

Abstract
ICI 56,780 (5) displayed causal prophylactic and blood schizonticidal activity (ED50 = 0.05 mg/kg) in rodent malaria models, but produced rapid acquisition of parasitological resistance in P. berghei infected mice.1 Herein we describe the synthesis of analogs of 5 with EC50s as low as 0.15 nM against multi-drug resistant P. falciparum. Optimal activity with low cross resistance indexes (RI) to atovaquone were achieved by introducing ortho-substituted aryl moieties at the 3-position of the 7-(2-phenoxyethoxy)-4(1H)-quinolone core.
SYNOPSIS TOC

Introduction
Malaria is amongst the most significant public health problems in the world. The disease occurs in tropical and sub-tropical climates and affects over 243 million people annually while claiming nearly one million lives in 2009.2, 3 P. falciparum and P. vivax are the two most prevalent species responsible for causing disease in humans.4 The development of curative antimalarial agents is difficult due to the various developmental stages of the parasite within the host. Following inoculation of sporozoites by an infected female Anopheles mosquito, the parasite must first undergo a proliferation period within the liver before the pathogenic infection of red blood cells ensues. The most effective drug for liver stage infections is primaquine, an 8-aminoquinoline that acts on actively growing liver stages and on the dormant forms known as hypnozoites. Hypnozoites of P. vivax can lay dormant in a host for weeks to years and upon reactivation cause a relapse. Discovery and development of drugs active against hypnozoites is limited by the lack of reliable high or medium throughput assays.5 In 2007 the Bill and Melinda Gates Foundation set an agenda for the elimination of malaria.2 Before this lofty goal can be achieved, new drugs are required that are safe and effective against liver and blood stage parasites simultaneously within the same host.
An additional difficulty for malaria drug development is the rapid emergence of multi-drug resistance. Many of the common antimalarials such as atovaquone, chloroquine, and more recently the artemisinin combination therapies (ACTs) have suffered from parasitological resistance being developed in many regions of the world, especially in Southeast Asia.6 Advances in drug discovery such as high-throughput screening, physicochemical property assessment, synthetic methodologies and improved in vivo efficacy protocols have allowed for re-examining old chemotypes or hits and for optimizing them to a more appropriate lead clinical candidate.7–11 Recently, endochin (1), a 4(1H)quinolone, and its related tetrahydroacridone analog (THA) floxacrine (2), were successfully optimized for antimalarial activity by substituting various benzenoid ring features and aryl moieties (3 and 4) while simultaneously assessing the physicochemical properties (Figure 1).9 Another such example is the 4(1H)-quinolone ester ICI 56,780 (5) which was found in 1970 to have antimalarial activity by Ryley and Peters (Figure 1).1 This compound possesses blood schizontocidal activity against P. berghei as well as prophylactic activity against P. cynomolgi sporozoite challenge assays. It was shown that Rhesus monkeys inoculated intravenously with P. cynomologi sporozoites and subsequently treated with compound 5 for 7 consecutive days had no relapse after 120 days of exposure, confirming potency against hypnozoites.12 Compound 5 was found to be curative at 15 mg/kg. Unfortunately, rapid selection of resistance was obtained after one passage in P. berghei infected mice leading to an abandonment of the compound.1
Figure 1.

