

Abstract
BACKGROUND
Uterine leiomyomas, also known as uterine fibroids or myomas, are the most common benign gynecological tumors and are found in women of reproductive and postmenopausal age. There is an exceptionally high prevalence of this tumor in women by the age of 50 years. Black women are particularly affected, with an increased incidence, earlier age of onset, larger and faster growing fibroids and greater severity of symptoms as compared to White women. Although advances in identifying genetic and environmental factors to delineate these fibroids have already been made, only recently has the role of epigenomics in the pathogenesis of this disease been considered.
OBJECTIVE AND RATIONALE
Over recent years, studies have identified multiple epigenomic aberrations that may contribute to leiomyoma development and growth. This review will focus on the most recent discoveries in three categories of epigenomic changes found in uterine fibroids, namely aberrant DNA methylation, histone tail modifications and histone variant exchange, and their translation into altered target gene architecture and transcriptional outcome. The findings demonstrating how the altered 3D shape of the enhancer can regulate gene expression from millions of base pairs away will be discussed. Additionally, translational implications of these discoveries and potential roadblocks in leiomyoma treatment will be addressed.
SEARCH METHODS
A comprehensive PubMed search was performed to identify published articles containing keywords relevant to the focus of the review, such as: uterine leiomyoma, uterine fibroids, epigenetic alterations, epigenomics, stem cells, chromatin modifications, extracellular matrix [ECM] organization, DNA methylation, enhancer, histone post-translational modifications and dysregulated gene expression. Articles until September 2021 were explored and evaluated to identify relevant updates in the field. Most of the articles focused on in the discussion were published between 2015 and 2021, although some key discoveries made before 2015 were included for background information and foundational purposes. We apologize to the authors whose work was not included because of space restrictions or inadvertent omission.
OUTCOMES
Chemical alterations to the DNA structure and of nucleosomal histones, without changing the underlying DNA sequence, have now been implicated in the phenotypic manifestation of uterine leiomyomas. Genome-wide DNA methylation analysis has revealed subsets of either suppressed or overexpressed genes accompanied by aberrant promoter methylation. Furthermore, differential promoter access resulting from altered 3D chromatin structure and histone modifications plays a role in regulating transcription of key genes thought to be involved in leiomyoma etiology. The dysregulated genes function in tumor suppression, apoptosis, angiogenesis, ECM formation, a variety of cancer-related signaling pathways and stem cell differentiation. Aberrant DNA methylation or histone modification is also observed in altering enhancer architecture, which leads to changes in enhancer–promoter contact strength, producing novel explanations for the overexpression of high mobility group AT-hook 2 and gene dysregulation found in mediator complex subunit 12 mutant fibroids. While many molecular mechanisms and epigenomic features have been investigated, the basis for the racial disparity observed among those in the Black population remains unclear.
WIDER IMPLICATIONS
A comprehensive understanding of the exact pathogenesis of uterine leiomyoma is lacking and requires attention as it can provide clues for prevention and viable non-surgical treatment. These findings will widen our knowledge of the role epigenomics plays in the mechanisms related to uterine leiomyoma development and highlight novel approaches for the prevention and identification of epigenome targets for long-term non-invasive treatment options of this significantly common disease.
Keywords: epigenomics, uterine leiomyoma, enhancer architecture, histone modification, DNA methylation, 3D-chromatin structure, stem cells
Introduction
Uterine leiomyomas [LMs], or fibroids, are benign tumors of the uterus. The estimated prevalence of this tumor in Black (persons who identified as having African, African American or Black ancestry) and White (persons who identified as having European, Middle Eastern or Northern African ancestry) women by the age of 50 years is above 80% and ∼ 70%, respectively (Islam et al., 2013). Fibroids are typically round in shape and their size can range from a few millimeters to over 20 cm in diameter (Williams, 2017). A pelvic exam is often sufficient in identifying the presence of these tumors; however, imaging techniques may be used to detect smaller fibroids and to determine a more precise size, quantity, location and impact on surrounding anatomical structures (Dagur et al., 2016). Ultrasonography is a rapid, cost-effective first-line imaging test used to confirm the clinical detection of LM and allows for the differentiation of LM from other gynecological diseases (Wozniak and Wozniak, 2017; Florence and Fatehi, 2021). Confident differentiation is important to ensure correct management and to prevent a misdiagnosis of adenomyosis, endometriosis, pregnancy, leiomyosarcoma [LMS], uterine carcinosarcoma or endometrial cancer since these can present with similar symptoms to LM (Fleischer et al., 1978; Florence and Fatehi, 2021). Additionally, this differentiation is important for developing an accurate prognosis as LM has been associated with a potentially increased risk of developing endometrial cancer (Lagana and Scioscia, 2020). LM usually appear as a well-defined solid, spherical hypoechoic mass, but the presence of calcification and necrosis may make determining a diagnosis difficult (Wozniak and Wozniak, 2017). This is especially true when differentiating LMS from atypical LM since their potential overlapping features may challenge the use of ultrasonography as a reliable imaging technique (Sun et al., 2019). Consequently, when ultrasound findings are not clear, MRI may be utilized as a further diagnostic tool (Wozniak and Wozniak, 2017; Sam et al., 2019; Suzuki et al., 2019).
