonal tumors) are now counted among the family of ependymomas due to the presence of molecular features that have been assured to be typical for ependymoma. On the other hand, tumors with molecular characteristics suggesting the affiliation to a distinct (mostly novel) WHO tumor entity will not be defined as ependymal tumors anymore, even though they show the morphological appearance of ependymoma. A second, major improvement presented by the 2021 WHO Classification is the taxonomy within the tumor family of ependymoma. Whereas the previous edition primarily defined ependymoma subtypes based on clinico‐pathological characteristics (with the exception of RELA‐fusion positive ependymoma), the current update mainly comprises molecular subtypes instead. More precisely, the types of subependymomas (SE), myxopapillary ependymomas (MPE), and RELA‐fusion positive (now: ZFTA‐fusion positive) ependymomas have been maintained in the 2021 classification, although some changes have been applied. The previously used terms of ependymoma and anaplastic ependymoma are not used to define an entity anymore. Instead, PFA and PFB ependymoma in the posterior fossa, YAP1‐fusion positive ependymoma in the cerebrum, and spinal ependymoma as well as spinal ependymoma, MYCN‐amplified, in the spinal cord have been newly defined. This not only provides an objective molecular basis for the diagnosis and classification of ependymomas, but is also intended to better predict the clinical outcome of the patients. Notably, first studies on tumor relapse samples indicate that this molecular classification might be more stable in the course of the disease than histology alone [3, 4]. Histopathological variants that had been separately described in the 2016 version are no longer listed as subtypes of ependymoma, after it had been recognized that papillary, tanycytic, or clear cell morphology has no clinical value by itself. Rather, tumors with the described pattern had been shown to frequently belong to other tumor families after thorough molecular work‐up [5]. A final, but important change in the 2021 update of ependymoma classification is the grading system, which was still crucial in the 2016 version, but is less prominent or not defined for many ependymoma types in the 2021 edition. In this review, we intend to summarize and discuss the changes in the classification of ependymoma, highlight clinical implications, and outline future directions.
2. NEW DEFINITION OF EPENDYMOMA TYPES
The new WHO classification defines eight specific types of ependymoma and two additional types that comprise tumors with specific locations, but that cannot be assigned to another, molecularly more specific type. Together, ten different ICD‐O diagnoses now exist to classify ependymal tumors (Figure 1).
FIGURE 1.

Overview of key characteristics and diagnostic criteria of the distinct ependymoma tumor types as proposed by the 2021 WHO Classification of CNS tumors. Range of age (in years) at onset of disease is indicated in black, the median age of onset is highlighted with a red triangle. PFS and OS are rated on a scale of green (very low progression rate and good OS), orange (intermediate PFS and OS) to red (high progression rate and dismal survival prognosis). For SE, we highlighted that tumors with supratentorial or spinal location have a low progression rate. In contrast, SE of the PF have a higher tendency to progress. Typical molecular features described to date are indicated for each tumor type. Obligatory criteria for the diagnosis of all ependymoma types are morphological and immunohistological features of ependymoma. Additional obligatory criteria are listed for each tumor type. Also, the 2021 WHO Classification provides desirable criteria for the diagnosis of each tumor type that can support the diagnosis. CNP, copy number profile; MPE, myxopapillary ependymoma; NEC, not elsewhere classified; NOS, not otherwise specified; OS, overall survival; PF, posterior fossa ependymoma; PF‐A, group A posterior fossa ependymoma; PF‐B, group B posterior fossa ependymoma; PFS, progression free survival; SE, subependymoma; SP‐EPN, spinal ependymoma; SP‐MYCN, MYCN‐amplified spinal ependymoma; ST, supratentorial ependymoma; ST‐YAP1, YAP1‐fusion positive ependymoma; ST‐ZFTA, ZFTA‐fusion positive ependymoma
Prerequisite for the diagnosis of all ependymoma is the presence of typical morphological and immunohistopathological features of ependymal tumors. Localization in one of the three neuroanatomical compartments, specific molecular and, in some cases, immunohistochemical criteria define the different tumor types. Exceptions regarding localization are subependymoma and myxopapillary ependymoma, where no specific anatomical localization is required as an obligatory criterion. Subependymomas can occur in all three anatomical compartments. The vast majority of myxopapillary ependymomas occurs in lower parts of the spinal cord, but very rare cases have been described to occur cranially or even outside of the CNS, although most of them have not been confirmed on a molecular level (Figure 2) [6, 7]. Also, DNA methylation profiling has become an integral part of classifying the tumor type. Confirmation of the tumor type by a match to the respective reference group is a desirable criterion in almost all tumor types and a mandatory criterion in PF‐B. In this section, we briefly summarize each ependymoma type as proposed by the WHO classification 2021. Key features and diagnostic criteria are depicted in Figure 1.
FIGURE 2.
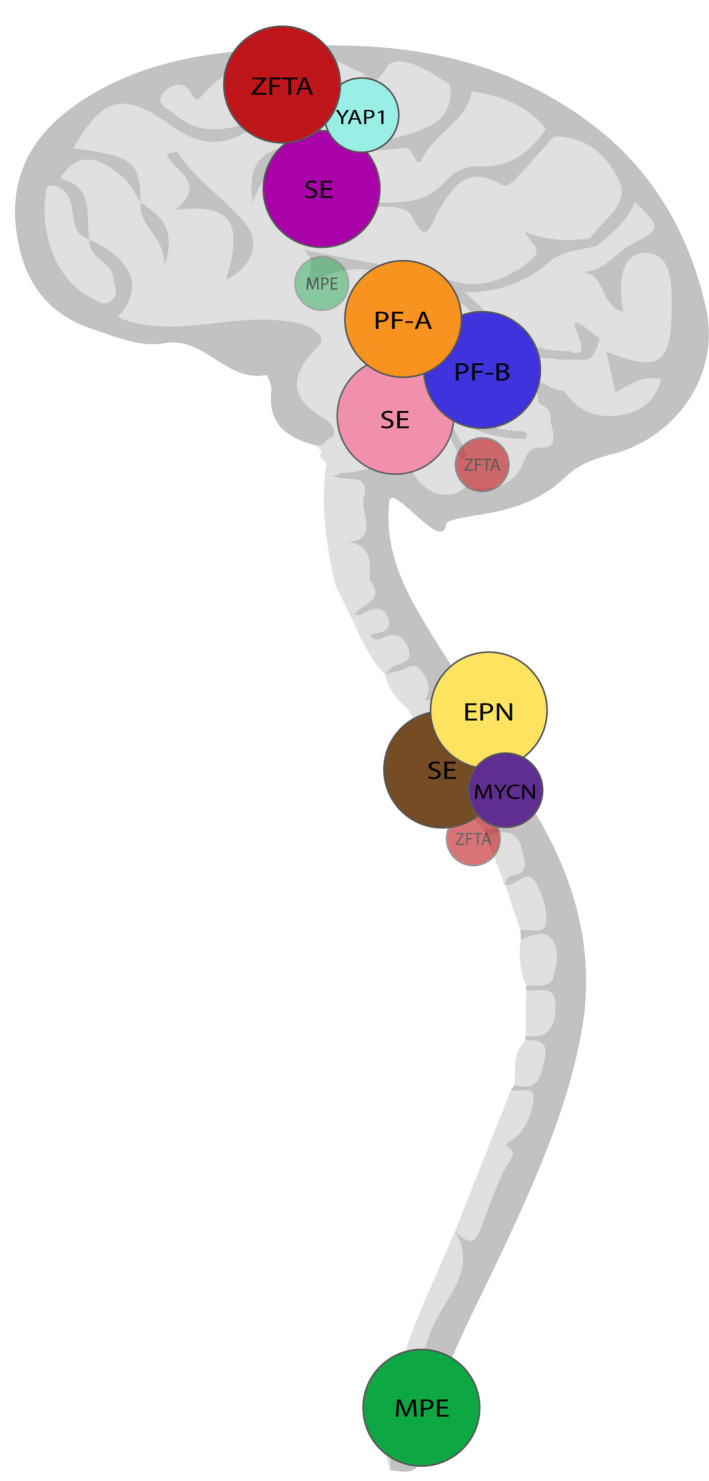
