

Abstract
The prognostic value of peripheral blood mononuclear cell (PBMC) expression profiles, when used in patients with chronic hypersensitivity pneumonitis (CHP), as an adjunct to traditional clinical assessment is unknown. RNA-seq analysis on PBMC from 37 patients with CHP at initial presentation determined that (1) 74 differentially expressed transcripts at a 10% false discovery rate distinguished those with (n=10) and without (n=27) disease progression, defined as absolute FVC and/or diffusing capacity of the lungs for carbon monoxide (DLCO) decline of ≥10% and increased fibrosis on chest CT images within 24 months, and (2) classification models based on gene expression and clinical factors strongly outperform models based solely on clinical factors (baseline FVC%, DLCO% and chest CT fibrosis).
INTRODUCTION
Hypersensitivity pneumonitis (HP) is an immunologically mediated form of lung disease resulting from inhalational exposure to a large variety of antigens. A subgroup of patients with HP develop chronic HP (CHP) and progressive pulmonary fibrosis, a leading cause of death in HP.1
The current traditional clinical assessment does not include the molecular attributes that are prototypical of CHP progression, and that could prove vital in terms of prognosis. Thus far, no study has attempted to use low-risk peripheral blood (safe and accessible alternative to bronchoscopy or lung biopsy) transcriptional data of affected patients to create CHP prognostic molecular signatures to enhance the prognostic accuracy of current CHP clinical risk stratification. Therefore, we aimed to determine if a risk indicative, transcriptomic signature in peripheral blood mononuclear cells (PBMCs) from patients with CHP can be used to predict disease progression within 2 years of presentation.
METHODS
Expression data were generated for peripheral blood RNA specimens from adult subjects with CHP enrolled in the National Jewish Health interstitial lung disease (NJH ILD) research programme.2 All participants in this study provided written institutional review board-approved informed consent (HS-2946). All subjects had a multidisciplinary consensus diagnosis of HP3 at initial clinic presentation (time of blood draw), and were evaluated for evidence of disease progression within the first 24 months of follow-up, defined as absolute FVC and/or diffusing capacity of the lungs for carbon monoxide (DLCO) decline of ≥10% and ≥10% increase in reticulation and/or honeycombing on chest CT.
PBMCs extracted from frozen cell pellets in whole blood were subjected to mRNA bead capture, quality control (Qiagen) and RNA-seq library build (Kapa Biosystems Inc). Libraries were QCed on the Bioanalyzer (Agilent Technologies) and sequenced on the Illumina 2500 at 1×50 bp (mean: 40 million standard error 12 million reads/sample).
Demographics, smoking history, occupational and environmental history, pulmonary function, high-resolution CT (HRCT) scan and treatment data were collected at the time of blood draw. HRCT scans were reviewed in a blinded fashion by a thoracic radiologist at baseline and 24 months. Percentage of lung fibrosis was scored to the nearest 5%.4 To evaluate the importance of baseline measures of disease severity in predicting disease progression, patients were also stratified into one of three levels: mild (FVC≥80%, CT without fibrosis), moderate (FVC: 70%–79%, CT fibrosis) and severe (FVC≤69%, CT fibrosis). Three cases were exceptions to the FVC thresholds because they had no CT fibrosis (mild FVC=75%, moderate FVC=68%, severe FVC=49%).
Differential expression (DE) analysis
FASTQ files from the Illumina bcl2fastq V.2.17 converter were adapter trimmed using skewer V.0.2.2 and aligned to the hg19 human reference genome with the STAR aligner V.2.4.15 using Ensembl V.75 (http://ensembl.org) gene models. Counts of uniquely mapped reads per gene were quantified using the Subread V.1.5.1 software. Subject data were scaled using the variance stabilising transform in the DESeq2 package and visualised using the pheatmap package and PC analysis princomp package (http://www.R-project.org/) Differential gene expression between progressors/non-progressors was performed with DESeq2 for transcripts with ≥10 reads, using the Wald test and a 10% false discovery rate (FDR). Pathway analysis used Gene Ontology terms in DAVID (https://david.ncifcrf.gov/).
