

Abstract
Metabolic engineering and multisubunit protein production necessitate the expression of multiple genes at coordinated levels. In bacteria, genes for multisubunit proteins or metabolic pathways are often expressed in operons under the control of a single promoter; expression of the genes is coordinated by varying transcript stability and the rate of translation initiation. We have developed a system to place multiple genes under the control of a single promoter and produce proteins encoded in that novel operon in different ratios over a range of inducer concentrations. RNase E sites identified in the Rhodobacter capsulatus puf operon and Escherichia coli pap operon were separately placed between the coding regions of two reporter genes, and novel secondary structures were engineered into the 5′ and 3′ ends of the coding regions. The introduced RNase E site directed cleavage between the coding regions to produce two secondary transcripts, each containing a single coding region. The secondary transcripts were protected from exonuclease cleavage by engineered 3′ secondary structures, and one of the secondary transcripts was protected from RNase E cleavage by secondary structures at the 5′ end. The relative expression levels of two reporter genes could be varied up to fourfold, depending on inducer concentration, by controlling RNase cleavage of the primary and secondary transcripts. Coupled with the ability to vary translation initiation by changing the ribosome binding site, this technology should allow one to create new operons and coordinate, yet separately control, the expression levels of genes expressed in that operon.
In prokaryotes, expression of genes for multistep pathways or for production of multisubunit proteins is often controlled by regulating the posttranscriptional processing of a polycistronic mRNA containing the coding regions for all the enzymes in that pathway or all subunits in a multisubunit protein. This type of control eliminates the need for multiple promoters of different strengths for each gene. To produce enzymes at appropriate levels, the cell balances translation efficiency with the rate that the message is inactivated by RNases.
These nucleases can be classified into two categories by comparing their mechanisms of action and role in mRNA decay. Two endonucleolytic activities are responsible for bulk mRNA processing—RNase III and RNase E (3, 10). RNase III cleaves mRNA at a weak consensus sequence within double-stranded regions of an RNA (9, 31). This indicates the role of RNase III in RNA processing, as it can cleave stem regions of RNA secondary structures (30, 31). RNase E is responsible for bulk inactivation of mRNA (14). The nuclease scans the mRNA transcript in a 5′-to-3′ direction and cleaves within AU-rich segments (1, 23, 24, 34). It is generally recognized that RNase E requires a free 5′ end to bind to the mRNA before scanning the transcript for cleavage sites (11–13, 15). The two exonucleases responsible for bulk mRNA degradation into mononucleotides are RNase II and polynucleotide phosphorylase (25). These enzymes degrade mRNA in a processive 3′-to-5′ direction, which indicates their role in degradation rather than inactivation (1).
For the majority of mRNA species, the initial cleavage within a transcript functionally inactivates the mRNA and is followed by 3′-to-5′ exonuclease activity (2). It has been shown that hairpin structures at the 3′ end protect the mRNA from degradation by exoribonucleases (1, 10, 26), and hairpins at the 5′ end protect from initial inactivation by endoribonucleases (11–13). Although there are a number of commercial products that contain 3′ hairpins to protect mRNA from exonucleases, there have been few attempts to design 5′ hairpins and recognition sites for endoribonucleases to alter mRNA stability and affect gene expression (2, 6, 7, 12, 13).
An expression system was developed that allows for the introduction of synthetic DNA cassettes into the region between the transcription and translation start sites of a gene of interest. Upon transcription, the 5′ end of the mRNA corresponding to the synthetic cassette forms a hairpin that protects the mRNA from endonucleolytic cleavage (6, 7). Depending on the structure of the inserted hairpin, the hairpin-containing mRNA exhibits half-lives between 3 and 10 times that of the mRNA with no hairpin, resulting in increases in mRNA and protein levels (8). These results indicate that it is possible to engineer mRNA stability as an additional means of controlling gene expression.
In this work, we constructed an expression system that allows one to vary the expression levels of two genes, both of which are under the control of the same promoter, by introducing DNA cassettes encoding mRNA secondary structures and RNase cleavage sites at various locations in the operon. The design for this decoupled, dual-gene expression system was derived from two native systems: the puf operon of Rhodobacter capsulatus (1, 21, 22) and the pap operon of Escherichia coli (5, 29). The expression system was tested with a combination of mRNA secondary structures at the 5′ and 3′ ends of the two coding regions and putative RNase E cleavage sites between the coding regions to determine if it is possible to independently vary expression of the two genes. Northern blotting and primer extension analysis indicate that the primary transcript was cleaved into two secondary transcripts, each containing a coding region, when a putative RNase E site was placed between the coding regions. Northern blotting and enzyme assays indicate that expression of the two genes was effectively decoupled and that the relative expression levels of the two genes varied with the mRNA secondary structures and RNase E sites in the operon.
MATERIALS AND METHODS
Bacterial strains, media, chemicals, and enzymes.
E. coli DH10B (Gibco BRL) was used for all cloning steps. E. coli JS4 (Bio-Rad) was used for Northern blot analysis and enzyme assays (Table 1).
TABLE 1.