In addition, it has been estimated that 25% of women with LM experience symptoms severe enough to seek treatment within the first year (Marsh et al., 2018). Although LMs do not cause mortality, they are associated with significant morbidity related to abnormal uterine bleeding, pelvic pressure and pain, backache and leg pains, infertility and reproductive dysfunction (Bukulmez and Doody, 2006; Carranza-Mamane et al., 2015). As a result, LMs account for a significant number of all hysterectomies performed annually (De La Cruz and Buchanan, 2017; Stewart et al., 2017). The societal and financial costs of LM are also significant. It was reported that the annual cost of healthcare for women in the USA who suffer from this disease ranges from $5.89 to $34.37 billion; with direct costs (medications, physician visits and hospital admissions) running at $4.1–9.4 billion and indirect expenses (lost work and obstetric complications related to uterine fibroids) scaling $1.79–24.97 billion (Cardozo et al., 2012). Furthermore, in a national survey of 968 women, 28% reported missing work owing to symptoms, while 24% expressed that they believed their symptoms prevented them from reaching their career potential (Borah et al., 2013). Unfortunately, LM remains understudied despite the high prevalence among women globally and the associated negative impact on a woman’s quality of life. Consequently, curative non-invasive treatment options are still lacking.
These monoclonal neoplasms originate from the smooth muscle cells of the myometrium and are complemented by an accumulation of extracellular matrix [ECM] components—a hallmark of the disease. Sex steroid hormones have been discovered to play an important role in LM development and growth (Marsh and Bulun, 2006), possibly because of its tissue of origin. Furthermore, NADPH oxidase-derived reactive oxygen species have been implicated in LM growth by serving as critical intermediates in the mitogenic signaling pathways of epidermal growth factor [EGF] and platelet-derived growth factor (Mesquita et al., 2010). The main molecular/genetic subtypes of LM include: mediator complex subunit 12 [MED12] mutations, high mobility group AT-hook 1 [HMGA1] or high mobility group AT-hook 2 [HMGA2] translocations, biallelic inactivation of fumarate hydratase [FH], collagen (COL4A5–COL4A6) deletions and mutations in genes encoding the Snf2-related CREBBP activator protein [SRCAP] complex subunits (Mehine et al., 2013; Berta et al., 2021). However, a comprehensive understanding of the exact pathogenesis of LM is lacking and requires attention as it can produce clues for future prevention and viable non-surgical treatment.
For most of history, scientists have looked at genetic and environmental factors to delineate disease, but recently, the role of epigenomics in biology and human disease has been considered. Chemical alterations to the DNA structure have now been implicated in the phenotypic manifestation of many disorders, including autoimmune diseases, various cancers and neurodegenerative disorders (Moosavi and Ardekani, 2016). Over the years, literature has identified multiple epigenomic aberrations that may contribute to LM development and growth (Karmon et al., 2014; Commandeur et al., 2015; Styer and Rueda, 2016; Yang et al., 2016; Lagana et al., 2017; Falahati et al., 2021). This review will focus on the most recent discoveries in the following categories of epigenomic changes found in LM: aberrant DNA methylation and histone tail modifications. Furthermore, these will be translated into the context of altered enhancer architecture and 3D chromatin reorganization. The findings demonstrating how this altered 3D shape of the enhancer can regulate gene expression from millions of base pairs away will be discussed. These epigenomic features of LM construct a plausible explanation for the pathways leading to altered gene expression and manifestation into the LM phenotype. The enormous potential of drugs targeting the epigenome in LM treatment and understanding will be highlighted.
