

Abstract
Nucleic-acid aptamers are bio-molecular recognition agents that bind to their targets with high specificity and affinity and hold promise in a range of biosensor and therapeutic applications. In the case of small-molecule targets, their small size and limited number of functional groups constitute challenges for their detection by aptamer-based biosensors because bio-recognition events may both be weak and produce poorly transduced signals. The binding affinity is principally used to characterize aptamer-ligand interactions; however, a structural understanding of bio-recognition is arguably more valuable in order to design a strong response in biosensor applications. Using a combination of nuclear magnetic resonance, circular dichroism, and isothermal titration calorimetry, we propose a binding model for a new methamphetamine aptamer and determine the main interactions driving complex formation. These measurements reveal only modest structural changes to the aptamer upon binding and are consistent with a conformational-selection binding model. The aptamer-methamphetamine complex formation was observed to be entropically driven, apparently involving hydrophobic and electrostatic interactions. Taken together, our results exemplify a means of elucidating small molecule-aptamer binding interactions, which may be decisive in the development of aptasensors and therapeutics and may contribute to a deeper understanding of interactions driving aptamer selection.
Graphical abstract
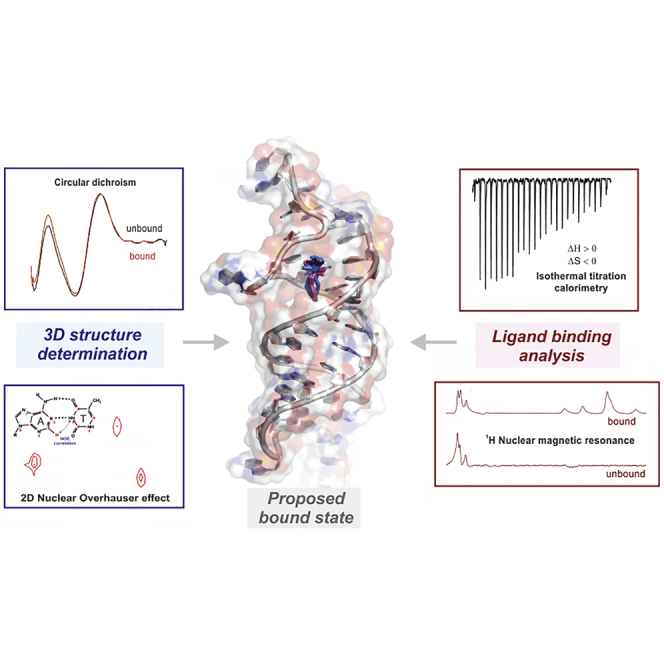
Significance
Here, we present a strategy combining spectroscopy, calorimetry, and molecular-docking simulations to interrogate the nature of ligand binding interactions, focusing on a novel methamphetamine-binding aptamer. We show that this system exhibits the hallmarks of a “conformational selection” binding model, in which different aptamer conformations exist in a dynamic equilibrium, and the ligand binds preferably to one of these conformations. However, we find that these conformations are structurally very similar, each retaining a stem-loop structure, and differing only by the loop conformation. We also find that ligand binding is entropically driven in this case, which is unlikely to produce the desired combination of high-affinity and distinct structural switching.
The implications of this work are far reaching. By focusing on building a structural binding model, the dissociation constant KD becomes secondary for the development of aptamers specific to a small-molecule ligand. The characterization approach we present here is suitable for studying other aptamer-small molecule complexes, and we suggest that such work should always inform the engineering of aptasensors. Only by doing so can signal-transduction strategies be chosen to generate strong binding signals.
Introduction
Nucleic-acid aptamers are functional bio-molecular recognition agents that bind to their targets with high specificity and affinity (1). They mostly originate from in vitro selection experiments involving the systematic evolution of ligands by exponential enrichment (SELEX) method, which, starting from random sequence libraries, optimizes the nucleic-acid sequences for high-affinity binding to the presented target ligands (2,3). Aptamers have proven to be highly stable, readily adaptable to chemical modification, and exhibit reversible binding. As a consequence, aptamer-based biosensors (aptasensors) are promising replacements for antibody-based biosensors in many applications (4,5).
An understanding of the aptamer-ligand binding domain and mechanism is valuable for sensing applications. With this knowledge, the design of the specific binding pocket by truncation or the addition of nucleotides, or hybridization with a complementary nucleotide sequence, can result in enhancement of specificity and the signal transduction and, thus, the sensitivity of the sensor (6, 7, 8, 9). Moreover, many sensing schemes rely on substantial target-induced structural changes to the aptamer. In this case, careful placement of a label can induce an electrochemical or fluorescent signal (10,11). On the other hand, even an aptamer-ligand complex with a strong binding constant may not make a good sensor if a lack of structural change prevents strong signal transduction. To a large extent, the degree of structural change upon ligand binding is expected on thermodynamic grounds to be negatively correlated with the binding constant. In any case, the most appropriate transduction system for a given aptamer-ligand pair can be selected according to the thermodynamic and structural properties of its complex (10,12).
The binding dynamics of aptamer-ligand systems can be described by three binding models, derived from the framework of protein-ligand interactions that describe folding tunnels and free-energy basins (Fig. 1) (6,13, 14, 15).
Figure 1.

Binding models of bioreceptors in the context of aptamer-ligand binding. (A–C) Lock and key (LAK) (A), conformational-selection (CS) (B), and induced-fit (C) binding models. Representation relates to the interaction of the Aptamer-2-40mer with methamphetamine. Binding constant equations are found in Eq. S1. To see this figure in color, go online.
The lock-and-key (LAK) binding model (Fig. 1 A) simplifies the binding interaction as between a rigid bio-receptor and ligand (16). Consequently, their binding interfaces are assumed to be perfectly matched. Accounting for this assumption in the case of aptamer-ligand interactions, the specific nucleotide strand is modeled as though it undergoes no structural change upon ligand addition. π stacking, van der Waals interactions, and hydrogen bonding between the aptamer and the ligand stabilize the complex formed (17).
The conformational-selection (CS) binding model (Fig. 1 B) acknowledges the inherent dynamic behavior of the aptamer in solution. Numerous conformational states coexist in equilibrium with different population distributions; the ligand can bind selectively to one of the conformational states, ultimately shifting the equilibrium toward this state (18).