Strains and plasmids used
| Strain or plasmid | Phenotype/description | Reference or source |
|---|---|---|
| DH10B | F−mcrA Δ(mrr-hsdRMS-mcrBC) φ80dlacZΔM15 ΔlacX74 deoR recA1 endA1 araD139 Δ(ara, leu)7697 galU galK λ−rpsL nupG | Gibco BRL |
| JS4 | F−araD139 Δ(ara, leu)7697 Δ(lac)χ74galU galK hsdR2 (rk− mk−) mcrA mcrBC rpsL (Strr) thi recA1 | Bio-Rad |
| pGFPuv | pUC-based plasmid (pUC19 ori); gfpuv gene under lac promoter control; Ampr | Clontech |
| pGFPuvm | pGFPuv with silent mutation in coding region to remove SalI restriction site and improve translation efficiency | This study |
| pGFPuvm2 | pGFPuvm with silent mutation in coding region to remove XhoI restriction site and improve translation efficiency | This study |
| pTC40 | pBAD24-based plasmid (pBR322 ori); lacZ under araC/PBAD promoter control; Ampr | 6 |
| pTC40m | pTC40 with mutation in pretranslation region to replace Asp718 with NheI restriction site | This study |
| p50gl | pTC40 with coding region from pGFPuvm2 and pretranslation region from pTC40m inserted 5′ of lacZ untranslated region | This study |
| p50gΔl | p50gl with lacZ coding region removed | This study |
| p50HP4gΔl | p50gΔl with HP4 inserted 5′ of gfp | This study |
| p50HP17gΔl | p50gΔl with HP17 inserted 5′ of gfp | This study |
| p60gl | p50gl with HP1 inserted 3′ of gfp and RNase E site (E1) inserted 5′ of lacZ | This study |
| p60gHP4l | p60gl with HP4 inserted 5′ of lacZ | This study |
| p70gl | p50gl with HP1 inserted 3′ of gfp and RNase E site (E2) inserted 5′ of lacZ | This study |
| p70gHP4l | p70gl with HP4 inserted 5′ of lacZ | This study |
| pGEM-4z | Commercial transcription vector (SP6/T7 promoters); multicloning site within lacZ′; Ampr | Promega |
| pTC01 | pGEM-4z with 5′ section of lacZ coding region from pTC40 inserted into multicloning site | This study |
| pCS01 | pGEM-4z with gfp coding region from pGFPuvm2 inserted into multicloning site | This study |
| pCS02 | pGEM-4z with coding region from p60gHP4l inserted into multicloning site | This study |
Luria-Bertani (LB) medium was made as described by Maniatis et al. (32). C medium (18) was supplemented with 3.4% glycerol, 1.0% Casamino Acids, and micronutrients (28). Ampicillin, used at a concentration of 100 μg/ml, and arabinose were purchased from Fisher Scientific. Diethyl pyrocarbonate and rifampin were obtained from Sigma.
Restriction enzymes were purchased from Roche and New England Biolabs. T4 DNA ligase and High-Fidelity PCR enzyme mix were obtained from Roche. Pfu Turbo polymerase, DNase, ribosomal RNasin, SP6 RNA polymerase, and avian myeloblastosis virus reverse transcriptase were purchased from Promega. T4 polynucleotide kinase was obtained from New England Biolabs. DNA sequencing kits were purchased from USB.
Plasmid construction.
The base dual-gene plasmid containing both gfp and lacZ (p50gl) was constructed in several cloning steps. Primers used in each PCR step were synthesized by Genemed Synthesis, Inc. DNA amplification was performed with High-Fidelity PCR enzyme mix. All PCRs were performed with 20 mM Tris-HCl (pH 8.3), 30 mM KCl, 1.5 mM MgCl2, 200 μM deoxynucleoside triphosphates (dNTPs), 300 μM primers, 0.5 μg of template, and 2.6 U of enzyme. Cycle times and temperatures followed those suggested by the enzyme manufacturer. All plasmids used in this study are described in Table 1.
PCR was performed on pGFPuv (Clonetech) using primer 1 (5′-TGCCTGCAGGTCGACTCTAGAGGATCCCC-3′) and primer 2 (5′-TCTTTCGAAAGGGCAGATTGTGTGGACAGGTAATGGTTGTC-3′) to remove the SalI restriction site in gfpuv and to replace codons to improve translation efficiency. The 669-bp PCR product and plasmid pGFPuv were digested with PstI and BstBI and ligated to form pGFPuvm by using T4 DNA ligase. The ligation products were electroporated into E. coli DH10B. The transformation products were plated onto LB agar plates containing ampicillin, arabinose, and 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) (Gibco BRL). To determine if the SalI site was present in pGFPuvm, plasmid was isolated from various colonies (32), digested with SalI, and analyzed by agarose gel electrophoresis.
A second round of PCR was performed on pGFPuvm using primers 1 and 10 (5′-GATGTATACATTGTGTGAGTTATAGTTGTACTCCAGTTTGTGTCCGAGAA-3′) to remove the XhoI restriction site in gfpuvm and to replace codons to improve translation efficiency. The 496-bp PCR product was substituted for the XhoI-containing fragment in pGFPuvm to form pGFPuvm2 by digesting both vector and insert with PstI and Bst1107I and ligating the two DNA fragments. Screening for pGFPuvm2 was performed by digesting with XhoI.
PCR was also used to replace the Asp718 restriction site in pTC40 with NheI using primer 11 (5′-CTCCATACGTCGACAGCTAGCGTATTTTGGATG-3′) and primer 12 (5′-GTCGGTTTATGCAGCAACGAGACGTCAC-3′). The 675-bp PCR product was cloned into pTC40 to form pTC40m by digesting both vector and insert with NheI and AatII and ligating the two fragments. Screening for pTC40m was performed by digesting with NheI.