Structures of historic and modern antimalarial compounds 1-6.
The in vivo anti-relapse activity in combination with the excellent blood stage activity of 5 shows great promise in developing a viable multi-stage antimalarial agent.1, 12A related set of 4-oxo-3-carboxyl analogs (6) were recently developed by using a parallel approach of SAR and pharmacologic characterization to design quinolones that were less prone to cross-resistance with atovaquone.11 Given the sparse number of chemotypes with proven anti-relapse activity we have explored the 7-(2-phenoxyethoxy)-4(1H)-quinolones (PEQs) scaffold to optimize SPR and blood stage antimalarial activity. Since the rapid induction of resistance reported in P. berghei was likely due to cytochrome b mutations, we also optimized the scaffold for potency against clinically relevant atovaquone resistant P. falciparum.
Results and Discussion
Synthetic Chemistry.
Compound 5 was first synthesized in order to obtain preliminary data on the PEQ scaffold. This compound was patented in 1968 by Bowie as an anti-malarial13 and in 1970 reported to have coccidiostatic activity by Mizzoni et al.14 The goal was to prepare more than 100 g of the intermediate aniline 12 to funnel into Conrad Limpach reaction sequences to generate analogs of 5. Iodination of a PEQ scaffold to prepare 3-aryl analogs would be achieved using our recently developed protocol.15 Furthermore, aniline precursors would be used in several iterative sequences to prepare structurally diverse analogs in the 6- and 7-positions.
The route to generate compound 5 began with 450 g of commercially available N-(3-hydroxyphenyl)acetamide that was transformed to 7 with butyryl chloride in pyridine (Scheme 1). Next a Fries-rearrangement was employed to arrive at compound 8 using AlCl3. Compound 8 was directly reduced to the butyl analog 10 in the original report, however, without access to a large-scale 500 psi hydrogenation apparatus and the reaction failing to work at 75 psi, an alternative route was employed.14 Conjugated ketones are much more stable to hydrogenolysis as compared to a benzylic alcohol.16 Therefore, compound 8 was reduced using NaBH4 to obtain benzylic alcohol 9 in moderate yield and high purity. Compound 9 was now much more prone to hydrogenolysis as compared to the conjugated ketone 8 and the hydrogenation in acetic acid at 60 psi occurred relatively smoothly to yield compound 10. Compound 11 was prepared through a simple alkylation using (2-bromoethoxy)benzene in high yield. The key aniline intermediate 12 was prepared via hydrolysis of the acetamido moiety in 11. Finally, compound 5 was synthesized in a two-step Gould-Jacobs sequence from 12.17 The compound was isolated via precipitation; however, the resulting material was not pure enough for accurate in vitro and in vivo tests. The solid was recrystallized in a mixture of DMF:methanol (4:1).
Scheme 1.

Synthesis of the PEQ 5 via key intermediate aniline 11.
aReaction conditions: (a) butyryl chloride, pyr. r.t. (b) AlCl3, 150–175°C 45min then 3h; (c) NaBH4, THF anhy., 0°C; (d) AcOH, 10% Pd/C, 60 psi, 36h; (e) NaH, DMF 30 min then (2bromoethoxy)benzene, 3h; (f) KOH (14 eq.), EtOH:H2O (9:1), reflux, 4h; (g) dimethyl 2(methoxymethylene)malonate, EtOH, reflux; (h) Ph2O, reflux, 12 min
Starting from aniline 13, prepared as in a previous report, a 6-chloro-7-methoxy-4(1H)quinolone bearing the β-dicarbonyl moiety (14) was synthesized using 1-ethyl 3-methyl 2acetylmalonate.9 A subset of 5 was prepared to determine the necessity of the methyl 3-benzoate substituent (Scheme 2, Table 1). The placement of a proton or ethyl group in the 3-postion as compared to a methyl 3-benzoate substituent would determine the importance of the β-dicarbonyl motif present in 5. Compound 12 was subjected to Conrad-Limpach conditions using two different 2-substituted βketoesters to generate compounds 15 and 16.
Scheme 2.

Synthesis of 4(1H)-quinolones 13-27.
a(a) ethyl acetoacetate or 2-ethyl acetoacetate, AcOH, benzene, Dean-Stark trap, reflux, overnight then Ph2O, reflux, 15min; (b) 1-ethyl 3-methyl 2-acetylmalonate, AcOH, benzene, Dean-Stark trap, reflux, overnight then Ph2O, reflux, 15min (c) Ac2O, AcOH; (d) corresponding alkyl halide, Cs2CO3, DMF, 48h; (e) KOH, EtOH:H2O (9:1), reflux; (f) BBr3; (g) Zn, AcOH, r.t., 4h; (h) 1,3-dibromo propane, Cs2CO3, r.t.; (i) N-Phenylpiperazine, K2CO3, DMF, r.t.; (j) Pd2(dba)3, SPHOS, K3PO4, DMF, Mesitylboronic acid, 130oC
Table 1.
EC50s of PEQ analogs

