

Abstract
Despite advances in precision medicine, the clinical prospects for patients with ovarian and uterine cancers have not substantially improved. Here, we analyzed genome-scale CRISPR/Cas9 loss-of-function screens across 851 human cancer cell lines and found that frequent overexpression of SLC34A2 – encoding a phosphate importer – is correlated to sensitivity to loss of the phosphate exporter XPR1 in vitro and in vivo. In patient-derived tumor samples, we observed frequent PAX8-dependent overexpression of SLC34A2, XPR1 copy number amplifications, and XPR1 mRNA overexpression. Mechanistically, in SLC34A2-high cancer cell lines, genetic or pharmacologic inhibition of XPR1-dependent phosphate efflux leads to the toxic accumulation of intracellular phosphate. Finally, we show that XPR1 requires the novel partner protein KIDINS220 for proper cellular localization and activity, and that disruption of this protein complex results in acidic vacuolar structures preceding cell death. These data point to the XPR1:KIDINS220 complex and phosphate dysregulation as a therapeutic vulnerability in ovarian cancer.
Editorial Summary
Golub and colleagues identify the phosphate exporter XPR1 as a therapeutic vulnerability in ovarian and uterine cancers, and show that phosphate efflux inhibition reduces tumor cell viability through accumulation of intracellular phosphate.
Introduction
An emerging paradigm in cancer medicine is the tailoring of a therapeutic strategy with the specific molecular profile of a patient’s tumor. Despite remarkable advances in ‘precision medicine’ in other cancer types, outcomes for ovarian and uterine cancers have not improved substantially in the past 20 years1–3. Accordingly ovarian and uterine cancers remain among the most deadly, globally killing over 300,000 women in 2020 alone4. New therapeutic strategies are needed.
We and others have demonstrated that novel cancer vulnerabilities can be discovered from genome-scale loss-of-function cell viability screens5. From these data, both biological insights6,7 and therapeutic hypotheses8,9 can be developed. Here, we systematically analyze CRISPR/Cas9 loss-of-function screens across 851 human cancer cell lines to identify novel candidate therapeutic targets in ovarian cancer. We identified an unexpected synthetic lethal relationship between increased expression of the phosphate importer - SLC34A2 - and loss of the phosphate exporter XPR1. We provide compelling evidence for the therapeutic development of XPR1 inhibitors through extensive in vitro and in vivo validation, analysis of patient samples, and proof-of-concept pharmacologic inhibition. The relationship between increased phosphate import and reliance on phosphate export suggests intracellular phosphate accumulation is toxic to cancer cells, and we leverage co-essentiality data to elucidate XPR1 as a member of a phosphate efflux protein complex also containing KIDINS220. Together, these data highlight the power of functional genomics screens to identify compelling therapeutic targets and elucidate their biological function.
Results
Loss of XPR1 is toxic to gynecological cancers that express SLC34A2
To identify novel therapeutic targets for ovarian and uterine cancers, we analyzed genome-scale, pooled CRISPR/Cas9 loss of viability screens in 851 genomically characterized human cancer cell lines as part of the Cancer Dependency Map10–12. We focused on genes which, when inactivated, selectively led to loss of viability in ovarian or uterine cancer cell lines, since a broad killing pattern is more likely to represent mechanisms that would be poorly tolerated if pharmacologically inhibited. This analysis (Figure 1a) yielded ‘selective dependencies’ such as the transcription factor PAX88, a known lineage-restricted transcription factor. This analysis also revealed that inactivation of the phosphate exporter XPR1 has a cell-killing pattern which was highly selective and enriched in ovarian and uterine cancers (Extended Data Figure 1a). XPR1 is a transmembrane protein13 and the only phosphate exporter annotated in human biology14.
Figure 1 -. Functional genomics identifies XPR1 loss as a cancer vulnerability in SLC34A2high ovarian and uterine cancers.
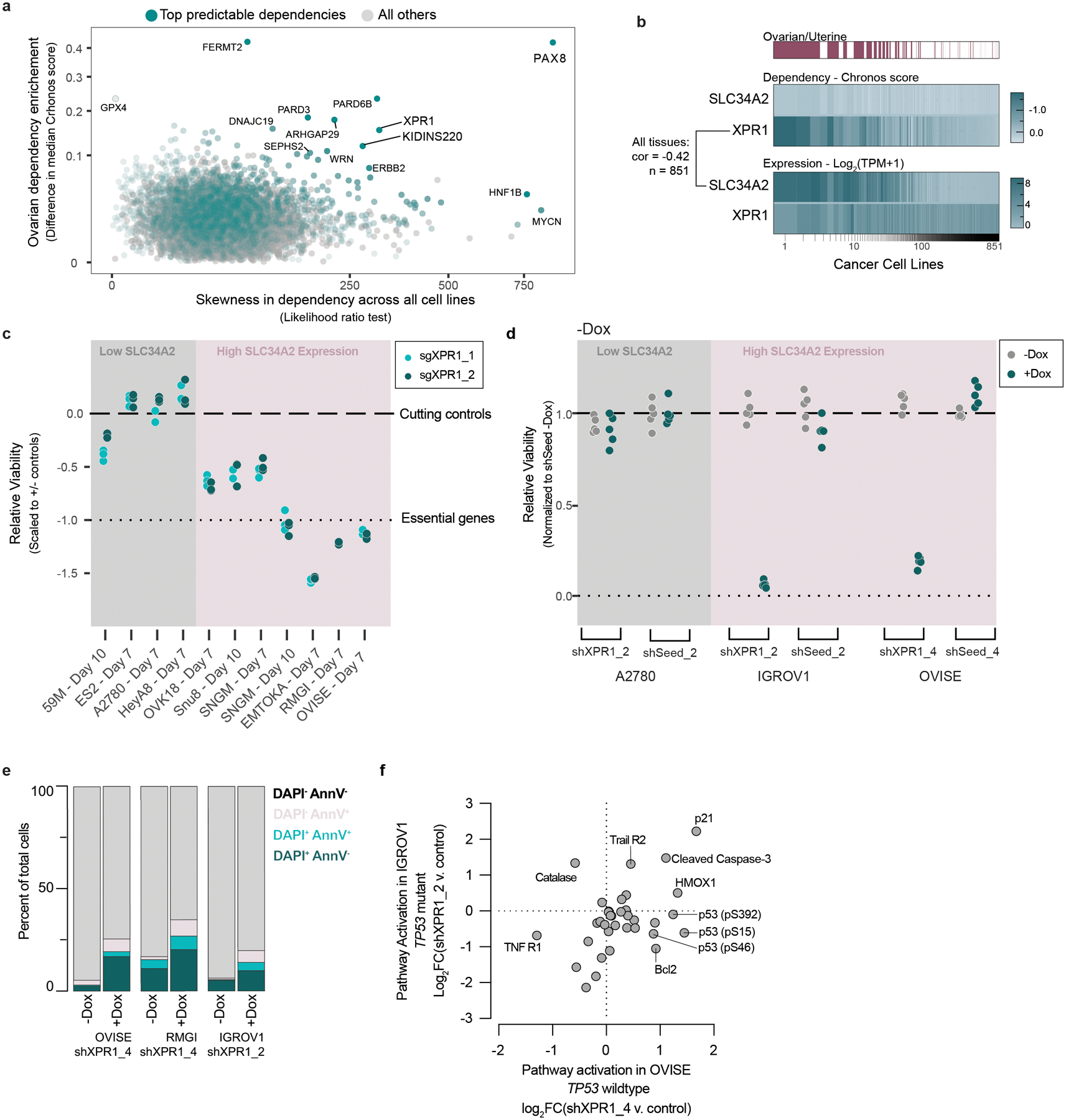
a) For the >18,000 genes tested in CRISPR/Cas9 loss of viability screens, the selectivity of the killing profile across all 851 cell lines (X-axis, likelihood ratio test, see methods) and the enrichment of that gene’s dependency (Chronos score) in ovarian and uterine cancers (Y-Axis) is plotted. The top 5% most predictable dependencies are highlighted in teal, where a random forest model using the genomic and molecular features of cancer cell lines can predict the strength of dependency.
b) Heatmap indicating XPR1 and SLC34A2 expression (Log2(TPM+1)) and dependency (CERES) values across all cell lines, approximately ranked by decreasing dependency on XPR1. The pearson correlation coefficient across all 851 cell lines is indicated.
c) Across a panel of ovarian and uterine cancer cell lines, viability effects after inactivation of XPR1 were evaluated by comparison to negative control sgRNA and sgRNA targeting pane-essential genes (N=3 independent transductions representative of at least N=2 independent experiments). Note that A2780 is not considered ovarian cancer despite its historical annotation. The data is scaled such that a value of 0 represents the viability effect of CRISPR/Cas9 genome editing and −1 represents loss of an essential gene. “High SLC34A2 expression” indicates mRNA expression greater than 3 TPM.
d) Viability assessment seven days after suppression of XPR1 using the indicated shRNA or seed-matched controls (N=5 technical replicates representative of at least N=3 independent experiments per cell line).
e) Six days after induction of shXPR1 in the indicated cell lines, cells were stained with DAPI to distinguish live and dead cells and Annexin V to distinguish non- and pre-apoptotic cells (N=1 flow cytometric analyses of at least 10,000 cells, representative of N=2 independent experiments). Right, quantitation of the percentage of cells in the indicated quadrants.
f) Analysis of cell death pathways in OVISE and IGROV1 five days after suppression of XPR1 by shRNA using protein arrays (N=1). Note that OVISE has wildtype (WT) TP53, while IGROV1 has an inactivating mutation in TP53.
We next pursued the molecular basis of the selective dependency on XPR1. Using more than 100,000 molecular features of cancer cell lines15, we built multivariate models - potential “biomarkers” of response - to predict XPR1 dependency16,17. Remarkably, the feature that most robustly predicted XPR1 dependency was expression of the phosphate importer SLC34A2 (Figure 1b, pearson coefficient = −0.42 in all cell lines). SLC34A2 overexpression in ovarian cancer is well documented18,19 and was highly correlated with XPR1 dependency in cell lines from the ovarian clear cell, high grade serous, and endometrial adenocarcinoma lineages (Extended Data Figure 1b–c).
We initially validated the pooled screening results in a total of 7 SLC34A2 high- and 3 low-expressing cancer cell lines, confirming that XPR1 is a selective and strong dependency in the context of SLC34A2 overexpression (Figure 1c–d and Extended Data Figure 2). Loss of XPR1 profoundly slows cell growth and leads to an increase in growth inhibitory and pro-apoptotic markers (Figure 1e–f, Extended Data Figure 2e–f). We next assessed the XPR1 dependency in a CRISPR/Cas9-based tumor formation competition assay with 74 total sgRNA, and observed XPR1 sgRNA depletion in SLC34A2-high tumors (Figure 2 and Extended Data Figure 3). In contrast, sgRNA targeting other metabolic dependencies, such as the ferroptosis regulator GPX4, were depleted in vitro but not in vivo, as has been previously reported20,21. These results indicate that the XPR1 dependency is retained in vivo.
Figure 2 -. XPR1 inactivation prevents tumor formation in vivo.
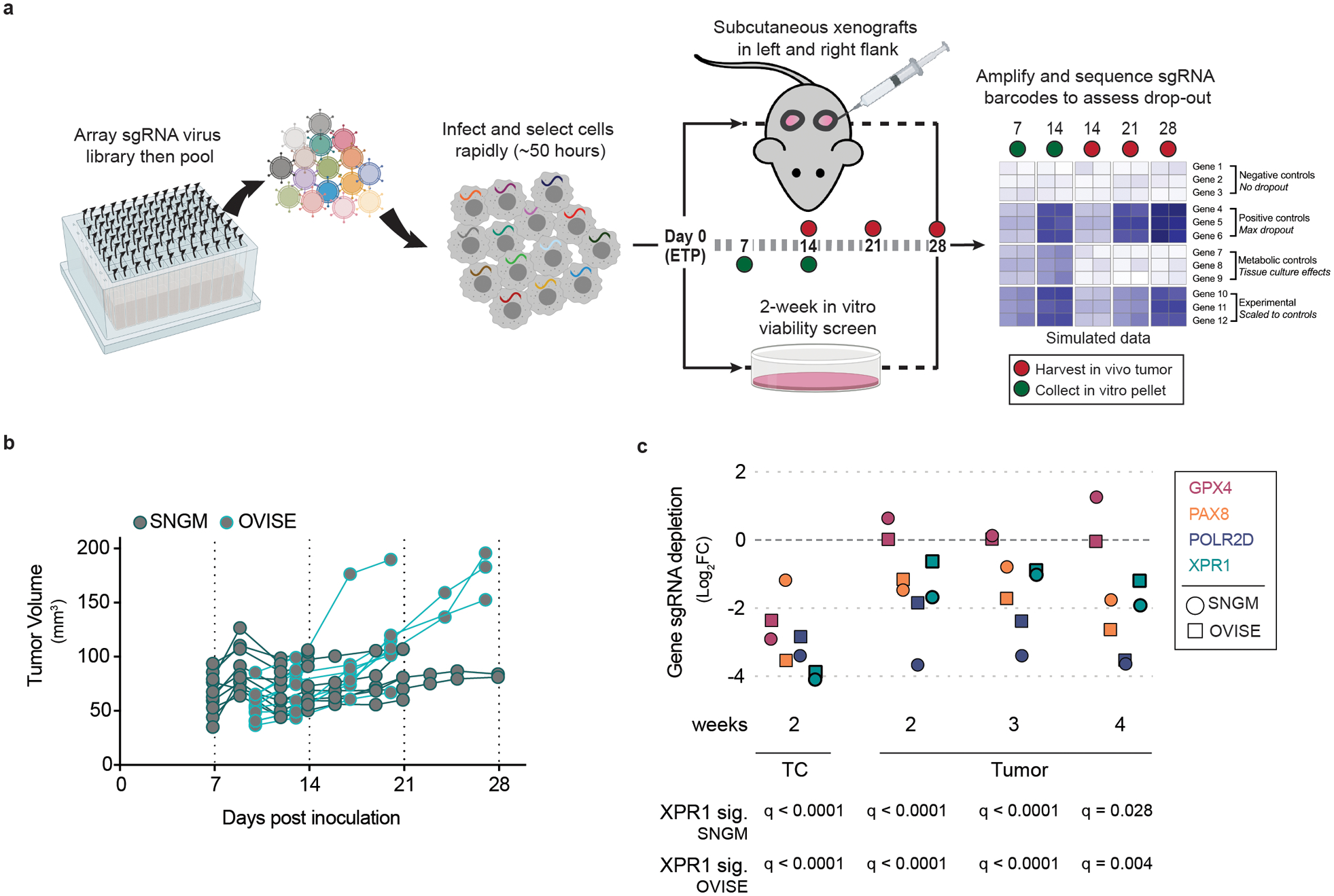
a) Experimental design for in vivo competition assays. Using a rapid infection and selection protocol, pooled sgRNA can be introduced via lentivirus into cancer cell lines and inoculated as subcutaneous xenografts and the effect of gene inactivation can be evaluated in a more physiologically-relevant environment than tissue culture.
b) After rapid infection with pooled sgRNA, 8 million SNGM or OVISE cells were inoculated as subcutaneous xenografts and allowed to grow. Tumor tissue was harvested at the indicated time points.
c) Evaluation of sgRNA targeting XPR1 and other cancer vulnerabilities in a tumor formation competition assay, as described in a for the OVISE (squares) and SNGM (circles) cancer cell lines (N=2–3 independent tumors derived from the same transduction per cell line per timepoint.). GPX4: glutathione peroxidase 4, a metabolic dependency reliant on the amount of peroxidated lipids in the metabolic environment of the cancer cells. PAX8: paired box 8 is a transcription factor dependency in many ovarian cancer cell lines. POLR2D: RNA Polymerase II Subunit D is a pan-essential gene and is used as a positive control. Below, the significance of depletion of three sgRNA targeting XPR1 relative to 7 control sgRNA was calculated via t-test and corrected for multiple comparisons with the Holm-Sidak method.
Evidence of phosphate dysregulation in primary patient samples
To extend the relevance of XPR1 and SLC34A2 beyond cell lines, we evaluated the relationship between XPR1 and SLC34A2 in primary patient samples from The Cancer Genome Atlas (TCGA)22,23 and normal samples from the Genotype-Tissue Expression project (GTEx)24–26. Ovarian tumors, on average, expressed SLC34A2 at levels 16-fold higher than normal fallopian tube epithelium (q = 0.006), which is thought to be the cell of origin of ovarian cancers27–30 (Figure 3a). Uterine cancers similarly overexpressed SLC34A2 relative to normal tissue. Interestingly, ovarian and uterine cancers were among the few tissues with high levels of SLC34A2 expression (Extended Data Figure 4a). We hypothesized that SLC34A2 expression may be maintained at high levels in ovarian cancer because of its regulation by the transcription factor PAX8, the expression of which is elevated in ovarian cancer and which is required for ovarian cancer cell survival8,31–33. In support of this, we found a strong correlation between PAX8 and SLC34A2 expression in patient samples (Extended Data Figure 4b,c). Furthermore, CRISPR-mediated inactivation or transcriptional repression of PAX8 led to a significant loss of SLC34A2 expression, as previously reported32–34 (Figure 3b,c and Extended Data Figure 4d,e).
Figure 3 -. XPR1 and SLC34A2 expression in patient samples indicate cancer-specific phosphate dysregulation caused by the lineage survival transcription factor PAX8.