The final step in the construction of the dual-gene operon was to place lacZ after gfp using splicing by overlap extension (19, 20). DNA amplification was performed with Pfu Turbo polymerase. Primer 13 (5′-TGGATGATAACGAGGCGCAAAAAATGAGTAAAGGAGAAGAACTTTTCACT-3′) and primer 4 (5′-AGCGGTACCAGCAGATCTTATTTGTAGAGCTCATCCATGCC-3′) were designed to amplify gfp from template pGFPuvm2, while primers 14 (5′-CGCTAACCAAACCGGTAACC-3′) and 15 (5′-TTTTTGCGCCTCGTTATCATCCA-3′) were designed to amplify lacZ from template pTC40m. During the splicing by overlap extension, the two amplified segments connected to form a single 999-bp product. This product was cloned into pTC40 to form p50gl by digesting both vector and insert with BstEII and Asp718 and ligating the two fragments. Cells that fluoresced were screened for p50gl.
The gfp single-gene control plasmid (p50gΔl) was constructed by digesting p50gl with PvuII and purifying by agarose gel electrophoresis and self-ligating the 5,559-bp product. The correct plasmid was found using blue/white screening by plating transformation products on LB agar plates containing arabinose and X-Gal and selecting white colonies.
The plasmid used to produce the gfp probe (pCS01) was constructed by digesting pGFPuvm2 and pGEM-4z (Promega) with EcoRI and Asp718. The 550-bp insert was ligated into pGEM-4z. The correct plasmid was found using blue/white screening.
The control plasmid for the primer extension studies (pCS02) was constructed by digesting p60gHP41 and pGEM-4z with PstI and SalI. The 4,000-bp insert containing gfp and lacZ was ligated into pGEM-4z. The correct plasmid was found using blue/white screening.
DNA cassettes.
The various DNA cassettes were synthesized (Genemed Synthesis, Inc.) as two complementary DNA oligonucleotides. These oligonucleotides were annealed at high concentration by heating to above their annealing temperature and ramping the temperature down to 20°C. The DNA cassettes were inserted into the plasmid using unique restriction sites at the ends of the cassettes and within the plasmid itself. The plasmid and the cassettes were digested and ligated together. The ligation products were electroporated into E. coli DH10B. The transformed cells containing the plasmid with the cassette insert were screened using a restriction site within the cassette.
Enzyme assays.
To determine β-galactosidase and green fluorescent protein (GFP) activities, C medium was inoculated with a stock culture to an optical density at 600 nm (OD600) of 0.0015 and grown at 30°C. At an OD600 of 0.20, samples were removed and placed on ice. β-Galactosidase assays were performed as described previously (6, 27). β-Galactosidase activity is reported in Miller units. GFP activity was determined by measuring the relative fluorescence of a 2-ml sample in a VersaFluor fluorometer (Bio-Rad). This fluorescent reading was divided by the OD600 reading for the sample to obtain the GFP specific activity.
Northern blot analysis.
Northern blot analysis was performed to determine transcript stabilities. C medium was inoculated with a stock culture of E. coli JS4 containing the various plasmids to an OD600 of 0.016 and grown at 30°C. At an OD600 of 0.05, arabinose was added to a final concentration of 0.1%. At an OD600 of 0.20, rifampin was added to a final concentration of 2 mg/ml. RNA extraction was performed on samples as described previously at an OD600 of 0.20 (6).
The lacZ and gfp probes were synthesized by digesting pTC01 and pCS01 with PvuII and NcoI. The fragments were separately combined with SP6 polymerase and radiolabeled [α-32P]CTP (Amersham) and nonradiolabeled ribonucleotide triphosphates (Promega).
Prior to the Northern blotting, all equipment was treated with 3% hydrogen peroxide and all solutions were treated with dimethyl pyrocarbonate. Northern blot analysis was conducted as described previously (2). Labeled probe was added to the hybridization solution at a concentration of 106 cpm/ml. Membranes were hybridized with 5 ml of the probe hybridization solution. The blots were visualized using a PhosphorImager (Molecular Dynamics). The intensities were recorded using IPLab Gel software and were plotted as a function of time for several different exposure times to ensure linearity of the band intensity with exposure time. Northern blots were repeated at least twice on samples to ensure reproducibility. Uncertainties in half-life calculations were determined according to the method described by Taylor (33).
Half-life calculations.
Half-lives of the various transcripts were determined according to the following model:
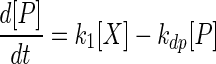 |
1 |
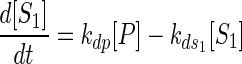 |
2 |
 |
3 |
where [P] is the concentration of the primary transcript, [S1] and [S2] are the concentrations of the two secondary transcripts, kdp is the decay constant for the primary transcript, kds1 and kds2 are the decay constants of the secondary transcripts, and k1[X] is the rate of synthesis of the primary transcript. This model assumes that the processing of the primary transcript is the only source of the secondary transcripts and that decay of the transcripts is proportional to the concentrations of the transcripts present. In the experiments reported here, k1[X] is set equal to zero, because transcription is stopped with rifampin. Thus, the half-life of the primary transcript can be calculated from the simplified form of equation 1.
We determined the half-lives of the secondary transcripts using two different methods and assumptions. The first method assumes that kdp is much larger than kds1 and kds2 or that the primary transcript is degraded fast relative to the secondary transcripts. With this assumption, the source term for the secondary transcripts in equations 2 and 3 can be ignored relative to the decay term. The half-lives of the secondary transcripts can be calculated from the simplified form of equations 2 and 3.
The assumptions made in the first method are correct only when no message is being produced (that is, for stable secondary transcripts and relatively unstable primary transcripts). The second method used to determine half-lives of the secondary transcripts makes no assumptions about the stability of the primary transcript and solves the equations using forward differences. This method provides more accurate half-life values for secondary transcripts arising from primary transcripts that have approximately the same half-life.
Primer extension analysis.