Typical and atypical localizations of the distinct ependymoma types. In the 2021 WHO Classification, anatomical localization is an essential or desirable criterion for all ependymoma types, with the exception of SE which can occur in all three compartments – supratentorial, infratentorial and spinal. ZFTA‐fusion positive ependymomas and YAP1‐fusion positive ependymomas are normally located supratentorially, whereas Group A and Group B posterior fossa ependymoma are found infratentorially. The cervical and thoracic spinal cord is the main localization of spinal ependymoma, spinal subependymoma, and MYCN‐amplified spinal ependymoma, whereas myxopapillary ependymomas predominantly arise in the most caudal part of the spinal cord. Rare cases of infratentorial and spinal ZFTA‐fusion positive ependymoma have been reported. In addition, the localization of myxopapillary ependymoma is not limited to the spinal cord since primary intracranial cases of ependymoma type have been described. EPN, (spinal) ependymoma; PF‐A, group A posterior fossa ependymoma; PF‐B, group B posterior fossa ependymoma; MYCN, MYCN‐amplified spinal ependymoma; YAP1, YAP1‐fusion positive ependymomas; ZFTA, ZFTA‐fusion positive ependymomas
2.1. Supratentorial ependymoma
Supratentorial ependymomas (ST‐EPN) now comprise two molecularly defined tumor types: ST‐ZFTA and ST‐YAP1. If no pathogenic gene fusion of ZFTA (formerly known as C11orf95) or YAP1 can be detected, the diagnosis supratentorial ependymoma NEC (not elsewhere classified) should be used. If molecular diagnosis was not feasible or successful, the tumor classifies as supratentorial ependymoma NOS (not otherwise specified). Subependymoma (SE) also occur supratentorially, but the current classification does not provide separate diagnoses for subependymoma of specific locations. Instead, subependymoma of all three anatomical departments are subsumed as the tumor type of subependymoma. This will be discussed separately below.
Up to now, the majority of ST‐EPN was classified as ST‐RELA ependymoma with gene fusions being present between ZFTA and RELA. In the classification of 2021, the newly defined ependymoma type ZFTA fusion‐positive (ST‐ZFTA) replaces the former ST‐RELA tumor type. This pays tribute to the fact that fusions of the ZFTA gene were shown to not only involve RELA, but also other fusion partners, such as MAML2/3, NCOA1/2, MN1, or CTNNA2 [8, 9, 10]. For these non‐ZFTA‐RELA‐fused ST‐EPN, DNA methylation profiling confirmed epigenetic proximity to the methylation class ST‐EPN‐RELA. However, looking at diagnostically ambiguous ST‐EPN without YAP1 or ZFTA‐RELA fusions and/or a mismatch between methylation class and histological diagnosis, Zheng et al. recently described that these tumors formed 4 discrete satellite clusters adjacent to the established methylation class of ST‐RELA [10]. Tumors in these 4 satellite clusters exhibit a broad spectrum of histological characteristics, some of them appearing as poorly differentiated tumors with features of sarcoma, diffuse high‐grade glioma, CNS embryonal tumors, or other primitive tumors [9, 10]. Notably, first retrospective studies imply that progression‐free survival (PFS) in non‐ZFTA‐RELA‐fused EPN might be worse than in ZFTA‐RELA fused ependymoma [9].
Mandatory criteria for the diagnosis of ST‐ZFTA are now supratentorial localization and evidence of a ZFTA fusion. Of note, rare cases of ZFTA‐fusion positive ependymoma with infratentorial or spinal localization have also been described (Figure 2) [3, 11]. To support the diagnosis further, alignment with the methylation class ST‐ZFTA and immunoreactivity for nuclear p65, the protein encoded by RELA, or LICAM, a characteristic histopathological marker for ST‐RELA, are desirable criteria [12]. ST‐ZFTA tumors frequently display dramatic copy number changes, reminiscent of chromothripsis, mostly involving chromosome 11. Additional recurrent alterations other than focal CDKN2A/B deletions have not yet been identified [3]. In summary, the new nomenclature emphasizes that fusions involving the ZFTA gene with or without the RELA gene occur in tumors with the same histomolecular characteristics. However, special attention should be paid to the fusion variant present in each individual case with respect to the divergent histomorphology and potentially altered clinical outcome. In mouse models, potential therapeutic targets shared by ZFTA fusion‐positive tumors, such as GLI2, were recently identified and might help to advance the translation of the expanding molecular knowledge into novel treatment approaches [10].
The second new supratentorial tumor type is YAP1 fusion‐positive ependymoma (ST‐YAP1). Such tumors typically harbor YAP1‐MAMLD1 fusions, but YAP1‐FAM118B fusions have also been described [13, 14]. Most ST‐YAP1 share similar histopathological features including areas of high cellularity, perivascular pseudo‐rosettes, and abundant cells with dot‐like cytoplasmic expression of epithelial membrane antigen [14]. Obligatory criteria for the diagnosis of ST‐YAP1 are supratentorial location and presence of a YAP1 fusion. In addition, confirmed matching to the methylation class of ST‐YAP1, lack of immunoreactivity for p65 and LICAM, and presence of PAS‐positive eosinophilic granular bodies can support the diagnosis.
Fusions involving ZFTA or YAP1 can be detected by molecular testing including several sequencing strategies or interphase FISH. Also, RT‐PCR can detect the most frequent fusions. Finally, methylation‐based classification complements these diagnostic assays.