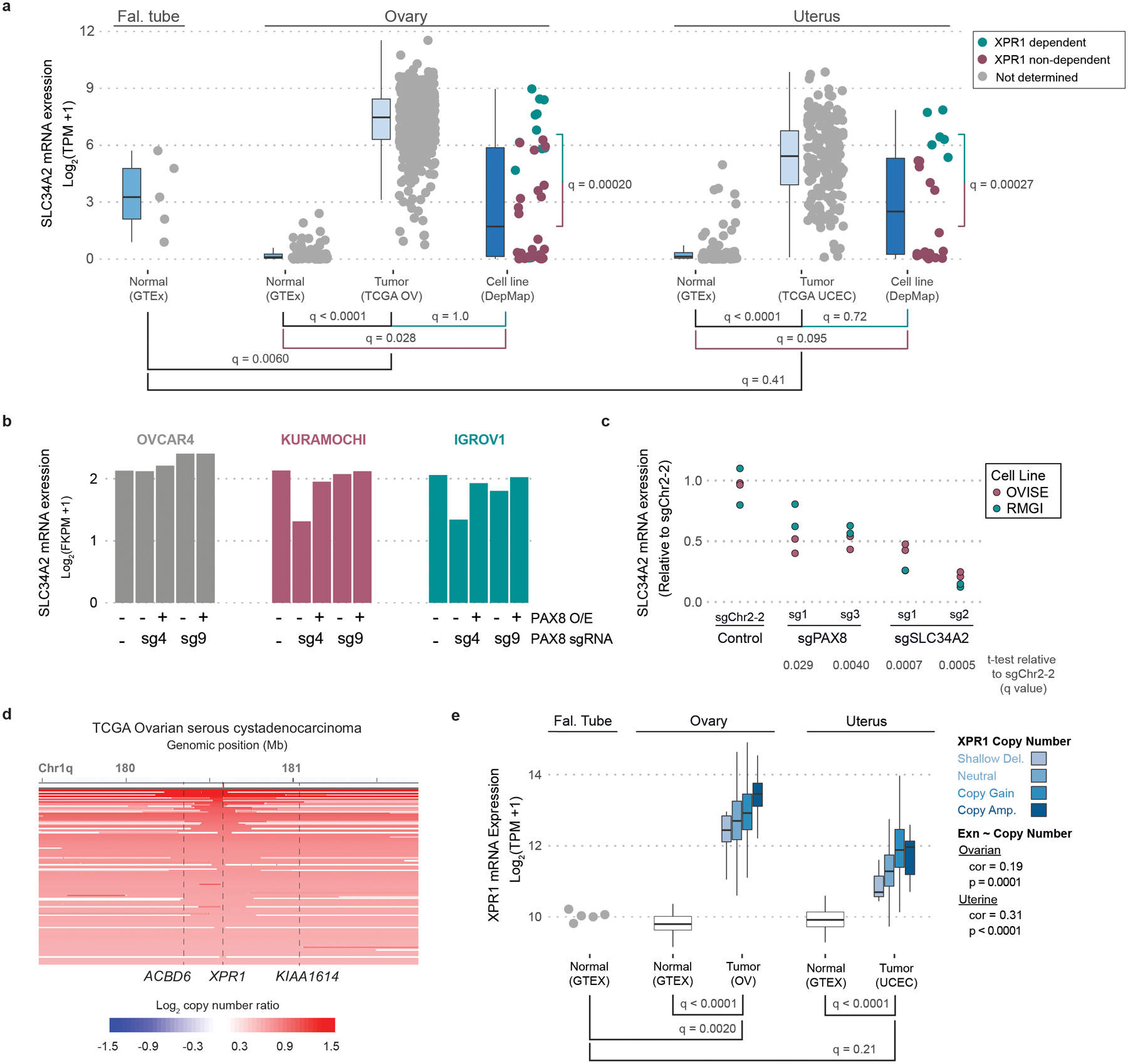
a) SLC34A2 is expressed in ovarian and uterine tumor samples at sufficient levels to predict dependency on XPR1. RNA expression values for SLC34A2 were compared within the idicated lineages. XPR1 dependency status is indicated by color where applicable (CERES < −0.5). q-values indicate the likelihood of the indicated populations having the same SLC34A2 expression according to a two-sided Wilcoxon ranked sums test with Bonferroni correction for multiple comparisons. Boxplots are drawn indicating the first and third quartiles, and whiskers span to the largest value within 1.5x the interquartile range. See methods for exact N values.
b) The expression of SLC34A2 was measured using RNAseq (N=1) after stable overexpression of PAX8 as indicated (PAX8 O/E), and induction of a PAX8-targeting (sg4) or control (sg9) sgRNA and dCas9-KRAB.
c) Seven days after transduction with the indicated sgRNA (N=2, separate transductions), RNA was extracted, converted to cDNA, and the expression of SLC34A2 was measured using RT-PCR. Significance was assessed by comparing the expression relative to sgChr2-2 across two cell lines with a one-tailed t-test, and corrected for multiple comparisons using the Bonferroni method. Data are representative of N=2 independent experiments.
d) XPR1 copy number heatmap for a ~2.5 Mb region of chromosome 1 indicating XPR1 amplification in TCGA serous ovarian cancer22. Each patient sample is represented by a horizontal line. Red indicates copy gain and blue indicates copy loss. Dashed vertical lines are the location of indicated genes. Data are a subset of the 489 samples, rank ordered by highest copy gain to indicate both focal and chromosome arm variants.
e) XPR1 mRNA expression is increased in ovarian and uterine cancer. XPR1 mRNA expression values from the same sources as panel a are compared for the indicated tissues, with TCGA OV and TCGA UCEC color-coded by XPR1 copy number status as determined by GISTIC analysis. Boxplots are drawn as in a. Statistical differences between tissues were determined as in a. The correlation of XPR1 copy number and expression was performed using spearman’s ranked correlation test. See methods for exact N values.
In contrast to the more restricted pattern of expression of SLC34A2 and PAX8, XPR1 is widely expressed in normal and cancer tissues (Extended Data Figure 4f). Nevertheless, we found strong evidence for positive selection of XPR1 copy number amplifications and enhanced mRNA expression in ovarian and uterine cancer, consistent with its dependency in these tissues (Figure 3d,e). In ovarian cancer, these amplifications were often focal, involving only the XPR1 gene (Figure 3d, q = 0.001535) whereas in uterine cancer, broader and less significant amplifications were observed (Extended Data Figure 4g, q = 0.568). XPR1 mRNA expression levels were correlated with XPR1 copy number alterations, but other mechanisms likely also contribute to XPR1 mRNA expression (Figures 3e). Thus, the dysregulated expression of the essential transcription factor PAX8 results in enhanced expression of SLC34A2, thereby creating dependency on XPR1.
XPR1 loss causes toxic phosphate accumulation
To further understand the mechanism by which XPR1 loss-of-function results in cancer cell death, we performed a genome-wide CRISPR/Cas9 rescue screen to determine which genes, when inactivated, were capable of rescuing XPR1-mediated loss of viability (Figure 4a, Extended Data Figure 5)36. Remarkably, the top rescuing gene of >18,000 tested was also the top predictive biomarker: SLC34A2. We further demonstrated that SLC34A2 is both necessary and sufficient to confer XPR1 dependency in three ovarian and uterine cell lines (Figure 4b).
Figure 4 -. XPR1 inactivation in SLC34A2-high ovarian cancer causes loss of cell viability via dysregulated intracellular phosphate homeostasis.
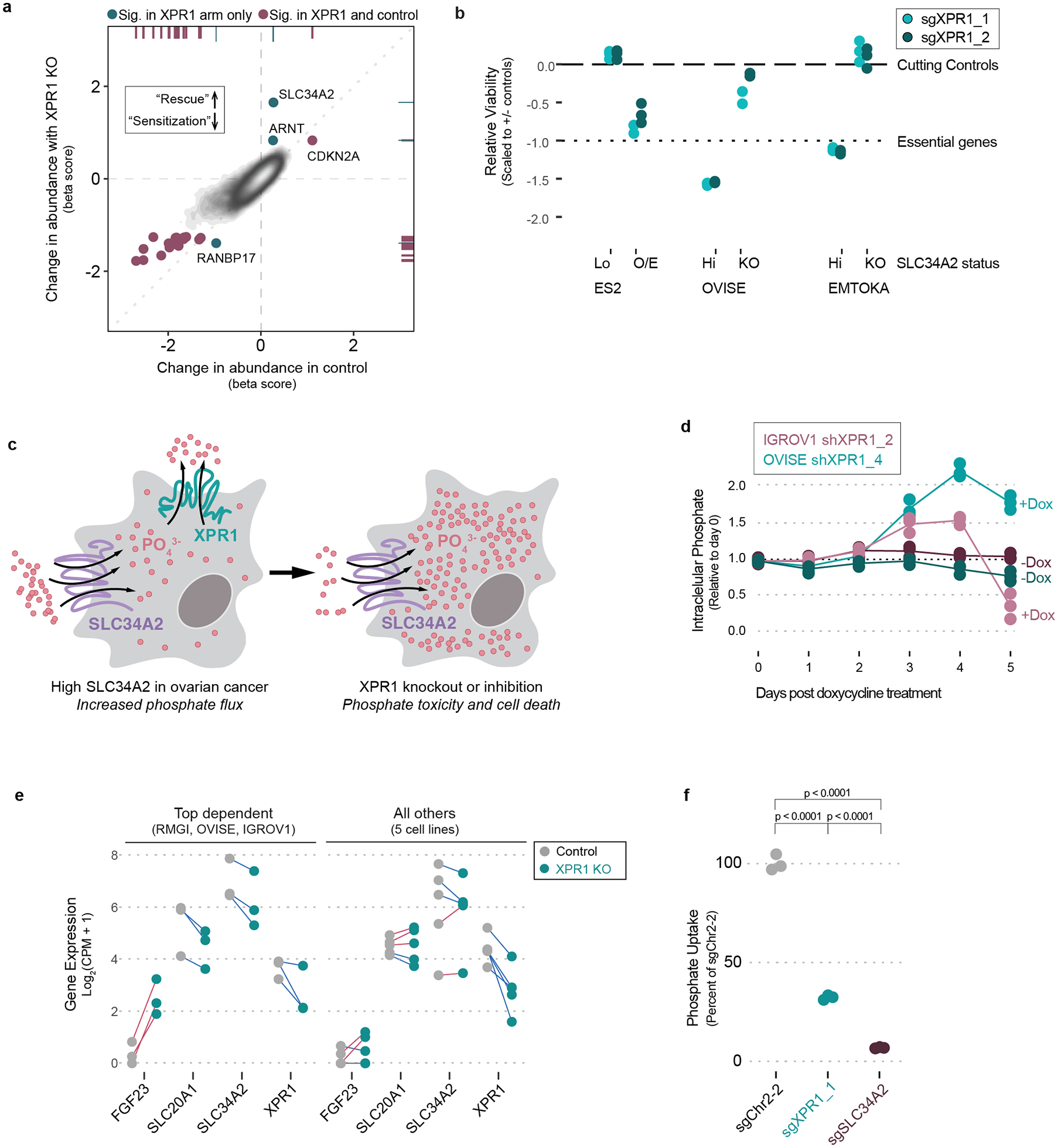
a) Identification of rescue genes that protect OVISE ovarian cancer cells from XPR1 dependency. Beta scores (determined by MaGeCK MLE) represent the change in representation for each gene from the initial library to the final timepoint for the control condition (X-axis) or combined with XPR1 inactivation (Y-axis). See methods for full experimental and analytical details. N=1 transduction per cell line, expanded and cultured as N=2 independent cultures.
b) The SLC34A2 status of normally XPR1-resistant (ES2, SLC34A2-low [lo]) or XPR1-sensitive (EMTOKA and OVISE, SLC34A2-high [hi]) cell lines was modified by overexpression (O/E) or inactivation (KO) of SLC34A2, and the XPR1 dependency was evaluated as in Figure 1c (N=3 separate transductions, representative of at least N=2 independent experiments per cell line).
c) Because of their relative directionalities of phosphate transport, we hypothesize that XPR1 perturbation is toxic because of intracellular phosphate accumulation in SLC34A2-high ovarian and uterine cancers.
d) At various time-points after treatment with doxycycline and induction of shRNA, the intracellular phosphate was measured in OVISE and IGROV1 cell lines (N=3 separate measurements per condition, representative of at least N=3 separate experiments).
e) A pool of 8 cancer cell lines were transduced with lentivirus to inactivate XPR1, and 4 days later the cells were subjected to 10X single cell transcriptomic measurement (N=1 transduction). The measured transcriptional change (relative to control sgRNA infection) in the indicated genes are plotted for the three cell lines with the largest and most correlated transcriptional change (see Extended Data Figure 7) on the left and the other five cell lines on the right. Blue lines connect cell lines displaying decreased expression upon XPR1 inactivation, red lines indicate increased expression.
f) XPR1 perturbation causes compensatory inhibition of phosphate uptake, measured by incubating OVISE ovarian cancer cells with medium supplemented with 32PO43- phosphate for 30 minutes prior to washing away excess medium and lysing the cells (N=1 transduction measured in technical duplicate, representative of N=2 independent experiments). Significance was assessed by one-way ANOVA and corrected for multiple comparisons using Bonferroni’s method.
The observation that high expression of the phosphate importer SLC34A2 is required for loss of cell viability after inactivation of the phosphate exporter XPR1 led us to hypothesize that accumulation of intracellular phosphate is toxic to ovarian and uterine cancer cells (Figure 4c). XPR1 is the only known phosphate exporter in humans14, suggesting that in the context of increased phosphate import, XPR1-dependent phosphate efflux would be in higher demand, consistent with frequent copy number amplifications observed in patient samples (Figure 3e).
Although the extracellular availability of phosphate in typical tissue culture medium far exceeds what is physiologically relevant, we found no correlation between the phosphate content of growth medium and XPR1 dependency across the Cancer Dependency Map dataset (Extended Data Figure 6a). Furthermore, XPR1 dependency was retained when cells were adapted to growth medium with near-physiological phosphate concentrations (reduced by ~90% from 72.8 to 7.8 mg/dL, Extended Data Figure 6b–c), indicating that XPR1 dependency is not an artifact of the high concentrations of extracellular phosphate.
Consistent with the phosphate accumulation hypothesis, we observed 2–4 fold increased intracellular phosphate following XPR1 suppression (Figure 4d). These large fluctuations in intracellular phosphate co-occur with loss of cell viability (Extended Data Figure 6d)37. To understand the cellular response to phosphate accumulation, we used single-cell RNA sequencing of 2,501 cells across 8 ovarian and uterine cancer cell lines at an early time-point following XPR1 inactivation (Figure 4e and Extended Data Figure 7a–g). The resulting transcriptional signature reflected cellular attempts to restore phosphate homeostasis, including the up-regulation of FGF23. This critical phosphate homeostatic hormone is typically expressed in osteogenic bone cells, and its expression in ovarian cancer cells - although not represented at the protein level (Extended Data Figure 7h) - is consistent with sensing elevated phosphate38. We also observed downregulation of phosphate importers at both the mRNA (Figure 4e) and protein level (Extended Data Figure 2c and 7i) after XPR1 inactivation or suppression. Consistent with this, XPR1 inactivation led to a 60% decrease in phosphate uptake (Figure 4f). This compensatory mechanism is also observed in XPR1-non-dependent cancer cell lines with known regulatory mechanisms for SLC34A2 (e.g. lung39,40, Extended Data Figure 1a,b, and 7j). These data suggest a potential protective mechanism to down-regulate phosphate uptake in response to toxic levels of inorganic phosphate41,42.
The phosphate efflux activity of XPR1 is required for cancer cell survival
We next confirmed that the phosphate efflux function of XPR1 was critical for cell survival. Expression of a naturally occurring hypomorphic XPR1 mutation (L218S), associated with a rare brain calcification disorder37, failed to rescue endogenous XPR1 inactivation, whereas wild-type XPR1 fully restored cell viability (Figure 5a and Extended Data Figure 8). In addition, we pharmacologically inhibited XPR1 using a previously reported protein inhibitor (XRBD, Figure 5b–c) and found that its cancer cell line growth-inhibitory effects paralleled inactivation of XPR1 (Figure 5d). Together, these results indicate that inhibition of the phosphate efflux capacity of XPR1 in SLC34A2-high cells is sufficient for loss of cancer cell viability.
Figure 5 -. Phosphate efflux activity by XPR1 is required for SLC34A2high cancer cell survival.
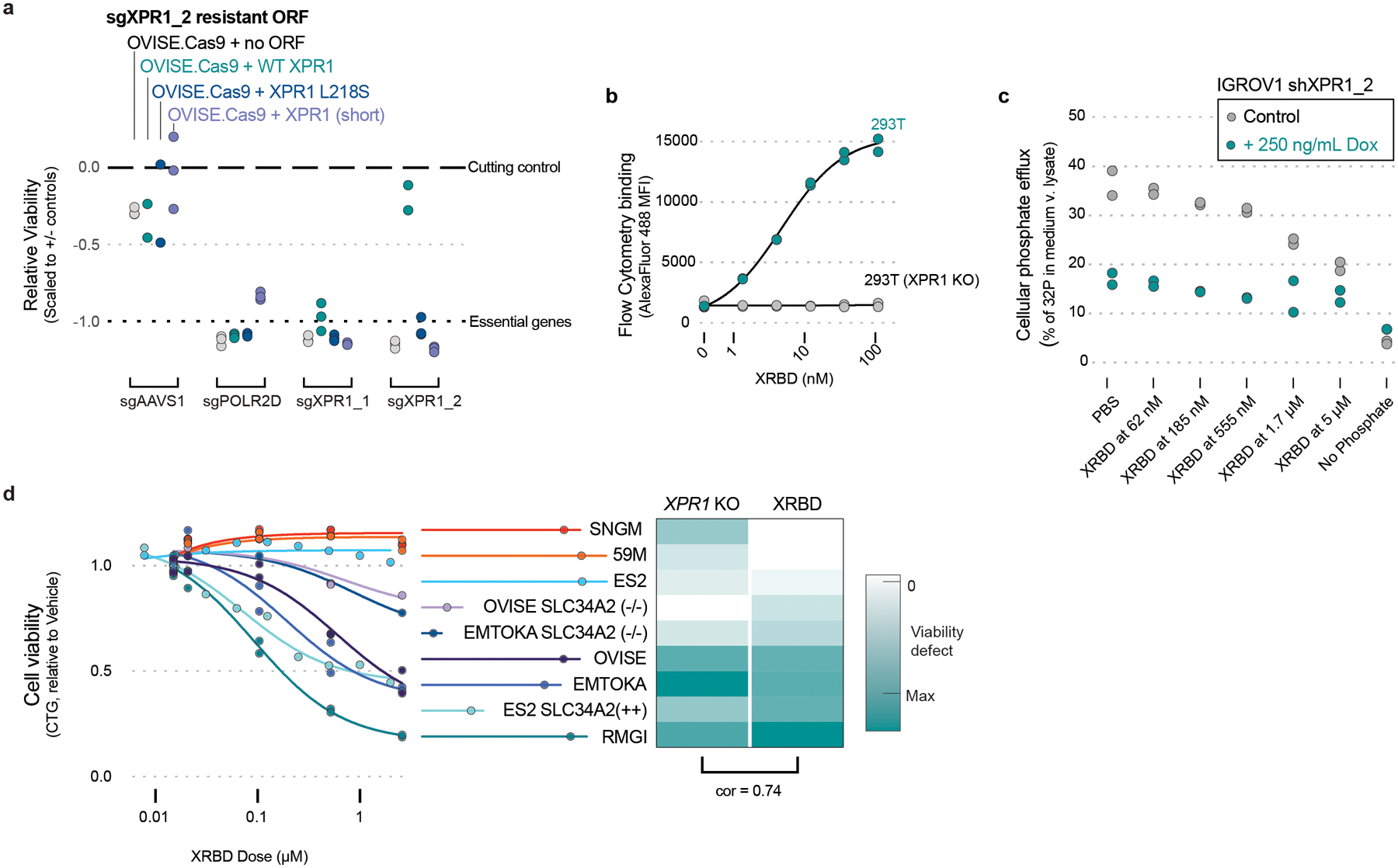
a) The indicated XPR1 open reading frames were tested for their ability to rescue inactivation of endogenous XPR1 (N=3 independent transductions, representative of N=2 independent experiments). The L218S mutation in XPR1 has previously been shown to only have ~50% the phosphate efflux function of wild-type XPR1 (see main text).
b) Purified XRBD (the soluble receptor binding domain of the NZB strain of xenotropic and polytropic murine leukemia virus) was incubated with 293T cells or 293T with XPR1 inactivation for 30 minutes at the indicated concentrations, washed, and stained with AlexaFluor488-conjugated anti-mouse secondary to detect the Fc tag on XRBD (N=2 flow cytometric analysis of at least 10,000 cells, representative of N=2 independent experiments).
c) Phosphate efflux was measured in the presence of XRBD or XPR1 suppression in IGROV1 mixed lineage ovarian cancer cell lines. IGROV1 were treated with doxycline (where indicated) to induce expression of shXPR1_2 three days prior to evaluating phosphate efflux by loading cells with 32P-labeled phosphate, washing the cells to remove excess 32P, and measuring the percent of total 32P in the conditioned medium after 30 minutes (N=2 technical replicates representative of N=3 independent experiments). Where indicated, XRBD was added to the cells during both the 32P loading and efflux portions of the experiment. Note that medium without phosphate does not stimulate phosphate efflux, and was used as a control.
d) Treatment of various cancer cell lines with the XPR1 inhibitor XRBD (the soluble receptor binding domain of the NZB strain of xenotropic and polytropic murine leukemia virus). Left, cells were incubated for 5 days with the indicated concentrations of XRBD and viability was assessed by Cell Titer Glo (N=2 independent treatments, representative of at least N=2 experiments per cell line.). Right, heatmap comparison of the viability defect after XPR1 inactivation (XPR1 KO) or XRBD treatment (5 μM dose), with pearson correlation coefficient indicated.
