

Abstract
During the past decade, artemisinin as an antimalarial has been in the spotlight, in part due to the Nobel Prize in Physiology or Medicine awarded to Tu Youyou. While many studies have been completed detailing the significant increase in activity resulting from the dimerization of natural product artemisinin, activity increases unaccounted for by the peroxide bridge have yet to be researched. Here we outline the synthesis and testing for antimalarial activity of artemisinin dimers in which the peroxide bridge in one half of the dimer is reduced, resulting in a dimer with one active and one deactivated artemisinin moiety.
Keywords: antiplasmodium, antimalarial, artemsinin, endoperoxide, artemisinin dimers
Table of Contents/Abstract Graphics
Artemisinin (1) was isolated in 1972 from Artemisia annua which has been used in Chinese herbal medicine for over 2000 years.1 Since then, various derivatives of the compound have been synthesized, including dihydroartemisinin (DHA) (2a), artemether (2b), arteether (2c) and artesunate (2d),2 all of which retain activity against drug-resistant strains of Plasmodium falciparum (Figure 1).3 For this reason, Artemisinin, or one of its derivatives, are one component of artemisinin-based combination therapies (ACTs) which currently stand as frontline treatment for acute, uncomplicated P. falciparum malaria.4 Artemisinins also have been a source of inspiration for the trioxolane antimalarial arterolane, also known as OZ277 and OZ439, that were discovered and developed by Vennerstrom and colleagues.5, 6 Significant interest has been generated over the outstanding activity of artemisinin dimers (Figure 1). Posner, O’Neill and others have produced dozens of analogues of the C-10 deoxoartemisinin dimer displaying impressive potency.7–11 The noteworthy increase in activity of some of the dimers over artemisinin monomers has led our research to investigate the activity of dimers in which one peroxide bridge is intact while the other is deactivated by reduction. For comparative studies, we synthesized a set of symmetric artemisinin dimers containing two endoperoxide bridges and an analogous dimer series containing only one peroxide bridge, to which we refer herein as the asymmetric artemisinin dimers. In order to study the additive effect to activity of dimerized artemisinin, our laboratory optimized methodology to covalently dimerize these molecules utilizing EDCI coupling, click chemistry, or Mitsunobu reactions. Using these methods, we were able to synthesize artemisinin dimers in a controlled manner, thereby allowing us to ascertain the impact of the peroxide bridge number, the linker polarity, and/or linker length on the potency of the dimers against asexual blood stages of the human malaria parasite Plasmodium falciparum.
Figure 1.

Artemisinin (1), its derivatives (2), and trioxolanes OZ277 and OZ439
Synthetic Chemistry
Previous research notes the short half-life and limited solubility of artemisinin derivatives is primarily due to the instability of the C-10 acetal linkage. Metabolism of artemisinin derivatives, mediated by cytochrome P450, identified the dealkylation to DHA as a major pathway,12 suggesting that our analogues should focus on C-10 deoxoartemisinin monomers and dimers to improve stability.13, 14
The synthesis of 10β-deoxoartemisinin (4) is shown in Scheme 1. Commercially available artemisinin was reduced to DHA (2a) with DIBAL, followed by acylation with benzoyl chloride to give ester 3. Reaction of 3 with allyl trimethylsilane and zinc chloride gave 10β-allyldeoxoartemisinin (4) (Scheme 1).15
Scheme 1.

Synthesis of 10β-allyldeoxoartemisinina
aReagents and conditions: (i) DIBAL, DCM, –78 °C, 2 h, quant.; (ii) BzCl, py, DCM, 0 °C, 16 h, 97%; (iii) ally-TMS, ZnCl2, 1,2-DCE, 4Å MS, 0 °C, 3 h, 81%.
A significant amount of optimization was required for the synthesis of 10β-allyldeoxoartemisinin due to the competing elimination reaction. Initially we followed a published protocol15, and when any versions of anhydrous zinc chloride that was not sealed in an ampoule were used, the major product was 5 as a result of the elimination pathway (Scheme 1).
Alkene 4 was diverged into 3-carbon chain lengths by submitting to hydroboration of the alkene with borane dimethyl sulfide followed by reduction of the organoborane with sodium perborate to give 6a. Compound 6a was further subjected to reduction of the endoperoxide bridge with zinc powder and acetic acid to give deoxyartemisinin alcohol 6b (Scheme 2).15
Scheme 2.

Synthesis of deactivated deoxoartemisinina
aReagents and conditions: (i) 1. BH3·SMe2, t-butyl methyl ether, 0 °C, 24 h; 2. NaBO3·4H2O, t-butyl methyl ether/H2O, rt, 24 h, 69%; (ii) Zn, AcOH, rt. 72 h, 90%.
One of our first goals was to synthesize artemisinin monomers functionalized with clickable azide and alkyne handles in order to utilize copper- or ruthenium-catalyzed azide-alkyne cycloaddition (CuAAC16 or RuAAC17, 18) to effectively dimerize artemisinin. In order to synthesize artemisinin monomers with azide functionality, alcohols 6a and 6b were subjected to mesylation to give 7a and 7b, respectively.15 Nucleophilic substitution with sodium azide gave artemisinin-azide monomer 8a and 8b, which were reduced with triphenylphosphine to give amines 9a and 9b, respectively. Two series of clickable alkyne handles were synthesized, also utilizing alcohols 6a and 6b as a diverging point. Mesyl esters 7a and 7b were substituted with propargyl alcohol to give propargyl ether monomers 11a and 11b. Alcohols 6a and 6b were oxidized to the corresponding carboxylic acids 10a and 10b19 (Scheme 3).
Scheme 3.

Synthesis of alkyne and azide-substituted artemisinin monomersa
aReagents and conditions: (i) MsCl, NEt3, CH2Cl2, 0 °C, 1.5 h; (ii) NaN3, DMF, 80 °C, 16 h; (iii) PPh3, MeOH, reflux 2 h; (iv) RuCl3, NaIO4, ACN, CCl4, H2O, rt, 30 min; (v) propargyl alcohol, NaH, anh DMF, rt, 3 h.
The first set of dimers was designed based on previously reported syntheses10 and ease of producing starting materials. Amine-substituted artemisinin 9 was coupled with carboxylic acid-substituted artemisinin 10 using EDCI to give all possible dimer combinations, symmetric artemisinin dimer 12a, symmetric deactivated deoxyartemisinin dimer 12c and asymmetric artemisinin-deoxyartemisinin dimer 12b. Following the promising results of the amide dimers, synthesis of the triazole dimers was initiated by way of copper-catalyzed azide-alkyne cycloaddition (CuAAC).16 It was quickly determined that the peroxide bridge was not compatible with copper(I) and further experimentally confirmed that artemisinin containing a reduced peroxide bridge is capable of reacting successfully in CuAAC with approximately 90% yield. Previous research has shown that the peroxide bridge is susceptible to Fenton chemistry in which copper(I) catalyzes peroxide reduction.20 Because of this ruthenium was used in place of copper and Cp*RuCl(PPh3)2 was used to form a triazole between azide 8 and alkyne 11. The reaction produced a variety of side products making the purification not straightforward. Furthermore, all attempts to prepare the symmetric bis-peroxide dimer failed to generate the desired product, and therefore the RuAAC dimerization approach was abandoned after the synthesis of asymmetric mono-peroxide dimers 13a and 13b and symmetric deactivated deoxoartemisinin dimer 13c (Scheme 4).
Scheme 4.

Synthesis of amide and triazole artemisinin dimersa
aReagents and conditions: (i) EDCI·HCl, NMM, HOBt, DCM, 0 °C to rt, 18 h; (ii) Cp*RuCl(PPh3)2, DMF, MW, 110 °C, 20 min.
Alternatively, a third set of dimers was synthesized utilizing sequential Mitsunobu21 reactions of alcohols 6a and 6b with mono-TBS-protected hydroquinone, catechol and resorcinol. Following the initial Mitsunobu reaction to form 14a – 14d, the protecting group was removed and a second Mitsunobu reaction was used to complete the symmetric and asymmetric dimers 16a – 16g (Scheme 5).
Scheme 5.

Synthesis of bis(3-deoxoartemisininpropoxy)benzenesa
aReagents and conditions: (i) mono-TBS-protected hydroquinone, catechol or resorcinol, PPh3, DIAD, anh THF, 0 °C to rt, 3 h; (ii) TBAF, THF, rt, 15 min; (iii) 6a or 6b, PPh3, DIAD, anh THF, 0 °C to rt, 3 h.
Results and Discussion
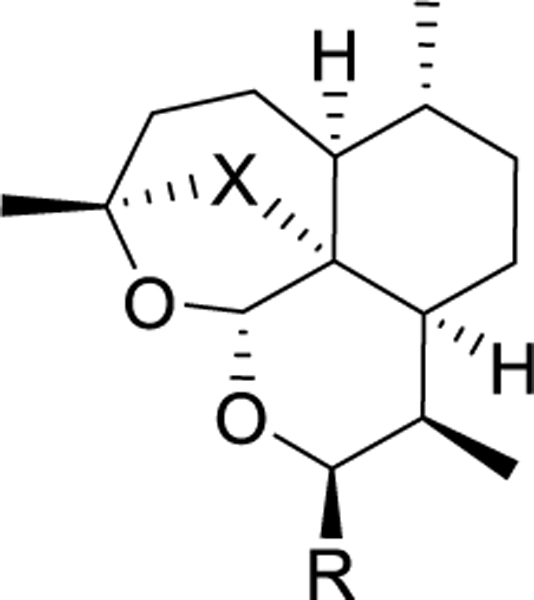
We determined the potency of all synthesized monomers and dimers against the P. falciparum asexual blood stages (PfABS) using slight modifications to a previously-reported, 72-hour, 384-well high content imaging microtiter plate assay22. We also measured cytotoxicity against the HepG2 human hepatocellular carcinoma cell line to characterize selectivity23. The overall trend among the monomers was that the compounds which maintained the peroxide bridge preserved activity within an approximate pEC50 of 9 for the artemisinin and artesunate controls (Table 1). Among the monomers bearing the peroxide bridges, alcohol 6a was the most active one followed by azide 8a and alkyne 11a. Monomers bearing the ether bridges, alcohol 6b, azide 8b, and alkyne 11b were almost 1000-times less potent than their analogues bearing the peroxide bridges. These three monomers also exhibited a three-fold increase in potency compared to artemisinin and artesunate. (Table 1).
Table 1.