Clinical characteristics of ZFTA‐ and YAP1‐ altered EPN differ regarding age at onset and prognosis: Both tumor types predominantly affect infants and small children, but also occur in adult patients. However, the median age at onset of disease is 8 years for ZFTA‐RELA‐fused and thereby considerably older than for YAP1‐fused EPN with a median age of 1.4 years [3, 9].
Available data suggest that the clinical prognosis for ST‐ZFTA is poor. A 5‐year PFS of 29% and a 5‐year overall survival (OS) of 75% were previously reported for ST‐RELA, and the addition of non‐ZFTA‐RELA‐fused tumors might contribute to an even worse outcome [3, 9]. In contrast, ST‐YAP1 tumors show satisfying OS despite occurrence of progression in a fraction of tumors (5‐year PFS 66%) [3].
2.2. Posterior fossa ependymoma
Ependymoma occurring in the posterior fossa now comprise the two molecularly defined tumor types PF‐A and PF‐B as well as the diagnosis of PF NEC/NOS, similar to the ST NEC/NOS described above. Subependymomas also occur in the posterior fossa, but will be discussed separately below. This classification reflects the advances in transcriptional and epigenetic subgrouping that emerged and gained importance in the last decade: In 2011, Witt et al. compared the transcriptome of grade 2 and 3 ependymoma and identified two robust, molecularly and clinically distinct subgroups of posterior fossa ependymoma, which were then called PF‐A and PF‐B EPN [15]. Following investigations in larger cohorts confirmed that PF‐A and PF‐B EPN form distinct molecular subgroups supported by hierarchical clustering of DNA methylation data [3, 16].
Obligatory criteria for the diagnosis PF‐A are localization in the posterior fossa and evidence of global reduction of K27me3 in tumor cell nuclei. If a global H3K27me3 reduction cannot be detected, a DNA methylation profile aligned with PF‐A can still secure this diagnosis according to the current WHO classification. Hypermethylation of CpG islands and global DNA hypomethylation are characteristic of PF‐A. Global H3K27me3 reduction has proven to be a reliable biomarker for PF‐A and can be conveniently assessed by immunohistochemistry. A cutoff value of 80% positively stained tumor cell nuclei has been recommended in the new WHO classification, above which PF‐B ependymoma is the more likely diagnosis [17, 18], although our personal experience is that PF‐A usually display a complete loss. H3K27me3 reduction is strongly associated with EZHIP (CXorf67) overexpression. EZHIP overexpression occurs at high levels in PF‐A, but not in other molecular groups of ependymoma [19, 20]. Previous investigations suggest that EZHIP binds to Polycomb Repressive Complex 2 (PRC2) and leads to the reduction of H3K27me3 levels [19, 21]. EZHIP (CXorf67) mutations have been described to be present in 9.4% and H3K27 M mutations in 4.2% of PF‐A with the two mutations occurring mutually exclusive [19].
For PF‐A, the most frequently observed copy number aberration is gain of 1q (60 of 240; 25%), which was also seen in PF‐B (9 of 51; 18%), and ST‐RELA tumors (21 of 88; 24%) [3]. Gain of 1q has been shown to be an independent marker of a poor outcome within PF‐A [22].
Recently, an ultra‐high‐risk subset of PF‐A was identified, harboring chromosome 6q loss. Approximately 9% of PFA tumors harbored chromosome 6q loss, which was associated with a very poor prognosis independent of 1q status and which was highly predictive of a low progression free‐ and overall survival [22]. Pajtler et al. further identified two molecular PF‐A subgroups and nine molecular subtypes of PF‐A. However, the clinical implications of these subgroups and subtypes are yet to be confirmed and are not incorporated in the current WHO classification [19].
PF‐A typically occur in young children with a median age of 3 years and are slightly more prevalent in males [3]. PF‐A have a dismal outcome with a 10‐year OS of 56% and a PFS of 24% [3].
On the contrary, PF‐B occur most commonly in adults and adolescents, have a median age of onset of 30 years, and are slightly more prevalent in females. PF‐B show a comparably good outcome with 10‐year PFS of 56% and OS 88% [3]. Obligatory criteria for the diagnosis of PF‐B are localization in the posterior fossa and a methylation profile aligned with the methylation class PF‐B. With only few exceptions, PF‐B show retained H3K27me3, which can be easily assessed by immunohistochemistry [23, 24]. PF‐B exhibit a variety of chromosomal aberrations with the most frequent being monosomy 6 (61.3%), 22q loss (48.1%), and trisomy 18 (51.9%), but also monosomy 10 (38.7%), monosomy 17 (33.5%), trisomy 5 (31.1%), trisomy 8 (23.5%), and enrichment of 1q gain (12%) [25]. Cavalli et al. distinguished five molecular subtypes of PF‐B by spectral clustering and t‐SNE analysis of genome‐wide methylation data. The subtypes PF‐B 1–5 showed distinct demographics, copy‐number alterations, and gene expression profile, suggesting significant biological heterogeneity within the group of PF‐B [25]. Again, these subtypes have not yet been incorporated in the 2021 WHO update.
2.3. Spinal cord ependymoma
Ependymal tumors of the spinal cord comprise four distinct tumor types: spinal ependymoma (SP‐EPN), spinal ependymoma with MYCN amplification (SP‐MYCN), myxopapillary ependymoma (MPE), and subependymoma (SE). An overarching group of spinal ependymomas similar to ST and PF NEC/NOS (for example ‘spinal ependymoma NEC/NOS’) is not designated in the current WHO classification. The tumor types SP‐EPN and SP‐MYCN require spinal localization as a mandatory diagnostic criterion, whereas MPE and SE may occur in other anatomical compartments, too. While SP‐MYCN, which was only recently identified as a distinct molecular type, shows a rather aggressive behavior, the remaining tumors with spinal location show more benign characteristics. All spinal ependymoma types predominantly occur in adult patients.
SP‐EPN are defined by their spinal localization and the absence of morphological features of MPE or SE. SP‐EPN occur predominantly in the cervical spine followed by localization in the thoracic spine and lumbar spine [26] (Figure 2). Median age of diagnosis is 41 years with a range between 11–59 years [3]. Most SP‐EPN harbor chromosomal losses on chromosome 22q, where the NF2 gene is located. Germline mutations of NF2 cause the tumor predisposition syndrome Neurofibromatosis type 2 (NF2). While little information is available about the frequency of SP‐EPN patients harboring NF2 (germline) mutations, 33%–53% of NF2 patients show evidence of spinal ependymomas in imaging [27, 28]. So far, it remains unclear, if and how NF2‐associated SP‐EPN differ from NF2 wild type cases apart from the NF2 mutation per se. Another open question is, which other driver mutations might play a role in the development of sporadic non‐NF2‐mutated SP‐EPN. Even though recurrences and progression of SP‐EPN can occur, overall survival is excellent [3]. Still, patients with NF2 syndrome often suffer from an overall high tumor burden, making safe and minimal invasive therapy especially desirable.