Predictive modelling
Logistic regression models used the R glm function to predict progressor or non-progressor status, controlling for age, gender and smoking status. Models with baseline measurements of clinical variables (FVC%, DLCO%, and CT presence of fibrosis–reticulation and/or honeycombing) were compared with models with gene expression alone or in combination with these clinical variables. Models were also made using expression data for sets of genes defined by three published signatures of idiopathic pulmonary fibrosis (IPF) in PBMCs (mild vs severe IPF, top 10 genes as in Yang et al6 from their table 5) and lung (IPF vs control, top 10 or 74 genes by p value as in Yang et al7 from their table S2; or HP vs IPF, top 10 or 74 genes by TNoM, Selman et al8 from their table E2). Expression data were included in the models using the first three principal components (PCs) of the data. Prediction performance was measured by leave-one-out cross-validation to compute area under the receiver operating characteristics curve (AUC), with 95% CIs using the cvAUC R package. The pROC package was used to compute Delong’s one-sided p value between two AUCs, at 0.05 significance.
As a secondary analysis, the transcriptomic signature data were hierarchically clustered (Ward’s linkage on correlation) to identify subclusters of subjects. The number of clusters was determined by the elbow method on tree-branch heights.
RESULTS
Compared with non-progressors, progressors were more likely to have lower FVC%, chest CT features of fibrosis and less likely to have mild disease severity at presentation (table 1). Clinical disease severity stratification at presentation did not perfectly correlate with disease course (table 1). Among progressors, 40% (4/10) had moderate disease at presentation. Among non-progressors, 15% (4/27) had severe disease. The majority of non-progressors cluster with the second PC (figure 1). Two of the non-progressors grouped with progressors along the first PC had mild disease. Statistical tests for DE between progressors and non-progressors revealed 74 DE transcripts, many on pathways relevant to lung fibrosis (figure 2A).9 10 11
Table 1.
Clinical variables of patients with chronic hypersensitivity pneumonitis with and without disease progression
| All patients |
Progressors |
Non-progressors |
||
|---|---|---|---|---|
| Characteristics | (N=37) | (n=10) | (n=27) | P value |
|
| ||||
| Demographics | ||||
| Age, years | 62±10 | 64±10 | 61±10 | 0.4897 |
| Male sex, n (%) | 17 (45) | 8 (80) | 12 (44) | 0.0425 |
| Former smoker, n (%) | 23 (62) | 6 (60) | 17 (63) | 0.8724 |
| Exposure, n (%) | 0.0814 | |||
| Known inciting antigen | 16 (43) | 2 (20) | 14 (52) | |
| Avian | 7 (44) | 0 | 7 (50) | |
| Mould/bacteria | 9 (56) | 2 (100) | 7 (50) | |
| Lung function testing | ||||
| FVC %-predicted at presentation | 75±15 | 62±12 | 76±11 | 0.0031 |
| 12-Month change | −4.6±7 | −7.6±5 | −0.6±8 | 0.015 |
| 24-Month change | −8.7±12 | −14.3±8.2 | −1.9±10 | 0.0041 |
| DLCO %-predicted at presentation | 57±19 | 54±24 | 58±18 | 0.5246 |
| 12-Month change | −3.1±11 | −12.8±9 | 0.4±10 | 0.0124 |
| 24-Month change | −6.3±13 | −19.3±14 | −0.1±10 | 0.0485 |
| HRCT features of fibrosis, n (%) | ||||
| Reticulation | 25 (67) | 10 (100) | 15 (55) | 0.0085 |
| Honeycombing | 12 (32) | 8 (80) | 4 (15) | 0.0002 |
| Fibrosis score | 28±19 | 37±25 | 25±15 | 0.2189 |
| 24-Month change | 3.9±5.4 | 11±5 | 0.2±4 | 0.0457 |
| Disease severity at presentation, n (%)* | 0.0062 | |||
| Mild (FVC≥80%, CT without fibrosis) | 12 (32) | 0 | 12 (44) | |
| Moderate (FVC 70%–79%, CT fibrosis) | 15 (40) | 4 (40) | 11 (41) | |
| Severe (FVC≤69%, CT fibrosis) | 10 (27) | 6 (60) | 4 (15) | |
| Absolute FVC and/or DLCO decline ≥10% and CT progression† | ||||
| Within 24 months, n (%) | 10 (27) | 10 (100) | 0 | 0.0000 |
| Within 12 months, n (%) | 4 (11) | 4 (40) | 0 | |
| Immunomodulary treatment at presentation, n (%)‡ | 13 (35) | 3 (30) | 7 (26) | 0.6859 |
| 5-Year mortality, n (%) | 3 (8) | 3 (30) | 0 | 0.0000 |
| ILD-GAP index, n (%) | 0.0978 | |||
| 0–1 | 27 (73) | 6 (60) | 21 (78) | |
| 2–3 | 9 (24) | 4 (40) | 5 (18) | |
| 4–5 | 1 (0.03) | 1 (10) | 0 | |
Data are presented as mean with SD or number (%) for categorical variables. Wilcoxon or χ2 tests were utilised for univariate analysis depending on the type of data. JMP V.13 was used for statistical analysis. Values of p≤0.01 indicate statistically significant differences between groups. The variable contains no missing values.