An oligonucleotide, RT-primer 1 (5′-CGACGGGATCTGCGATAGCTGTC-3′), was synthesized (Genemed Synthesis, Inc.) to bind 50 nucleotides downstream of the suspected RNase E cleavage site location. RT-primer 1 was labeled at its 5′ end with [γ-32P]ATP (ICN Biochemicals) using T4 polynucleotide kinase. The labeled oligonucleotide, at a concentration of 106 cpm/μl, was annealed to 25 μg of the RNA isolated from the cellular extract (6). RNA samples were taken at an OD600 of 0.20. Primer extension was performed using avian myeloblastosis virus reverse transcriptase and 5 μg of total RNA in 50 mM KCl, 12 mM DTT, 3 mM spermidine, 10 mM MgCl2, and 1.1 mM dNTPs. The reactions were stopped after 30 min with an 8 M urea–0.1% sodium dodecyl sulfate stop buffer-denaturing buffer solution. To determine the locations of the stop bands, a DNA sequencing ladder was generated by annealing the labeled oligonucleotide at a concentration of 5 × 105 cpm/μl to 5 μg of the corresponding denatured plasmid. Plasmid DNA was denatured in a 0.2 M NaOH–0.2 mM EDTA solution at 37°C for 30 min. Denatured DNA was neutralized by adding sodium acetate to a final concentration of 0.3 M. The dideoxynucleotide (Sanger) method of sequencing was performed using Sequenase Kit Version 2.0. Protocol for sequencing followed that provided by the manufacturer. These samples were separated in individual lanes on a denaturing 7 M urea–6% polyacrylamide gel in Tris-borate-EDTA (Stratagene CastAway Sequencing System). This 16- by 7-in. gel was run for 1.5 h at 40 W and then dried for 30 min.
The sequence was read from the gel by exposing it to film for an appropriate amount of time and developing the film. The film was scanned into Adobe Photoshop using VistaScan software. Because the amount of RNA that could be added to the reactions was limited by poisoning of the primer extension reaction from the total RNA present in the samples, it was not possible to vary the intensity of the primer extension bands. Therefore, bands for primer extension and DNA sequencing differed greatly in intensity, and the images were adjusted separately. Image intensities were adjusted, first to read the sequence ladders. The intensities of the bands in the lanes containing the three reverse transcriptase reactions were adjusted equally to visualize the bands in these lanes. These increased intensity lanes were placed next to their corresponding DNA sequencing ladder and lower intensity lanes on the original image for comparison.
Cleavage locations were determined where reverse transcriptase would no longer extend the transcript. To ensure that the stop bands were not a result of secondary structure, the procedure was repeated with 10 ng of RNA synthesized in vitro. In vitro-synthesized RNA was obtained by digesting pCS02 with PvuII. The fragments were combined with SP6 RNA polymerase and ribonucleotide triphosphates.
RESULTS AND DISCUSSION
Design of dual- and single-gene systems.
A synthetic operon containing two genes was designed to allow introduction of secondary structures and RNase cleavage sites in the form of DNA cassettes at various locations in the operon and easy evaluation of the effects of these changes on gene expression. The system was constructed in a high-copy-number plasmid that has an arabinose-inducible araBAD promoter (PBAD) and ampicillin resistance marker (Fig. 1A). The gfp gene was placed upstream of the lacZ gene. Unique restriction sites were placed at the 5′ end of each untranslated region for the introduction of DNA cassettes that, when transcribed, would form secondary structures. These sites were placed out of range of the ribosome binding sites so that secondary structures would not interfere with translation efficiency. To ensure that there would be no differences in translation initiation, the regions between the unique restriction sites and the translation start sites in the two genes were identical. The restriction sites at the 5′ end of gfp allow one to introduce the hairpin at the very beginning of the transcript, as it has been reported that more than five unpaired bases at the 5′ end of a transcript will relieve the stability enhancements of the hairpins (4). Hairpins at the 3′ end of lacZ protect the mRNA against degradation by 3′-to-5′ exonucleases and terminate transcription.
FIG. 1.

Plasmid and DNA cassette design. (A) Backbone structure of plasmid. All plasmids in this study have the same backbone and differ only in the variable region. Some examples of RNA representations of the variable region for various plasmids are shown in panel C. (B) RNase E sites obtained from literature. E1 was taken from the puf operon of R. capsulatus and was used in the p60 constructs, and E2 was taken from the pap operon of E. coli and was used in the p70 constructs. (C) Schematic for inserting control elements. RNA representations of the variable regions for several plasmids are shown, including steps taken to insert control elements. p50gl is the basic dual-gene plasmid to which stabilizing elements were added. p50gΔl lacks the lacZ coding region. p60gl and p70gl differ in the RNase E site that was added 5′ of lacZ. p50HP4gΔl, p50HP17gΔl, p60gHP4l, and p70gHP4l are the previously mentioned constructs with hairpins inserted 5′ of the various genes.
The decay characteristics of lacZ are known from previous work (6, 7). An understanding of the decay characteristics for gfp alone was required. By removing the lacZ coding region from the construct described above, an expression system with the gfp gene alone was developed. This single-gene system had 5′ restriction sites for the insertion of DNA cassettes and the same transcription terminators positioned at the 3′ end of gfp.
Design of DNA cassettes.