The nomenclature SP‐EPN runs a minor risk to suggest that this might represent an overarching tumor type similar to ST NEC/NOS or PF NEC/NOS. Also, the mandatory diagnostic criteria for SP‐EPN by now only require spinal localization and absence of morphological features of MPE or SE. It will therefore be exciting to see, which additional tumor characteristics will be identified in the future and whether a general tumor type of SP NEC/NOS could help to clarify the nomenclature in addition to more specific types (e. g. ‘SP‐NF2’).
SP‐MYCN were recently described as a novel, clinically aggressive type of ependymoma, typically showing early metastases, rapid progression after relapse, leptomeningeal dissemination, and poor response to multimodal treatment strategies [29]. SP‐MYCN are defined by their spinal localization and presence of MYCN amplification. To date, only few cases have been reported [29, 30, 31, 32]. Histologically, SP‐MYCN show pseudorosettes and have a papillary or pseudopapillary architecture. They typically display high‐grade features including microvascular proliferation, necrosis, and high mitotic cell count. MYCN amplification can, for instance, be detected by immunohistochemistry or FISH. Tumors occur predominantly in the cervical or thoracic spinal cord and grow as intramedullary or, more often, extramedullary large tumors with a high rate of leptomeningeal dissemination upon diagnosis (Figure 2). Of the 27 cases reported to date, gender distribution was balanced, and the age at onset ranged between 12 and 56 years with a median age of 32 [29]. SP‐MYCN show a high recurrence rate of 75–100% [31, 32] and a median PFS of 17 months was reported from a case series of 13 tumors. Median overall survival in this cohort was 87 months [29]. All tumors have high‐level MYCN amplifications in common, which remain stable at relapse [29]. In some tumors, additional chromosomal copy number alterations, such as loss of chromosome 10 and focal losses on chromosome 11q were reported [29]. Identification of this especially aggressive tumor type among the generally more benign spinal ependymoma underlines the importance of reevaluating existing tumor classifications by careful molecular and clinical examinations.
2.4. Myxopapillary ependymoma
Myxopapillary ependymoma are morphologically defined tumors that predominantly arise in the most caudal part of the spinal cord. As for other spinal ependymomas, the majority of patients with MPE are adults, with a median age of 39 years [33, 34]. By far, the most common localizations of MPE are the conus medullaris and the filum terminale, but rare cases with occurrence in the brain and in the upper spinal cord or even outside the CNS have been described [6, 7] (Figure 2). The clinical outcome of patients with MPE is favorable, with long‐term overall survival rates exceeding 90 % after total or partial surgical resection [33]. However, in around 20 % of MPE, relapses occur, either locally or at a distant site. Recurrence is associated with several clinical parameters, such as subtotal resection, tumor size, and multifocal lesions [34]. MPE might also present as disseminated disease with distant spinal or intracranial metastases, both in pediatric and adult patients [34, 35]. Acknowledging this partially aggressive clinical behavior with recurrences and dissemination, the grading of MPE was changed from WHO grade I to CNS WHO grade 2 in the current version of the WHO classification. Histologically, MPE are characterized by immunoreactivity for GFAP and by the presence of papillary structures (radial arrangement of spindled or epitheloid tumor cells around hyalinized fibrovascular cores) and myxoid changes around blood vessels or in microcysts. Of note, myxoid changes are already sufficient for the diagnosis of MPE, even if they occur only focally (Figure 3A,B). Studies have demonstrated a distinct DNA methylation profile for MPEs, and numerous chromosomal gains and losses, predominantly affecting large regions, including whole chromosomes or chromosomal arms [36]. For unresolved lesions, the alignment of global DNA methylation with the molecular subgroup of SP‐MPE is a required criterion. In this regard, it needs to be mentioned that the molecular subgroup of SP‐MPE does not show complete concordance with the morphological group of MPE, since it additionally includes cases of histologically classical ependymoma [3, 36] (Figure 3C,D). Future studies need to clarify, whether histopathological or molecular classification is more suitable to describe a group of myxopapillary ependymoma that is both biologically and clinically as homogeneous as possible.
FIGURE 3.
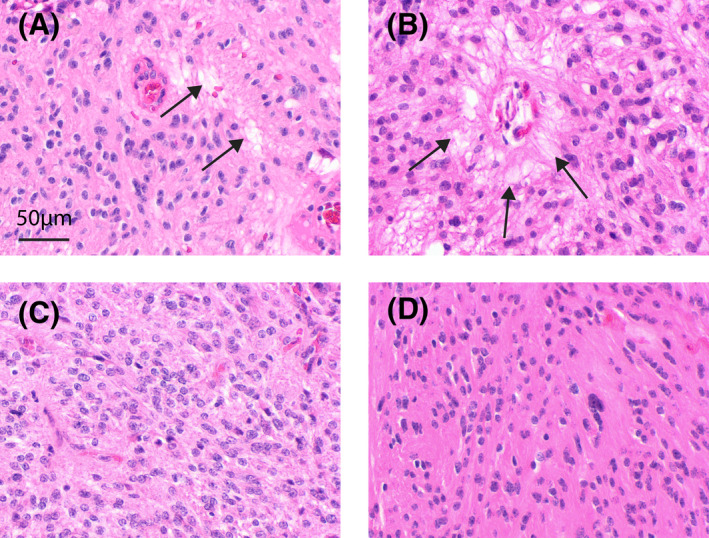
Morphology of myxopapillary ependymoma with focal or absent myxoid changes. Examples given in panels A and B represent tumors with focal myxoid changes (arrows) that qualify for the diagnosis of myxopapillary ependymoma according to the updated WHO classification. Tumors or tumor regions as exemplarily shown in C and D may show a significant match to the methylation group of myxopapillary ependymoma without overt myxoid or papillary morphology. Scale bar in a corresponds to 50 µm in A–D
2.5. Subependymoma
Subependymoma (SE) arise in all compartments of the central nervous system (Figure 2). Most frequently, they originate from the fourth ventricle and lateral ventricles. Histologically, the tumors appear as circumscribed glioma with absent or minimal mitotic activity and lack of conspicuous nuclear atypia. Unresolved lesions require a DNA methylation profile aligned with ST‐SE, PF‐SE, or SP‐SE. Clinically, SE often remain asymptomatic and mainly occur in adults with a median age of approximately 50 years [3, 37]. Overall, the prognosis is excellent, and recurrence is extremely rare, even after subtotal resection. However, SE located in the posterior fossa seem to behave slightly more aggressive with a 5‐year PFS of only 83% compared to 100% in supratentorial and spinal SE [3]. In this context, a subgroup of subependymoma in the posterior fossa harboring TERT promoter mutations and/or loss of chromosome 6 were identified as biologically more aggressive with a significantly shorter progression‐free survival [38]. Therefore, even though not intended in the current WHO classification, localization and molecular characteristics may be important to consider.