The activity of XPR1 is entirely dependent on a novel partner protein KIDINS220
To gain further insight into the mechanism by which XPR1 regulates phosphate homeostasis, we analyzed the Cancer Dependency Map for genes with highly correlated dependency profiles to XPR1. These “co-dependencies” often indicate proteins that are part of the same protein complex43,44. Of the ~18,000 genes analyzed, XPR1 dependency is most strongly correlated with KIDINS220 dependency, a gene with no known connection to phosphate homeostasis45–48 (pearson correlation = 0.81, Figure 6a and Extended Data Figure 9a). Given the strength of this correlation, we extensively validated KIDINS220 dependency (Extended Data Figure 9b–c), and hypothesized that KIDINS220 might be part of an XPR1 phosphate export complex.
Figure 6 -. KIDINS220 is a critical component of the phosphate efflux protein complex.

a) Across 851 cancer cell lines, the viability defects of XPR1 and KIDINS220 inactivation in each cell line is plotted and the Pearson correlation is indicated. A chronos value of −1 is the median viability defect of inactivating pan-essential genes in a given cell line.
b) The interaction between the V5-tagged XPR1 mutant and KIDINS220 was evaluated using co-immunoprecipitation. XPR1 WT corresponds to isoform NM_004736, while XPR1 (Short) corresponds to isoform NM_001135669. Green arrows indicate the expected molecular weight. N=1 experiment representative of N=3 independent transfections.
c) After XPR1-V5 immunoprecipitation, interacting proteins were identified using in-gel tryptic digestion followed by mass spectrometry. The X and Y axes show the total number of peptides per protein detected specifically in the XPR1 immunoprecipitation for N=2 independent transfection and immunoprecipitations. Higher abundance proteins (>10 peptides detected in IP:XPR1) are highlighted in teal.
d) Glycerol gradient sedimentation analysis of XPR1-containing native protein complexes with or without KIDINS220 inactivation. The crude lysate of the indicated cell lines was layered onto 10–30% glycerol gradients and centrifuged to fractionate protein complexes by molecular weight, followed by immunoblot analysis (N=1 centrifugation representative of N=3 independent experiments). The elution profile of protein standards is indicated below the immunoblot.
e) Localization of XPR1-V5 proteins after inactivation of KIDINS220. Scale bar = 100 μm. N=1 technical replicate of N=2 independent experiments.
f) Evaluation of XPR1 cell-surface localization after KIDINS220 inactivation. N=1 flow cytometric analysis of 10,000 cells, representative of N=4 independent experiments.
g) Cellular phosphate efflux after KIDINS220 inactivation. Three days after genetic inactivation of XPR1 or KIDINS220, cellular phosphate efflux was assessed. Cells were loaded with 32P-labeled phosphate, washed extensively to remove excess 32P, and then phosphate efflux was measured at the indicated times by isolating conditioned medium and cellular lysates (N=3 technical replicates of the same transduction, representative of N=3 independent transductions). Phosphate efflux is calculated as the percentage of 32P in the conditioned medium relative to the total 32P measured for that sample. Note that medium without phosphate does not stimulate phosphate efflux, and was used as a control.
In support of an XPR1:KIDINS220 protein complex, protein interaction databases indicate XPR1 and KIDINS220 interact with each other (Extended Data Figure 9d). Further, their gene expression was highly correlated across diverse tissues (Extended Data Figure 9e), suggesting co-function and co-regulation. To confirm this interaction, we performed co-immunoprecipitation experiments and found that XPR1 and KIDINS220 indeed interact with few each other and few other partner proteins (Figure 6b–c, Extended Data Figure 9f–g). Native XPR1:KIDINS220 is consistent with a highly oligomerized protein complex (Figure 6d). We mapped the XPR1:KIDINS220 interaction to the C-terminus of XPR1 containing the EXS domain (Figure 6b, Extended Data Figure 9g), an evolutionarily conserved domain known to be required for XPR1 trafficking between the golgi apparatus and the plasma membrane in order to achieve phosphate efflux49–51. In contrast, the N-terminal SPX domain of XPR1, which has been implicated in phosphate efflux and regulation42,52, was neither necessary nor sufficient to bind KIDINS220 (Figure 6b).
Further supporting an XPR1: KIDINS220 protein complex, we found dramatically decreased KIDINS220 protein levels following XPR1 genetic inactivation or suppression, but not inhibition with XRBD (Extended Data Figures 2c, 7i, and 9h). In addition, KIDINS220 inactivation decreased XPR1 cell-surface localization and dramatically changed the sedimentation pattern of XPR1, indicating the native localization and conformation of XPR1 requires KIDINS220 (Figure 6d–f, Extended Data Figure 9h). Finally, we directly measured phosphate efflux and found that inactivation of either XPR1 or KIDINS220 impaired phosphate efflux to a similar degree (Figure 6g) and resulted in increased intracellular phosphate (Extended Data Figure 9i). These results, taken together, indicate that phosphate efflux is achieved by the XPR1:KIDINS220 protein complex, and that loss of either complex member leads to a disruption in phosphate efflux which is required for cancer cell survival.
A striking feature of XPR1- or KIDINS220-mediated loss of cell viability is the formation of large, cytoplasmic, ‘vacuolar’ structures preceding loss of cell viability (Figure 7 and Supplemental Movie). Co-localization with the acidic dye Lysotracker and the lysosomal marker LAMP1 (Figure 7b–c) suggested they may be related to the lysosomal system. Ultrastructural analysis by transmission electron microscopy found these structures to be bound by a double-membrane that was often fenestrated (Figure 7d–e). Although they lack the electron-dense appearance typical of lysosomes, we did note their fusion with lysosomes (Figure 7d–e).
Figure 7 -. Vacuole structures precede loss of cell viability and are not derived from many common organelles.
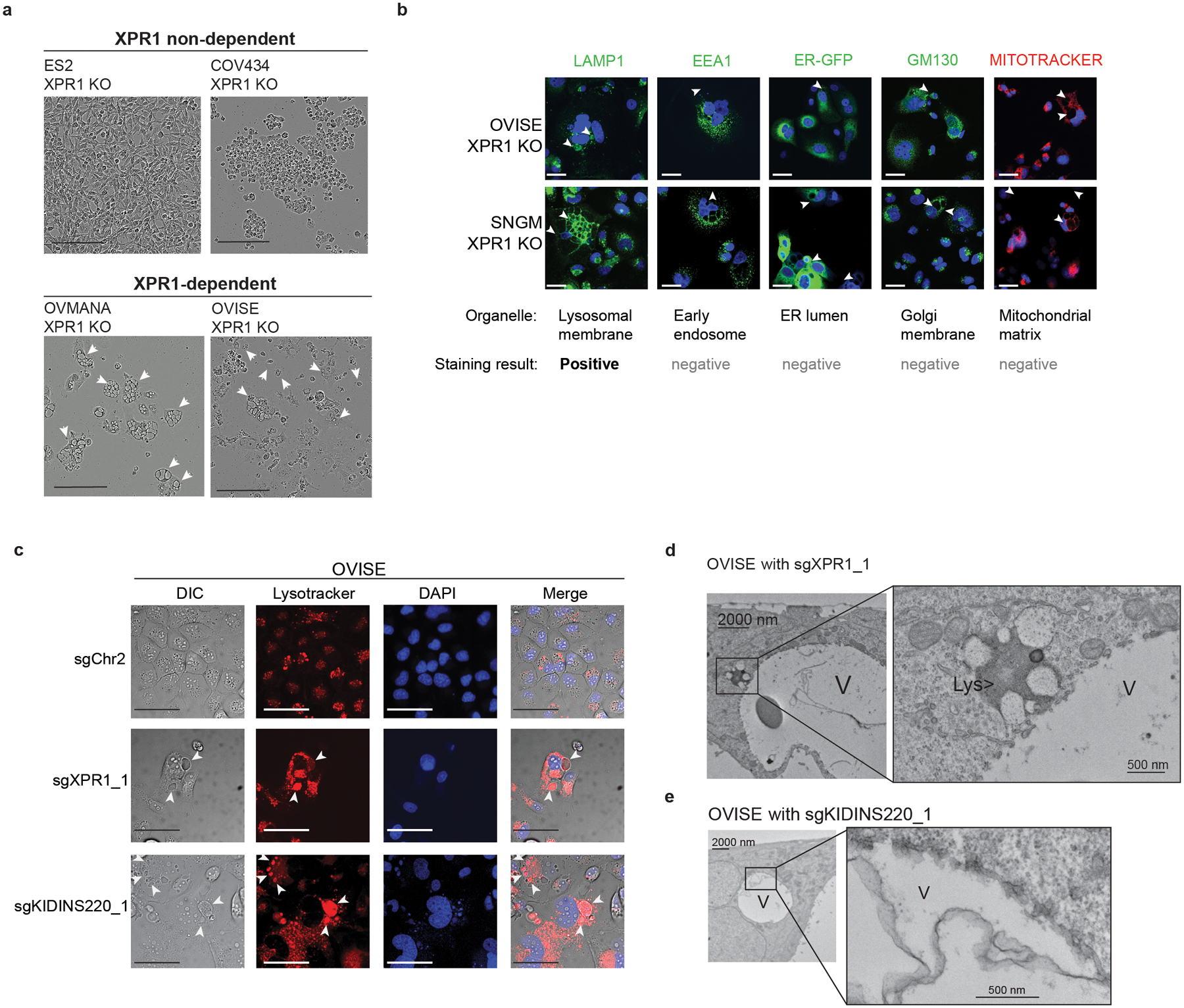
a) Phase-contrast images of ‘vacuole-like’ phenotype 4–5 days after XPR1 inactivation. Arrowheads indicate the location of ‘vacuole-like’ structures. Scale bars = 200 μm. Data are representative of N=2 independent transductions.
b) 6 days after infection with lentivirus encoding sgXPR1_2, OVISE and SNGM cell lines were stained and imaged using the indicated dyes and stains. Arrowheads indicate the location of vacuole structures by phase contrast (not pictured). Positive staining was only observed for the lysosomal dye LAMP1. Scale bars = 100 μm. Data are representative of N=2 independent transductions.
c) The acidic dye Lysotracker was used to stain live cells five days after inactivation of XPR1. Scale bars = 100 μm. Data are representative of N=2 independent transductions.
d) Transmission electron micrographs of “vacuole-like” structures (labeled V) or lysosomes (Lys) in OVISE cancer cells after XPR1 inactivation. N=1 experiment.
e) Same as d, but with KIDINS220 inactivation. N=1 experiment.
Discussion
This study highlights a previously unappreciated strategy to kill cancer cells: the disruption of phosphate homeostatic mechanisms that are normally tightly regulated. Interestingly, antibody-drug conjugates targeting the phosphate importer SLC34A253 have been explored for the past decade, but this approach exploits SLC34A2 simply as a biomarker of ovarian cancer, rather than serving as a mechanism to disrupt phosphate homeostasis. Furthermore, these strategies may have alternate mechanisms of resistance and/or on-target toxicity due to the physiological roles for SLC34A2 in intestinal54 and lung39,40 tissues. Nevertheless, the expression and regulation of SLC34A2 within fallopian tube epithelial cells – the likely cell-of-origin of ovarian and uterine cancers – has not been extensively studied. We observed elevated SLC34A2 expression in normal fallopian tube samples (Figure 3a), and hypothesize that SLC34A2 may play a similar role as in the lung for uptake of inorganic phosphate derived from surfactant metabolism.
In cancer contexts, it is not clear whether enhanced SLC34A2 expression is required for ovarian and uterine carcinogenesis or survival in vivo. Our data clearly indicate that SLC34A2 is not required for cancer cell survival in vitro (Figure 1b, 4a–b) and that these cell lines display a profound ability to down-regulate SLC34A2 in response to XPR1 inhibition (Extended Data Figure 2c and 7i), suggesting that strong overexpression of SLC34A2 is not necessarily required for ovarian cancer cell survival. We hypothesize that elevated activity of the lineage-survival transcription factor PAX8 drives SLC34A2 expression, and that PAX8-driven SLC34A2 expression is sufficient to engender XPR1 sensitivity. Further work is needed to elucidate the many mechanisms by which SLC34A2 is regulated in normal and cancer cells.
This study also highlights fundamental gaps in our understanding of how intracellular phosphate is sensed, regulated, and stored. Although excessive phosphate has previously been shown to be toxic55,56, the exact mechanism of this toxicity is unknown. Our transcriptional profiling experiments failed to identify a clear ‘phosphate stress response,’ and yet XPR1 perturbation causes a profound delay in cell growth. Along with cell-cycle arrest, we also observed large, acidic, ‘vacuolar’ structures (Figure 7). Whether these structures reflect a compensatory cell survival mechanism involving the sequestration of potentially toxic phospho-metabolites57,58, or are themselves the cause of cell death remains to be determined.
Finally, this study uses the power of large-scale functional genomics to elucidate biological processes: we show that the phosphate efflux activity of XPR1 is entirely dependent on KIDINS220. The KIDINS220 gene essentiality profile (Figure 6a) is both selective and highly-correlated with XPR1, suggesting a specific role for these two proteins in phosphate efflux, as opposed to a more general or pleiotropic role. We show that the XPR1:KIDINS220 protein complex is likely oligomeric (Figure 6d) and trafficks between multiple subcellular compartments, an activity previously attributed to both proteins45,50. Interestingly, we found that XPR1 inactivation or suppression invariably caused loss of KIDINS220 protein. Nevertheless, KIDINS220 protein loss is not required for cell death, as the XRBD phosphate efflux inhibitor did not decrease KIDINS220 protein levels (Extended Data Figure 7i). The exact mechanism of the XPR1:KIDINS220 phosphate efflux complex requires further study, as does reconciling cellular phosphate efflux with other activities attributed to KIDINS220, such as neurotrophin signaling47,48 and genetic associations between XPR1, KIDINS220, and various diseases59.
Together, this study establishes the XPR1:KIDINS220 protein complex as a previously unrecognized therapeutic target in ovarian and uterine cancer. Moreover, the work highlights disruption of phosphate homeostasis as a possible new anti-cancer strategy.
Methods
For in vivo studies, these studies were used under the Institutional Animal Care and Use Committee (IACUC) of the Broad Institute under animal protocol 0194- 01-18.
Statistics and Reproducibility
In general, tissue culture experiments were conducted with cells grown in parallel vessels to evaluate the reproducibility of numeric data (e.g. 2 wells treated with the identical dose of XRBD in Figure 5d). In most cases, experiments were always conducted at least twice to confirm effect sizes; in most situations only representative experiments are shown. This is the case for data from each cell line in Figures 1c–e, 3c, 4b, 4d, 4f, 5, 6b, 6c (both replicates are shown), 6d–g, and 7, and Extended Data Figure 2b–f, 4d, 5b, 6c–d, 7h–j, 8, 9b, 9c, 9g, 9i. The experiment presented in Extended Data Figure 2a was only conducted once. The cell lines presented in Figure 6f and Extended Data Figure 9h are representative of at least two clones derived by single-cell isolation. For the in vivo competition assay (Figure 2), cells were infected with the small library of sgRNA only once, and parallel cultures or different subcutaneous xenografts were treated as ‘technical replicates.’ Sample sizes were chosen (8 mice, 2 tumors per mouse) to account for the penetrance of tumor development, to evaluate multiple timepoints, and to have at least 2 animals per timepoint. The animals with the largest tumor burden were euthanized at the indicated timepoints (i.e. no randomization was employed). No statistical method was used to predetermine sample size. For cell death pathway profiling after XPR1 suppression (Figure 1f), RNAseq after PAX8 suppression (Figure 3b), genome-scale modifier screen of XPR1 dependency (Figure 4a), and MixSeq transcriptional profiling (Figure 4e), experiments were only conducted once. Statistical significance testing is discussed in more detail in the relevant Methods section.
Genetic Dependency Data
The dependency data used in this manuscript come from the Public Avana 21Q2 dataset consisting of dependency data for 18,025 genes across 859 cancer cell lines from 26 lineages. Expression data from the Cancer Cell Line Encyclopedia was also used. These data are available online at depmap.org/downloads. In Figure 1a, the selectivity (NormLRT60) and predictability12,17 was determined as previously reported. “Highly Predictable” genes are indicated if the pearson correlation coefficient between the experimental data and the top predictive model is greater than 0.4. The median dependency for each gene in ovarian/uterine cancers (n = 62) was subtracted from the median dependency in all other cancer cell lines (n = 671) to calculate the ovarian/uterine genetic dependency enrichment on the Y-axis of Figure 1a. In Figure 1b and Extended Data Figure 1b, the correlation of SLC34A2 expression and XPR1 dependency was performed using a two-tailed pearson correlation test. To compare XPR1 co-dependencies (Extended Data Figure 9a), a two-tailed pearson correlation test was performed for XPR1 versus all other genes (k = 18,025 genes, n = 859 cell lines although some cell line:gene pairs are not represented), and p-values are reported after correcting for multiple comparisons with the Benjamini-Hochberg method.