Values of p≤0.01 indicate statistically significant differences between groups.
The variable contains no missing values.
Three patients without CT fibrosis were exceptions based on FVC (mild FVC=75%, moderate FVC=68%, severe FVC=49%).
Chest CT progression ≥10% increase in reticulation and/or honeycombing.
Prednisone, azathioprine and mycophenolate mofetil.
DLCO, diffusing capacity of the lungs for carbon monoxide; ILD-GAP, interstitial lung disease-gender, age, physiology.
Figure 1. PC analysis of chronic hypersensitivity pneumonitis gene expression data.
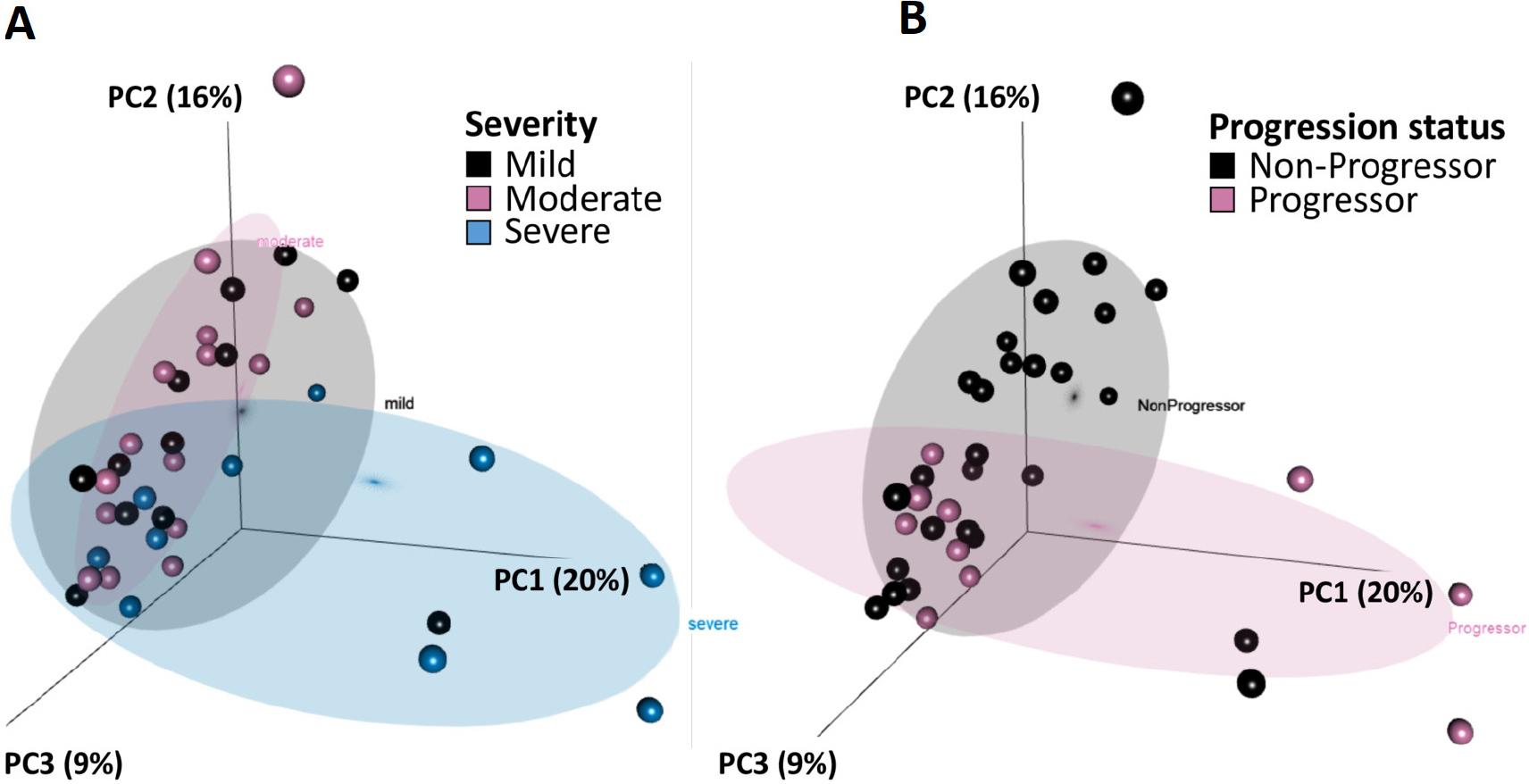
Using expression data normalised for sequencing depth, we generated a PC analysis using the top 15% highly variable genes and labelled samples by (A) severity or (B) progression status. Variability in genes was determined using the IQR divided by the median for each gene across samples, a non-parametric analogue to coefficient of variation. Though perfect separation of clusters is not expected when no phenotype information is used for gene selection a priori, the stronger clustering by progression status than by disease severity in this unbiased analysis suggests that the expression data would be a useful predictor of progression. PC, principal component.