An overview of genetic mutations in uterine leiomyoma
Decades ago, researchers began identifying genetic events that play important roles in LM pathogenesis. In the recent past, with technological advances, several key genetic mutations are found to be mutually exclusive and major drivers of the distinct gene expression changes in LM. These include FH biallelic inactivation, MED 12 mutations, HMGA2 overexpression, deletion of COL4A5 and COL4A6, and mutation in SCRAP subunits (Makinen et al., 2011; Mehine et al., 2013, 2016; Berta et al., 2021).
Biallelic inactivation in the FH gene was linked to predisposition to LM along with cutaneous LM and renal cell cancer in an inherited tumor susceptibility syndrome, known as hereditary leiomyomatosis and renal cell cancer (Launonen et al., 2001; Tomlinson et al., 2002). Dominant inheritance of the mutated copy followed by alterations in the wild-type FH allele cause loss in the activity of FH, a known tumor suppressor protein and a key enzyme of the tricarboxylic acid cycle, which may promote the accumulation of fumarate, and to a lesser extent succinate, in the affected tissues (Lehtonen et al., 2004; Pollard et al., 2005). Pathways that are specifically altered in this subtype of LM are nuclear factor erythroid 2-related factor 2-dependent oxidative stress and pentose phosphate metabolic pathway (Mehine et al., 2016). This subtype is considered rarer, affecting an estimated 0.4–1.6% of all patients with LM (Popp et al., 2020). However, how FH inactivation and the metabolites fumarate and succinate contribute to LM development and progression is not clear.
With the utilization of exome sequencing, and in a breakthrough discovery, frequent mutations in exon 2 of MED12 and to some extent intron 1- exon 2 junction and exon 1 alterations were observed in LM (Makinen et al., 2011). This molecular subtype is the most common with about 70% of LMs displaying this mutation (Mehine et al., 2014). MED12 is a part of the transcriptional mediator complex involved in relaying signals between enhancer-bound transcription factors [TFs] and promoter-bound RNA polymerase II transcription assembly (Borggrefe and Yue, 2011).
In ∼ 10% of LM cases, overexpression of HMGA2 is found that is mainly contributed by rearrangements of 12q15 (Quade et al., 2003; Lagana et al., 2017). HMGA2 is a chromatin-binding non-histone protein and is pro-tumorigenic (Fusco and Fedele, 2007). In these LM, insulin-like growth factor 2 mRNA-binding protein 2 [IGF2BP2] along with PLAG1 zinc finger [PLAG1], a proto-oncogene, were among the significantly deregulated genes (Mehine et al., 2016). In the fourth subtype, where deletions of COL4A5 and COL4A6 have been found, LM had characteristic upregulation of insulin receptor substrate 4 [IRS4] (Mehine et al., 2016).
Most recently, researchers identified another subtype of LM that shows mutation in SCRAP subunits (Berta et al., 2021), discussed in more detail in section ‘Histone Variants, Chromatin Remodelers and a New Subtype of Uterine LM’. As has been observed before by Mehine et al. (2016), Berta et al. found that their RNA-seq samples formed separate clusters for MED12 mutant, HMGA2 overexpressing, loss of FH, and additionally there was a separate cluster for SCRAP mutant LM based on the differential gene expression (Berta et al., 2021).
The different subtypes of LM have unique gene expression changes based on the specific mutation type, but they also show features common to all LMs, which possibly converge and lead to LM formation. Owing to the affordability of high throughput screenings and the sensitivity of next-generation sequencing [NGS], many of the epigenomic modalities, which may contribute to gene expression changes observed in subtypes of LM when compared to normal myometrium (or myometrium proximal to LMs), can now be analyzed on patient samples, which together with genetic characteristics may provide a more comprehensive understanding of LM, its subtypes and its genesis. Epigenomics may also offer attractive targets for the treatment of LM. Therefore, in this review, we have discussed the progress made so far in this specific area of research.
