

Abstract
With the rapid development of society, the incidence of metabolic syndrome (MS) is increasing rapidly. Evidence indicated that patients diagnosed with MS usually suffered from cardiomyopathy, called metabolic syndrome–associated cardiomyopathy (MSC). The clinical characteristics of MSC included cardiac hypertrophy and diastolic dysfunction, followed by heart failure. Despite many studies on this topic, the detailed mechanisms are not clear yet. As the center of cellular metabolism, mitochondria are crucial for maintaining heart function, while mitochondria dysfunction plays a vital role through mechanisms such as mitochondrial energy deprivation, calcium disorder, and ROS (reactive oxygen species) imbalance during the development of MSC. Accordingly, in this review, we will summarize the characteristics of MSC and especially focus on the mechanisms related to mitochondria. In addition, we will update new therapeutic strategies in this field.
1. Introduction
Along with the changes in lifestyle and population aging, the incidence of a series of metabolic disorders, such as obesity, diabetes mellitus (DM), insulin resistance, dyslipidemia, and hypertension, is increasing worldwide [1, 2]. People suffering from these disorders are diagnosed with metabolic syndrome [3]. Current evidence indicated that these patients are twice as at risk to develop cardiovascular diseases (CVD) as those without. Metabolic syndrome–associated cardiac diseases (MSCDs) include cardiomyopathy, coronary artery disease (CAD), left ventricular hypertrophy (LVH), and systolic and/or diastolic contractile dysfunction with an increased incidence of heart failure (HF) [4]. However, the underlying mechanisms of this pathological process are not clear yet.
Mitochondria is the most important organelle involved in cellular or tissue metabolic physiological processes [5]. Since mitochondrial homeostasis is vital to maintaining physical cellular activity, mitochondrial quality control disorders through various processes such as proteostasis, mitochondrial dynamics, and mitophagy could lead to the development of metabolic syndrome–related cardiomyopathy (MSC) [6]. In this review, we would first introduce the changes in MSC, and then we will review the role of dysregulated mitochondrial homeostasis in cardiomyopathy induced by metabolic syndrome. Finally, as healthy mitochondria may be essential for normal cardiovascular function, we will focus on the underlying therapeutic strategies targeting mitochondria in the development of MSC.
2. Definition of Metabolic Syndrome–Related Cardiomyopathy
Patients suffering from obesity, insulin resistance, dyslipidemia, and diabetes are at high risk to develop cardiomyopathy [7, 8]. If patients with diabetes or obesity suffered cardiomyopathy, independently of hypertension, or CAD, it can be termed as metabolic syndrome-associated cardiomyopathy. We will discuss obesity-related and diabetes-related cardiomyopathy as the most-studied situations in detail in this section. Diabetic cardiomyopathy (DCM) is first described in the 1970s in some DM patients with no attributable factors [9]. The following Framingham study identified DM as an independent risk factor for cardiomyopathy regardless of comorbidities such as hypertension, CAD, and dyslipidemia [10]. Lately, clinical physicians found that many patients develop DCM with well-controlled DM [11, 12]. It's reported that about 60% diabetic patients suffered unexplained myocardial hypertrophy and fibrosis, which is characterized by left ventricular diastolic dysfunction. This situation emphasized the urgency to elucidate the underlying molecular mechanisms driving this pathological condition [13, 14, 15]. Obesity, insulin resistance, and dyslipidemia could also initiate similar cardiomyopathy even in the absence of DM, which is defined as obesity-related cardiomyopathy [16, 17, 18]. Although there may be distinct underlying mechanisms of cardiac structural and functional abnormality between obesity and DM, they shared similar dysregulated metabolism in the development of cardiomyopathies [16].
3. Heart Changes in MSC Structure, Function, and Metabolism
3.1. Structural Change
The Framingham study found that diabetes may be an independent factor of LVH and cardiac stiffness. Lately, another study found that increased left ventricular mass was only identified in diabetic patients but not in those with impaired fasting glucose concentrations [19], indicating that cardiac dysfunction did not present in the early stage but was a consequence of chronic hyperglycemia. In the following research, Eguchi and colleagues also uncovered the close interrelation between DM and propensity of LVH [20]. Similarly, alterations in the left ventricular structure are also found in obese patients without hypertension and diabetes, in which condition the heart may develop a thicker left ventricular wall, elevated left ventricular mass, left ventricular dilatation, and significant left ventricular remodeling (Figure 1), [21, 22].
Figure 1.
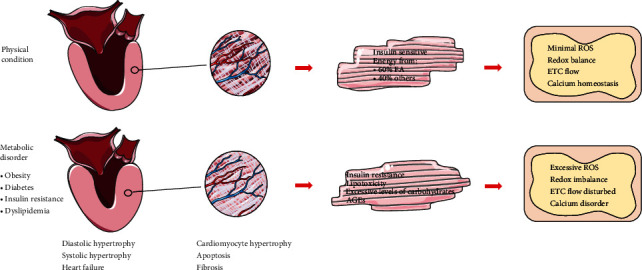
The structural changes of MSC and the mitochondrial changes. Under physical condition, the cardiomyocytes generate energy about 40% via FA metabolism, and mitochondrial hemostasis is regulated via various mechanisms. Obesity, insulin resistance, diabetes, and dyslipidemia cause cardiomyopathy, characterized by thickness and stiffness of the ventricular wall (interstitial fibrosis and cardiomyocyte hypertrophy), cell death, and eventually contractile dysfunction. The accumulation of toxic metabolism intermediates and leads to decreased oxygen utilization induced by mitochondrial dysfunction (reactive oxygen species (ROS) generation, altered calcium, and ETC distribution).
Fibrosis is also a key characteristic in MSC [15, 23], characterized by diastolic restriction, other features including decreased early diastolic filling and increased atrial filling, and isovolumetric relaxation time [24, 25]. One of the dominant features of the diabetic rat model is the increased expression levels of TGF-β1 and connective tissue growth factor, which could attribute to the deposition of collagen in the heart [26, 27]. According to Shimizu et al., diabetes-associated myocardial fibrosis comes with increased deposition of both types I and III collagen. In DM patients presenting diastolic dysfunction, the deposit of procollagen type I carboxyl-terminal peptide levels increased significantly [28]. Myocardial tissue from diabetic patients showed dominant interstitial fibrosis and perivascular fibrosis [9]. In diabetic hearts, the increased matrix metalloproteinase (MMPs) also causes cardiac extracellular matrix disorder and increases connective tissue content [27]. Left ventricular remodeling, including increased wall thickness and ventricular dilatation, is a common result of hemodynamic changes induced by obesity [29, 30]. Both eccentric and concentric LV hypertrophy were found in obese individuals. In addition, the right ventricle is also affected adversely by obesity; the manifestation of right ventricular hypertrophy and final dilatation were reported in heart failure induced by obesity [31, 32]. Histologic findings revealed that increased heart weight and left and right ventricular hypertrophy are proportional to the development of obesity [33].
3.2. Functional Change
Diastolic dysfunction is a significant clinical characteristic of MSC. Schannwell et al. [34] indicated that patients with diabetes demonstrated cardiac diastolic dysfunction without known coronary artery diseases [35]. According to previous evidence, the incidence of diastolic dysfunction varies from 30% to 75% in patients with well-controlled DM but without coronary artery diseases [36].
In addition to diabetes, there is also evidence finding that LV diastolic dysfunction is associated with obesity. Berkalp et al. [37] reported diastolic dysfunction diagnosed by load-dependent spectral Doppler in young obese women. Besides, body mass index (BMI) was an independent predictor of diastolic dysfunction according to a specific Doppler-derived load-independent measurement [38, 39, 40]. Various studies demonstrated that patients with MSCs were usually characterized by increased stiffness of ventricular myocardium and developed systolic dysfunction followed by diastolic dysfunction [41, 42, 43].
3.3. Metabolic Change
The heart is a high-energy-consuming organ. In physical conditions, FA (FA) contributes 60% of energy for the adult heart, with the rest 40% supported by the oxidation of other fuel substrates (Figure 1), [44]. The cardiac mitochondria are the energy house. Thus, the heart muscle is vulnerable to mitochondria damage. Sarah et al. first reported the increased mitochondrial fission in the myocardium of diabetic patients [45]. Lately, other studies found that FA metabolism in the hearts of asymptomatic diabetic patients significantly increased to compensate for the decreased respiratory capacity, suggesting that changes in mitochondrial metabolism are detrimental to the heart [46, 47].
3.4. Lipotoxicity
Deleterious effects of lipid accumulation in non-adipose tissues are usually termed lipotoxicity [47], which may be caused by the accumulation of products from incomplete oxidation, such as acylcarnitines and ROS (Figure 2(a))[22]. Kim and colleagues [48] found that the level of even-chain acylcarnitines increased in skeletal muscle, correlated with increased mitochondria-mediated incomplete oxidation. In the following analysis, Palomer et al. found that PPAR, a mitochondrial biogenic receptor, was involved in lipid metabolism [49], and increased PPAR expression reduced the level of incomplete oxidation of fatty acids in myoblasts. PPAR is also vital to regulating the transcription of FA metabolism via stimulating related transcriptional genes, such as CD36 [50] and pyruvate dehydrogenase kinase 4 [51]. In addition, glycogen synthase kinase 3α (GSK-3α) is a sensor, inducing lipid uptake and accumulation via PPAR activation [52]. Patients with diabetic cardiomyopathy received agents targeted at GSK-3α or PPAR could demonstrate alleviated cardiac lipotoxicity. Ceramides belong to the lipid molecule families, including sphingosine and fatty acid [53]. PPARα-related agents such as K-877, a novel selective PPARα modulate, have a great triglyceride-lowering activity [54]. K-877 could upregulate fibroblast growth factor 21 and lipoprotein lipase levels, which have been identified in various clinical trials [55, 56, 57, 58].
Figure 2.
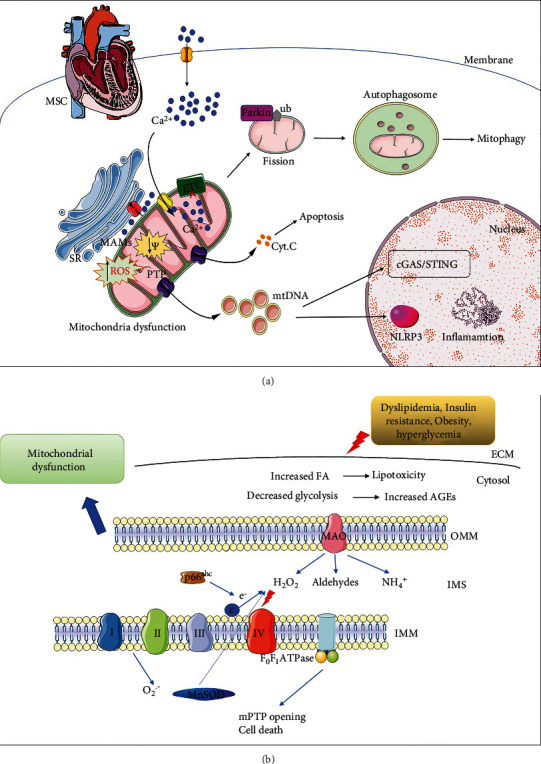
Mitochondrial signaling in metabolic syndrome-related cardiomyopathy. (a) Dyslipidemia, insulin resistance, obesity, and hyperglycemia could cause cardiomyocytes altered calcium homeostasis, including the cytoplasmic calcium, SR, and mitochondrial calcium. Damaged mitochondria undergo fission and are finally cleaned by mitophagy. In addition, the damaged mitochondria exert mPTP opening and cytochrome c release, inducing apoptosis. (b) Dyslipidemia, insulin resistance, obesity, and hyperglycemia lead to lipotoxicity and accumulation of AGEs, leading to the activation of mitochondrial ROS-producing enzymes. p66Shc is activated and catalyzes the formation of hydrogen peroxide (H2O2). Increased ROS could disturb the mitochondrial oxidation respiratory chain, followed by cell death.
Ceramides could activate mitochondria-related apoptotic pathways via increased mitochondrial membrane permeability, leading to increased ROS levels through mitochondrial electron transport distributions, followed by apoptosis and insulin resistance [16]. As a saturated long-chain fatty acid, palmitate could induce apoptosis of rat neonatal cardiomyocytes via the decrease of mitochondrial cardiolipin [59]. Calcium and ROS disorders could initiate long-chain FA accumulation and alter mitochondrial membranes by changing phospholipid composition, followed by the release of cytochrome c from the mitochondria and final apoptosis [60, 61]. To protect the heart from lipotoxicity damage, mitochondrial uncoupling and futile cycling may work as major adaptive mechanisms [22, 62]. Uncoupling proteins (UCPs) work as FA anion exporters, exporting fatty acids out of the mitochondrial matrix by a flip-flop mechanism, reducing the proton gradient, followed by a subsequent decrease in oxidative stress [63]. Similarly, mitochondrial thioesterase 1 could hydrolyze fatty acyl-CoA in the mitochondrial matrix and release FA anion and CoASH, which is essential for the increased oxidation states [64, 65, 66].
