

Abstract
Oxidative stress is a defining feature of most cancers, including those that stem from carcinogenic infections1. Reactive oxygen species (ROS) can drive tumor formation2–4, yet the molecular oxidation events that contribute to tumorigenesis are largely unknown. Here we show that inactivation of a single, redox-sensitive cysteine in the host protease legumain, which is oxidized during infection with the gastric cancer-causing bacterium Helicobacter pylori, accelerates tumor growth. By using chemical proteomics to map cysteine reactivity in human gastric cells, we determined that H. pylori infection induces oxidation of legumain at Cys219. Legumain oxidation dysregulates intracellular legumain processing and decreases the activity of the enzyme in H. pylori-infected cells. We further show that the site-specific loss of Cys219 reactivity increases tumor growth and mortality in a xenograft model. Our findings establish a link between an infection-induced oxidation site and tumorigenesis while underscoring the importance of cysteine reactivity in tumor growth.
Graphical Abstract
Introduction
Nearly one-sixth of all human cancers are caused by microbes that induce chronic inflammation and oxidative stress5. The stomach pathogen Helicobacter pylori, which is the strongest risk factor for gastric cancer6, enhances the accumulation of reactive oxygen species (ROS) in human gastric tissues7,8 by stimulating ROS-generating inflammatory pathways9, secreting virulence factors that induce ROS production10, and depleting host cells of the antioxidant glutathione (GSH)11. As in other tumorigenic infections2,3, ROS generated during H. pylori infection can drive tumor formation in vivo4, yet the cellular targets of ROS that contribute to cancer pathogenesis are underexplored. Prior studies of pathogen-induced ROS have predominantly focused on oxidative damage at the DNA level, which is a widely appreciated cause of genomic instability in cancer1. However, ROS can also enhance cell proliferation through the site-specific oxidation of proteins that regulate cell metabolism12–14 and growth factor signaling15–17. Although tumorigenic infections are often associated with increased protein oxidation in human tissues18,19, it is unknown whether the infection-induced oxidation of host proteins can directly contribute to tumor growth.
In this paper, we use chemical proteomics to identify Cys219 of the lysosomal protease legumain as a specific site of oxidation during H. pylori infection. We demonstrate that Cys219 reactivity is required for the intracellular activation of legumain, which is markedly reduced in H. pylori-infected cells. Furthermore, we show that mutating Cys219 accelerates tumor growth in a xenograft model. Taken together, these findings provide evidence that oxidation induced by a carcinogenic microbe can alter the reactivity of a host cysteine that regulates tumor growth.
Results
Mapping cysteine reactivity in infected cells
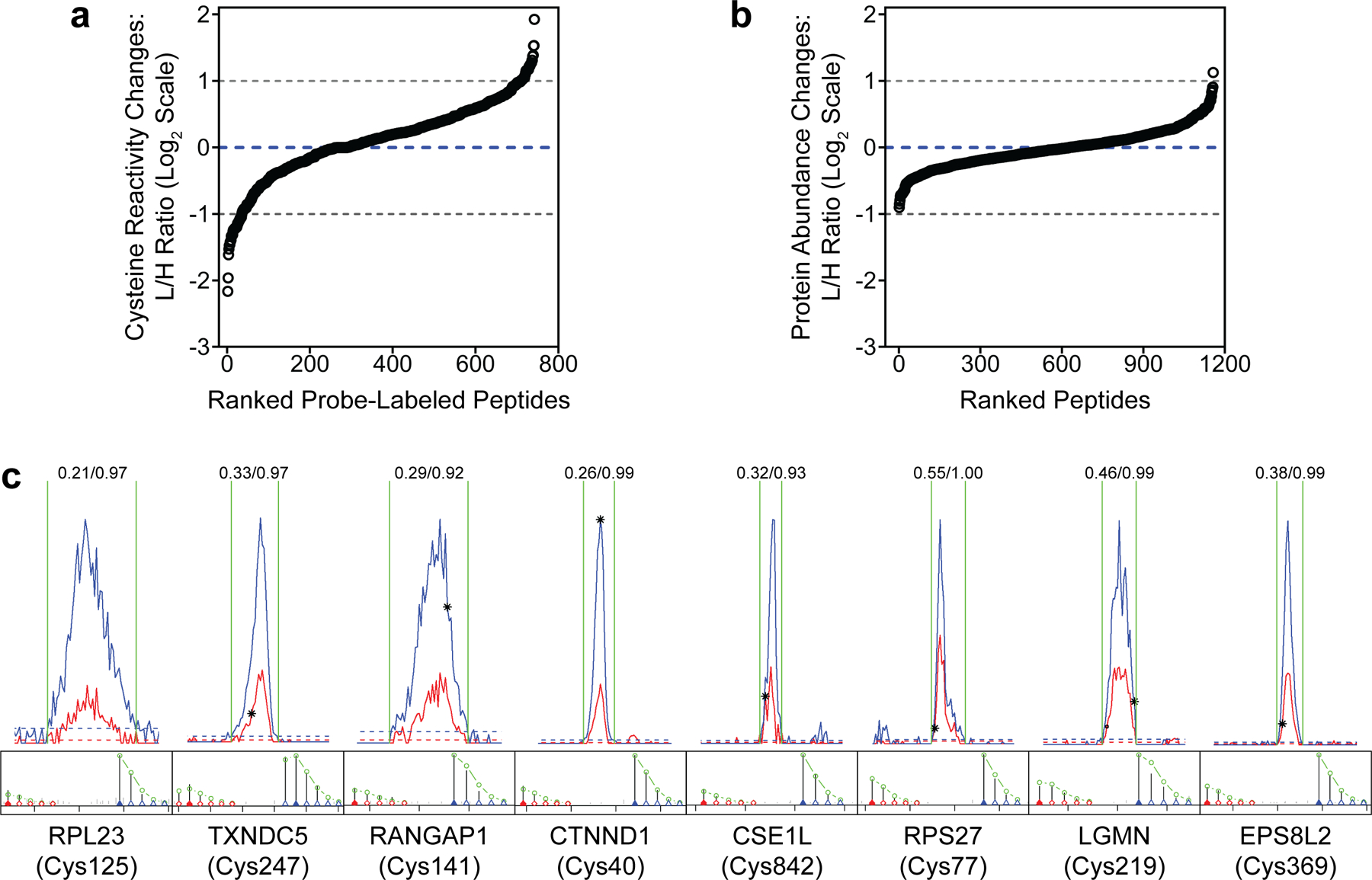
Recently developed chemical proteomic techniques have made it possible to identify specific sites of oxidation in the proteome by quantifying changes in the reactivity of cysteine thiols, which are major targets of ROS20,21. Cysteine oxidation results in a decrease in cysteine reactivity upon ROS exposure. To identify proteins that are oxidized during H. pylori infection, we generated isotopically light (L) and heavy (H) AGS human gastric adenocarcinoma cells via stable isotope labeling by amino acids in cell culture (SILAC) for quantitative mass spectrometry (MS)-based analyses of cysteine reactivity using isotopic tandem orthogonal proteolysis–activity-based protein profiling (isoTOP–ABPP)22 (Fig. 1a). We used infection conditions that induced the production of intracellular ROS (Extended Data Fig. 1a) and a decrease in host GSH (Extended Data Fig. 1b) comparable to the amount of GSH depletion observed in human gastric tissues infected with H. pylori11, while minimizing AGS cell death. Under these conditions, we also detected a marked increase in the labeling of H. pylori-infected versus uninfected cell lysates using a probe specific for a subclass of oxidized thiols (Extended Data Fig. 1c), consistent with elevated oxidative stress during infection. The isoTOP–ABPP analysis was performed by labeling the lysates of H. pylori-infected (L) and uninfected (H) cells with the thiol-reactive probe iodoacetamide alkyne (IA). IA-labeled proteins from L and H samples were covalently linked to a biotinylated tag with a cleavable linker, combined and enriched on streptavidin-agarose beads, and digested with trypsin. The IA-labeled peptides were chemically cleaved from the beads and analyzed by tandem liquid chromatography–tandem mass spectrometry (LC/LC–MS/MS). The relative abundance of each IA-labeled peptide was quantified in H. pylori-infected versus uninfected cells. We identified 35 cysteines from host proteins with reduced IA labeling in H. pylori-infected cells (i.e., ≥2-fold decrease in IA labeling; L/H ratio ≤0.5 with SD <1, n=3), including cancer-relevant proteins such as the tumor suppressor p120-catenin23 and the ribosomal protein L23, which moonlights as a positive regulator of the tumor suppressor p5324 (Fig. 1b, Extended Data Fig. 2a, and Supplementary Dataset 1). In addition, we identified 34 cysteines from host proteins with enhanced IA labeling in H. pylori-infected cells (i.e., ≥2-fold increase in IA labeling; L/H ratio ≥2 with SD <1, n=3), including a few residues that are known to be redox-sensitive (e.g., Cys47 and Cys91 of peroxiredoxin-6 (PRDX6)25) (Supplementary Dataset 1). Although the increase in IA labeling of such cysteines was unexpected, we speculate that factors specific to our infection model, like the depletion of host GSH (Extended Data Fig. 1b), could influence the cysteine reactivity of GSH-utilizing enzymes like PRDX6 and other host proteins during H. pylori infection. In tandem, we performed a quantitative LC/LC–MS/MS analysis of unenriched cell lysates to determine the relative abundance of proteins in H. pylori-infected versus uninfected cells (Fig. 1a, Extended Data Fig. 2b, and Supplementary Dataset 1). We selected proteins that exhibited substantially reduced cysteine reactivity, but not abundance, in H. pylori-infected cells for further analysis. By correcting for changes in protein abundance, we aimed to identify cysteines that were directly oxidized during infection, as opposed to cysteines with indirect changes in reactivity due to secondary effects like transcriptional regulation. We identified eight cysteines with L/H ratios ≤0.5 (Table 1 and Extended Data Fig. 2c); of these, we identified five cysteines for which the L/H ratio was significantly different (P <0.05, n=3) from the corresponding protein abundance ratio (Fig. 1c). To validate our proteomic data, we analyzed selected hits by immunoblotting, wherein IA-labeled proteins from H. pylori-infected versus uninfected cell lysates were enriched, and the degree of enrichment assessed by immunoblot. These immunoblotting analyses confirmed the reduced degree of IA labeling determined by isoTOP–ABPP (Fig. 1d).
Figure 1. Legumain Cys219 is oxidized during H. pylori infection.
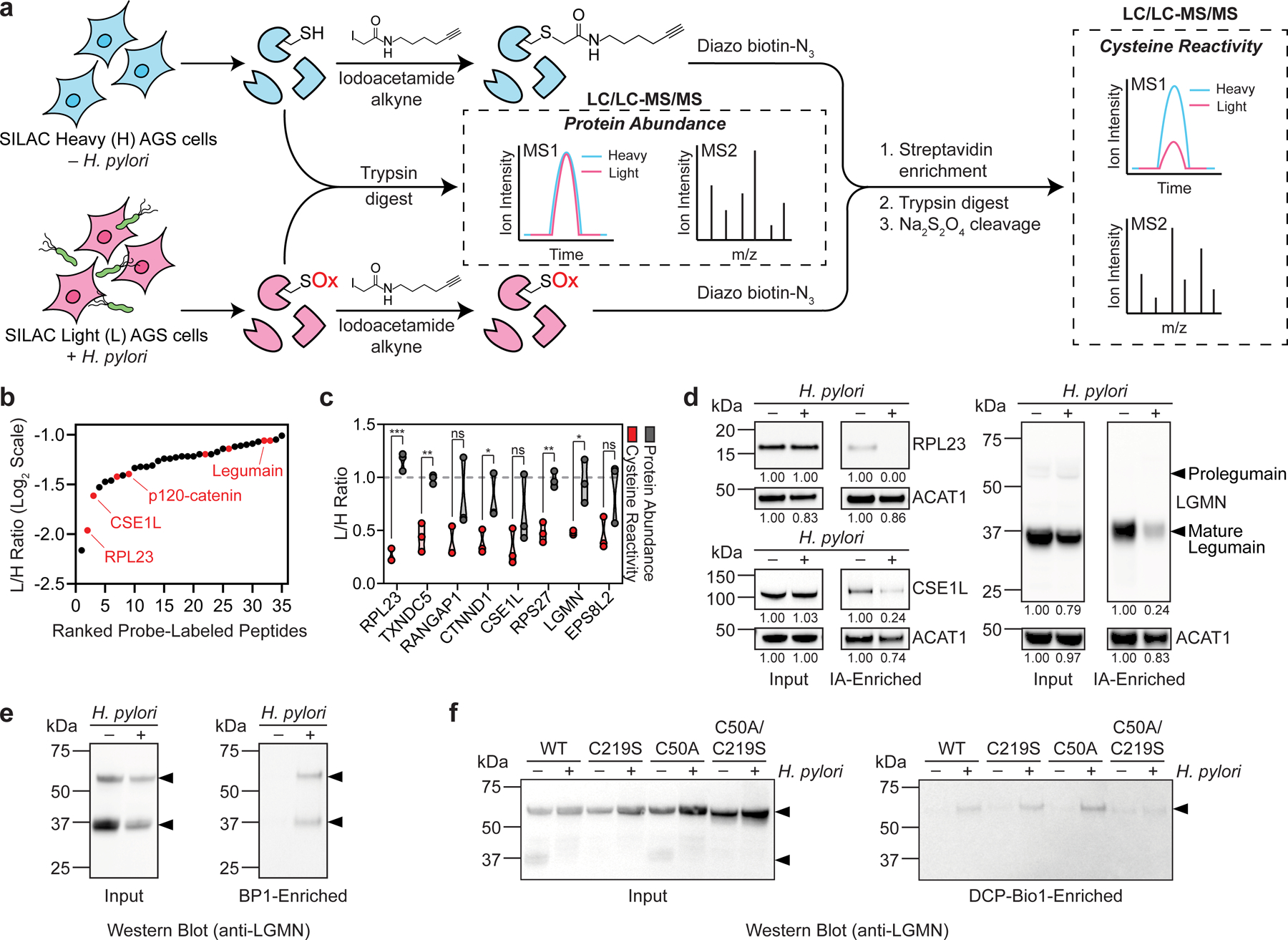
(a) isoTOP–ABPP method for quantifying cysteine reactivity in SILAC-labeled, H. pylori-infected (“light”, L) and uninfected (“heavy”, H) AGS cells. In parallel, relative protein abundance of L and H cells was quantified by mass spectrometry prior to labeling cell lysates with iodoacetamide alkyne (IA). (b) Peptides containing cysteines with reduced IA labeling (reactivity) in L versus H cells (L/H ratio ≤0.5, SD <1, n=3). Peptides with cysteine reactivity ratios (L/H) ≤0.5 after correcting for changes in protein abundance are highlighted in red. (c) Plot of cysteine reactivity and protein abundance ratios for peptides in Table 1. *P=0.04 (CTNND1); *P=0.02 (LGMN); **P=0.003 (TXNDC5); **P=0.002 (RPS27); ***P=0.00012 (RPL23); n.s., not significant, by two-tailed unpaired t-test. (d) Western blot analysis of RPL23, CSE1L, and legumain (LGMN) in H. pylori-infected and uninfected AGS cells before (input) and after IA enrichment. Acetyl-CoA Acetyltransferase 1 (ACAT1), which exhibited little change in protein abundance and cysteine reactivity by isoTOP–ABPP, was used as a loading control. (e) Western blot analysis of legumain in H. pylori-infected and uninfected AGS cells before (input) and after BP1 enrichment. (f) Western blot analysis of legumain in H. pylori-infected and uninfected KO cells transiently transfected with WT LGMN, LGMNC219S, LGMNC50A, or LGMNC50A/C219S before (input) and after DCP-Bio1 enrichment. H. pylori infections were performed using H. pylori G27MA at MOI 50 for 18 h unless stated otherwise. Band intensities of RPL23, CSE1L, mature legumain, and ACAT1 were normalized by the corresponding band intensities in the uninfected sample and are indicated below each blot. Arrowheads denote prolegumain (top) and mature legumain (bottom). Data (b and c) represent three biological replicates. Average L/H ratios (n=3) are shown in (b). Each circle represents an individual replicate in (c). Western blot analyses were performed two (f) or three times (d and e) with consistent results.
Table 1.
Cysteines with abundance-corrected L/H ratio ≤0.5.
| Protein Description | Residue | Cys Reactivity L/H Ratio (Abundance corrected) |
|---|---|---|
| 60S Ribosomal Protein L23 (RPL23) | 125 | 0.22 |
| TRX Domain-containing Protein 5 (TXNDC5) | 247 | 0.44 |
| Ran GTPase-activating Protein 1 (RANGAP1) | 141 | 0.46 |
| p120-catenin (CTNND1) | 450 | 0.48 |
| CSE1L Exportin-2 | 842 | 0.49 |
| 40S Ribosomal Protein S27 (RPS27) | 77 | 0.49 |
| Legumain (LGMN) | 219 | 0.50 |
| EGFR Kinase Substrate EPS8L2 | 369 | 0.50 |
Infection-induced oxidation of a host protease
The lysosomal protease legumain, which contains one of the five high-confidence cysteines with reduced reactivity identified by our quantitative proteomic analyses (Fig. 1c), presented a possible link between cysteine oxidation and tumorigenic signaling. Numerous reports have implicated legumain expression and activity in tumorigenesis26–29. Administration of a legumain inhibitor decreased tumor growth and metastasis in a murine orthotopic model of gastric cancer27. In addition, the expression of legumain by tumor-associated macrophages enhanced tumor growth and angiogenesis in a xenograft model30. Because legumain expression is upregulated in a variety of human tumors27, including those from gastric cancer patients, legumain is considered a potential cancer biomarker and has been targeted for the development of tumor-specific probes and prodrugs28,31. Furthermore, unlike the other top hits from our screen, legumain is also known for its role in antigen processing32 and could presumably affect immunomodulatory responses to H. pylori infection. For these reasons, we decided to focus our initial studies on legumain Cys219. Given that cysteine oxidation has previously been shown to regulate the activity of other lysosomal proteases33,34, we hypothesized that oxidation of legumain Cys219 could potentially influence legumain activity and, in turn, tumor growth.