Two DNA cassettes encoding previously designed hairpin structures (HP17 and HP4) (8) were inserted into the single-gene gfp system (Fig. 1). Three DNA cassettes were inserted sequentially into the dual-gene system. A DNA cassette encoding a 3′ hairpin for gfp and containing an intercistronic region with a putative RNase E site immediately 5′ of lacZ was inserted into the dual-gene plasmid p50gl, making plasmid p60gl. This cassette was based on the sequence between the pufQ and pufB genes of R. capsulatus, which has been shown to contain an RNase E site (16). The second DNA cassette encoded a 5′ hairpin for lacZ and was inserted immediately downstream of the putative RNase E site, making plasmid p60gHP4l. The design of this hairpin was based upon HP4 (8). The third DNA cassette encoded a known RNase E site found between the papA and papB regions of the E. coli pap operon (29) and was inserted to replace the first intercistronic region, making plasmid p70gHP4l (Fig. 1).
Transcript stability analysis.
Northern blot analysis performed on the single-gene gfp systems (p50gΔl, p50HP4gΔl, and p50HP17gΔl) revealed that the half-life of the gfp transcript was unaffected by 5′ hairpins (Table 2). This result indicates that gfp is not susceptible to inactivation by an endoribonuclease, such as RNase E. In contrast, similar hairpins placed at the 5′ end of lacZ significantly affected its stability (6).
TABLE 2.
Primary and secondary transcript half-lives for tested plasmidsa
| Plasmid | Half-life (min)
|
||
|---|---|---|---|
| Primary transcript | Secondary transcript (lacZ) | Secondary transcript (gfp) | |
| p50gl | 2.0 (±0.1) | ||
| p50gΔl | 8.0 (±0.3) | ||
| p50HP4gΔl | 7.6 (±0.3) | ||
| p50HP17gΔl | 7.5 (±0.5) | ||
| p60gHP4l | 3.5 (±0.2) | 3.5 (±0.2) [2.9 (±0.2)]b | 2.9 (±0.1) [2.9 (±0.1)] |
| p70gHP4l | 4.0 (±0.2) | 6.9 (±0.4) [5.4 (±0.4)] | 3.9 (±0.4) [3.1 (±0.4)] |
Values for half-lives are given as mean values and standard errors obtained by method 1.
Values in brackets are mean values and standard errors obtained by method 2.
Northern blot analysis conducted on the dual-gene systems revealed useful qualitative and quantitative information. No stable intermediate transcripts were detected for p50gl when probed with either gfp or lacZ (Fig. 2B and 3B). The primary transcript containing the lacZ and gfp coding regions (Fig. 2B and 3B, band a) was slightly larger (by the length of gfp) than the lacZ-only transcript of pHP4l (Fig. 2A, band b). As there was no 5′ hairpin to protect the primary transcript, it was rapidly degraded. As there were no internal hairpins (5′ to lacZ), no stable secondary transcripts resulted or can be seen. The primary transcript had a short half-life of approximately 2 min, since the 5′ end of this transcript was not protected by secondary structures (Table 2).
FIG. 2.

Northern blot analysis of lacZ mRNA stability. Each lane has 5 μg of total RNA loaded for each sample taken at a particular time after addition of rifampin. Lane 1, 0 min; lane 2, 2.5 min; lane 3, 5 min; lane 4, 10 min; lane 5, 20 min. Markers: a, primary transcript; b, secondary lacZ transcript; c, 23S rRNA; d, 16S rRNA. (A) Blot of pHP4l (single-gene lacZ system) probed with lacZ. (B) Blot of p50gl probed with lacZ. There is a gradual smearing under the primary band, indicating that there is no stable secondary lacZ transcript. (C) Blot of p60gHP4l with lacZ. In contrast to panels A and B, there are a dark primary band and a secondary band running under the primary transcript (corresponding to a stable secondary lacZ transcript).
FIG. 3.

Northern blot analysis of gfp mRNA stability. Each lane has 5 μg of total RNA loaded for a sample taken at a particular time after addition of rifampin: lane 1, 0 min; lane 2, 2.5 min; lane 3, 5 min; lane 4, 10 min; lane 5, 20 min. Markers: a, primary transcript; b, secondary gfp transcript; c, 23S rRNA; d, 16S rRNA. (A) Blot of p50HP4gΔl (single-gene gfp system) probed with gfp. (B) Blot of p50gl probed with gfp. There is a dark smearing where the gfp secondary transcript should run, indicating that there is no stable secondary gfp transcript. (C) Blot of p60gHP4l probed with gfp. In contrast to panels A and B, there are a dark primary band (a) and a secondary band (b) corresponding to a stable secondary gfp transcript.
The results of the Northern blot analysis for p50gl can be compared to those obtained with p60gHP4l. The blots for this system revealed bands for both the primary and secondary transcripts for lacZ and gfp (Fig. 2C and 3C), indicating that after the initial RNase cleavage between the genes, the resulting transcripts were protected at vulnerable ends from RNases. The lacZ secondary transcript of p60gHP4l (Fig. 2C, band b) was darker and more distinct than that of p50gl (Fig. 2B, band b). Given the size of the bands, cleavage must occur approximately upstream of the lacZ 5′ hairpin. The 3′ hairpin for gfp protects against exonuclease activity. (Note that the secondary gfp transcript from p60gHP4l is smaller than that from p50gΔl because it lacks the 5′ end of lacZ and the 3′ hairpins on p50gΔl.) The half-life of the primary transcript was approximately 3.5 min. The half-lives of the lacZ and gfp secondary transcripts were approximately 3.5 and 3 min, respectively, from method 1 and 2.9 min for both from method 2 (Table 2). Note that the half-lives for the gfp secondary transcript from p60gHP4l and p70gHP4l were significantly shorter than those for the transcripts of p50gΔl, p50HP4gΔl, and p50HP17gΔl. The secondary gfp transcripts of p60gHP4l and p70gHP4l carry smaller hairpins 3′ of gfp, in contrast to p50gΔl, p50HP4gΔl, and p50HP17gΔl, which carry the large hairpins. The differences in the hairpins at the 3′ end could significantly affect 3′-to-5′ processing and thus stability.