Cell Lines
ES2, HeyA8, A2780, 59M, SNU8, OVK18, SNGM, OVISE, EMTOKA, IGROV1, OVCAR4, KURAMOCHI, RMGI, COV413a, JHOS4, HEC6 and JHUEM1 cancer cell lines were collected by the CCLE before distribution for our use. The sources of the aforementioned cell lines can be found at DepMap.org. All cell lines were adapted to growth in RPMI 1640 (Corning) + 10% FBS before use. All cell lines are routinely validated using short-tandem repeat profiling.
sgRNA sequences
The negative control guides sgChr2 and sgAAVS1 were designed to cleave a gene desert and an intronic region in PPP1R12C, respectively, to control for the effects of DNA double-strand breaks. sgLacZ targets a sequence not found in the human genome. Positive control sRNA target common-essential splicing factors (SF3B1), ribosomal subunits (POLR2D), or kinesin motor proteins (KIF11). The 20 bp targeting sequences can be found in the Supplementary Table.
Lentiviral production
Lentiviral production was performed using HEK293T cells as described on the Broad Institute Genetic Perturbation Web Portal (https://portals.broadinstitute.org/gpp/public/).
Plasmids, overexpression constructs, site directed mutagenesis
Open reading frames of the following genes were obtained from a genome-scale library of annotated genes61. SLC34A2 (NM_006424) was isolated from this library in pDONR223 and was transferred into the expression vector pLX-TRC313 (similar to Addgene #118017) using gateway cloning. The resultant construct has a C-terminal V5 tag, and after stable integration into cell lines using lentiviral infection, the proper protein product with a V5 tag was detected using western blot (not shown). XPR1 constructs (both isoforms NM_004736 and NM_001135669) were obtained in a similar way. Only the NM_004736 isoform was observed using isoform-agnostic PCR primers and cDNA generated from the OVISE cancer cell line. Mutations were introduced with PCR based methods, either the Q5 Site-directed Mutagenesis kit (NEB cat# E0554S) for large deletions or the QuickChange II XL (Agilent cat# 200521) for point mutations, and were confirmed using sanger sequencing. The XPR1 ORFs were then transferred to pLX-TRC313 or the same expression vector with a weaker, PGK promoter. For co-immunoprecipitation experiments in HEK-293T cells, the stronger promoter XPR1 mutants were used to maximize expression levels. The weaker promoter construct was used for stable expression in ovarian cancer cell lines and immunofluorescence and mutant-rescue experiments.
Immunohistochemistry
Cell pellets were fixed using paraformaldyhde and then paraffin embedded. Immunohistochemistry was performed on the Leica Bond RX automated staining platform using the Leica Biosystems Refine Detection Kit. The antibody for SLC34A2/MX35 (Creative Biolabs, cat# TAB-467MZ, recombinant) was run at 1:400 dilution with citrate antigen retrieval.
CRISPR Viability Assays
CRISPR viability assays were performed in 96-well plates with cells seeded at a low density to allow for logarithmic growth throughout the entire assay. For 7 day assays, cells were seeded and infected with lentivirus expressing the sgRNA in pXPR-BRD003 on Day 0. The next day, the infection media was replaced with 100 uL of media. On Day 7 post-infection, viability was evaluated by addition of 25 uL of Cell Titer Glo (Promega) reagent per well, and luminescence was measured. Infection efficiency was determined by comparing the viability of cells with and without puromycin after infection, and assays were repeated if less than 80% of cells were infected with every sgRNA. The data were normalized such that the cutting control sgRNA (targeting Chr2-2 and AAVS1) is 0 and positive control sgRNA (targeting the common essential genes KIF11, SF3B1, and POLR2D) is −1.0. For 10 day assays, infections were carried out in 6-well plates. Three days post-infection, the cells were lifted and seeded into replicate 96-well plates. On Days 3, 7 and 10 post-infection, viability was evaluated by addition of 50 uL of Cell Titer Glo (Promega) reagent per well and monitoring the luminescence. The fold change viability was calculated by comparing Days 7 or 10 to Day 3, and the data was normalized as above.
shRNA sequences
shRNA sequences for XPR1 were selected from project DRIVE’s sub-genome scale shRNA library60 using DEMETER2 estimates for on-target and off-target seed effects62. A detailed protocol for selecting shRNA using these datasets is available online (https://protocols.io/view/shrna-selection-and-quality-control-for-cancer-tar-bfmnjk5e). Doxycycline inducible shRNA expression was accomplished by cloning these sequences into the pRSITEP-U6Tet-(shRNA)-EF1-TetRep-2A-Puro vector (Cellecta #SVSHU6TEP-L). shRNA seed matched negative control sequences62 were generated by substituting complementary base pair sequences into positions 9–11 bp of the target shRNA using the web-based tool (https://web.archive.org/web/20180605134130/http://rnai.nih.gov/haystack/C911Calc2.html).
shXPR1 and seed matched control sequences were rigorously tested for on-target XPR1 suppression as well as off target cell viability effects. Off target cell viability effects were determined when a given shSeed control sequence did not knockdown XPR1 but produced strong loss of cell viability, regardless of whether a cell line was predicted to be XPR1 dependent or non-dependent. shRNA target sequences are provided below:
Antibodies
A full list of Antibodies and their respective dilutions is included in the Reporting Summary accompanying this paper.
Protein analysis of cell lysates by immunoblotting.
For protein analyses, cells were grown in 6-well dishes. The cells were harvested by washing with PBS, incubating with 0.5 mL of TrypLE until all cells had lifted and diluting to 1.5 mL with PBS. The cells were then centrifuged and washed once with PBS, then lysed with Radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 1.0% IGEPAL® CA-630, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0) supplemented with cOmplete, Mini Protease and Phosphatase Inhibitor Cocktail Tablets (Roche). Cell extracts were cleared by spinning at 15000 rpm for 10 minutes at 4°C. Protein content was quantified by BCA analysis and 4x LDS reducing sample buffer was added. We found boiling the lysates led to a loss of XPR1 protein by western blot, and so the lysates were incubated at 37°C for 30 minutes to denature proteins. For standard western blots (i.e. Extended Data Figure 5b and Extended Data Figure 6e), equal amounts of protein (typically 25 μg) were resolved by SDS-PAGE, transferred to nitrocellulose and incubated with the indicated primary and secondary antibodies were used to visualize protein levels. Images were obtained using a Licor Odyssey CLx system.
To quantitatively determine cellular proteins (i.e. Figure 6b), lysates were analyzed using the Protein Simple: an automated capillary-based protein separation and immuno-blotting assay. Lysates were prepared as above and diluted to 0.1 mg/mL with sample buffer and 40 mM DTT prior to loading 3 μL of sample onto each plate.
Foci formation
Cells stably expressing doxycycline inducible short hairpins against XPR1 or the corresponding seed controls were plated at three different densities (18000, 12000, and 6000 cells/well) to determine optimal seeding density. Six replicates of each cell line were plated in 24 well plates, and half the wells were treated with 0.5 μg/mL doxycycline. Media and doxycycline were refreshed every two to three days until untreated wells reached 100 percent confluence. The cells were then washed with 1X PBS and fixed with 4% PFA in 1X PBS for 15 minutes. Fixation was quenched with deionized water and cells were stained with 0.1% crystal violet in deionized water for 20 minutes. Cells were then washed with deionized water to remove residual crystal violet and left to dry overnight. To quantify crystal violet staining, 10% acetic acid was incubated in each well for 20 minutes, diluted 1:3 with deionized water, and re-plated in quadruplicate in a 96 well plate. Absorbance was measured at 590 nm.
Time course analysis of cell growth
To measure cell growth over time after XPR1 suppression, cells were seeded in a 96-well plate, treated with doxycycline to induce shXPR1, and imaged every 6 hours (Essen Incucyte S3). Images were quantified using the built-in software.
Proteome Profiler for cells death markers
To evaluate markers associated with different cell death pathways, cells were treated with doxycycline for five days to induce shXPR1, cells were washed with PBS, lysed, and processed according to the instructions for the Proteome Profiler Human Apoptosis Array Kit (R&D Systems cat# ARY009). The resulting dot blot array was quantified using ImageJ, and values were normalized to the -shXPR1 condition per cell line.
Annexin and DAPI staining
To evaluate the extent of cell death, cells were treated for six days with doxycycline to induce shXPR1, and then non-attached and adherent cells were collected and pooled, then stained for 40 minutes at room temperature with DAPI, washed with Annexin Staining Buffer, and then incubated for 15 minutes at room temperature with Annexin V-APC (Thermo Fisher cat# A35110). Cells were then washed, and then >10,000 single cells were analyzed by flow cytometry. Data were analyzed using FlowJo, gating against smaller cellular debris as well as events consistent with more than one cell per droplet.
In vivo sgRNA competition assay
These studies were used under the Institutional Animal Care and Use Committee (IACUC) of the Broad Institute under animal protocol 0194-01-18. A detailed protocol for the tumor formation competition assay is available online (https://www.protocols.io/view/in-vivo-nanopool-pooled-sgrna-competition-assays-t-bfsbjnan). A small lentiviral library of 74 total sgRNA (including non-targeting, negative controls targeting gene deserts or introns, positive controls targeting pan-essential genes, and experimental sgRNA) was made in an arrayed format and then pooled together. To minimize time in tissue culture, we optimized the infection and puromycin conditions to achieve roughly 30–50% infection efficiency and nearly 100% selection 50 hours after infection. OVISE cells were selected with 8 μg/mL Puromycin, and SNGM cells were selected with 4 μg/mL puromycin.
50 hours after infection, the cells were lifted, counted, and diluted in 50% matrigel at a final concentration of 8 million cells per 100 uL. Some cells were frozen to determine the early representation of the library. For in vitro experiments, the cells were re-plated and grown in standard conditions for 2 weeks. In total, this study used 26 female, 7-week-old, Rag1−/− Il2rg−/− (NRG) mice obtained from The Jackson Laboratories. For in vivo experiments, mice were anesthetized under isoflurane gas, and two bilateral subcutaneous xenografts were inoculated in each of five mice (10 tumors per experiment). Tumors were measured twice weekly with calipers and tumor volumes were calculated using the formula: pi/6 × (width2 × length). Animal body weights were recorded weekly or twice weekly during the course of all studies. Mice were euthanized and tumors harvested on days 14, 21, and 28 after inoculation, ensuring they had not reached endpoints (tumor size > 2500 mm3, no visual distress, and no evidence of ascites development). After harvesting and weighing, tumors were flash frozen in liquid nitrogen until genomic DNA isolation.
At the end of the study, (28 days post inoculation; 30 days post-infection), the tumors were thawed, minced, and genomic DNA was extracted from all samples using the Qiagen DNeasy Blood and Tissue kits. Sample barcodes were sequenced by Illumina Next-Generation sequencing then deconvoluted with Broad Genetic Perturbation Platform’s PoolQ software for sgRNA read counts. Data from all animals included in the study are displayed.
Statistical Analysis for in vivo competition assay
The gene-level effect was determined by comparing normalized sgRNA read counts of the early time point (two days after infection, the day of inoculation) compared to the indicated time-points. If normalized read counts at the early time points were significantly different for a particular sgRNA, that sgRNA was not included in downstream analyses (as a sign that the plasmid did not produce lentivirus). sgRNA read counts were normalized such that the 7 “cutting-control” sgRNA (representing viability effects from CRISPR/Cas9 genome editing) had a median depletion of 0. Next, the median fold change of all sgRNA targeting a particular gene was calculated, and only genes with 2-fold depletion or greater relative to the control sgRNA are reported per replicate in Extended Data Figure 3. The different time-point replicates were median-averaged and are reported in Figure 2c. Statistical significance of XPR1 depletion was calculated by comparing the gene-level depletion of XPR1 (3 different sgRNA per replicate) compared to cutting control sgRNA (7 different sgRNA per replicate) using a t-test to compare all replicates at each time-point. The test was conducted in GraphPad Prism 8.0 and Holm-Sidak multiple comparisons correction was applied. Corrected p-values (q-values) are reported in Figure 2c. Test statistics are as follows: OVISE TC 2 weeks (n = 2, t-value = 29.6, 14 degrees of freedom); OVISE tumor 2 weeks (n = 4, t-value = 8.9, 30 degrees of freedom); OVISE tumor 3 weeks (n = 4, t-value = 7.1, 30 degrees of freedom); OVISE tumor 4 weeks (n = 2, t-value = 4.6, 30 degrees of freedom); SNGM TC 2 weeks (n = 2, t-value = 19.1, 14 degrees of freedom); SNGM tumor 2 weeks (n = 2, t-value = 5.6, 16 degrees of freedom); SNGM tumor 3 weeks (n = 4, t-value = 2.3, 28 degrees of freedom); SNGM tumor 4 weeks (n = 4, t-value = 5.0, 22 degrees of freedom).
Comparing expression of genes across normal and tumor tissues
We compiled log2(TPM + 1) gene expression data for normal fallopian tube (GTEx, n = 5), normal ovary (GTEx, n = 88), normal uterus (GTEx, n = 78), ovarian cancer (TCGA OV, n = 426) and uterine cancer (TCGA UCEC, n = 238) from the TOIL RSEM log2(TPM + 0.01) data at Xena Browser (https://xenabrowser.net/) and then converted the data to log2(TPM + 1). RNAseq gene expression for ovarian cancer cell lines (CCLE n = 40), and uterine cancer cell lines (CCLE, n = 22) were downloaded from the Cancer Cell Line Encyclopedia (https://depmap.org) as Log2(TPM + 1). Because most TCGA ovarian and uterine samples have relatively high purity (>80%)64, we used these data directly for the following comparisons. In Figure 3a and 3e and Extended Data Figures 4c and 4f, boxplots are drawn using the “geom_boxplot” command in the R package ggplot2, such that the box spans the 1st and 3rd quartiles of values with the median indicated by a line. The whiskers extend 1.5x the interquartile range, and outlier’s beyond this range are excluded. In Figure 3a and 3e and Extended Data Figure 4c, Wilcoxon ranked sum test was employed using the R package ‘rstatix’ to compare the distribution of expression of the indicated gene between the indicated tissues, and p-values were corrected for multiple comparisons using Bonferoni’s method. In Extended Data Figure 4a a pairwise Wilcoxon ranked sum test was used to compare the expression of SLC34A2 in each tissue relative to all other tissues and p-values were corrected for multiple comparisons using Bonferoni’s method and are reported on the Y-axis. The difference in median SLC34A2 expression (tissue - expression across all tissues) is plotted on the X-axis of Extended Data Figure 4a. In Extended Data Figure 4b, the correlation between SLC34A2 and PAX8 mRNA was tested using a two-tailed pearson correlation test across the indicated tissues (n = 897). In Extended Data Figure 9e, the correlation between XPR1 and KIDINS220 mRNA was tested using a two-tailed pearson correlation test for the indicated tissue groups (all 60 tissues, n = 17,194; top 15 correlated, n = 2,799, for all other tissues, listed from top to bottom, n = 337, 55, 172, 173, 36, 47, 182, 520, 66, 182, 496, 154, 119, 181, and 79).
RT-PCR
After the indicated perturbations, cells were lysed and RNA was extracted using Qiazol and phenol-chloroform extraction. Total RNA was determined spectrophotometrically and normalized amounts of RNA as converted to cDNA using the iScript kit (Bio-Rad cat#1708890). Diluted cDNA was then mixed with gene-specific primers and Power Sybr Green Master Mix (Thermo Fisher cat# 4367659) and analyzed on a Quant Studio 7 RT-PCR instrument. A full list of RT-PCR primers used can be found in the Supplementary Table. The quality of the RNA extraction was evaluated by comparing CT values of cDNA samples with control samples treated in the same way but without addition of Reverse Transcriptase. Gene expression values were corrected for loading with a house-keeping gene (VCL). In Figure 3c, SLC34A2 gene expression values were compared using a one-tailed t-test corrected for multiple comparisons with the bonferroni method. Test statistics are as follows: sgPAX8_1 (n = 4, t-value = 3.54, df = 5.46); sgPAX8_3 (n = 4, t-value = 5.66, df = 5.19); sgSLC34A2_1 (n = 4, t-value = 7.24, df = 5.94); sgSLC34A2_2 (n = 4, t-value = 11.45, df = 4.20).
Analysis of XPR1 copy number in TCGA
To evaluate the frequency of XPR1 amplification, we evaluated precomputed GISTIC235 analyses for recurrent copy number alterations in ovarian and uterine TCGA datasets22,23. To compare the expression of XPR1 with its copy-number status, XPR1 copy number thresholds - as determined by GISTIC - were downloaded from CBIoPortal78,79 and then matched to the corresponding TCGA samples. 410 ovarian cancer and 171 uterine cancer samples were included in this analysis. In Figure 3d and Extended Data Figure 4g, each patient sample is represented by a horizontal line. Red indicates copy gain and blue indicates copy loss. Dashed vertical lines are the location of indicated genes. The samples are rank-ordered by highest copy gain to indicate both focal and chromosome arm variants. In Figure 3e, we tested whether there was a significantly non-zero correlation between XPR1 copy number and XPR1 mRNA expression using a two-tailed spearman correlation test, and reported Spearman’s rho and the p-value. Also in figure 3e, the expression of XPR1 between tissue categories was compared using Wilcoxon ranked sums test with Bonferroni correction.
Modifier Screen
The anchor modifier screen was performed as described previously36. OVISE cells stably expressing sgRNA targeting Chr2-2, XPR1_1, or XPR1-2 in the lentiviral guide-only vector pXPR-BRD016 were infected with the Brunello “All-in-one” vector (pXPR-BRD023) in a format so that each cell received a maximum of one sgRNA. 24 hours post infection, the cells were split into two replicates and treated with 2 ug/mL puromycin to select for cells with stable integration. Every 3–4 days, the cells were trypsinized and re-plated to maintain a minimum representation of 500x library representation per replicate. After 15 days post-infection, all cells were collected and counted. The final representations for each replicate were as follows: 533x and 533x for sgChr2-2, 287x and 231x for the sgXPR1_1, and 81x and 83x for the sgXPR1_2. Genomic DNA was then extracted from each cell line and sgRNA barcodes were amplified by PCR.
Modifier Screen Analysis
The representation of each sgRNA in each arm of the experiment was determined by next generation sequencing, and compared with plasmid DNA representation of the entire library. Using MAGeCK-MLE65, this change in representation was converted to gene-level beta-values, representing the variability of each gene relative to all other genes. A permutation test (10 different permutations) on the sgRNA labels was used to empirically determine p-values, which were then corrected using the Benjamini-Hochberg Procedure. Any gene with an FDR < 0.1 was considered a potential hit in that screen. SLC34A2 was the only statistically significant “rescue” gene (i.e. a beta value > 0) for both sgXPR1_1 and sgXPR1_2 arms that was not significant in the control arm. There were several genes with beta-values significantly less than 0. Comparing the beta-values for this list of potential “sensitizer” genes between the sgXPR1_1/2 arms and the sgChr2-2 arm, we note that all of these are significantly depleted across every arm, with little difference in beta-value. This indicates these are essential genes, and their depletion observed in every arm of the experiment is likely not due to a relationship with XPR1.