In vitro antimalarial activity of artemisinin derivatives against the P. falciparum asexual blood stages (PfABS) and cytotoxicity against HepG2.
| |||||
|---|---|---|---|---|---|
|
| |||||
| Compound | X | R | PfABS pEC50 (±S.D.) | Cytotoxicity pCC50 (±S.D.) | Selectivity Index |
|
| |||||
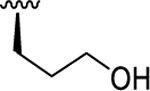
| 6a | O-O |
|
7.95 ( 0.14) | < 5.00 (0.00) | > 891 |
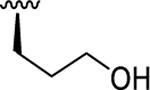
| 6b | O |
|
< 5.10 (0.21) | < 5.00 (0.00) | N.D. |
| 8a | O-O |
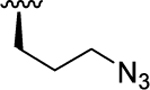
|
8.53 (0.34) | < 5.32 (0.28) | > 1620 |
| 8b | O |
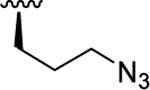
|
< 5.00 (0.00) | < 5.00 (0.00) | N.D. |
| 11a | O-O |
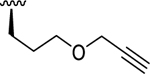
|
9.22 (0.16) | < 5.00 (0.00) | > 16600 |
| 11b | O |
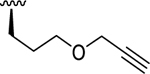
|
5.40 (0.14) | < 5.00 (0.00) | > 2.51 |
| artesunate | 8.95 (0.34) | 5.45 (0.34) | 3160 | ||
| artemisinin | 8.47 (0.32) | < 5.00 (0.00) | > 2950 | ||
| DHA | 9.06 (0.24) | < 5.07 (0.18) | > 9770 | ||
| puromycin | 7.33 (0.22) | 6.39 (0.24) | 8.71 | ||
Values represent mean (± standard deviation) from all independent experiments (n≥4 for PfABS, n≥3 for cytotoxicity). A value of <5.00 indicates inactivity at all concentrations tested; N.D. indicates not determined due to inactivity. If one or more result was <5, then pEC50 = 5 was used to determine the S.D..
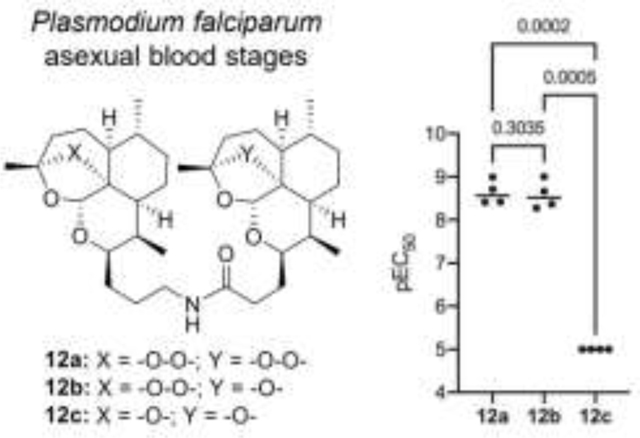
In order to determine how the number of peroxide bridges affects potency of the dimers, structurally related dimers were tested together in the same assay plate and repeated in four or more independent experiments. A one-way ANOVA with Tukey’s multiple comparisons was performed on matching (based on independent experiments) potency values obtained between groups of three members (group 1: 12a, 12b, and 12c, group 2: 13a, 13b, and 13c, group 3: 16a, 16b, and 16c) or a student’s t-test was performed on matching potency values (based on independent experiments) obtained for groups of two members (group 4: 16d and 16e, group 5: 16f and 16g). As expected, dimers containing two peroxide bridges were among the most potent dimers with pEC50 values in the range of 7.56 – 8.63, whereas dimers without a peroxide bridge were devoid of activity (Table 2, Figure 2). Amide-linked dimer 12a with a pEC50 of 8.63 was equipotent to the alcohol or azide monomers 6a and 8a, from which it was synthesized. Bis-peroxide-artemisinin dimers derived from hydroquinone (16a), resorcinol (16d), or catechol (16f) derivatives displayed equal or slightly decreased activity compared to their alcohol building block 6a, but their potency was reduced by a factor of ten in comparison to amide-linked bis-peroxide dimer 12a. These results are in agreement with previous studies reporting antimalarial activity of dimerized artemisinin to depend on the length and the nature of the linker connecting the two artemisinin monomers.7–11 In particular, dimers containing polar groups in the linker have been reported to be more potent than lipophilic dimers.8, 9, 19, 24 A similar activity trend was observed when comparing amide-linked bis-peroxide 12a with the more hydrophobic ethers of hydroquinone (16a), resorcinol (16d), and catechol (16f).
Table 2.
In vitro antimalarial activity of artemisinin dimers against P. falciparum asexual blood stages (PfABS) and cytotoxicity against HepG2.
| Compound | Structure | PfABS pEC50 (±S.D.) | Cytotoxicity pCC50 (±S.D.) | Selectivity Index |
|---|---|---|---|---|
|
| ||||
| 12a |
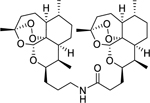
|
8.63 (0.28) | 5.51 (0.21) | 1320 |
| 12b |
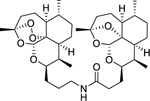
|
8.58 (0.33) | 5.49 (0.35) | 1230 |
| 12c |
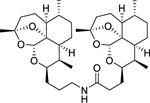
|
< 5.00 (0.00) | < 5.00 (0.00) | N.D. |
| 13a |
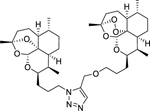
|
8.43 (0.19) | 6.12 (0.17) | 204 |
| 13b |
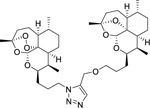
|
8.39 (0.14) | 6.04 (0.06) | 224 |
| 13c |
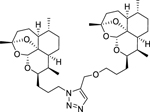
|
5.67 (0.19) | < 5.00 (0.00) | > 4.68 |
| 16a |
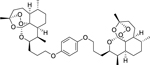
|
7.69 (0.11) | 6.88 (0.19) | 6.46 |
| 16b |
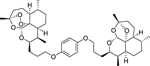
|
6.71 (0.13) | 6.44 (0.10) | 1.86 |
| 16c |
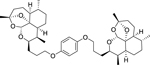
|
< 5.19 (0.26) | < 5.00 (0.00) | N.D. |
| 16d |
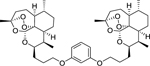
|
7.56 (0.13) | 7.10 (0.28) | 2.88 |
| 16e |
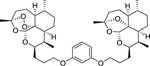
|
6.93 (0.18) | 6.66 (0.07) | 1.86 |
| 16f |
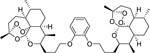
|
7.99 (0.16) | 7.00 (0.34) | 9.78 |
| 16g |
|
7.19 (0.16) | 6.77 (0.21) | 2.63 |
Values represent mean (± standard deviation) from all independent experiments (n≥4 for PfABS, n≥3 for cytotoxicity). A value of <5.00 indicates inactivity at all concentrations tested; N.D. indicates not determined due to inactivity. If one or more result was <5, then pEC50 = 5 was used to determine the S.D..
Figure 2.

Structurally related groups of dimers were assayed together with ≥ 4 independent experiments and potency compared by either a one-way ANOVA with matching potency values (based on independent experiments) followed by Tukey’s multiple comparisons or a student’s t-test with matching potency values (based on independent experiments). Individual data points represent the pEC50 calculated from an independent experiment, bar indicated average, and P-value is indicated above respective comparisons; a P-value <0.05 is considered significant.
In contrast, the mono-peroxide-bridged artemisinin dimers, namely the amide 12b, the triazolo-linked dimers 13a and 13b, and the dihydroxyphenyls 16b, 16g and 16e possessed structural features that previously have not been tested for antimalarial activity. Surprisingly, amide 12b as well as triazoles 13a and 13b displayed pEC50 values comparable to the pEC50 of bis-peroxide-bridged amide 12a. The equipotency of the pair of triazole dimers 13a and 13b also suggests that antimalarial activity is retained irrespective of which of the two artemisinin moieties is substituted with the peroxide bridge. Quite different results were obtained with dihydroxyphenyl 16b, 16g and 16e. These three mono-peroxide-bridged dimers were approximately 10-fold less potent than their bis-peroxide congeners 16a, 16d and 16f. At the same time, mono-peroxide dimer 16b was almost a 100-fold more potent than ether-bridged, deactivated derivative 16c.
Cytotoxocity of the artemisinin dimers was dependent primarily on the linkage and secondarily on the number of peroxide bridges. Dimers were shown to be 5- to 15-fold more cytotoxic compared to the monomeric artemisinin building blocks (Table 2). Furthermore, mono-peroxide-bridged molecules appeared to be marginally less toxic compared to the bis-peroxide analogues. Bis-peroxide- and mono-peroxide-bridged amides 12a and 12b were slightly less cytotoxic than mono-peroxide-bridged triazoles 13a and 13b, which were less toxic than bis-hydroxyphenyls.
Resistance to artemisinin has emerged globally and is expressed as reduced susceptibility at the early ring stage of P. falciparum asexual blood stage development.25, 26 The relative potency of a set of structurally related set of dimers (12a, 12b, and 12c) were assessed for an artemisinin-resistant clone (4G) and compared to an artemisinin-susceptible clone (W2) in both the standard 72-hour drug susceptibility assay and an extended ring stage survival assay (eRSA). We found 12a and 12b inhibited 4G with pEC50’s of 7.94 (± 0.15, n=3) and 7.81 (± 0.26, n=3), respectively, while 12c was inactive. The slightly reduced potency of 12a and 12b against 4G versus W2 (4.9–5.8 fold) was statistically significant (P = 0.0120 for 12a, P = 0.0209 for 12b, unpaired student’s t-test) and in agreement with our previous report with the clones.27 For the eRSA, tightly synchronized ring stages were exposed to 700 nM DHA, 12a, 12b, or 12c for 6 hr, the drug was washed out, and percent parasitemia was determined daily for 5 days. The symmetric artemisinin dimer 12a and asymmetric artemisinin-deoxyartemisinin dimer 12b both produced responses similar to DHA (Figure 3). Upon exposure to drug, parasitemia (presence of morphologically normal P. falciparum blood stages) drops to 0 and only the artemisinin-resistant clone 4G begins to recover on day 3 when treated with 12c, similar to control. By day 5, we observed recovery of the drug susceptible W2 clone in 12a and DHA treated parasites. Interestingly, in the eRSA assay we observed no recovery by day 5 after exposure to 700 nM 12b for both artemisinin-susceptible and -resiststant clones. As we expected, the symmetric deactivated deoxyartemisinin dimer 12c did not affect growth of either the resistant or susceptible clones of P. falciparum (Table 2). These data demonstrate that artemisnin-resistant P. falciparum has reduced susceptibility not only to the artemisinin monomers, but to symmetric artemisinin dimer 12a and asymmetric artemisinindeoxyartemisinin dimer 12b. Although there appears to be no major benefit of the dimers over the monomers, the curious result in the eRSA where we observed no recovery by day 5 after exposure to the asymmetric artemisinin-deoxyartemisinin dimer 12b warrants further studies with extended recovery periods.