The induced-fit (IF) binding model (Fig. 1 C) considers the aptamer to have a flexible binding site able to adopt the optimal binding conformation (or structural “switch”) for a ligand (19). Therefore, this model does not require the initial binding interfaces between the aptamer and ligand to match. The structural switch causes the formation of Watson-Crick or non-Watson-Crick base pairings in the aptamer structure, which enables the arrangement of a specific binding interface for the ligand. In addition to the aptamer-target interactions considered in the LAK binding model, the formation of new intra-aptamer hydrogen bonds is possible (13).
In reality, aptamers are known to retain structural plasticity and bind to their targets by changing their structure in accordance with the target structures (17). In seeking to develop aptamers for small-molecule sensing applications, a significant and measurable bio-recognition event is required. In this context, it is important to know whether the aptamer-ligand interaction is necessarily accompanied by a structural change. If so, the timing of the conformational change in relation to the binding event must be determined. Furthermore, small-molecule ligands, by virtue of their negligible size and few functional groups, may not induce a significant structural change. Finally, the focus on the dissociation constant (KD) for sensing experiments remains valuable, but the crucial requirement for detection by sensors is the measurable transduction of the bio-recognition event. Without specific and significant structural change, a reliable signal may not be detected.
Here, we propose an aptamer characterization workflow using spectroscopic and calorimetric methods coupled with in silico predictions to get a more complete understanding of the interaction between an aptamer and its small-molecule ligand. This workflow starts with the determination of the overall complex tertiary-structure parameters and concludes with an in-depth analysis of the specific region and binding interactions between the aptamer and the ligand. Altogether, this information allows us to propose a detailed binding mechanism for the interaction between a small molecule and a SELEX-generated aptamer.
We focus on an aptamer selected to bind methamphetamine (Meth), a controlled substance that produces a psychological and physical stimulant effect and can lead to dependency. The simple structure of Meth makes it relatively easy to produce illegally, which can also lead to significant issues of contamination in properties used for clandestine manufacturing (20).
The combination of nuclear magnetic resonance, circular dichroism, and isothermal titration calorimetry measurements allows us to propose a binding model for the new Meth aptamer and determine the main interactions during complex formation. Little structural change in the aptamer was detected, and a CS binding model was proposed. Entropically driven formation of the aptamer-Meth complex, combining hydrophobic and electrostatic interactions, was observed. Molecular-docking simulations support the findings made with nuclear magnetic resonance (NMR) spectroscopy and isothermal titration calorimetry (ITC).
This approach should be applicable to any aptamer selected for binding a small molecule, enabling a better structural understanding of the complex formed. In the context of aptasensor development, the structural information collected will help in the design and choice of the most suitable transduction platform.
Materials and methods
Materials
AuramerBio (Wellington, New Zealand) selected the 75-nucleotide-long Meth aptamer family via “Affinity Matrix Selex” (21). The parent aptamer 75-mer was produced by the SELEX technique, which was conducted via similar methods reported for the generation of an E2 binding aptamer (22). An affinity-based SELEX approach was used in which the Meth target was tethered to agarose beads. The selection buffer used for the SELEX had the same composition as binding washing buffer except for the presence of 0.05% Pluronic. Synthetic oligonucleotides were obtained from AlphaDNA (Montreal, Canada). DNA strands were purified by SDS-PAGE. The binding washing buffer (BWB) was tris(hydroxymethyl)aminomethane hydrochloride buffer (2 mM Tris-HCl, 10 mM NaCl, 0.5 mM KCl, 0.2 mM MgCl2, and 0.1 mM CaCl2; pH 7.5). Sodium 3-(trimethylsilyl)-1-propanesulfonate was used as an internal standard for NMR experiments (23). Before any experimentation, the aptamer was denatured at 95°C for 5 min, followed by cooling in an ice bath for 10 min.
SYBR I green assay
Each aptamer was mixed in BWB solution with the fluorescent dye SYBR I green (SG), then titration with Meth was conducted for each of them; the fluorescence signal was monitored during the Meth titration. Ten μL SG (7×) and 10 μL aptamer (10 μM) were mixed. A range of Meth concentrations varying from 0 to 1 μM was prepared in BWB and added directly to the SG-aptamer solution to deliver a final volume of 1000 μL. Fluorescence emission spectra were recorded from 500 to 650 nm using an excitation wavelength of 497 nm. The fluorescence emission of SG-stained DNA was measured at 520 nm.
Circular dichroism (CD) analysis
BWB solutions containing each aptamer with and without Meth were analyzed with a CD instrument. BWB solutions of each aptamer (1.5 μM) were prepared with and without Meth (3 μM). CD analyses were performed on a Chirascan Spectrometer in a 1-mm-path-length quartz cuvette. The spectra were recorded from 380 to 180 nm at 20°C at a rate of 200 nm/min and were corrected by subtraction of the background scan of the buffer.
1H NMR and nuclear Overhauser enhancement (NOESY) experiments
The aptamer sample was analyzed by 1H NMR and two-dimensional (2D) NOESY. DNA samples were dissolved in 527 μL BWB containing 10% D2O. The final concentration of DNA was 1 mM. An internal standard sodium 3-(trimethylsilyl)-1-propanesulfonate was added to the sample, and its final concentration was 1 mM. For spectra in the presence of Meth, the Meth concentration was fixed to 1 mM. NMR experiments were performed on a JEOL 600 MHz Nuclear Magnetic Resonance Spectrometer type JNM-ECZ600R with data recorded at 20°C. 1D proton NMR spectra were acquired with 3500 data points using 1280 scans due to low sample concentration. NOESY spectra were obtained using a mixing time of 200 ms (24). Solvent suppression was achieved by the “Watergate sequence” (25). Acquired data were processed and analyzed using MestReNova software (26).
ITC assay
Calorimetric experiments were conducted with the aptamer and a stem-mutated version. ITC assays were performed on an Affinity ITC system (TA Instruments, New Castle, DE, USA). One μL injections of a 2.5 mM Meth solution in BWB were added by a computer-controlled microsyringe at an interval of 240 s into the DNA aptamer solution (50 μM in BWB, cell volume 200 μL) with 100 Rpm stirring at 20°C. Heats produced by Meth dilution and aptamer dilution were evaluated and subtracted. The experimental data were fitted to a theoretical titration curve using software supplied by TA, with Gibbs free energy (ΔG), KD, and number of binding sites per monomer (n) as adjustable parameters.