Comparison of XPR1 dependency and tissue culture medium
Information on the growth medium is available for cell lines at depmap.org/portal. As each cell line is grown in a mixture of several medium types (e.g. 90% RPMI 1640 + 10% FBS), we estimated the concentration of phosphate by using a weighted average of phosphate concentrations of each component (see the Supplementary Table and ref. 66). The XPR1 dependency score was then compared to the concentration of phosphate using a one-tailed pearson correlation test in which we expected to find that more dependent cell lines were grown in higher concentrations of phosphate, and exact p-values and sample sizes are reported in Extended Data Figure 6a.
Cell competition assays in low phosphate media
A competition experiment was designed using isogenic cell lines expressing either Firefly or Renilla luciferase; the Firefly luciferase expressing version of the cell line was also engineered to express Cas9.
For the competition assay in low phosphate, luciferase expressing cells were adapted from normal growth media (RPMI-1640 +10% Fetal Bovine Serum) to low phosphate media for 7 days. The concentration of phosphate in low phosphate media was empirically determined as the minimal phosphate concentration to support sustained cell viability for 3 weeks in culture. Low phosphate media was a 9:1 ratio of (RPMI-1640 without L-Glutamine and Phosphate +10% FBS; MP Biomedicals cat#: 09162975) with normal growth media (RPMI-1640 +10% Fetal Bovine Serum). We estimate 100% FBS to have 5.3 mg/dL of inorganic phosphate, though this value is likely to vary72. Therefore, this low-phosphate RPMI would have a concentration of 7.8 mg/dL.
After cells were adapted to low phosphate media for 7 days, Renilla luciferase expressing cells were mixed at a 1:1 ratio with Firefly luciferase +Cas9 expressing cells and infected with the indicated sgRNAs on day 8. Cells were selected with puromycin 24 hours after infection. On day 11, cells were split and half were re-plated to propagate the cultured mixtures while the other half were subjected to Dual-Glo Luciferase Assay (Promega) to set baseline signal. The final luciferase assay was performed on day 16 after initial culture in low phosphate media.
XRBD protein purification
We kindly thank Jean Luc Battini for providing the sequence for the XRBD-mFc construct published previously14. The plasmid encoding XRBD (strain NZB) was synthesized and cloned into pcDNA3.4 by Thermo Fisher Scientific GENEART GmbH. XRBD protein was expressed in CHO cells and purified using Protein A affinity chromatography by Thermo Fisher Scientific GENEART GmbH.
XRBD flow cytometry
Cells were lifted from culture vessels using TrypLE Express (Thermo Fisher cat#12604013) and then diluted in PBS + 2% FBS. 300,000 cells were transferred to a U-bottom 96-well plate in 50 uL, followed by addition of 50 uL of XRBD staining solution. In Figure 5b, the indicated doses were tested and in Figure 6f, XRBD was used at 100 nM. Cells were incubated at 37c for 40 minutes, washed once, and then incubated with an anti-mouse secondary antibody conjugated to AlexaFluor488 (ThermoFisher cat# A-11004). Cells were then incubated on ice for 40 minutes, washed four times in PBS + 2% FBS, and then analyzed on a CytoFlex LX instrument. At least 10,000 single cell events were recorded for each condition.
XRBD viability assays
To assess viability defects after exposure to XRBD, dilutions of the XRBD were made in PBS and then added to cells plated in 96-well plates 24 hours prior. Five days after addition of XRBD, cellular viability was assessed by Cell Titer Glo.
Measuring Intracellular Phosphate
To measure intracellular phosphate, cells were plated in 6-well plates as described above to perturb XPR1, KIDINS220, or SLC34A2. After the indicated times, the cells were washed three times with tris-buffered saline to remove residual phosphate from the media. The cells were then lysed in 1% NP-40, 50 mM Tris pH 7.5, and protease inhibitors, and the cellular debris was cleared by centrifugation. The cell lysate, or a dilution series of a phosphate standard, was then diluted in water to 50 uL in a 96-well microtiter plate and 10 uL Malachite Green Reagent A was added per manufacturer instructions (R&D Systems cat# DY966). After 10 minutes, 10 uL of Malachite Green Reagent B was added to the samples and the absorbance at 620 nm was immediately read. If any sample was not within the linear range of the assay, then the samples were iteratively re-diluted and re-analyzed. Intracellular phosphate levels were calculated by dividing the interpolated phosphate concentration by the concentration of protein (determined by BCA assay) in each sample. In Figure 4d, Extended Data Figure 6d, and Extended Data Figure 9i, technical triplicates representative of at least 3 experiments are displayed.
Immunofluorescence and fixed cell compatible dyes
Cells stably expressing V5-tagged XPR1 and KIDINS constructs were plated at a density of 10,000 – 20,000 cells per well in Nunc™ Lab-Tek™ II CC2-™ 8-well Chamber Slides (Thermo Fisher). When investigating localization upon knockdown, cells were plated in chamber slides after 5 days of lentiviral transduction of sgRNA targeting XPR1, KIDINS220, or a non-coding portion of Chromosome 2. The following day, cells were washed with 1X Phosphate Buffered Saline (PBS) (Corning) and fixed in 4% paraformaldehyde (PFA) (Electron Microscopy Sciences) in 1X PBS for 15 minutes. Cells were washed twice with 1X PBS to quench fixation, permeabilized with 0.1% Triton X-100 in 1X PBS for 15 minutes, and were blocked in 1% bovine serum albumin (BSA) in 1X PBS for 1 hour. Cells were probed overnight at 4 °C with primary antibody diluted in 0.1% BSA in 1X PBS according to the table below. The following day, cells were washed three times with 0.1% Triton in 1X PBS and stained for 1 hour at 25 °C with Alexa-fluor conjugated secondary antibodies (Molecular Probes, ThermoFisher) diluted in the blocking buffer according to the table below. The wells were then washed with 1X PBS three times and counterstained with 4′,6-diamidino-2-phenylindole (DAPI) in 1X PBS at 2 μg/mL for 20 minutes. The wells were then washed twice with deionized water, and cells were mounted in ProLong Gold AntiFade Mountant (ThermoFisher).
For determining the organelle source of “vacuole-like” structures, the same general immunofluorescent staining protocol was used as above with the following changes. SNGM and OVISE Cas9 stable cell lines were plated at 10,000–20,000 cells per well on μ-Slide 8-well coated chamber slides (IBIDI, cat# 80826) and simultaneously infected with lentivirus expressing sgRNAs. The next day transduced cells were selected with 2 ug/mL puromycin for 24 hours, removed from puro selection and were fixed 6 days after infection.
For ER labelling, cells were transduced with 24 uL of CellLight™ ER-GFP, BacMam 2.0 in 200 uL culture media 24 hours before imaging (Thermo-Fisher, cat# C10590) and no permeabilization step was performed. For mitochondrial imaging, cells were treated with 100 nM MitoTracker Red CMXRos (Thermo-Fisher, cat# M7512) in serum-free RPMI for 30 min at 37C, then MitoTracker dye media was replaced with normal growth media (RPMI with 10% FBS) and incubated for 1 hour at 37C before fixation. All other antibody-based stains were treated as described above. Information on the antibodies used and their concentrations for staining are provided in the antibodies Methods section.
Multiplexed Transcriptional profiling
Multiplexed Transcriptional profiling (MixSeq67) was performed using custom pools of ovarian and uterine cancer cell lines. Cancer cell lines were pooled together (5–7 cell lines per mini pool) based on doubling time and frozen. To initiate the experiment, the cells were thawed and plated in 12-well dishes. The next day, virus encoding mixtures of sgRNAs (sgLacZ/sgChr2-2 or sgXPR1_1/sgXPR1_2) under conditions in which each cell received both sgRNA to increase the penetrance of inactivation. Cells were treated with 2 ug/mL puromycin 24 hours after infection. Four days after infection, the cells were lifted with TrypLE, spun down, resuspended in cell-staining buffer (PBS + 2%BSA + 0.02%Tween) and counted.
Perturbations were multiplexed for 10x sequencing using Cell Hashing68. Equal numbers of each ‘mini-pool’ were then pooled together, blocked with FcX blocking buffer (BioLegend) for 10 minutes on ice and then incubated with hash-antibodies (TotalSeq from BioLegend) for 30 minutes on ice. The cells were then washed thrice with cell-staining buffer and resuspended in Cell Capture buffer (PBS + 0.04% BSA), filtered with a 40 μm filter, and diluted to ~1,500 cells per μL. A detailed protocol can be found online (https://www.protocols.io/view/cell-hashing-zn9f5h6). Approximately 40,000 cells were then loaded onto a 10x Chromium controller using v3. Single Cell 3’ reagent chemistries. Library preparation and next generation sequencing were performed as before68.
Sequencing data was processed using 10x Cell Ranger software (v3, hg19 reference genome), run with the ‘Cumulus’ cloud-based analysis framework69. SNP-based cell line classification and quality control was performed according to the methods described in reference 70. In brief, for each cell line the allelic fractions across a predefined 100,000 SNP reference set was estimated from bulk RNA-seq data using Freebayes70. A logistic regression model was then used to estimate the likelihood of the observed SNP reads for an individual cell having come from each cell line given the allelic fractions across the SNP reference set. A similar model was used to detect doublets where allelic fractions were modeled as a mixture of the allelic fractions from two different cell lines. After SNP-based classification, low quality cells and doublets were removed according to a set of stringent filters. These include the proportion of UMIs from mitochondrial genes being between 0.25 and 0.01, the number of reads at reference SNP sites being greater than 50, and the likelihood that a cell matches a particular cell line being much higher than the likelihood that it matches a different cell line or doublet.
MixSeq single-cell data was analyzed using the Seurat R package (v377. The data was first normalized and scaled using the NormalizeData and ScaleData functions (default parameters). To generate the UMAP embedding (Extended Data Figure 7d), the top 5,000 most variable genes were selected using the FindVariableFeatures function, principal components were computed using the RunPCA function, and the embedding was generated using the RunUMAP function with 20 principal components, 10 nearest neighbors, and a minimum distance of 0.3 (default parameters otherwise). Cell cycle phase classification was performed with the CellCycleScoring function, using the S- and G2M-phase marker gene sets reported previously. ΔG0/G1 (Extended Data Figure 7e) was calculated for each cell line as the fraction of cells in G0/G1 in the XPR1 sgRNA condition minus the fraction of cells in G0/G1 in the control condition.
MixSeq differential expression analysis was performed using the “limma-voom” pipeline71,72. First, per cell normalization factors were calculated using the “TMMwzp” method from the edgeR R package73. Then, counts were converted into logCPM and the mean-variance relationship estimated using the voom function from the limma R package80. To identify each cell lines’ transcriptional response to XPR1 knockout removing the effect of cell cycle (Extended Data Figure 7e) a linear model was used with the ‘S.Score’ and ‘G2M.Score’ values from Seurat CellCycleScoring as covariates to regress out the effect of cell cycle. The top 500 differentially expressed genes displayed in the heatmap were identified based on the average log-fold-change across the 8 cell lines. To identify the average transcriptional response within the three highly correlated cell lines (RMGI, IGROV1, and OVISE) and five less correlated (JHUEM1,OVCAR4,COV413A, JHOS4, and HEC6) cell lines (Extended Data Figure 7f,g) a similar linear model was used, but cell line identify was added as a covariate to account for differences in baseline gene expression between the cell lines. P-values were derived from empirical-Bayes moderated t-statistics, and FDR adjusted Q-values were obtained using the Benjamini-Hochberg method.
FGF23 enzyme-linked immuno-assay
To measure levels of secreted FGF23 from cancer cell lines, the indicated cell lines were plated in 6-well dishes and treated with doxycycline for four days to induce shRNA targeting XPR1. The conditioned medium was then collected, centriguged to remove any debris, and then FGF23 levels were measured according to the manufacturer’s instructions (R&D Systems DY2604-05). Similar results were obtained using a second manufacturer’s ELISA (Quidel Corporation 606600)
Phosphate uptake and efflux assays
To determine phosphate uptake or efflux, OVISE cells were infected in 6-well plates with lentivrius encoding sgRNA targeting XPR1, KIDINS220. Stable cell lines with SLC34A2 knockout were also used. IGROV1 cells three days after doxycycline treatment to suppress XPR1 using shRNA were also used. The day before the experiment, the cells were split into 96-well plates such that the cells would be confluent the following day. Cells were first “pulsed” using low-phosphate RPMI 1640 (a 1:9 ratio of standard RPMI to no-phosphate RPMI, see the lower phosphate competition assay above) supplemented with 10 μci/mL 32PO4 (Perkin Elmer NEX053001MC) and incubated at room temperature for 30 minutes. The cells were then washed with “no phosphate” RPMI 1640. To determine phosphate uptake, and initial phosphate levels for time course efflux experiments, cells were lysed with 1% Triton X-100 and the amount of intracellular 32P was measured using a liquid scintillation counter. For efflux time course experiments, the “chase” of phosphate efflux was then measured using high-phosphate RPMI (i.e. standard RPMI 1640). When incubated without phosphate in the medium (0 mM phosphate RPMI 1640), phosphate efflux is far lower (as has been reported before in reference 14 and was used as a control. At each timepoint, the conditioned medium was taken, the cells were washed thrice with no-phosphate RPMI 1640 (to remove any radioactivity outside of the cells) and then lysed with 1% Triton X-100. Conditioned medium and cell lysates were analyzed for 32P using liquid scintillation counter. The extent of phosphate efflux was determined by dividing the 32P measured in the conditioned medium with the total 32P measured for that sample (in cell lysates and in the conditioned medium). For experiments using XRBD, the protein was diluted in PBS and added to the medium during both the uptake/pulse and efflux/chase portions of the experiment.
High-throughput Mass Spectrometry analysis
High throughput protein interaction databases BioPlex74 and BioGRID75 were used to search and download lists of primary physical interactors of both XPR1 and KIDINS220. Interactions were identified by affinity capture-mass spectrometry, where epitope tags on target ‘bait’ proteins act as affinity capture probes for identifying ‘prey’ interactor proteins. Common interactors were found by comparing the four gene lists then visualizing the overlap.
Co-Immunoprecipitation of V5-tagged proteins
HEK293T cells were transiently transfected with 40 μg of pLXTRC313 ORF vectors expressing XPR1, GFP, or Luciferase for 24 hours. Cells were then washed twice with 1X TBS and lysed with 0.4% NP40, 50 mM Tris, 150 mM NaCl, supplemented with Halt Protease Inhibitor Cocktail (ThermoScientific). Cell extracts were cleared by spinning at 15000 rpm for 15 minutes at 4 °C on a table top centrifuge, and quantified by BCA. 50 uL of V5-tagged immuno magnetic beads (MBL International, cat# M167-11) was incubated with one milligram of protein lysate overnight at 4°C. The next day, beads were washed five times with 0.2% NP40 50mM Tris 150mM NaCl. Bound protein was then eluted by adding 10X sample buffer (Protein Simple) and incubating the beads for 30 minutes at 37°C. Eluate was removed by beads and was probed alongside whole cell lysates by Protein Simple Automated Western or standard immunoblotting.
Protein ID by Mass Spectrometry
Eluate from the V5 CoIP were run on 4–12% PAGE, stained with Safe Stain (Invitrogen cat#LC6065) and cut into 8 pieces that span the entire mW range of the gel. These excised gel bands were cut into approximately 1 mm 3 pieces and then subjected to a modified in-gel trypsin digestion procedure76. Gel pieces were washed and dehydrated with acetonitrile for 10 minutes followed by removal of acetonitrile. Pieces were then completely dried in a speed-vac. Rehydration of the gel pieces was with 50 mM ammonium bicarbonate solution containing 12.5 ng/μl modified sequencing-grade trypsin (Promega, Madison, WI) at 4°C. After 45 min., the excess trypsin solution was removed and replaced with 50 mM ammonium bicarbonate solution to just cover the gel pieces. Samples were then placed in a 37°C room overnight. Peptides were later extracted by removing the ammonium bicarbonate solution, followed by one wash with a solution containing 50% acetonitrile and 1% formic acid. The extracts were then dried in a speed-vac (~1 hr) and stored at 4°C until analysis. On the day of analysis the samples were reconstituted in 5 – 10 μl of HPLC solvent A (2.5% acetonitrile, 0.1% formic acid). A nano-scale reverse-phase HPLC capillary column was created by packing 2.6 μm C18 spherical silica beads into a fused silica capillary (100 μm inner diameter x ~30 cm length) with a flame-drawn tip. After equilibrating the column each sample was loaded via a Famos autosampler (LC Packings, San Francisco CA) onto the column. A gradient was formed and peptides were eluted with increasing concentrations of solvent B (97.5% acetonitrile, 0.1% formic acid).
As peptides eluted they were subjected to electrospray ionization and then entered into an LTQ Orbitrap Velos Pro ion-trap mass spectrometer (Thermo Fisher Scientific, Waltham, MA). Peptides were detected, isolated, and fragmented to produce a tandem mass spectrum of specific fragment ions for each peptide. Peptide sequences (and hence protein identity) were determined by matching protein databases with the acquired fragmentation pattern by the software program, Sequest (Thermo Fisher Scientific, Waltham, MA)77. All databases include a reversed version of all the sequences and the data was filtered to between a one and two percent peptide false discovery rate. Proteins identified and displayed in Figure 6c met the following criteria: Less than 10 total peptides identified in IP:Luciferase and 10x more peptides in IP:XPR1 than IP:Luciferase.
Live cell lysotracker staining of acidic organelles
Similar to the immunofluorescence methods above, OVISE Cas9 stable cells were plated at 10,000 cells per well in μ-Slide 8 Well coated chamber slides (IBIDI, cat# 80826) and simultaneously infected with lentivirus expressing sgRNAs. 24 hours after infection, cells were selected with 2ug/mL puromycin for 2 days and then replaced with RPMI-1640 without phenol red supplemented with 10% FBS. Live cells were incubated with dyes and imaged 5 days after infection.
For dye staining, LysoTracker Red DND-99 (Invitrogen cat#: L7528) was resuspended as a 1 mM stock in DMSO. Cells were stained in phenol-red free RPMI growth media 50 nM Lysotracker for 45 min at 37C in the dark. The dye was then washed out once with dye free growth media and then incubated with 1 ug/mL Hoechst 33342, (Invitrogen cat#: H3570) in phenol red free growth media at 37C for 30 min and remained in Hoechst containing media during imaging. Cells were imaged immediately after on a Nikon Eclipse Ti microscope with a Yokogawa Life Sciences CSU-W1 spinning disc confocal system.