3.5. Mitochondria-Mediated Redox Status in DCM
In mitochondrial electron transfer chain (ETC), electrons transfer from reduced to oxidized submits, finally forming oxygen, which is a classic example of redox reactions [11]. NAD+/NADH and NADP+/NADPH, TrxSS/TrxSH2, and GSSG/GSH are important redox couples to maintain mitochondria function [67]. The NADPH/NADP+ redox couple is an important antioxidant defense mechanism by transferring electrons to glutathione and thioredoxin systems, both of which are scavengers of hydrogen peroxide (H2O2) [68]. The mitochondrial redox states of the NADH/NAD+ and NADPH/NADP+ redox couples have different metabolic roles [69]. ETC (via complex I) and the antioxidant system received electrons from fuel substrates with support from NADH/NAD+ [11]. The NADPH/NADP+ couple offers electrons to GSH reductase and thioredoxin reductase 2, maintaining their reduced states [11]. NAD+ participates in cardiac mitochondrial fitness by regulating mitochondrial biogenesis and dynamics. In addition, NAD+ is also a substrate of various enzymes, such as the family of sirtuins (SIRTs) [70].
The interaction between NAD+/NADH redox and mitochondria bioenergetics is critical to maintaining normal cardiac function as an essential regulator in the heart [71]. The early stage of obesity- or diabetes-induced cardiomyopathy is usually accompanied by highly reduced NAD+/NADH redox couple. Furthermore, the well-controlled glucose levels in diabetic patients usually have lower cardiac mortality, indicating that hyperglycemia is a detrimental factor. Previous evidence proved that some non-energy pathways, such as the formation of the late glycation end products, activation of the hexosamine pathway, and activation of the polyol pathway, are central factors contributing to diabetic chronic microvascular complications, mediating cellular redox changes [72, 73]. Aldose reductase converted glucose to sorbitol via NAD+/NADH redox, which has been extensively studied in microvascular complications [74]; however, its impact on diabetic myocardium was not fully elucidated. In high glucose conditions, increased diacylglycerol caused protein kinase C activation and final cardiomyopathy [75]. Glucose-derived methylglyoxal could target mitochondrial proteins and components of calcium cycling, causing metabolic disorders in the heart [76]. O-glcNAcylation is an important post-translational modification via O-GlcNAc transferase and substrate from the hexosamine pathway [77], causing contractile dysfunction, epigenetic modifications, and genetic changes in the diabetic heart.
Compared with glucose, excessive FA damaged mitochondrial energy via increasing NADH, which promoted ETC electron flow, mitochondrial membrane hyperpolarization, and superoxide formation [70]. Excessive circulating FA seems to be the trigger of the change of mitochondrial NAD+/NADH redox state during energetic challenges [11]. Decreased circulating FA could activate NAD+-dependent mitochondrial SIRTs to deacetylate ETC proteins and reverse diastolic dysfunction in DCM [78]. Besides the regulation of the energetic metabolism of NAD+/NADH redox couple, various researches focused on the role of increased acetyl-CoA derived from the oxidation of excessive fuels [79]. In diabetic conditions, increased mitochondrial FA oxidation is usually accompanied by a high acetylation level of related enzymes regardless of unchanged SIRT3 protein abundance [80]. Similarly, a high-fat diet (HFD) led to increased FA oxidation enzyme acetylation and long-chain acyl-CoA dehydrogenase (LCAD) activation, enhancing cardiac FA oxidation [81]; thus, the increased NAD+/NADH redox state and decreased FA oxidation enzyme acetylation could improve heart function in diabetes [82]. SIRT3 is downregulated followed by decreased NAD+/NADH ratio in HFD, the acetylation levels of heat shock protein 10 increased, and induced mitochondria FA oxidation [83]. In the diabetic heart, chronic hyperglycemia could create an excessive oxidative redox environment and disturb mitochondrial antioxidant scavengers via non-mitochondrial mechanisms [84]. In physical conditions, the glutathione and thioredoxin redox couples are the major scavengers of H2O2. However, during hyperglycemia, this scavenger system becomes more oxidized, and short-term mitochondrial palmitate oxidation could provide reduced NADH, normalizing the redox status and providing a more energetic budget to improve the function of the diabetic heart [85]. Chronic exposure to hyperglycemia and excessive FA exhibit multiple metabolic effects. Excessive FA oxidation could generate a high level of acetyl-CoA and NADH, which could activate PDK4, inhibiting pyruvate oxidation [86]. Nicotinamide mononucleotide (NMN) is a kind of intermediate in NAD+ biosynthesis, which was reported to improve multi-organ insulin sensitivity [87]. NMN can improve various metabolic complications related with obesity, although this effect on many other obesity-related metabolomic complications is yet to be explored [88]. Keio University and Washington University School of Medicine of St. Louis carried out a collaborative preclinical trial of NMN to observe the efficacy of NMN on aging [89], hoping that there will be evidence in the field of cardiovascular diseases in the future.
4. Mitochondrial Signaling in Metabolic Syndrome-Related Cardiomyopathy
4.1. Mitochondrial Biogenesis
The biogenesis of mitochondria is associated with rapid cell growth and proliferating cells [90]. It is reported that peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α) is an important transcriptional activator for the regulation of mitochondrial function [91, 92], via the activation of various transcription factors that mediated the gene expression both in the nucleus and mitochondria [93]. PGC-1α-driven mitochondrial biogenesis is indispensable for normal cardiac and skeletal muscle contraction and relaxation by regulating mitochondrial biogenesis, such as regulating fatty acid metabolism and oxidative phosphorylation, increasing mitochondrial number, and enhancing mitochondrial respiration ability [94]. EET analog is an activator of PGC-1α pathway; various evidence indicated that increased levels of EETs are beneficial in the prevention of cardiac consequences besides diabetic treatment [95, 96].
Changes in mitochondrial biogenesis have been reported in patients diagnosed with metabolic syndrome and diabetes [97, 98]. Studies have shown that diabetic cardiomyopathy is characterized by the accumulation of toxic materials, such as long-chain acyl-CoA and acylinosine, which influence the mitochondrial ATP/ADP ratio and reduce mitochondrial metabolic rates [99]. A reduction in mitochondrial biogenesis is observed in metabolic syndrome models, which is consistent with low ATP levels and faulty electron transport. Increased cardiac energy demand modulates gene expression in nuclear and mitochondrial DNA and maximizes the ability of mitochondria to perform oxidative phosphorylation. In the setting of obesity and insulin resistance, nutrient excess, coupled with reduced energy requirements, leads to reduced mitochondrial DNA gene expression, which in turn leads to reduced oxidative phosphorylation [100].
4.2. Mitophagy
It is necessary to remove dysfunctional mitochondria from cardiomyocytes to maintain myocyte survival. Mitophagy is an efficient way to remove damaged mitochondria. Many pathways have been reported to regulate mitophagy (Figure 2(a)). The PINK/Parkin pathway is the first pathway reported to clean damaged mitochondria. As a serine/threonine kinase, PINK1 is imported to healthy mitochondria and degraded by related proteases [101]. With the decrease of mitochondrial membrane potential, PINK1 could accumulate on the outer membrane of mitochondria, followed by the phosphorylation of the membrane proteins. Mitofusin 2 is a mitochondrial dynamic protein, which could recruit Parkin to the mitochondria [102]. Parkin is an E3 ubiquitin ligase; upon translocating to mitochondria, Parkin could ubiquitinate mitochondrial proteins, promoting the recruitment of autophagosome and initiating mitophagy [103]. Studies including obese or diabetic patients found that the transcript levels of PINK1 are decreased in skeletal muscle, suggesting that mitophagy regulated by the PINK1/Parkin pathway is inhibited [104]. In addition, Parkin dysfunction may lead to the loss of pancreatic β cells and the development of insulin deficiency [105]. Bharath and colleagues found that in diabetic mice, the level of p53 increased in β-cells, inhibiting Parkin-mediated mitophagy [106]. However, in p53 deficient mice, restored mitophagy protected β-cell loss induced by streptozotocin (STZ). While the PINK1/Parkin pathway plays an important role in mitochondrial homeostasis in the heart, its role in diabetic cardiomyopathy remains unclear.
The role of mitophagy and its underlying mechanisms in the diabetic heart remain largely unknown [107]. PINK1/Parkin pathway is inhibited in the hearts of various diabetic animals, indicating that mitophagy is closely related to diabetic hearts [108]. Beclin 1 or Atg 16L1 deficient mice could restore the PINK1/Parkin pathway, alleviating diabetic heart function [109]. Autophagy and mitophagy performed differently in the pathologic process of diabetic hearts. In the early stage of the diabetic heart, autophagy is inhibited, but mitophagy decreased in the advanced stage [110]. Thus, it is important to separate mitophagy from autophagy to uncover the underlying mechanisms of diabetic hearts. Xu et al. found that the expression of Rab 9 increased in diabetes, mediating damaged mitochondria degradation [111]. This result indicated that alternative autophagy may be increased to initiate mitophagy, compensating canonical autophagy and finally protecting the diabetic heart. However, other autophagic pathways, such as Beclin-independent and Atg7-dependent pathways, are necessary to be explored.
4.3. Mitochondrial-Derived ROS
The generation and clearance of ROS are important in various pathological conditions [112]. Current evidence found that ROS could damage numerous molecules including proteins, amino acids, lipids, and DNA (Figure 2(a)). Hyperglycemia produced mitochondrial ROS in various ways [113], such as GAPDH inhibition, activation of the polyol pathway, formation of AGEs, and glucose auto-oxidation; in turn, these pathways could exacerbate ROS generation. For example, in the polyol pathway, NAPDH is indispensable for GSH generation, and the increased AGEs lead to ROS formation [114]. Inhibition of the formation of AGEs or blocking its receptors significantly improved the cardiac function of DCM. In addition, Kaludercic and colleagues found that in diabetic mice, increased expression of p53 led to disturbed mitochondrial respiration and increased oxidative stress, followed by cytochrome c release from mitochondria [113]. In addition to hyperglycemia, lipotoxicity could also lead to oxidative stress via the oxidation of free fatty acids [115]. Mitochondrial ROS could activate the NLRP3 inflammasome, promoting cardiac fibrosis [116]. Moreover, hyperglycemia, as well as inflammation, exhibits synergistic effects, enhancing ROS generation and final cell death [113].
In the diabetic heart, ROS is involved in various processes, such as inflammation, angiogenesis, and apoptosis to mediate fibrosis and hypertrophy [117]. In diastolic cardiac dysfunction, ROS could target sarcomere proteins, leading to the accumulation of oxidative thick and thin filaments, demonstrating LV stiffness [118, 119, 120, 121]. Evidence indicated that oxidation and disulfide bridge formation led to increased cardiac stiffness [122]. Recently, Grutzner et al. found that ATP synthase was cleaved by calpain-1 induced by hyperglycemia, resulting in increased ROS generation and deteriorated DCM [123]. Moreover, mitochondrial ROS could increase the propensity of mitochondrial permeability transition pore (mPTP) opening and apoptosis [124].
P66Shc is another important source of ROS in mitochondria (Figure 2(b)) [125, 126]. Epigenetic modification of the p66Shc promoter is important for its expression [127]. Hyperglycemia could induce p66Shc phosphorylation and translocate to mitochondrial intermembrane space, resulting in the formation of H2O2 [128, 129]. Meanwhile, p66Shc could also amplify oxidative stress by activating mitochondrial NOX or by downregulating the synthesis of other antioxidant enzymes [130]. Accordingly, cells and mice lacking p66Shc show reductions in markers of oxidative stress [131]. Phosphorylation by PKC is required for p66Sshc translocating to mitochondria in hyperglycemia conditions [132], indicating that p66Shc may play a role in DCM [132]. Diabetic p66Shc−/− hearts displayed preserved cardiac progenitor cell replication and LV end-diastolic pressure [133]. Szeto-Schiller peptide SS31 is a free-radical scavenger that protects from cardiac hypertrophy, via preventing cardiolipin from converting cytochrome c into a peroxidase or protecting mitochondrial cristae structure [134, 135].
Mitochondria dynamics are also reported to be associated with ROS formation that may reciprocally modulate each other. In diabetic mice and diabetic patients, cardiomyocytes demonstrated increased ROS, mitochondrial fragmentation, cristae disruption, and swelling [136]. Dillmann et al. found that c ronic-hyperglycemia–induced mitochondrial fragmentation could be reversed by antioxidants, indicating that ROS are causally related to mitochondrial morphology. This partially explained the underlying mechanism of antioxidants treatment in DCM [137]. Mitochondrial ROS reduction via cardiac-specific Mn-SOD overexpression or stimulation of the AMPK pathway will rescue mitochondria from fatty acid- or hyperglycemia-induced events [138]. Brownlee's group reported that high intracellular glucose disturbed mitochondrial respiration, leading to mitochondrial membrane hyperpolarization, and ETC superoxide production [139, 140]. Interventions such as ETC complex II inhibition could decrease the levels of mitochondrial ROS and prevent the activation of the hexosamine pathway, AGEs formation, and sorbitol accumulation [112]. Additionally, Brownlee et al. also found that hyperglycemia could not lead to increased ETC superoxide production in Rho endothelial cells lacking ETC function [141].