Molecular modeling
Three plausible 2D structures of Aptamer-2-40mer were generated using Mfold and RNAstructure (27,28). The 2D structure, related to the lowest ΔG, was then imported to RNAComposer (29) to generate a 3D model, which was used in subsequent docking studies. The 2D structure of Meth was sketched on Maestro and prepared for docking by generating a low-energy 3D conformer using LigPrep (30). The program Genetic Optimization for Ligand Docking (31) was used for all molecular-docking analyses. The aptamer binding site was defined as a 100 Å sphere around the coordinates x = 0.3155, y = -26.7188, and z = 6.1465. Because Genetic Optimization for Ligand Docking’s genetic algorithm is stochastic in nature, an exhaustive approach was adopted such that 100 genetic-algorithm runs were set on the input conformation, giving 100 poses for further analysis. Search efficiency was set to 200% (highest accuracy). The ChemPLP (32) scoring function was used to score and rank poses, with a higher score corresponding to a pose having a relatively better “fit” to the cavity. The resulting 100 poses were clustered according to their respective geometries using the cluster_conformers.py script implemented in the Schrödinger 2019-2 package (33). The average linkage method was used for cluster generation, and the optimal number of clusters was automatically determined using the Kelley index (34). Finally, clusters were visually compared and merged based on their conformational similarity.
Results and discussion
The 75-nucleotide-long Meth aptamer family developed via Affinity Matrix SELEX comprised four structures: Aptamer-1, Aptamer-2, Aptamer-3, and Aptamer-4 (Table S1). At pH 7.5, the cationic form of Meth dominates (Fig. 2), making the ammonium part of the structure hydrophilic and electron deficient (red shading), while the phenylpropyl moiety is hydrophobic and electron rich (gray shading). The hypothetical interactions with the DNA strand of the aptamer will be electrostatic attractions with the cationic nitrogen, hydrogen-bond formation between the protonated amine and an atom of oxygen or nitrogen in the aptamer, and hydrophobic and π stacking with the phenyl ring (17,35).
Figure 2.

Skeletal formula of protonated methamphetamine (Meth) as dominant at pH 7.5. To see this figure in color, go online.
A fluorescence experiment (SG assay) was undertaken to compare the binding affinity of the four aptamers with Meth and to guide the choice between the four aptamers for further structural investigations (Fig. S1). The SG molecule emits a fluorescent light by interacting non-covalently with the duplex part of a DNA strand (36). The addition of the target molecule to the SG-aptamer complex causes the release of the SG molecule from the DNA structure induced by the interaction between the target and the DNA strand. As a consequence, a decrease of intensity of the fluorescent light can be monitored, and the interaction event between the aptamer and its target, as well as the resulting KD, can be deduced (37). Based on the best KD found with the SG assay (KD = 244.2 nM) and on the structural information acquired with CD experiments (Figs. 3 and S2), Aptamer-2 was chosen for a more thorough structural investigation.
Figure 3.

CD experiments for Aptamer-2 upon Meth addition. The positive ellipticity peak at 277 nm and the negative ellipticity peak at 249 nm are characteristic of a B-form structure. To see this figure in color, go online.
CD shows that Aptamer-2, in the presence or absence of Meth, has a B-form structure
The CD spectrum (Fig. 3) of Aptamer-2 reveals a positive ellipticity peak at 277 nm and a negative ellipticity peak at 249 nm, characteristic of a B-form structure containing a duplex region (38, 39, 40). This is assumed to be the most stable form of the aptamer at the conditions used for the experiment (41,42). Heterogeneous nucleotide DNA sequences typically adopt the B-form structure, in contrast to RNA strands that adopt an A-form structure (which produces a positive band at 260 nm and a negative band at 210 nm), or guanine-rich sequences that will preferentially adopt G-quadruplex structures (producing two positive bands at 215 and 295 nm and a negative band at 260 nm) (43). In our case, the addition of Meth to Aptamer-2 caused no shift or significant changes in peak intensity for the two distinguishable B-form peaks (Fig. 3). This indicates that Aptamer-2 retains its B-form conformation upon target addition. Regarding the binding models, this could indicate that the major secondary structural motif is preformed and is not dependent on the presence of the target, meaning the induced-fit binding model could be ruled out.
Computational predictions of the truncated Meth aptamer, Aptamer-2-40mer, disclose a potential secondary structure
To obtain simplified NMR spectra for analysis, a truncated aptamer variant (reduced from 75 to 40 nucleotides) was used: Aptamer-2-40mer. The non-essential parts (5′ and 3′ primers) of the original Aptamer-2 were removed. The primers are necessary to amplify target-bound sequences by PCR during the SELEX process. They do not belong to the randomized part of the aptamer and thus can be removed without significant effect on ligand binding (44). Additionally, they can convolute results by interacting non-specifically with the ligand, and so their removal is desirable for our subsequent studies.
For Aptamer-2-40mer, three secondary structures were predicted computationally by Mfold and RNAstructure (27,28). All three structures contained a hairpin fold with a duplex region. The form represented in Fig. 4 was calculated by Mfold to have the lowest ΔG at 20°C and at the specific buffer ionic strength used. Moreover, the structure shown in Fig. 4 is consistent with that predicted by Mfold for the original 75-mer version of the Aptamer-2. NMR analysis was undertaken to confirm the presence of the hairpin-loop structure.
Figure 4.

Representation of the Mfold-calculated lowest-energy Aptamer-2-40mer structure with the hairpin structure circled. To see this figure in color, go online.
NMR experiments with Aptamer-2-40mer partially identify the nucleotides belonging to the stem
The 1H NMR spectrum of Aptamer-2-40mer (Fig. S3) is typical of a partially double-stranded DNA sequence. Peaks observed in the 9–15 ppm region correspond to imino protons in the thymine and guanine bases (45,46). Due to proton exchange between imino protons and bulk solvent, the only imino protons typically observed are those involved in hydrogen bonding (H-bonding) through nucleotide base pairing, which can include Watson-Crick (Fig. 5) or non-Watson-Crick interactions, or are otherwise protected by structural folding (45, 46, 47). These types of interactions influence the tertiary structure that may contain a solvent-exposed pocket capable of binding proteins and small molecules (48,49). The resonance region of the Watson-Crick imino protons in an 1H NMR spectrum is 12–15 ppm, and the resonance region of non-Watson-Crick imino protons is 9–12 ppm (45,46).
Figure 5.

Watson-Crick basepairs A-T and G-C. To see this figure in color, go online.