Figure 3.

The potency of 12a, 12b, and 12c for artemisinin-susceptible (W2)(A) and -resistant (4G)(B) P. falciparum clones was assessed by using an extended ring-stage survival assay. Parasites were exposed to 700 nM of each drug for 6 hours and parasitemia assessed every 24 hours for five days. (W2, n=4; 4G, n=2)
Conclusions
Potent antimalarial artemisinin dimers containing two peroxide bridges or inactivated analogues, in which the two peroxide bridges have been replaced by two ether bridges, have been synthesized and tested for antimalarial potency and selectivity.7–11 Synthetic routes were developed for novel asymmetric artemisin dimers designed with one peroxide bridge and an ether bridge to assess their antimalarial activity and selectivity. In order to utilize the copper-catalyzed azide-alkyne cycloaddition to form a variety of symmetric and asymmetric artemisinin dimers, four alkyne and azide functionalized artemisinin-derived analogues were synthesized. Although we quickly determined that copper(I) would not be a viable option, ruthenium catalysis gave enabled three artemisinin dimers to be synthesized. Alternative routes were established utilizing an amidation or Mitsunobu reactions to dimerize artemisinins, which differ from the triazole dimers. Monomeric artemisinin building blocks as well as dimeric artemisinins were tested for their antimalarial activity and for selectivity. Of the synthesized artemisinin dimers, the amide- and triazolo-linked dimers 12a, 13a and 13b were more potent than dimers 16a, 16d, and 16f derived from bis-hydroxyphenyl. With this set of artemisinin dimers, experimental data was obtained suggesting for the first time that bis-peroxide-derived artemisinin dimers are equally or slightly more potent than mono-peroxide analogs. This surprising result suggests that previously reported potent artemisinin dimers may derive their potency not only by the number of peroxide bridges, but also by the increased lipophilicity due to the dimerization process. Simultaneously, cytotoxicity studies confirmed that bis-peroxide analogues are equally or slightly more cytotoxic than mono-peroxide bridged artemisinin dimers. This study demonstrates that instead of two peroxide bridges, only one is necessary to preserve antimalarial activity and maintain potency comparable to artemisinin controls.
Methods
Synthetic Chemistry
General.
Unless otherwise noted all reactions were performed in flame-dried round bottom flasks under argon atmosphere. Tetrahydrofuran was distilled from benzophenone and sodium metal before use. Anhydrous diethyl ether and dichloromethane were purchased from EMD Millipore. All other reagents were purchased from Aldrich Chemical Co. and used without further purification.
Analytical thin layer chromatography was performed on 0.25 mm silica gel 60 F254 precoated plates from EMD Millipore. Plates were visualized with ultraviolet light (254 nm) or by treatment with iodine, cerium ammonium molybdate, potassium permanganate or ninhydrin stain followed by heating. EMD silica gel 230–400 (particle size 40–63 μm) mesh was used for all flash column chromatography.
NMR spectra were recorded at ambient temperature on a 400 MHz or 500 MHz Varian NMR spectrometer in the solvent indicated. All 1H NMR experiments are reported in δ units, parts per million (ppm) downfield of TMS, and were measured relative to the signals of chloroform (7.26 ppm) and dimethyl sulfoxide (39.5 ppm) with 1H decoupled observation. Data for 1H NMR are reported as follows: chemicals shift (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), integration and coupling constant (Hz) whereas 13C NMR analyses were reported in terms of chemical shift. NMR data was analyzed by using MestReNova Software version 6.0.2-5475.
The purity of the final compounds was determined to be ≥95% by high-performance liquid chromatography (HPLC) using an Agilent 1100 LC/MSD-VL with electrospray ionization. Low-resolution mass spectra were performed on an Agilent 1100 LC/MSD-VL with electrospray ionization. Microwave heating was performed in a single-mode Anton Paar Monowave 300 and all microwave-irradiated reactions were conducted in heavy-walled glass vials sealed with Teflon septa.
General Procedure A: To a flame-dried round bottom flask was added 6a (202 mg, 0.619 mmol) and anhydrous DCM (9 mL). The solution was cooled on an ice bath to 0 °C followed by the addition of triethylamine (0.17 mL, 1.24 mmol) and mesyl chloride (90 μL, 1.14 mmol). The reaction was stirred at 0 °C for 1.5 hours. Upon completion by TLC, 10 mL DI water was added and extracted with DCM (3 × 10 mL). The organic extracts were combined and dried over Na2SO4, filtered and concentrated under reduced pressure. Purification by flash column chromatography (5:1 Hex:EtOAc) gave 7a as a white solid (242 mg) in quantitative yield.15
General Procedure B: NaN3 (180 mg, 2.78 mmol) was added to a stirring solution of 7a (449 mg, 1.12 mmol) in dimethylformamide (4.3 mL). The reaction mixture was heated to 80 °C and allowed to stir 18 hours. After 18 hours, DI water (3 mL) was added and extracted with tert-butyl methyl ether (3 × 10 mL). The organic extracts were combined and dried over Na2SO4, filtered and concentrated under reduced pressure to give an orange oil. Purification by flash column chromatography (20:1 Hex:EtOAc) gave 8a as a white solid (283 mg, 72%).
General Procedure C: PPh3 (155 mg, 0.591 mmol) was added to a stirring solution of 8a (139 mg, 0.394 mmol) in anhydrous MeOH (4 mL) and refluxed under inert atmosphere for 1.5 hours. The reaction mixture was concentrated under reduced pressure, the residue dissolved in 1.0 M HCl (5 mL) and extracted with CH2Cl2 (2 × 10 mL). The aqueous phase was then treated with 2.0 M NaOH until basic and extracted with CH2Cl2 (2 × 10 mL). The organic extracts were dried over Na2SO4, filtered and concentrated under reduced pressure to give 9a as a crude oil (89 mg, 70%).
General Procedure D: To a stirring solution of 6a (200 mg, 0.613 mmol) in CCl4 (0.2 mL), acetonitrile (0.2 mL) and DI water (0.3 mL) was added NaIO4 (275 mg, 1.287 mmol), followed by ruthenium(III) chloride hydrate (catalytic amount). The reaction was stirred for at room temperature for 30 minutes, then 15 mL of diethyl ether was added and stirred an additional 10 minutes. The reaction mixture was dried over Na2SO4, filtered through a pad of Celite and concentrated under reduced pressure to give a white foam which was used in the next step without further purification.8
General Procedure E: To a flame-dried round bottom flask was added 60% sodium hydride (11 mg, 0.272 mmol), anhydrous DMF (2.5 mL) and propargyl alcohol (21.4 μL, 0.371 mmol). The reaction mixture was allowed to stir at room temperature 15 minutes before adding 7a (100 mg, 0.247 mmol) and continuing to stir for 1 hour before adding 60% sodium hydride (5 mg) and propargyl alcohol (7.1 μL). Upon disappearance of starting material as determined by TLC, the reaction was quenched with DI H2O (4 mL) and extracted with tert-butyl methyl ether (3 × 5 mL). The organic layers were then dried over Na2SO4, filtered and concentrated under reduced pressure. Purification by flash column chromatography (10:1, Hex:EtOAc) gave 11a (74 mg, 82%).
General Procedure F: To a flame-dried round bottom flask was added carboxylic acid 10a (94 mg, 0.279 mmol), DCM (3.0 mL), amine 9a (90 mg, 0.277 mmol) and N-methylmorpholine (91 μL, 0.830 mmol). Reaction mixture was cooled to 0 °C and stirred for 10 minutes before adding EDC·HCl (63.6 mg, 0.332 mmol) and HOBt (45mg, 0.333 mmol). The reaction was allowed to warm to room temperature and stirred overnight. Dilute with DCM (30 mL) and was with 2 M HCl (3 × 15 mL), saturated bicarbonate (3 × 15 mL) and brine (15 mL). The organic layers were then dried over Na2SO4, filtered and concentrated under reduced pressure. Purification by flash column chromatography (95:5, DCM:MeOH) gave 12a (103 mg, 57%).15
General Procedure G: To a solution of azide 8b (20 mg, 0.060 mmol) and alkyne 12a (20 mg, 0.054 mmol) in anhydrous DMF (0.5 mL), was added Cp*RuCl(PPh3)2 (4 mg, 10 mol%). Microwave vial was evacuated and backfilled with argon then run in microwave reactor at 110 °C for 20 minutes. Solvent was evaporated and 13a was isolated by column chromatography in 42% yield.
General Procedure H: To a flame-dried round bottom flask was added PPh3 (22 mg, 0.084 mmol) and anhydrous THF (0.2 mL). The stirring solution was cooled to 0 °C before adding DIAD (16.6 μL, 0.084 mmol) dropwise, followed by 4-((tert-butyldimethylsilyl)oxy)phenol (19 mg, 0.084 mmol) and finally 6a (25 mg, 0.077 mmol). The reaction was allowed to warm to room temperature and stirred for 3 hours. When no further reaction can be seen by TLC, the reaction was poured over DI water and DCM (1 mL: 2 mL). The aqueous layer was extracted with DCM (3 × 2 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. Purification by flash column chromatography (50:1, Hex:EtOAc) gave 14a (35 mg, 86%).
General Procedure I: To a stirring solution of 14a (150 mg, 0.284 mmol) in THF (1 mL mL), was added TBAF (1M in THF, 0.3 mL) dropwise at 0 °C. The reaction was allowed to warm to room temperature and stir for 15 minutes or until completion by TLC. The reaction mixture was poured over DI water and DCM (1 ml: 2 mL) and then the aqueous layer was extracted with DCM (3 × 2 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. Purification by flash column chromatography (20:1, Hex:EtOAc) gave 15a (110 mg, 93%) as a white foamy solid.