High glucose induced cardiomyocyte damage via a similar mechanism [142, 143, 144]. Current evidence proved that mitochondrial ROS could be generated from cardiac lipotoxicity [145]. Palmitate stimulated ROS generation, leading to mitochondrial fission via Drp1 phosphorylation and OPA1 degradation [145]. This process could be amplified by increased ROS levels and vice versa [146]. In addition, mitochondrial oxidative stress promoted the occurrence of post-translational modifications, such as methylglyoxal formation and O-GlcNAcylation, contributing to the impairment of heart function [147, 148]. O-GlcNAc transferase enzyme located in the IMM could interact with complex IV of the respiratory chain in physical conditions. Hyperglycemia could alter the function of the respiratory chain in mitochondria via dysregulation of O-GlcNAcylation [147].
In the diabetic rat model, the interaction between O-GlcNAc transferase and complex IV is impaired, leading to the disorder of complex IV activity and reduction of mitochondrial membrane potential. Many mitochondrial proteins are O-GlcNAcylated, such as Drp1 and OPA1, resulting in mitochondrial dysfunction [149]. On the other hand, methylglyoxal modifications also cause calcium disturbance, followed by mitochondrial damage [150], calcium-related proteins including ryanodine receptor 2 and SERCA2a that may transform to methylglyoxal forms and cause mitochondrial dysfunction [76]. Collectively, these studies emphasized the critical role of ETC-derived superoxide in diabetic conditions. TACT (trial to assess chelation therapy) is a clinical trial which uses edetate disodium to inhibit conversion of H2O2 to peroxynitrite. It came out that the chelators could significantly reduce cardiac events especially in diabetic patients [151].
4.4. Monoamine Oxidases
Monoamine oxidases (MAOs) are flavoenzymes localized at the outer mitochondrial membrane [152]. There are two isoforms of MAOs (MAO-A and MAO-B), with different characteristics of structures, substrate inclination, and tissue distribution (Figure 2(b)) [153]. Over the last decade, many studies demonstrated that the redox unbalance induced by MAOs promoted the development of cardiovascular diseases [154, 155, 156, 157]. MAOs have been reported to be involved in diabetic complications; Emory et al. found that MAO inhibitors could reverse hyperglycemia conditions and improve cardiac function [158]. Additionally, pioglitazone could function as a specific and reversible MAO-B inhibitor in diabetic patients [159]. In diabetic animal models including mice and rats, MAO inhibition was also proved to reverse DCM function [160, 161]. MAO inhibitors can prevent oxidative changes, followed by diastolic dysfunction, and myocardial fibrosis.
Besides, MAO inhibitors could prevent mast cell degranulation, alleviating cardiac fibrosis in diabetic hearts. Similar pathological processes were also found in cardiomyocytes and mesenchymal stromal cells [160, 161]. According to current studies, the underlying mechanism of MAO toxicity was due to the high level of H2O2 and aldehyde formation, leading to impaired mitochondrial function. Increased MAO activity induced inflammasome activation and promoted the inflammatory process, while MAO inhibition could also cause mitochondrial dysfunction and ER stress, contributing to DCM [160]. Mitochondria and ER communicate a lot in physical and functional aspects; there is a special domain named mitochondrial associated membranes (MAMs) function as a platform to regulate calcium transport, lipid metabolism, and autophagy flow (Figure 2(a)). MAO is localized at the outer mitochondrial membrane and could interact with proteins in MAMs to regulate ER function [113]. Mitochondrial aldehyde dehydrogenase 2 (ALDH2) is important to mediate mitochondrial metabolism; MAO-related oxidative stress inhibits ALDH2 activity, resulting in the accumulation of toxic aldehydes [162], while increasing ALDH2 activity protects cardiac damage induced by streptozotocin. Some evidence supports that there is a ROS amplification mechanism or so-called the ROS-induced ROS release mechanism, where initial stress triggers ROS production that, in turn, leads to more ROS formation, setting up a positive feedback loop for the ROS-induced ROS release [163, 164]. An example of this is that high glucose in adult cardiomyocytes increased MAO-dependent ROS formation, leading to mPTP opening and ER stress, which may take other processes into this amplification loop [165, 166]. MAO inhibitors, such as iproniazid and nialamide, have been evaluated for their potential effects in patients with heart diseases [167]. In the future, hoping there would be some clinical trials focusing on the applications of MAO inhibitors in metabolic cardiomyopathy.
4.5. Calcium Homeostasis
Calcium is required for maintaining the proper function of the heart; abnormal mitochondrial calcium handling contributes to mitochondrial dysfunction in diabetic cardiomyopathy (Figure 2(a))[168]. Diabetic cardiomyopathy could present various degrees of calcium abnormalities; the decreased activity of sarcolemma Na-Ca exchanger in the diabetic heart can decrease the removal of calcium, leading to the increase of intracellular calcium concentration. MCU is an integral membrane protein mediating mitochondrial calcium across the inner mitochondrial membrane [169]. Previous evidence found that MCU protein levels decreased in the heart of diabetic mice; similar results could be observed in mouse neonatal cardiomyocytes under hyperglycemia. The decrease of MCU expression usually coexisted with impaired mitochondria calcium uptake and release in high glucose conditions. Similar results were observed in mitochondria from diabetic hearts [170]. Diaz-Juarez et al. found that hyperglycemia impaired both mitochondrial calcium uptake and release in living cardiomyocytes detected by calcium sensor Pericam [171]. Furthermore, the fact that cardiomyocytes from diabetic mouse hearts or neonatal cardiomyocytes exposed to high glucose and high fat inducing type 2 diabetes demonstrated a significant decrease in mitochondrial calcium [168, 172]. Mitochondrial Ca2+ is an important signaling mechanism to regulate mitochondrial energetic activity. Mitochondrial calcium is an important co-factor to regulate the activity of ATP formation and mitochondrial ETC [173]. It is clear that the heart mainly metabolizes fatty acids under physiological conditions [174, 175]. However, in diabetic hearts, the FA oxidation increased with diminished glucose oxidation and decreased mitochondrial calcium. Decreased calcium further caused the decreased activity of PDC [168]. Calcium/calmodulin dependent protein kinase II (CaMKII) is a multifunctional serine/threonine kinase that mediates physiological responses upon acute βadrenergic activation [176]. KN-93 is a CaMKII inhibitor; Sommese et al. reported that KN-93 could prevent spontaneous SR calcium release and the following arrhythmias in diabetic rats.
More evidence also found PDC activity decreased in leptin receptor-deficient mice and increased PDC phosphorylation in cardiomyocytes under hyperglycemia. Impaired glucose oxidation will impair cardiomyocytes' contractile function in the following ways: (1) ATP generated from glycolysis is necessary to promote calcium uptake and maintain SR, sarcolemma, and mitochondria dynamic homeostasis [177, 178, 179]; (2) the “oxygen-wasting” effect caused by fatty acids would induce a higher ratio between cardiac oxygen consumption and heart work, especially following β-adrenergic stimulation [180]; and (3) decreased ATP generation from glycolysis would damage the integrity of cellular membranes.
4.6. Mitochondria Dynamics
Mitochondrial dynamics (Figure 2(a)) is important to maintain mitochondrial homeostasis. The myocardia cell H9C2 exposed to hyperglycemia demonstrated a significant mitochondrial fragmentation [181], leading to mitochondrial ROS accumulation and final cell apoptosis [182]. Furthermore, these events could be restored by the inhibition of Drp1, indicating that cell apoptosis is mitochondrial dynamics dependent. Moreover, Watanabe et al. reported that Drp1 could interact with ROS, inducing mitochondrial dysfunction and inhibiting the insulin signaling pathway. Antioxidants could partially overcome this process [183]. Also, Makino et al. found mitochondrial fission in endothelial cells from diabetic hearts; further exploration displayed reduced OPA1 and increased Drp1 [184], which could be abolished by antioxidants treatment. This evidence suggested a closed relationship between oxidative stress and mitochondrial dynamics in endothelial cells [185].
Consistently, neonatal cardiomyocytes suffered high glucose presenting mitochondrial fission and decreased mitochondrial membrane potential [186]. In this process, the expression of mitochondrial fusion proteins (OPA1 and Mfn1/2) decreased, and the fission-related proteins (Fis 1 and Drp1) increased significantly. This situation could be restored via overexpression of OPA1 or decreased expression of Drp1[184]. Recently, Parra et al. reported the relationship between mitochondrial dynamics and insulin's impact on cardiomyocyte metabolism [187]. Meanwhile, treating cardiomyocytes with insulin could increase OPA1 levels and promote mitochondrial fusion, increasing the production of ATP [185]. These findings are consistent with those reported by Quirós et al.; they found that OPA1 disturbance causes insulin resistance, impaired glucose homeostasis, and disordered thermogenesis in Oma1 knockout mice [188]. Oma1–/– mice developed metabolic disorders after HFD, highlighting the role of OPA1 in regulating mitochondrial function [189]. And Parra et al. also reported that the impact of insulin on mitochondrial metabolism was impaired in OPA1−/− and Mfn2−/− cells [187]. Clinically, in diabetic individuals, impaired mitochondrial functions and diminished levels of OPA1 and Mfn2 were also observed [190, 191, 192]. However, whether mitochondrial dynamic regulation contributes to the cardiac function in diabetic cardiomyopathy is unclear so far. However, current evidence indicates that mitochondrial fragmentation may be a “starting point” of pathological processes involved in cardiac metabolic diseases [193]. Based on this, we hypothesize that ventricular dysfunction occurring in the transition from obesity to DM is caused, at least in part, by the deterioration of cardiac myocyte mitochondrial function. Further studies are necessary to explore the impact of mitochondrial dynamic targets on cardiac performance.
4.7. Potential Therapeutic Options
With insight into the mechanisms of MSC, studies were focused on developing novel strategies to protect the heart from obesity and diabetes and to improve the clinical outcomes of these patients. In this section, we will discuss the targets of mitochondria in animal studies and clinical trials.
A series of evidence linked oxidative stress to cardiovascular diseases [194, 195]. Various researches hold the idea that reducing ROS formation could protect the heart from damage caused by diabetes [113]. Nevertheless, many clinical trials failed to identify the protective effects of antioxidants in human subjects. When exploring the certain reason for this situation, it is found that ROS is a double-edged sword; a high level of ROS is deleterious; however, a certain level of ROS is beneficial and required for maintaining physical function. SGLT2 inhibitors are novel antidiabetic agents, exhibiting cardiac protective effects in patients with or without diabetes [196, 197, 198]. Interestingly, human and rodent hearts do not express SGLT2, indicating that the direct cardioprotective effects of SGLT2 inhibitors are independent of their action on SGLT2 [199]. Uthman and colleagues reported that SGLT2 inhibitors could reduce intracellular calcium and sodium levels, improving cardiac mitochondrial function [200]. Empagliflozin could reduce oxidative stress in diabetic rats by interfering with NADPH oxidase activity [201]. In addition, Chenguang et al. also found that empagliflozin suppressed oxidative stress and fibrosis via the inhibition of the transforming growth factor β/Smad pathway and activation of Nrf2/ARE signaling [202]. Furthermore, in diabetic mice, the GC-cGMP-PKG pathway was activated by empagliflozin, coming with alleviation of cardiac hypertrophy, reduced cardiac fibrosis, oxidative stress, and cell death [203]. Increased cGMP levels could help protect against cardiac fibrosis and hypertrophy and various agents such as cGMP-binding phosphodiesterase type 5 (PDE5) [204, 205].
Another FDA-approved agent related to oxidative stress is the soluble guanylate cyclase stimulator vericiguat [206]. Substantial evidence indicated that diastolic cardiac dysfunction is associated with excessive ROS production from the coronary microvasculature, decreasing cGMP levels, and PKG activity [207]. A recent clinical trial found that patients with diastolic cardiac dysfunction receiving vericiguat have an improvement in quality of life [208]. In the future, more studies should be carried out to identify ROS targets and develop successful therapies for the diseased heart. More details are necessary to uncover the mechanisms of SGLT2 inhibitors and vericiguat in MSC.
Animal experiments identified NOX4 as a potential therapeutic target [209]. NOX regulation could maintain the mitochondrial ROS homeostasis. Various NOX4 inhibitors such as GKT-831 are developed [210], although the effects of NOX inhibitors in reducing cardiovascular complications in diabetic patients need more tests. It is clear that mitochondria are the major ROS producers, Ting et al. employed mito-TEMPO, a mitochondria-targeted antioxidant, in the mice model of DCM and found the cardioprotective effects of mito-TEMPO [211]. Metformin is a widely used antidiabetic agent, which is also identified as a potent autophagy inducer. It has been proved in OVE26 mice that metformin could activate the AMPK pathway, enhancing mitochondrial autophagy and preventing cardiomyopathy [212]. Thus, the activation of AMPK may act as a novel approach for DCM treatment. Several molecules designed by targeting heme oxygenase-1 and mitochondrial aldehyde dehydrogenase (ALDH2) may serve as activators of AMPK, recovering normal autophagic activity and protecting from cardiomyopathy [162]. AICAR is an AMPK activator which was found to reduce blood glucose concentration and increase insulin sensitivity; however, AICAR was reported not suitable for clinical application [213, 214].