3. CHALLENGES IN THE GRADING OF EPENDYMOMA
Historically, histological grades have been assigned to brain tumors in order to characterize their biological behaviour and to estimate their intrinsic aggressiveness. In several CNS tumor entities, the histological grade is a required criterion for the diagnosis, or at least a valuable addition to the diagnosis and is used for risk‐adapted therapy stratification. The current grading system for ependymoma corresponds to the general WHO grading system, which is applied across most of the CNS tumor entities and which is based on various histological features, such as cellularity, mitotic activity, pleomorphism, necrosis, and vascular proliferation.
The family of ependymoma generally comprises tumors ranging from CNS WHO grade 1 to grade 3. However, histological grading of ependymomas has long been the subject of critical discussion. Statements on the correlation between histological grade and clinical outcome have been contradictory between studies over the past decades [39, 40, 41, 42]. Specific pathological features, such as the mitotic index were identified as a valid prognostic factors in some studies, whereas other studies did not confirm this correlation [43, 44]. Another major issue of the grading system is the unreliable distinction of grade 2 and grade 3 ependymomas, which shows high interobserver variability, even when assessed by experienced neuropathologists [45]. Given these controversies regarding the limited clinical impact of histological grading and the highly variable distinction of grade 2 and grade 3 tumors, therapeutic stratification should not be made based on the current histopathological grading system. Yet, current recommendations on treatment stratification for adult patients with intracranial ependymoma still rely on the differentiation between histological grade 2 or 3 [46].
In the current update of the WHO classification, the histological grading of ependymoma is basically retained, yet some minor changes were applied. SE are still classified as histological grade 1, whereas the grading of MPE was changed from grade 1 to grade 2 due to its clinical behaviour, including possible dissemination and recurrence, following the recommendation of the cIMPACT‐7 group [13]. Other ependymomas, which are now defined by anatomical location and molecular profiles, show histological features equivalent to WHO grade 2 or 3. According to the current update of the WHO classification, for cases of supratentorial, posterior fossa, and spinal ependymoma without any molecular specification, histological grading (grade 2 or 3) should still be part of the diagnosis. However, the molecularly defined subtypes of ZFTA‐fusion positive, YAP1‐fusion positive, PF‐A, and PF‐B ependymomas, as well as MYCN‐amplified spinal ependymomas were not assigned to a grade in the current update of the WHO classification. According to the WHO classification, more meaningful data on the outcome of molecularly defined ependymoma types are necessary in order to define a new grading system for these tumors. Still, the clinical prognosis of patients with PF‐A, PF‐B, and ZFTA‐fusion positive supratentorial ependymoma has been consistently described in large cohorts, and for the rare molecular subtypes YAP1‐fusion positive supratentorial ependymoma and spinal ependymoma with MYCN‐amplifications, clinical data from smaller cohorts have been reported [3, 14, 19, 25, 29]. The editors of the WHO classification argue that data from prospective clinical trials are required for the implementation of a new grading system for ependymoma subgroups. While this appears reasonable and desirable, it might still be useful to provide a (provisional) grading in order to guide clinicians into a rather benign or a rather malignant direction. Of note, this has already been practiced in the past, even for rare entities without prospective clinical data (e. g. angiocentric glioma or ETMR). Also, myxopapillary ependymomas have now been assigned to the CNS WHO grade 2 for good reasons, even if large prospective trials are missing.
The WHO classification still recommends to include information on the histological features, especially on the extent of anaplasia into an integrated diagnosis, even though the clinical utility of the histological grading is controversial. In fact, risk stratification based on distinct molecular ependymoma subtypes has been shown to be superior to histopathological grading [3]. In ZFTA‐fusion positive supratentorial ependymoma, the histological grade is not associated with the outcome, which further puts the clinical utility of the histological grading into question [19]. The assignment of histological grades to tumors of a molecularly defined subgroup can even be misleading, as tumors of the prognostically unfavorable subgroups PF‐A, ZFTA‐fusion positive supratentorial ependymoma, and MYCN‐altered spinal ependymoma are sometimes classified as histological grade 2, erroneously indicating a rather benign clinical behavior [3, 19, 29].
For the future, it is therefore highly desirable to establish a coherent grading system that is valid for all (often novel and molecularly defined) ependymoma types. Even if new data and future prospective clinical trials will slightly change this picture at some point and require an adaption of such a grading system, this should not prevent us from constantly translating current knowledge into a clinically helpful grading system.
4. POTENTIAL LIMITATIONS OF THE CURRENT WHO DEFINITION OF EPENDYMOMA
Whether a CNS tumor shall be defined as ependymoma is no longer solely dependent on the tumor morphology. For the diagnosis of many ependymoma types, the presence of a certain molecular feature or profile is now essential. Examples for required molecular criteria are ZFTA fusions for supratentorial ependymomas with ZFTA fusion, a global reduction of H3K27me3 in tumour cell nuclei for the diagnosis of PF‐A, or a global DNA methylation profile matching with the respective reference cohort for PF‐B. The definition of molecularly homogenous tumor types clearly is a valuable addendum, providing many advantages for translational studies and clinical trials.
However, the novel molecular criteria necessarily raise the question how to classify tumors carrying the mentioned molecular features without presenting an ependymoma‐like morphology. This applies particularly to tumors that may appear with a rather small‐, blue‐, and round‐cell character and that had previously been named primitive neuroectodermal tumor (PNET) [47]. Many of these tumors might not show typical features of ependymomas, such as perivascular pseudorosettes, ependymal linings, or true ependymal rosettes. On the other hand, these tumors may carry ZFTA fusions or global DNA methylation profiles perfectly matching to a given references group of ependymomas (Figure 4). Additional reports describe ZFTA fusion‐positive tumors exhibiting a broad spectrum of mainly high‐grade and undifferentiated histological features including characteristics of sarcomas or embryonal tumors, astroblastoma‐like features, or signs of myogenic differentiation [9, 10, 48]. Similarly, PF‐A tumors have been reported to display histomorphological features compatible with medulloblastoma [49]. Hence, in some cases, morphological features do not necessarily match the molecular criteria of the new WHO classification. However, the new WHO classification still requires “morphological and immunohistochemical features of ependymoma” as an essential criterion for the diagnosis of ependymoma. It remains unclear, whether this holds true for totally undifferentiated tumors, but it seems clear from this that – for now – tumors with morphological and immunohistochemical features that are incompatible with the diagnosis of an ependymoma should not be diagnosed as such. For instance, this may be the case for neoplasms that display a purely mesenchymal morphology. A discrepancy between histology and the molecular background of a tumor may also appear for tumors that show perivascular pseudorosettes or other morphological hallmarks of ependymoma, but present a distinct molecular profile not matching to one of the ependymoma types. Such a profile may either fit to a different CNS tumor entity described by the WHO classification, or to a molecularly homogeneous group of tumors that has not yet been defined as a separate tumor type by the WHO. For example, CNS high grade tumors with BCOR alterations have been described to carry typical ependymoma‐like features and have also been diagnosed as such in the past [47]. Also, only recently described supratentorial neuroepithelial tumors with recurrent fusion in PLAG1 or PATZ1 frequently show ependymoma‐like morphology [50, 51]. For these cases, it is the neuropathologist's responsibility to evaluate, whether the overall characteristics of a tumor, including histomorphology and molecular biology, fit the diagnosis of ependymoma or the diagnosis of a different tumor entity.