Northern blot analysis for p70gHP4l showed qualitative trends similar to those of p60gHP4l. The half-lives of the primary transcript and the gfp secondary transcript were similar (within experimental error) to those for p60gHP4l (Table 2). In contrast, the half-life of the lacZ secondary transcript was approximately 7 min according to method 1 for determining half-lives and 5.4 min according to method 2, nearly double that for p60gHP4l. The higher stability of the lacZ secondary transcript in p70gHP4l may be the result of the putative RNase E site in p70gHP4l being more susceptible to cleavage by E. coli RNase E than the one in p60gHP4l (17).
The stability analysis for these systems is complicated by the fact that the lacZ secondary transcript is close in size to the primary transcript and runs directly beneath the primary band on the gel, making the two bands appear as one longer band on the Northern blot. The break between the two bands can be determined by comparing it to the single-gene blots (Fig. 2A and 3A).
Primer extension analysis.
To confirm that the mRNAs of p60gHP4l and p70gHP4l were cleaved between the two coding regions and that two secondary transcripts were present, the nuclease cleavage site location between the two genes in p60gHP4l and p70gHP4l was determined using primer extension analysis (Fig. 4). For p60gHP4l, cleavage occurred approximately 10 bases from the base of the hairpin stem 5′ of the lacZ coding region in the mRNA (indicated by band 2 in Fig. 4A). For p70gHP4l, cleavage occurred approximately 5 bases from the base of the hairpin stem 5′ of the lacZ coding region in the mRNA (band 2 in Fig. 4A). For p50gl, no band appeared on the gel, indicating that cleavage does not occur between the two coding regions. This analysis indicates that the system is working as designed and suggests that a stable secondary transcript containing lacZ is formed when the primary transcript of p60gHP4l and p70gHP4l is cleaved by an RNase. Presumably this RNase is RNase E, as the regions were designed with previously identified RNase E sites. However, the actual identity of the RNase responsible for the processing is not critical. What is critical is that cleavage occurs between the two coding regions, thereby stabilizing the coding regions for the downstream gene. The higher stability of the lacZ secondary transcript from p70gHP4l than that for the transcript from p60gHP4l may be due to the number of the 5′ unpaired bases. The p60gHP4l lacZ secondary transcript contained approximately 10 unpaired bases, whereas the p70gHP4l transcript had approximately 5 unpaired bases. It has been shown that having more than 5 unpaired bases at the 5′ end of a transcript alleviates the protective effect of the secondary structure (4). Cleavage closer to the hairpin 5′ of lacZ results in a more stable lacZ transcript and more β-galactosidase produced from p70gHP4l than from p60gHP4l.
FIG. 4.

Primer extension analysis of mRNA from p50gl, p60gHP4l, and p70gHP4l. (A) Sequencing ladders and primer extension lanes are shown for each plasmid. The first four lanes of each set are the plasmid sequencing results. G, ddGTP included in the reaction; A, ddATP; T, ddTTP; C, ddCTP. The two lanes to the right of the sequencing ladder are the primer extension results for the in vivo mRNA. N, no dideoxynucleotides included in the reaction; *, lane N with intensity magnified to visualize bands. Bands 1 and 2 are reverse transcriptase stop sites. (B) Sequence of p50gl in this region (no secondary structures or cleavage sites). (C) Location of the cleavage site relative to the predicted hairpin structure in p60gHP4l. (D) Location of the cleavage site relative to the predicted hairpin structure in p70gHP4l. Note that the sequence read from the gel is the complement to that shown in panels B to D.
Note that the larger bands (indicated by the numeral 1 in Fig. 4A) in the primer extension lanes for p60gHP4l and p70gHP4l are due to reverse transcriptase stopping at the secondary structure 3′ of the gfp coding region; p50gl lacks this secondary structure and this band. It should also be pointed out that the intensity of this band, resulting from the secondary structure, was higher in both systems than that of the band corresponding to the cleavage site. These relative intensities indicate relative abundance of mRNA transcripts; more full-length transcript than processed transcript was present in the sample.
Protein production.
Enzyme assays were conducted on the extracts of cells carrying the dual-gene operon to determine whether the changes in transcript stability resulted in changes in protein production. Expression was induced over a range of inducer concentrations (Fig. 5A). Cells harboring p60gHP4l produced twice as much β-galactosidase as cells harboring p50gl across all inducer concentrations. Cells harboring p70gHP4l had approximately 50 times more β-galactosidase at low inducer concentrations and 2 times more β-galactosidase at high inducer concentrations than did p50gl.
FIG. 5.

Enzyme activities at various inducer concentrations. (A and B) Triangles, p70gHP4l; squares, p60gHP4l; diamonds, p50gl. (A) β-Galactosidase activity. (B) GFP activity. (C) Relative ratios. Black bars, β-galactosidase/GFP enzyme activity ratio for each construct relative to p50gl; gray bars, ratio of lacZ to gfp secondary transcript half-lives for p60gHP4l and p70gHP4l calculated using method 1; white bars, ratio of lacZ to gfp secondary transcript half-lives for p60gHP4l and p70gHP4l calculated using method 2. Since p50gl has no secondary transcripts, half-life ratios could not be provided.
Assays of GFP showed a trend similar to that found in the β-galactosidase assays (Fig. 5B). At inducer concentrations below 0.001%, cells harboring p70gHP4l produced approximately 10-fold more GFP than cells harboring p60gHP4l and 20-fold more than cells harboring p50gl. At inducer concentrations above 0.001%, cells harboring p70gHP4l produced about the same amount of GFP as cells harboring p60gHP4l, which was approximately twofold greater than cells harboring p50gl.