| ||||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Compound | R1 | R2 | R3 | R4 | EC50 W2 (nM) | EC50 TM90-C2B (nM) | RI | EC50 J774 (mM) |
|
| ||||||||
| 5 | -CO2Me |

|
-Bu | -H | 0.05 | 11.2 | 223 | 46 |
| 14 | -CO2Me | -MeO | -Cl | -H | 134 | 765 | 5.7 | 37 |
| 15 | -H |

|
-Bu | -Me | 216 | 44.1 | 0.20 | 28 |
| 16 | -Et |
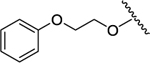
|
-Bu | -Me | 1.92 | 0.15 | 0.08 | 1 |
| 27 | -Et |

|
-H | -Me | 104 | 71.3 | 0.79 | 4 |
| 17 | -Et |

|
-Cl | -Me | 256 | 72.1 | 0.28 | >28 |
| 25 | -Et |

|
-Br | -Me | 203 | 43.5 | 0.21 | >25 |
| 18 | -Et |

|
-Me | -Me | 255 | 27.7 | 0.11 | >30 |
| 19 | -Et |

|
-MeO | -Me | 184 | 19.5 | 0.11 | >29 |
| 20 | -Et |

|
-Cl | -Me | 908 | 79.7 | 0.09 | 19 |
| 21 | -Et |
|
-Cl | -Me | 8450 | 776 | 0.09 | N.D. |
| 22 | -Et |

|
-Cl | -Me | 6130 | 6130 | 1.0 | N.D. |
| 23 | -Et |
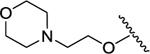
|
-Cl | -Me | 5090 | 726 | 0.14 | N.D. |
| 24 | -Et |

|
-Cl | -Me | 5050 | 604 | 0.12 | N.D. |
| 26 | -Et |

|
-Cl | -Me | 976 | 91.7 | 0.09 | >23 |
Dihydroartemisinin (DHA), chloroquine (CQ), and atovaquone (ATO) are internal controls for each in vitro assay. DHA (1.8 nM W2 and 0.9 nM TM90-C2B), CQ (131 nM for TM90-C2B and 162 nM for W2) and ATO (0.53 nM W2 and >170 nM TM90-C2B). N.D. not determined.
Next, a set of 3-ethyl-4(1H)-quinolones substituted at 6- or 7-positions were prepared. In contrast, the 3-benzoate 4(1H)-quinolones were not prepared to avoid structural overlap with antimicrobial β-dicarbonyl containing quinolones,18 which have shown to possess weak intrinsic potency as antimalarials.19 The 6- or 7-substitutions contained various solubilizing groups with different linker lengths. Starting from 5-amino-2-chlorophenol, 4(1H)-quinolones 17-24 were prepared (Scheme 2, Table 1). N-Acylation of 5-amino-2-substituted-phenols produced an intermediate acetamide, which could be alkylated using various alkyl halides. These intermediates were then hydrolyzed using KOH to arrive at the necessary anilines. The anilines were then cyclized using either 2-ethyl-β-ketoester to yield the corresponding 4(1H)-quinolones 17-24 (Scheme 2). An alternative route was used to prepare compounds 25-27 utilizing a commercially available di-substituted nitro precursor (Scheme 2). Compound 26 was initially synthesized via an acetamide intermediate as in Scheme 2 (17-24). Unfortunately, usage of 1,3-dibromopropane (conditions h) led to inseparable mixtures of the aniline required to prepare 26 as well as an O-allyl side product generated from elimination using Cs2CO3. Employment of 2-chloro-5-nitrophenol as the starting material led to overall improved yields and easier separation of elimination side products. The synthesis of compound 27 was initially attempted starting from 17 using several standard Pd hydrogenation conditions, however, a mixture consisting of 27 and a 4(1H)-quinolone product containing a partially reduced benzenoid ring was obtained. Previously, we observed that cross-couplings of 3-halo-4(1H)-quinolone with mesitylboronic acid yield mainly the proto-dehalogenated 4(1H)-quinolone.15 Based on these findings, 17 was heated in a Schlenk tube for 36 hours with several additions of mesitylboronic acid until the chlorine was all consumed generating compound 27 in high yield.
A small library of 3-aryl PEQs 31-44 was prepared by cyclizing aniline 12 using a Conrad-Limpach protocol followed by a regioselective iodination to generate 4(1H)-quinolone 28 (Scheme 3, Table 1), which was subjected to Suzuki-Miyaura cross-coupling conditions. The 3-halo-4(1H)-quinolone core can tolerate couplings with or without alkyl protection, however, depending on the nature of the boronic acid used, better yields are achieved starting from alkylated quinolones.15 Therefore, using O-methyl or O-ethyl-3-iodo-quinolines 29 or 30, the coupling could be employed successfully followed by chemoselective de-alkylation using HBr in refluxing acetic acid.20 It is worthy to note that this de-alkylation is a time sensitive reaction and unwanted bromination or de-alkylations may occur depending on the substituents of the 6- or 7-position. For example, when the 7-(2-phenoxyethoxy) group is present, a bromine replaces the phenoxy moiety when the reaction is left to reflux too long. A variety of boronic acids including ones containing an ortho substituent were utilized to generate 3-aryl quinolones.
Scheme 3.