DeoxyART-alcohol, 3C 6b. Zinc dust (1 g) was activated by washing with 5% HCl, DI water, EtOH, and diethyl ether, then dried in vacuo. Activated zinc dust (75 mg, 0.722 mmol) was added to a stirring solution of 6a (500 mg, 1.556 mmol) in acetic acid (52 mL). The reaction mixture was stirred at room temperature for 72 hours, with more zinc dust (75 mg) being added at 24 and 48 hours. After 72 hours, the reaction mixture was filtered through celite and washed with DCM. The filtrate was neutralized with saturated sodium bicarbonate, then the organic layer was separated and washed with saturated sodium bicarbonate, brine and water. The organic extracts were combined and dried over Na2SO4, filtered and concentrated under reduced pressure. Purification by flash column chromatography (2:1 Hex:EtOAc) gave 6b as a white solid (417 mg) in 86% yield. Rf = 0.34 (1:1, Hex:EtOAc). 1H NMR (399 MHz, CDCl3) δ 5.28 (s, 1H), 4.12 (ddd, J = 10.3, 7.1, 3 Hz, 1H), 3.68 (s, 2H), 2.21 (m, 1H), 1.98 – 1.74 (m, 5H), 1.73 – 1.65 (m, 4H), 1.65 – 1.52 (m, 3H), 1.51 (s, 3H), 1.27 – 1.14 (m, 4H), 0.89 (d, J = 5.8 Hz, 3H), 0.87 (d, J = 7.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 107.21, 97.37, 82.63, 68.71, 62.82, 45.51, 40.47, 35.74, 34.71, 34.67, 30.34, 30.01, 28.08, 25.37, 282, 22.33, 18.95, 12.21.
ART-mesyl ester, 3C 7a was synthesized following general procedure A in quantitative yield. Rf = 0.56 (1:1, Hex:EtOAc). 1H NMR (399 MHz, CDCl3) δ 5.26 (s, 1H), 4.33 – 4.22 (m, 2H), 4.21 – 4.12 (m, 1H), 2.98 (s, 3H), 2.60 (m, 1H), 2.28 (td, J = 14.3, 6 Hz, 1H), 2.09 – 1.94 (m, 2H), 1.94 – 1.71 (m, 3H), 1.70 – 1.42 (m, 5H), 1.36 (s, 3H), 1.31 – 1.17 (m, 4H), 0.92 (d, J = 5.7 Hz, 3H), 0.83 (d, J = 7.5 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 103.09, 89.28, 81.14, 74.34, 70.18, 52.22, 44.14, 37.47, 37.27, 36.57, 34.40, 30.38, 27.30, 26.10, 25.38, 24.89, 24.75, 20.19, 12.8.
DeoxyART-mesyl ester, 3C 7b was synthesized following general procedure A in 90% yield. Rf = 0.67 (1:1, Hex:EtOAc). 1H NMR (399 MHz, CDCl3) δ 5.25 (s, 1H), 4.35 – 4.21 (m, 2H), 4.13 – 4.03 (m, 1H), 2.99 (s, 3H), 2.27 – 2.14 (m, 1H), 2.01 – 1.51 (m, 10H), 1.49 (s, 3H), 1.37 – 1.09 (m, 5H), 0.90 – 0.82 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 107.16, 97.26, 82.58, 70.34, 67.95, 45.42, 40.41, 37.40, 35.70, 34.63, 34.62, 29.79, 27.08, 26.68, 25.31, 23.85, 22.27, 18.90, 12.23.
ART-azide, 3C 8a was synthesized following general procedure B in 72% yield. Rf = 0.71 (2:1, Hex:EtOAc). 1H NMR (399 MHz, CDCl3) δ 5.29 (s, 1H), 4.21 – 4.14 (m, 1H), 3.35 (m, 2H), 2.64 (m, 1H), 2.31 (td, J = 14.3, 4.0 Hz, 1H), 2.05 – 1.99 (m, 1H), 1.94 – 1.86 (m, 2H), 1.78 (ddd, J = 13, 7.3, 5 Hz, 1H), 1.68 – 1.57 (m, 4H), 1.52 – 1.43 (m, 2H), 1.40 (s, 3H), 1.33 – 1.22 (m, 4H), 0.95 (d, J = 5.9 Hz, 3H), 0.86 (d, J = 7.5 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 103.22, 89.27, 81.23, 74.75, 52.38, 51.41, 44.33, 37.58, 36.67, 34.53, 30.42, 27.07, 26.64, 26.23, 25.02, 24.83, 20.29, 12.99.
DeoxyART-azide, 3C 8b was synthesized following general procedure B in 94% yield. Rf = 0.81 (2:1, Hex:EtOAc). 1H NMR (500 MHz, CDCl3) δ 5.18 (d, J = 5.5 Hz, 1H), 4.00 (t, J = 12.9 Hz, 1H), 32 – 17 (m, 2H), 2.20 – 2.07 (m, 1H), 1.91 – 1.82 (m, 1H), 1.82 – 1.66 (m, 3H), 1.66 – 1.58 (m, 2H), 1.58 – 1.46 (m, 2H), 1.46 – 1.36 (m, 5H), 1.25 – 1.05 (m, 4H), 0.95 – 0.85 (m, 1H), 0.85 – 0.75 (m, 6H). 13C NMR (126 MHz, CDCl3) δ 106.79, 97.10, 82.32, 67.66, 51.14, 45.28, 40.30, 35.50, 34.53, 34.49, 29.62, 28.08, 26.05, 25.14, 23.66, 22.13, 18.74, 12.04.
ART-amine, 3C 9a was synthesized following general procedure C in 70% crude yield. The crude oil was carried on to the next step without further purification.
DeoxyART-amine, 3C 9b was synthesized following general procedure C in 98% crude yield. The crude oil was carried on to the next step without further purification.
ART-carboxylic acid, 3C 10a was synthesized following general procedure D in quantitative crude yield. The crude was carried on to the next step without further purification.
DeoxyART-carboxylic acid, 3C 10bwas synthesized following general procedure D in 72% crude yield. The crude was carried on to the next step without further purification.
ART-propargyl ether, 3C 11a was synthesized following general procedure E in 82% yield as a white solid. Rf = 0.50 (2:1, Hex:EtOAc). 1H NMR (399 MHz, CDCl3) δ 5.27 (s, 1H), 4.16 – 4.12 (m, 1H), 4.11 (d, J = 2.2 Hz, 2H), 3.61 – 3.48 (m, 2H), 2.64 (m, 1H), 2.39 (t, J = 2.1 Hz, 1H), 2.29 (td, J = 14.0, 9 Hz, 1H), 2.04 – 1.96 (m, 1H), 1.95 – 1.83 (m, 2H), 1.80 – 1.72 (m, 1H), 1.68 – 1.41 (m, 6H), 1.38 (s, 3H), 1.36 – 1.17 (m, 4H), 0.93 (d, J = 6.0 Hz, 3H), 0.84 (d, J = 7.5 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 103.21, 89.05, 81.23, 80.18, 75.36, 74.18, 69.88, 58.06, 52.45, 44.48, 37.52, 36.67, 34.56, 30.41, 27.59, 26.24, 25.89, 24.97, 24.80, 20.30, 13.12.
DeoxyART-propargyl ether, 3C 11b was synthesized following general procedure E in 89% yield as a white solid. Rf = 0.68 (2:1, Hex:EtOAc). 1H NMR (399 MHz, CDCl3) δ 5.21 (s, 1H), 4.08 (d, J = 2.2 Hz, 2H), 4.06 – 99 (m, 1H), 3.58 – 3.43 (m, 2H), 2.36 (t, J = 2.1 Hz, 1H), 2.15 (m, 1H), 1.93 – 1.84 (m, 1H), 1.83 – 1.68 (m, 3H), 1.64 (dd, J = 12, 5.2 Hz, 2H), 1.60 – 1.47 (m, 3H), 1.45 (s, 3H), 1.30 – 1.06 (m, 5H), 0.96 – 0.86 (m, 1H), 0.86 – 0.79 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 106.84, 97.25, 82.44, 80.12, 74.10, 69.89, 68.14, 57.97, 45.42, 40.40, 35.60, 34.63, 34.60, 29.73, 27.79, 26.67, 25.26, 23.76, 22.22, 18.86, 12.13.
ART-ART amide dimer 12a was synthesized following general procedure F in 57% yield as a white foam. Rf = 0.25 (95:5 DCM:MeOH). 1H NMR (500 MHz, CDCl3) δ 5.90 (t, J = 5.0 Hz, 1H), 5.29 (d, J = 2 Hz, 2H), 4.18 – 4.13 (m, 1H), 4.05 (dd, J = 9.6, 6.1 Hz, 1H), 3.34 (tt, J = 13, 6.5 Hz, 1H), 3.24 (dq, J = 10, 6.4 Hz, 1H), 2.76 – 2.68 (m, 1H), 2.64 (dt, J = 14.1, 7.0 Hz, 1H), 2.44 (ddd, J = 14.6, 8.8, 5.7 Hz, 1H), 2.37 – 2.28 (m, 2H), 2.24 (dt, J = 14.9, 7.6 Hz, 1H), 2.05 – 1.97 (m, 2H), 1.96 – 1.84 (m, 3H), 1.84 – 1.71 (m, 5H), 1.69 – 1.51 (m, 8H), 1.40 (d, J = 9 Hz, 6H), 1.31 (ddd, J = 21, 13, 4.0 Hz, 8H), 0.98 – 0.93 (m, 6H), 0.87 (d, J = 7.6 Hz, 3H), 0.85 (d, J = 7.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 173.02, 103.40, 130.21, 89.09, 88.68, 81.18, 81.12, 76.21, 75.39, 52.49, 52.33, 44.54, 44.32, 39.44, 37.48, 37.38, 36.59, 36.56, 34.82, 34.50, 34.47, 30.38, 30.21, 29.74, 27.76, 26.96, 26.21, 26.16, 25.12, 24.91, 24.75, 24.68, 20.27, 20.24, 13.28, 13.02. HRMS: m/z calcd for C36H58NO9 [M+H]+ 648.4107; found 648.4108.
ART-DeoxyART amide dimer 12b was synthesized following general procedure F in 78% yield as a white foam. Rf = 0.23 (1:1, Hex:EtOAc). 1H NMR (500 MHz, CDCl3) δ 5.87 (t, J = 5.2 Hz, 1H), 5.29 (s, 1H), 5.24 (s, 1H), 4.17 – 4.11 (m, 1H), 4.06 – 3.99 (m, 1H), 3.45 – 3.36 (m, J = 15, 6.7 Hz, 1H), 3.18 – 3.10 (m, 1H), 2.65 (m, 1H), 2.35 – 2.27 (m, 2H), 2.22 (m, 2H), 2.05 – 1.97 (m, 1H), 1.95 – 1.82 (m, 5H), 1.81 – 1.73 (m, 3H), 1.71 – 1.61 (m, 5H), 1.58 (dd, J = 12.3, 5.6 Hz, 4H), 1.46 (s, 3H), 1.40 (s, 3H), 1.30 (dd, J = 14, 2 Hz, 2H), 1.27 – 1.21 (m, 4H), 1.21 – 1.14 (m, 3H), 0.95 (d, J = 6.1 Hz, 3H), 0.88 (d, J = 5.9 Hz, 3H), 0.86 (d, J = 7.9 Hz, 3H), 0.84 (d, J = 7.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 173.20, 106.95, 103.17, 97.11, 89.02, 82.37, 81.10, 75.38, 67.92, 52.32, 45.27, 44.32, 40.44, 39.32, 37.44, 36.58, 35.61, 34.55, 34.46, 33.82, 30.36, 29.71, 29.59, 27.82, 27.30, 26.85, 26.12, 25.14, 24.86, 24.74, 23.76, 22.19, 20.22, 18.81, 13.02, 12.16. HRMS: m/z calcd for C36H58NO8 [M+H]+ 632.4157; found 632.4163.