To judge how the secondary structures and putative RNase E cleavage sites affected mRNA stability and protein production, the ratios of half-lives and enzyme activities for one construct relative to another are presented. At concentrations of inducer of less than 0.001% (in the linear range of inducer concentrations), the ratios of β-galactosidase-to-GFP activities for p70gHP4l and p60gHP4l relative to the same for p50gl, (β-gal/GFP)p70/(β-gal/GFP)p50, and (β-gal/GFP)p60/(β-gal/GFP)p50 were consistently 3.5 and 1.7, respectively (Fig. 5C). The ratios of the lacZ secondary transcript half-life to gfp secondary transcript half-life (based on values obtained using method 1 of the half-life calculations) for these systems were 1.8 and 1.2, respectively. (The secondary transcripts of p50gl were nonexistent.) This can be compared to the ratios for the values obtained using method 2 of the half-life calculations, 1.7 and 1.0, respectively. As the only difference between these two constructs was the putative RNase E site, this result may reflect the relative efficiency with which RNase E cleaved the RNase E site (16, 29). Indeed, the results of the enzyme assay indicate that p70gHP4l contains an RNase site upstream of the lacZ gene that is more susceptible to cleavage by an RNase than that in p60gHP4l. The secondary lacZ transcripts, and to some extent the gfp transcripts, from p60gHP4l and p70gHP4l are more stable than the primary transcript of p50gl. Cleavage by an RNase directly 5′ of the 5′ hairpin of lacZ results in a secondary transcript with hairpins at the 5′ and 3′ ends to protect against further immediate RNase cleavage. While larger than the lacZ secondary transcript by the addition of gfp at the 5′ end, the primary transcript of p50gl is not protected from RNase attack by any 5′ hairpins. This stabilization results in more β-galactosidase production relative to GFP production from p60gHP4l and p70gHP4l than from p50gl. These results show that this technology can be used to differentially control the stabilities of secondary transcripts, resulting in differential gene expression. More importantly, the relative ratios of the enzymes produced can be affected with this type of control.
ACKNOWLEDGMENTS
We thank N. Pace and S. Kustu for their help with the primer extension work and C. Bertozzi and P. Schultz for use of their PhosphorImager.
This research was supported in part by the ERC Program of the National Science Foundation under award number EEC-9731725, National Science Foundation grants BES-9502495 and BES-9906405, and a National Science Foundation graduate fellowship to C. D. Smolke.
REFERENCES
- 1.Alifano P, Bruni C, Carlomagno M. Control of mRNA processing and decay in prokaryotes. Genetica. 1994;94:157–172. doi: 10.1007/BF01443430. [DOI] [PubMed] [Google Scholar]
- 2.Arnold T E, Yu J, Belasco J. mRNA stabilization by the ompA 5′ untranslated region: two protective elements hinder distinct pathways for mRNA degradation. RNA. 1998;4:319–330. [PMC free article] [PubMed] [Google Scholar]
- 3.Belasco J, Brawerman G. Control of messenger RNA stability. New York, N.Y: Academic Press; 1993. [Google Scholar]
- 4.Bouvet P, Belasco J. Control of RNase E-mediate RNA degradation by 5′-terminal base pairing of E. coli. Nature. 1992;360:488–491. doi: 10.1038/360488a0. [DOI] [PubMed] [Google Scholar]
- 5.Bricker A, Belasco J. Importance of a 5′ stem-loop for longevity of papA mRNA in Escherichia coli. J Bacteriol. 1999;181:3587–3590. doi: 10.1128/jb.181.11.3587-3590.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carrier T A, Keasling J D. Engineering mRNA stability in E. coli by the addition of synthetic hairpins using a 5′ cassette system. Biotechnol Bioeng. 1997;55:577–580. doi: 10.1002/(SICI)1097-0290(19970805)55:3<577::AID-BIT16>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 7.Carrier T A, Keasling J D. Controlling messenger RNA stability in bacteria: strategies for engineering gene expression. Biotechnol Prog. 1997;13:699–708. doi: 10.1021/bp970095h. [DOI] [PubMed] [Google Scholar]
- 8.Carrier T A, Keasling J D. Library of synthetic 5′ secondary structures to manipulate mRNA stability in Escherichia coli. Biotechnol Prog. 1999;15:58–64. doi: 10.1021/bp9801143. [DOI] [PubMed] [Google Scholar]
- 9.Chelladuri B, Li H, Nicholson A. A conserved sequence element for ribonuclease III processing signals is not required for accurate in vitro enzymatic cleavage. Nucleic Acids Res. 1991;19:1759–1766. doi: 10.1093/nar/19.8.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duetscher M, Zhang J. Ribonucleases, diversity and regulation. In: McCarthy J, Tuite M, editors. Post-transcriptional control of gene expression. Berlin, Germany: Springer-Verlag; 1990. pp. 1–11. [Google Scholar]
- 11.Ehrettsmann C, Carpousis A, Krisch H. mRNA degradation in prokaryotes. FASEB J. 1992;6:3186–3192. doi: 10.1096/fasebj.6.13.1397840. [DOI] [PubMed] [Google Scholar]