FIGURE 4.
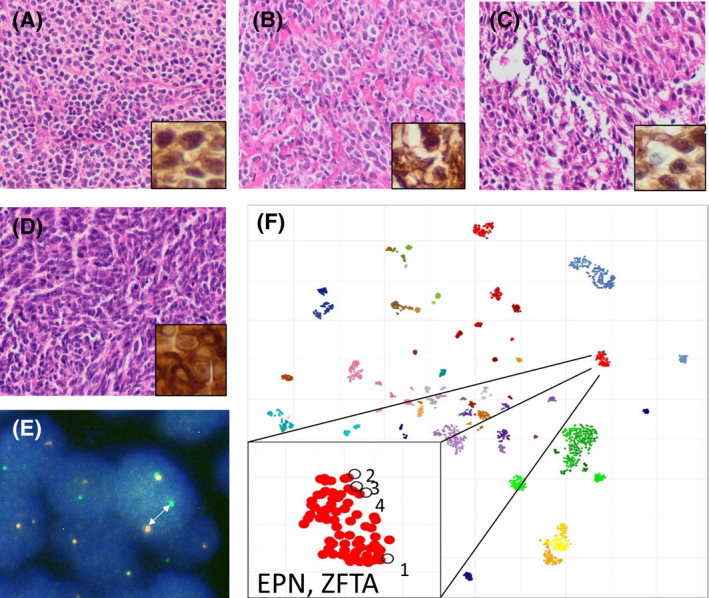
Undifferentiated brain tumors with ZFTA fusion. Histomorphology of four examples (cases 1–4) is shown in A–D, respectively, with insets demonstrating nuclear expression of p65 as a marker for the activation of NFκB signaling. Break‐apart of RELA (arrow in E) as demonstrated by FISH analysis (exemplarily shown for case 1) as well as t‐SNE analysis after methylation profiling (F) further suggest the diagnosis of a supratentorial ependymoma with ZFTA fusion. F shows reference data from 2800 brain tumors previously published by Capper et al. as well as the four cases shown in A–D
The use of global DNA methylation profiling has gained increasing importance in the classification und identification of subgroups of CNS tumors and marks a major change in the current definition of ependymoma. Without any doubt, the discovery of distinct methylation signatures in biologically and clinically homogeneous CNS tumor types was a major breakthrough in the field and represents an extremely useful diagnostic tool in neuropathology [52]. Thereby, ten molecular types of ependymoma and a constantly increasing number of subtypes have been identified over the last years [3, 19, 25, 29]. Acknowledging these findings, the updated WHO classification has included the alignment of the global DNA methylation profile with a reference cohort of the respective ependymoma type as a desirable diagnostic criterion for many ependymoma types. Performing DNA methylation analyses is now even mandatory for histologically unresolved cases of MPE and SE, and for the diagnosis of all cases of PF‐B. While this approach appears helpful and pragmatic, researchers may still need to find solutions for how epidemiological, clinical, and bioinformatic data of such reference cohorts shall be stored, cured and made available in a centralized manner. Also, the alignment to a specific reference group certainly depends on the bioinformatical algorithm (classifier version) that is used. This is not specifically mentioned by the WHO, although, for good reasons, algorithms are constantly updated by researchers responsible for the online platform (www.molecularneuropathology.org), e.g. if new methylation classes are discovered.
Overall, the new WHO classification has changed the definition of whether a tumor classifies as ependymoma and it has become more precise in many cases. For instance, if a supratentorial tumor with close contact to the lateral ventricle has a primitive neuroectodermal appearance, the updated WHO classification clearly suggests defining this tumor as an ependymoma, if OLIG2 is negative, a ZFTA fusion is present, and DNA methylation profiling reveals a significant match to the reference group of ZFTA ependymomas. Nevertheless, some cases display a discrepancy between histomorphology and molecular biology or belong to potentially novel groups with a homogeneous genetic profile, but heterogeneous morphology and unknown clinical behavior. For others, molecular analyses may not be available. These cases still require the assessment by an experienced neuropathologist and an interdisciplinary discussion to assign the most suitable diagnosis.
5. CONCLUSION
The 2021 revision of the WHO classification of CNS tumors presents a major change from the previous histomorphological classification of ependymal tumors towards a classification of ten ependymoma types based on anatomical localization and molecular features. With the emergence of these new molecular types, the general definition, whether or not a tumor belongs to the family of ependymomas has changed. Another important change has been applied to the grading system of ependymoma: The previous grades have only been retained for the types of subependymoma (grade 1) and spinal ependymoma (grade 2), whereas myxopapillary ependymomas are now designated grade 2. For all other ependymoma subtypes, a grade should only be assigned to morphologically‐defined cases (“NEC/NOS”), but not to cases with a known molecularly‐defined subgroup.
The 2021 WHO Classification for ependymoma undoubtedly presents a major improvement in the field of neurooncology and neuropathology and serves as a good example for the successful translation of current molecular findings into a new classification system and more precise diagnostic criteria. Nevertheless, some challenges in the diagnosis and classification of ependymoma still remain. For instance, it might still be difficult to classify cases that show discrepancy between histological and molecular features. Another limitation is the lack of a uniform grading system that could be applied to all subtypes and that could reliably predict a patients’ clinical outcome rather than describing histological markers for biological aggressiveness. Still, the new update of the WHO classification of ependymal tumors is a great advance in the era of precision medicine and for sure paves the way for future progress, both regarding a new molecular grading system as well as clinical trials that are based on the updated classification system.
AUTHOR CONTRIBUTIONS
Figures have been created by C.K. and S.N. The manuscript has been written by all authors.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
ACKNOWLEDGEMENTS
We thank the Deutsche Forschungsgemeinschaft and the Fördergemeinschaft Kinderkrebszentrum Hamburg for continuous generous support.