Legumain is an asparagine-specific endopeptidase that exists as both a glycosylated, 56-kDa zymogen (prolegumain; inactive form) and a mature, 36-kDa active form that is generated in part by autocatalytic cleavage of prolegumain35 (Fig. 1d). As in H. pylori-infected AGS cells (Fig. 1d), and compared to corresponding uninfected controls, we detected decreased levels of IA-labeled, mature legumain in H. pylori-infected KATO III gastric cancer cells (Extended Data Fig. 3a), in AGS cells infected with other H. pylori strains (Extended Data Fig. 3b), and in AGS cells infected with H. pylori at a lower multiplicity of infection (MOI 25; Extended Data Fig. 3c), which was used in subsequent experiments to better normalize cell viability across experimental conditions (Extended Data Fig. 3d). While the mature form of legumain consistently exhibited reduced IA labeling in infected cells, it was also present at lower levels (see Fig. 1d, uninfected versus infected input). To rule out the possibility that the reduced IA labeling of legumain was solely due to the decreased abundance of the mature form in H. pylori-infected cells, we took a complementary approach to determine whether legumain is directly oxidized during infection. We labeled and enriched proteins from H. pylori-infected versus uninfected cell lysates with the biotinylated probe biotin-1,3-cyclopentanedione (BP1), which selectively labels sulfenic acids, a specific class of reversibly oxidized thiols, and assessed the degree of enrichment by Western blot. We detected increased levels of BP1-labeled mature and unprocessed legumain in H. pylori-infected cells, indicating that legumain exhibits enhanced sulfenylation during infection (Fig. 1e). To determine whether legumain is sulfenylated at Cys219, legumain-deficient AGS cells (KO cells; Extended Data Fig. 4) were transfected with a plasmid encoding wild-type (WT) legumain or a C219S mutant (legumainC219S), and proteins from H. pylori-infected and uninfected cell lysates were labeled and enriched with the biotinylated probe 3-(2,4-dioxocyclohexyl)propyl 5-((3aR,6S,6aS)-hexahydro-2-oxo-1H-thieno[3,4-d]imidazol-6-yl)pentanoate (DCP-Bio1; Fig. 1f). Like BP1, DCP-Bio1 covalently modifies cysteine sulfenic acids, but with a chemically distinct reactive group. We detected increased DCP-Bio1-based enrichment of WT prolegumain in H. pylori-infected cells, consistent with infection-induced sulfenylation. However, we also observed an infection-dependent increase in the levels of DCP-Bio1-labeled legumainC219S. Given that legumain contains seven cysteines, we hypothesized that the enzyme might be sulfenylated at multiple sites during H. pylori infection, which could limit the ability to detect changes in the probe-based enrichment of single cysteine mutants by Western blot. Indeed, legumain Cys50 and Cys219 were previously shown to be reversibly oxidized in a mouse model of aging36, suggesting both residues may be susceptible to sulfenylation. Accordingly, we repeated our analysis using lysates from KO cells transfected with LGMNC50A or LGMNC50A/C219S. Like the WT enzyme and C219S mutant, legumainC50A exhibited increased DCP-Bio1-based enrichment during H. pylori infection; however, we observed a marked decrease in the probe-based enrichment of the C50A/C219S double mutant, demonstrating that both Cys50 and Cys219 are sulfenylated during H. pylori infection. Altogether, these results indicate that H. pylori infection decreases the reactivity of legumain Cys219, most likely via sulfenylation.
Infection inhibits prolegumain processing
We next investigated whether H. pylori infection influences legumain activity. Using a fluorogenic, legumain-selective activity-based probe (LE28; gift of Matthew Bogyo, Stanford University)37, we determined that legumain activity is substantially reduced during H. pylori infection, consistent with the decreased abundance of the mature enzyme in H. pylori-infected cells (Fig. 2a). Similarly, we observed decreased levels of mature legumain in the lysosomal fraction of H. pylori-infected cells (Fig. 2b and Extended Data Fig. 5a). Autocatalytic cleavage of prolegumain occurs at pH ~4–5, which enables processing of the inactive enzyme to the mature form in the lysosome38 (Fig. 2c). Endolysosomal trafficking is inhibited by H. pylori infection39, which could contribute to the decreased abundance of mature legumain in H. pylori-infected cells; however, we did not observe a comparable reduction in the lysosomal localization or processing of cathepsins B or D (Extended Data Fig. 5b), suggesting that another mechanism may account for the decreased abundance of mature legumain in H. pylori-infected cells. Prolegumain localization is known to be dysregulated in various disease states, including cancer40. Cancer cells secrete prolegumain into the extracellular space, where enzyme activation is thought to be triggered by the acidic pH and/or other proteases of the tumor microenvironment41. We analyzed prolegumain levels in the culture supernatants of H. pylori-infected versus uninfected AGS cells and observed a significant increase in extracellular prolegumain following H. pylori infection (Fig. 2d). The abundance of extracellular prolegumain increased over time and coincided with an infection-dependent decrease in the abundance of the fully cleaved, intracellular enzyme (Fig. 2e and 2f). Although cell lysis was more pronounced at later time points, we detected comparable levels of the cytosolic marker glyceraldehyde-3-phosphate (GAPDH) in the conditioned media of H. pylori-infected and uninfected cells (Fig. 2d and 2f), suggesting that the increased abundance of extracellular prolegumain during infection was not solely a product of lysed cells. Prolegumain secretion is regulated by K63-linked polyubiquitination of the enzyme’s activation peptide, which is catalyzed by the ubiquitin ligase TRAF642. TRAF6 binds to prolegumain immediately upstream of Cys219 and ubiquitinates Lys318, which lies within the activation peptide. Lys318 is required for the ubiquitination and secretion of prolegumain. Indeed, transfection of KO cells with a plasmid encoding legumainK318R blocked prolegumain secretion during H. pylori infection and increased intracellular accumulation of the unprocessed enzyme (Fig. 2g). Notably, H. pylori can induce TRAF6-mediated ubiquitination in infected cells43, providing a possible mechanism for the increase in prolegumain secretion during infection. Consistent with this hypothesis, we observed an infection-dependent increase in legumain ubiquitination using HEK293T cells transfected with plasmids encoding FLAG-tagged legumain, hemagglutinin (HA)-tagged ubiquitin, and Myc-tagged TRAF6 (Fig. 2h and Extended Data Fig. 6). Together, these data suggest that H. pylori infection inhibits the intracellular processing of prolegumain and enhances the ubiquitin-mediated secretion of the unprocessed enzyme, which reduces the abundance of active legumain in infected cells.
Figure 2. H. pylori infection alters prolegumain processing, localization, and ubiquitination.
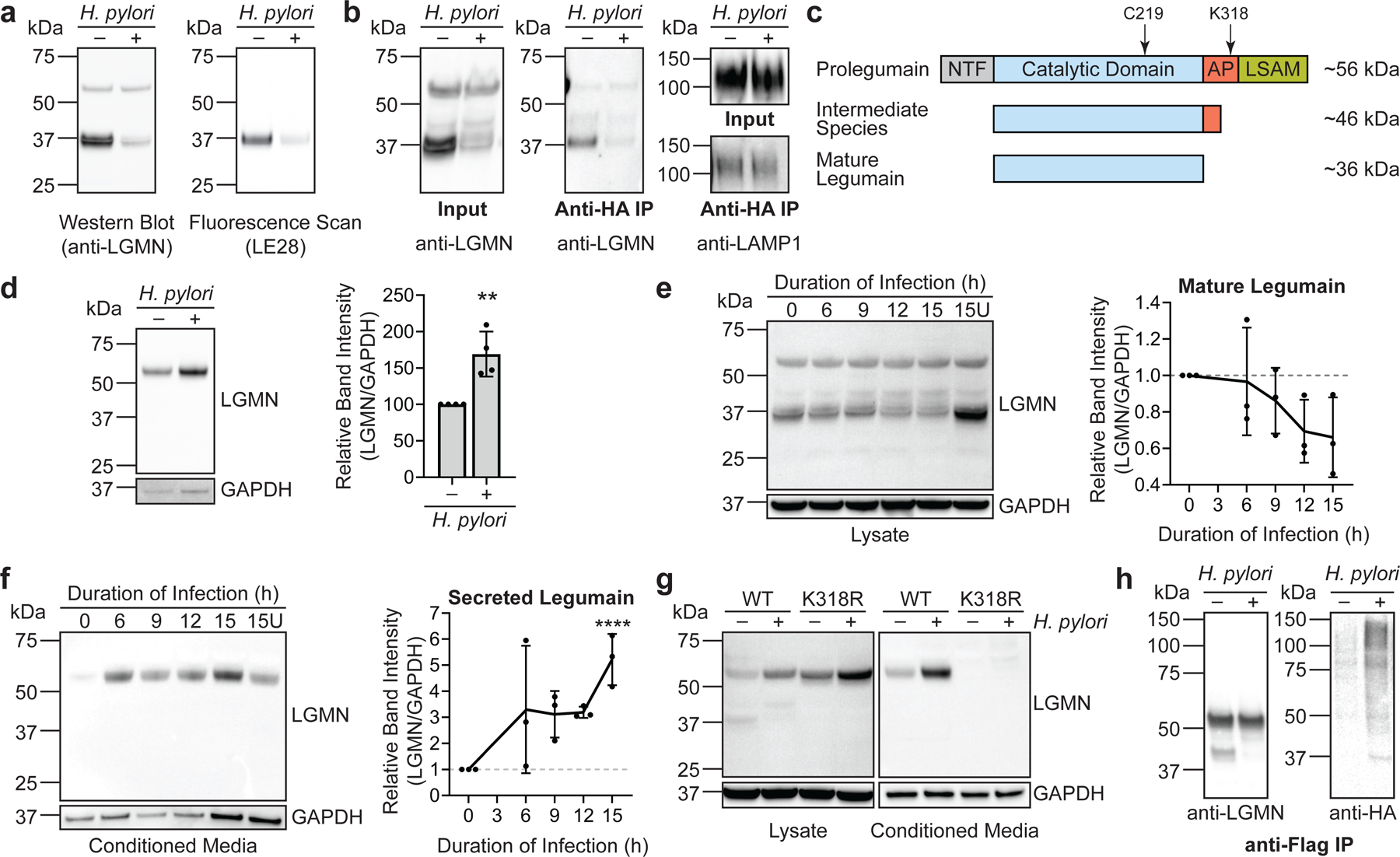
(a) Western blot (left) and in-gel fluorescence (right) analysis of legumain in H. pylori-infected and uninfected AGS cells labeled with LE28. (b) Western blot analysis of legumain and lysosomal-associated membrane protein 1 (LAMP1) in lysosomes isolated via anti-HA immunoprecipitation (IP) of HA-tagged lysosomal transmembrane protein TMEM192 from H. pylori-infected and uninfected AGS cells. (c) Simplified diagram of prolegumain processing. Prolegumain undergoes autocatalytic processing at acidic pH (pH ~4–5) to an intermediate species, followed by in trans processing by an unknown protease to mature legumain40. Additional intermediates are not shown. Approximate locations of residues Cys219 and Lys318 are noted. NTF, N-terminal fragment; AP, activation peptide; LSAM, Legumain Stabilization and Activity Modulation domain. (d) Western blot analysis of legumain and GAPDH in conditioned media from H. pylori-infected and uninfected AGS cells (left). Band intensity of prolegumain relative to the corresponding intensity of GAPDH was quantified across four biological replicates (right). **P=0.004, by two-tailed unpaired t-test. Western blot analysis of legumain and GAPDH in cell lysates (e) and conditioned media (f) from H. pylori-infected AGS cells at various times post infection (left), quantified as in (d) across three biological replicates (right). U, uninfected. ****P <0.0001 by two-tailed unpaired t-test compared to corresponding value at 0 h. (g) Western blot analysis of legumain and GAPDH in cell lysates and conditioned media from H. pylori-infected and uninfected legumain-deficient AGS (KO) cells transfected with WT LGMN or LGMNK318R. (h) Western blot analysis of legumain and HA-tagged ubiquitin following anti-FLAG IP of lysates from H. pylori-infected and uninfected HEK293T cells transiently transfected with FLAG-tagged LGMN, HA-tagged UBB, and Myc-tagged TRAF6. H. pylori infections were performed using H. pylori G27MA at MOI 25 for 18 h (a and b) or for 15 h followed by a 3-h incubation in serum-deficient medium (d-g). Each circle (d) represents an independent experiment. Error bars represent means ± SD. Western blot analyses were performed three (a, b, e-h) or four (d) times with consistent results.
Oxidized cysteine regulates prolegumain processing
We hypothesized that oxidation of legumain Cys219 could directly inhibit prolegumain processing during H. pylori infection. To test this, we transfected KO cells with a plasmid encoding either WT legumain or legumainC219S, as serine substitution is routinely used to block cysteine reactivity while preserving the steric architecture of the native thiol12,17,34. As in WT AGS cells, we detected the fully cleaved and unprocessed forms of legumain in KO cells transfected with the WT enzyme (Fig. 3a, lanes 1 and 5). Prolegumain processing was markedly reduced in both AGS cells and KO cells expressing WT legumain during H. pylori infection (Fig. 3a, lanes 2 and 6), and the amount of active legumain detected in these cells by LE28 labeling was less than in uninfected controls (Fig. 3b, lanes 2 and 6). In contrast, we were unable to detect fully cleaved, active legumain in KO cells expressing legumainC219S (Fig. 3b); prolegumain was the major species observed in both H. pylori-infected and uninfected cells (Fig. 3a, lanes 7 and 8). Thus, introducing a serine at Cys219, a non-catalytic residue with no known role in prolegumain processing, largely restricts the intracellular enzyme to its pro-form. Notably, the C219S mutation recapitulates key biochemical properties of Cys219 oxidation: the LGMNC219S transgene is expressed and encodes a stable protein (Fig. 3a), and the C219S mutation phenocopies the reduced levels of mature legumain and legumain activity observed in WT AGS cells during H. pylori infection (Fig. 3a and 3b). We were also unable to detect mature legumain in KO cells expressing a legumainC219A mutant (Extended Data Fig. 7), suggesting a reactive thiol is required at Cys219 for intracellular processing of prolegumain. To gain further insight into how Cys219 influences prolegumain maturation, we examined the crystal structure of prolegumain38. Legumain’s activation peptide, which is cleaved during protease maturation to generate the mature enzyme35, binds to Cys219 and neighboring residues within the catalytic domain (Fig. 3c)38. To determine whether other residues along this interface are important for prolegumain processing, we transfected KO cells with plasmids encoding either legumainY217A or legumainY220A (alanine is natively encoded at position 218). Mutating Tyr220, but not Tyr217, blocked prolegumain processing like the C219S mutant (Fig. 3d). Cys219 and Tyr220 are similarly oriented with respect to the legumain activation peptide, whereas Tyr217 is oriented in the opposite direction (Fig. 3c). These findings suggest that molecular interactions between Cys219, Tyr220, and the activation peptide may influence prolegumain maturation. We next assessed whether the C219S mutation plays a role in prolegumain secretion. Using KO cells transduced with either LGMN or LGMNC219S, we observed an infection-dependent increase in the extracellular localization of both WT prolegumain and prolegumainC219S (Fig. 3e). Notably, similar levels of the WT and mutant enzyme were detected in the conditioned medium of uninfected cells, indicating that the C219S mutation alone is insufficient to enhance prolegumain secretion. Acidification of conditioned medium containing either secreted prolegumain or prolegumainC219S activated autocatalytic processing of both enzymes to a ~46-kDa species (Fig. 3f), which corresponds to the molecular weight of a well-characterized legumain intermediate that can be processed to the 36-kDa mature form by other protease(s)35,44. Collectively, these results support that the loss of Cys219 reactivity inhibits intracellular processing of prolegumain, yet still permits acid-catalyzed activation of the secreted enzyme.
Figure 3. Legumain Cys219 is required for intracellular prolegumain processing.