Aptamer-2-40mer contains five Watson-Crick basepair interactions (two G-C and three A-T) according to the predicted Mfold structure (Fig. 4). Only thymine and guanine imino protons involved in this H-bonding are expected to be visible in the 1H NMR spectrum (Fig. 5). The three peaks observed between 13.2 and 13.8 ppm (Fig. 6 A) correspond closely in chemical shift to those observed previously for thymine imino protons (50) and could therefore represent the three thymine imino protons involved in Watson-Crick H-bondings in Aptamer-2-40mer (46). Furthermore, NOE correlations were observed between resonances 13.37 and 7.30, 13.49 and 7.82, and 13.54 and 7.26 ppm (Fig. 6 B). These are proposed to correspond to through-space interactions between the thymine N3H and the adenine C2H, which, in Watson-Crick base pairing, is known to be significant (45,51). Therefore, the 1H NMR and NOE results are consistent with the three A-T base pairs proposed by Mfold to be present in the Aptamer-2-40mer stem in the favored structural form (Fig. 4). Due to the similar chemical environment of thymines T2 and T4, their imino chemical shifts are expected to be very similar. By deduction, the two imino protons observed at 13.49 and 13.54 ppm are tentatively assigned as those of thymines T2 and T4 and, at 13.37 ppm, the imino proton of thymine T8.
Figure 6.

(A) Expansion of 1H NMR spectrum of Aptamer-2-40mer showing three thymine imino-proton peaks. (B) 2D NOESY correlations between the three imino protons and the three adenine C2 protons showing A-T base pairing. To see this figure in color, go online.
The two G-C pairs in the structure predicted by Mfold are not clearly visible in the 1H NMR spectrum. This may be due to their position at the ends of the stem, meaning they are more prone to solvent exchange than the three NH protons of the thymines positioned in the middle of the stem. A weak resonance observed at 12.97 ppm may represent one imino NH from a guanine in the stem. This cannot be confirmed by NOE correlations in G-C basepairs due to the lack of a formamidine proton (or other similarly sited proton) in cytosine (45). The other chemical groups involved in H-bonding are the amino groups of adenine, guanine, and cytosine (Fig. 5), and these are observed in the 7.5–8.5 ppm region (Fig. S5) (45,46).
This NMR-based structural assignment partially confirms the secondary structure predicted by Mfold in that the stem composition of three adenine-thymine Watson-Crick basepairs correspond to the spectral observations.
1H NMR spectroscopy reveals new imino-proton signals upon Meth binding to Aptamer-2-40mer
1H NMR spectroscopy was used to investigate the binding mechanism and elucidate which part of Aptamer-2-40mer is involved in target recognition.
A comparison of the 1H NMR spectra of Aptamer-2-40mer in the absence and presence of Meth (saturated, 1 M equivalent) shows the formation of four new imino resonances (peaks 5, 6, 7, and 8 in Fig. 7) in the chemical-shift region of H-bonding for non-Watson-Crick basepairs (46). This is in line with studies by Neves et al. (50) and Jenison et al. (1) in which ligand addition caused additional resonances to appear, indicating that binding of Meth either induces a conformational change in the aptamer or stabilizes a Meth-bound aptamer conformation that otherwise exists in dynamic equilibrium with other unbound aptamer conformations.
Figure 7.

1H NMR spectra of Aptamer 2-40mer with molar loadings of Meth 0% and 100%. To see this figure in color, go online.
Upon addition of 1 mole equivalent Meth, there was an apparent reduction in the intensity of peak 1 (assigned to the thymine imino proton involved in A-T2 or A-T4) relative to the other A-T resonances. This could be interpreted as a weakening of one of the two A-T interactions in the stem caused by Meth addition.
The appearance of peaks 5, 6, 7, and 8 (Fig. 7), residing in the resonance region of imino protons not involved in Watson-Crick basepairs, leads to several hypotheses (vide infra). ITC was used, as described in the next section, to provide thermodynamics parameters to enable a better understanding about the creation of these new peaks and to propose a binding model related to the Meth-Aptamer-2-40mer complex formation.
ITC confirms the importance of the stem and entropically driven binding
ITC was used for collecting thermodynamic properties and binding affinities related to Aptamer-2-40mer-Meth binding. Comparison of the original Aptamer-2-40mer with a mutated Aptamer-2-40mer sequence, where the base-paired stem is not able to form, highlights the importance of the stem and the resulting loop for binding. As shown in Fig. 8 A, the reaction is exothermic (heat release upon target addition) for the Aptamer-2-40mer, whereas the mutant lacking the ability to form a stem shows no variation of the calorimetric signal upon target addition in Fig. 8 B.
Figure 8.

ITC results for the Aptamer-2-40mer and the mutated Aptamer-2-40mer. (A and B) The heat profiles for (A) Aptamer-2-40mer and (B) mutated Aptamer-2-40mer are represented. (C and D) The integrated heat profiles are represented for (C) Aptamer-2-40mer and (D) mutated Aptamer-2-40mer. The heat profiles presented have been corrected for the heat of dilution of the titrant and for the heat of dilution of the aptamer. To see this figure in color, go online.
The mutated Aptamer-2-40mer was unable to bind with Meth (Figs. 8 B and D), which was attributable to the fact that the two remaining Watson-Crick basepairs cannot stabilize the stem formed in Aptamer-2-40mer. This experiment shows that disrupting the stem led to a full loss of binding, allowing us to conclude that the stem is important to the specific binding event.
The enthalpic signal is weak and slightly exothermic (favorable contribution to the overall free energy of the aptamer-Meth complex), indicating enthalpic competition between the disruption of non-covalent bonds (hydrogen bonds between the surrounding water and the aptamer surface) and the formation of new hydrogen bonds during the interaction between the ligand and the aptamer (13). The weak calorimetric signal makes it hard to reliably extract KD and entropy change (ΔS), but in the absence of a strong favorable enthalpic term, binding between Aptamer-2-40mer and Meth must be entropically driven.
The entropic component makes a favorable contribution to the overall free energy of the aptamer-Meth complex. The dominant contributors to the entropic effect could be the release of ordered solvated water molecules and counter ions during ligand binding (52,53). A large favorable entropy component has previously been observed in hydrophobic binding interactions (13,54, 55, 56). Indeed, when ligand binding is associated with a large water-gain entropy, the energy decrease (related to the attractive interactions between the aptamer and the Meth) is canceled out by the energy increase originating from the energetic dehydration effect upon binding (52). Given the complementary charges of the cationic Meth ligand and the anionic phosphate backbone of Aptamer-2-40mer, we can suggest that an electrostatic attraction may help to capture Meth molecules and position them near the stem region, where hydrophobic interactions can be manifest. A related thermodynamic model (unfavorable binding enthalpy with a favorable binding entropy) has been previously proposed for the adsorption of the anionic surface of cytochrome b5 on a positively charged anion-exchange chromatography (57). In both cases, an unfavorable overall binding enthalpy is compensated by a strong entropic contribution, and electrostatic interactions influence the nature of binding.