Figure 2. Analyses with differentially expressed genes.
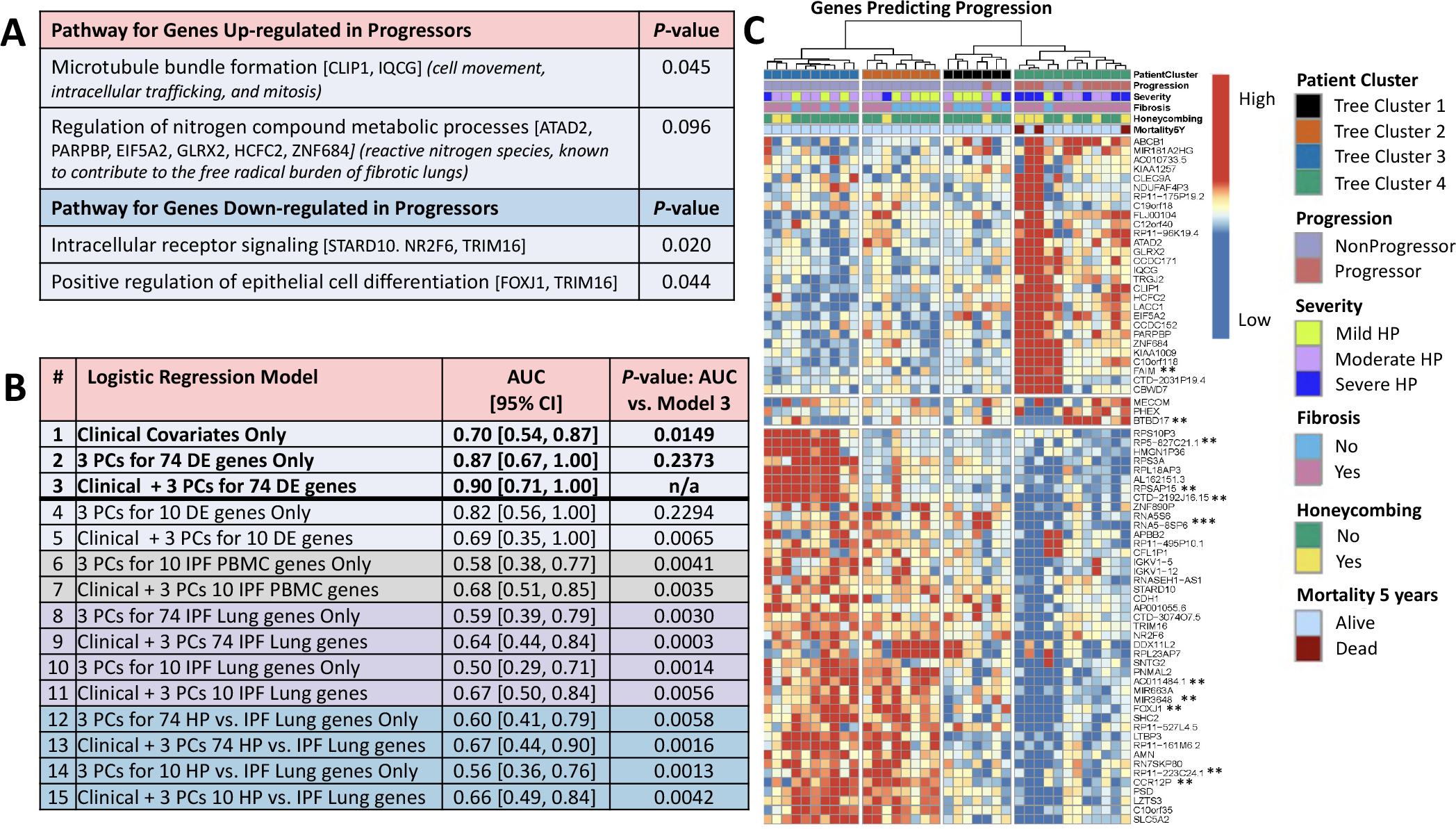
(A) Pathway analysis using gene ontology GO_FAT_BP terms in DAVID (https://david.ncifcrf.gov/). Next to the terms, genes annotated to the term are shown in brackets and the significance of the upregulated genes to HP biology is shown in parentheses. (B) Predictive performance by leave-one-out cross-validation of logistic regression classifiers of progressors versus non-progressors (adjusted for age, gender and smoking status) using only baseline clinical parameters (FVC%, DLCO% and CT presence of fibrosis–reticulation and/or honeycombing); using only expression data; or using clinical data in combination with expression data. Shown is AUC, with 95% CIs in brackets, and the p value for the one-sided Delong test of significant difference (p<0.05) between AUC of a given model and the best AUC among all models. Gene expression data were included in a model using the first three PCs of the data for a given set of genes. Gene sets were either taken from among the 74 DE genes for HP progressors versus non-progressors (all 74 or top 10 by FDR value, models 2–5) or from three published gene signatures of IPF in PBMCs (mild vs severe IPF genes, top 10 genes as in Yang et al6 from their table 5; models 6–7) and lung (IPF vs control, top 74 or 10 genes by p value as in Yang et al7 from their table S2; models 8–11; or HP vs IPF, top 74 or 10 genes by TNoM as in Selman et al8 from their table E2, models 12–15). The original gene signatures varied greatly in size across the IPF studies (from 13 genes to 5465 genes). To make comparisons unbiased by signature size, we used the ranking established by the original authors and considered only the top 74 genes of each, where possible, to be comparable to our signature of size 74, or the top 10, as limited by the smallest signature in IPF PBMCs, where only 10 of the 13 original publication’s genes had data in our dataset (CCDC18-AS1 and the two unnamed transcripts were not used). None of the top 10 or top 74 genes listed in the published signatures were found among our 74 DE genes. Performance for predicting CHP progression using only clinical data (model 1 AUC=0.70) was significantly improved when adding the first three PCs of the 74 DE genes combined with clinical features (model 3 AUC=0.90, Delong one-sided test of the two AUCs p=0.0149). The combined model (model 3) was also a significant improvement over using a signature of just the top 10 DE genes in combination