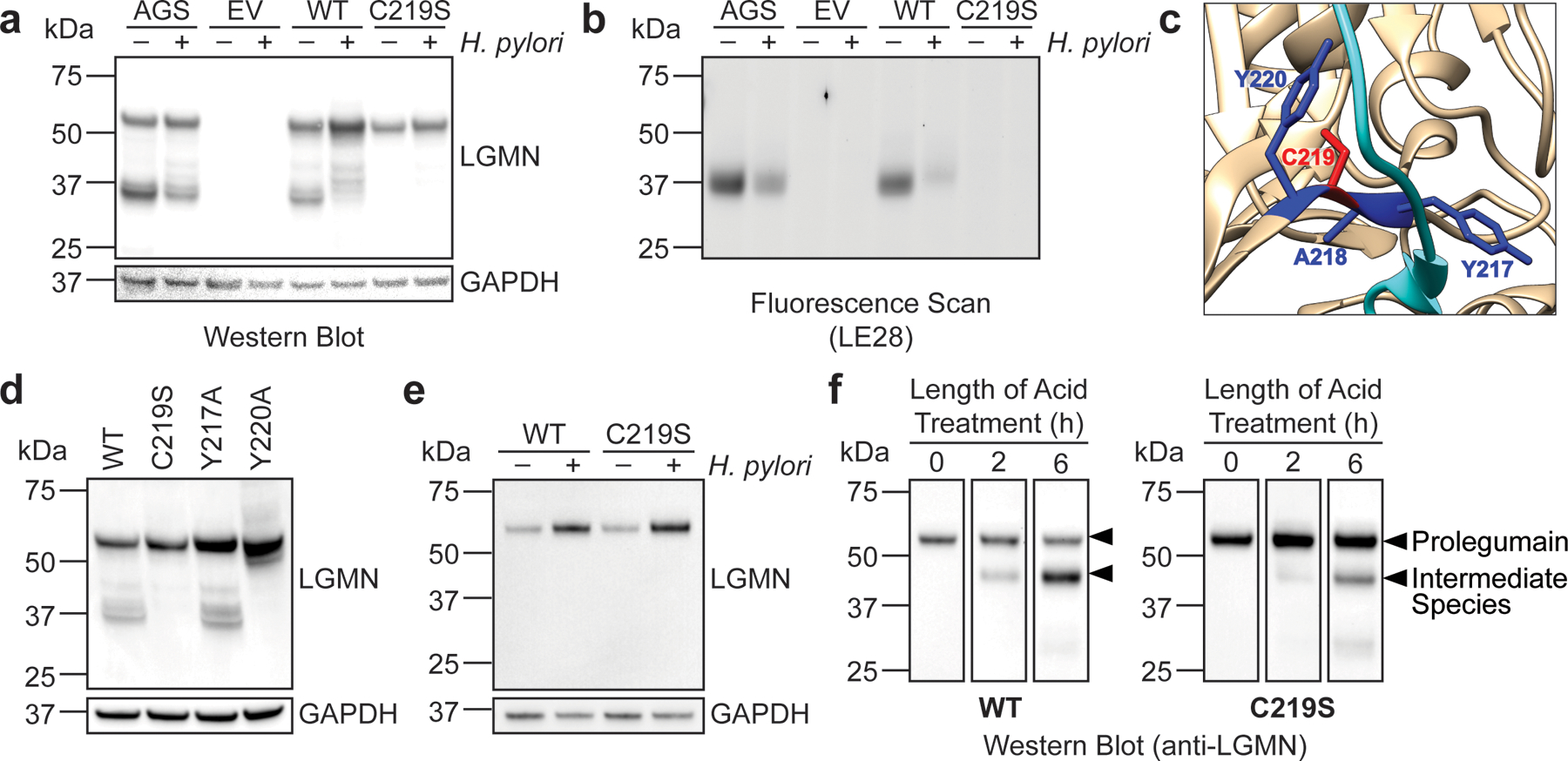
(a) Western blot analysis of legumain and GAPDH in H. pylori-infected and uninfected AGS or KO cells transiently transfected with empty vector (EV), WT LGMN, or LGMNC219S. (b) In-gel fluorescence analysis of legumain in H. pylori-infected and uninfected AGS or KO cells transiently transfected with empty vector (EV), WT LGMN, or LGMNC219S and labeled with LE28. (c) Crystal structure of prolegumain (PDB code 4fgu38) with activation peptide (cyan), Cys219 (red), and neighboring residues (Tyr217, Ala218, and Tyr220; dark blue) highlighted using the UCSF Chimera software program49. (d) Western blot analysis of legumain and GAPDH in KO cells transiently transfected with WT LGMN, LGMNC219S, LGMNY217A, or LGMNY220A. (e) Western blot analysis of legumain and GAPDH in conditioned media from H. pylori-infected and uninfected KO cells transduced with WT LGMN or LGMNC219S. (f) Western blot analysis of legumain in acidified conditioned media from H. pylori-infected KO cells transduced with WT LGMN or LGMNC219S. H. pylori infections were performed using H. pylori G27MA at MOI 25 for 18 h (a, b, d) or for 15 h followed by a 3-h incubation in serum-deficient medium (e and f). Western blot analyses were performed three times with consistent results.
Cysteine mutation accelerates tumor growth
Given the importance of Cys219 to prolegumain processing and activity, we examined how the C219S mutation influences the major biological functions of legumain. Since legumain can cleave antigens, Toll-like receptors, and other endo-lysosomal proteases that influence immune signaling40, we hypothesized that the C219S mutation might affect immunomodulatory responses to H. pylori infection, such as NF-κB activation and/or the production of IL-8, the major proinflammatory cytokine produced by H. pylori-infected gastric cells45. However, H. pylori-infected KO cells transduced with either LGMN or LGMNC219S exhibited similar levels of NF-κB activity (Extended Data Fig. 8a) and IL-8 expression (Extended Data Fig. 8b), suggesting legumain does not significantly regulate the cell-autonomous immune response to H. pylori infection.
Since legumain activity has also been shown to promote tumorigenesis26–29, we next tested whether the legumain C219S mutation influences tumor growth in vivo. Notably, we detected legumain expression in the gastric tissues of H. pylori-infected Mongolian gerbils (Extended Data Fig. 9), which can develop gastric cancer lesions46. Furthermore, legumain Cys219 was reversibly oxidized in a mouse model of aging36, demonstrating that this amino acid is redox-active in vivo. However, because the contribution of specific host factors to cancer development cannot be readily investigated using the gerbil model of H. pylori infection, which relies on outbred rodents that are genetically intractable46, we used a murine xenograft model to directly assess the role of legumain Cys219 reactivity in tumor growth. AGS-derived LGMN KO cells transduced with LGMN or LGMNC219S (Extended Data Fig. 10a), which exhibited uniform growth kinetics in vitro (Extended Data Fig. 10b), were subcutaneously implanted into immunodeficient Rag2-/- IL2RG-/- mice, and tumor growth was monitored until endpoint (tumor volume ≥1 cm3). LGMNC219S-expressing tumors exhibited significantly accelerated growth relative to tumors expressing WT legumain (Fig. 4a and Extended Data Fig. 10c). The median survival time of legumainC219S tumor-bearing mice (55.5 d) was also significantly shorter than that of mice bearing WT legumain tumors (72 d; Fig. 4b and Extended Data Fig. 10d). Notably, we detected fully cleaved, active legumain in LGMNC219S-expressing tumors by immunoblotting and LE28 labeling (Fig. 4c), suggesting that prolegumainC219S may be activated in vivo. Together, these data support that mutating a single, redox-sensitive amino acid in legumain dramatically accelerates tumor growth and mortality in a murine xenograft model.
Figure 4. Legumain C219S enhances tumor growth.
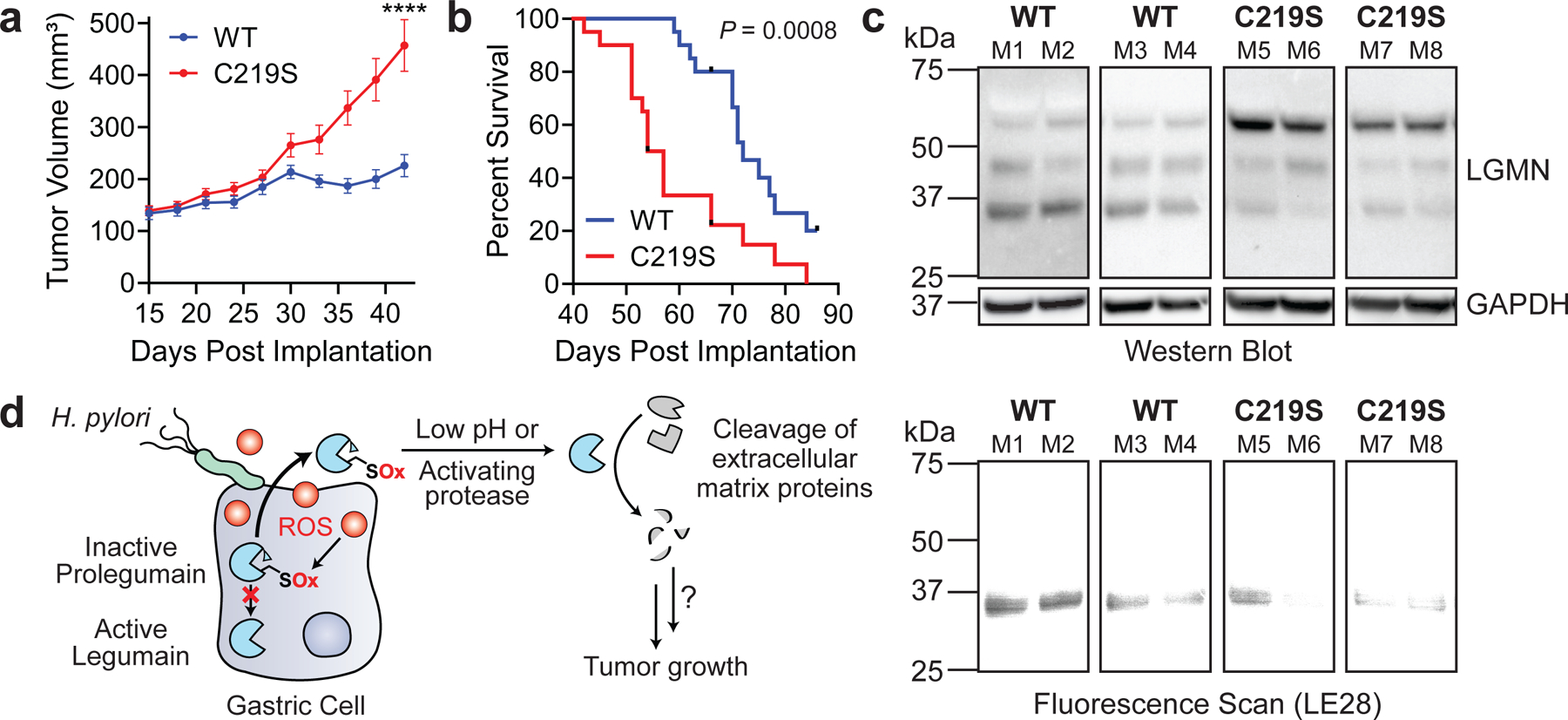
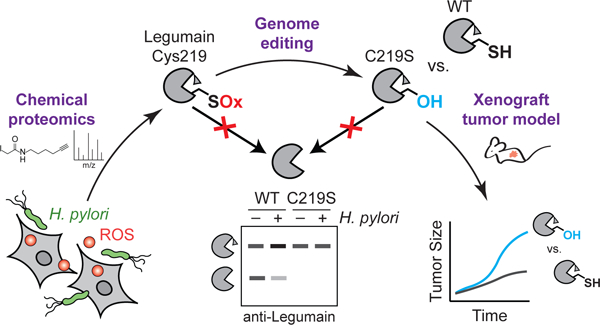
(a) Average tumor volume of Rag2−/− IL2RG−/− mice subcutaneously implanted with KO cells transduced with WT LGMN or LGMNC219S. n=20 mice per condition, using two independently derived clonal cell lines (n=10 mice per clonal line). ****P < 0.0001 by two-tailed unpaired t-test, error bars represent means ± SEM. (b) Survival curve of mice in 4A. ***P = 0.0008 by Mantel-Cox test. Tick marks represent censored events. A second independent experiment was performed using one clone per transduced line with consistent results (Extended Data Fig. 10c–d). (c) Western blot (top) and in-gel fluorescence (bottom) analysis of legumain in representative tumors from 4A labeled with LE28. Each blot represents tumors derived from independent cell lines. Analyses were performed on tumors from eight mice per clonal line with consistent results. (d) Cartoon depiction of working model. ROS, reactive oxygen species; SOx, oxidized cysteine.
Discussion
Identifying redox-regulated proteins that contribute to tumorigenesis is important for understanding how infection-associated oxidative stress promotes cancer development. We found that legumain Cys219, which is oxidized during H. pylori infection, regulates prolegumain processing and tumor growth. Cys219 oxidation inhibits intracellular cleavage of prolegumain, which is secreted by cancer cells via a ubiquitin-dependent mechanism during H. pylori infection (Fig. 4d). ProlegumainC219S, which phenocopies the defect in intracellular processing associated with Cys219 oxidation, is activated in vivo and accelerates tumor growth.
In contrast to prior studies linking legumain expression and activity to tumorigenesis26–29, our data unexpectedly show that genetic inactivation of intracellular prolegumain processing enhances tumor growth in vivo. We speculate that secretion and extracellular activation of prolegumainC219S in the tumor microenvironment accounts for the accelerated growth of C219S-expressing tumors in our xenograft model. Prolegumain secretion by cancer cells is believed to facilitate tumor growth by promoting cleavage of extracellular matrix components such as fibronectin and pro-matrix metalloproteinase-241. Although secreted prolegumain is inactive, extracellular processing of the enzyme by the acidic pH of the tumor microenvironment and/or accessory proteases is likely responsible for the high levels of secreted legumain activity reported in tumor tissues47. Given that legumain is only secreted in its proform, and the C219S mutation inhibits intracellular processing of the enzyme (Fig. 3a), prolegumainC219S may be secreted at greater levels by tumor cells than its WT counterpart. Moreover, because the C219S mutation does not inhibit acid-catalyzed activation of the secreted enzyme (Fig. 3f), extracellular activation of the C219S mutant could increase the cleavage of extracellular matrix components that facilitate tumorigenesis. Although our data suggest that prolegumainC219S is activated in vivo (Fig. 4c), additional studies are needed to elucidate the relative importance of intracellular versus extracellular legumain activity to tumor growth. Mutating legumain Cys219 may also affect other aspects of cancer cell signaling that contribute to tumorigenesis in our xenograft model. For example, the C219S mutation could inhibit the intracellular cleavage of legumain substrates that regulate tumor growth. Such signaling events may be independent of Cys219 oxidation and H. pylori infection; thus, further studies are needed to dissect the pathways that drive tumor growth in this model system.
Given that Cys219 is found in many animal legumains38, this residue may be a conserved oxidation site that redirects prolegumain for secretion under conditions of oxidative stress. Cys219 is reversibly oxidized in selected tissues of aged mice36, and our data are consistent with sulfenylation of Cys219 in human gastric cells during H. pylori infection; however, the precise oxidative modification on Cys219 remains unknown and may well include other oxoforms that stem from sulfenylation, such as sulfinic acid or glutathione disulfide. Although our data suggest that Cys219 oxidation alone is most likely insufficient to enhance prolegumain secretion (Fig. 3e), we hypothesize that the oxidation-induced inhibition of intracellular prolegumain processing, together with an increase in prolegumain ubiquitination via a secondary mechanism, promotes extracellular localization of the enzyme. Unlike other mammalian proteases, legumain exhibits highly specific endopeptidase activity towards asparagine residues48; thus, prolegumain secretion could facilitate the selective cleavage of extracellular substrates that are not targeted by other proteases. Similarly, prolegumain secretion may permit access to host degradation products that are otherwise inaccessible to H. pylori.
Together, our data establish legumain Cys219 as an infection-induced oxidation site that regulates tumor growth and suggest a new mechanism by which H. pylori may mediate cancer development. Given that increased extracellular legumain activity has previously been associated with gastrointestinal tumors28, we hypothesize that secreted prolegumain could be a potential indicator of cancer risk in H. pylori-infected tissues, and that small-molecule inhibitors of legumain activity may be a useful strategy for inhibiting tumor progression. More broadly, our work underscores the value of chemical proteomics in uncovering reactive cysteines that promote tumor growth.
Materials and Methods
H. pylori strains and culture conditions
A complete list of strains, plasmids, and primers can be found in Supplementary Tables 1–3. H. pylori cultures were grown on Columbia blood agar (Difco) plates with 5% (v/v) defibrinated horse blood (Hemostat Labs), 50 μg/mL cycloheximide (Sigma), 10 μg/mL vancomycin (Sigma), 5 μg/mL cefsulodin (Sigma), 2.5 U/mL polymyxin B (Sigma), 5 μg/mL trimethoprim (Sigma), 8 μg/mL amphotericin B (Fisher), and 0.2% (w/v) β-cyclodextrin (Sigma) at 37 °C in a humidified 10% CO2 incubator for 2–3 days. Prior to infection, H. pylori was cultured in dialyzed Brucella broth with 10% (v/v) dialyzed fetal bovine serum (FBS; Gibco, US origin) in vented T25 flasks (Falcon) with shaking at 100 rpm for 16–18 h at 37 °C under microaerophilic conditions maintained using Oxoid CampyGen Sachets (Thermo). Given that H. pylori grows more slowly in dialyzed media than in regular media, dialyzed media was used across all experiments for consistency. The bacterial cultures were either diluted to OD600 ~0.3 and shaken under the same conditions for an additional 1 h, or used directly for infection. Dialyzed Brucella broth (Difco) was prepared by dialysis in 3.5-kDa molecular weight cut-off SnakeSkin Dialysis Tubing (Thermo) in autoclaved 1X phosphate-buffered saline (PBS; Fisher) to reduce the concentration of free amino acids for compatibility with SILAC. A ratio of 67:1 PBS:Brucella broth was used, and the buffer was changed every 4 h for 12 h.
Mammalian cell lines and cell culture conditions
Human gastric epithelial AGS cells (ATCC CRL-1739) and human embryonic kidney (HEK) 293T cells (ATCC CRL-3216) were cultured in flasks containing Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS-HI; Gibco) at 37 °C in a humidified 5% CO2 incubator. KATO III cells (ATCC HTB-103) were grown in Iscove’s Modified Dulbecco’s Medium (IMDM; ATCC) supplemented with 10% (v/v) FBS-HI. Both adherent and non-adherent KATO III cells were collected and pooled for all experiments.
To quantify cell growth kinetics, cells were plated in black-walled, clear-bottom tissue-culture 96-well plates (Corning) at 1x104 cells per well. alamarBlue (Thermo) was added to 10% (v/v) final volume at the indicated times. After 4 h of treatment with alamarBlue, fluorescence (λex = 560 nm, λem = 590 nm) was measured using the SpectraMax i3x Multi-Mode Microplate Reader (Molecular Devices).