DeoxyART-DeoxyART amide dimer 12c was synthesized following general procedure F in 52% yield as a white foam. Rf = 0.32 (1:1, Hex:EtOAc). 1H NMR (500 MHz, CDCl3) δ 5.90 (t, J = 5.1 Hz, 1H), 5.25 (s, 1H), 5.22 (s, 1H), 4.07 – 3.97 (m, 2H), 3.40 – 3.31 (m, 1H), 3.10 (td, J = 12.7, 6.9 Hz, 1H), 2.35 – 2.25 (m, 1H), 2.25 – 2.12 (m, 3H), 1.95 – 1.87 (m, 2H), 1.87 – 1.80 (m, 3H), 1.79 – 1.72 (m, 2H), 1.70 – 1.51 (m, 10H), 1.48 (s, 3H), 1.44 (s, 3H), 1.33 – 1.08 (m, 11H), 0.89 – 0.78 (m, 12H). HRMS: m/z calcd for C36H58NO7 [M+H]+ 616.4208; found 616.4210.
DeoxyART-ART triazole dimer 13a was synthesized following general procedure G in 42% yield. 1H NMR (500 MHz, CDCl3) δ 7.59 (s, 1H), 5.27 (s, J = 10.5 Hz, 1H), 5.25 (s, 1H), 4.55 (d, J = 9 Hz, 2H), 4.46 – 4.32 (m, 2H), 4.18 – 4.08 (m, 2H), 3.55 – 3.45 (m, 2H), 2.70 – 2.59 (m, 1H), 2.31 (td, J = 14.0, 9 Hz, 1H), 2.24 – 2.14 (m, 1H), 2.13 – 1.98 (m, 3H), 1.98 – 1.81 (m, 6H), 1.81 – 1.73 (m, 3H), 1.71 – 1.53 (m, 9H), 1.50 (s, J = 2.9 Hz, 3H), 1.39 (s, 3H), 1.32 – 1.20 (m, 8H), 0.95 (d, J = 5.9 Hz, 3H), 0.88 (d, J = 5.5 Hz, 3H), 0.86 – 0.81 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 139.80, 133.90, 107.10, 1025, 97.29, 89.19, 82.53, 81.28, 75.26, 70.45, 67.99, 60.84, 52.45, 48.23, 45.46, 44.45, 40.45, 37.59, 36.71, 35.71, 34.67, 34.57, 30.47, 29.83, 29.75, 28.20, 27.68, 27.64, 26.28, 25.92, 25.34, 25.03, 24.85, 23.94, 22.31, 20.34, 18.93, 13.12, 12.25. HRMS: m/z calcd for C39H62N3O8 [M+H]+ 700.4532; found 700.4544.
ART-DeoxyART triazole dimer 13b was synthesized following general procedure G in 35% yield. 1H NMR (399 MHz, CDCl3) δ 7.60 (s, 1H), 5.24 (d, J = 7.3 Hz, 2H), 4.56 (s, 1H), 4.47 – 4.38 (m, 1H), 4.25 – 4.14 (m, 1H), 4.10 – 4.00 (m, 1H), 3.56 – 3.40 (m, 2H), 2.69 – 2.56 (m, 1H), 2.39 – 2.25 (m, 1H), 2.25 – 2.13 (m, 2H), 2.12 – 1.97 (m, 3H), 1.97 – 1.81 (m, 4H), 1.81 – 1.53 (m, 11H), 1.48 (s, 3H), 1.47 – 1.41 (m, 3H), 1.39 (s, J = 12.0 Hz, 3H), 1.33 – 1.12 (m, 9H), 0.95 (d, J = 5.9 Hz, 3H), 0.88 (d, J = 5.5 Hz, 3H), 0.85 (d, J = 7.6 Hz, 3H), 0.82 (d, J = 7.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 134.04, 133.40, 107.01, 103.24, 97.35, 89.26, 82.57, 81.24, 74.83, 70.67, 68.27, 60.85, 52.42, 48.33, 45.51, 44.36, 40.48, 37.56, 36.71, 35.72, 34.70, 34.68, 34.56, 30.41, 29.85, 28.13, 27.90, 26.84, 26.47, 26.27, 25.36, 25.00, 24.85, 289, 22.32, 20.33, 18.95, 13.02, 12.26. HRMS: m/z calcd for C39H62N3O8 [M+H]+ 700.4532; found 700.4543.
DeoxyART-DeoxyART triazole dimer 13c was synthesized following general procedure G in 54% yield. 1H NMR (500 MHz, CDCl3) δ 7.58 (s, 1H), 5.24 (s, 2H), 4.54 (s, 2H), 4.48 – 4.31 (m, 2H), 4.15 – 4.08 (m, 1H), 4.08 – 4.01 (m, 1H), 3.54 – 3.47 (m, 1H), 3.47 – 3.39 (m, 1H), 2.26 – 2.14 (m, 2H), 2.13 – 2.02 (m, 1H), 2.00 – 1.73 (m, 10H), 1.69 (d, J = 9 Hz, 2H), 1.66 (d, J = 4.4 Hz, 2H), 1.61 – 1.53 (m, 3H), 1.50 (s, 3H), 1.47 (s, 3H), 1.35 – 1.16 (m, 10H), 0.90 – 0.80 (m, 14H). HRMS: m/z calcd for C39H62N3O7 [M+H]+ 684.4583; found 684.4591.
ART-OPhOPG para 14a was synthesized following general procedure H in 86% yield. Rf = 0.77 (2:1, Hex:EtOAc). 1H NMR (399 MHz, CDCl3) δ 6.81 – 6.70 (m, 4H), 5.31 (s, 1H), 4.25 – 4.18 (m, 1H), 4.03 – 88 (m, 2H), 2.75 – 2.63 (m, 1H), 2.39 – 2.27 (m, 1H), 2.17 – 1.98 (m, 2H), 1.96 – 1.86 (m, 1H), 1.86 – 1.74 (m, 2H), 1.74 – 1.56 (m, 5H), 1.41 (s, 3H), 1.38 – 1.26 (m, 4H), 0.96 (d, J = 6.6 Hz, 12H), 0.87 (d, J = 7.5 Hz, 3H), 0.15 (d, J = 1 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 153.74, 149.34, 120.72, 115.35, 103.32, 89.18, 81.32, 75.36, 68.27, 52.50, 44.52, 37.62, 36.73, 34.61, 30.47, 27.58, 26.31, 26.15, 25.87, 25.08, 24.86, 20.37, 18.32, 13.19, −4.35.
DeoxyART-OPhOPG para 14b was synthesized following general procedure H in 58% yield. Rf = 0.78 (2:1, Hex:EtOAc). 1H NMR (399 MHz, CDCl3) δ 6.78 – 6.70 (m, 4H), 5.28 (s, J = 0 Hz, 1H), 4.18 – 4.09 (m, 1H), 3.94 (dtd, J = 15.5, 9.1, 6.3 Hz, 2H), 2.29 – 2.15 (m, 1H), 2.03 – 1.90 (m, 2H), 1.89 – 1.73 (m, 3H), 1.73 – 1.50 (m, 6H), 1.48 (s, 3H), 1.31 – 1.17 (m, 4H), 0.96 (s, 9H), 0.91 – 0.84 (m, 6H), 0.15 (s, 6H).
ART-OPhOPG meta 14c was synthesized following general procedure H in 62% yield. Rf = 0.81 (2:1, Hex:EtOAc). 1H NMR (500 MHz, CDCl3) δ 7.09 (t, J = 8.1 Hz, 1H), 6.51 (dd, J = 8.1, 1.9 Hz, 1H), 6.44 – 6.38 (m, 2H), 5.32 (s, 1H), 4.28 – 4.18 (m, 1H), 4.05 – 90 (m, 2H), 2.76 – 2.63 (m, 1H), 2.32 (td, J = 14.0, 9 Hz, 1H), 2.16 – 2.06 (m, 1H), 2.02 (dt, J = 7.5, 4.3 Hz, 1H), 1.96 – 1.87 (m, 1H), 1.87 – 1.77 (m, 2H), 1.77 – 1.57 (m, 5H), 1.41 (s, 3H), 1.37 – 1.25 (m, 4H), 0.98 (s, J = 20.4 Hz, 9H), 0.96 (d, J = 6.0 Hz, 3H), 0.87 (d, J = 7.6 Hz, 3H), 0.19 (s, J = 15 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 160.33, 156.81, 129.68, 112.46, 107.59, 106.97, 103.24, 89.13, 81.24, 75.21, 67.67, 52.44, 44.46, 37.56, 36.68, 34.56, 30.42, 27.38, 26.26, 26.08, 25.79, 25.03, 24.82, 20.32, 18.28, 13.12, −4.30.
ART-OPhOPG ortho 14d was synthesized following general procedure H in 68% yield. Rf = 0.84 (2:1, Hex:EtOAc). 1H NMR (399 MHz, CDCl3) δ 6.92 – 6.74 (m, 4H), 5.31 (s, 1H), 4.26 – 4.17 (m, 1H), 4.08 – 3.92 (m, 2H), 2.74 – 2.63 (m, 1H), 2.32 (td, J = 14.0, 8 Hz, 1H), 2.22 – 2.09 (m, 1H), 2.07 – 1.97 (m, 1H), 1.95 – 1.56 (m, 8H), 1.41 (d, J = 7.3 Hz, 3H), 1.37 – 1.14 (m, 4H), 1.00 (s, 9H), 0.95 (d, J = 5.9 Hz, 3H), 0.87 (d, J = 7.5 Hz, 3H), 0.16 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 150.69, 144.93, 121.83, 121.08, 120.65, 1109, 1019, 89.10, 81.22, 75.38, 68.21, 52.39, 44.42, 37.51, 36.65, 34.53, 30.44, 27.70, 26.27, 26.22, 25.82, 24.97, 24.80, 20.29, 18.44, 13.12, −4.50, −4.52.