Ultrastructural analysis by transmission electron microscopy
After infection with cutting control (sgChr2-2) or experimental sgRNA (sgXPR1_1 and sgKIDINS220_1), OVISE.Cas9 and EMTOKA cell lines were grown for 5 days in 6-well dishes. The cells were washed once with PBS and then fixed in 2.5% Glutaraldehyde and 2.5% Paraformaldehyde in 100 mM sodium cacodylate buffer, pH 7.4. Cells were then washed in 100 mM Sodium cacodylate buffer pH 7.4, postfixed for 30 min in a solution of 1% Osmium Tetroxide and 1.5% Potassium Ferrocyanide, washed in water thrice and incubated in 1% aqueous uranyl acetate for 30 minutes followed by two washes in water and subsequent dehydration in grades of alcohol (5 minutes each; 50%, 70%, 95%, twice in 100%).
Cells were removed from the dish in propylene oxide, pelleted at 3000 rpm for 3 minutes and infiltrated for 2 – 16 hours in a 1:1 mixture of propylene oxide and TAAB Epon (TAAB Laboratories Equipment Ltd, https://taab.co.uk). The samples were subsequently embedded in TAAB Epon and polymerized at 60 °C for 48 hours. Ultrathin sections (about 60 nm) were cut on a Reichert Ultracut-S microtome, picked up on to copper grids stained with lead citrate and examined in a JEOL 1200EX Transmission electron microscope and images were recorded with an AMT 2k CCD camera.
Extended Data
Extended Data Fig. 1. XPR1 dependency is observed selectively in SLC34A2-high cancer cell lines.
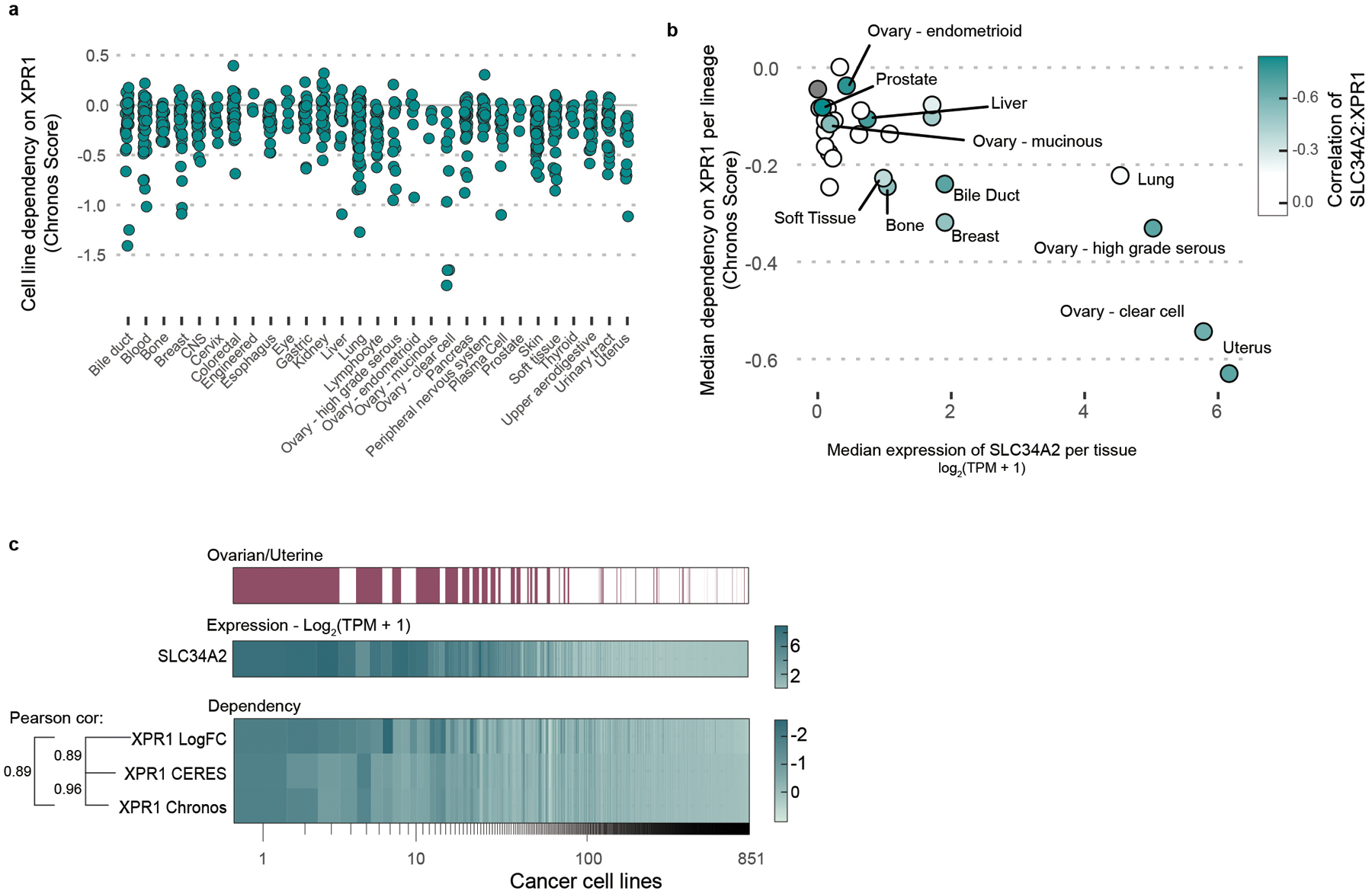
a) For every cell line profiled in the Cancer Dependency Map dataset (N=851 cancer cell lines), the degree of XPR1 essentiality is plotted on the Y-axis. The Chronos score is a scaled value of gene essentiality, where 0 is the effect of CRISPR/Cas9 genome editing and −1 is the effect of inactivation of pan-essential genes. Note that the ovarian lineage is separated into cancer subtypes.
b) For every tissue type, the 10 highest SLC34A2 expressing cell lines were analyzed for their median expression of SLC34A2 (X-axis) and dependency on XPR1 (Y-axis). Note that some lineages may have less than 10 cell lines. Color encodes the correlation of SLC34A2 expression and XPR1 dependency across all cell lines within that lineage.
c) Comparison of analytical methods for CRISPR/Cas9 genome-scale loss of function screens.
Extended Data Fig. 2. Validation of SLC34A2 and XPR1 protein levels and viability defects upon shRNA induction.

a) Validation of SLC34A2 in cell lines using immunohistochemistry. N=1 experiment.
b) Five days after viral transduction of the indicated sgRNA in the indicated cell lines stably expressing Cas9, cells were harvested and XPR1 levels were analyzed by immunoblotting. Note that irrelevant lanes were cropped out for clarity, but that this image represents a single blot at a single exposure. N=1 technical replicate of at least N=5 representative experiments.
c) Three days after induction of shRNA, protein levels were measured in cellular lysates by protein simple. Protein levels normalized to vinculin and the untreated (-Dox) conditions are expressed below each band. Note that shXPR1 reagents effectively suppress XPR1 protein levels but shSeed reagents do not. N=1 technical replicate of at least N=5 representative experiments.
d) Colony formation assay to measure the long-term (14 day) penetrance and viability effect of suppression of XPR1 using shRNA in IGROV1 and OVISE cells . N=3 technical replicates of at least N=2 representative experiments.
d) Colony formation assay to measure the long-term (14 day) penetrance and viability effect of suppression of XPR1 using shRNA in IGROV1 and OVISE cells . N=3 technical replicates of at least N=2 representative experiments.
e) Growth curves of SLC34A2-expressing cell lines after suppression of XPR1. In 96-well plates, confluency of the indicated cell lines was assessed every 4 hours. N=3 technical replicates of at least N=2 representative experiments.
f) Six days after induction of shXPR1 in the indicated cell lines, cells were stained with DAPI to distinguish live and dead cells and Annexin V to distinguish non- and pre-apoptotic cells. N=2 flow cytometric analyses of at least 10,000 cells, representative of N=2 experiments.
Extended Data Fig. 3. In vivo CRISPR/Cas9 competition assays for target validation in mouse xenografts.

a) sgRNA abundance in SNGM tumor xenografts was evaluated by PCR and next-generation sequencing analysis, and the fold change compared to the early time point is shown as a heatmap for all of the negative control genes as well as any gene with a >4 fold change in abundance in any of the screens. Each tumor/replicate is shown as an individual column, N=1 transduction.
b) Same as in d, but with the OVISE cancer cell line.
Extended Data Fig. 4. SLC34A2 in ovarian cancer is likely driven by PAX8.
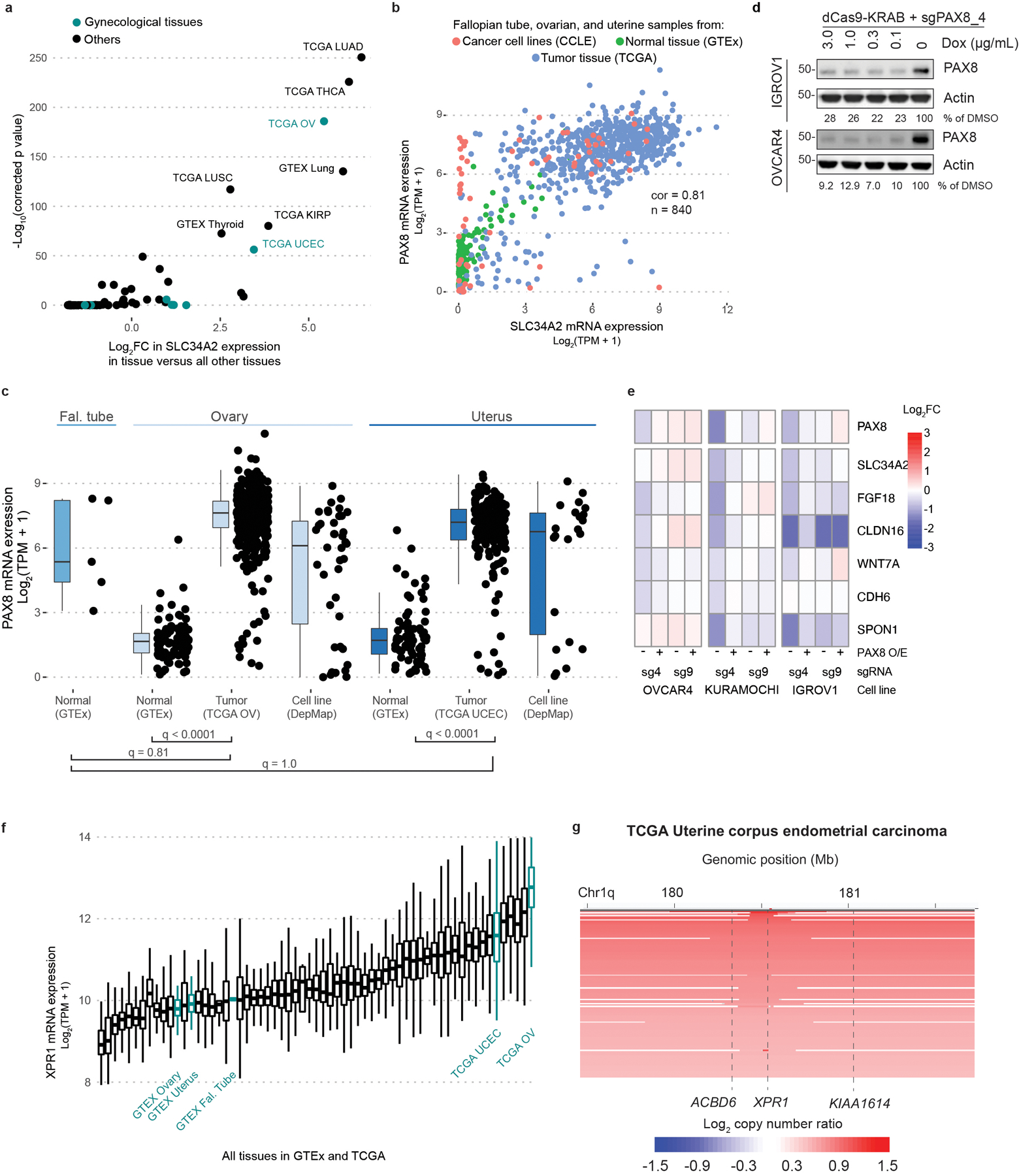
a) Using the combined GTEx, TCGA, and CCLE dataset, the differential expression of SLC34A2 in each tissue relative to the average of all tissues is compared. The relevant gynecological tissues (fallopian tube, ovary, and uterus) are highlighted in teal. The false discovery rate (FDR) was calculated using a two-sided Wilcoxon ranked sum test comparing each group to the average expression across all groups and correcting for multiple comparisons using Bonferoni’s method. The Cancer Genome Atlas abbreviations used include: LUAD = Lung adenocarcinoma; THCA = Thyroid carcinoma; KRP = Kidney renal cell papillary carcinoma ; LUSC = Lung squamous cell carcinoma; OV = Ovarian serous cystadenocarcinoma; UCEC = Uterine corpus endometrial carcinoma.
b) The expression of PAX8 and SLC34A2 mRNA in the indicated tissues is plotted. The pearson correlation within these samples is indicated.
c) Expression of PAX8 across the indicated tissues was compared as in Figure 3a. See methods for exact N values. Boxplots are drawn indicating the first and third quartiles, and whiskers span to the largest value within 1.5x the interquartile range.
d) Immunoblot validation of CRISPR-interference mediated suppression of PAX8. N=1 technical replicate, representative of N=2 independent experiments.
e) Gene expression - relative to un-perturbed, parental cell lines profiled in parallel - of reported PAX8 target genes (see main text) after stable overexpression of WT-PAX8 (“PAX8 O/E”) and/or induction of PAX8-target (sg4) or control (sg9) sgRNA and dCas9-KRAB. Data represents a single experiment. N=1 replicate.
f) XPR1 expression across all tissues in TCGA and GTEx, with ovarian and uterine tissues highlighted in teal. Boxplots are drawn as in b.
g) XPR1 copy number heatmap for a ~2.5 Mb region of chromosome 1 indicating XPR1 amplification in TCGA Uterine Corpus Endometrial Carcinoma 20. Each patient sample is represented by a horizontal line. Red indicates copy gain and blue indicates copy loss. Data are a subset of the samples rank-ordered by highest copy gain to indicate both focal and chromosome arm-level gains.
Extended Data Fig. 5. A genome-scale CRISPR/Cas9 screen validates the relationship between XPR1 dependency in the context of high expression of SLC34A2.
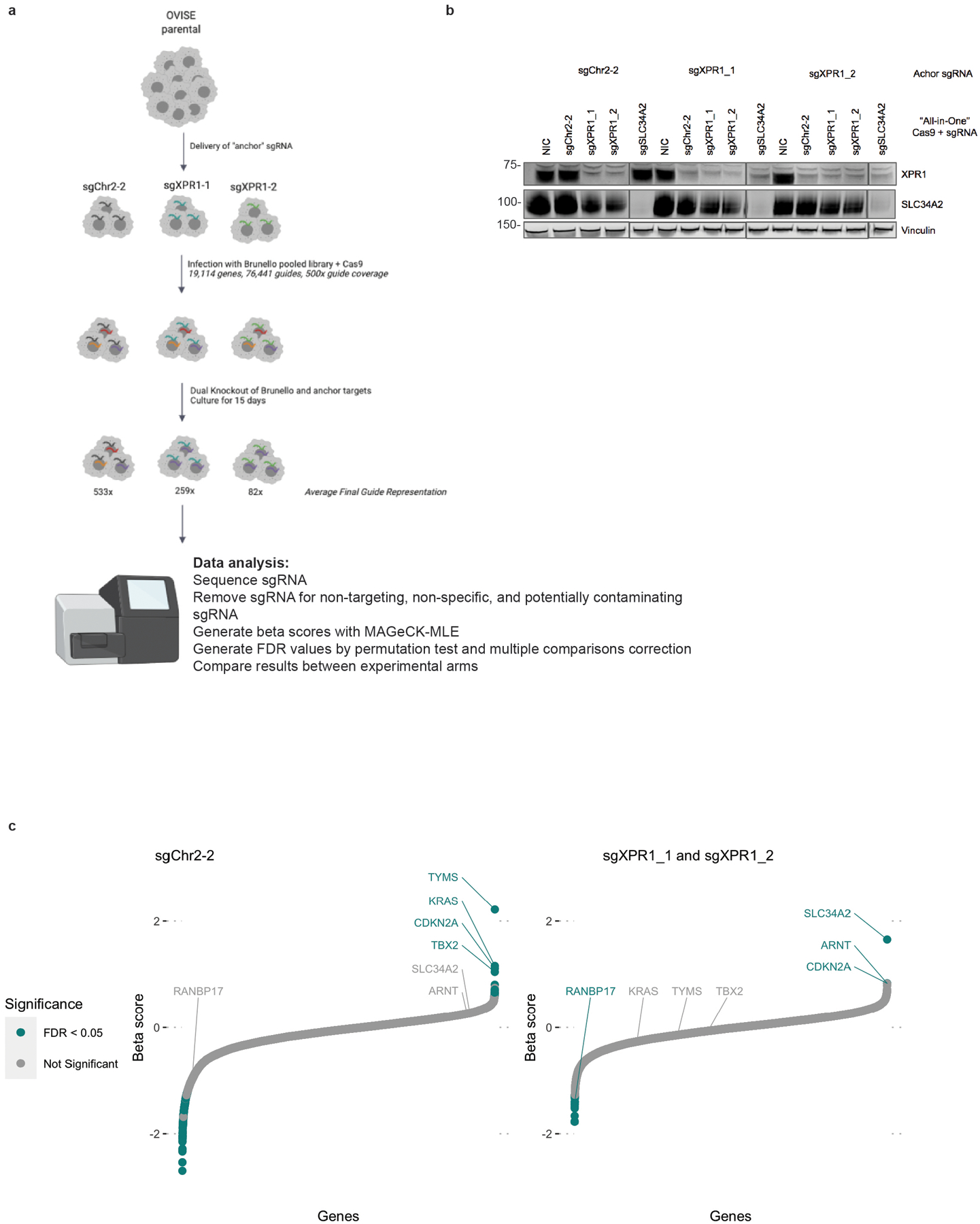
a) Outline of the experimental method and analysis for a genome-scale, dual-knock-out modifier screen. OVISE (without Cas9 expression) is engineered to stably express sgRNA targeting XPR1 (the “anchor” sgRNA). Upon introduction of “all-in-one” lentivirus, containing both Cas9 ORF and a second sgRNA, both genes are simultaneously inactivated by Cas9. We used three achor sgRNA: one targeting a gene desert on chromosome 2 (sgChr2-2) and two targeting XPR1 (sgXPR1_1 and sgXPR1_2) and infecting the cells with the Brunello genome-scale sgRNA library. 15 days after infection, cells were harvested, genomic DNA was isolated, and sgRNA barcodes were quantified with next generation sequencing. See methods for full experimental and analytical details.
b) Western confirmation of dual-knock-out of XPR1 and SLC34A2. The three cell lines used in the genome-scale screen were infected with “all-in-one” lentivirus expressing control-, XPR1-, or SLC34A2-targeting sgRNA. Note that in the sgXPR1 “anchor” cell lines, XPR1 is suppressed with the control virus, indicating that the first infection provides XPR1-targeting sgRNA and the second infection provides Cas9 protein. NIC stands for “no-infection control’. N=1 technical replicate representative of N=3 independent transductions.
c) Arm-level results of the genome-scale modifier screen. See methods for full analysis details. Beta-scores represent the extent to which a gene was enriched or depleted relative to the initial plasmid representation. An XPR1-positive and control-neutral score represents a likely rescue gene (i.e. SLC34A2 and ARNT). XPR1-positive and control-positive scores represent genes with profound viability defects without specificity for XPR1 (e.g. RANBP17). N=1 transduction per anchor condition, expanded and cultured as N=2 independent replicates.