Generation of LGMN knockout cells
An sgRNA (5’-ATCACAACGATCTGTTCGTC-3’) targeting the LGMN gene was cloned into the PX459 plasmid (Addgene plasmid #62988 from Feng Zheng50) encoding Cas9 and puromycin resistance. To establish the LGMN knockout (KO) clonal cell line, AGS cells were transiently transfected for 24 h with the sgRNA-containing pX459 vector using polyethylenimine (PEI) at a 4:1 PEI:DNA ratio in Opti-MEM I Reduced Serum Media (OptiMEM; Gibco). Puromycin treatment (2 µg/mL; Sigma) was used to select for successfully transfected cells. After a 48-h treatment with puromycin, single cells were isolated, expanded, and assessed for legumain expression by Western blot to identify clonal lines.
Generation of plasmid constructs for transient transfection and lentiviral transduction
A pOTB7 vector containing the full-length human LGMN gene was obtained from Horizon Discovery (Clone ID: 3504506). Mutant LGMN genes were generated using site-directed mutagenesis (QuikChange II kit; Agilent). Genes were amplified by PCR and inserted into the lentiviral vector pLVX-Hhi3 (gift from Sanford M. Simon51) at the XhoI restriction site via Gibson cloning. All constructs were confirmed by DNA sequencing.
LGMN KO cells were transiently transfected with 2.5 µg (100-mm dishes) or 10 µg (150-mm dishes) pLVX-Hhi3 encoding WT or mutant LGMN genes using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions ~24 h prior to infection or collection.
To generate lentivirus, HEK293T cells were grown to ~60–80% confluence in 100-mm dishes and transfected with 2 µg pLVX-Hhi3 encoding LGMN or LGMNC219S, 2 µg pCRV1 NL GagPol packaging plasmid, and 400 ng pHCMV VSV-G using PEI at a 4:1 PEI:DNA ratio in OptiMEM. After 24 h, the lentivirus-containing conditioned medium was passed through a 0.45-µm polyethersulfone syringe filter and added to LGMN KO cells for 48–72 h. Hygromycin B (2 mg/mL; Invitrogen) was used to select for successfully transduced cells. After 48 h, single cells were isolated, expanded, and assessed for legumain expression by Western blot. Successfully transduced clonal lines were maintained in DMEM supplemented with 10% (v/v) FBS-HI and 20 µg/mL Hygromycin B.
H. pylori infection of mammalian cells
Cells were seeded in the appropriate medium (DMEM or IMDM) supplemented with 10% (v/v) FBS-HI 24–72 h prior to infection using 150-mm tissue culture-treated plates (Corning) at 6x106 cells per dish for AGS cells or 1.8x107 cells per dish for HEK293T cells (for mass-spectrometry (MS) analyses, enrichment of probe-labeled proteins, lysosome isolation, and ubiquitination experiments), 100-mm tissue culture-treated plates (Corning) at 2x106 cells per dish (for preparation of conditioned media, LE28 labeling, and qRT-PCR experiments), 12-well flat-bottom tissue culture-treated plates (Corning) at 4x105 cells per dish (for ROS measurement experiments), or 24-well flat-bottom tissue culture-treated plates (Corning) at 2x105 cells per dish (for GSH and NF-κB activity measurement experiments). On the day of infection, the cell culture medium was replaced with co-culture medium (DMEM or IMDM supplemented with 10% (v/v) dialyzed Brucella broth and 5% (v/v) dialyzed FBS). Cultured H. pylori was added to the cells at a multiplicity of infection (MOI) of 25 or 50 as noted, and the infection was allowed to proceed at 37 °C in a humidified 5% CO2 incubator for 18 h. H. pylori G27MA was used for all infection experiments except where indicated. An equivalent volume of 10% (v/v) dialyzed FBS in dialyzed Brucella broth was used as a mock infection control (i.e., for uninfected cells). A portion of the H. pylori-containing co-culture medium was serially diluted at the outset of each infection to enumerate bacterial colony-forming units (CFU) for confirmation of the MOI. Following infection, cells were washed with Dulbecco’s PBS (DPBS; HyClone), treated with 400 µg/mL kanamycin (Fisher) in the appropriate medium (DMEM or IMDM) for 1 h, and washed once more with DPBS. Cells were either collected via dissociation by TrypLE Express (Gibco) and pelleted by centrifugation (300 rcf, 3 min, room temperature (RT) in 15-mL or 50-mL conical tubes (Corning); then 21,000 rcf, 2 min, RT in Eppendorf tubes) for immediate use (i.e., in lysosome isolation, GSH measurement, ROS measurement, or NF-κB activity measurement experiments), or collected via scraping, pelleted, and stored at −80 °C prior to further analysis (all other experiments). To quantify cell viability, cell pellets were resuspended in PBS and analyzed via the Trypan Blue (Thermo) exclusion test using a Countess II FL automated cell counter (Applied Biosystems).
Preparation of conditioned media
AGS cells were plated in 100-mm tissue culture-treated dishes, as described above, and infected with H. pylori at MOI 25 for 15 h unless stated otherwise, washed with DPBS, and incubated with DMEM for an additional 3 h. The conditioned medium was collected and stored at 4 °C for no more than 24 h, while cells were collected via scraping, pelleted by centrifugation (300 rcf, 3 min, RT in 15-mL conical tubes; then 21,000 rcf, 2 min, RT in Eppendorf tubes), and stored at −80 °C until further analysis. Conditioned medium was centrifuged (3,000 rcf, 3 min, 4 °C) to pellet cell debris, then concentrated in 3-kDa molecular weight cutoff 15-mL centrifugal filter tubes (Amicon Ultra) by centrifugation (3,000 rcf, 2 h, 4 °C). Protein concentration was quantified using the Coomassie Plus Protein Assay (Pierce) and normalized across samples. Samples were boiled in LDS sample buffer (NuPAGE) containing 25 mM DTT (Bolt Sample Reducing Buffer; Thermo) for 5 min at 95 °C and analyzed by Western blot. For comparative analyses of prolegumain levels in conditioned medium from H. pylori-infected and uninfected cells, the background-subtracted band intensity of prolegumain in each sample was quantified using ImageJ Version 1.52p and divided by the background-subtracted band intensity of the corresponding GAPDH loading control. Where indicated, conditioned medium was acidified by treating protein-normalized, concentrated conditioned medium (50 µL) with 2 µL 1 N HCl at 37 °C for the specified time. Reactions were quenched with LDS sample buffer containing 25 mM DTT, boiled for 5 min at 95 °C, and analyzed by Western blot. All analyses of conditioned media were performed a total of three times with consistent results.
H. pylori infection of gerbils and processing of stomach tissue
Gerbil infection protocols were reviewed and approved by the Vanderbilt University Institutional Animal Care and Use Committee. Experiments were performed as previously described52. H. pylori strain 7.13 was cultured in Brucella broth with 10% (v/v) FBS for ~16 h under microaerophilic conditions. The bacteria were spun down and resuspended to a concentration of 1x109 CFU/mL. After fasting overnight, male and female Mongolian gerbils (35–49 g; Charles River) were infected via oral gavage with 5x108 CFU of bacteria on days 0 and 2. After 12 weeks of infection, gerbil stomachs were excised, and the nonglandular portion of the stomach (forestomach) was removed. The glandular portion of the stomach (corpus, transition zone, and antrum) was clipped along the major curvature and cut open along the minor curvature. Longitudinal strips of stomach tissue were fixed in 10% (v/v) formalin and embedded in paraffin.
Xenograft tumor model
Xenograft experiments were performed by the Yale Center for Precision Cancer Modeling. Animal protocols were reviewed and approved by the Yale University Institutional Animal Care and Use Committee. LGMN KO cells transduced with either WT LGMN or LGMNC219S (1x107 cells per mouse) were implanted subcutaneously into the right flank of immunodeficient Rag2−/− IL2RG−/− double knockout female mice (Envigo) in a volume of 100 µL 1:1 mix of DMEM and Matrigel (Corning). Tumor dimensions were recorded by caliper measurements every three days, and the tumor volume was calculated using the formula 0.5 x length x width2. The mice were euthanized when the tumor reached a predetermined endpoint volume of 1,000 mm3. Tumors were excised from 3–8 mice from each arm at the end of treatment and frozen at −80 °C for further analysis.
Xenograft experiments were performed twice with consistent results: 1) n=12 mice per arm, using a single clonal cell line for each arm (i.e., LGMN KO cells transduced with either WT LGMN or LGMNC219S); 2) n=20 mice per arm, using two independently derived clonal cell lines for each arm (i.e., n=10 mice per clonal line).
Measurement of intracellular GSH concentration
Cell pellets were resuspended in PBS, transferred to white-walled 96-well plates (Corning), and processed using the GSH-Glo Glutathione Assay kit (Promega) according to the manufacturer’s instructions. Luminescence was measured using the SpectraMax i3x Multi-Mode Microplate Reader. In parallel, a portion of cells from each sample was used to measure cell viability. GSH concentrations were quantified across three independent experiments, with three technical replicates per experiment.
Measurement of intracellular ROS levels
AGS cells were either mock-infected or infected with H. pylori, or treated with 5 mM hydrogen peroxide or an equivalent volume of water (untreated control) for 1 h. Cell pellets were resuspended in PBS, transferred to black-walled, clear-bottom 96-well plates (Corning), and incubated with 10 µM CM-H2DCFDA (Invitrogen) at 37 °C in a humidified 5% CO2 incubator for 30 min. Fluorescence (λex = 495 nm, λem = 520 nm) was measured using the SpectraMax i3x Multi-Mode Microplate Reader. In parallel, a portion of cells from each sample was used to measure cell viability. ROS levels were quantified across three independent experiments, with four technical replicates per experiment.
Preparation of isotopically labeled AGS cells
Heavy medium (HM) was prepared by adding 146 mg/mL [13C6,15N2]L-Lysine-2HCl (Cambridge Isotope Laboratories) and 84 mg/mL [13C6,15N4]L-Arginine-HCl (Cambridge Isotope Laboratories) to DMEM Medium for SILAC (Thermo). For light medium (LM), 146 mg/mL L-lysine (Sigma) and 84 mg/mL L-arginine (Sigma) were added to DMEM Medium for SILAC. HM and LM were additionally supplemented with 10% (v/v) dialyzed FBS. AGS cells were cultured in light (“light” cells) or heavy (“heavy” cells) medium for nine cell doublings to allow full incorporation of the stable isotope-containing amino acids. Enzyme-free cell dissociation buffer (Gibco) was used to dissociate cells for passaging. Complete isotopic incorporation was confirmed by MS.
Preparation of proteomes for isoTOP-ABPP and LC/LC–MS/MS analysis of unenriched cell lysates
“Heavy” and “light” cells were plated in the appropriate medium (HM or LM) supplemented with 10% (v/v) dialyzed FBS in 150-mm tissue culture-treated dishes. On the day of infection, the cell culture medium was replaced with co-culture medium (HM or LM supplemented with 10% (v/v) dialyzed Brucella broth and 5% (v/v) dialyzed FBS), and “light” cells were infected with H. pylori at MOI 50 for 18 h, while “heavy” cells were treated with an equivalent volume of 10% (v/v) dialyzed FBS in dialyzed Brucella broth. The infection was repeated twice more on separate days to generate three biological replicates for MS analyses. Notably, CFU enumeration revealed the actual MOIs for all three infections to be slightly lower than 50, with an average MOI ~20. Cells were washed and treated with kanamycin as described above, scraped, pelleted by centrifugation (300 rcf, 3 min, RT in 50-mL conical tubes; then 21,000 rcf, 2 min, RT in Eppendorf tubes), and stored at −80 °C. Frozen pellets were thawed on ice and resuspended in chilled PBS. The samples were sonicated (1 s on, 1 s off for 40 s total, 35% amplitude, repeated 5 times, 4 °C; Ultrasonic Cell Disruptor 500W, 110V (Qsonica)) to obtain whole-cell lysates, then centrifuged (21,000 rcf, 30 min, 4 °C) to pellet insoluble debris. The protein concentrations of the resulting supernatants were determined using the Coomassie Plus Protein Assay and normalized across samples.
Probe labeling and enrichment of labeled proteins for isoTOP-ABPP
To quantify differences in cysteine reactivity between H. pylori-infected and uninfected cells, protein labeling, enrichment, and analysis by isoTOP-ABPP were performed as previously described22. In brief, cell lysates from uninfected (“heavy”) or infected (“light”) cells (500 μL, 2 mg/mL total protein) were labeled with 100 μM N-(hex-5-ynyl)-2-iodoacetamide (IA-alkyne, synthesized as previously described22; characterization matched literature values) for 1 h at RT. Samples were subsequently incubated with 100 μM diazo biotin azide (azo-tag; Click Chemistry Tools), 1 mM tris(2-carboxyethyl)phosphine (TCEP; Sigma), 100 μM tris((1-benzyl-4-triazolyl)methyl)amine (TBTA; Sigma), and 1 mM CuSO4 (Sigma) for 1 h at RT to conjugate probe-labeled proteins with the chemically cleavable azo-tag via Cu(I)-catalyzed [3 + 2] cycloaddition (CuAAC). The proteins precipitated during the reaction, and equal volumes of the resulting “light” and “heavy” protein samples were combined and centrifuged (21,000 rcf, 4 min, 4 °C) to collect the precipitated protein. The supernatant was discarded, and the protein pellet was resuspended via quick sonication (1 s, 35% amplitude, 4 °C) in methanol that was pre-chilled on dry ice. Following centrifugation (21,000 rcf, 4 min, 4 °C), the resulting pellet was resuspended once more in methanol, sonicated, and centrifuged as described, then dissolved in 1.2% (w/v) sodium dodecyl sulfate (SDS; Fisher) in PBS. Sonication (1 s, 35% amplitude, 4 °C) followed by heating at 80–95 °C for 5 min was used to ensure complete solubilization. Samples were cooled to RT, diluted to 0.2% (w/v) SDS in PBS, and incubated overnight with streptavidin-agarose beads (200 μL of 50% aqueous slurry per sample; Thermo) at 4 °C on a tube rotator. Samples were then transferred to RT and rotated for an additional 3 h, pelleted by centrifugation (1,400 rcf, 3 min, RT), and the supernatant was discarded. Beads were then sequentially washed with 0.2% (w/v) SDS in PBS (5 mL x 1), PBS (5 mL x 3), and water (distilled and deionized, ddH2O; 5 mL x 3) for a total of seven washes.
On-bead trypsin digestion and chemical cleavage of probe-labeled peptides
The washed beads were resuspended in 500 μL 6 M urea (Sigma) in PBS and incubated with 10 mM DTT (GoldBio) for 20 min at 65 °C. Each sample was then treated with 20 mM iodoacetamide (IA; Sigma) for 30 min at 37 °C, diluted with 950 μL PBS, and pelleted by centrifugation (1,400 rcf, 3 min, RT). The supernatant was discarded, and the beads were incubated with 200 μL of a pre-mixed solution containing 2 M urea in PBS, 1 mM CaCl2, and 2 μg Sequencing Grade Trypsin (Promega). The beads were shaken overnight at 37 °C and pelleted by centrifugation (1,400 rcf, 3 min, RT). The beads were then washed sequentially with PBS (500 μL x 3) and ddH2O (500 μL x 3), resuspended in sodium dithionite (50 μL of a 50 mM stock solution in ddH2O; Sigma), and incubated at RT for 1 h. The beads were pelleted by centrifugation (1,400 rcf, 3 min, RT), and the supernatant was transferred to a new tube. The remaining beads were twice incubated with sodium dithionite (75 μL of 50 mM in ddH2O) and pelleted as before, and the resulting supernatants were transferred to the same tube. The beads were then washed twice with PBS (100 μL), and each wash was combined with the previous supernatants to give a total volume of ~350 μL. The cleaved peptides were treated with 1/20 volume of formic acid (Sigma), and the samples were stored at −20 °C until LC/LC–MS/MS analysis.
In-solution trypsin digestion of unenriched proteins from cell lysates
To quantify differences in protein abundance between H. pylori-infected and uninfected cells, in-solution trypsin digestion was performed as previously described53. In brief, cell lysates from uninfected (“heavy”) or infected (“light”) cells (25 μL, 2 mg/mL total protein each) were combined, vortexed with 1/10 volume of 100% (w/v) trichloroacetic acid (TCA; Sigma), and incubated overnight at −80 °C. Proteins were then thawed, pelleted by centrifugation (21,000 rcf, 10 min, 4 °C), washed with ice-cold acetone, sonicated (1 s, 35% amplitude, 4 °C), and pelleted by centrifugation once more. The supernatant was removed, and the pellet was air dried and resuspended in 30 μL 8 M urea in PBS. The sample was diluted with PBS (70 μL), incubated with 1 M DTT for 15 min at 65 °C, and then treated with 400 mM IA for 30 min at 25 °C. PBS (120 μL) was added to the sample, which was subsequently incubated with 2 μg Sequencing Grade Trypsin and 1 mM CaCl2 overnight at 37 °C. Digested peptides were treated with 1/20 volume of formic acid and centrifuged (21,000 rcf, 10 min, RT) to remove particulates. Peptides were stored at −20 °C until LC/LC–MS/MS analysis.