Aberrant DNA methylation in uterine leiomyoma
Background
The modification of chromatin structure, through DNA methylation, influences TFs’, cofactors’ and enzymes’ access to DNA. One of the most common epigenomic regulation mechanisms that exist in eukaryotes is the methylation of cytosine residues in CpG dinucleotides, which are embedded with the genome. The process of DNA methylation is catalyzed by a family of DNA methyltransferases [DNMTs], which work to transfer a methyl group from a S-adenosyl methionine [SAM] donor to the 5′ position of cytosine’s heterocyclic aromatic ring, resulting in a 5-methylcytosine residue [5mC] (Moore et al., 2013). The formation of 5mC during germ cell development and early embryogenesis is established via de novo DNMT subtypes DNMT3A and DNMT3B, while the maintenance of specific DNA methylation patterns over replication is mediated via DNMT1 (Chen and Li, 2004; Kato et al., 2007; Ren et al., 2018). It has been found that 70–80% of CpG sites in the genome are normally methylated with the exception of gene regulatory regions such as CpG islands [CPGIs] (Jabbari and Bernardi, 2004). CPGIs are defined as stretches of DNA consisting of a large number of 5′-cytosine-phosphate-guanosine-3′ repeats. Generally, CPGIs are rarely methylated since they often house promoter sequences necessary for TF and accessory protein binding for the transcription of housekeeping genes (Aoto et al., 2020). Although the addition of a methyl group is not associated with changes to the primary sequence of DNA, the presence of 5mC regions within CPGIs of promoters can result in the stable silencing of gene expression (Mohn et al., 2008).
Previously, DNA methylation was believed to be an irreversible epigenomic event leading to gene repression. However, an enzyme termed ten-eleven translocation protein 1 [TET1] was discovered to potentially erase DNA methylation (Tahiliani et al., 2009). It has now been shown that the family of TET enzymes—consisting of TET1, TET2 and TET3—function to catalyze the oxidation reactions of 5mC to 5-hydroxymethylcytosine [5-hmC], 5-formylcytosine and 5-carboxylcytosine (Rasmussen and Helin, 2016). These oxidation products are viewed as intermediates in the process of DNA demethylation, which converts 5mC to unmodified cytosines. The regulation of DNA methylation, via TET enzymes removing the epigenomic mark, allows for the specific hypomethylated DNA regions to colocalize with TFs and other transcriptional machinery to initiate gene transcription. However, it is interesting to note that tissue-specific DNA hypomethylation occurs more frequently in intragenic or intergenic enhancers as well as in actively transcribed gene bodies than at promoters (Kundaje et al., 2015; Ehrlich et al., 2016). The outcome of hypermethylation within the promoter region is generally a repression of gene transcription, while the result of hypomethylation is generally active transcription leading to gene expression (Smith et al., 2020).
Normally during development, the genome undergoes complex changes in CG methylation. This is thought to contribute to development- and tissue-specific gene expression, genomic imprinting and X chromosome inactivation (Laurent et al., 2010; Moore et al., 2013; Elhamamsy, 2017; George et al., 2019). Consequently, aberrant methylation in CPGIs can manifest themselves in serious disease phenotypes. Aberrant DNA methylation has been associated with various diseases including diabetes mellitus, various neurological disorders, immunological diseases, cancers, atherosclerosis and osteoporosis (de Mello et al., 2014; Armstrong et al., 2019; Ehrlich, 2019). Altered DNA methylation has also been reported in LM, but its role in the development and growth of LM remains not entirely clear.
Hypermethylation and hypomethylation of gene promoters
The observation of deregulated DNMTs in LM has since prompted many researchers to perform genome-wide DNA methylation studies (Yamagata et al., 2009). To investigate the role of DNA methylation events in this disease, various researchers explored the methylation profiling arrays to identify the promoter proximal sites showing aberrant methylation patterns in LM (summarized in Tables I and II). Navarro et al. (2012) compared DNA methylation and mRNA expression profiles in LM and matched adjacent normal myometrium tissues from 18 Black women. However, it is noteworthy that these results might not be specific to this ethnic group alone, as discussed later. Navarro et al. (2012) found differential promoter methylation and mRNA expression of genes believed to have tumor suppressor functions, namely, Krüppel-like factor 11 [KLF11], DLEC1 cilia and flagella associated protein [DLEC1] (previously named deleted in lung and esophageal cancer 1) and keratin 19 [KRT19]. These genes displayed more significant levels of methylated CpG dinucleotides in their promoter region, and their associated mRNA levels were lower in LM than in normal myometrium (Navarro et al., 2012). It has already been found that the silencing of tumor suppressor genes is correlated with tumor initiation in cancer and other tumor-bearing diseases (Kazanets et al., 2016; Wang et al., 2018). As such, this was the first time the repression of genes with associated tumor suppressor functions, via promoter DNA methylation, was determined in the context of LM. Another independent study identified KLF11 as being downregulated in LM tissue, and accordingly, this study provided insight into the mechanism around KLF11-regulated cell proliferation (Yin et al., 2010). Other genes, such as those involved in the retinoic acid pathway (alcohol dehydrogenase 1A [Class I], alpha polypeptide [ADH1]); in the WNT pathway (Wnt family member 2B [WNT2B]); and the developmental TFs (GATA binding protein 2 [GATA2] and Krüppel-like factor 4) (Ray, 2016; Katerndahl et al., 2021), were identified in both Black and White patient LM samples to be hypermethylated in their promoter regions and were consequently transcriptionally downregulated (George et al., 2019). The aberrant expression of these key tumor suppressor and developmental genes may partly be involved in the pathogenesis of the benign tumors found in LM. As LM is characterized by the accumulation of ECM, determining whether ECM gene promoters also show dysregulated promoter methylation may be more helpful in better understanding this disease.