Extended Data Fig. 6. The XPR1 dependency is not affected by phosphate levels in the tissue culture.
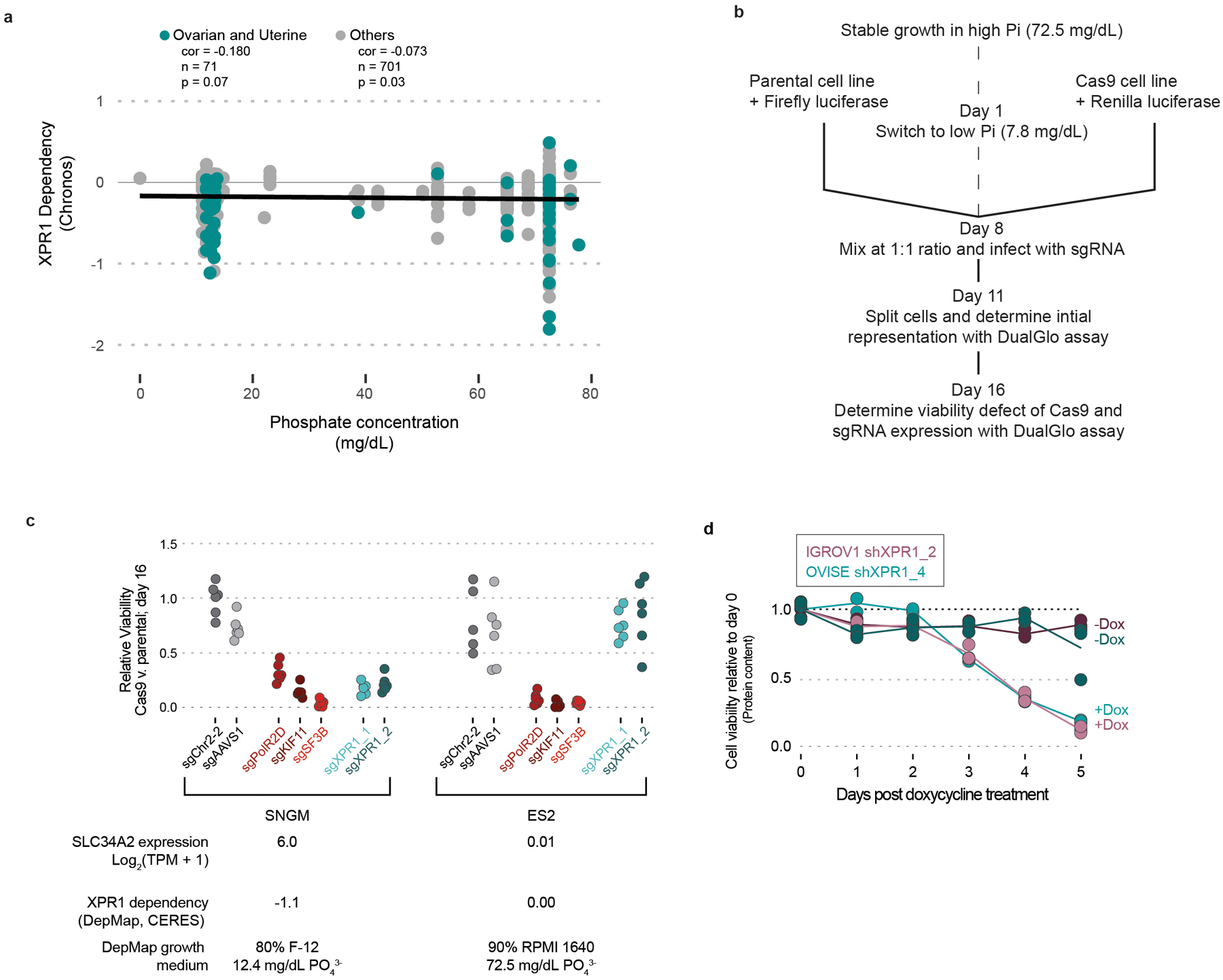
medium a) The concentration of phosphate in the growth medium of DepMap cell lines does not determine the extent of XPR1 dependency. Concentrations of phosphate were estimated from manufacturer formulations (see methods) and the pearson correlation between growth medium phosphate and XPR1 dependency is indicated.
b) Experimental procedure for manipulating tissue culture medium and assessing its effect on XPR1 dependency. The same parental cancer cell line was engineered to express firefly luciferase and Cas9, or Renilla luciferase alone. After a one-week adaptation to lowered phosphate, the two variants were mixed together and infected with sgRNA-encoding lentivirus. After selection for lentivirus-infected cell lines, the initial representation of Cas9:parental cells was determined by measuring the ratio of Firefly:Renilla luciferase using a DualGlo assay (Promega). One week after infection (Day 16 of the protocol), the extent to which genetic perturbation was detrimental to cell viability was determined using the DualGlo assay.
c) The XPR1 dependency is maintained in a low phosphate medium condition. SNGM and ES2 were profiled in the assay outlined in panel b. Note that the CERES score - displayed below the plot - represents the viability defect of the cell line 21 days after knock-out of XPR1 and growth in the indicated growth medium. N=5 technical replicates representative of N=2 experiments.
d) The viability of cells (as measured by total protein content) was measured in parallel with total phosphate as in Figure 4d. N=3 technical replicates representative of N=4 independent experiments per cell line.
Extended Data Fig. 7. Transcriptional profiling reveals a phosphate-related homeostatic response after XPR1 inactivation.
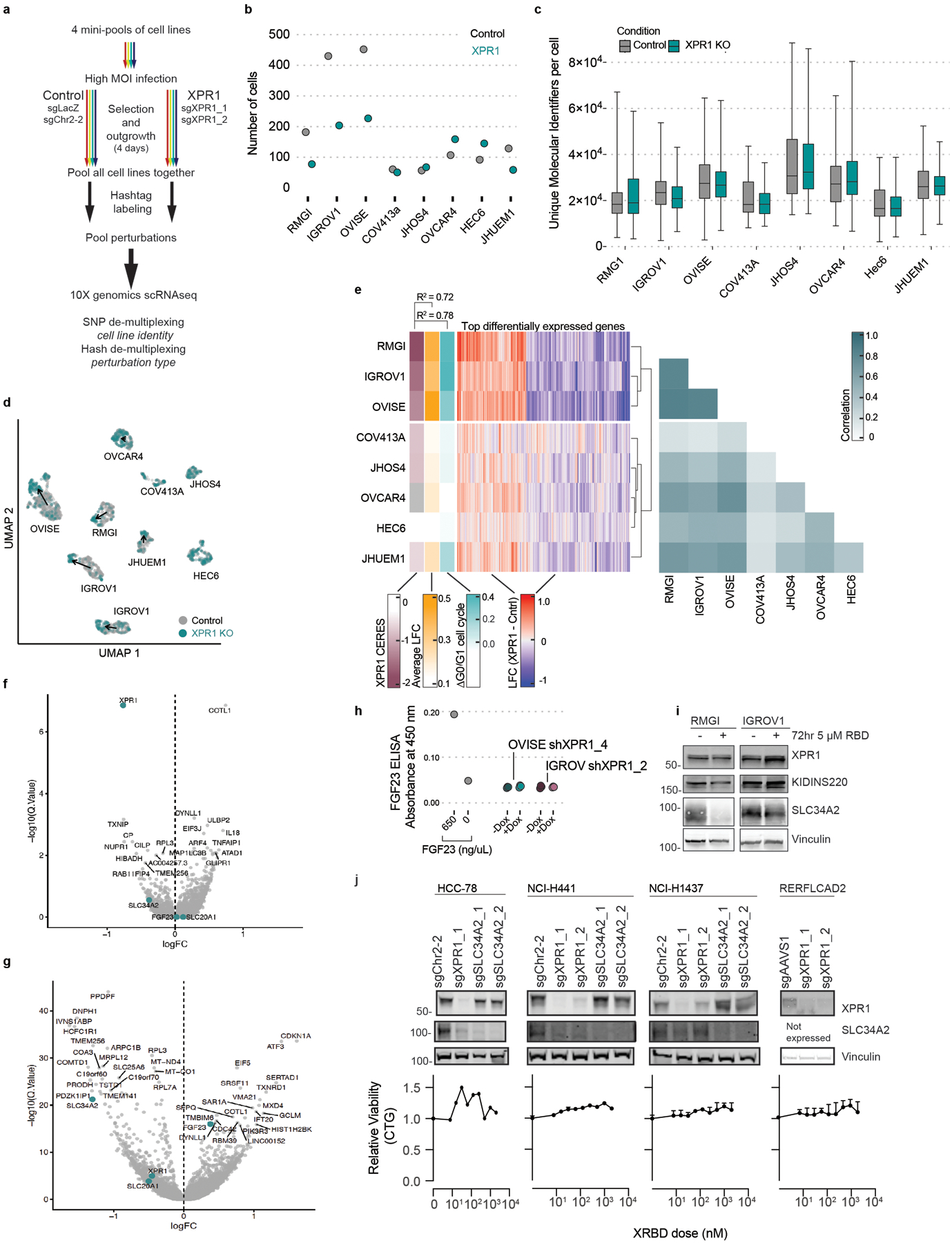
a) Experimental workflow to determine the transcriptional profile of XPR1 inactivation across many different cancer cell lines. See methods for full details; N=1 transduction event for panels b-g.
b) The total number of cells per cell line de-multiplexed from the 10X scRNAseq library.
c) The total number of unique transcripts measured for each cell, as measured by unique molecular identifiers (UMIs). Box plots represent the 1st – 3rd quartiles, with whiskers representing the minimum and the maximum. The exact N for each sample in c is indicated in panel b.
d) UMAP projection of the 2,501 cells from the indicated cell lines (determined by SNP profiles) and perturbations (indicated by cell-surface antibody ‘hash-tag’ labeling). Arrows indicate the degree to which the average transcriptional profile changes between the control sgRNA and the sgXPR1 infection condition.
e) Summary of transcriptional effects across cell lines after inactivation of XPR1. Middle, the log-fold change of the top 500 differentially expressed genes after regressing out the effect of cell cycle. Left, summary annotations for each cell line include XPR1 dependency (XPR1 CERES), the overall transcriptional change (average log2 fold-change), and the degree of cell cycle arrest observed in the single-cell data (ΔG0/G1). The pearson correlation of these values is shown above the heatmap. Right, the pearson correlation of the top 500 differentially expressed genes between each cell line.
f) Differentially expressed genes - after correcting for cell cycle - in the less sensitive cell lines (COV413a, JHOS4, OVCAR4, HEC6, and JHUEM1). Significance was assessed by the limma-voom pipeline using a two-tailed statistical test (see methods).
g) Same as in f, but for the highly correlated cell lines RMG1, IGROV1, and OVISE.
h) Four days after induction of shXPR1_2 (IGROV1) or shXPR1_4 (OVISE) using doxycycline, the amount of secreted FGF23 was measured in the conditioned medium using ELISA. N=2 technical replicates representative of N=3 independent experiments.
i) 72 hours after treatment with the XPR1 inhibitor XRBD, the indicated proteins were detected using immunoblot. N=1 technical replicate representative of N=2 independent experiments.
j) Top, western blot analysis of SLC34A2 and XPR1 in the SLC34A2-high yet XPR1-insensitive lung cancer cell lines, five days after infection with lentivirus expressing the indicated sgRNA. Bottom, viability of the indicated cancer cell lines was assessed using Cell Titer Glo after a five day XRBD treatment to inhibit XPR1. Points represent the mean of N=3 technical replicates; error bars represent standard error of the mean. Data are representative of N=2 independent experiments.
Extended Data Fig. 8. Open-reading frames of XPR1 resistant to CRISPR/Cas9-mediated gene editing.
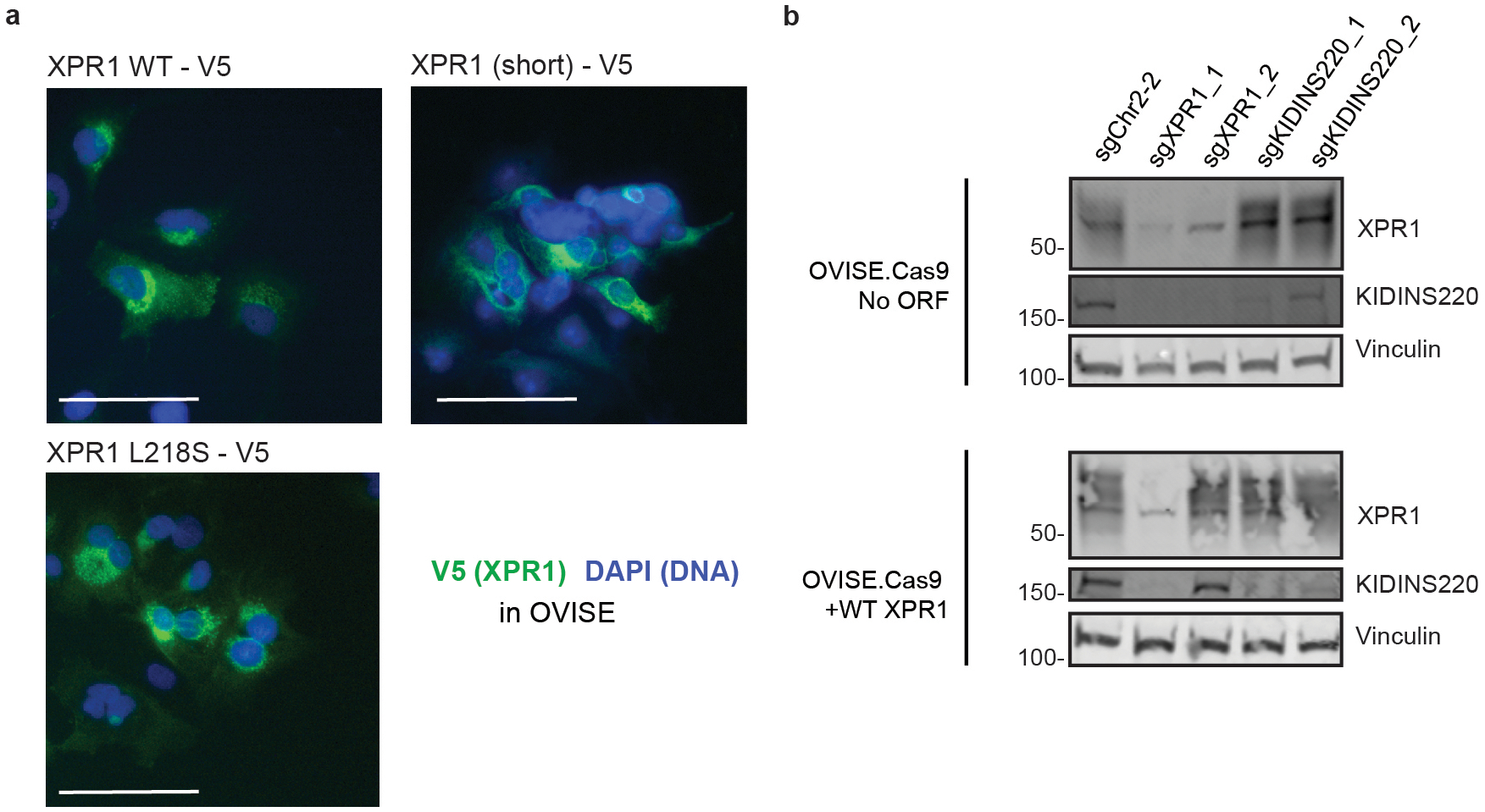
a)Immunofluorescent localization of XPR1 mutants using the V5 epitope tag. Left, WT XPR1 localizes to the secretory pathways as well as puncta within the cytoplasm. Middle, XPR1 (short) staining appears more diffuse. Note similar localization patterns between L218S and wildtype XPR1 alleles. Scale bar = 200 μm. N=1 experiment representative of N=2 independent transductions.
b) Western blot validation of guide-resistant ORF. OVISE.Cas9 cells (parental, left, or overexpressing the WT XPR1 ORF, right, used in Figure 3e) were infected with the indicated sgRNA and harvested 5 days after infection. The XPR1 ORF includes a mutation to block binding of sgXPR1_2 but not sgXPR1_1. Note the inactivation of both endogenous and overexpression ORF with sgXPR1_1 and only endogenous XPR1 with sgXPR1_2. N=1 experiment representative of N=2 independent transductions.
Extended Data Fig. 9. KIDINS220 is a unique partner protein of the XPR1 phosphate efflux complex.