LC/LC–MS/MS analysis
LC/LC–MS/MS analysis was performed using a Thermo LTQ Orbitrap Discovery mass spectrometer coupled to an Agilent 1200 series HPLC. Peptide samples were pressure loaded onto a 250-mm fused silica desalting column packed with 4 cm of Aqua C18 reverse phase resin (Phenomenex). Peptides were eluted onto a 100-mm fused silica biphasic column packed with 10 cm C18 resin and 4 cm Partisphere strong cation exchange resin (SCX; Whatman) using a five-step multidimensional LC–MS protocol (MudPIT). Each of the five steps used a salt push (0%, 50%, 80%, 100%, and 100% for probe-enriched peptide samples; 0%, 25%, 50%, 80%, and 100% for unenriched peptide samples), followed by a gradient of Buffer B in Buffer A (Buffer A: 95% water, 5% acetonitrile, 0.1% formic acid; Buffer B: 20% water, 80% acetonitrile, 0.1% formic acid) as outlined previously22. The flow-rate through the column was ~0.25 μL/min, with a spray voltage of 2.75 kV. One full MS1 scan (400–1800 MW) was followed by eight data-dependent scans of the nth most intense ion. Dynamic exclusion was enabled. The tandem MS data generated by the five MudPIT runs were analyzed using the SEQUEST algorithm54. For probe-enriched peptides, both static (+57.0215 m/z, IA alkylation) and differential (+258.1481 m/z, IA-alkyne labeling) modifications on cysteine residues were specified. For unenriched samples, only the static modification on cysteine residues was specified. The precursor–ion mass tolerance was set at 50 ppm while the fragment–ion mass tolerance was set to 0 (default setting). Data were searched against a combined human and H. pylori reverse-concatenated non-redundant FASTA database containing UniProt identifiers (H. pylori UniProt proteome ID UP000000429). MS datasets were independently searched with light and heavy SILAC parameter files; for these searches, static modifications on lysine and arginine for either light (+0.0 and +0.0) or heavy (+8.0142 and +10.0083) peptides were used. MS2 spectra matches were assembled into protein identifications and filtered using DTASelect2.055 to generate a list of protein hits with a peptide false-discovery rate of <5%, with the –trypstat options applied.
For probe-enriched samples, with the additional –modstat option applied, peptides were restricted to fully tryptic (-y 2) with a found modification (-m 0) and a delta-CN score greater than 0.06 (-d 0.06). Single peptides per locus were also allowed (-p 1) as were redundant peptide identifications from multiple proteins, but the database contained only a single consensus splice variant for each protein.
Quantification of peptide “light” to “heavy” (L/H) ratios were calculated using the cimage quantification package described previously22. Protein L/H ratios were calculated as the average of all peptide L/H ratios quantified for that protein. Annotation of protein subcellular localization as well as cysteine function and conservation were generated from the UniProt Protein Knowledgebase (UniProtKB) as described previously56. The data were analyzed across three biological replicates. Stringent filtering criteria were applied to identify proteins of interest: (i) probe-enriched peptides and their corresponding proteins identified in the unenriched samples (i.e., protein abundance) must have quantifiable L/H ratios in all three replicates with SD <1, (ii) the L/H ratio (average across all three replicates) of enriched peptides, normalized to the L/H ratio (average across all three replicates) of their protein abundance, must be ≤0.5, and (iii) the average L/H ratios of the enriched peptides and their protein abundance must be significantly different based on a two-tailed unpaired t-test.
The mass-spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE57 partner repository with the dataset identifier PXD025841.
Western blot analyses
Protein samples were resolved by SDS-PAGE using 4–12% Bis-Tris NuPAGE precast gels with MES running buffer alongside the Precision Plus prestained protein standard (Bio-Rad). Proteins were transferred to nitrocellulose membranes following SDS-PAGE using an iBlot 2 Dry Blotting System (Thermo). Membranes were blocked with 3% (w/v) dry milk in Tris-buffered saline (150 mM NaCl (Fisher), 50 mM Tris HCl (Sigma), pH 7.5) containing 1% (v/v) Tween 20 (Fisher) (TBST) prior to incubation with a primary antibody for 1 h at RT or overnight at 4 °C. Following 3 x 5 min washes with TBST, membranes were incubated for 1 h at RT in 10% (w/v) dry milk in TBST with a peroxidase-conjugated secondary antibody. Membranes were again washed 3 x 5 min with TBST and then incubated with SuperSignal West Pico PLUS or SuperSignal West Femto Maximum Sensitivity chemiluminescent substrates (Thermo). Protein bands were detected using a ChemiDoc Gel Imaging System (Bio-Rad). All Western blot analyses were performed at least twice with consistent results.
The following antibodies were used in this study: goat polyclonal anti-legumain (R&D Systems AF2199; 1:2,000 dilution), mouse monoclonal anti-GAPDH (VWR 1E6D9; 1:5,000 dilution), rabbit polyclonal anti-ACAT1 (Cell Signaling 44276; 1:1,000 dilution), rabbit polyclonal anti-RPL23 (Thermo A305–010A-M; 1:500 dilution), rabbit polyclonal anti-CSE1L (Abcam ab189180; 1:1,000 dilution), goat polyclonal anti-cathepsin B (R&D Systems AF953; 1:800 dilution), goat polyclonal anti-cathepsin D (R&D Systems AF1014; 1:200 dilution), mouse monoclonal anti-HA (Thermo 26183; 1:10,000 dilution), rabbit monoclonal anti-HA (Cell Signaling 3724; 1:1,000 dilution), mouse monoclonal anti-LAMP1 (Santa Cruz sc-20011; 1:500 dilution), rabbit polyclonal anti-S6K1 (Cell Signaling 9202; 1:1,000 dilution), rabbit monoclonal anti-VDAC (Cell Signaling 4661; 1:1,000 dilution), rabbit monoclonal anti-calreticulin (Cell Signaling 12238; 1:1,000 dilution), rabbit polyclonal anti-actin (Sigma A2066; 1:100 dilution), mouse monoclonal anti-Myc (Cell Signaling 2276; 1:1,000 dilution), Pierce High Sensitivity Streptavidin-HRP (Thermo 21130; 1:2,500 dilution), donkey anti-goat IgG-HRP (Novus HAF109; 1:1,000 dilution), goat anti-rabbit IgG-HRP (Sigma A4914; 1:5,000 dilution), and goat anti-mouse IgG-HRP (Promega W4021; 1:5,000 dilution).
Probe labeling and enrichment of labeled proteins for Western blot analysis
Frozen pellets of H. pylori-infected and uninfected cells were prepared as described above and thawed on ice prior to probe labeling. For IA labeling, pellets were resuspended in chilled PBS containing 1% (v/v) Triton X-100 (Sigma). Following a 10-min incubation on ice, the samples were centrifuged (21,000 rcf, 30 min, 4 °C) to pellet insoluble debris, and the resulting supernatant was collected. Protein concentrations were determined using the BCA Protein Assay (Pierce) and normalized across samples. A small portion of each cell lysate was treated with LDS sample buffer containing 25 mM DTT, boiled for 5 min at 95 °C, and reserved for Western blot analysis (‘input’). Lysate containing 1–2 mg of protein was treated with 100 µM Iodoacetyl-PEG2-Biotin (Thermo) for 1 h at RT. Following probe labeling, protein was precipitated with methanol (pre-chilled on dry ice) and centrifuged (21,000 rcf, 4 min, 4 °C) to collect the precipitated protein.
For BP1 and DCP-Bio1 labeling, pellets were resuspended in chilled PBS supplemented with 1% (v/v) Triton X-100, 4 M urea, and 1 mM biotin-1,3-cyclopentanedione (BP1; Kerafast) or chilled PBS supplemented with 1% (v/v) Triton X-100 and 1 mM 3-(2,4-dioxocyclohexyl)propyl 5-((3aR,6S,6aS)-hexahydro-2-oxo-1H-thieno[3,4-d]imidazol-6-yl)pentanoate (DCP-Bio1; Kerafast). The samples were incubated for 2 h at 4 °C on a tube rotator, then centrifuged (21,000 rcf, 30 min, 4 °C) to pellet insoluble debris, and the resulting supernatant was collected. Protein concentrations were determined using the BCA Protein Assay and normalized across samples. A small portion of each cell lysate was treated with LDS sample buffer containing 25 mM DTT, boiled for 5 min at 95 °C, and reserved for Western blot analysis (‘input’). BP1-labeled lysate containing 1–2 mg of protein was precipitated with methanol (pre-chilled on dry ice) and centrifuged (21,000 rcf, 4 min, 4 °C) to collect the precipitated protein.
For DYn-2 labeling, pellets were resuspended in chilled PBS containing 1 mM 4-(pent-4-yn-1-yl)cyclohexane-1,3-dione (DYn-2; Cayman). The samples were sonicated (1 s on, 1 s off for 40 s total, 35% amplitude, repeated 5 times, 4 °C), incubated for 2 h at 4 °C on a tube rotator, then centrifuged (21,000 rcf, 30 min, 4 °C) to pellet insoluble debris, and the resulting supernatant was collected. Protein concentrations were determined using the Coomassie Plus Protein Assay and normalized across samples. A small portion of each cell lysate was treated with LDS sample buffer containing 25 mM DTT, boiled for 5 min at 95 °C, and reserved for Western blot analysis (‘input’). DYn-2-labeled lysate containing 1–2 mg of protein was then incubated with 100 μM azo-tag, 1 mM TCEP, 100 μM TBTA, and 1 mM CuSO4 for CuAAC as described above. The protein precipitated during the reaction, and the samples were centrifuged (21,000 rcf, 4 min, 4 °C) to collect the precipitated protein. The supernatant was discarded, and the protein pellet was resuspended via sonication (1 s, 35% amplitude, 4 °C) in methanol that was pre-chilled on dry ice. The precipitated protein was collected via centrifugation (21,000 rcf, 4 min, 4 °C).
For all probe-labeled samples, protein pellets were resuspended in chilled methanol via sonication (1 s, 35% amplitude, 4 °C) and centrifuged (21,000 rcf, 4 min, 4 °C). The resulting pellets were redissolved in 1 mL of 1.2% (w/v) SDS in PBS via sonication (1 s, 35% amplitude, 4 °C) and then incubated at 80–95 °C for 5 min to ensure complete solubilization. Samples were cooled to RT, diluted to 0.2% (w/v) SDS in PBS, and incubated overnight with streptavidin-agarose beads (200 μL of 50% aqueous slurry per sample) at 4 °C on a tube rotator. Samples were transferred to RT for 3 h, pelleted by centrifugation (1,400 rcf, 3 min, RT), and the resulting supernatants were discarded. Beads were then sequentially washed with 0.2% SDS in PBS (5 mL x 1), PBS (5 mL x 3), and ddH2O (5 mL x 3) for a total of seven washes. Following the final wash, the protein-bound streptavidin beads were resuspended in 500 μL 6 M urea in PBS, transferred to Eppendorf tubes, and pelleted by centrifugation (1,400 rcf, 3 min, RT); the supernatant was discarded. Proteins were eluted by boiling the beads in LDS sample buffer containing 100 mM DTT for 10 min at 95 °C, followed by centrifugation (1,400 rcf, 3 min, RT). The resulting supernatants were then analyzed by Western blot. To quantify protein levels in input and probe-enriched samples, background-subtracted band intensities were measured in ImageJ Version 1.52p. All analyses of probe-enriched proteins were performed at least twice with consistent results.
LE28 labeling and in-gel fluorescence analysis
Frozen cell pellets were thawed on ice and resuspended in 50 mM citrate buffer (pH 4.5) containing 1% (v/v) Triton X-100. Following a 10-min incubation on ice, the samples were centrifuged (21,000 rcf, 30 min, 4 °C) to pellet insoluble debris, and the resulting supernatants were collected. Protein concentrations were determined using the BCA protein assay and normalized across samples.
Tumor tissue was thawed on ice and homogenized in acidic buffer (50 mM sodium citrate, pH 5, 250 mM NaCl) for 3 min using 3.2-mm stainless steel beads and a Mini-Beadbeater (BioSpec). The samples were centrifuged (21,000 rcf, 30 min, 4 °C) to pellet insoluble debris. The resulting supernatants were collected, and the protein concentrations were determined using the Coomassie Plus Protein Assay and normalized across samples.
Samples (50 µL) were treated with 1 µM LE28 (gift of Matthew Bogyo37) for 1 h at 37 °C. Reactions were quenched with LDS sample buffer containing 25 mM DTT and boiled for 5 min at 95 °C. Proteins were resolved by SDS-PAGE, and in-gel fluorescence (λex=633 nm, λem=670 nm) was detected using a Typhoon FLA 9000 imager (GE Healthcare). The proteins were then transferred to nitrocellulose membranes and analyzed by Western blot. All analyses of LE28 labeling of cell culture lysates were performed three times with consistent results. LE28 labeling was used to analyze 3–8 tumors per implanted clonal cell line (19 total tumors per condition, expressing either WT LGMN or LGMNC219S) across two independent experiments with consistent results.
Lysosome isolation
AGS cells in 150-mm dishes were transiently transfected 24 h prior to H. pylori infection with 10 µg pLJC5-Tmem192–3xHA (Addgene plasmid #102930 from David Sabatini58) using Lipofectamine 3000 (Thermo) according to the manufacturer’s instructions. Cells were infected as described above, pelleted by centrifugation (300 rcf, 3 min, RT in 50-mL conical tubes; then 21,000 rcf, 2 min, RT in Eppendorf tubes), and sonicated in chilled PBS (1 s on, 1 s off for 20 s total, 20% amplitude, 4 °C). The cell lysates were centrifuged (1,000 rcf, 2 min, 4 °C). The supernatant was collected and centrifuged once more (1000 rcf, 2 min, 4 °C). The resulting supernatant containing the cellular organelles, including lysosomes, was collected. Protein concentration was quantified using the Coomassie Plus Protein Assay and normalized across samples. A small portion of each sample was diluted 1:2 in PBS containing 1% (v/v) Triton X-100, incubated at RT for 10 min, boiled in LDS sample buffer containing 25 mM DTT for 5 min at 95 °C, and reserved for Western blot analysis (‘input’). Lysates containing 1–2 mg protein were incubated with prewashed anti-HA agarose beads (100 µL of 50% aqueous slurry per sample; Thermo) for 1 h at RT. The beads were washed 3x with PBS, resuspended in 50 µL 0.1 M glycine (Sigma), pH 2.5, and centrifuged (21,000 rcf, 2 min, RT). The supernatant was transferred to a fresh tube, and the elution with glycine was repeated twice more. The combined supernatants (150 µL total) were then diluted with 20 µL 0.1 M Tris-HCl, pH 8.5. The samples were treated with LDS sample buffer containing 25 mM DTT, boiled for 5 min at 95 °C, and analyzed by Western blot. Lysosome isolation and Western blot analyses were performed three separate times with consistent results.
Immunoprecipitation of ubiquitinated legumain
HEK293T cells in 150-mm dishes were transiently transfected 24-h prior to H. pylori infection with 5 µg pLVX-Hhi3-FLAG-LGMN, 5 µg HA-Ubiquitin (Addgene plasmid #18712 from Edward Yeh59), and/or 5 µg Myc-TRAF6 using Lipofectamine 3000 (Thermo) according to the manufacturer’s instructions. Cells were infected as described above, pelleted by centrifugation (300 rcf, 3 min, RT in 50-mL conical tubes; then 21,000 rcf, 2 min, RT in Eppendorf tubes), and frozen at −80 °C until further analysis. Pellets were thawed on ice and resuspended in chilled PBS supplemented with 150 mM NaCl, 1 mM EDTA (ethylenediaminetetraacetic acid; Sigma), and 1% (v/v) Triton X-100. Following a 10-min incubation on ice, the samples were centrifuged (21,000 rcf, 30 min, 4 °C) to pellet insoluble debris, and the resulting supernatants were collected. Protein concentrations were determined using the BCA protein assay and normalized across samples. A small portion of each sample was diluted 1:2 in PBS, boiled in LDS sample buffer containing 25 mM DTT for 5 min at 95 °C, and reserved for Western blot analysis (‘input’). Lysates containing 1–2 mg protein were incubated with prewashed M2 anti-FLAG affinity resin (40 µL of 50% aqueous slurry per sample; Sigma) overnight at 4 °C. The beads were washed 3 times with PBS, resuspended in 40 µL PBS supplemented with 150 ng/µL 3X FLAG peptide (APExBIO), and incubated for 1 h at 4 °C. The beads were centrifuged (21,000 rcf, 2 min, RT) and the supernatant was transferred to a fresh tube, treated with LDS sample buffer containing 25 mM DTT, boiled for 5 min at 95 °C, and analyzed by Western blot. Western blot analyses were performed three separate times with consistent results.
Measurement of NF-κB activity
Transduced LGMN KO cells stably expressing LGMN or LGMNC219S in tissue culture-treated 24-well plates were transiently transfected 24 h prior to H. pylori infection with 500 ng pNiFty2-Luc (InvivoGen) using PEI at a 4:1 PEI:DNA ratio in Opti-MEM. Cells were infected as described above, pelleted by centrifugation, and resuspended in PBS. The cells were transferred to white-walled 96-well plates and processed using the Bright-Glo Luciferase Assay System (Promega) according to the manufacturer’s instructions. Luminescence was measured using the SpectraMax i3x Multi-Mode Microplate Reader. In parallel, a portion of cells from each sample was used to measure cell viability. NF-κB activity was quantified across three independent experiments.
Quantitative Reverse-Transcription PCR
RNA was extracted from cells using the Direct-zol RNA Miniprep Plus kit (Zymo Research). Quantitative reverse-transcription polymerase chain reaction (qRT-PCR) of the RNA was performed using a CFX96 Real-Time PCR Detection System (Bio-Rad) using the KAPA SYBR FAST One-Step qRT-PCR kit (Kapa Biosystems) according to the manufacturer’s instructions. GAPDH was used to normalize mRNA expression. Relative changes in gene expression between H. pylori-infected and uninfected cells were analyzed using the ΔΔCt method60. qRT-PCR analysis was performed three separate times, with three technical replicates per experiment.