ART-OPhOH para 15a was synthesized following general procedure I in 93% yield as a white foamy solid. Rf = 0.60 (3:1, Hex:EtOAc). 1H NMR (399 MHz, CDCl3) δ 6.76 (s, 4H), 5.33 (s, 1H), 4.18 (d, J = 6.3 Hz, 1H), 3.92 (dt, J = 22.0, 7.8 Hz, 2H), 2.77 – 2.64 (m, 1H), 2.33 (td, J = 14.0, 8 Hz, 1H), 2.13 – 1.97 (m, 2H), 1.90 (d, J = 10.9 Hz, 1H), 1.85 – 1.44 (m, 7H), 1.41 (s, 3H), 1.38 – 1.21 (m, 5H), 0.96 (d, J = 5.8 Hz, 3H), 0.87 (d, J = 7.5 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 153.22, 149.74, 116.12, 115.73, 103.48, 89.12, 81.34, 75.53, 68.39, 52.49, 44.53, 37.60, 36.69, 34.58, 30.42, 27.49, 26.24, 25.98, 25.05, 24.83, 20.36, 13.22.
DeoxyART-OPhOH para 15b was synthesized following general procedure I in quantitative yield. Rf = 0.32 (3:1, Hex:EtOAc). 1H NMR (399 MHz, CDCl3) δ 6.80 – 6.69 (m, 4H), 5.30 (s, 1H), 4.19 – 4.08 (m, 1H), 4.01 – 83 (m, 2H), 2.29 – 2.15 (m, 1H), 2.04 – 1.90 (m, 2H), 1.90 – 1.83 (m, 1H), 1.83 – 1.75 (m, 2H), 1.75 – 1.65 (m, 3H), 1.65 – 1.51 (m, 3H), 1.49 (s, 3H), 1.35 – 1.13 (m, 5H), 0.93 – 0.83 (m, 6H).
ART-OPhOH meta 15c was synthesized following general procedure I in 86% yield. Rf = 0.16 (3:1, Hex:EtOAc). 1H NMR (399 MHz, CDCl3) δ 7.07 (t, J = 8.0 Hz, 1H), 6.51 – 6.39 (m, 3H), 5.34 (s, 1H), 4.20 (dd, J = 11.8, 4.8 Hz, 1H), 4.02 – 3.87 (m, 2H), 2.76 – 2.65 (m, 1H), 2.33 (td, J = 14.0, 8 Hz, 1H), 2.18 – 1.98 (m, 3H), 1.97 – 1.86 (m, 1H), 1.85 – 1.74 (m, 2H), 1.73 – 1.44 (m, 5H), 1.41 (s, 3H), 1.36 – 1.16 (m, 4H), 0.95 (d, J = 5.7 Hz, 3H), 0.86 (d, J = 7.5 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 160.37, 157.46, 130.03, 107.84, 106.80, 103.60, 102.11, 89.08, 81.31, 75.62, 67.57, 52.41, 44.49, 37.51, 36.62, 34.50, 30.34, 27.14, 26.10, 25.80, 24.97, 24.75, 20.31, 13.19.
ART-OPhOH ortho 15d was synthesized following general procedure I in quantitative yield. Rf = 0.55 (3:1, Hex:EtOAc). 1H NMR (500 MHz, CDCl3) δ 6.94 (d, J = 6.5 Hz, 1H), 6.91 – 6.79 (m, 3H), 5.34 (s, 1H), 4.37 – 4.26 (m, 1H), 4.20 – 4.08 (m, 2H), 2.74 – 2.64 (m, 1H), 2.35 (td, J = 14.0, 8 Hz, 1H), 2.18 – 2.01 (m, 3H), 2.01 – 1.89 (m, 2H), 1.87 – 1.71 (m, 3H), 1.71 – 1.57 (m, 3H), 1.44 (s, 3H), 1.39 – 1.28 (m, 4H), 0.98 (d, J = 5.8 Hz, 3H), 0.89 (d, J = 7.5 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 146.13, 146.09, 121.43, 120.04, 114.69, 112.11, 103.18, 89.29, 81.18, 74.75, 68.93, 52.30, 44.29, 37.52, 36.63, 34.49, 30.42, 27.28, 26.29, 26.13, 24.99, 24.79, 20.23, 12.92.
ART-OPhO-ART para 16a was synthesized following general procedure H in 51% yield as a white solid. Rf = 0.58 (2:1, Hex:EtOAc). 1H NMR (399 MHz, CDCl3) δ 6.81 (s, 4H), 5.31 (s, 2H), 4.24 – 4.16 (m, 2H), 4.02 – 3.86 (m, 4H), 2.75 – 2.62 (m, 2H), 2.32 (td, J = 14.1, 8 Hz, 2H), 2.16 1.96 (m, 5H), 1.95 – 1.85 (m, 2H), 1.85 – 1.73 (m, 5H), 1.72 – 1.55 (m, 8H), 1.40 (s, 6H), 1.37 1.21 (m, 8H), 0.95 (d, J = 5.8 Hz, 6H), 0.86 (d, J = 7.5 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 153.24, 115.47, 103.28, 89.09, 81.27, 75.35, 68.34, 52.44, 44.46, 37.55, 36.66, 34.54, 30.41, 27.52, 26.28, 26.04, 25.01, 24.80, 20.34, 13.18. HRMS: m/z calcd for C42H63O10 [M+H]+ 727.4416; found 727.4411.
ART-OPhO-DeoxyART para 16b was synthesized following general procedure H in 67% yield as a white solid. Rf = 0.68 (2:1, Hex:EtOAc). 1H NMR (399 MHz, CDCl3) δ 6.81 (s, 4H), 5.30 (s, 1H), 5.27 (s, 1H), 4.23 – 4.16 (m, 1H), 4.15 – 4.08 (m, 1H), 4.02 – 3.85 (m, 4H), 2.75 – 2.61 (m, 1H), 2.31 (td, J = 14.0, 9 Hz, 1H), 2.27 – 2.15 (m, 1H), 2.10 – 1.73 (m, 11H), 1.73 – 1.50 (m, 10H), 1.47 (s, 3H), 1.40 (s, 3H), 1.35 – 1.12 (m, 8H), 0.95 (d, J = 5.9 Hz, 3H), 0.90 – 0.84 (m, 9H). 13C NMR (100 MHz, CDCl3) δ 153.28, 153.17, 115.48, 115.36, 106.98, 103.26, 97.35, 89.09, 82.53, 81.26, 75.33, 68.36, 68.19, 68.18, 52.44, 45.46, 44.47, 40.44, 37.55, 36.67, 35.67, 34.67, 34.65, 34.55, 30.41, 29.81, 27.80, 27.52, 26.68, 26.27, 26.04, 25.33, 25.01, 24.80, 23.84, 22.28, 20.33, 18.93, 13.16, 12.25. HRMS: m/z calcd for C42H63O9 [M+H]+ 711.4467; found 711.4460.
DeoxyART-OPhO-DeoxyART para 16c was synthesized following general procedure H in 58% yield. Rf = 0.73 (2:1, Hex:EtOAc). 1H NMR (399 MHz, CDCl3) δ 6.81 (s, 4H), 5.28 (s, 2H), 4.12 (ddd, J = 10.5, 7.1, 4 Hz, 2H), 3.94 (ddt, J = 31.8, 9.2, 6.3 Hz, 4H), 2.28 – 2.15 (m, 2H), 2.02 – 1.89 (m, 4H), 1.89 – 1.73 (m, 6H), 1.73 – 1.65 (m, 4H), 1.65 – 1.50 (m, 6H), 1.48 (s, 6H), 1.32 – 1.12 (m, 10H), 0.92 – 0.83 (m, 12H). 13C NMR (100 MHz, CDCl3) δ 153.28, 115.39, 107.01, 97.38, 82.56, 68.23, 68.21, 45.49, 40.47, 35.70, 34.69, 34.67, 29.83, 27.82, 26.70, 25.36, 286, 22.30, 18.95, 12.27. HRMS: m/z calcd for C42H62NaO8 [M+Na]+ 717.4337; found 717.4330.
ART-OPhO-ART meta 16d was synthesized following general procedure H in 48% yield. Rf = 0.51 (2:1, Hex:EtOAc). 1H NMR (399 MHz, CDCl3) δ 7.14 (t, J = 8.1 Hz, 1H), 6.53 – 6.42 (m, 3H), 5.31 (s, 2H), 4.26 – 4.16 (m, 2H), 4.07 – 3.88 (m, 4H), 2.75 – 2.61 (m, 2H), 2.32 (td, J = 14.0, 7 Hz, 2H), 2.19 – 1.54 (m, 19H), 1.41 (s, 6H), 1.37 – 1.25 (m, 9H), 0.95 (d, J = 5.8 Hz, 6H), 0.87 (d, J = 7.5 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 160.42, 129.85, 106.73, 103.31, 101.55, 89.10, 81.29, 75.38, 67.74, 52.45, 44.48, 37.57, 36.67, 34.56, 30.42, 27.44, 26.30, 26.11, 25.03, 24.82, 20.36, 13.20. HRMS: m/z calcd for C42H63O10 [M+H]+ 727.4416; found 727.4419.
ART-OPhO-DeoxyART meta 16e was synthesized following general procedure H in 62% yield. Rf = 0.59 (2:1, Hex:EtOAc). 1H NMR (399 MHz, CDCl3) δ 7.13 (t, J = 8.0 Hz, 1H), 6.54 – 6.42 (m, 3H), 5.31 (s, 1H), 5.28 (s, 1H), 4.25 – 4.16 (m, 1H), 4.16 – 4.07 (m, 1H), 4.07 – 3.90 (m, 4H), 2.75 – 2.63 (m, 1H), 2.39 – 2.27 (m, 1H), 2.27 – 2.15 (m, 1H), 2.16 – 1.75 (m, 12H), 1.73 – 1.56 (m, 9H), 1.49 (s, 3H), 1.41 (s, 3H), 1.37 – 1.21 (m, 8H), 0.95 (d, J = 5.8 Hz, 3H), 0.92 – 0.83 (m, 9H). 13C NMR (100 MHz, CDCl3) δ 160.43, 129.82, 107.01, 106.69, 106.65, 103.30, 101.46, 97.35, 89.07, 82.54, 81.27, 75.42, 68.20, 67.71, 67.60, 52.44, 45.45, 44.48, 40.43, 37.55, 36.66, 35.68, 34.66, 34.65, 34.54, 30.41, 29.82, 27.83, 27.45, 26.62, 26.29, 26.08, 25.34, 25.02, 24.80, 286, 22.28, 20.36, 18.95, 13.21, 12.27. HRMS: m/z calcd for C42H63O9 [M+H]+ 711.4467; found 711.4466.