Synthesis of 3-aryl PEQs
aReaction conditions: (a) KI (20% aq.), I2, 2M NaOH, MeOH, r.t. 2h (b) Cs2CO3, DMF, EtI / MeI, 0°C to r.t. 5h (c) Pd2(dba)3, SPHOS, K3PO4, DMF, ArB(OH)2, 110oC or Pd(PPh3)4, 2M Na2CO3, DMF, ArB(OH)2, 110°C (MW) (e) HBr / AcOH, reflux 1–2h
Antimalarial Activity and Cytotoxicity.
All synthesized quinolones were tested as previously reported for in vitro antimalarial activity against the clinically relevant multi-drug resistant malarial strains W2 (chloroquine and pyrimethamine resistant) and TM90-C2B (chloroquine, mefloquine, pyrimethamine and atovaquone resistant) and for cytotoxicity against J774 mammalian cells.9, 21 Generally, the PEQs do not display signs of cytotoxicity at concentrations lower than 20 µM rendering cytotoxicity indices (CI=EC50(J774)/EC50(TM90-C2B)) of hundred or more. These results indicate that the majority of the PEQs are selective and nontoxic agents.
The emergence of resistance and cross resistance with atovaquone is a concern for new antimalarials that target the parasite’s mitochondria (e.g., atovaquone). For the structure-activity relationship study, the resistance index (RI), calculated as the ratio of the effective concentrations for TM90-C2B and W2 (RI = EC50(TM90-C2B)/EC50(W2)), was also taken into account.9 Compounds with an RI = 0.3 – 3.0 are considered acceptable in regards to risk of cross resistance with atovaquone, whereas compounds with an RI > 10 or < 0.1 are likely to have clinically relevant levels of cross resistance with atovaquone.22, 23
Structure-Activity Studies.
First, compound 5 was shown to have excellent activity against W2 and TM90-C2B with EC50s of 0.05 nM and 11.17 nM respectively (Table 1). However, the potency difference between the two strains yielding an RI = 223 is a concern as the atovaquone resistance mutations in cytochrome b can be rapidly selected in vivo.22 Therefore a small series of PEQs were designed to examine the 3-, 6- and 7-susbtitutents of compound 5 with the aim to improve potency against both atovaquone sensitive and resistant P. falciparum. Interestingly, the 6-chloro-7-methoxy analog 14 lead to a 70-fold or larger decrease in antimalarial activity for both strains, while PEQ 15 displayed a 4000-fold loss in activity for W2 as compared to only a 4-fold potency decrease for TM90C2B. Conversely, introduction of an ethyl group at the 3-position in 15 provided PEQ 16 with restored EC50 values of 1.92 nM against W2 and 150 pM against TM90-C2B. The potency of compound 16 shows a reversed preference for TM90-C2B with a RI = 0.08, which stands in sharp contrast to compound 5 that inhibits W2 approximately 220-fold more than TM90-C2B. This result led us to prepare a series of 6- and 7-substituted 2-methyl-4(1H)-quinolones containing an ethyl group in the 3position.