Combination of spectroscopic and calorimetric data to establish a CS binding model
By linking the results obtained from CD, NMR, and ITC measurements, a binding model can be used to explain the Aptamer-2-40mer interaction with Meth. The three main binding models related to a bio-receptor-ligand interaction are displayed in Fig. 1.
From the 1H NMR experiment, the creation of four imino-proton peaks not involved in Watson-Crick basepairs upon Meth addition was observed (peaks 5, 6, 7, and 8 in Fig. 7). The formation of the new peaks in the 1H NMR spectrum could result from intra-aptamer H-bonding (consistent with IF or CS binding models) or from other imino-proton resonances such as H-bond of Meth-Aptamer, NH of Aptamer only, or NH2 of Meth only (consistent with IF, CS, and LAK binding models). Two scenarios for the formation of these peaks can be considered.
-
(1)
Meth is a hydrogen-bond donor (Fig. 2), and, thus, one hydrogen bond (H-bond) may be formed between Meth and an oxygen or a nitrogen atom of a nucleotide base. This H-bond may have a chemical shift in the region of 10–11 ppm, which is the region of the imino protons not involved in Watson-Crick basepairs. In the case of a theophylline aptamer, Zimmermann et al. (58) observed the formation of a pseudo basepair between the cytosine and the purine-like theophylline in the appearance of an H-bonding resonance in the region of 15 ppm. The Meth H-bonding interaction with an aptamer nucleotide will resonate in a more upfield region due to the lower electronegative environment provided by the Meth molecule. In addition, one of the Watson-Crick basepairs from the stem might be disrupted upon Meth addition, and, therefore, one of the imino protons remains free in solution. It is still structurally protected by the stem from solvent-exchange effects; thus, it is visible in the 1H NMR spectrum and resonates in the non-Watson-Crick region. Two signals are expected to appear in the 10.0–11.5 ppm region.
-
(2)
No H-bond is formed between the Meth and the aptamer; however, the amine group can be resolved because Meth may interact with the aptamer in a specific cavity where the protonated amine group of Meth is protected from solvent exchange. The cationic amine group of Meth has two hydrogens that are not visible in the 1H NMR spectrum of the unbound ligand due to proton exchange with the aqueous environment but may become visible in the bound form (Fig. S4). Neves et al. (50) showed that, when bound with its aptamer, the cocaine imino proton became visible in the 10 ppm region of the 1H NMR spectrum. A similar effect may also occur with Meth amino protons. Indeed, if Meth interacts with a specific cavity of the aptamer, proton exchange with the aqueous environment would similarly decrease due to the structural protection of the aptamer and possibly resolve the two amino proton resonances. The electron demand is similar for Meth and cocaine; nevertheless, as Meth is a secondary amine and cocaine a tertiary amine, the chemical shift will differ slightly. One of the Watson-Crick basepairs from the stem may be disrupted, and, therefore, one of the imino protons is free in solution (visible as explained in the first scenario) and resonates in this region. Three signals are expected to appear in the 10.0–11.5 ppm region.
Neither of these scenarios explain the four new NMR peaks observed. Consequently, some of the four peaks observed may be related to intra-aptamer H-bonds, and, thus, this indicates that different structures of the aptamer are present in solution before and after target addition. Consequently, the LAK binding model is ruled out. The structural observations obtained with NMR are in line with the CS and IF binding models.
A main difference between the CS and IF binding models is the number of conformational states present initially in solution (6,13, 14, 15). The IF binding model assumes an initial non-binding conformation present in solution; by adding the target to a 1:1 molar ratio (aptamer:target), it is considered that a new binding state is present and that no non-binding conformation remains in solution. From a thermodynamic perspective, the IF binding model would require a strong attractive interaction (i.e., enthalpic) to drive this structural change (13,59,60). This is inconsistent with the lack of enthalpic attraction observed in the ITC experiment, so we can suppose that the IF binding model for the Aptamer-2-40mer-Meth interaction can be ruled out.
Moreover, the CD experiments support this suggestion because no significant change in the duplex structure upon ligand addition is observed (Fig. 3). Furthermore, in the 1H NMR experiment, we observed that the three peaks (1, 2, and 3 in Fig. 6) related to the stem are present before the target addition. Despite the modest decrease in intensity of peak 1, all are still present after the target addition. No new resonance peaks related to Watson-Crick basepair formation are visible after Meth addition. These two observations provide additional evidence that no major aptamer structural changes occur upon Meth addition.
Finally, the CS binding model is considered. Different conformations of the aptamer exist in a dynamic equilibrium in solution as shown in Fig. 1 B (13,14,61). Meth binds specifically to one of these conformations. At 0% Meth, the aptamer conformations are in a dynamic equilibrium where k′rev > k′for (unbound conformation is favored and thus is observed at 0% Meth). The addition of Meth causes the binding state to be favored in solution k′on > k′rev; therefore, the 1H NMR spectrum, upon addition of one equivalent (saturated), represents the binding state with additional non-Watson-Crick resonances. Because the loop region contains enough guanine or thymine residues to form non-Watson-Crick basepairs, it is hypothesized that these interactions are situated in the loop region. ITC experiments support this hypothesis, as no signal is measured for the mutated Aptamer-2-40mer where the loop is not formed. Furthermore, in support of the thermodynamic data collected, a mixture of binding and non-binding state conformations is present in solution. By adding the target, the equilibrium between the two states shifts to the binding state conformation. Compared with the IF binding mode, the equilibrium shift is less significant due to the initial presence of a binding state aptamer. Thermodynamically, this equilibrium switch may not afford a strong attractive interaction (enthalpic in contrast to the IF binding mode) and, thus, is consistent with an entropically driven binding event.
Aptamers following the IF binding model have already been published in scientific literature, e.g., the cocaine aptamer MN19 (ligand-induced folding process) shows great imino-proton chemical-shift differences among cocaine addition on NMR data combined with a significant enthalpically driven reaction (50). Another example is the ATP aptamer presented by Lin et al. (62). They showed that the ATP aptamer binds two AMP molecules following the adaptive binding model. Significant imino-proton-intensity differences have been observed. These two examples contrast with our Meth aptamer, where the prominent unbound aptamer form is different than the bound aptamer form.
Molecular modeling supports the proposed binding structure
Following the experimental determination of Meth binding to the Aptamer-2-40mer, molecular docking was used to rationalize the proposed binding hypotheses.