Many studies have investigated the efficacy of agents to prevent adverse cardiac remodeling in diabetic models. Cinnamoyl anthranilate could inhibit the TGFβ pathway [215]. FT23 and FT011 are agents derived from cinnamoyl anthranilate, which could attenuate cardiac dysfunction in mice with DCM [216, 217]. cGMP homeostasis is a key factor to maintain PKG activity and thereby prevent cardiac stiffness [218]. Relaxin is a kind of antifibrotic hormone and exerts antifibrotic properties [219]. It has been reported that relaxin plays a critically protective role in various cardiovascular diseases, including suppression of inflammation [220]. Particularly, relaxin alleviated cardiac fibrosis by inhibiting collagen production from cardiac fibroblasts, fibroblast to myofibroblast transition, and increasing MMP levels [221]. Relaxin−/− mice exerted relief of ventricular chamber stiffness and improved diastolic function after receiving recombinant human relaxin [222, 223]. A clinical trial of PDE5A carried out in patients with diabetes and diastolic cardiac dysfunction [224] found that sildenafil could improve LV torsion, strain, and contraction, reducing the levels of the pro-inflammatory factors C- X-C motif chemokine 10 and IL-8 in plasma [225].
GLP1 is an incretin secreted from intestinal L-type cells and reported to reduce blood glucose levels in a glucose-dependent manner by promoting insulin secretion. Zhou et al. reported that liraglutide, a GLP1 analog, is protected against the development of diabetic cardiomyopathy via inhibiting ER stress [226]. In addition, liraglutide could protect cardiomyocytes from IL-1β-induced mitochondrial membrane potential reduction and reduced ATP production [227]. In 2019, a clinical trial enrolled patients with diabetes treated with liraglutide demonstrated reduced early LV diastolic filling and LV filling pressure [228]. Conclusively, GLP1 receptor agonists are recommended as the first-line injectable agent for patients with diabetes and cardiovascular diseases [229].
In summary, various agents were developed as potential therapeutic strategies for MSC, and some have been proved to regulate mitochondrial function. However, there are still many unsolved questions needed to answer. More efforts are needed to facilitate the translation of basic research to the clinic.
5. Conclusion
Mitochondrial homeostasis is important for heart function. In this review, we summarized the evidence that mitochondrial disorder is a major cause of MSC. Many mechanisms including mitochondrial ROS, mitochondrial dynamics, and calcium disorder play great roles in MSC. Accordingly, many targets involved in mitochondrial homeostasis provide opportunities for the development of novel therapeutics. Although there are currently few drugs with successful clinical transformations, more researches are necessary to carry out and elucidate more promising targets in the future.
Acknowledgments
This work was supported by the National Key R&D Program of China (Grant No.2021ZD0111004), the National Natural Science Foundation of China (Grant No.82070357), and the Beijing Key Clinical Subject Program.
Abbreviations
- AGEs:
Advanced glycation end products
- ALDH2:
Aldehyde dehydrogenase
- AMPK:
Adenosine monophosphate activated protein kinase
- ARE:
Antioxidant response element
- BMI:
Body mass index
- CAD:
Coronary artery disease
- CVD:
Cardiovascular diseases
- DCM:
Diabetic cardiomyopathy
- DM:
Diabetes mellitus
- ETC:
Electron transfer chain
- ER:
Endoplasmic reticulum
- FA:
Fatty acid
- GLP-1:
Glucagon-like peptide-1
- GSK-3α:
Glycogen synthase kinase 3α
- HF:
Heart failure
- HFD:
High fat diet
- H2O2:
Hydrogen peroxide
- IMM:
Inner mitochondrial membrane
- LCAD:
Long chain acyl-CoA dehydrogenase
- LVH:
Left ventricular hypertrophy
- MAOs:
Monoamine oxidases
- MAMs:
Mitochondrial associated membranes
- MCU:
Mitochondrial calcium uniporter
- MMPs:
Matrix metalloproteinase
- mPTP:
Mitochondrial permeability transition pore
- MSCDs:
Metabolic syndrome–associated cardiac diseases
- MSC:
Metabolic syndrome–associated cardiomyopathy
- NOX4:
NADPH oxidase 4
- Nrf2:
Nuclear factor 2 related factor 2
- PDC:
Pyruvate dehydrogenase complex
- PDK4:
Pyruvate dehydrogenase kinase 4
- PKC:
Protein kinase C
- PINK:
PTEN-induced putative kinase 1
- PPAR:
Peroxisome proliferators activate receptors
- ROS:
Reactive oxygen species
- SGLT2:
Sodium-glucose cotransporter-2
- SIRTs:
Sirtuins
- STZ:
Streptozotocin
- UCPs:
Uncoupling proteins.
Contributor Information
Yanguo Xin, Email: xinyanguo77@163.com.
Hongwei Li, Email: lhw19656@sina.com.
Data Availability
All related materials are available from Hongwei Li.
Consent
All authors offer consent for publication.
Conflicts of Interest
All authors declare that there are no conflicts of interest.
Authors' Contributions
Jiayu Li and Yanguo Xin wrote the main manuscript. Jingye Li and Yijun Chen prepared the figures. Wenyu Hu gave the frame of the manuscript. Hui Qiu, Xuhe Gong, Hui Chen, and Hongwei Li reviewed the manuscript. Hongwei Li offered the idea of the manuscript. Jiayu Li and Jingye Li contributed to equally to this work.
References
- 1.Piché M. E., Tchernof A., Després J. P. Obesity phenotypes, diabetes, and cardiovascular diseases. Circulation Research . 2020;126:1477–1500. doi: 10.1161/CIRCRESAHA.120.316101. [DOI] [PubMed] [Google Scholar]
- 2.Lim S. S., Vos T., Flaxman A. D., et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet . 2012;380:2224–2260. doi: 10.1016/S0140-6736(12)61766-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zafar U., Khaliq S., Ahmad H. U., Manzoor S., Lone K. P. Metabolic syndrome: an update on diagnostic criteria, pathogenesis, and genetic links. Hormones (Athens) . 2018;17:299–313. doi: 10.1007/s42000-018-0051-3. [DOI] [PubMed] [Google Scholar]
- 4.Mottillo S., Filion K. B., Genest J., et al. The metabolic syndrome and cardiovascular risk a systematic review and meta-analysis. Journal of the American College of Cardiology . 2010;56:1113–1132. doi: 10.1016/j.jacc.2010.05.034. [DOI] [PubMed] [Google Scholar]
- 5.Federico M., De la Fuente S., Palomeque J., Sheu S. S. The role of mitochondria in metabolic disease: a special emphasis on heart dysfunction. J Physiol . 2021;599:3477–3493. doi: 10.1113/JP279376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Del Campo A., Perez G., Castro P. F., Parra V., Verdejo H. E. Mitochondrial function, dynamics and quality control in the pathophysiology of HFpEF. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease . 2021;1867, article 166208 doi: 10.1016/j.bbadis.2021.166208. [DOI] [PubMed] [Google Scholar]
- 7.Gargiulo P., Marsico F., Renga F., et al. The metabolic syndrome in heart failure: insights to specific mechanisms. Heart Failure Reviews . 2020;25:1–7. doi: 10.1007/s10741-019-09838-6. [DOI] [PubMed] [Google Scholar]
- 8.Kain V., Halade G. V. Metabolic and biochemical stressors in diabetic cardiomyopathy. Frontiers in Cardiovascular Medicine . 2017;4, article 31 doi: 10.3389/fcvm.2017.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rubler S., Dlugash J., Yuceoglu Y. Z., Kumral T., Branwood A. W., Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. The American Journal of Cardiology . 1972;30:595–602. doi: 10.1016/0002-9149(72)90595-4. [DOI] [PubMed] [Google Scholar]
- 10.Kannel W. B., Hjortland M., Castelli W. P. Role of diabetes in congestive heart failure: the Framingham study. The American Journal of Cardiology . 1974;34:29–34. doi: 10.1016/0002-9149(74)90089-7. [DOI] [PubMed] [Google Scholar]
- 11.Berthiaume J. M., Kurdys J. G., Muntean D. M., Rosca M. G. Mitochondrial NAD(+)/NADH redox state and diabetic cardiomyopathy. Antioxid Redox Signal . 2019;30:375–398. doi: 10.1089/ars.2017.7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poirier P., Bogaty P., Garneau C., Marois L., Dumesnil J. G. Diastolic dysfunction in normotensive men with well-controlled type 2 diabetes: importance of maneuvers in echocardiographic screening for preclinical diabetic cardiomyopathy. Diabetes Care . 2001;24:5–10. doi: 10.2337/diacare.24.1.5. [DOI] [PubMed] [Google Scholar]
- 13.Di Bonito P., Moio N., Cavuto L., et al. Early detection of diabetic cardiomyopathy: usefulness of tissue Doppler imaging. Diabetic Medicine . 2005;22:1720–1725. doi: 10.1111/j.1464-5491.2005.01685.x. [DOI] [PubMed] [Google Scholar]
- 14.Ernande L., Rietzschel E. R., Bergerot C., et al. Impaired myocardial radial function in asymptomatic patients with type 2 diabetes mellitus: a speckle-tracking imaging study. Journal of the American Society of Echocardiography . 2010;23:1266–1272. doi: 10.1016/j.echo.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 15.Jia G., Hill M. A., Sowers J. R. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circulation Research . 2018;122:624–638. doi: 10.1161/CIRCRESAHA.117.311586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakamura M., Sadoshima J. Cardiomyopathy in obesity, insulin resistance and diabetes. The Journal of Physiology . 2020;598:2977–2993. doi: 10.1113/JP276747. [DOI] [PubMed] [Google Scholar]
- 17.Chrysant S. G., Chrysant G. S. New insights into the true nature of the obesity paradox and the lower cardiovascular risk. Journal of the American Society of Hypertension . 2013;7:85–94. doi: 10.1016/j.jash.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 18.Mandavia C. H., Pulakat L., DeMarco V., Sowers J. R. Over-nutrition and metabolic cardiomyopathy. Metabolism . 2012;61:1205–1210. doi: 10.1016/j.metabol.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Packer M. Autophagy-dependent and -independent modulation of oxidative and organellar stress in the diabetic heart by glucose-lowering drugs. Cardiovascular Diabetology . 2020;19:p. 62. doi: 10.1186/s12933-020-01041-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eguchi K., Boden-Albala B., Jin Z., et al. Association between diabetes mellitus and left ventricular hypertrophy in a multiethnic population. The American Journal of Cardiology . 2008;101:1787–1791. doi: 10.1016/j.amjcard.2008.02.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y., Ren J. Epigenetics and obesity cardiomyopathy: from pathophysiology to prevention and management. Pharmacology & Therapeutics . 2016;161:52–66. doi: 10.1016/j.pharmthera.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 22.Harmancey R., Wilson C. R., Taegtmeyer H. Adaptation and maladaptation of the heart in obesity. Hypertension . 2008;52:181–187. doi: 10.1161/HYPERTENSIONAHA.108.110031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun N., Finkel T. Cardiac mitochondria: a surprise about size. Journal Of Molecular and Cellular Cardiology . 2015;82:213–215. doi: 10.1016/j.yjmcc.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamblin M., Friedman D. B., Hill S., Caprioli R. M., Smith H. M., Hill M. F. Alterations in the diabetic myocardial proteome coupled with increased myocardial oxidative stress underlies diabetic cardiomyopathy. Journal Of Molecular and Cellular Cardiology . 2007;42:884–895. doi: 10.1016/j.yjmcc.2006.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.von Bibra H., St John Sutton M. Diastolic dysfunction in diabetes and the metabolic syndrome: promising potential for diagnosis and prognosis. Diabetologia . 2010;53:1033–1045. doi: 10.1007/s00125-010-1682-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qin W. D., Liu G. L., Wang J., et al. Poly(ADP-ribose) polymerase 1 inhibition protects cardiomyocytes from inflammation and apoptosis in diabetic cardiomyopathy. Oncotarget . 2016;7:35618–35631. doi: 10.18632/oncotarget.8343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frangogiannis N. G. The extracellular matrix in myocardial injury, repair, and remodeling. The Journal of Clinical Investigation . 2017;127:1600–1612. doi: 10.1172/JCI87491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shimizu M., Umeda K., Sugihara N., et al. Collagen remodelling in myocardia of patients with diabetes. Journal of Clinical Pathology . 1993;46:32–36. doi: 10.1136/jcp.46.1.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lakhani M., Fein S. Effects of obesity and subsequent weight reduction on left ventricular function. Cardiology in Review . 2011;19:1–4. doi: 10.1097/CRD.0b013e3181f877d2. [DOI] [PubMed] [Google Scholar]