First, a subset was prepared to probe the role of the 6-butyl group of 16. Complete removal of the butyl group generated compound 27 with EC50s of 104 nM and 71.3 nM against W2 and TM90-C2B respectively. In comparison to 27, 6-chloro- or 6-bromo-substituted PEQs 17 and 25 displayed slightly reduced activities against W2, while their potencies for TM90-C2B were unaffected or slightly improved. PEQs 18 and 19 substituted with a methyl or a methoxy group in 6-position were also shown to be less active against W2, but approximately 3-fold more potent against TM90-C2B as compared to compound 27. Next, a subset of 2-methyl-3-ethyl-6-chloro-substituted PEQs 20-26 were examined, in which the group at the 7-position was varied. With the exception methoxymethyl ether 20 and piperazine 26, all others 21-25 were 5–30 times less active against W2 and TM90-C2B in comparison to their reference compound 17. PEQs 20 and 26 were similar to 17 in potency against TM90-C2B and approximately four-fold less active against W2. These results indicate that the 7-(2-phenoxyethoxy) moiety greatly affects antimalarial activity and that the 3-ethyl-susbtituted PEQs display a more favorable RI values in comparison to methyl carboxylate 5.
While retaining the 6-butyl-7-(2-phenoxyethoxy) moiety, a small series of 3-aryl analogs (compounds 31-43) was prepared and tested against W2 and TM90-C2B (Table 2). Generally, PEQs 38–43 containing an ortho-substituted aromatic ring in 3-position were approximately 10-fold more potent compared to the 3-aryl-substituted analogs 31-37. Ortho-substituted 3-aryl-analogs 38-43 were also more potent against the W2 strain whereas the 3-aryl PEQs 31-37 were more potent against TM90-C2B. Initially, the 3-phenyl analog 31 was prepared, which displayed poor EC50s of 1072 nM against W2 and 764 nM against TM90-C2B. Next, 3-para- and 3-meta-pyridyl analogs 32 and 33 were shown to have moderate activity in low μM or high nM range. Trifluoromethyl phenyl and trifluoromethoxy phenyl substituted PEQs 34 and 35 were similar to compounds 32 and 33 in activity. The bi-aryl and benzyl aryl analogs 36 and 37 were inactive. Of the ortho-substituted 3-aryl analogs, compounds 38, 40 and 42 were the least promising with high EC50s against one or both strains rendering poor RI values. Fluorotrifluoromethyl-phenyl-substituted PEQ 41 displayed the best antimalarial activities with EC50s of 27.9 nM and 31.0 nM against W2 and TM90-C2B yielding an RI = 1.1. Interestingly, analog 43 substituted with 3,5-dimethylisoxazolyl in 3-position was also very potent against W2 with an EC50 of 27.0 nM and approximately five times less potent against TM90-C2B. Overall, though the 3-aryl series was less potent compared to compound 5, several analogs such as fluoro-trifluoromethyl-phenyl-substituted PEQ 41 or isoxazole 43 showed promise in terms of antimalarial activity and acceptable RI value. These findings are similar to our endochin optimization studies, in which the aryl substituent of a different 4(1H)-quinolone pharmacophore has been shown to be the best suited for the development of a lead devoid of cross resistance with atovaquone.9 Although these new PEQ analogs show promise, additional studies will be required to assess their potential for rapid resistance selection which has been a concern for several cytochrome b inhibitors. Additional studies also are required to determine if the improved PEQs maintain the potent activity against liver stages as the lead compound 5.
Table 2.
EC50s of 3-aryl PEQs
| Compound | Ar | EC50 W2 (nM) | EC50 TM90-C2B (nM) | RI | EC50 J774 (μM) |
|---|---|---|---|---|---|
|
| |||||
| 31 |
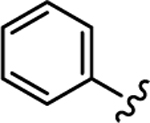
|
1070 | 764 | 0.71 | 23 |
| 32 |