First, a 3D model of the Aptamer-2-40mer was constructed using RNAcomposer (29). Mfold (27) and RNAstructure (28) provided a 2D structure corresponding to experimental observations (27,28). A stem composed of the three A-T base pairs is predicted and conforms to the NMR experimental evidence. Additionally, with the RNAstructure software, the partition-function calculation (63) predicted basepair probabilities. By using the partition function for the Aptamer-2-40mer, it appears that the five basepairs predicted by Mfold and represented in the Aptamer-2-40mer structure (Fig. 4) have a high probability of formation (Fig. S5). Finally, the 2D structure and DNA sequence have been used to generate the 3D structure with RNAcomposer.
Exhaustive docking of Meth revealed several binding poses homogeneously distributed within the major groove of the stem region (Fig. 9 A). All poses predict the placement of the cationic-amine group of Meth near C5, T4, A′ (a nucleotide within the loop), T′′ (a nucleotide within the loop), T‴ (a nucleotide within the loop), G6, and the aromatic ring near T2, A3, and A7. Conformational clustering of the 100 poses gave two distinct clusters: the highest scored was least populated, with 12 poses (C1), while the most populated (C2) contained 88 poses that scored slightly worse (see Fig. 9 B for score distributions). Analysis of C1 (Fig. 9 C) shows the cationic amine of Meth forming a salt bridge with the T″ phosphate backbone. The remainder of the Meth framework (from methyl-9 to the aromatic ring) resides along a column of polar moieties from the surrounding bases (G6 carbonyl, C5 amine, T4 carbonyl, A7 amine, A3 amine, T8 carbonyl). For C2 (Fig. 9 D), the cationic amine retains the ionic bond with the T″ phosphate backbone, but the methyl-9 of Meth is positioned toward the T‴ thymine-methyl group rather than the T4 thymine methyl, as seen in C1. Overall, the docking study suggests that Meth could favorably occupy the pocket by forming ionic and hydrophobic interactions with the surrounding environment. These findings are in line with those experimentally deduced, and both highlight the importance of the stem region for Meth binding. The predicted binding modes may also support the hypothesis that the magnitude change of peak 1 in the 1H NMR spectrum upon Meth treatment (in Fig. 7) corresponds to partial disruption of H-bonding between A-T2 or A-T4 basepairs.
Figure 9.

Molecular docking of Meth with Aptamer-2-40mer. (A) All 100 docking poses of Meth in the hydrophobic pocket are shown in stick form, and the aptamer is shown in cartoon form with a transparent surface rendered. (B) A violin plot of the docking scores of each cluster. (C) The 12 poses of cluster_1 C1 (Meth in stick form) in the pocket, with surrounding nucleotides shown in wire form. (D) The 88 poses of cluster_2 C2 (Meth in stick form) in the pocket with surrounding nucleotides shown in wire form. To see this figure in color, go online.
Furthermore, the hydrophobic and ionic interactions between the phosphate backbone and Meth correlate with the thermodynamic parameters obtained by ITC (Fig. 8).
Conclusions
Through a combination of CD, NMR, and ITC experiments, a binding mechanism for the Meth-Aptamer-2-40mer complex is proposed and supported by molecular-modeling simulations. Specifically, no significant structural change has been detected with CD experiments upon addition of Meth. NMR experiments indicate a change in the imino region for Aptamer-2-40mer in solution upon Meth addition that is proposed to consist of a form where new non-Watson-Crick basepairs are present in the loop (potentially arising through a CS binding process). Moreover, a partial weakening of one of the A-T2 or A-T4 Watson-Crick pairs is tentatively interpreted from the NMR spectra to occur upon target addition. ITC experiments demonstrate the importance of the stem for the binding event. Indeed, by removing the stem, no binding event occurs. Thermodynamic parameters collected with ITC provided information suggesting an entropically driven binding event. Molecular modeling supports the proposed hypotheses in the preferred positions of Meth calculated to be within the major groove of the stem and with interactions to the phosphate backbone of the loop.
This approach may be suited to study of other aptamer-small molecule complexes as a characterization protocol prior to development of an aptasensor platform. Following the findings related to the binding region localization as well as the structural-switch magnitude of the aptamer upon the target addition, the best transduction strategy needs to be considered. The small magnitude of the conformational switch and accompanying enthalpic change indicate that Affinity Matrix SELEX may not provide the optimal aptamer structures for detection of monofunctional small-molecule targets. Other SELEX protocols must be investigated for this purpose.
Author contributions
C.S. wrote the paper, conceived, designed, and collected the data for the SG, CD, and ITC analyses; J.A.J.M. conceived, designed, and collected the data for the molecular-modeling prediction; A.S. contributed to interpretation and revised the manuscript; I.V. performed and contributed to the NMR data analysis; J.E.H. contributed to the NMR data interpretation and the writing of the paper; J.M.H. supervised the project and contributed to the writing and revision of the manuscript.
Acknowledgments
This work was supported through a grant from the New Zealand’s Ministry of Business, Innovation and Employment (grant number E2933/3487).
Declaration of interests
The authors declare no competing interests.
Editor: Elsa C.Y. Yan.
Footnotes
Supporting material can be found online at https://doi.org/10.1016/j.bpj.2022.04.027.
Supporting material
Data availability
All aptamer sequences used in the study are reported in the supporting material.