Immunofluorescence microscopy
Paraffin-embedded stomach tissue sections from six H. pylori-infected gerbils, representing two independent infection experiments, were analyzed by immunofluorescence microscopy. Tissue sections were deparaffinized by incubation at 55 °C for 30 min, followed by xylene washes (2 x 5 min). Sections were rehydrated via multiple ethanol washes (100% for 10 min, 95%, 75%, 50%, 25% for 2 min each) and then subjected to antigen retrieval by boiling for 20 min in 10 mM sodium citrate buffer, pH 6. After cooling to RT, sections were blocked for 1 h in PBS containing 0.2% (v/v) Triton X-100 (PBST), 5% (v/v) donkey serum (Sigma), 3% (w/v) bovine serum albumin (BSA; Fisher), and 0.05 M glycine. Sections were then incubated with goat anti-legumain (R&D Systems AF2199; 1:100 dilution) or PBST (negative control) overnight in a humidified chamber at 4 °C. All subsequent washes and stainings were performed at RT in the dark. Sections were washed 3x with PBST, incubated with Alexa Fluor 647 AffiniPure Donkey Anti-Goat IgG (Jackson Immuno Research 705–605-003; 1:500 dilution) for 30 min, washed 3x with PBST, incubated with Wheat Germ Agglutinin, Alexa Fluor 555 Conjugate (WGA; Thermo Fisher W32464; 1:500 dilution) for 20 min, washed once more with PBST, and incubated with 4’, 6-diamidino-2-phenylindole dihydrochloride (DAPI; Thermo Fisher D1306; 1:1,000 dilution) for 20 min. After a final wash with PBST, sections were treated with ProLong Diamond Antifade Mountant with DAPI (Thermo Fisher P36966) and sealed with a cover slip.
Images were acquired at the Yale West Campus Imaging Core using a Nikon TiE Inverted Spinning Disk Confocal Microscope equipped with a Yokogawa CSU-W1 spinning disk with a 50-µm disk pattern, a Nikon Plan APO 100X oil, N.A. 1.45 objective, and an Andor iXon Ultra888 EMCCD camera with 13-µm pixel size and no binning. The samples were excited and imaged with the following lasers (MLC400, Agilent Technologies) and filter sets (Chroma, Bellow Falls VT, USA), respectively: 405 nm with Chroma ET 455/50 nm, 561 nm with ET 605/70 nm, 640 nm with ET 700/75 nm, and a 405/488/561/647 nm dichroic mirror. Images were assembled, color intensities and profiles were adjusted, and scale bars were added in ImageJ Version 1.53. At least two different fields of view per section were analyzed with consistent results.
Statistical analyses
GraphPad Prism (Version 9) and Microsoft Excel (Professional Plus 2016) were used to perform statistical analyses. Differences between two groups of data were analyzed by a two-tailed unpaired t-test. Differences between multiple groups of data were assessed by analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test (comparisons across all datasets) or Šídák’s multiple comparisons test (comparisons across specified datasets). Comparisons of Kaplan-Meier survival curves were analyzed by the Mantel-Cox test. Differences with P <0.05 were regarded as statistically significant.
Extended Data
Extended Data Fig. 1.
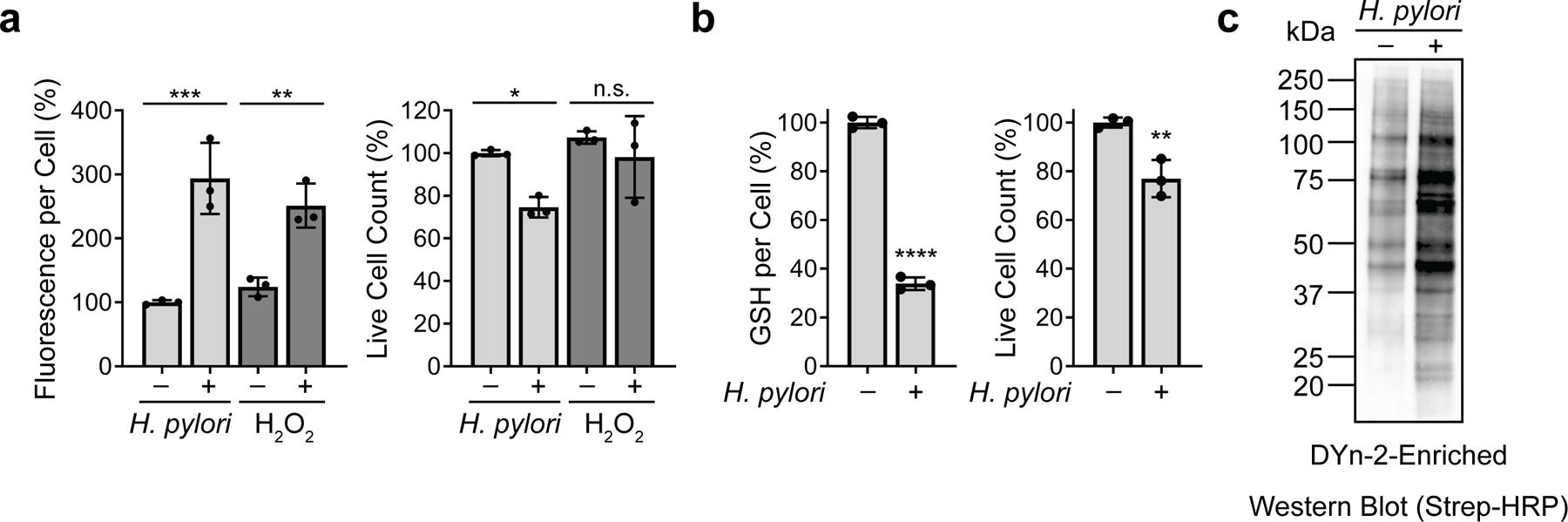
H. pylori infection increases ROS accumulation, decreases GSH levels, and enhances protein cysteine sulfenylation in AGS cells. (a) Intracellular ROS in AGS cells incubated with H. pylori G27MA (MOI 50, 18 h), 5 mM hydrogen peroxide for 1 h, or media alone were quantified using the fluorogenic ROS indicator CM-H2DCFDA (left). Fluorescence measurements were normalized by the total number of live cells per condition (right). (b) Intracellular GSH levels in H. pylori-infected (H. pylori G27MA, MOI 50, 18 h) and uninfected AGS cells (left), normalized by the total number of live cells per condition (right). Data represent three independent experiments. Each circle represents an independent experiment. Bars represent means ± SD. *P=0.03; **P=0.007; ***P=0.005; ****P <0.0001; n.s., not significant. Two-way analysis of variance (ANOVA) with Šídák’s multiple comparisons test was used for (a); two-tailed unpaired t-test was used for (b). (c) Western blot analysis of biotinylated proteins in H. pylori-infected (H. pylori G27MA, MOI 25, 18 h) and uninfected AGS cells. Proteins labeled with the sulfenic acid-specific probe DYn-2 were conjugated with diazo biotin azide and enriched on streptavidin beads prior to Western blot analysis. Western blot analysis was performed twice with consistent results.
Extended Data Fig. 2.
Supplementary data for Fig. 1b and 1c. (a) L/H ratios of peptides identified in IA-enriched samples from H. pylori-infected (H. pylori G27MA, MOI 50, 18 h) and uninfected AGS cells. (b) L/H ratios of peptides identified in unenriched lysates from H. pylori-infected (H. pylori G27MA, MOI 50, 18 h) and uninfected AGS cells. (c) Representative light (red) and heavy (blue) extracted ion chromatographs (EICs) (top) and isotopic envelopes (bottom) of cysteine-containing peptides from IA-enriched samples with protein abundance-corrected L/H ≤0.5. The L/H ratio of each peptide is shown above the corresponding EIC. The average L/H ratios (n=3) of peptides are shown in (a) and (b). Representative L/H ratios of peptides are shown in (c).
Extended Data Fig. 3.
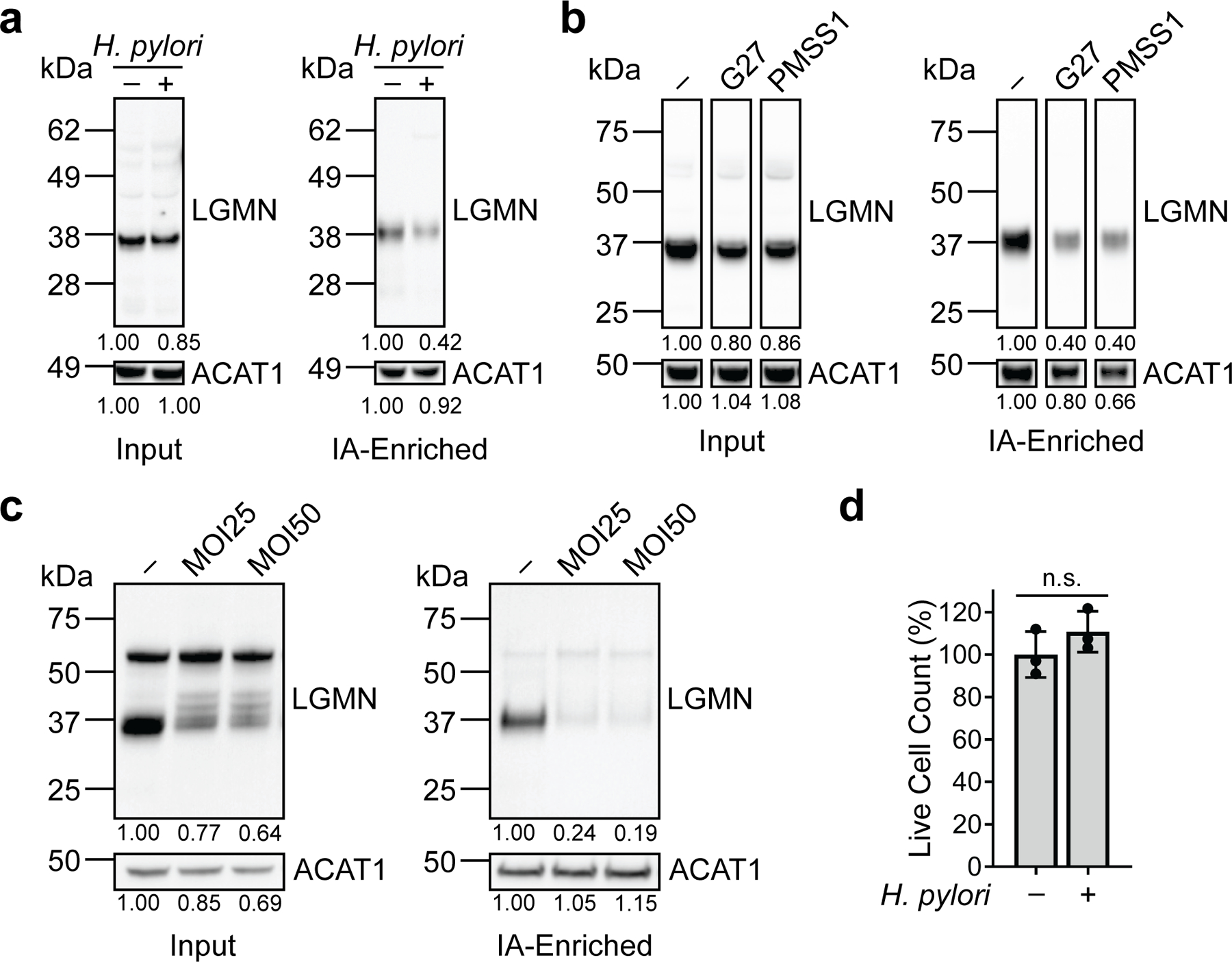
Legumain consistently exhibits reduced cysteine reactivity in H. pylori-infected cells under various infection conditions. (a) Western blot analysis of legumain in H. pylori-infected (H. pylori G27MA, MOI 50, 18 h) and uninfected KATO III cells before (input) and after IA enrichment. (b) Western blot analysis of legumain in uninfected AGS cells and AGS cells infected with the H. pylori strains G27 or PMSS1 (MOI 50, 18 h) before (input) and after IA enrichment. (c) Western blot analysis of legumain in uninfected AGS cells and AGS cells infected with H. pylori G27MA for 18 h at MOI 25 or 50 before (input) and after IA enrichment. (d) Quantification of AGS cell viability following infection with H. pylori G27MA (MOI 25, 18 h) or incubation with media alone for 18 h. Data represent three independent experiments. Bars represent means ± SD. Each circle represents an independent experiment. n.s., not significant, by two-tailed unpaired t-test. Western blot analyses were performed two (c) or three (a, b) times with consistent results. Band intensities of mature legumain and ACAT1 were normalized by the corresponding band intensities in the uninfected sample.
Extended Data Fig. 4.
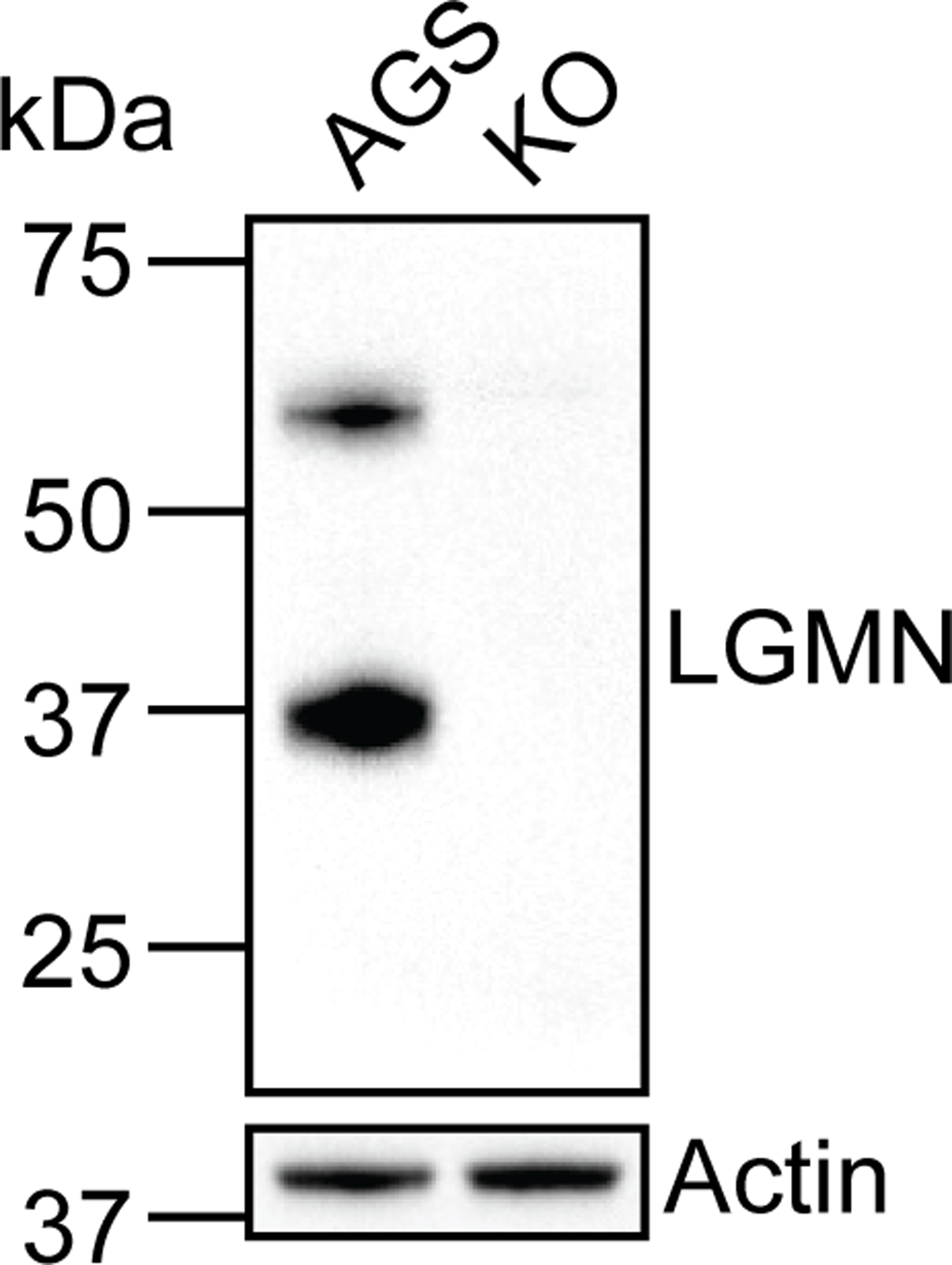
Biochemical validation of legumain-deficient KO cells generated via CRISPR/Cas9 genome editing. Western blot analysis of legumain and actin in WT AGS and KO cells. Western blot analysis was performed three times with consistent results.
Extended Data Fig. 5.