|
3150 | 1016 | 0.32 | 23 |
| 33 |
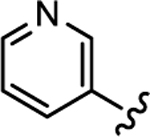
|
1350 | 588 | 0.44 | 23 |
| 34 |

|
509 | 2450 | 4.82 | 20 |
| 35 |

|
3220 | 2510 | 0.78 | 20 |
| 36 |

|
1390 | 1002 | 0.72 | 19 |
| 37 |
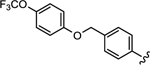
|
4050 | 4050 | 1.00 | 16 |
| 38 |

|
58.5 | 475 | 8.12 | 16 |
| 39 |

|
71.8 | 140 | 1.95 | 20 |
| 40 |

|
907 | 1630 | 1.79 | 22 |
| 41 |

|
27.9 | 30.9 | 1.11 | 19 |
| 42 |
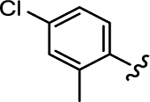
|
68.7 | 542 | 7.89 | >21 |
| 43 |

|
27.0 | 128 | 4.73 | 22 |
Dihydroartemisinin (DHA), chloroquine (CQ), and atovaquone (ATO) are internal controls for each in vitro assay. DHA (1.8 nM W2 and 0.9 nM TM90-C2B), CQ (131 nM W2 and 162 nM TM90-C2B), and ATO (0.53 nM W2 and >170 nM TM90-C2B).
Conclusions.
A series of 29 novel PEQ analogs with varying substitution at the 2-,3-,6-, and 7- positions were synthesized and assessed for anti-malarial activity against the clinically relevant strains TM90-C2B and W2, with the objective to improve SAR and reduce cross resistance with atovaquone. The most potent anti-malarial activities were obtained when the 3-position contained an ethyl group or a fluoroaryl moiety. With the exception of the original compound 5, most of the PEQ analogs lacking the 7-(2-phenoxyethoxy) substituent showed significant differences in EC50s against the two strains providing unfavorable RIs. For 3-ethyl-substituted PEQs, the best activities and RI values were obtained with compounds containing a 2-phenoxyethoxy moiety in 7-position, whereas the group in 6-position produced the activity order Bu>>MeO>Me>Br>Cl>H for TM90-C2B and Bu>H>MeO>Br>Me>Cl for W2 providing a strain preference that had minor dependence on the moiety in 6-position. Similarly, 3-aryl-substituted PEQs displaying good potencies against both strains and acceptable RIs contained the butyl in 6-position and the 2-phenoxyethoxy group in 7-position. Best activities and acceptable RIs were obtained with PEQs 41 and 43 containing in 3-position an ortho-substituted aromatic ring such as a fluoro-trifluoromethyl-phenyl or a 3,5-dimethylisoxazolyl.
In summary the 3-aryl or 3-ethyl-substituted PEQs (e.g., 14, 41, and 43) are less potent against W2 and TM90-C2B than the original compound 5, nevertheless these compounds have been improved significantly by reducing cross resistance in the clinically relevant atovaquone resistant TM90-C2B parasite. Our data therefore suggests these compounds have potential for further optimization to identify PEQs optimal for in vivo liver stage and blood stage efficacy studies.
Supplementary Material
ACKNOWLEDGMENT:
We thank the Medicines for Malaria Venture (MMV) and the Florida Center of Excellence for Drug Discovery and Innovation (CDDI) for financial support.
a Abbreviations:
- EC50
half maximal effective concentration
- ACT
artemisinin combination therapy
- WRAIR
Walter Reed Army Institute of Research
- SAR
structure-activity relationship
- SPR
structure-property relationship
- SPHOS
dicyclohexyl(2’,6’-dimethoxybiphenyl-2-yl)phosphine
- DCM
dichloromethane
- Pd2(dba)3