References
- 1.Jenison R.D., Gill S.C., et al. Polisky B. High-resolution molecular discrimination by RNA. Science. 1994;263:1425–1429. doi: 10.1126/science.7510417. [DOI] [PubMed] [Google Scholar]
- 2.Tuerk C., Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 3.Ellington A.D., Szostak J.W. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 4.Jayasena S.D. Aptamers: an emerging class of molecules that rival antibodies in diagnostics. Clin. Chem. 1999;45:1628–1650. doi: 10.1093/clinchem/45.9.1628. [DOI] [PubMed] [Google Scholar]
- 5.Zhou J., Rossi J. Aptamers as targeted therapeutics: current potential and challenges. Nat. Rev. Drug Discov. 2017;16:181–202. doi: 10.1038/nrd.2016.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Munzar J.D., Ng A., Juncker D. Comprehensive profiling of the ligand binding landscapes of duplexed aptamer families reveals widespread induced fit. Nat. Commun. 2018;9:343. doi: 10.1038/s41467-017-02556-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feagin T.A., Maganzini N., Soh H.T. Strategies for creating structure-switching aptamers. ACS Sens. 2018;3:1611–1615. doi: 10.1021/acssensors.8b00516. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Z., Oni O., Liu J. New insights into a classic aptamer: binding sites, cooperativity and more sensitive adenosine detection. Nucleic Acids Res. 2017;45:7593–7601. doi: 10.1093/nar/gkx517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilson B.D., Hariri A.A., et al. Soh H.T. Independent control of the thermodynamic and kinetic properties of aptamer switches. Nat. Commun. 2019;10:5079. doi: 10.1038/s41467-019-13137-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiao Y., Lubin A.A., et al. Plaxco K.W. Label-free electronic detection of thrombin in blood serum by using an aptamer-based sensor. Angew. Chem. 2005;117:5592–5595. doi: 10.1002/ange.200500989. [DOI] [PubMed] [Google Scholar]
- 11.Stojanovic M.N., de Prada P., Landry D.W. Aptamer-based folding fluorescent sensor for cocaine. J. Am. Chem. Soc. 2001;123:4928–4931. doi: 10.1021/ja0038171. [DOI] [PubMed] [Google Scholar]
- 12.Baker B.R., Lai R.Y., et al. Plaxco K.W. An electronic, aptamer-based small-molecule sensor for the rapid, label-free detection of cocaine in adulterated samples and biological fluids. J. Am. Chem. Soc. 2006;128:3138–3139. doi: 10.1021/ja056957p. [DOI] [PubMed] [Google Scholar]
- 13.Du X., Li Y., et al. Liu S.-Q. Insights into protein-ligand interactions: mechanisms, models, and methods. Int. J. Mol. Sci. 2016;17:144. doi: 10.3390/ijms17020144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma B., Kumar S., et al. Nussinov R. Folding funnels and binding mechanisms. Protein Eng. 1999;12:713–720. doi: 10.1093/protein/12.9.713. [DOI] [PubMed] [Google Scholar]
- 15.Csermely P., Palotai R., Nussinov R. Induced fit, conformational selection and independent dynamic segments: an extended view of binding events. Trends Biochem. Sci. 2010;35:539–546. doi: 10.1038/npre.2010.4422.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer E. Einfluss der Configuration auf die Wirkung der Enzyme. Berichte der deutschen chemischen Gesellschaft. 1894;27:2985–2993. doi: 10.1002/cber.18940270364. [DOI] [Google Scholar]
- 17.Hermann T., Patel D.J. Adaptive recognition by nucleic acid aptamers. Science. 2000;287:820–825. doi: 10.1126/science.287.5454.820. [DOI] [PubMed] [Google Scholar]
- 18.Monod J., Wyman J., Changeux J.-P. On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 19.Koshland D.E. Application of a theory of enzyme specificity to protein synthesis. Proc. Natl. Acad. Sci. U S A. 1958;44:98–104. doi: 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gluckman P. (2018). Office of the Prime Minister's Chief Science Advisor. Methamphetamine contamination in residential properties: Exposures, risk levels, and interpretation of standards.
- 21.Li S., Clarkson M., McNatty K. Selection and characterisation of triclosan-specific aptamers using a fluorescence microscope-imaging assay. Anal. Bioanal. Chem. 2020;412:7285–7294. doi: 10.1007/s00216-020-02863-7. [DOI] [PubMed] [Google Scholar]
- 22.Alsager O.A., Kumar S., et al. Hodgkiss J.M. Ultrasensitive colorimetric detection of 17β-estradiol: the effect of shortening DNA aptamer sequences. Anal Chem. 2015;87:4201–4209. doi: 10.1021/acs.analchem.5b00335. [DOI] [PubMed] [Google Scholar]
- 23.Wishart D.S., Bigam C.G., et al. Sykes B.D. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR. 1995;6:135–140. doi: 10.1007/bf00211777. [DOI] [PubMed] [Google Scholar]
- 24.Jeener J., Meier B.H., et al. Ernst R.R. Investigation of exchange processes by two-dimensional NMR spectroscopy. J. Chem. Phys. 1979;71:4546–4553. doi: 10.1063/1.438208. [DOI] [Google Scholar]
- 25.Piotto M., Saudek V., Sklenář V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR. 1992;2:661–665. doi: 10.1007/bf02192855. [DOI] [PubMed] [Google Scholar]
- 26.Willcott M.R. MestRe Nova. J. Am. Chem. Soc. 2009;131:13180. doi: 10.1021/ja906709t. [DOI] [Google Scholar]
- 27.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reuter J.S., Mathews D.H. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 2010;11:129. doi: 10.1186/1471-2105-11-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Popenda M., Szachniuk M., et al. Adamiak R.W. Automated 3D structure composition for large RNAs. Nucleic Acids Res. 2012;40:e112. doi: 10.1093/nar/gks339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schrödinger . Schrödinger Release 2019-2: LigPrep. Schrödinger, LLC; New York, NY: 2019. [Google Scholar]
- 31.Jones G., Willett P., et al. Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 32.Korb O., Stützle T., Exner T.E. Empirical scoring functions for advanced Protein−Ligand docking with PLANTS. J. Chem. Inf. Model. 2009;49:84–96. doi: 10.1021/ci800298z. [DOI] [PubMed] [Google Scholar]
- 33.Schrödinger . Schrödinger Release 2019-2: Maestro. Schrödinger, LLC; New York, NY: 2019. [Google Scholar]
- 34.Kelley L.A., Gardner S.P., Sutcliffe M.J. An automated approach for clustering an ensemble of NMR-derived protein structures into conformationally related subfamilies. Protein Eng. Des. Selection. 1996;9:1063–1065. doi: 10.1093/protein/9.11.1063. [DOI] [PubMed] [Google Scholar]
- 35.Cai S., Yan J., et al. Liu Z. Investigations on the interface of nucleic acid aptamers and binding targets. Analyst. 2018;143:5317–5338. doi: 10.1039/c8an01467a. [DOI] [PubMed] [Google Scholar]