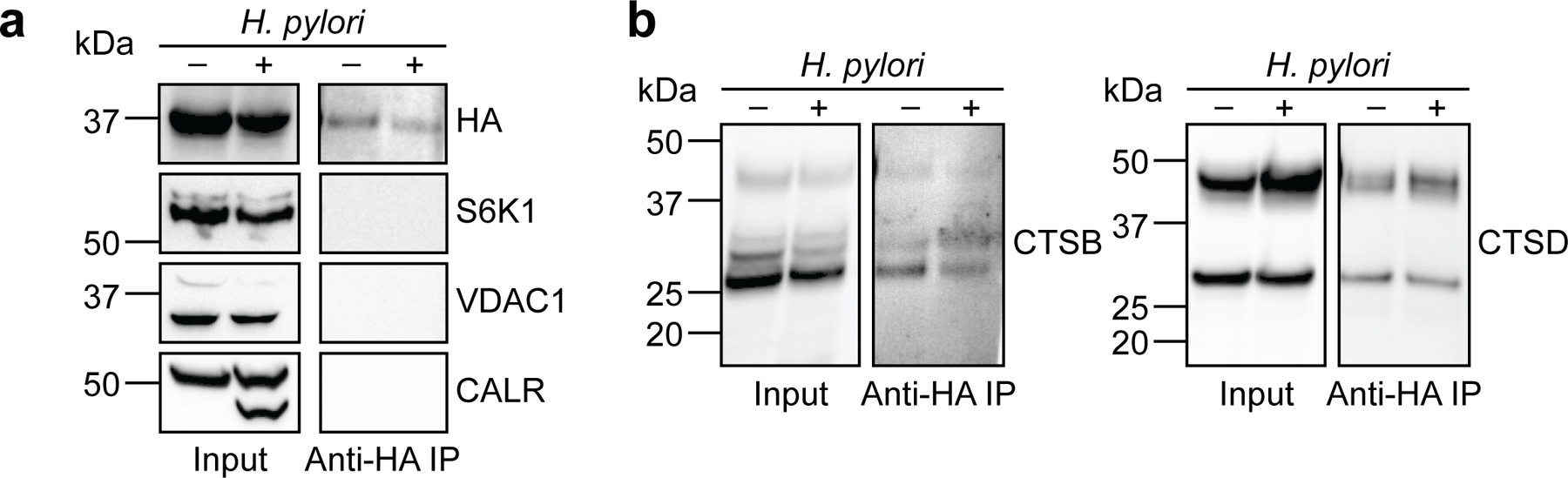
Supplementary data for Fig. 2b. Western blot analysis of (a) HA (top), protein markers of various subcellular compartments (S6K1, cytosol; VDAC1, mitochondria; CALR, endoplasmic reticulum), and (b) cathepsins B (left) and D (right) before (input) and after (anti-HA IP) lysosome enrichment of H. pylori-infected (H. pylori G27MA, MOI 25, 18 h) and uninfected AGS cells. Western blot analyses were performed three times with consistent results.
Extended Data Fig. 6.
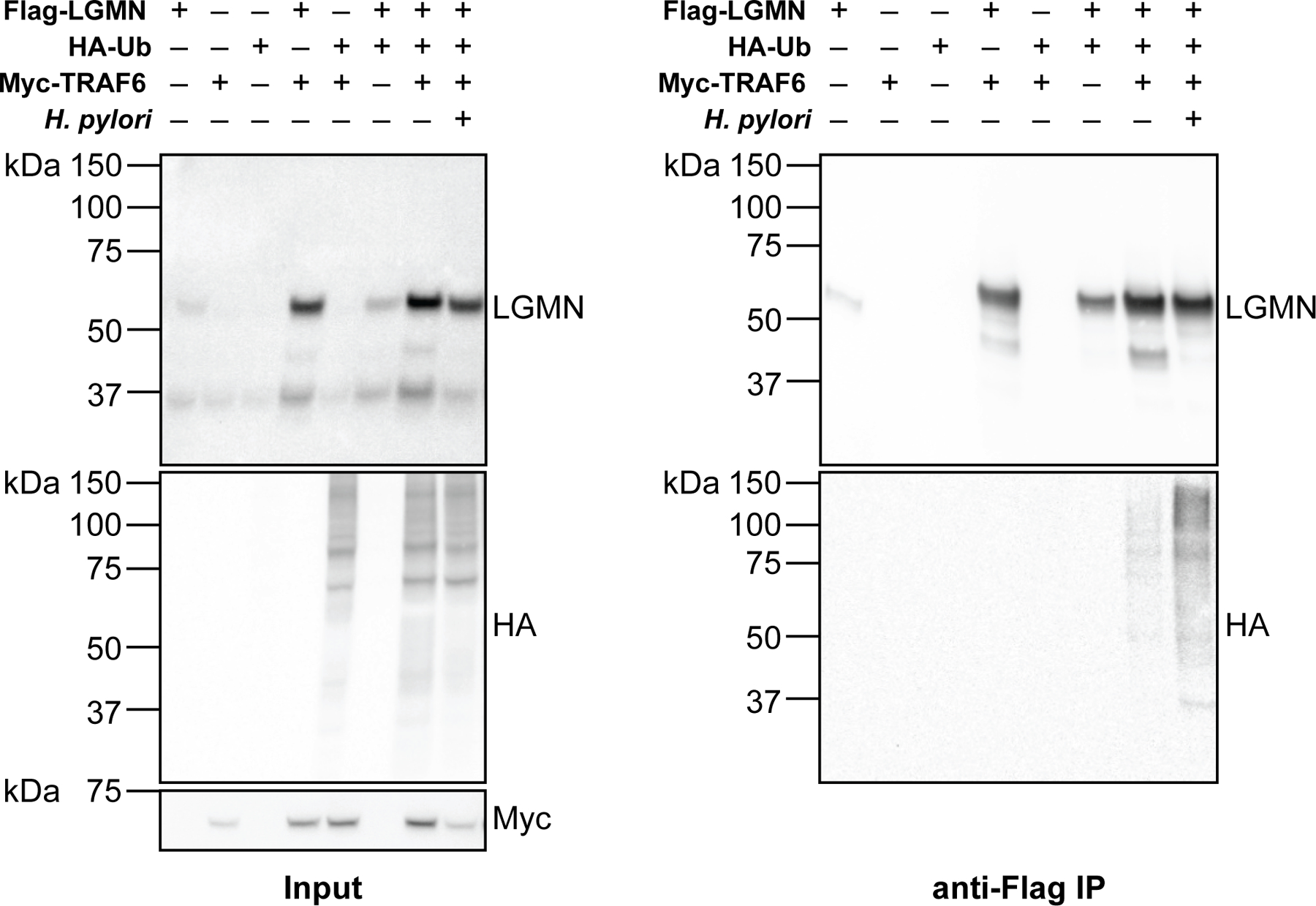
Supplementary data for Fig. 2h. Western blot analysis of legumain, HA-tagged ubiquitin, and Myc-tagged TRAF6 before (left, input) and after (right, IP) anti-FLAG IP of lysates from H. pylori-infected and uninfected HEK293T cells transiently transfected with FLAG-tagged LGMN, HA-tagged UBB, and/or Myc-tagged TRAF6. Cells were infected with H. pylori G27MA at MOI 25 for 15 h followed by a 3-h incubation in serum-deficient medium. Western blot analyses were performed three times with consistent results.
Extended Data Fig. 7.
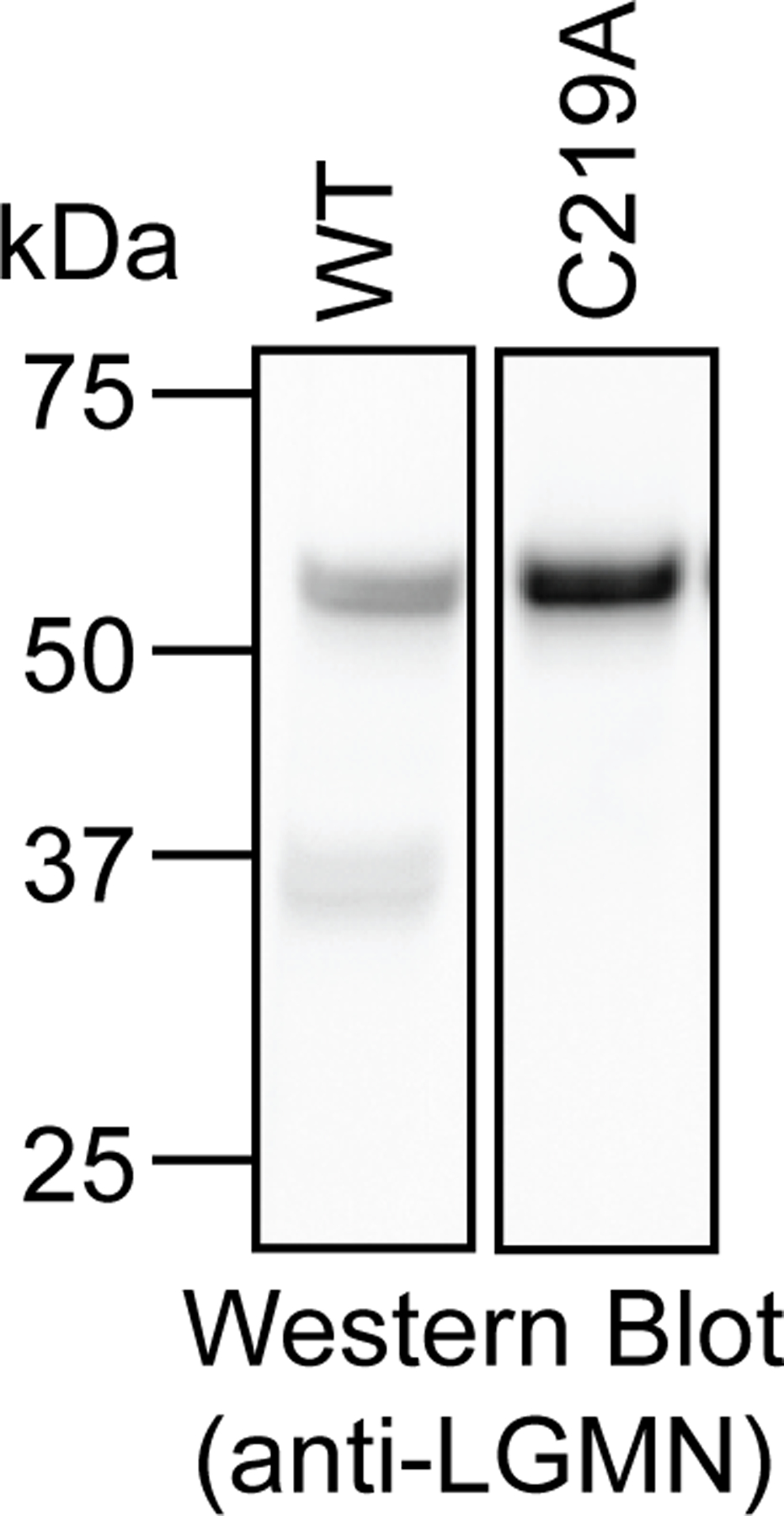
Intracellular legumainC219A is not processed to the mature enzyme. Western blot analysis of legumain in KO cells transfected with WT LGMN or LGMNC219A. Western blot analysis was performed three times with consistent results.
Extended Data Fig. 8.
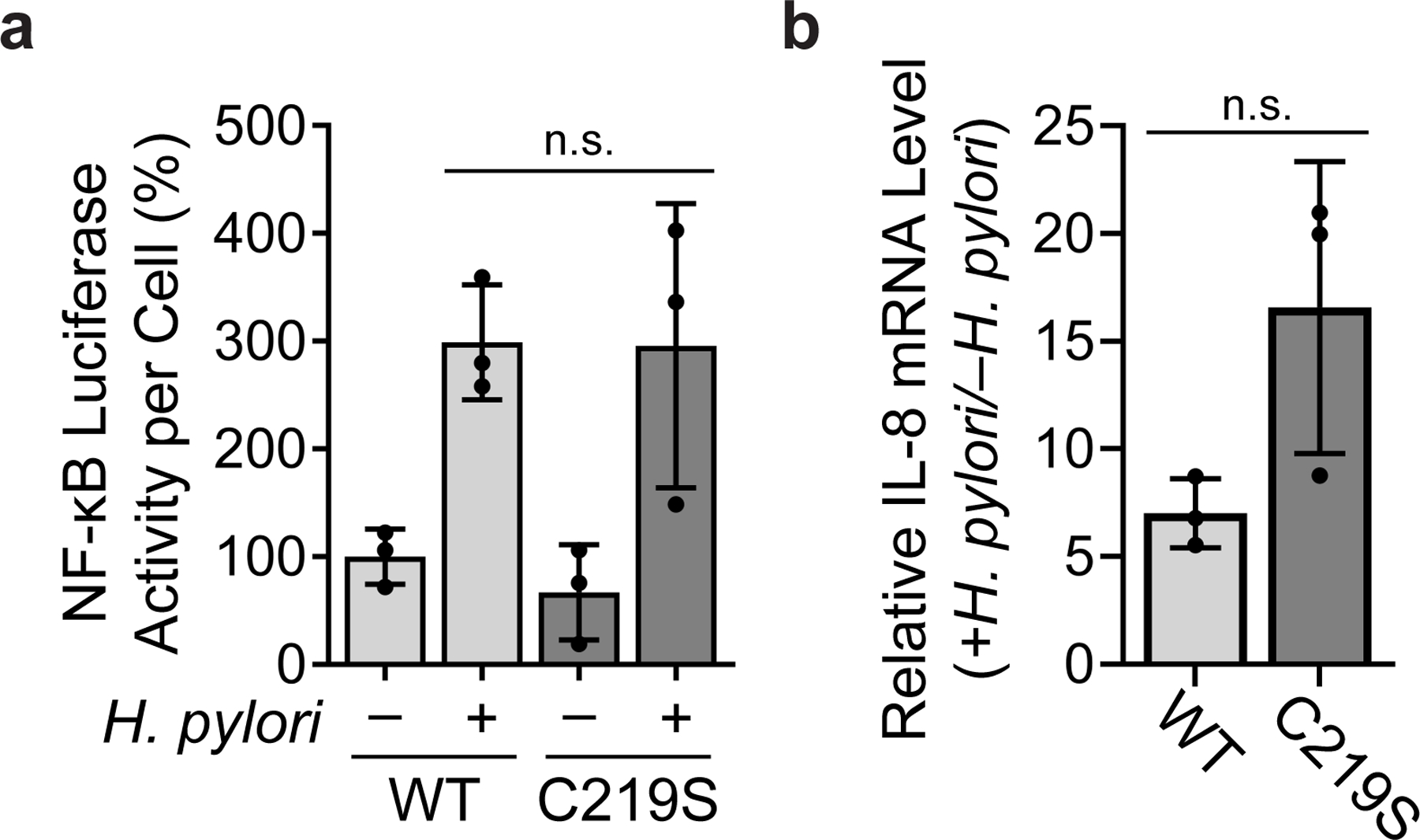
KO cells expressing WT legumain or legumainC219S exhibit similar NF-κB activity and IL-8 expression during H. pylori infection. (a) Luminescence of H. pylori-infected (H. pylori G27MA, MOI 25, 18 h) and uninfected KO cells transduced with WT LGMN or LGMNC219S and transiently transfected with the NF-κB-inducible luciferase reporter pNiFty2-Luc. (b) RT-qPCR analysis of IL-8 expression normalized to GAPDH expression in H. pylori-infected (H. pylori G27MA, MOI 25, 18 h) and uninfected KO cells transduced with WT LGMN or LGMNC219S. Data represent three independent experiments. Each circle represents an independent experiment. Bars represent means ± SD. n.s., not significant. Two-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test was used in (a); two-tailed unpaired t-test was used in (b).
Extended Data Fig. 9.
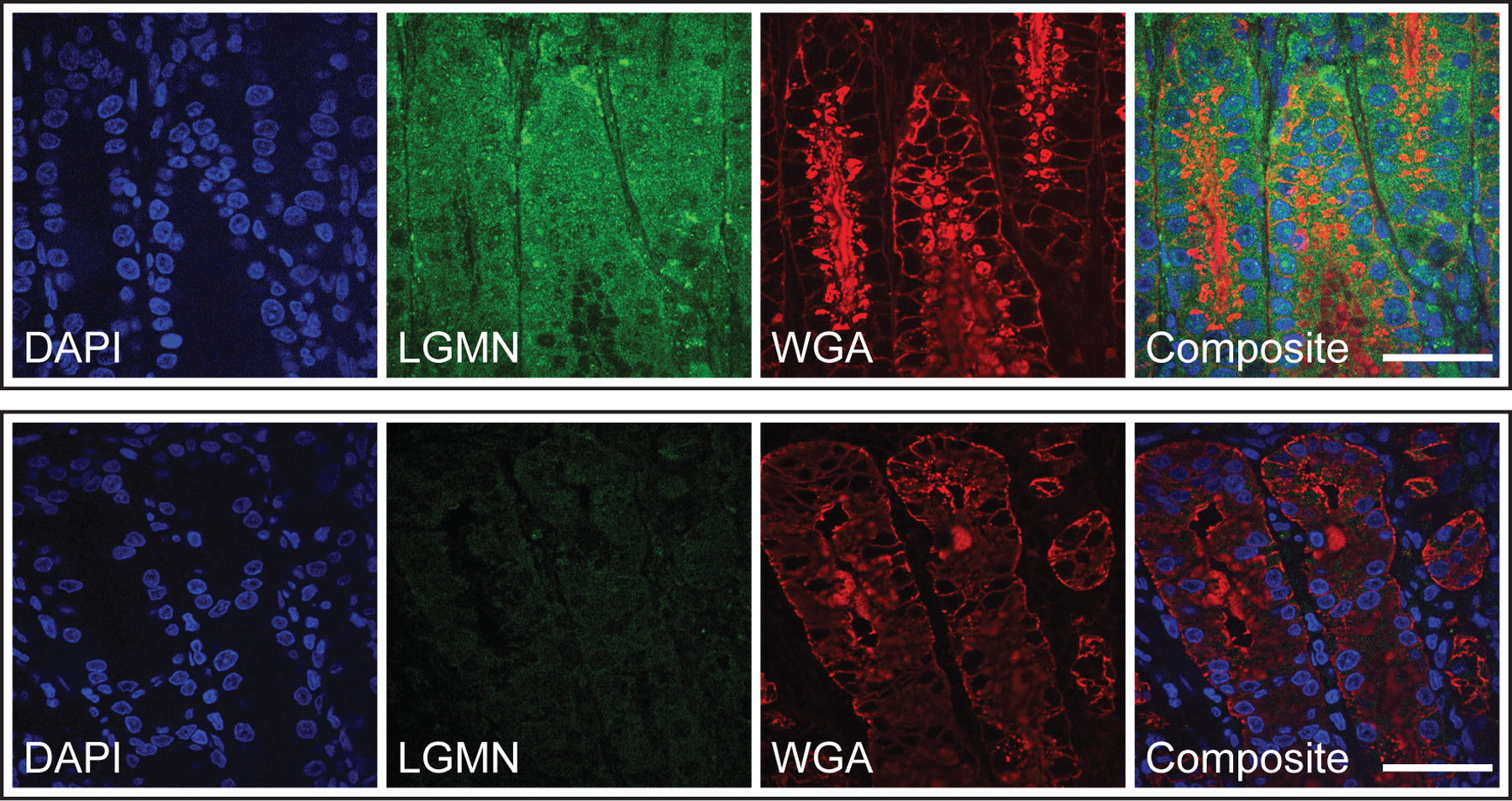
Legumain is expressed in the stomach tissue of H. pylori-infected gerbils. Representative confocal micrographs with immunofluorescent detection of legumain (LGMN; green) in gastric tissue sections from H. pylori-infected gerbils. Sections with labeled legumain (top) or stained with secondary antibody alone (bottom) are shown. Tissue sections were counterstained with DAPI (blue) and wheat germ agglutinin (WGA; red) to detect nuclei and cellular glycoconjugates, respectively. Sections from six gerbils, using at least two different fields of view per section, were analyzed with consistent results. Scale bar, 40 µm.