Kresbach C, Neyazi S, Schüller U. Updates in the classification of ependymal neoplasms: The 2021 WHO Classification and beyond. Brain Pathol. 2022;32:e13068. 10.1111/bpa.13068
REFERENCES
- 1. Louis DN, Ohgaki H, Wiestler OD, Ellison DW, Figarella‐Branger D, Perry A, et al. WHO classification of tumours of the central nervous system (revised 4th ed.). IARC: Lyon, 2016. [Google Scholar]
- 2. WHO Classification of Tumours Editorial Board . Central nervous system tumours [Internet]. Lyon (France): International Agency for Research on Cancer; 2021. [cited 2021 11 29]. (WHO classification of tumours series, 5th ed.; vol. 6). Available from: https://tumourclassification.iarc.who.int/chapters/45 [Google Scholar]
- 3. Pajtler K, Witt H, Sill M, Jones D, Hovestadt V, Kratochwil F, et al. Molecular classification of ependymal tumors across All CNS compartments, histopathological grades, and age groups. Cancer Cell. 2015;27(5):728–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yang D, Holsten T, Börnigen D, Frank S, Mawrin C, Glatzel M, et al. Ependymoma relapse goes along with a relatively stable epigenome, but a severely altered tumor morphology. Brain Pathol. 2021;31(1):33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Neumann JE, Spohn M, Obrecht D, Mynarek M, Thomas C, Hasselblatt M, et al. Molecular characterization of histopathological ependymoma variants. Acta Neuropathol. 2020;139(2):305–18. [DOI] [PubMed] [Google Scholar]
- 6. Tseng YC, Hsu HL, Jung SM, Chen CJ. Primary intracranial myxopapillary ependymomas: report of two cases and review of the literature. Acta Radiol. 2004;45(3):344–7. [DOI] [PubMed] [Google Scholar]
- 7. Yust Katz S, Cachia D, Kamiya‐Matsuoka C, Olar A, Theeler B, Penas Prado M, et al. Ependymomas arising outside of the central nervous system: a case series and literature review. J Clin Neurosci. 2018;47:202–7. [DOI] [PubMed] [Google Scholar]
- 8. Tamai S, Nakano Y, Kinoshita M, Sabit H, Nobusawa S, Arai Y, et al. Ependymoma with C11orf95‐MAML2 fusion: presenting with granular cell and ganglion cell features. Brain Tumor Pathol. 2021;38(1):64–70. [DOI] [PubMed] [Google Scholar]
- 9. Tauziède‐Espariat A, Siegfried A, Nicaise Y, Kergrohen T, Sievers P, Vasiljevic A, et al. Supratentorial non‐RELA, ZFTA‐fused ependymomas: a comprehensive phenotype genotype correlation highlighting the number of zinc fingers in ZFTA‐NCOA1/2 fusions. Acta Neuropathol Commun. 2021;9(1):135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zheng T, Ghasemi DR, Okonechnikov K, Korshunov A, Sill M, Maass KK, et al. Cross‐species genomics reveals oncogenic dependencies in ZFTA/C11orf95 fusion‐positive supratentorial ependymomas. Cancer Discov. 2021;11(9):2230–47. [DOI] [PubMed] [Google Scholar]
- 11. Lim KY, Lee KH, Phi JH, Yun H, Won JK, Choi SH, et al. ZFTA‐YAP1 fusion‐positive ependymoma can occur in the spinal cord: Letter to the editor. Brain Pathol. 2022;32:e13020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chavali P, Rao S, Palavalasa S, Bevinahalli N, Muthane YTC, Sadashiva N, et al. L1CAM immunopositivity in anaplastic supratentorial ependymomas: correlation with clinical and histological parameters. Int J Surg Pathol. 2019;27(3):251–8. [DOI] [PubMed] [Google Scholar]
- 13. Ellison DW, Aldape KD, Capper D, Fouladi M, Gilbert MR, Gilbertson RJ, et al. cIMPACT‐NOW update 7: advancing the molecular classification of ependymal tumors. Brain Pathol. 2020;30(5):863–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Andreiuolo F, Varlet P, Tauziède‐Espariat A, Jünger ST, Dörner E, Dreschmann V, et al. Childhood supratentorial ependymomas with YAP1‐MAMLD1 fusion: an entity with characteristic clinical, radiological, cytogenetic and histopathological features. Brain Pathol. 2019;29(2):205–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Witt H, Mack S, Ryzhova M, Bender S, Sill M, Isserlin R, et al. Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell. 2011;20(2):143–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wani K, Armstrong TS, Vera‐Bolanos E, Raghunathan A, Ellison D, Gilbertson R, et al. A prognostic gene expression signature in infratentorial ependymoma. Acta Neuropathol. 2012;123(5):727–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bayliss J, Mukherjee P, Lu C, Jain SU, Chung C, Martinez D, et al. Lowered H3K27me3 and DNA hypomethylation define poorly prognostic pediatric posterior fossa ependymomas. Sci Transl Med. 2016;8(366):366ra161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xi S, Sai KE, Hu W, Wang F, Chen Y, Wang J, et al. Clinical significance of the histological and molecular characteristics of ependymal tumors: a single institution case series from China. BMC Cancer. 2019;19(1):717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pajtler KW, Wen JI, Sill M, Lin T, Orisme W, Tang BO, et al. Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol. 2018;136(2):211–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nambirajan A, Sharma A, Rajeshwari M, Boorgula MT, Doddamani R, Garg A, et al. EZH2 inhibitory protein (EZHIP/Cxorf67) expression correlates strongly with H3K27me3 loss in posterior fossa ependymomas and is mutually exclusive with H3K27M mutations. Brain Tumor Pathol. 2021;38(1):30–40. [DOI] [PubMed] [Google Scholar]
- 21. Jain SU, Do TJ, Lund PJ, Rashoff AQ, Diehl KL, Cieslik M, et al. PFA ependymoma‐associated protein EZHIP inhibits PRC2 activity through a H3 K27M‐like mechanism. Nat Commun. 2019;10(1):2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baroni LV, Sundaresan L, Heled A, Coltin H, Pajtler KW, Lin T, et al. Ultra high‐risk PFA ependymoma is characterized by loss of chromosome 6q. Neuro Oncol. 2021;23(8):1360–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Panwalkar P, Clark J, Ramaswamy V, Hawes D, Yang F, Dunham C, et al. Immunohistochemical analysis of H3K27me3 demonstrates global reduction in group‐A childhood posterior fossa ependymoma and is a powerful predictor of outcome. Acta Neuropathol. 2017;134(5):705–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Routman DM, Raghunathan A, Giannini C, Mahajan A, Beltran C, Nagib MG, et al. Anaplastic ependymoma and posterior fossa grouping in a patient with H3K27ME3 loss of expression but chromosomal imbalance. Adv Rad Oncol. 2019;4:466–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cavalli FMG, Hübner J‐M, Sharma T, Luu B, Sill M, Zapotocky M, et al. Heterogeneity within the PF‐EPN‐B ependymoma subgroup. Acta Neuropathol. 2018;136(2):227–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Plotkin SR, O’Donnell CC, Curry WT, Bove CM, MacCollin M, Nunes FP. Spinal ependymomas in neurofibromatosis Type 2: a retrospective analysis of 55 patients. J Neurosurg Spine. 2011;14(4):543–7. [DOI] [PubMed] [Google Scholar]