ART-OPhO-ART ortho 16f was synthesized following general procedure H in 44% yield. Rf = 0.63 (2:1, Hex:EtOAc). 1H NMR (399 MHz, CDCl3) δ 6.95 – 6.80 (m, 4H), 5.30 (s, 2H), 4.25 – 4.15 (m, 2H), 4.14 – 3.95 (m, 4H), 2.74 – 2.61 (m, 2H), 2.31 (td, J = 14.0, 8 Hz, 2H), 2.19 – 2.05 (m, 2H), 2.06 – 1.97 (m, 3H), 1.95 – 1.74 (m, 6H), 1.74 – 1.54 (m, 8H), 1.40 (s, 6H), 1.35 – 1.23 (m, 9H), 0.95 (d, J = 5.8 Hz, 6H), 0.87 (d, J = 7.5 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 149.18, 121.15, 114.24, 103.27, 89.08, 81.27, 75.28, 68.87, 52.44, 44.48, 37.56, 36.67, 34.57, 30.41, 27.49, 26.29, 25.93, 25.03, 24.81, 20.36, 13.21. HRMS: m/z calcd for C42H63O10 [M+H]+ 727.4416; found 727.4423.
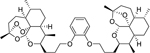
ART-OPhO-ART ortho 16g was synthesized following general procedure H in 44% yield. Rf = 0.66 (2:1, Hex:EtOAc). 1H NMR (399 MHz, CDCl3) δ 6.95 – 6.80 (m, 4H), 5.30 (s, 1H), 5.27 (s, 1H), 4.24 – 4.15 (m, 1H), 4.15 – 4.04 (m, 3H), 4.04 – 3.94 (m, 2H), 2.76 – 2.62 (m, 1H), 2.32 (td, J = 14.0, 8 Hz, 1H), 2.26 – 2.05 (m, 3H), 2.03 – 1.75 (m, 9H), 1.75 – 1.64 (m, 6H), 1.64 – 1.49 (m, 4H), 1.47 (s, 3H), 1.40 (s, 3H), 1.35 – 1.20 (m, 8H), 0.95 (d, J = 5.9 Hz, 3H), 0.91 – 0.81 (m, 9H). 13C NMR (100 MHz, CDCl3) δ 149.25, 149.06, 121.14, 120.97, 114.23, 113.89, 106.98, 103.29, 97.34, 89.02, 82.52, 81.26, 75.45, 68.89, 68.68, 68.04, 52.46, 45.45, 44.51, 40.44, 37.54, 36.66, 35.68, 34.66, 34.65, 34.57, 30.39, 29.79, 27.71, 27.51, 26.55, 26.29, 25.90, 25.34, 25.01, 24.80, 23.84, 22.27, 20.36, 18.95, 13.22, 12.30. HRMS: m/z calcd for C42H62NaO9 [M+Na]+ 733.4287; found 733.4288.
Parasitology
Plasmodium falciparum Cultivation:
P. falciparum strain W228, 29 was continuously cultured in RPMI media (Gibco) supplemented with 10% inactivated human plasma (Interstate Blood Bank) and 5% hematocrit (Interstate Blood Bank) based on methods previously described.30 Forty-eight hours before assay initiation, parasites were synchronized using filter-sterilized 5% D-sorbitol (Millipore-Sigma) in water such that assays were started with >90% rings as previously described.31
Drug plate preparation and compound treatment of P. falciparum assay plates:
Following synthesis, compound powder was diluted to 10 mM in dehydrated, sterile DMSO (Tocris) and a duplicate-well, 12-point, 3-fold semi log dilution series was prepared at 1000x final concentration in 384 well plates (Greiner Bio-one) in DMSO using a Biomek 4000 (Beckman Coulter). Plates were sealed using foil sealing tape (VWR) and kept in a desiccator until used to inoculate P. falciparum assay plates. Assay plates were prepared by plating 20 μL of culture media to pre-wet all wells, followed by addition of compound using a 40-nL pin tool (V&P Scientific) as previously described.32 Plates were then inoculated with 20 μL P. falciparum culture at 2% parasitemia and 0.75% hematocrit, leading to dilution of all test compounds to 1x and DMSO to 0.1% in complete media. Assay plates were maintained in bioassay dishes with water cups to prevent edge effect due to evaporation for 72–96 hours as previously described.22,25
Imaging and Data Analysis:
After 72–96 hours of incubation, assay plates were simultaneously stained and fixed by adding to each well 40 μL of PBS containing 20 μg/mL Hoechst 33342 (Thermo Fisher Scientific) and 0.1% glutaraldehyde (Electron Microscopy Resources), similar to that previously described.22 Plates were maintained for 24 hours in 4 °C and imaged the following day using the Lionheart FX high content imager (Biotek). Using a 4x objective, a single field of view was captured for each well using the DAPI filter and a background flattening algorithm to reduce Hoechst 33342 autofluorescence to identify Hoeschst 33342-stained parasite DNA. The net DNA-area data was then exported to CDD Vault (Collaborative Drug Discovery) and inhibition values normalized using the DMSO negative control and the dihydroartemsinin positive control wells whereby:
Compound potency was determined by constructing a dose-response curve fit using the Levenberg–Marquardt algorithm33, 34 to calculate pEC50’s. Outliers, if any, were identified by comparing inhibition values across replicate wells and were manually removed using CDD Vault’s user interface. Each grouping of compounds was tested in 4 or 5 independent experiments and the presented data is the average and standard deviation from all independent experiments.
Cytotoxicity Assessment:
The HepG2 human hepatocyte line from hepatocellular carcinoma (ATCC HC-8065) were cultured in rat collagen I-coated (5 μg/cm2) flasks (Corning) at 20–90% confluence in EMEM (Lonza) supplemented with 1 mM Sodium Pyruvate (Lonza), 2 mM L-glutamine (Gibco) and 10% fetal bovine serum (Hyclone) in a cell culture incubator at 37 °C and 5% CO2. Cells were passed by treating with Trypsin LE (Gibco) for 7 minutes at 37 °C. Toxicity assays were started by harvested cells from a flask, counting viability by trypan blue, and seeding 2000 live cells in 40 μL per well into rat-tail collagen I-coated 384-well plates (Greiner Bio-one) using a Biomek NX (Beckman Coulture). Compounds were then added using the 40 nL pin tool as above, using the same source plate used for P. falciparum assay as above. After 72 hours, media was removed and cells were fixed with 4% paraformaldehyde (Thermo Fisher Scientific) in PBS and then stained with 10 ug/mL Hoechst 33342 for 1 hour. The entire culture area of each well of assay plates were imaged with a Lionheart FX with a 4x objective, and net hepatic nuclei per well quantified. Data were loaded into CDD Vault for normalization, curve fitting, and pCC50 calculation as described above, but using puromycin as the positive control instead of 2a. All compounds were tested in 3 or 4 independent experiments, and the presented data is the average and standard deviation from all independent experiments.
Extended Ring Stage Survival Assay (eRSA)
Tightly synchronized ring stages of artemisinin-susceptible (W2) and -resistant (4G clone of ARC08–22)27 P. falciparum strains were exposed to 700 nM DHA, 12a, 12b or 12c in a tissue culture treated flat bottom 48 well plate (Corning Inc) for 6 hours at 1% parasitemia and 3% hematocrit. After 6 hours of drug exposure, infected erythrocytes were washed 3 times with RPMI medium and returned to standard growing conditions. The eRSA assay was performed as previously described with modification to extend the recovery observation period to day 5.35, 36 Daily blood smears were prepared, stained with Giemsa, and parasitemia quantified by microscopical observations. Parasitemia was assessed for morphologically normal asexual blood stages of P. falciparum.
Supplementary Material
Funding Sources
We thank the National Institutes of Health (R01AI090662 and R01AI144464) and the Georgia Research Alliance for financial support of the herein presented studies.