Extended Data Fig. 10.
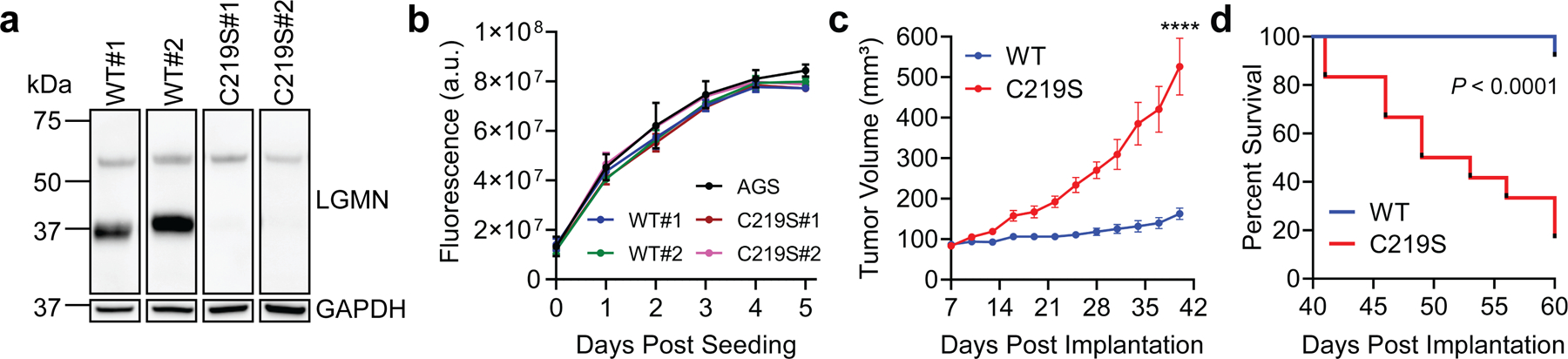
Supplementary data for Fig. 4. (a) Western blot analysis of legumain in two independently derived clonal lines of KO cells transduced with WT LGMN (WT#1 and WT#2) or LGMNC219S (C219S#1 and C219S#2). Western blot analysis was performed three times with consistent results. (b) Cell viability of WT AGS cells and WT#1, WT#2, C219S#1, and C219S#2 cells quantified by alamarBlue fluorescence. Data represent three independent experiments. Error bars represent means ± SD. (c) Average tumor volume of Rag2−/− IL2RG−/− mice subcutaneously implanted with KO cells transduced with WT LGMN or LGMNC219S. These data represent a second independent experiment performed using n=12 mice and a single clonal cell line per condition (WT#1 and C219S#1). Error bars represent means ± SEM. ****P <0.0001 by two-tailed unpaired t-test. (d) Survival curve of mice in (c). Tick marks represent censored events. ****P <0.0001 by Mantel-Cox test.
Supplementary Material
Acknowledgments:
We thank V. Muthusamy (Center for Precision Cancer Modeling, Yale Cancer Center) and J. Nikolaus (West Campus Imaging Core, Yale University) for experimental assistance. We also thank R. Gaudet, J. MacMicking, R. Morris, R. Wasko, C. Takacs, S. Simon, and M. Bogyo for reagents and/or technical support. We are grateful to the Hatzios lab, C. Crews, and A. Goodman for comments on the manuscript, and to D. Monack and M. Waldor for helpful discussions. This work was supported by NIH Training Grant T32 GM067543 to Y.K. and E.M.G. and T32 AI007281 to J.H.B.S.; a Gruber Science Fellowship to E.M.G; NSF Graduate Research Fellowship DGE 1147470 and a Stanford Graduate Fellowship to C.F.; NIH AI118932, CA116087, and Department of Veterans Affairs BX004447 to T.L.C.; Novo Nordisk Foundation Challenge Programme NNF19OC0056411 to M.R.A.; NIH R35GM134964 to E.W.; NIH R35GM137952, American Cancer Society Institutional Research Grant #IRG 17–172-57, a Pilot Grant from the Yale Cancer Center, and a Conquer Cancer Now Award from the Concern Foundation to S.K.H; and NIH Research Grant CA-16359 from the National Cancer Institute.
Footnotes
Competing interests: The authors declare no competing interests.
Data availability
Supplementary Information is available for this paper. Mass-spectrometry proteomics data that support the findings of this study have been deposited in ProteomeXchange via the PRIDE partner repository with the dataset identifier PXD025841.
References
- 1.Hussain SP, Hofseth LJ & Harris CC Radical causes of cancer. Nat Rev Cancer 3, 276–285, doi: 10.1038/nrc1046 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Liu Y et al. Nuclear lactate dehydrogenase A senses ROS to produce alpha-hydroxybutyrate for HPV-induced cervical tumor growth. Nat Commun 9, 4429, doi: 10.1038/s41467-018-06841-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Teoh ML, Turner PV & Evans DH Tumorigenic poxviruses up-regulate intracellular superoxide to inhibit apoptosis and promote cell proliferation. J Virol 79, 5799–5811, doi: 10.1128/JVI.79.9.5799-5811.2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chaturvedi R et al. Increased Helicobacter pylori-associated gastric cancer risk in the Andean region of Colombia is mediated by spermine oxidase. Oncogene 34, 3429–3440, doi: 10.1038/onc.2014.273 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Plummer M et al. Global burden of cancers attributable to infections in 2012: a synthetic analysis. Lancet Glob Health 4, e609–616, doi: 10.1016/S2214-109X(16)30143-7 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Robinson K, Letley DP & Kaneko K The human stomach in health and disease: infection strategies by Helicobacter pylori. Curr Top Microbiol Immunol 400, 1–26, doi: 10.1007/978-3-319-50520-6_1 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Davies GR et al. Helicobacter pylori stimulates antral mucosal reactive oxygen metabolite production in vivo. Gut 35, 179–185 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Katsurahara M et al. Reactive nitrogen species mediate DNA damage in Helicobacter pylori-infected gastric mucosa. Helicobacter 14, 552–558, doi: 10.1111/j.1523-5378.2009.00719.x (2009). [DOI] [PubMed] [Google Scholar]
- 9.Butcher LD, den Hartog G, Ernst PB & Crowe SE Oxidative stress resulting from Helicobacter pylori infection contributes to gastric carcinogenesis. Cell Mol Gastroenterol Hepatol 3, 316–322, doi: 10.1016/j.jcmgh.2017.02.002 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hardbower DM, de Sablet T, Chaturvedi R & Wilson KT Chronic inflammation and oxidative stress: the smoking gun for Helicobacter pylori-induced gastric cancer? Gut Microbes 4, 475–481, doi: 10.4161/gmic.25583 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shirin H et al. Helicobacter pylori decreases gastric mucosal glutathione. Cancer Lett 164, 127–133 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Anastasiou D et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 334, 1278–1283, doi: 10.1126/science.1211485 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bell EL et al. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J Cell Biol 177, 1029–1036, doi: 10.1083/jcb.200609074 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horak P et al. Negative feedback control of HIF-1 through REDD1-regulated ROS suppresses tumorigenesis. Proc Natl Acad Sci U S A 107, 4675–4680, doi: 10.1073/pnas.0907705107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao J et al. Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. EMBO J 28, 1505–1517, doi: 10.1038/emboj.2009.101 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Irani K et al. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 275, 1649–1652, doi: 10.1126/science.275.5306.1649 (1997). [DOI] [PubMed] [Google Scholar]
- 17.Truong TH et al. Molecular basis for redox activation of Epidermal Growth Factor Receptor Kinase. Cell Chem Biol 23, 837–848, doi: 10.1016/j.chembiol.2016.05.017 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Marco F et al. Oxidative stress in HPV-driven viral carcinogenesis: redox proteomics analysis of HPV-16 dysplastic and neoplastic tissues. PLoS One 7, e34366, doi: 10.1371/journal.pone.0034366 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li CQ, Pignatelli B & Ohshima H Increased oxidative and nitrative stress in human stomach associated with cagA+ Helicobacter pylori infection and inflammation. Dig Dis Sci 46, 836–844 (2001). [DOI] [PubMed] [Google Scholar]
- 20.Gordon EM & Hatzios SK Chemical tools for decoding redox signaling at the host-microbe interface. PLoS Pathog 16, e1009070, doi: 10.1371/journal.ppat.1009070 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi Y & Carroll KS Activity-based sensing for site-specific proteomic analysis of cysteine oxidation. Acc Chem Res 53, 20–31, doi: 10.1021/acs.accounts.9b00562 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weerapana E et al. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468, 790–795, doi: 10.1038/nature09472 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stairs DB et al. Deletion of p120-catenin results in a tumor microenvironment with inflammation and cancer that establishes it as a tumor suppressor gene. Cancer Cell 19, 470–483, doi: 10.1016/j.ccr.2011.02.007 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meng X et al. RPL23 links oncogenic RAS signaling to p53-mediated tumor suppression. Cancer Res 76, 5030–5039, doi: 10.1158/0008-5472.CAN-15-3420 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou S et al. Peroxiredoxin 6 homodimerization and heterodimerization with glutathione S-transferase pi are required for its peroxidase but not phospholipase A2 activity. Free Radic Biol Med 94, 145–156, doi: 10.1016/j.freeradbiomed.2016.02.012 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.D’Costa ZC et al. TBX2 represses CST6 resulting in uncontrolled legumain activity to sustain breast cancer proliferation: a novel cancer-selective target pathway with therapeutic opportunities. Oncotarget 5, 1609–1620, doi: 10.18632/oncotarget.1707 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li N et al. Effects of legumain as a potential prognostic factor on gastric cancers. Med Oncol 30, 621, doi: 10.1007/s12032-013-0621-9 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Liu C, Sun C, Huang H, Janda K & Edgington T Overexpression of legumain in tumors is significant for invasion/metastasis and a candidate enzymatic target for prodrug therapy. Cancer Res 63, 2957–2964 (2003). [PubMed] [Google Scholar]
- 29.Shen L et al. Legumain-deficient macrophages promote senescence of tumor cells by sustaining JAK1/STAT1 activation. Cancer Lett 472, 40–49, doi: 10.1016/j.canlet.2019.12.013 (2020). [DOI] [PubMed] [Google Scholar]
- 30.Wang H, Chen B, Lin Y, Zhou Y & Li X Legumain promotes gastric cancer progression through tumor-associated macrophages in vitro and in vivo. Int J Biol Sci 16, 172–180, doi: 10.7150/ijbs.36467 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang DS et al. Legumain protease-sheddable PEGylated, tuftsin-modified nanoparticles for selective targeting to tumor-associated macrophages. J Drug Target, 1–25, doi: 10.1080/1061186X.2021.1906886 (2021). [DOI] [PubMed]
- 32.Manoury B et al. An asparaginyl endopeptidase processes a microbial antigen for class II MHC presentation. Nature 396, 695–699, doi: 10.1038/25379 (1998). [DOI] [PubMed] [Google Scholar]
- 33.Lalmanach G et al. Regulation of the proteolytic activity of cysteine cathepsins by oxidants. Int J Mol Sci 21, 1944, doi: 10.3390/ijms21061944 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou Y et al. Chemoproteomic strategy to quantitatively monitor transnitrosation uncovers functionally relevant S-nitrosation sites on Cathepsin D and HADH2. Cell Chem Biol 23, 727–737, doi: 10.1016/j.chembiol.2016.05.008 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dall E & Brandstetter H Activation of legumain involves proteolytic and conformational events, resulting in a context- and substrate-dependent activity profile. Acta Crystallogr Sect F Struct Biol Cryst Commun 68, 24–31, doi: 10.1107/S1744309111048020 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiao H et al. A quantitative tissue-specific landscape of protein redox regulation during aging. Cell 180, 968–983 e924, doi: 10.1016/j.cell.2020.02.012 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edgington LE et al. Functional imaging of legumain in cancer using a new quenched activity-based probe. J Am Chem Soc 135, 174–182, doi: 10.1021/ja307083b (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dall E & Brandstetter H Mechanistic and structural studies on legumain explain its zymogenicity, distinct activation pathways, and regulation. Proc Natl Acad Sci U S A 110, 10940–10945, doi: 10.1073/pnas.1300686110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Capurro MI et al. VacA generates a protective intracellular reservoir for Helicobacter pylori that is eliminated by activation of the lysosomal calcium channel TRPML1. Nat Microbiol 4, 1411–1423, doi: 10.1038/s41564-019-0441-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dall E & Brandstetter H Structure and function of legumain in health and disease. Biochimie 122, 126–150, doi: 10.1016/j.biochi.2015.09.022 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Lunde NN, Bosnjak T, Solberg R & Johansen HT Mammalian legumain - A lysosomal cysteine protease with extracellular functions? Biochimie 166, 77–83, doi: 10.1016/j.biochi.2019.06.002 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Lin Y et al. Functional role of asparaginyl endopeptidase ubiquitination by TRAF6 in tumor invasion and metastasis. J Natl Cancer Inst 106, dju012, doi: 10.1093/jnci/dju012 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Lamb A et al. Helicobacter pylori CagA activates NF-kappaB by targeting TAK1 for TRAF6-mediated Lys 63 ubiquitination. EMBO Rep 10, 1242–1249, doi: 10.1038/embor.2009.210 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen JM, Fortunato M & Barrett AJ Activation of human prolegumain by cleavage at a C-terminal asparagine residue. Biochem J 352 Pt 2, 327–334 (2000). [PMC free article] [PubMed] [Google Scholar]
- 45.Lee KE et al. Helicobacter pylori and interleukin-8 in gastric cancer. World J Gastroenterol 19, 8192–8202, doi: 10.3748/wjg.v19.i45.8192 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Noto JM, Romero-Gallo J, Piazuelo MB & Peek RM The Mongolian gerbil: A robust model of Helicobacter pylori-induced gastric inflammation and cancer. Methods Mol Biol 1422, 263–280, doi: 10.1007/978-1-4939-3603-8_24 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu W et al. Targeting cell-impermeable prodrug activation to tumor microenvironment eradicates multiple drug-resistant neoplasms. Cancer Res 66, 970–980, doi: 10.1158/0008-5472.CAN-05-2591 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Chen JM et al. Cloning, isolation, and characterization of mammalian legumain, an asparaginyl endopeptidase. J Biol Chem 272, 8090–8098, doi: 10.1074/jbc.272.12.8090 (1997). [DOI] [PubMed] [Google Scholar]
- 49.Pettersen EF et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612, doi: 10.1002/jcc.20084 (2004). [DOI] [PubMed] [Google Scholar]
References for Materials and Methods
- 50.Ran FA et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308, doi: 10.1038/nprot.2013.143 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takacs CN et al. Green fluorescent protein-tagged apolipoprotein E: A useful marker for the study of hepatic lipoprotein egress. Traffic 18, 192–204, doi: 10.1111/tra.12467 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin AS et al. Temporal control of the Helicobacter pylori Cag Type IV Secretion System in a Mongolian gerbil model of gastric carcinogenesis. mBio 11, doi: 10.1128/mBio.01296-20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hatzios SK et al. Chemoproteomic profiling of host and pathogen enzymes active in cholera. Nat Chem Biol 12, 268–274, doi: 10.1038/nchembio.2025 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eng JK, McCormack AL & Yates JR An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5, 976–989, doi: 10.1016/1044-0305(94)80016-2 (1994). [DOI] [PubMed] [Google Scholar]
- 55.Tabb DL, McDonald WH & Yates JR 3rd. DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J Proteome Res 1, 21–26, doi: 10.1021/pr015504q (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bak DW, Pizzagalli MD & Weerapana E Identifying functional cysteine residues in the mitochondria. ACS Chem Biol 12, 947–957, doi: 10.1021/acschembio.6b01074 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Perez-Riverol Y et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47, D442–D450, doi: 10.1093/nar/gky1106 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abu-Remaileh M et al. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 358, 807–813, doi: 10.1126/science.aan6298 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kamitani T, Kito K, Nguyen HP & Yeh ET Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein. J Biol Chem 272, 28557–28562, doi: 10.1074/jbc.272.45.28557 (1997). [DOI] [PubMed] [Google Scholar]
- 60.Livak KJ & Schmittgen TD Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408, doi: 10.1006/meth.2001.1262 (2001). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Supplementary Information is available for this paper. Mass-spectrometry proteomics data that support the findings of this study have been deposited in ProteomeXchange via the PRIDE partner repository with the dataset identifier PXD025841.