- 36.Zipper H., Brunner H., et al. Vitzthum F. Investigations on DNA intercalation and surface binding by SYBR Green I, its structure determination and methodological implications. Nucleic Acids Res. 2004;32:e103. doi: 10.1093/nar/gnh101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kong L., Xu J., et al. Chai Y. A universal and label-free aptasensor for fluorescent detection of ATP and thrombin based on SYBR Green I dye. Biosens. Bioelectron. 2013;42:193–197. doi: 10.1016/j.bios.2012.10.064. [DOI] [PubMed] [Google Scholar]
- 38.Watson J.D., Crick F.H.C. Molecular structure of nucleic acids: a structure for deoxyribose nucleic acid. Nature. 1953;171:737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- 39.Gray D.M., Ratliff R.L., Vaughan M.R. Circular dichroism spectroscopy of DNA. Methods Enzymol. 1992;211:389–406. doi: 10.1016/0076-6879(92)11021-a. [DOI] [PubMed] [Google Scholar]
- 40.Bishop G.R., Chaires J.B. Characterization of DNA structures by circular dichroism. Curr. Protoc. Nucleic Acid Chem. 2002;11:7–11. doi: 10.1002/0471142700.nc0711s11. [DOI] [PubMed] [Google Scholar]
- 41.Gondeau C., Maurizot J.C., Durand M. Circular dichroism and UV melting studies on formation of an intramolecular triplex containing parallel T∗A:T and G∗G:C triplets: netropsin complexation with the triplex. Nucleic Acids Res. 1998;26:4996–5003. doi: 10.1093/nar/26.21.4996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bing T., Zheng W., et al. Shangguan D. Triplex-quadruplex structural scaffold: a new binding structure of aptamer. Sci. Rep. 2017;7:15467. doi: 10.1038/s41598-017-15797-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vorlickova M., Kejnovska I., et al. Kypr J. Circular dichroism spectroscopy of DNA: from duplexes to quadruplexes. Chirality. 2012;24:691–698. doi: 10.1002/chir.22064. [DOI] [PubMed] [Google Scholar]
- 44.Li H.-H., Wen C.-Y., et al. Lai J.-C. Evaluation of aptamer specificity with or without primers using clinical samples for C-reactive protein by magnetic-assisted rapid aptamer selection. RSC Adv. 2017;7:42856–42865. doi: 10.1039/c7ra07249j. [DOI] [Google Scholar]
- 45.Fürtig B., Richter C., et al. Schwalbe H. NMR spectroscopy of RNA. ChemBioChem. 2003;4:936–962. doi: 10.1002/cbic.200300700. [DOI] [PubMed] [Google Scholar]
- 46.Wijmenga S.S., van Buuren B.N.M. The use of NMR methods for conformational studies of nucleic acids. Prog. Nucl. Magn. Reson. Spectrosc. 1998;32:287–387. doi: 10.1016/s0079-6565(97)00023-x. [DOI] [Google Scholar]
- 47.Mirau P.A., Smith J.E., et al. Naik R. Structured DNA aptamer interactions with gold Nanoparticles. Langmuir. 2018;34:2139–2146. doi: 10.1021/acs.langmuir.7b02449. [DOI] [PubMed] [Google Scholar]
- 48.Leontis N.B., Stombaugh J., Westhof E. The non-Watson-Crick base pairs and their associated isostericity matrices. Nucleic Acids Res. 2002;30:3497–3531. doi: 10.1093/nar/gkf481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hermann T., Westhof E. Non-Watson-Crick base pairs in RNA-protein recognition. Chem. Biol. 1999;6:R335–R343. doi: 10.1016/s1074-5521(00)80003-4. [DOI] [PubMed] [Google Scholar]
- 50.Neves M.A.D., Slavkovic S., et al. Johnson P.E. Salt-mediated two-site ligand binding by the cocaine-binding aptamer. Nucleic Acids Res. 2017;45:1041–1048. doi: 10.1093/nar/gkw1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin C.H., Wang W., et al. Patel D.J. Formation of an amino-acid-binding pocket through adaptive zippering-up of a large DNA hairpin loop. Chem. Biol. 1998;5:555–572. doi: 10.1016/s1074-5521(98)90114-4. [DOI] [PubMed] [Google Scholar]
- 52.Hayashi T., Oshima H., et al. Kinoshita M. Binding of an RNA aptamer and a partial peptide of a prion protein: crucial importance of water entropy in molecular recognition. Nucleic Acids Res. 2014;42:6861–6875. doi: 10.1093/nar/gku382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Potty A.S.R., Kourentzi K., et al. Willson R.C. Biophysical characterization of DNA and RNA aptamer interactions with hen egg lysozyme. Int. J. Biol. Macromolecules. 2011;48:392–397. doi: 10.1016/j.ijbiomac.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 54.Sakamoto T., Ennifar E., Nakamura Y. Thermodynamic study of aptamers binding to their target proteins. Biochimie. 2018;145:91–97. doi: 10.1016/j.biochi.2017.10.010. [DOI] [PubMed] [Google Scholar]
- 55.Fisher H.F., Singh N. Vol. 259. Academic Press; 1995. Methods in Enzymology; pp. 194–221. [Google Scholar]
- 56.Archer W.R., Schulz M.D. Isothermal titration calorimetry: practical approaches and current applications in soft matter. Soft Matter. 2020;16:8760–8774. doi: 10.1039/d0sm01345e. [DOI] [PubMed] [Google Scholar]
- 57.Gill D.S., Roush D.J., Willson R.C. Presence of a preferred anion-exchange binding site on cytochrome b5: structural and thermodynamic considerations. J. Chromatogr. A. 1994;684:55–63. doi: 10.1016/s0021-9673(94)89132-x. [DOI] [PubMed] [Google Scholar]
- 58.Zimmermann G.R., Wick C.L., et al. Pardi A. Molecular interactions and metal binding in the theophylline-binding core of an RNA aptamer. RNA. 2000;6 doi: 10.1017/s1355838200000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li H., Xie Y., et al. Liu S. Physicochemical bases for protein folding, dynamics, and protein-ligand binding. Sci. China Life Sci. 2014;57:287–302. doi: 10.1007/s11427-014-4617-2. [DOI] [PubMed] [Google Scholar]
- 60.Chang C.-E., Gilson M.K. Free energy, entropy, and induced fit in Host−Guest recognition: calculations with the second-generation mining minima algorithm. J. Am. Chem. Soc. 2004;126:13156–13164. doi: 10.1021/ja047115d. [DOI] [PubMed] [Google Scholar]
- 61.Paul F., Weikl T.R. How to distinguish conformational selection and induced fit based on chemical relaxation rates. PLoS Comput. Biol. 2016;12:e1005067. doi: 10.1371/journal.pcbi.1005067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lin C.H., Patei D.J. Structural basis of DNA folding and recognition in an AMP-DNA aptamer complex: distinct architectures but common recognition motifs for DNA and RNA aptamers complexed to AMP. Chem. Biol. 1997;4:817–832. doi: 10.1016/s1074-5521(97)90115-0. [DOI] [PubMed] [Google Scholar]
- 63.Mathews D.H. Using an RNA secondary structure partition function to determine confidence in base pairs predicted by free energy minimization. RNA. 2004;10:1178–1190. doi: 10.1261/rna.7650904. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All aptamer sequences used in the study are reported in the supporting material.