with clinical features (model 5 AUC=0.69; the 10 genes are starred ** in C, only 11th ranked AC011484.1 was not used), or any of the models using genes from published signatures of IPF (all models 6–15 with AUCs ranging from 0.50 to 0.68 had one-sided pairwise Delong tests against model three with p≤0.0065), indicating our 74 DE signature combined with clinical features is specific to predicting CHP progression. The combined model (model 3) was not statistically better than either expression only model, using 74 DE genes (model 2, AUC=0.87, Delong p=0.24) or 10 DE genes (model 4, AUC=0.82, Delong p=0.23), indicating that expression was a major contributor to predictive accuracy. (C) Hierarchical clustering of 74 DE genes (FDR=0.1). Data were scaled per gene (row) to have mean zero and SD 1 and clustered using Ward’s linkage on correlation. Eleven genes were DE at FDR=0.05 (**) and one at FDR=0.01 (***). AUC, area under the curve; CHP, chronic hypersensitivity pneumonitis; DE, differential expression; DLCO, diffusing capacity of the lungs for carbonmonoxide; FDR, false discovery rate; IPF, idiopathic pulmonary fibrosis; PC, principal component.
The prediction performance by leave-one-out cross-validation of a logistic regression model of progression using baseline clinical parameters (AUC=0.70) was significantly improved when adding the first three PCs of the 74 DE gene data in combination with baseline clinical variables (AUC=0.90, pairwise Delong p=0.0149) (figure 2B). The combined model also outperformed models using only the top 10 DE genes and clinical data or any model using published IPF signature genes (all Delong p≤0.0065), indicating the 74 DE signature is specific to CHP progression.
Hierarchical clustering of the 74 DE transcripts further subclustered samples (figure 2C). Clinical characteristics of patients showed patient tree cluster 4 to be enriched for patients with severe disease at presentation, CT reticulation and honeycombing, though not all patients with advanced disease fell into this cluster. Five-year mortality was also confined to tree cluster 4 (three patients). The majority of the non-progressors were further subdivided by gene expression into three subgroups, distinct from cluster 4. Compared with the logistic regression models, 10/13 predicted as progressors using only the 74 DE genes and all nine predicted as progressors by combined clinical and 74 DE genes fall in cluster 4, reinforcing that prediction performance was driven largely by the molecular data.