- 27. Patronas NJ, Courcoutsakis N, Bromley CM, Katzman GL, MacCollin M, Parry DM. Intramedullary and spinal canal tumors in patients with neurofibromatosis 2: MR imaging findings and correlation with genotype. Radiology. 2001;218(2):434–42. [DOI] [PubMed] [Google Scholar]
- 28. Mautner VF, Tatagiba M, Lindenau M, Fünsterer C, Pulst SM, Baser ME, et al. Spinal tumors in patients with neurofibromatosis type 2: MR imaging study of frequency, multiplicity, and variety. Am J Roentgenol. 1995;165(4):951–5. [DOI] [PubMed] [Google Scholar]
- 29. Ghasemi DR, Sill M, Okonechnikov K, Korshunov A, Yip S, Schutz PW, et al. MYCN amplification drives an aggressive form of spinal ependymoma. Acta Neuropathol. 2019;138(6):1075–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Scheil S, Brüderlein S, Eicker M, Herms J, Herold‐Mende C, Steiner H‐H, et al. Low frequency of chromosomal imbalances in anaplastic ependymomas as detected by comparative genomic hybridization. Brain Pathol. 2001;11(2):133–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Swanson AA, Raghunathan A, Jenkins RB, Messing‐Jünger M, Pietsch T, Clarke MJ, et al. Spinal cord ependymomas with MYCN amplification show aggressive clinical behavior. J Neuropathol Exp Neurol. 2019;78(9):791–7. [DOI] [PubMed] [Google Scholar]
- 32. Raffeld M, Abdullaev Z, Pack SD, Xi L, Nagaraj S, Briceno N, et al. High level MYCN amplification and distinct methylation signature define an aggressive subtype of spinal cord ependymoma. Acta Neuropathol Commun. 2020;8(1):101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bates JE, Choi G, Milano MT. Myxopapillary ependymoma: a SEER analysis of epidemiology and outcomes. J Neurooncol. 2016;129(2):251–8. [DOI] [PubMed] [Google Scholar]
- 34. Montero A‐S, Tran S, Amelot A, Berriat F, Lot G, Gaillard S, et al. Clinical characteristics and long‐term surgical outcome of spinal myxopapillary ependymoma: a French cohort of 101 patients. J Neurooncol. 2021;152(3):491–9. [DOI] [PubMed] [Google Scholar]
- 35. Fassett DR, Pingree J, Kestle JRW. The high incidence of tumor dissemination in myxopapillary ependymoma in pediatric patients. Report of five cases and review of the literature. J Neurosurg. 2005;102(1 Suppl):59–64. [DOI] [PubMed] [Google Scholar]
- 36. Witt H, Gramatzki D, Hentschel B, Pajtler KW, Felsberg J, Schackert G, et al. DNA methylation‐based classification of ependymomas in adulthood: implications for diagnosis and treatment. Neuro Oncol. 2018;20(12):1616–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rushing EJ, Cooper PB, Quezado M, Begnami M, Crespo A, Smirniotopoulos JG, et al. Subependymoma revisited: clinicopathological evaluation of 83 cases. J Neurooncol. 2007;85(3):297–305. [DOI] [PubMed] [Google Scholar]
- 38. Thomas C, Thierfelder F, Träger M, Soschinski P, Müther M, Edelmann D, et al. TERT promoter mutation and chromosome 6 loss define a high‐risk subtype of ependymoma evolving from posterior fossa subependymoma. Acta Neuropathol. 2021;141(6):959–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ernestus RI, Schröder R, Stützer H, Klug N. The clinical and prognostic relevance of grading in intracranial ependymomas. Br J Neurosurg. 1997;11(5):421–8. [DOI] [PubMed] [Google Scholar]
- 40. Korshunov A, Golanov A, Sycheva R, Timirgaz V. The histologic grade is a main prognostic factor for patients with intracranial ependymomas treated in the microneurosurgical era: an analysis of 258 patients. Cancer. 2004;100(6):1230–7. [DOI] [PubMed] [Google Scholar]
- 41. Tihan T, Zhou T, Holmes E, Burger PC, Ozuysal S, Rushing EJ. The prognostic value of histological grading of posterior fossa ependymomas in children: a Children’s Oncology Group study and a review of prognostic factors. Mod Pathol. 2008;21(2):165–77. [DOI] [PubMed] [Google Scholar]
- 42. Lopez‐Rivera V, Dono A, Abdelkhaleq R, Sheth SA, Chen PR, Chandra A, et al. Treatment trends and overall survival in patients with grade II/III ependymoma: the role of tumor grade and location. Clin Neurol Neurosurg. 2020;199:106282. [DOI] [PubMed] [Google Scholar]
- 43. Godfraind C, Kaczmarska JM, Kocak M, Dalton J, Wright KD, Sanford RA, et al. Distinct disease‐risk groups in pediatric supratentorial and posterior fossa ependymomas. Acta Neuropathol. 2012;124(2):247–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kurt E, Zheng P‐P, Hop WCJ, van der Weiden M, Bol M, van der Bent MJ, et al. Identification of relevant prognostic histopathologic features in 69 intracranial ependymomas, excluding myxopapillary ependymomas and subependymomas. Cancer. 2006;106(2):388–95. [DOI] [PubMed] [Google Scholar]
- 45. Ellison DW, Kocak M, Figarella‐Branger D, Felice G, Catherine G, Pietsch T, et al. Histopathological grading of pediatric ependymoma: reproducibility and clinical relevance in European trial cohorts. J Negat Results Biomed. 2011;10:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rudà R, Reifenberger G, Frappaz D, Pfister SM, Laprie A, Santarius T, et al. EANO guidelines for the diagnosis and treatment of ependymal tumors. Neuro Oncol. 2018;20(4):445–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sturm D, Orr B, Toprak U, Hovestadt V, Jones D, Capper D, et al. New brain tumor entities emerge from molecular classification of CNS‐PNETs. Cell. 2016;164(5):1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Oon ML, Hendriansyah L, Pratiseyo PD, Wahjoepramono EJ, Goh JY, Kuick CH, et al. The multifaceted appearance of supratentorial ependymoma with ZFTA‐MAML2 fusion. Free Neuropathol. 2021;2(24):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ruf VC, Schöler A, Capper D, Arzberger T, Herms J, Schüller U. An 8‐year‐old girl with posterior fossa mass. Brain Pathol. 2020;30(3):713–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sievers P, Stichel D, Sill M, Schrimpf D, Sturm D, Selt F, et al. GOPC:ROS1 and other ROS1 fusions represent a rare but recurrent drug target in a variety of glioma types. Acta Neuropathol. 2021;142:1065–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Alhalabi KT, Stichel D, Sievers P, Peterziel H, Sommerkamp AC, Sturm D, et al. PATZ1 fusions define a novel molecularly distinct neuroepithelial tumor entity with a broad histological spectrum. Acta Neuropathol. 2021;142(5):841–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, et al. DNA methylation‐based classification of central nervous system tumours. Nature. 2018;555(7697):469–74. [DOI] [PMC free article] [PubMed] [Google Scholar]