- 30.Obokata M., Reddy Y. N. V., Pislaru S. V., Melenovsky V., Borlaug B. A. Evidence supporting the existence of a distinct obese phenotype of heart failure with preserved ejection fraction. Circulation . 2017;136:6–19. doi: 10.1161/CIRCULATIONAHA.116.026807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Obokata M., Reddy Y. N. V., Melenovsky V., Pislaru S., Borlaug B. A. Deterioration in right ventricular structure and function over time in patients with heart failure and preserved ejection fraction. European Heart Journal . 2019;40:689–697. doi: 10.1093/eurheartj/ehy809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mohammed S. F., Hussain I., AbouEzzeddine O. F., et al. Right ventricular function in heart failure with preserved ejection fraction: a community-based study. Circulation . 2014;130:2310–2320. doi: 10.1161/CIRCULATIONAHA.113.008461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alpert M. A. Obesity cardiomyopathy: pathophysiology and evolution of the clinical syndrome. The American Journal of the Medical Sciences . 2001;321:225–236. doi: 10.1097/00000441-200104000-00003. [DOI] [PubMed] [Google Scholar]
- 34.Schannwell C. M., Schneppenheim M., Perings S., Plehn G., Strauer B. E. Left ventricular diastolic dysfunction as an early manifestation of diabetic cardiomyopathy. Cardiology . 2002;98:33–39. doi: 10.1159/000064682. [DOI] [PubMed] [Google Scholar]
- 35.Bugger H., Abel E. D. Mitochondria in the diabetic heart. Cardiovascular Research . 2010;88:229–240. doi: 10.1093/cvr/cvq239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ha J. W., Lee H. C., Kang E. S., et al. Abnormal left ventricular longitudinal functional reserve in patients with diabetes mellitus: implication for detecting subclinical myocardial dysfunction using exercise tissue Doppler echocardiography. Heart . 2007;93:1571–1576. doi: 10.1136/hrt.2006.101667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berkalp B., Cesur V., Corapcioglu D., Erol C., Baskal N. Obesity and left ventricular diastolic dysfunction. International Journal of Cardiology . 1995;52:23–26. doi: 10.1016/0167-5273(95)02431-u. [DOI] [PubMed] [Google Scholar]
- 38.Rozenbaum Z., Topilsky Y., Khoury S., Pereg D., Laufer-Perl M. Association of body mass index and diastolic function in metabolically healthy obese with preserved ejection fraction. International Journal of Cardiology . 2019;277:147–152. doi: 10.1016/j.ijcard.2018.08.008. [DOI] [PubMed] [Google Scholar]
- 39.Musaeus K. D., Pareek M. Body mass index, type 2 diabetes, and left ventricular function. Cardiovascular Diabetology . 2018;17:p. 3. doi: 10.1186/s12933-017-0649-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mehta S. K., Richards N., Lorber R., Rosenthal G. L. Abdominal obesity, waist circumference, body mass index, and echocardiographic measures in children and adolescents. Congenital Heart Disease . 2009;4:338–347. doi: 10.1111/j.1747-0803.2009.00330.x. [DOI] [PubMed] [Google Scholar]
- 41.Wong C. Y., O'Moore-Sullivan T., Leano R., Byrne N., Beller E., Marwick T. H. Alterations of left ventricular myocardial characteristics associated with obesity. Circulation . 2004;110:3081–3087. doi: 10.1161/01.CIR.0000147184.13872.0F. [DOI] [PubMed] [Google Scholar]
- 42.Peterson L. R., Waggoner A. D., Schechtman K. B., et al. Alterations in left ventricular structure and function in young healthy obese women: assessment by echocardiography and tissue Doppler imaging. Journal of the American College of Cardiology . 2004;43:1399–1404. doi: 10.1016/j.jacc.2003.10.062. [DOI] [PubMed] [Google Scholar]
- 43.Ferraro S., Perrone-Filardi P., Desiderio A., et al. Left ventricular systolic and diastolic function in severe obesity: a radionuclide study. Cardiology . 1996;87:347–353. doi: 10.1159/000177118. [DOI] [PubMed] [Google Scholar]
- 44.Lopaschuk G. D., Ussher J. R., Folmes C. D., Jaswal J. S., Stanley W. C. Myocardial fatty acid metabolism in health and disease. Physiological Reviews . 2010;90:207–258. doi: 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- 45.Kassab S., Albalawi Z., Daghistani H., Kitmitto A. Mitochondrial arrest on the microtubule highway-a feature of heart failure and diabetic cardiomyopathy? Frontiers in Cardiovascular Medicine . 2021;8, article 689101 doi: 10.3389/fcvm.2021.689101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harayama T., Riezman H. Understanding the diversity of membrane lipid composition. Nature Reviews Molecular Cell Biology . 2018;19:281–296. doi: 10.1038/nrm.2017.138. [DOI] [PubMed] [Google Scholar]
- 47.Labbé S. M., Grenier-Larouche T., Noll C., et al. Increased myocardial uptake of dietary fatty acids linked to cardiac dysfunction in glucose-intolerant humans. Diabetes . 2012;61:2701–2710. doi: 10.2337/db11-1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huffman K. M., Koves T. R., Hubal M. J., et al. Metabolite signatures of exercise training in human skeletal muscle relate to mitochondrial remodelling and cardiometabolic fitness. Diabetologia . 2014;57:2282–2295. doi: 10.1007/s00125-014-3343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Palomer X., Barroso E., Zarei M., Botteri G., Vázquez-Carrera M. PPARβ/δ and lipid metabolism in the heart. Biochim Biophys Acta . 2016;1861:1569–1578. doi: 10.1016/j.bbalip.2016.01.019. [DOI] [PubMed] [Google Scholar]
- 50.Maréchal L., Laviolette M., Rodrigue-Way A., et al. The CD36-PPARγ pathway in metabolic disorders. International Journal of Molecular Sciences . 2018;19 doi: 10.3390/ijms19051529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Watt M. J., Southgate R. J., Holmes A. G., Febbraio M. A. Suppression of plasma free fatty acids upregulates peroxisome proliferator-activated receptor (PPAR) alpha and delta and PPAR coactivator 1alpha in human skeletal muscle, but not lipid regulatory genes. Journal of Molecular Endocrinology . 2004;33:533–544. doi: 10.1677/jme.1.01499. [DOI] [PubMed] [Google Scholar]
- 52.Nakamura M., Liu T., Husain S., et al. Glycogen synthase kinase-3α promotes fatty acid uptake and lipotoxic cardiomyopathy. Cell Metabolism . 2019;29:1119–1134.e12. doi: 10.1016/j.cmet.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gaggini M., Pingitore A., Vassalle C. Plasma ceramides pathophysiology, measurements, challenges, and opportunities. Metabolites . 2021;11 doi: 10.3390/metabo11110719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamashita S., Masuda D., Matsuzawa Y. Pemafibrate, a new selective PPARα modulator: drug concept and its clinical applications for dyslipidemia and metabolic diseases. Current Atherosclerosis Reports . 2020;22:1–17. doi: 10.1007/s11883-020-0823-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Takei K., Nakagawa Y., Wang Y., et al. Effects of K-877, a novel selective PPARα modulator, on small intestine contribute to the amelioration of hyperlipidemia in low-density lipoprotein receptor knockout mice. Journal of Pharmacological Sciences . 2017;133:214–222. doi: 10.1016/j.jphs.2017.02.003. [DOI] [PubMed] [Google Scholar]
- 56.Ishibashi S., Yamashita S., Arai H., et al. Effects of K-877, a novel selective PPARα modulator (SPPARMα), in dyslipidaemic patients: a randomized, double blind, active- and placebo-controlled, phase 2 trial. Atherosclerosis . 2016;249:36–43. doi: 10.1016/j.atherosclerosis.2016.02.029. [DOI] [PubMed] [Google Scholar]
- 57.Arai H., Yamashita S., Yokote K., Araki E., Suganami H., Ishibashi S. Efficacy and safety of pemafibrate versus fenofibrate in patients with high triglyceride and low HDL cholesterol levels: a multicenter, placebo-controlled, double-blind, randomized trial. Journal of Atherosclerosis and Thrombosis . 2018;25:521–538. doi: 10.5551/jat.44412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ishibashi S., Arai H., Yokote K., Araki E., Suganami H., Yamashita S. Efficacy and safety of pemafibrate (K-877), a selective peroxisome proliferator-activated receptor α modulator, in patients with dyslipidemia: results from a 24-week, randomized, double blind, active-controlled, phase 3 trial. Journal of Clinical Lipidology . 2018;12:173–184. doi: 10.1016/j.jacl.2017.10.006. [DOI] [PubMed] [Google Scholar]
- 59.Ostrander D. B., Sparagna G. C., Amoscato A. A., McMillin J. B., Dowhan W. Decreased cardiolipin synthesis corresponds with cytochrome c release in palmitate-induced cardiomyocyte apoptosis. Journal of Biological Chemistry . 2001;276:38061–38067. doi: 10.1074/jbc.M107067200. [DOI] [PubMed] [Google Scholar]
- 60.Barayeu U., Lange M., Méndez L., et al. Cytochrome c autocatalyzed carbonylation in the presence of hydrogen peroxide and cardiolipins. Journal of Biological Chemistry . 2019;294:1816–1830. doi: 10.1074/jbc.RA118.004110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bayir H., Fadeel B., Palladino M. J., et al. Apoptotic interactions of cytochrome c: redox flirting with anionic phospholipids within and outside of mitochondria. Biochim Biophys Acta . 2006;1757:648–659. doi: 10.1016/j.bbabio.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 62.Akhmedov A. T., Rybin V., Marín-García J. Mitochondrial oxidative metabolism and uncoupling proteins in the failing heart. Heart Failure Reviews . 2015;20:227–249. doi: 10.1007/s10741-014-9457-4. [DOI] [PubMed] [Google Scholar]
- 63.Schrauwen P., Saris W. H., Hesselink M. K. An alternative function for human uncoupling protein 3: protection of mitochondria against accumulation of nonesterified fatty acids inside the mitochondrial matrix. The FASEB Journal . 2001;15:2497–2502. doi: 10.1096/fj.01-0400hyp. [DOI] [PubMed] [Google Scholar]
- 64.Tillander V., Alexson S. E. H., Cohen D. E. Deactivating fatty acids: Acyl-CoA thioesterase-mediated control of lipid metabolism. Trends in Endocrinology & Metabolism . 2017;28:473–484. doi: 10.1016/j.tem.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maloberti P., Castilla R., Castillo F., et al. Silencing the expression of mitochondrial acyl-CoA thioesterase I and acyl-CoA synthetase 4 inhibits hormone-induced steroidogenesis. The FEBS Journal . 2005;272:1804–1814. doi: 10.1111/j.1742-4658.2005.04616.x. [DOI] [PubMed] [Google Scholar]
- 66.Kang H. W., Niepel M. W., Han S., Kawano Y., Cohen D. E. Thioesterase superfamily member 2/acyl-CoA thioesterase 13 (Them2/Acot13) regulates hepatic lipid and glucose metabolism. The FASEB Journal . 2012;26:2209–2221. doi: 10.1096/fj.11-202853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chiao Y. A., Chakraborty A. D., Light C. M., et al. NAD(+) redox imbalance in the heart exacerbates diabetic cardiomyopathy. Circulation: Heart Failure . 2021;14, article e008170 doi: 10.1161/CIRCHEARTFAILURE.120.008170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Prigge J. R., Coppo L., Martin S. S., et al. Hepatocyte hyperproliferation upon liver-specific co-disruption of thioredoxin-1, thioredoxin reductase-1, and glutathione reductase. Cell Reports . 2017;19:2771–2781. doi: 10.1016/j.celrep.2017.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xiao W., Wang R. S., Handy D. E., Loscalzo J. NAD(H) and NADP(H) redox couples and cellular energy metabolism. Antioxid Redox Signal . 2018;28:251–272. doi: 10.1089/ars.2017.7216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xie N., Zhang L., Gao W., et al. NAD(+) metabolism: pathophysiologic mechanisms and therapeutic potential. Signal Transduction and Targeted Therapy . 2020;5:1–37. doi: 10.1038/s41392-020-00311-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goodman R. P., Calvo S. E., Mootha V. K. Spatiotemporal compartmentalization of hepatic NADH and NADPH metabolism. Journal of Biological Chemistry . 2018;293:7508–7516. doi: 10.1074/jbc.TM117.000258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Paul S., Ali A., Katare R. Molecular complexities underlying the vascular complications of diabetes mellitus - a comprehensive review. Journal of Diabetes and its Complications . 2020;34, article 107613 doi: 10.1016/j.jdiacomp.2020.107613. [DOI] [PubMed] [Google Scholar]
- 73.Kitada M., Zhang Z., Mima A., King G. L. Molecular mechanisms of diabetic vascular complications. Journal of Diabetes Investigation . 2010;1:77–89. doi: 10.1111/j.2040-1124.2010.00018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Setter S. M., Campbell R. K., Cahoon C. J. Biochemical pathways for microvascular complications of diabetes mellitus. Annals of Pharmacotherapy . 2003;37:1858–1866. doi: 10.1345/aph.1D002. [DOI] [PubMed] [Google Scholar]
- 75.Hiramatsu Y., Sekiguchi N., Hayashi M., et al. Diacylglycerol production and protein kinase C activity are increased in a mouse model of diabetic embryopathy. Diabetes . 2002;51:2804–2810. doi: 10.2337/diabetes.51.9.2804. [DOI] [PubMed] [Google Scholar]
- 76.Shah M. S., Brownlee M. Molecular and cellular mechanisms of cardiovascular disorders in diabetes. Circulation Research . 2016;118:1808–1829. doi: 10.1161/CIRCRESAHA.116.306923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ducheix S., Magré J., Cariou B., Prieur X. Chronic O-GlcNAcylation and diabetic cardiomyopathy: the bitterness of glucose. Frontiers in Endocrinology . 2018;9:p. 642. doi: 10.3389/fendo.2018.00642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Parodi-Rullán R. M., Chapa-Dubocq X. R., Javadov S. Acetylation of mitochondrial proteins in the heart: the role of SIRT3. Frontiers in Physiology . 2018;9:p. 1094. doi: 10.3389/fphys.2018.01094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fukushima A., Lopaschuk G. D. Acetylation control of cardiac fatty acid β-oxidation and energy metabolism in obesity, diabetes, and heart failure. Biochim Biophys Acta . 2016;1862:2211–2220. doi: 10.1016/j.bbadis.2016.07.020. [DOI] [PubMed] [Google Scholar]