Tris(dibenzylideneacetone)dipalladium (0)
- Ph
phenyl
- DMF
N,N-dimethylformamide
- RPMI
Roswell Park Memorial Institute medium
- RI
resistance index
- Ac
acetyl
- r.t.
room temperature
- CI
cytotoxicity index
- MW
microwave
- ED50
half maximal effective dose
- PEQ
7-(2-Phenoxyethoxy)-4(1H)-quinolones
Footnotes
Experimental Section Compounds were prepared using General Procedures A-J (see supporting information). The purity of each compound was ≥95% via HPLC analysis that was synthesized and tested for antimalarial activity.
Supporting Information Available: Experimental details of the synthesis of compounds 7-43 including all general procedures as well as 1H NMR, 13C NMR, and 19F NMR characterizations for all tested compounds as well as HRMS. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Ryley JF; Peters W Antimalarial activity of some quinolone esters. Ann. Trop. Med. Parasitol. 1970, 64, 209–222. [DOI] [PubMed] [Google Scholar]
- 2.Wells TNC; Alonso PL; Gutteridge WE New medicines to improve control and contribute to the eradication of malaria. Nat. Rev. Drug Discovery 2009, 8, 879–891. [DOI] [PubMed] [Google Scholar]
- 3.World Malaria Report 2009; The World Health Organization: Geneva, S., 2008; http://www.who.int/whosis/whostat/EN_WHS09_Full.pdf. [Google Scholar]
- 4.White NJ; Nosten F; Looareesuwan S; Watkins WM; Marsh K; Snow RW; Kokwaro G; Ouma J; Hien TT; Molyneux ME; Taylor TE; Newbold CI; Ruebush TK 2nd; Danis M; Greenwood BM; Anderson RM; Olliaro P Averting a malaria disaster. Lancet 1999, 353, 1965–1967. [DOI] [PubMed] [Google Scholar]
- 5.Mazier D; Renia L; Snounou G A pre-emptive strike against malaria’s stealthy hepatic forms. Nat. Rev. Drug Discovery 2009, 8, 854–864. [DOI] [PubMed] [Google Scholar]
- 6.Dondorp AM; Nosten F; Yi P; Das D; Phyo AP; Tarning J; Lwin KM; Ariey F; Hanpithakpong W; Lee SJ; Ringwald P; Silamut K; Imwong M; Chotivanich K; Lim P; Herdman T; Sam An S; Yeung S; Singhasivanon P; Day NPJ; Lindegardh N; Socheat D; White NJ Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2009, 361, 455467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vennerstrom JL; Arbe-Barnes S; Brun R; Charman SA; Chiu FCK; Chollet J; Dong Y; Dorn A; Hunziker D; Matile H; McIntosh K; Padmanilayam M; Santo Tomas J; Scheurer C; Scorneaux B; Tang Y; Urwyler H; Wittlin S; Charman WN Identification of an antimalarial synthetic trioxolane drug development candidate. Nature (London, U. K.)) 2004, 430, 900–904. [DOI] [PubMed] [Google Scholar]
- 8.O’Neill PM; Amewu RK; Nixon GL; El Garah FB; Mungthin M; Chadwick J; Shone AE; Vivas L; Lander H; Barton V; Muangnoicharoen S; Bray PG; Davies J; Park BK; Wittlin S; Brun R; Preschel M; Zhang K; Ward SA Identification of a 1,2,4,5-Tetraoxane Antimalarial Drug-Development Candidate (RKA 182) with Superior Properties to the Semisynthetic Artemisinins. Angew. Chem., Int. Ed. 2010, 49, 5693–5697, S5693/1-S5693/10. [DOI] [PubMed] [Google Scholar]
- 9.Cross RM; Monastyrskyi A; Mutka TS; Burrows JN; Kyle DE; Manetsch R Endochin Optimization: Structure-Activity and Structure-Property Relationship Studies of 3-Substituted 2-Methyl-4(1H)-quinolones with Antimalarial Activity. J. Med. Chem. 2010, 53, 7076–7094. [DOI] [PubMed] [Google Scholar]