- 12.Emory S, Belasco J. The ompA 5′ untranslated RNA segment functions in E. coli as a growth-rate-regulated mRNA stabilizer whose activity is unrelated to translational efficiency. J Bacteriol. 1990;172:4472–4481. doi: 10.1128/jb.172.8.4472-4481.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emory S, Bouvet P, Belasco J. A 5′ terminal stem-loop structure can stabilize mRNA in E. coli. Genes Dev. 1992;6:135–148. doi: 10.1101/gad.6.1.135. [DOI] [PubMed] [Google Scholar]
- 14.Fritsch J, Rothfuchs R, Rauhut R, Klug G. Identification of an mRNA element promoting rate-limiting cleavage of the polycistronic puf mRNA in Rhodobacter capsulatus by an enzyme similar to RNase E. Mol Microbiol. 1995;15:1017–1029. doi: 10.1111/j.1365-2958.1995.tb02277.x. [DOI] [PubMed] [Google Scholar]
- 15.Hansen M, Chen L, Fejzo M, Belasco J. The ompA 5′ untranslated region impedes a major pathway for mRNA degradation in E. coli. Mol Microbiol. 1994;12:707–716. doi: 10.1111/j.1365-2958.1994.tb01058.x. [DOI] [PubMed] [Google Scholar]
- 16.Heck C, Rothfuchs R, Jager A, Rauhut R, Klug G. Effect of the pufQ-pufB intercistronic region on puf mRNA stability in Rhodobacter capsulatus. Mol Microbiol. 1996;20:1165–1178. doi: 10.1111/j.1365-2958.1996.tb02637.x. [DOI] [PubMed] [Google Scholar]
- 17.Heck C, Evguenieva-Hackenberg E, Balzer A, Klug G. RNase E enzymes from Rhodobacter capsulatus and Escherichia coli differ in context- and sequence-dependent in vivo cleavage within the polycistronic puf mRNA. J Bacteriol. 1999;181:7621–7625. doi: 10.1128/jb.181.24.7621-7625.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Helmstetter C, Cooper S. DNA synthesis during the division cycle of rapidly growing Escherichia coli. J Mol Biol. 1968;31:507–510. doi: 10.1016/0022-2836(68)90424-5. [DOI] [PubMed] [Google Scholar]
- 19.Horton R M, Cai Z, Ho S N, Pease L R. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. BioTechniques. 1990;8:528–535. [PubMed] [Google Scholar]
- 20.Horton R M. In vitro recombination and mutagenesis of DNA. Methods Mol Biol. 1993;15:251–261. doi: 10.1385/0-89603-244-2:251. [DOI] [PubMed] [Google Scholar]
- 21.Klug G, Jock S, Rothfuchs R. The rate of decay in Rhodobacter capsulatus-specific puf mRNA segments is differentially affected by RNase E activity in E. coli. Gene. 1992;121:95–102. doi: 10.1016/0378-1119(92)90166-m. [DOI] [PubMed] [Google Scholar]
- 22.Klug G. The role of mRNA degradation in the regulated expression of bacterial photosynthesis genes. Mol Microbiol. 1993;9:1–7. doi: 10.1111/j.1365-2958.1993.tb01663.x. [DOI] [PubMed] [Google Scholar]
- 23.Lundberg U, von Gabain A, Melefors O. Cleavages in the 5′ region of the ompA and bla mRNA control stability: studies with an E. coli mutant altering mRNA stability and a novel endoribonuclease. EMBO J. 1990;9:2731–2741. doi: 10.1002/j.1460-2075.1990.tb07460.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McDowall K, Lin-Chao S, Cohen S. A+U content rather than a particular nucleotide order determines the specificity of RNase E cleavage. J Biol Chem. 1994;269:10790–10796. [PubMed] [Google Scholar]
- 25.McLauren R, Newbury S, Dance G, Causton H, Higgins C. mRNA degradation by processive 3′–5′ exoribonucleases in vitro and the implications for prokaryotic mRNA decay in vivo. J Mol Biol. 1991;221:81–95. [PubMed] [Google Scholar]
- 26.Mejia J, Burnett M, An H, Barnell W, Keshav K, Conway T, Ingram L. Coordination of expression of Zymomona mobilis glycolytic and fermentative enzymes: a simple hypothesis based on mRNA stability. J Bacteriol. 1992;174:6438–6443. doi: 10.1128/jb.174.20.6438-6443.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller J. A short course in bacterial genetics. Plainview, N.Y: Cold Spring Harbor Laboratory Press; 1992. [Google Scholar]
- 28.Neidhardt F, Bloch P, Smith P. Culture medium for enterobacteria. J Bacteriol. 1974;119:736–747. doi: 10.1128/jb.119.3.736-747.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nilsson P, Uhlin B E. Differential decay of a polycistronic Escherichia coli transcript is initiated by RNase E-dependent endonucleolytic processing. Mol Microbiol. 1991;5:1791–1799. doi: 10.1111/j.1365-2958.1991.tb01928.x. [DOI] [PubMed] [Google Scholar]
- 30.Portier C, Dondon L, Manago M, Regnier P. The first step in the functional inactivation of the E. coli polynucleotide phosphorylase messenger is a ribonuclease III processing at the 5′ end. EMBO J. 1987;6:2165–2176. doi: 10.1002/j.1460-2075.1987.tb02484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Regnier P, Grunberg-Manago M. RNase III cleavages in non-coding leaders of E. coli transcripts control mRNA stability and genetic expression. Biochimie. 1990;72:825–834. doi: 10.1016/0300-9084(90)90192-j. [DOI] [PubMed] [Google Scholar]
- 32.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 33.Taylor J R. An introduction to error analysis. 2nd ed. Sausalito, Calif: University Science Books; 1982. [Google Scholar]
- 34.Zubiaga A, Belasco J, Greenberg M. The nonamer UUAUUUAUU is the key AU-rich sequence motif that mediates mRNA degradation. Mol Cell Biol. 1995;15:2219–2230. doi: 10.1128/mcb.15.4.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]