- 80.Vazquez E. J., Berthiaume J. M., Kamath V., et al. Mitochondrial complex I defect and increased fatty acid oxidation enhance protein lysine acetylation in the diabetic heart. Cardiovascular Research . 2015;107:453–465. doi: 10.1093/cvr/cvv183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Alrob O. A., Sankaralingam S., Ma C., et al. Obesity-induced lysine acetylation increases cardiac fatty acid oxidation and impairs insulin signalling. Cardiovascular Research . 2014;103:485–497. doi: 10.1093/cvr/cvu156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kraus D., Yang Q., Kong D., et al. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature . 2014;508:258–262. doi: 10.1038/nature13198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hirschey M. D., Shimazu T., Jing E., et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Molecular Cell . 2011;44:177–190. doi: 10.1016/j.molcel.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fillmore N., Mori J., Lopaschuk G. D. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. British Journal of Pharmacology . 2014;171:2080–2090. doi: 10.1111/bph.12475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bhatt N. M., Aon M. A., Tocchetti C. G., et al. Restoring redox balance enhances contractility in heart trabeculae from type 2 diabetic rats exposed to high glucose. American Journal of Physiology-Heart and Circulatory Physiology . 2015;308:H291–H302. doi: 10.1152/ajpheart.00378.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zuurbier C. J., Bertrand L., Beauloye C. R., et al. Cardiac metabolism as a driver and therapeutic target of myocardial infarction. Journal of Cellular and Molecular Medicine . 2020;24:5937–5954. doi: 10.1111/jcmm.15180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stromsdorfer K. L., Yamaguchi S., Yoon M. J., et al. NAMPT-mediated NAD(+) biosynthesis in adipocytes regulates adipose tissue function and multi-organ insulin sensitivity in mice. Cell Reports . 2016;16:1851–1860. doi: 10.1016/j.celrep.2016.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Uddin G. M., Youngson N. A., Sinclair D. A., Morris M. J. Head to head comparison of short-term treatment with the NAD(+) precursor nicotinamide mononucleotide (NMN) and 6 weeks of exercise in obese female mice. Frontiers in Pharmacology . 2016;7, article 258 doi: 10.3389/fphar.2016.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tsubota K. The first human clinical study for NMN has started in Japan. NPJ Aging And Mechanisms of Disease . 2016;2, article 16021 doi: 10.1038/npjamd.2016.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang Y., Xu H. Translational regulation of mitochondrial biogenesis. Biochemical Society Transactions . 2016;44:1717–1724. doi: 10.1042/BST20160071C. [DOI] [PubMed] [Google Scholar]
- 91.Scarpulla R. C., Vega R. B., Kelly D. P. Transcriptional integration of mitochondrial biogenesis. Trends in Endocrinology & Metabolism . 2012;23:459–466. doi: 10.1016/j.tem.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Di W., Lv J., Jiang S., et al. PGC-1: the energetic regulator in cardiac metabolism. Current Issues in Molecular Biology . 2018;28:29–46. doi: 10.21775/cimb.028.029. [DOI] [PubMed] [Google Scholar]
- 93.Islam H., Edgett B. A., Gurd B. J. Coordination of mitochondrial biogenesis by PGC-1alpha in human skeletal muscle: a re-evaluation. Metabolism . 2018;79:42–51. doi: 10.1016/j.metabol.2017.11.001. [DOI] [PubMed] [Google Scholar]
- 94.Leone T. C., Kelly D. P. Transcriptional control of cardiac fuel metabolism and mitochondrial function. Cold Spring Harbor Symposia on Quantitative Biology . 2011;76:175–182. doi: 10.1101/sqb.2011.76.011965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Batchu S. N., Lee S. B., Qadhi R. S., et al. Cardioprotective effect of a dual acting epoxyeicosatrienoic acid analogue towards ischaemia reperfusion injury. British Journal of Pharmacology . 2011;162:897–907. doi: 10.1111/j.1476-5381.2010.01093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Waldman M., Bellner L., Vanella L., et al. Epoxyeicosatrienoic acids regulate adipocyte differentiation of mouse 3T3 cells, via PGC-1α activation, which is required for HO-1 expression and increased mitochondrial function. Stem Cells and Development . 2016;25:1084–1094. doi: 10.1089/scd.2016.0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nishio Y., Kanazawa A., Nagai Y., Inagaki H., Kashiwagi A. Mitochondrial Pathogenesis . Berlin, Heidelberg: Springer; 2004. Regulation and role of the mitochondrial transcription factor in the diabetic rat heart; pp. 78–85. [DOI] [PubMed] [Google Scholar]
- 98.Nisoli E., Clementi E., Carruba M. O., Moncada S. Defective mitochondrial biogenesis: a hallmark of the high cardiovascular risk in the metabolic syndrome? Circulation Research . 2007;100:795–806. doi: 10.1161/01.RES.0000259591.97107.6c. [DOI] [PubMed] [Google Scholar]
- 99.Lopaschuk G. D., Spafford M. Response of isolated working hearts to fatty acids and carnitine palmitoyltransferase I inhibition during reduction of coronary flow in acutely and chronically diabetic rats. Circulation Research . 1989;65:378–387. doi: 10.1161/01.res.65.2.378. [DOI] [PubMed] [Google Scholar]
- 100.Kim J. A., Wei Y., Sowers J. R. Role of mitochondrial dysfunction in insulin resistance. Circulation Research . 2008;102:401–414. doi: 10.1161/CIRCRESAHA.107.165472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Matsuda S., Kitagishi Y., Kobayashi M. Function and characteristics of PINK1 in mitochondria. Oxidative Medicine and Cellular Longevity . 2013;2013:6. doi: 10.1155/2013/601587.601587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Thomas R. E., Andrews L. A., Burman J. L., Lin W. Y., Pallanck L. J. PINK1-Parkin pathway activity is regulated by degradation of PINK1 in the mitochondrial matrix. PLoS Genetics . 2014;10, article e1004279 doi: 10.1371/journal.pgen.1004279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shires S. E., Quiles J. M., Najor R. H., et al. Nuclear Parkin activates the ERRα transcriptional program and drives widespread changes in gene expression following hypoxia. Scientific Reports . 2020;10, article 8499 doi: 10.1038/s41598-020-65438-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kubli D. A., Gustafsson A. B. Unbreak my heart: targeting mitochondrial autophagy in diabetic cardiomyopathy. Antioxidants & Redox Signaling . 2015;22:1527–1544. doi: 10.1089/ars.2015.6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lee Y. H., Kim J., Park K., Lee M. S. Lee, β-cell autophagy: mechanism and role in β-cell dysfunction. Molecular Metabolism . 2019;27:S92–s103. doi: 10.1016/j.molmet.2019.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bharath L. P., Rockhold J. D., Conway R. Selective autophagy in hyperglycemia-induced microvascular and macrovascular diseases. Cells . 2021;10 doi: 10.3390/cells10082114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Onishi M., Yamano K., Sato M., Matsuda N., Okamoto K. Molecular mechanisms and physiological functions of mitophagy. The EMBO Journal . 2021;40, article e104705 doi: 10.15252/embj.2020104705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Li Y., Zheng N., Ding X. Mitophagy disequilibrium, a prominent pathological mechanism in metabolic heart diseases. Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy . 2021;14:4631–4640. doi: 10.2147/DMSO.S336882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liang Q., Kobayashi S. Mitochondrial quality control in the diabetic heart. Journal of Molecular and Cellular Cardiology . 2016;95:57–69. doi: 10.1016/j.yjmcc.2015.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kobayashi S., Liang Q. Autophagy and mitophagy in diabetic cardiomyopathy. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease . 2015;1852:252–261. doi: 10.1016/j.bbadis.2014.05.020. [DOI] [PubMed] [Google Scholar]
- 111.Xu X., Kobayashi S., Chen K., et al. Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. Journal of Biological Chemistry . 2013;288:18077–18092. doi: 10.1074/jbc.M113.474650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Giacco F., Brownlee M. Oxidative stress and diabetic complications. Circulation Research . 2010;107:1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kaludercic N., Di Lisa F. Mitochondrial ROS formation in the pathogenesis of diabetic cardiomyopathy. Frontiers in Cardiovascular Medicine . 2020;7, article 12 doi: 10.3389/fcvm.2020.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hamada Y., Fujii H., Fukagawa M. Role of oxidative stress in diabetic bone disorder. Bone . 2009;45(Suppl 1):S35–S38. doi: 10.1016/j.bone.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 115.Ly L. D., Xu S., Choi S. K., et al. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Experimental & Molecular Medicine . 2017;49, article e291 doi: 10.1038/emm.2016.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yan Z., Qi Z., Yang X., et al. The NLRP3 inflammasome: Multiple activation pathways and its role in primary cells during ventricular remodeling. Journal of Cellular Physiology . 2021;236:5547–5563. doi: 10.1002/jcp.30285. [DOI] [PubMed] [Google Scholar]
- 117.Frati G., Schirone L., Chimenti I., et al. An overview of the inflammatory signalling mechanisms in the myocardium underlying the development of diabetic cardiomyopathy. Cardiovascular Research . 2017;113:378–388. doi: 10.1093/cvr/cvx011. [DOI] [PubMed] [Google Scholar]
- 118.Borlaug B. A. The pathophysiology of heart failure with preserved ejection fraction. Nature Reviews Cardiology . 2014;11:507–515. doi: 10.1038/nrcardio.2014.83. [DOI] [PubMed] [Google Scholar]
- 119.Santos C. X., Anilkumar N., Zhang M., Brewer A. C., Shah A. M. Redox signaling in cardiac myocytes. Free Radical Biology and Medicine . 2011;50:777–793. doi: 10.1016/j.freeradbiomed.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Steinberg S. F. Oxidative stress and sarcomeric proteins. Circulation Research . 2013;112(2):393–405. doi: 10.1161/CIRCRESAHA.111.300496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Breitkreuz M., Hamdani N. A change of heart: oxidative stress in governing muscle function? Biophysical Reviews . 2015;7:321–341. doi: 10.1007/s12551-015-0175-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Grützner A., Garcia-Manyes S., Kötter S., Badilla C. L., Fernandez J. M., Linke W. A. Modulation of titin-based stiffness by disulfide bonding in the cardiac titin N2-B unique sequence. Biophysical Journal . 2009;97:825–834. doi: 10.1016/j.bpj.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Arrington D. D., Van Vleet T. R., Schnellmann R. G. Calpain 10: a mitochondrial calpain and its role in calcium-induced mitochondrial dysfunction. American Journal of Physiology-Cell Physiology . 2006;291:C1159–C1171. doi: 10.1152/ajpcell.00207.2006. [DOI] [PubMed] [Google Scholar]
- 124.Kalani K., Yan S. F., Yan S. S. Mitochondrial permeability transition pore: a potential drug target for neurodegeneration. Drug Discovery Today . 2018;23:1983–1989. doi: 10.1016/j.drudis.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Di Lisa F., Giorgio M., Ferdinandy P., Schulz R. New aspects of p66Shc in ischaemia reperfusion injury and other cardiovascular diseases. British Journal of Pharmacology . 2017;174:1690–1703. doi: 10.1111/bph.13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Xiao Y., Xia J., Cheng J., et al. Inhibition of S-adenosylhomocysteine hydrolase induces endothelial dysfunction via epigenetic regulation of p66shc-mediated oxidative stress pathway. Circulation . 2019;139:2260–2277. doi: 10.1161/CIRCULATIONAHA.118.036336. [DOI] [PubMed] [Google Scholar]
- 127.Messina E., Giacomello A. Diabetic cardiomyopathy: a "cardiac stem cell disease" involving p66Shc, an attractive novel molecular target for heart failure therapy. Circulation Research . 2006;99:1–2. doi: 10.1161/01.RES.0000233141.65522.3e. [DOI] [PubMed] [Google Scholar]
- 128.Pinton P., Rimessi A., Marchi S., et al. Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science . 2007;315:659–663. doi: 10.1126/science.1135380. [DOI] [PubMed] [Google Scholar]
- 129.Giorgio M., Migliaccio E., Orsini F., et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell . 2005;122:221–233. doi: 10.1016/j.cell.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 130.Galimov E. R. The role of p66shc in oxidative stress and apoptosis. Acta Naturae . 2010;2:44–51. [PMC free article] [PubMed] [Google Scholar]