DISCUSSION
Using a cross-validation method, we demonstrate that including baseline gene expression signature data leads to a significant increase in the prediction accuracy and AUC compared with that by clinical parameters alone or compared with existing signatures of IPF. Hierarchical clustering applied to the 74 DE transcripts shows distinct subgroups among subjects, distinguishing patients with disease progression from patients with more stable disease regardless of baseline disease severity.
Prior observational studies have evaluated the value of gene expression profiling in HP.8 12 These studies were limited by specimen collection bias by only including subjects with surgical lung biopsy specimens (not a practical biomarker measurable in the clinic).
While this pilot study provides the first and the largest cohort evaluating PBMC expression profiles as a potential adjunct to traditional clinical assessment in providing HP-specific prognostic information, the dataset was not large enough to have a separate training and test set; thus, leave-one-out cross-validation was used to provide a more efficient use of limited data. We recognise that gene expression signatures may not overlap completely between blood and lung tissue in HP. In the future, once we establish a prognostic peripheral blood HP biomarker, we can determine whether that signal is present within the lungs consistently across all individuals. Also, despite an expert thoracic radiologist providing a visual estimation of fibrosis extent, potential observer variability might limit the reliability of CT scoring. Lastly, caution is needed in interpreting the prediction model’s performance since further independent external validations with a large sample size are warranted.
In conclusion, despite the differences in clinical and imaging features at the initial presentation, molecular phenotyping by gene expression can be a promising and valuable predictor of CHP disease progression and complement traditional clinical risk stratification.
Funding
Supported by NIH/NHLBI grant R01HL148437, a National Jewish Health grant as well as by a generous donation from Forrest Shook.
Footnotes
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
REFERENCES
- 1.Fernández Pérez ER, Swigris JJ, Forssén AV, et al. Identifying an inciting antigen is associated with improved survival in patients with chronic hypersensitivity pneumonitis. Chest 2013;144:1644–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chung JH, Zhan X, Cao M, et al. Presence of air trapping and mosaic attenuation on chest computed tomography predicts survival in chronic hypersensitivity pneumonitis. Ann Am Thorac Soc 2017;14:1533–8. [DOI] [PubMed] [Google Scholar]
- 3.Fernández Pérez ER, Travis WD, Lynch DA, et al. Diagnosis and Evaluation of Hypersensitivity Pneumonitis: CHEST Guideline and Expert Panel Report. Chest. 2021;160(2):e97–e156 [DOI] [PubMed] [Google Scholar]
- 4.Desai SR, Veeraraghavan S, Hansell DM, et al. Ct features of lung disease in patients with systemic sclerosis: comparison with idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia. Radiology 2004;232:560–7. [DOI] [PubMed] [Google Scholar]
- 5.Dobin A, Davis CA, Schlesinger F, et al. Star: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang IV, Luna LG, Cotter J, et al. The peripheral blood transcriptome identifies the presence and extent of disease in idiopathic pulmonary fibrosis. PLoS One 2012;7:e37708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang IV, Coldren CD, Leach SM, et al. Expression of cilium-associated genes defines novel molecular subtypes of idiopathic pulmonary fibrosis. Thorax 2013;68:1114–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Selman M, Pardo A, Barrera L, et al. Gene expression profiles distinguish idiopathic pulmonary fibrosis from hypersensitivity pneumonitis. Am J Respir Crit Care Med 2006;173:188–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sandbo N, Dulin N. Actin cytoskeleton in myofibroblast differentiation: ultrastructure defining form and driving function. Transl Res 2011;158:181–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stefanov AN, Fox J, Depault F, et al. Positional cloning reveals strain-dependent expression of Trim16 to alter susceptibility to bleomycin-induced pulmonary fibrosis in mice. PLoS Genet 2013;9:e1003203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anathy V, Lahue KG, Chapman DG, et al. Reducing protein oxidation reverses lung fibrosis. Nat Med 2018;24:1128–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horimasu Y, Ishikawa N, Iwamoto H, et al. Clinical and molecular features of rapidly progressive chronic hypersensitivity pneumonitis. Sarcoidosis Vasc Diffuse Lung Dis 2017;34:48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]