Table I.
Alterations in promoter methylation leading to the downregulated expression of various genes in uterine leiomyoma.
| Deregulated gene* | Gene function | Fibroid sample type | Source |
|---|---|---|---|
| KLF111 | Tumor suppressor (apoptosis-related) |
|
(Navarro et al., 2012) |
| DLEC12 | Tumor suppressor (inhibits cell proliferation) |
|
(Navarro et al., 2012) |
| KRT193 | Enables structural constituent of cytoskeleton and muscle, Tumor suppressor |
|
(Navarro et al., 2012) |
| ADH1B4 | Catalyzes the oxidation of all-trans-retinol and its derivatives (retinoic acid pathway) |
|
(George et al., 2019) |
| WNT2B5 | Ligand for members of the frizzled family of seven transmembrane receptors (WNT pathway) |
|
(George et al., 2019) |
| GATA26 | Transcription factor for genes involved in hematopoiesis (stem cell function), Tumor suppressor |
|
(George et al., 2019) |
| KLF47 | Transcription factor involved in embryonic stem cell self-renewal and maintenance, Tumor suppressor |
|
(George et al., 2019) |
| DAPK18 | Tumor suppressor (apoptosis-related) | Tissue (n = 10 Japanese) | (Maekawa et al., 2013) |
| NUAK19 | Tumor suppressor (apoptosis-related) | Tissue (n = 10 Japanese) | (Maekawa et al., 2013) |
| EFEMP110 | Targets metalloproteinase pathways; modulates cell morphology/adhesion |
|
(Marsh et al., 2016) |
*Genes which are downregulated and have hypermethylation at their promoter in uterine leiomyoma along with their gene function and fibroid sample type used in the respective studies are summarized.
Krüppel like Factor 11
DLEC1 cilia and flagella associated protein
keratin 19
alcohol dehydrogenase 1B (class I), beta polypeptide
Wnt family member 2B
GATA binding protein 2
Krüppel like Factor 4
death associated protein kinase 1
NUAK family kinase 1
EGF containing fibulin extracellular matrix protein 1
Table II.
Alterations in promoter methylation leading to the upregulated expression of various genes in uterine leiomyoma.
| Upregulated gene* | Gene function | Fibroid sample type | Source |
|---|---|---|---|
| COL4A111 | Collagen IV biosynthesis | Tissue (n = 10 Japanese) | (Maekawa et al., 2013) |
| COL4A212 | Collagen IV biosynthesis | Tissue (n = 10 Japanese) | (Maekawa et al., 2013) |
| COL6A313 | Collagen VI biosynthesis | Tissue (n = 10 Japanese) | (Maekawa et al., 2013) |
*Genes which are upregulated and have hypomethylation at their promoter in uterine leiomyoma along with their gene function and fibroid sample type used in the respective studies are summarized.