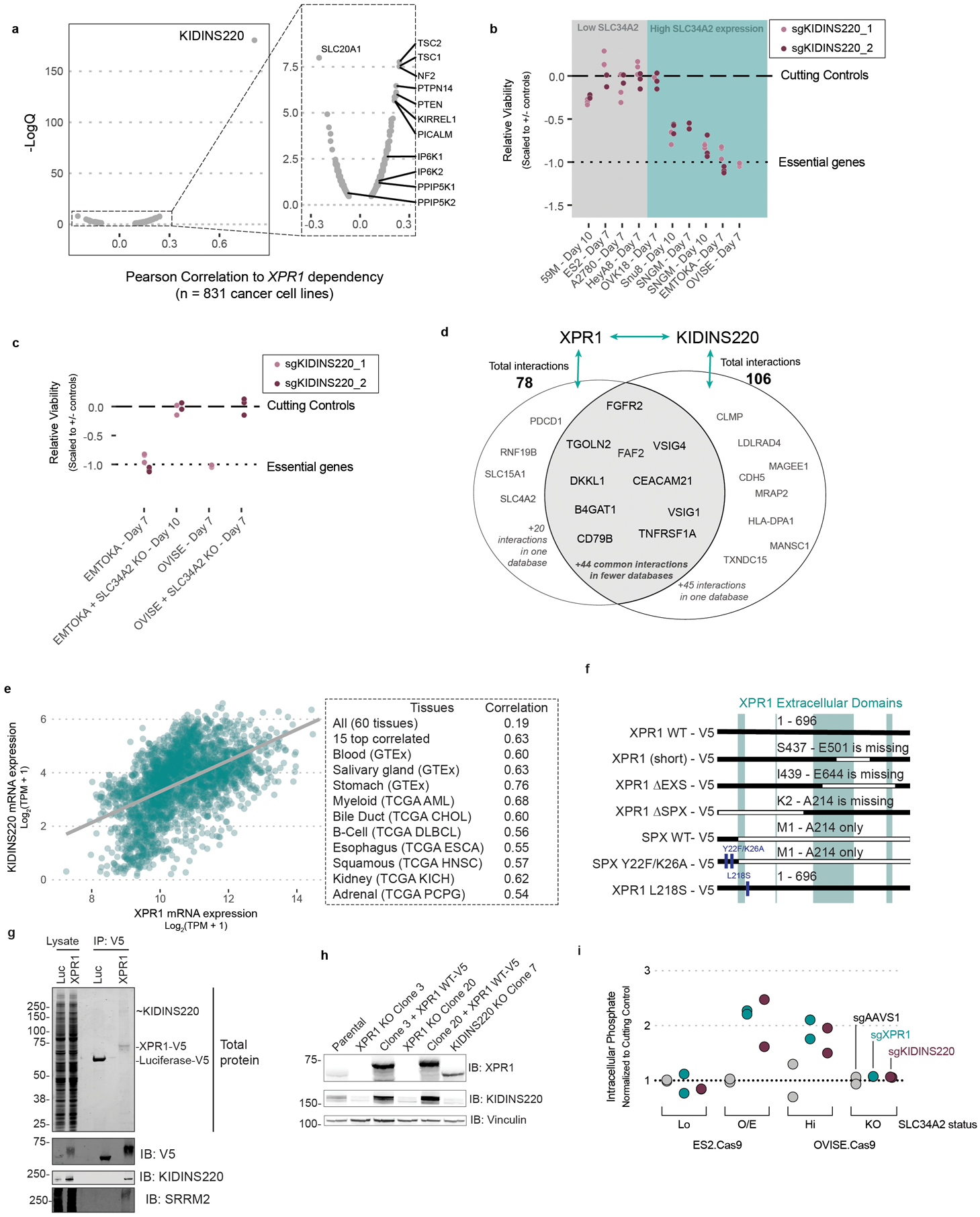
a) Genetic dependency correlations to XPR1 dependency across 851 cancer cell lines was assessed by pearson correlation test and corrected for multiple comparisons using the Benjamini-Hochberg method. Genes with significantly correlated dependency profiles are highlighted, as are proteins with known connection to XPR1 regulation.
b) The viability defect of the indicated cancer cell lines after KIDINS220 inactivation was evaluated as in Figure 1c. N=3 technical replicates representative of at least N=2 independent transductions per cell line.
c) SLC34A2 was inactivated in EMTOKA and OVISE, and the KIDINS220 dependency was evaluated as in b. N=3 technical replicates representative of at least N=2 independent transductions per cell line.
d) The interacting partners of XPR1 and KIDINS220 were downloaded from the BioGrid and Bioplex databases and compared. Proteins present in the XPR1 or KIDINS220 interactomes are highlighted as text.
e) Left, the mRNA expression of XPR1 and KIDINS220 is shown for the fifteen tissues with the highest correlation in expression. The line represents linear regression for these samples (N = 2,799). Right, the Pearson correlation for those tissues, highlighting the diverse tissues in which there is a high correlation between XPR1 and KIDINS220 expression.
f) Mutants of XPR1 used in this manuscript. XPR1 WT refers to the 696 amino acid protein produced by NM_004736 (the only isoform detected by RT-PCR of OVISE mRNA), while XPR1 (short) refers to the 631 amino acid product of NM_001135669. All constructs have C-terminal V5 tags for immunoprecipitation, western blotting, and immunofluorescent detection.
g) XPR1 or Luciferase (Luc) were overexpressed in 293T cells and immunoprecipitated using the V5 tag and analyzed by targeted immunoblot or for total protein. Proteins were extracted from this gel and identified using mass spectrometry, the results of which are shown in Figure 6c. N=1 replicate of N=2 independent transfections.
h) Cas9 + sgRNA targeting XPR1 or KIDINS220 were transfected into 293T cells, and clones were isolated that lacked expression of the target proteins. For XPR1 inactivated cells, the XPR1 ORF was re-expressed, and the relative levels of the indicated proteins were detected by immunoblot. At least N=2 clonal populations were profiled for each inactivation condition.
i) Five days after infection with the indicated sgRNA targeting XPR1 or KIDINS220, free inorganic intracellular phosphate was determined as in Figure 4d. N=3 technical replicates of N=3 independent transductions.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This work was funded in part by the Slim Initiative in Genomic Medicine for the Americas (SIGMA), a joint U.S-Mexico project funded by the Carlos Slim Foundation (TRG), grants from the National Cancer Institute CA242457 (TRG) and CA212229 (DPB). We thank Dana-Farber/Harvard Cancer Center in Boston, MA, for the use of the Specialized Histopathology Core, which provided histology and immunohistochemistry service and is supported in part by an NCI Cancer Center Support Grant CA06516. We thank J Barnett, B Buckley, M Veneskey, M Cecilia-Saberi, and D Tennen for technical support.
Footnotes
Competing Interests Statement
FV receives research funding from Novo Holdings. DER receives research funding from the Functional Genomics Consortium (Abbvie, Jannsen, Merck and Vir) and is a director of Addgene. TRG previously received cash and/or equity compensation for consulting to GlaxoSmithKline, Sherlock Biosciences and FORMA Therapeutics, currently is a paid consultant to Anji Pharmaceuticals, and receives research funding from Bayer HealthCare, Calico Life Sciences, and Novo Holdings. W.C.H. is a consultant for ThermoFisher, Solasta, MPM Capital, iTeos, Frontier Medicines, and Paraxel and is a Scientific Founder and serves on the Scientific Advisory Board (SAB) for KSQ Therapeutics. All other authors declare no competing interests.
Code Availability
Computer code to reproduce these results is available from the corresponding authors upon request.
Data Availability
Publicly available data used in this study include CRISPR/Cas9 loss of viability screens for 851 cancer cell lines10–12, cancer cell line RNAseq expression data15, harmonized gene expression data for GTEx and TCGA datasets24, and copy number alterations for Ovarian adenocarcinoma and Uterine Corpus Endometrial Carcinoma22,23. RNA sequencing data after PAX8 suppression and multiplexed transcriptional profiling are available from the corresponding author upon request. RNAseq data after PAX8 knockdown are available at https://doi.org/10.6084/m9.figshare.19125671.v1. MixSeq data after XPR1 knockdown are available at https://doi.org/10.6084/m9.figshare.19125677.v1.
References
- 1.Lord CJ & Ashworth A PARP inhibitors: Synthetic lethality in the clinic. Science 355, 1152–1158 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lheureux S, Braunstein M & Oza AM Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA Cancer J. Clin 69, 280–304 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Vaughan S et al. Rethinking ovarian cancer: recommendations for improving outcomes. Nat. Rev. Cancer 11, 719–725 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sung H et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin 71, 209–249 (2021). [DOI] [PubMed] [Google Scholar]
- 5.Boehm JS et al. Cancer research needs a better map. Nature 589, 514–516 (2021). [DOI] [PubMed] [Google Scholar]
- 6.Costanzo M et al. A global genetic interaction network maps a wiring diagram of cellular function. Science 353, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pan J et al. Sparse dictionary learning recovers pleiotropy from human cell fitness screens. Cell Syst (2022) doi: 10.1016/j.cels.2021.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheung HW et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc. Natl. Acad. Sci. U. S. A 108, 12372–12377 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Price C et al. Genome-Wide Interrogation of Human Cancers Identifies EGLN1 Dependency in Clear Cell Ovarian Cancers. Cancer Res. 79, 2564–2579 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meyers RM et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet 49, 1779–1784 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DepMap, Broad (2021): DepMap 21Q2 Public. figshare. Dataset. 10.6084/m9.figshare.14541774.v2 [DOI] [Google Scholar]
- 12.Dempster JM, Rossen J, Kazachkova M & Pan J Extracting Biological Insights from the Project Achilles Genome-Scale CRISPR Screens in Cancer Cell Lines. BioRxiv (2019). [Google Scholar]
- 13.Battini JL, Rasko JE & Miller AD A human cell-surface receptor for xenotropic and polytropic murine leukemia viruses: possible role in G protein-coupled signal transduction. Proc. Natl. Acad. Sci. U. S. A 96, 1385–1390 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giovannini D, Touhami J, Charnet P, Sitbon M & Battini J-L Inorganic phosphate export by the retrovirus receptor XPR1 in metazoans. Cell Rep. 3, 1866–1873 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Ghandi M et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsherniak A et al. Defining a Cancer Dependency Map. Cell 170, 564–576.e16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dempster JM et al. Gene expression has more power for predicting in vitro cancer cell vulnerabilities than genomics. bioRxiv 2020.02.21.959627 (2020) doi: 10.1101/2020.02.21.959627. [DOI] [Google Scholar]
- 18.Levan K et al. Immunohistochemical evaluation of epithelial ovarian carcinomas identifies three different expression patterns of the MX35 antigen, NaPi2b. BMC Cancer 17, 303 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yin BWT et al. Monoclonal antibody MX35 detects the membrane transporter NaPi2b (SLC34A2) in human carcinomas. Cancer Immun. 8, 3 (2008). [PMC free article] [PubMed] [Google Scholar]
- 20.Zou Y et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat. Commun 10, 1617 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Magtanong L et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem Biol 26, 420–432.e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cancer Genome Atlas Research Network et al. Integrated genomic characterization of endometrial carcinoma. Nature 497, 67–73 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldman M et al. The UCSC Xena platform for public and private cancer genomics data visualization and interpretation. bioRxiv 326470 (2019) doi: 10.1101/326470. [DOI] [Google Scholar]
- 25.Vivian J et al. Toil enables reproducible, open source, big biomedical data analyses. Nat. Biotechnol 35, 314–316 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Consortium GTEx. The Genotype-Tissue Expression (GTEx) project. Nat. Genet 45, 580–585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Köbel M Ovarian carcinoma subtypes are different diseases: implications for biomarker studies. PLoS Med 5, e232 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Piek JM et al. Dysplastic changes in prophylactically removed Fallopian tubes of women predisposed to developing ovarian cancer. J. Pathol 195, 451–456 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Jarboe E et al. Serous carcinogenesis in the fallopian tube: a descriptive classification. Int. J. Gynecol. Pathol 27, 1–9 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Hu Z et al. The Repertoire of Serous Ovarian Cancer Non-genetic Heterogeneity Revealed by Single-Cell Sequencing of Normal Fallopian Tube Epithelial Cells. Cancer Cell 37, 226–242.e7 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Mittag J, Winterhager E, Bauer K & Grümmer R Congenital hypothyroid female pax8-deficient mice are infertile despite thyroid hormone replacement therapy. Endocrinology 148, 719–725 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Elias KM et al. Epigenetic remodeling regulates transcriptional changes between ovarian cancer and benign precursors. JCI Insight 1, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adler EK et al. The PAX8 cistrome in epithelial ovarian cancer. Oncotarget 8, 108316–108332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi K et al. PAX8 regulon in human ovarian cancer links lineage dependency with epigenetic vulnerability to HDAC inhibitors. Elife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mermel CH et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeWeirdt PC et al. Genetic screens in isogenic mammalian cell lines without single cell cloning. Nat. Commun 11, 752 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson MS, Jessen HJ & Saiardi A The inositol hexakisphosphate kinases IP6K1 and −2 regulate human cellular phosphate homeostasis, including XPR1-mediated phosphate export. J. Biol. Chem 294, 11597–11608 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Minisola S et al. Tumour-induced osteomalacia. Nat Rev Dis Primers 3, 17044 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Whitsett JA, Wert SE & Weaver TE Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annu. Rev. Med 61, 105–119 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lituiev DS & Kiyamova RG Mutations in the gene of human type IIb sodium-phosphate cotransporter SLC34A2. Biopolymers and Cell = Biopolimery i Kletka 26, 13–22 (2010). [Google Scholar]
- 41.Li X et al. Control of XPR1-dependent cellular phosphate efflux by InsP8 is an exemplar for functionally-exclusive inositol pyrophosphate signaling. Proc. Natl. Acad. Sci. U. S. A (2020) doi: 10.1073/pnas.1908830117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.López-Sánchez U et al. Interplay between PFBC-associated SLC20A2 and XPR1 phosphate transporters requires inositol polyphosphates for control of cellular phosphate homeostasis. J. Biol. Chem (2020) doi: 10.1074/jbc.RA119.011376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Legati A et al. Mutations in XPR1 cause primary familial brain calcification associated with altered phosphate export. Nat. Genet 47, 579–581 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wainberg M et al. A genome-wide almanac of co-essential modules assigns function to uncharacterized genes. bioRxiv 827071 (2019) doi: 10.1101/827071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim E et al. A network of human functional gene interactions from knockout fitness screens in cancer cells. Life Sci Alliance 2, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cabrera-Poch N, Sánchez-Ruiloba L, Rodríguez-Martínez M & Iglesias T Lipid raft disruption triggers protein kinase C and Src-dependent protein kinase D activation and Kidins220 phosphorylation in neuronal cells. J. Biol. Chem 279, 28592–28602 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Iglesias T et al. Identification and cloning of Kidins220, a novel neuronal substrate of protein kinase D. J. Biol. Chem 275, 40048–40056 (2000). [DOI] [PubMed] [Google Scholar]
- 48.Arévalo JC, Yano H, Teng KK & Chao MV A unique pathway for sustained neurotrophin signaling through an ankyrin-rich membrane-spanning protein. EMBO J. 23, 2358–2368 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kong H, Boulter J, Weber JL, Lai C & Chao MV An evolutionarily conserved transmembrane protein that is a novel downstream target of neurotrophin and ephrin receptors. J. Neurosci 21, 176–185 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wege S et al. The EXS Domain of PHO1 Participates in the Response of Shoots to Phosphate Deficiency via a Root-to-Shoot Signal. Plant Physiol 170, 385–400 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arpat AB et al. Functional expression of PHO1 to the Golgi and trans-Golgi network and its role in export of inorganic phosphate: PHO1 localization to the Golgi/trans-Golgi network. Plant J. 5, no–no (2012). [DOI] [PubMed] [Google Scholar]
- 52.Snyder NA et al. H+ and Pi Byproducts of Glycosylation Affect Ca2+ Homeostasis and Are Retrieved from the Golgi Complex by Homologs of TMEM165 and XPR1. G3 7, 3913–3924 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gerber DE et al. Phase Ia Study of Anti-NaPi2b Antibody-Drug Conjugate Lifastuzumab Vedotin DNIB0600A in Patients with Non-Small Cell Lung Cancer and Platinum-Resistant Ovarian Cancer. Clin. Cancer Res 26, 364–372 (2020). [DOI] [PubMed] [Google Scholar]
- 54.Marks J The role of SLC34A2 in intestinal phosphate absorption and phosphate homeostasis. Pflugers Arch. 471, 165–173 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.He P, Mann-Collura O, Fling J, Edara N, Hetz R, & Razzaque MS High phosphate actively induces cytotoxicity by rewiring pro-survival and pro-apoptotic signaling networks in HEK293 and HeLa cells. FASEB J. 35, e20997 (2021). [DOI] [PubMed] [Google Scholar]
- 56.Di Marco GS et al. Increased inorganic phosphate induces human endothelial cell apoptosis in vitro. Am. J. Physiol. Renal Physiol 294, F1381–7 (2008). [DOI] [PubMed] [Google Scholar]
- 57.Marks MS, Heijnen HFG & Raposo G Lysosome-related organelles: unusual compartments become mainstream. Curr. Opin. Cell Biol 25, 495–505 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ruiz FA, Lea CR, Oldfield E & Docampo R Human platelet dense granules contain polyphosphate and are similar to acidocalcisomes of bacteria and unicellular eukaryotes. J. Biol. Chem 279, 44250–44257 (2004). [DOI] [PubMed] [Google Scholar]
- 59.Josifova DJ et al. Heterozygous KIDINS220/ARMS nonsense variants cause spastic paraplegia, intellectual disability, nystagmus, and obesity. Hum. Mol. Genet 25, 2158–2167 (2016). [DOI] [PubMed] [Google Scholar]
- 60.McDonald ER 3rd et al. Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell 170, 577–592.e10 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Yang X et al. A public genome-scale lentiviral expression library of human ORFs. Nat. Methods 8, 659–661 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McFarland JM et al. Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat. Commun 9, 4610 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Buehler E, Chen Y-C & Martin S C911: A bench-level control for sequence specific siRNA off-target effects. PLoS One 7, e51942 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aran D, Sirota M & Butte AJ Systematic pan-cancer analysis of tumour purity. Nat. Commun 6, 8971 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li W et al. Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol. 16, 281 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Phosphorus concentrations in blood, milk, feces, bone and selected fluids and tissues of growing heifers as affected by dietary phosphorus. http://www.lrrd.org/lrrd3/2/florida4.htm.
- 67.McFarland JM et al. Multiplexed single-cell profiling of post-perturbation transcriptional responses to define cancer vulnerabilities and therapeutic mechanism of action. bioRxiv 868752 (2019) doi: 10.1101/868752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stoeckius M et al. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 19, 224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li B et al. Cumulus: a cloud-based data analysis framework for large-scale single-cell and single-nucleus RNA-seq. bioRxiv 823682 (2019) doi: 10.1101/823682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Garrison E & Marth G Haplotype-based variant detection from short-read sequencing. arXiv [q-bio.GN] (2012). [Google Scholar]
- 71.Law CW, Chen Y, Shi W & Smyth GK voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ritchie ME et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Robinson MD, McCarthy DJ & Smyth GK edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huttlin EL et al. Dual Proteome-scale Networks Reveal Cell-specific Remodeling of the Human Interactome. bioRxiv 2020.01.19.905109 (2020) doi: 10.1101/2020.01.19.905109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Oughtred R et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 47, D529–D541 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shevchenko A, Wilm M, Vorm O & Mann M Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem 68, 850–858 (1996). [DOI] [PubMed] [Google Scholar]
- 77.Eng JK, McCormack AL & Yates JR An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom 5, 976–989 (1994). [DOI] [PubMed] [Google Scholar]
- 78.Cerami E et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gao J et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, l1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Publicly available data used in this study include CRISPR/Cas9 loss of viability screens for 851 cancer cell lines10–12, cancer cell line RNAseq expression data15, harmonized gene expression data for GTEx and TCGA datasets24, and copy number alterations for Ovarian adenocarcinoma and Uterine Corpus Endometrial Carcinoma22,23. RNA sequencing data after PAX8 suppression and multiplexed transcriptional profiling are available from the corresponding author upon request. RNAseq data after PAX8 knockdown are available at https://doi.org/10.6084/m9.figshare.19125671.v1. MixSeq data after XPR1 knockdown are available at https://doi.org/10.6084/m9.figshare.19125677.v1.