- 131.Trinei M., Giorgio M., Cicalese A., et al. A p53-p66Shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced apoptosis. Oncogene . 2002;21:3872–3878. doi: 10.1038/sj.onc.1205513. [DOI] [PubMed] [Google Scholar]
- 132.Francia P., Cosentino F., Schiavoni M., et al. p66(Shc) protein, oxidative stress, and cardiovascular complications of diabetes: the missing link. Journal of Molecular Medicine . 2009;87:885–891. doi: 10.1007/s00109-009-0499-3. [DOI] [PubMed] [Google Scholar]
- 133.Rota M., LeCapitaine N., Hosoda T., et al. Diabetes promotes cardiac stem cell aging and heart failure, which are prevented by deletion of the p66shc gene. Circulation Research . 2006;99:42–52. doi: 10.1161/01.RES.0000231289.63468.08. [DOI] [PubMed] [Google Scholar]
- 134.Xu Y. J., Tappia P. S., Neki N. S., Dhalla N. S. Prevention of diabetes-induced cardiovascular complications upon treatment with antioxidants. Heart Failure Reviews . 2014;19:113–121. doi: 10.1007/s10741-013-9379-6. [DOI] [PubMed] [Google Scholar]
- 135.Szeto H. H. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. British Journal of Pharmacology . 2014;171:2029–2050. doi: 10.1111/bph.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Galloway C. A., Yoon Y. Mitochondrial dynamics in diabetic cardiomyopathy. Antioxid Redox Signal . 2015;22:1545–1562. doi: 10.1089/ars.2015.6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dillmann W. H. Diabetic Cardiomyopathy. Circulation Research . 2019;124:1160–1162. doi: 10.1161/CIRCRESAHA.118.314665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pei Z., Deng Q., Babcock S. A., He E. Y., Ren J., Zhang Y. Inhibition of advanced glycation endproduct (AGE) rescues against streptozotocin-induced diabetic cardiomyopathy: role of autophagy and ER stress. Toxicology Letters . 2018;284:10–20. doi: 10.1016/j.toxlet.2017.11.018. [DOI] [PubMed] [Google Scholar]
- 139.Nishikawa T., Edelstein D., Du X. L., et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature . 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 140.Du X. L., Edelstein D., Rossetti L., et al. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proceedings of the National Academy of Sciences . 2000;97:12222–12226. doi: 10.1073/pnas.97.22.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes . 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 142.Zuo L., Youtz D. J., Wold L. E. Particulate matter exposure exacerbates high glucose-induced cardiomyocyte dysfunction through ROS generation. PLoS One . 2011;6, article e23116 doi: 10.1371/journal.pone.0023116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ye G., Metreveli N. S., Donthi R. V., et al. Catalase protects cardiomyocyte function in models of type 1 and type 2 diabetes. Diabetes . 2004;53:1336–1343. doi: 10.2337/diabetes.53.5.1336. [DOI] [PubMed] [Google Scholar]
- 144.Roul D., Recchia F. A. Metabolic alterations induce oxidative stress in diabetic and failing hearts: different pathways, same outcome. Antioxid Redox Signal . 2015;22:1502–1514. doi: 10.1089/ars.2015.6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tsushima K., Bugger H., Wende A. R., et al. Mitochondrial reactive oxygen species in lipotoxic hearts induce post-translational modifications of AKAP121, DRP1, and OPA1 that promote mitochondrial fission. Circulation Research . 2018;122:58–73. doi: 10.1161/CIRCRESAHA.117.311307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Newsholme P., Cruzat V. F., Keane K. N., Carlessi R., de Bittencourt P. I. H., Jr. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochemical Journal . 2016;473:4527–4550. doi: 10.1042/BCJ20160503C. [DOI] [PubMed] [Google Scholar]
- 147.Banerjee P. S., Ma J., Hart G. W. Diabetes-associated dysregulation of O-GlcNAcylation in rat cardiac mitochondria. Proceedings of the National Academy of Sciences . 2015;112:6050–6055. doi: 10.1073/pnas.1424017112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hu Y., Suarez J., Fricovsky E., et al. Increased enzymatic O-GlcNAcylation of mitochondrial proteins impairs mitochondrial function in cardiac myocytes exposed to high glucose. Journal of Biological Chemistry . 2009;284:547–555. doi: 10.1074/jbc.M808518200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Marín-García J., Akhmedov A. T. Mitochondrial dynamics and cell death in heart failure. Heart Failure Reviews . 2016;21:123–136. doi: 10.1007/s10741-016-9530-2. [DOI] [PubMed] [Google Scholar]
- 150.Chan C. M., Huang D. Y., Huang Y. P., et al. Methylglyoxal induces cell death through endoplasmic reticulum stress-associated ROS production and mitochondrial dysfunction. Journal of Cellular and Molecular Medicine . 2016;20:1749–1760. doi: 10.1111/jcmm.12893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lamas G. A., Navas-Acien A., Mark D. B., Lee K. L. Heavy metals, cardiovascular disease, and the unexpected benefits of chelation therapy. Journal of the American College of Cardiology . 2016;67:2411–2418. doi: 10.1016/j.jacc.2016.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Edmondson D. E., Binda C., Wang J., Upadhyay A. K., Mattevi A. Molecular and mechanistic properties of the membrane-bound mitochondrial monoamine oxidases. Biochemistry . 2009;48:4220–4230. doi: 10.1021/bi900413g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wouters J. Structural aspects of monoamine oxidase and its reversible inhibition. Curr Med Chem . 1998;5:137–162. [PubMed] [Google Scholar]
- 154.Kaludercic N., Carpi A., Nagayama T., et al. Monoamine oxidase B prompts mitochondrial and cardiac dysfunction in pressure overloaded hearts. Antioxid Redox Signal . 2014;20:267–280. doi: 10.1089/ars.2012.4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kaludercic N., Mialet-Perez J., Paolocci N., Parini A., Di Lisa F. Monoamine oxidases as sources of oxidants in the heart. Journal of Molecular and Cellular Cardiology . 2014;73:34–42. doi: 10.1016/j.yjmcc.2013.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Santin Y., Sicard P., Vigneron F., et al. Oxidative stress by monoamine oxidase-A impairs transcription factor EB activation and autophagosome clearance, leading to cardiomyocyte necrosis and heart failure. Antioxid Redox Signal . 2016;25:10–27. doi: 10.1089/ars.2015.6522. [DOI] [PubMed] [Google Scholar]
- 157.Kaludercic N., Carpi A., Menabò R., Di Lisa F., Paolocci N. Monoamine oxidases (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochim Biophys Acta . 2011;1813:1323–1332. doi: 10.1016/j.bbamcr.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Emory H., Mizrahi N. Glycaemic control by monoamine oxidase inhibition in a patient with type 1 diabetes. Diabetes and Vascular Disease Research . 2017;14:163–165. doi: 10.1177/1479164116675492. [DOI] [PubMed] [Google Scholar]
- 159.Binda C., Aldeco M., Geldenhuys W. J., Tortorici M., Mattevi A., Edmondson D. E. Molecular insights into human monoamine oxidase B inhibition by the glitazone anti-diabetes drugs. ACS Medicinal Chemistry Letters . 2011;3:39–42. doi: 10.1021/ml200196p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Deshwal S., Forkink M., Hu C. H., et al. Monoamine oxidase-dependent endoplasmic reticulum-mitochondria dysfunction and mast cell degranulation lead to adverse cardiac remodeling in diabetes. Cell Death & Differentiation . 2018;25:1671–1685. doi: 10.1038/s41418-018-0071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Umbarkar P., Singh S., Arkat S., Bodhankar S. L., Lohidasan S., Sitasawad S. L. Monoamine oxidase-A is an important source of oxidative stress and promotes cardiac dysfunction, apoptosis, and fibrosis in diabetic cardiomyopathy. Free Radical Biology and Medicine . 2015;87:263–273. doi: 10.1016/j.freeradbiomed.2015.06.025. [DOI] [PubMed] [Google Scholar]
- 162.Zhang Y., Babcock S. A., Hu N., Maris J. R., Wang H., Ren J. Mitochondrial aldehyde dehydrogenase (ALDH2) protects against streptozotocin-induced diabetic cardiomyopathy: role of GSK3β and mitochondrial function. BMC Medicine . 2012;10:1–17. doi: 10.1186/1741-7015-10-40. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 163.Shen E., Li Y., Li Y., et al. Rac1 is required for cardiomyocyte apoptosis during hyperglycemia. Diabetes . 2009;58:2386–2395. doi: 10.2337/db08-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hansen S. S., Aasum E., Hafstad A. D. The role of NADPH oxidases in diabetic cardiomyopathy. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease . 2018;1864:1908–1913. doi: 10.1016/j.bbadis.2017.07.025. [DOI] [PubMed] [Google Scholar]
- 165.Liang L., Shou X. L., Zhao H. K., et al. Antioxidant catalase rescues against high fat diet-induced cardiac dysfunction via an IKKβ-AMPK-dependent regulation of autophagy. Biochim Biophys Acta . 2015;1852:343–352. doi: 10.1016/j.bbadis.2014.06.027. [DOI] [PubMed] [Google Scholar]
- 166.Pajares M., Cuadrado A., Engedal N., Jirsova Z., Cahova M. The role of free radicals in autophagy regulation: implications for ageing. Oxidative Medicine and Cellular Longevity . 2018;2018:19. doi: 10.1155/2018/2450748.2450748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Oblath R. W., Griffith G. C. The aminase oxidase inhibitors. Their current place in clinical medicine. California Medicine . 1960;93:343–346. [PMC free article] [PubMed] [Google Scholar]
- 168.Suarez J., Cividini F., Scott B. T., et al. Restoring mitochondrial calcium uniporter expression in diabetic mouse heart improves mitochondrial calcium handling and cardiac function. Journal of Biological Chemistry . 2018;293:8182–8195. doi: 10.1074/jbc.RA118.002066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Diaz-Juarez J., Suarez J. A., Dillmann W. H., Suarez J. Mitochondrial calcium handling and heart disease in diabetes mellitus. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease . 2021;1867, article 165984 doi: 10.1016/j.bbadis.2020.165984. [DOI] [PubMed] [Google Scholar]
- 170.Flarsheim C. E., Grupp I. L., Matlib M. A. Mitochondrial dysfunction accompanies diastolic dysfunction in diabetic rat heart. American Journal of Physiology-Heart and Circulatory Physiology . 1996;271:H192–H202. doi: 10.1152/ajpheart.1996.271.1.H192. [DOI] [PubMed] [Google Scholar]
- 171.Diaz-Juarez J., Suarez J., Cividini F., et al. Expression of the mitochondrial calcium uniporter in cardiac myocytes improves impaired mitochondrial calcium handling and metabolism in simulated hyperglycemia. American Journal of Physiology-Heart and Circulatory Physiology . 2016;311:C1005–c1013. doi: 10.1152/ajpcell.00236.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ji L., Liu F., Jing Z., et al. MICU1 alleviates diabetic cardiomyopathy through mitochondrial Ca(2+)-dependent antioxidant response. Diabetes . 2017;66:1586–1600. doi: 10.2337/db16-1237. [DOI] [PubMed] [Google Scholar]
- 173.Glancy B., Balaban R. S. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry . 2012;51:2959–2973. doi: 10.1021/bi2018909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Belke D. D., Swanson E., Suarez J., Scott B. T., Stenbit A. E., Dillmann W. H. Increased expression of SERCA in the hearts of transgenic mice results in increased oxidation of glucose. American Journal of Physiology-Heart and Circulatory Physiology . 2007;292:H1755–H1763. doi: 10.1152/ajpheart.00884.2006. [DOI] [PubMed] [Google Scholar]
- 175.Neely J. R., Rovetto M. J., Oram J. F. Myocardial utilization of carbohydrate and lipids. Progress in Cardiovascular Diseases . 1972;15:289–329. doi: 10.1016/0033-0620(72)90029-1. [DOI] [PubMed] [Google Scholar]
- 176.Grimm M., Brown J. H. Beta-adrenergic receptor signaling in the heart: role of CaMKII. Journal of Molecular and Cellular Cardiology . 2010;48:322–330. doi: 10.1016/j.yjmcc.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Weiss J. N., Lamp S. T. Cardiac ATP-sensitive K+ channels. Evidence for preferential regulation by glycolysis. The Journal of General Physiology . 1989;94:911–935. doi: 10.1085/jgp.94.5.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Entman M. L., Bornet E. P., Van Winkle W. B., Goldstein M. A., Schwartz A. Association of glycogenolysis with cardiac sarcoplasmic reticulum: II. Effect of glycogen depletion, deoxycholate solubilization and cardiac ischemia: evidence for a phorphorylase kinase membrane complex. Journal of Biological Chemistry . 1977;9:515–528. doi: 10.1016/s0022-2828(77)80367-2. [DOI] [PubMed] [Google Scholar]
- 179.Weiss J. N., Lamp S. T. Glycolysis preferentially inhibits ATP-sensitive K+ channels in isolated guinea pig cardiac myocytes. Science . 1987;238:67–69. doi: 10.1126/science.2443972. [DOI] [PubMed] [Google Scholar]