ABBREVIATIONS
- ACTs
artemisinin-based combination therapies
- anh
anhydrous
- CDD
Collaborative Drug Discovery
- CuAAC
copper(I)-catalyzed azide-alkyne cycloaddition
- DHA
dihydroartemisinin
- pCC50
negative log of the extract concentration that reduced the cell viability by 50%
- pEC50
negative log of half maximal effective concentration
- PfABS
P. falciparum asexual blood stages
- RuAAC
ruthenium-catalyzed azide-alkyne cycloaddition
- TBSCl
tert-butyldimethylsilyl chloride
- TLC
thin layer chromatography
Footnotes
ASSOCIATED CONTENT
Supporting Information. Molecular Formula Strings are available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
References
- 1.Liu J, Structure and Reaction of Arteannuin. Huaxue Xuebao 1979, 37, 129–143. [Google Scholar]
- 2.Brossi A; Venugopalan B; Dominguez Gerpe L; Yeh HJ; Flippen-Anderson JL; Buchs P; Luo XD; Milhous W; Peters W, Arteether, a new antimalarial drug: synthesis and antimalarial properties. J. Med. Chem. 1988, 31, 645–650. [DOI] [PubMed] [Google Scholar]
- 3.Meshnick SR, Artemisinin: mechanisms of action, resistance and toxicity. Int. J. Parasitol. 2002, 32, 1655–1660. [DOI] [PubMed] [Google Scholar]
- 4.World Health Organization, W. H., World Malaria Report 2015. 2015. [Google Scholar]
- 5.Vennerstrom JL; Arbe-Barnes S; Brun R; Charman SA; Chiu FCK; Chollet J; Dong Y; Dorn A; Hunziker D; Matile H; McIntosh K; Padmanilayam M; Santo Tomas J; Scheurer C; Scorneaux B; Tang Y; Urwyler H; Wittlin S; Charman WN, Identification of an antimalarial synthetic trioxolane drug development candidate. Nature 2004, 430, 900–904. [DOI] [PubMed] [Google Scholar]
- 6.Charman SA; Arbe-Barnes S; Bathurst IC; Brun R; Campbell M; Charman WN; Chiu FC; Chollet J; Craft JC; Creek DJ; Dong Y; Matile H; Maurer M; Morizzi J; Nguyen T; Papastogiannidis P; Scheurer C; Shackleford DM; Sriraghavan K; Stingelin L; Tang Y; Urwyler H; Wang X; White KL; Wittlin S; Zhou L; Vennerstrom JL, Synthetic ozonide drug candidate OZ439 offers new hope for a single-dose cure of uncomplicated malaria. Proc. Natl. Acad. Sci. U S A 2011, 108, 4400–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Posner GH; Ploypradith P; Parker MH; O’Dowd H; Woo SH; Northrop J; Krasavin M; Dolan P; Kensler TW; Xie S; Shapiro TA, Antimalarial, antiproliferative, and antitumor activities of artemisinin-derived, chemically robust, trioxane dimers. J. Med. Chem. 1999, 42, 4275–4280. [DOI] [PubMed] [Google Scholar]
- 8.Posner GH; Paik IH; Sur S; McRiner AJ; Borstnik K; Xie S; Shapiro TA, Orally active, antimalarial, anticancer, artemisinin-derived trioxane dimers with high stability and efficacy. J. Med. Chem. 2003, 46, 1060–1065. [DOI] [PubMed] [Google Scholar]
- 9.Jeyadevan JP; Bray PG; Chadwick J; Mercer AE; Byrne A; Ward SA; Park BK; Williams DP; Cosstick R; Davies J; Higson AP; Irving E; Posner GH; O’Neill PM, Antimalarial and Antitumor Evaluation of Novel C-10 Non-Acetal Dimers of 10β-(2-Hydroxyethyl)deoxoartemisinin. J. Med. Chem. 2004, 47, 1290–1298. [DOI] [PubMed] [Google Scholar]
- 10.Chadwick J; Mercer AE; Park BK; Cosstick R; O’Neill PM, Synthesis and biological evaluation of extraordinarily potent C-10 carba artemisinin dimers against P. falciparum malaria parasites and HL-60 cancer cells. Bioorg. Med. Chem. 2009, 17, 1325–1338. [DOI] [PubMed] [Google Scholar]
- 11.Fröhlich T; Çapcı Karagöz A; Reiter C; Tsogoeva SB, Artemisinin-Derived Dimers: Potent Antimalarial and Anticancer Agents. J. Med. Chem. 2016, 59, 7360–7388. [DOI] [PubMed] [Google Scholar]
- 12.Grace JM; Aguilar AJ; Trotman KM; Peggins JO; Brewer TG, Metabolism of beta-arteether to dihydroqinghaosu by human liver microsomes and recombinant cytochrome P450. Drug Metab. Dispos. 1998, 26, 313–7. [PubMed] [Google Scholar]
- 13.O’Neill PM; Searle NL; Kan K-W; Storr RC; Maggs JL; Ward SA; Raynes K; Park BK, Novel, Potent, Semisynthetic Antimalarial Carba Analogues of the First-Generation 1,2,4-Trioxane Artemether. J. Med. Chem. 1999, 42, 5487–5493. [DOI] [PubMed] [Google Scholar]
- 14.O’Neill PM; Pugh M; Stachulski AV; Ward SA; Davies J; Park BK, Optimisation of the allylsilane approach to C-10 deoxo carba analogues of dihydroartemisinin: synthesis and in vitro antimalarial activity of new, metabolically stable C-10 analogues. J.e Chem. Soc., Perkin Trans. 2001, 2682–2689. [Google Scholar]
- 15.Chadwick J; Jones M; Mercer AE; Stocks PA; Ward SA; Park BK; O’Neill PM, Design, synthesis and antimalarial/anticancer evaluation of spermidine linked artemisinin conjugates designed to exploit polyamine transporters in Plasmodium falciparum and HL-60 cancer cell lines. Bioorg. Med. Chem. 2010, 18, 2586–97. [DOI] [PubMed] [Google Scholar]
- 16.Rostovtsev VV; Green LG; Fokin VV; Sharpless KB, A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [DOI] [PubMed] [Google Scholar]
- 17.Zhang L; Chen X; Xue P; Sun HHY; Williams ID; Sharpless KB; Fokin VV; Jia G, Ruthenium-Catalyzed Cycloaddition of Alkynes and Organic Azides. J. American Chem. Soc. 2005, 127, 15998–15999. [DOI] [PubMed] [Google Scholar]
- 18.Rasmussen LK; Boren BC; Fokin VV, Ruthenium-Catalyzed Cycloaddition of Aryl Azides and Alkynes. Organic Lett. 2007, 9, 5337–5339. [DOI] [PubMed] [Google Scholar]
- 19.Posner GH; McRiner AJ; Paik I-H; Sur S; Borstnik K; Xie S; Shapiro TA; Alagbala A; Foster B, Anticancer and Antimalarial Efficacy and Safety of Artemisinin-Derived Trioxane Dimers in Rodents. J. Med. Chem. 2004, 47, 1299–1301. [DOI] [PubMed] [Google Scholar]
- 20.Bousejra-El Garah F; Pitie M; Vendier L; Meunier B; Robert A, Alkylating ability of artemisinin after Cu(I)-induced activation. J. Biol. Inorg. Chem. 2009, 14, 601–610. [DOI] [PubMed] [Google Scholar]
- 21.Wada M; Mitsunobu O, Intermolecular dehydration between alcohols and active hydrogen compounds by means of diethyl azodicarboxylate and triphenylphosphine. Tetrahedron Lett. 1972, 13, 1279–1282. [Google Scholar]
- 22.Duffy S; Avery VM, Development and optimization of a novel 384-well anti-malarial imaging assay validated for high-throughput screening. Am. J. Trop. Med. Hyg. 2012, 86, 84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miret S; De Groene EM; Klaffke W, Comparison of in vitro assays of cellular toxicity in the human hepatic cell line HepG2. J. Biomol. Screen. 2006, 11, 184–193. [DOI] [PubMed] [Google Scholar]
- 24.Jung M; Lee S; Ham J; Lee K; Kim H; Kim SK, Antitumor Activity of Novel Deoxoartemisinin Monomers, Dimers, and Trimer. J. Med. Chem. 2003, 46, 987–994. [DOI] [PubMed] [Google Scholar]
- 25.Ashley EA; Dhorda M; Fairhurst RM; Amaratunga C; Lim P; Suon S; Sreng S; Anderson JM; Mao S; Sam B; Sopha C; Chuor CM; Nguon C; Sovannaroth S; Pukrittayakamee S; Jittamala P; Chotivanich K; Chutasmit K; Suchatsoonthorn C; Runcharoen R; Hien TT; Thuy-Nhien NT; Thanh NV; Phu NH; Htut Y; Han KT; Aye KH; Mokuolu OA; Olaosebikan RR; Folaranmi OO; Mayxay M; Khanthavong M; Hongvanthong B; Newton PN; Onyamboko MA; Fanello CI; Tshefu AK; Mishra N; Valecha N; Phyo AP; Nosten F; Yi P; Tripura R; Borrmann S; Bashraheil M; Peshu J; Faiz MA; Ghose A; Hossain MA; Samad R; Rahman MR; Hasan MM; Islam A; Miotto O; Amato R; MacInnis B; Stalker J; Kwiatkowski DP; Bozdech Z; Jeeyapant A; Cheah PY; Sakulthaew T; Chalk J; Intharabut B; Silamut K; Lee SJ; Vihokhern B; Kunasol C; Imwong M; Tarning J; Taylor WJ; Yeung S; Woodrow CJ; Flegg JA; Das D; Smith J; Venkatesan M; Plowe CV; Stepniewska K; Guerin PJ; Dondorp AM; Day NP; White NJ, Spread of Artemisinin Resistance in Plasmodium falciparum Malaria. N. Eng. J. Med. 2014, 371, 411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ariey F; Witkowski B; Amaratunga C; Beghain J; Langlois A-C; Khim N; Kim S; Duru V; Bouchier C; Ma L; Lim P; Leang R; Duong S; Sreng S; Suon S; Chuor CM; Bout DM; Ménard S; Rogers WO; Genton B; Fandeur T; Miotto O; Ringwald P; Le Bras J; Berry A; Barale J-C; Fairhurst RM; Benoit-Vical F; Mercereau-Puijalon O; Ménard D, A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 2014, 505, 50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hott A; Casandra D; Sparks KN; Morton LC; Castanares G-G; Rutter A; Kyle DE, Artemisinin-Resistant Parasites Exhibit Altered Patterns of Development in Infected Erythrocytes. Antimicrob. Agents Chemother. 2015, 59, 3156–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oduola AM; Weatherly NF; Bowdre JH; Desjardins RE, Plasmodium falciparum: cloning by single-erythrocyte micromanipulation and heterogeneity in vitro. Exp. Parasitol. 1988, 66, 86–95. [DOI] [PubMed] [Google Scholar]
- 29.Canfield CJ; Pudney M; Gutteridge WE, Interactions of atovaquone with other antimalarial drugs against Plasmodium falciparum in vitro. Exp Parasitol 1995, 80 (3), 373–81. [DOI] [PubMed] [Google Scholar]
- 30.Trager W; Jensen JB, Human malaria parasites in continuous culture. Science 1976, 193, 673–675. [DOI] [PubMed] [Google Scholar]
- 31.Lambros C; Vanderberg JP, Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 1979, 65, 418–420. [PubMed] [Google Scholar]
- 32.Roth A; Maher SP; Conway AJ; Ubalee R; Chaumeau V; Andolina C; Kaba SA; Vantaux A; Bakowski MA; Thomson-Luque R; Adapa SR; Singh N; Barnes SJ; Cooper CA; Rouillier M; McNamara CW; Mikolajczak SA; Sather N; Witkowski B; Campo B; Kappe SHI; Lanar DE; Nosten F; Davidson S; Jiang RHY; Kyle DE; Adams JH, A comprehensive model for assessment of liver stage therapies targeting Plasmodium vivax and Plasmodium falciparum. Nat. Commun. 2018, 9, 1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levenberg K, A Method for the Solution of Certain Non-Linear Problems in Least Squares. Q.Appl. Math. 1944, 2, 164–168. [Google Scholar]
- 34.Marquardt D, An Algorithm for Least-Squares Estimation of Nonlinear Parameters. SIAM J. Appl. Math. 1963, 11, 431–441. [Google Scholar]
- 35.Witkowski B; Amaratunga C; Khim N; Sreng S; Chim P; Kim S; Lim P; Mao S; Sopha C; Sam B; Anderson JM; Duong S; Chuor CM; Taylor WRJ; Suon S; Mercereau-Puijalon O; Fairhurst RM; Menard D, Novel phenotypic assays for the detection of artemisinin-resistant Plasmodium falciparum malaria in Cambodia: in-vitro and ex-vivo drug-response studies. Lancet Infect. Dis. 2013, 13, 1043–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davis SZ; Singh PP; Vendrely KM; Shoue DA; Checkley LA; McDew-White M; Button-Simons KA; Cassady Z; Sievert MAC; Foster GJ; Nosten FH; Anderson TJC; Ferdig MT, The extended recovery ring-stage survival assay provides a superior association with patient clearance half-life and increases throughput. Malar. J. 2020, 19 , 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.