- 10.Winter RW; Kelly JX; Smilkstein MJ; Dodean R; Hinrichs D; Riscoe MK Antimalarial quinolones: Synthesis, potency, and mechanistic studies. Exp. Parasitol. 2008, 118, 487497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y; Guiguemde WA; Sigal M; Zhu F; Connelly MC; Nwaka S; Guy RK Synthesis and structure-activity relationships of antimalarial 4-oxo-3-carboxyl quinolones. Bioorg. Med. Chem. 2010, 18, 2756–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Puri SK; Dutta GP Quinoline esters as potential antimalarial drugs: effect on relapses of Plasmodium cynomolgi infections in monkeys. Trans. R. Soc. Trop. Med. Hyg. 1990, 84, 759–760. [DOI] [PubMed] [Google Scholar]
- 13.Bowie RA; Grant MS; Jones WGM 4-Quinolinol-3-carboxylate coccidiostats. 1966309741120870, 19660711., 1968. [Google Scholar]
- 14.Mizzoni RH; Goble F; Konopka E; Gelzer J,; Szanto J; Maplesden DC; Brown JE; Boxer J; Zaunius G; Ziegler JB; DeStevens G Structure and anticoccidial activity among some 4hydroxyquinolinecarboxylates. J. Med. Chem. 1970, 13, 870–878. [DOI] [PubMed] [Google Scholar]
- 15.Cross RM; Manetsch R Divergent Route to Access Structurally Diverse 4-Quinolones via Mono or Sequential Cross-Couplings. J. Org. Chem. 2010, 75, 8654–8657. [DOI] [PubMed] [Google Scholar]
- 16.Hattori K; Sajiki H; Hirota K Chemoselective control of hydrogenation among aromatic carbonyl and benzyl alcohol derivatives using Pd/C(en) catalyst. Tetrahedron 2001, 57, 4817–4824. [Google Scholar]
- 17.Curran TT Gould-Jacobs reaction. Name React. Heterocycl. Chem. 2005, 423–436. [Google Scholar]
- 18.Mitscher LA Bacterial Topoisomerase Inhibitors: Quinolone and Pyridone Antibacterial Agents. Chem. Rev. (Washington, DC, U. S.) 2005, 105, 559–592. [DOI] [PubMed] [Google Scholar]
- 19.Huse H; Whiteley M 4-Quinolones: Smart Phones of the Microbial World. Chem. Rev. (Washington, DC, U. S.) 2011, 111, 152–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reid CS; Patrick DA; He S; Fotie J; Premalatha K; Tidwell RR; Wang MZ; Liu Q; Gershkovich P; Wasan KM; Wenzler T; Brun R; Werbovetz KA Synthesis and antitrypanosomal evaluation of derivatives of N-benzyl-1,2-dihydroquinolin-6-ols: Effect of core substitutions and salt formation. Bioorg. Med. Chem. 2011, 19, 513–523. [DOI] [PubMed] [Google Scholar]
- 21.Desjardins RE; Canfield CJ; Haynes JD; Chulay JD Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob. Agents Chemother. 1979, 16, 710–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Looareesuwan S; Viravan C; Webster HK; Kyle DE; Hutchinson DB; Canfield CJ Clinical studies of atovaquone, alone or in combination with other antimalarial drugs, for treatment of acute uncomplicated malaria in Thailand. Am. J. Trop. Med. Hyg. 1996, 54, 62–66. [DOI] [PubMed] [Google Scholar]
- 23.Milhous WK; Gerena L; Kyle DE; Oduola AMJ In vitro strategies for circumventing antimalarial drug resistance. Prog. Clin. Biol. Res. 1989, 313, 61–72. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.