- 180.Griffin T. M., Humphries K. M., Kinter M., Lim H. Y., Szweda L. I. Nutrient sensing and utilization: getting to the heart of metabolic flexibility. Biochimie . 2016;124:74–83. doi: 10.1016/j.biochi.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ding M., Feng N., Tang D., et al. Melatonin prevents Drp1-mediated mitochondrial fission in diabetic hearts through SIRT1-PGC1α pathway. Journal of Pineal Research . 2018;65, article e12491 doi: 10.1111/jpi.12491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hom J., Sheu S. S. Morphological dynamics of mitochondria--a special emphasis on cardiac muscle cells. Journal of Molecular and Cellular Cardiology . 2009;46:811–820. doi: 10.1016/j.yjmcc.2009.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Watanabe T., Saotome M., Nobuhara M., et al. Roles of mitochondrial fragmentation and reactive oxygen species in mitochondrial dysfunction and myocardial insulin resistance. Experimental Cell Research . 2014;323:314–325. doi: 10.1016/j.yexcr.2014.02.027. [DOI] [PubMed] [Google Scholar]
- 184.Makino A., Scott B. T., Dillmann W. H. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia . 2010;53:1783–1794. doi: 10.1007/s00125-010-1770-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Vásquez-Trincado C., García-Carvajal I., Pennanen C., et al. Mitochondrial dynamics, mitophagy and cardiovascular disease. The Journal of Physiology . 2016;594:509–525. doi: 10.1113/JP271301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Yu J., Maimaitili Y., Xie P., et al. High glucose concentration abrogates sevoflurane post-conditioning cardioprotection by advancing mitochondrial fission but dynamin-related protein 1 inhibitor restores these effects. Acta Physiologica . 2017;220:83–98. doi: 10.1111/apha.12812. [DOI] [PubMed] [Google Scholar]
- 187.Parra V., Verdejo H. E., Iglewski M., et al. Insulin stimulates mitochondrial fusion and function in cardiomyocytes via the Akt-mTOR-NFκB-Opa-1 signaling pathway. Diabetes . 2014;63:75–88. doi: 10.2337/db13-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Quirós P. M., Ramsay A. J., Sala D., et al. Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. The EMBO Journal . 2012;31:2117–2133. doi: 10.1038/emboj.2012.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Garcia I., Calderon F., la Torre P., et al. Mitochondrial OPA1 cleavage is reversibly activated by differentiation of H9c2 cardiomyoblasts. Mitochondrion . 2021;57:88–96. doi: 10.1016/j.mito.2020.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kelley D. E., He J., Menshikova E. V., Ritov V. B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes . 2002;51:2944–2950. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- 191.Lowell B. B., Shulman G. I. Mitochondrial dysfunction and type 2 diabetes. Science . 2005;307:384–387. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- 192.Zorzano A., Liesa M., Palacín M. Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. The International Journal of Biochemistry & Cell Biology . 2009;41:1846–1854. doi: 10.1016/j.biocel.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 193.Montaigne D., Marechal X., Coisne A., et al. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation . 2014;130:554–564. doi: 10.1161/CIRCULATIONAHA.113.008476. [DOI] [PubMed] [Google Scholar]
- 194.Zhang Y., Murugesan P., Huang K., Cai H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: novel therapeutic targets. Nature Reviews Cardiology . 2020;17:170–194. doi: 10.1038/s41569-019-0260-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Forrester S. J., Kikuchi D. S., Hernandes M. S., Xu Q., Griendling K. K. Reactive oxygen species in metabolic and inflammatory signaling. Circulation Research . 2018;122:877–902. doi: 10.1161/CIRCRESAHA.117.311401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Packer M., Anker S. D., Butler J., et al. Effect of empagliflozin on the clinical stability of patients with heart failure and a reduced ejection fraction: the EMPEROR-reduced trial. Circulation . 2021;143:326–336. doi: 10.1161/CIRCULATIONAHA.120.051783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Zannad F., Ferreira J. P., Pocock S. J., et al. SGLT2 inhibitors in patients with heart failure with reduced ejection fraction: a meta-analysis of the EMPEROR-Reduced and DAPA-HF trials. Lancet . 2020;396:819–829. doi: 10.1016/S0140-6736(20)31824-9. [DOI] [PubMed] [Google Scholar]
- 198.Kaneto H., Obata A., Kimura T., et al. Unexpected pleiotropic effects of SGLT2 inhibitors: pearls and pitfalls of this novel antidiabetic class. International Journal of Molecular Sciences . 2021;22 doi: 10.3390/ijms22063062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Kaplan A., Abidi E., El-Yazbi A., Eid A., Booz G. W., Zouein F. A. Direct cardiovascular impact of SGLT2 inhibitors: mechanisms and effects. Heart Failure Reviews . 2018;23:419–437. doi: 10.1007/s10741-017-9665-9. [DOI] [PubMed] [Google Scholar]
- 200.Uthman L., Baartscheer A., Schumacher C. A., et al. Direct cardiac actions of sodium glucose cotransporter 2 inhibitors target pathogenic mechanisms underlying heart failure in diabetic patients. Frontiers in Physiology . 2018;9:p. 1575. doi: 10.3389/fphys.2018.01575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Tahara A., Kurosaki E., Yokono M., et al. Effects of sodium-glucose cotransporter 2 selective inhibitor ipragliflozin on hyperglycaemia, oxidative stress, inflammation and liver injury in streptozotocin-induced type 1 diabetic rats. Journal of Pharmacy and Pharmacology . 2014;66:975–987. doi: 10.1111/jphp.12223. [DOI] [PubMed] [Google Scholar]
- 202.Li C., Zhang J., Xue M., et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovascular Diabetology . 2019;18:1–13. doi: 10.1186/s12933-019-0816-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Xue M., Li T., Wang Y., et al. Empagliflozin prevents cardiomyopathy via sGC-cGMP-PKG pathway in type 2 diabetes mice. Clinical Science . 2019;133:1705–1720. doi: 10.1042/CS20190585. [DOI] [PubMed] [Google Scholar]
- 204.Greene S. J., Gheorghiade M., Borlaug B. A., et al. The cGMP signaling pathway as a therapeutic target in heart failure with preserved ejection fraction. Journal of the American Heart Association . 2013;2, article e000536 doi: 10.1161/JAHA.113.000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Lukowski R., Krieg T., Rybalkin S. D., Beavo J., Hofmann F. Turning on cGMP-dependent pathways to treat cardiac dysfunctions: boom, bust, and beyond. Trends in Pharmacological Sciences . 2014;35:404–413. doi: 10.1016/j.tips.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 206.Armstrong P. W., Pieske B., Anstrom K. J., et al. Vericiguat in patients with heart failure and reduced ejection fraction. New England Journal of Medicine . 2020;382:1883–1893. doi: 10.1056/NEJMoa1915928. [DOI] [PubMed] [Google Scholar]
- 207.Kovács Á., Alogna A., Post H., Hamdani N. Is enhancing cGMP-PKG signalling a promising therapeutic target for heart failure with preserved ejection fraction? Netherlands Heart Journal . 2016;24:268–274. doi: 10.1007/s12471-016-0814-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Filippatos G., Maggioni A. P., Lam C. S. P., et al. Patient-reported outcomes in the SOluble guanylate cyclase stimulatoR in heArT failurE patientS with PRESERVED ejection fraction (SOCRATES-PRESERVED) study. European Journal of Heart Failure . 2017;19:782–791. doi: 10.1002/ejhf.800. [DOI] [PubMed] [Google Scholar]
- 209.Gorin Y., Cavaglieri R. C., Khazim K., et al. Targeting NADPH oxidase with a novel dual Nox1/Nox4 inhibitor attenuates renal pathology in type 1 diabetes. American Journal of Physiology-Renal Physiology . 2015;308:F1276–F1287. doi: 10.1152/ajprenal.00396.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Elbatreek M. H., Mucke H., Schmidt H. H. Reactive Oxygen Species . Cham: Springer; 2021. NOX inhibitors: From bench to naxibs to bedside; pp. 145–168. [DOI] [PubMed] [Google Scholar]
- 211.Cao T., Fan S., Zheng D., et al. Increased calpain-1 in mitochondria induces dilated heart failure in mice: role of mitochondrial superoxide anion. Basic Research in Cardiology . 2019;114:1–15. doi: 10.1007/s00395-019-0726-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.He C., Zhu H., Li H., Zou M. H., Xie Z. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes . 2013;62:1270–1281. doi: 10.2337/db12-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Sriwijitkamol A., Musi N. Advances in the development of AMPK-activating compounds. Expert Opinion on Drug Discovery . 2008;3:1167–1176. doi: 10.1517/17460441.3.10.1167. [DOI] [PubMed] [Google Scholar]
- 214.Lihn A. S., Jessen N., Pedersen S. B., Lund S., Richelsen B. AICAR stimulates adiponectin and inhibits cytokines in adipose tissue. Biochemical and Biophysical Research Communications . 2004;316:853–858. doi: 10.1016/j.bbrc.2004.02.139. [DOI] [PubMed] [Google Scholar]
- 215.Lee W. S., Kim J. Application of animal models in diabetic cardiomyopathy. Diabetes & Metabolism Journal . 2021;45:129–145. doi: 10.4093/dmj.2020.0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Zhang Y., Edgley A. J., Cox A. J., et al. FT011, a new anti-fibrotic drug, attenuates fibrosis and chronic heart failure in experimental diabetic cardiomyopathy. European Journal of Heart Failure . 2012;14:549–562. doi: 10.1093/eurjhf/hfs011. [DOI] [PubMed] [Google Scholar]
- 217.Tan S. M., Zhang Y., Wang B., et al. FT23, an orally active antifibrotic compound, attenuates structural and functional abnormalities in an experimental model of diabetic cardiomyopathy. Clinical and Experimental Pharmacology and Physiology . 2012;39:650–656. doi: 10.1111/j.1440-1681.2012.05726.x. [DOI] [PubMed] [Google Scholar]
- 218.Krüger M., Kötter S., Grützner A., et al. Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circulation Research . 2009;104:87–94. doi: 10.1161/CIRCRESAHA.108.184408. [DOI] [PubMed] [Google Scholar]
- 219.Ng H. H., Shen M., Samuel C. S., Schlossmann J., Bennett R. G. Relaxin and extracellular matrix remodeling: mechanisms and signaling pathways. Molecular and Cellular Endocrinology . 2019;487:59–65. doi: 10.1016/j.mce.2019.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Martin B., Romero G., Salama G. Cardioprotective actions of relaxin. Molecular and Cellular Endocrinology . 2019;487:45–53. doi: 10.1016/j.mce.2018.12.016. [DOI] [PubMed] [Google Scholar]
- 221.Zhou X., Chen X., Cai J. J., et al. Relaxin inhibits cardiac fibrosis and endothelial-mesenchymal transition via the Notch pathway. Drug Design, Development and Therapy . 2015;9:4599–4611. doi: 10.2147/DDDT.S85399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Lekgabe E. D., Kiriazis H., Zhao C., et al. Relaxin reverses cardiac and renal fibrosis in spontaneously hypertensive rats. Hypertension . 2005;46:412–418. doi: 10.1161/01.HYP.0000171930.00697.2f. [DOI] [PubMed] [Google Scholar]
- 223.Samuel C. S., Hewitson T. D., Zhang Y., Kelly D. J. Relaxin ameliorates fibrosis in experimental diabetic cardiomyopathy. Endocrinology . 2008;149:3286–3293. doi: 10.1210/en.2008-0250. [DOI] [PubMed] [Google Scholar]
- 224.Giannetta E., Isidori A. M., Galea N., et al. Chronic Inhibition of cGMP phosphodiesterase 5A improves diabetic cardiomyopathy: a randomized, controlled clinical trial using magnetic resonance imaging with myocardial tagging. Circulation . 2012;125:2323–2333. doi: 10.1161/CIRCULATIONAHA.111.063412. [DOI] [PubMed] [Google Scholar]
- 225.Tan Y., Zhang Z., Zheng C., Wintergerst K. A., Keller B. B., Cai L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence. Nature Reviews Cardiology . 2020;17:585–607. doi: 10.1038/s41569-020-0339-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Zhou Y., Wu W. The sodium-glucose co-transporter 2 inhibitor, empagliflozin, protects against diabetic cardiomyopathy by inhibition of the endoplasmic reticulum stress pathway. Cellular Physiology and Biochemistry . 2017;41:2503–2512. doi: 10.1159/000475942. [DOI] [PubMed] [Google Scholar]
- 227.Zhang L., Tian J., Diao S., Zhang G., Xiao M., Chang D. GLP-1 receptor agonist liraglutide protects cardiomyocytes from IL-1β-induced metabolic disturbance and mitochondrial dysfunction. Chemico-Biological Interactions . 2020;332, article 109252 doi: 10.1016/j.cbi.2020.109252. [DOI] [PubMed] [Google Scholar]
- 228.Bizino M. B., Jazet I. M., Westenberg J. J. M., et al. Effect of liraglutide on cardiac function in patients with type 2 diabetes mellitus: randomized placebo-controlled trial. Cardiovascular Diabetology . 2019;18:p. 55. doi: 10.1186/s12933-019-0857-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Davies M. J., D'Alessio D. A., Fradkin J., et al. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) Diabetologia . 2018;61(12):2669–2701. doi: 10.2337/dci18-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All related materials are available from Hongwei Li.