collagen type IV alpha 1 chain
collagen type IV alpha 2 chain
collagen type VI alpha 3 chain
Along these lines, Maekawa et al. (2013) conducted a more comprehensive study by utilizing Illumina Infinium HumanMethylation450 Bead Chip platform in Japanese women. In their study using DNA methylome and transcriptome data, the researchers uncovered that estrogen response elements [ERE] were present in the promoter region of ∼ 18% of altered genes. Collagen Type IV alpha 1 chain [COL4A1], collagen Type IV alpha 2 chain [COL4A2] and collagen type VI alpha 3 chain [COL6A3] were hypomethylated and had increased gene expression in LM compared to matched or normal myometrium obtained from patients presenting with LM or cervical cancer without LM (Maekawa et al., 2013). Reduced methylation at promoters of the above-mentioned collagen genes was proposed to allow estrogen receptor alpha [ERα] to bind to its respective promoter regions and initiate an overproduction of collagen, which is the major component of the accumulated ECM (Stewart, 2001). Other genes, such as death-associated protein kinase 1 [DAPK1] and NUAK family kinase 1 [NUAK1], were hypermethylated and had decreased gene expression (Maekawa et al., 2013). Increased methylation is thought to block ERα binding to these genes, and although the exact mechanism of gene inactivation remains unknown, it may play a role in inhibiting their functions of signaling apoptosis-related events (Steinmann et al., 2019). This finding led the researchers to conclude that aberrant DNA methylation at the promoter of ERα target genes is responsible, at least in part, for the aberrant response to estrogen, reinforcing the previous observations on the role of estrogen in the growth of LM. Considering that ERα binds to enhancers, it will be critical to determine whether promoter distal EREs show similar methylation-sensitive properties. Additionally, causal experiments will need to be performed to verify the proposed mode of action involving ERα and DNA methylation levels in regulating the above-mentioned potential target genes.
Collagens constitute an important part of the ECM; however, a recent study pursued fibulins, which are extracellular glycoproteins. Fibulins function to modulate cell morphology, growth, adhesion and motility (Gallagher et al., 2005). Studies conducted on solid malignant tumors have shown that EGF containing fibulin extracellular matrix protein 1 [EFEMP1], the gene that encodes fibulin-3, is downregulated and that this observation serves as a biomarker for prostate cancer and hepatocellular cancer (Nomoto et al., 2010; Kim et al., 2011). Furthermore, other studies have linked the loss of fibulin-3 with increased tumor angiogenesis (Sadr-Nabavi et al., 2009; Hwang et al., 2010; Kim et al., 2011). The study performed in LM found that fibulin-3 is repressed in LM compared to normal myometrium (Marsh et al., 2016). Although simple association is not sufficient to establish causality in LM genesis, the researchers also showed that treatment of primary LM cells with the DNMT inhibitor termed 5-Aza-2′-deoxycytidine [5-aza-dC] increased EFEMP1 expression significantly compared to vehicle, partly linking DNA methylation activity to its downregulation. However, the methylation profile at or near the EFEMP1 gene in LM compared to the normal condition needs to be validated. Various studies in both cancerous and non-cancerous conditions have shown that fibulin-3 acts by targeting metalloproteinase pathways (Klenotic et al., 2004; Kim et al., 2012). Further insight into the implications of fibulin-3 deregulation in LM and the mechanisms governing its altered expression may be beneficial toward its treatment.
In an effort to study the upstream regulatory genes that may play a role in LM pathogenesis, genes that were differentially methylated and altered in LM were analyzed. Ingenuity Pathway Analysis revealed the highly expressed transcription regulators, SATB homeobox 2 [SATB2] and neuregulin 1 [NRG1], as potential upstream regulatory genes. Interestingly, more than 50% of pathways perturbed in the immortalized uterine smooth muscle cell line, upon overexpression of these two genes independently, were common to altered pathways in LM (Sato et al., 2019). Those pathways included the WNT/β-catenin and the transforming growth factor [TGF]-β signaling pathways known to play an important role in LM (Borahay et al., 2015; Ciebiera et al., 2017; Sato et al., 2019). In addition, the researchers confoundingly found hypermethylation near one of their several transcription start sites [TSS] for both SATB2 and NRG1 genes (Sato et al., 2019). This discrepancy may be linked to the expression of variant forms from the TSS not analyzed by the DNA methylome study and/or related to other epigenomic events. Another limitation is that cell lines may not fully reflect in vivo conditions, such as those found in tissues. With the development of cell lines, passaging cells may cause them to lose characteristics that are found in situ (Kaur and Dufour, 2012). Therefore, identifying the exact mechanisms leading to higher expression of these regulatory genes in LM may expose processes crucial to its development.
It is important to mention and acknowledge that although the authors of the above-mentioned studies highlight specific genes and their associations with DNA methylation, the observations remain correlative; direct and causal experiments need to be performed to establish a mechanistic connection between promoter methylation level and gene expression changes.
Genome-wide DNA methylation and stem cell regulation in uterine leiomyoma
Goodell et al. (1996) were the first ones to identify a population of cells in bone marrow that can efflux fluorescent DNA-binding dye (Hoechst 33342). This subset of the cell population was observed aside from the main population at Hoechst blue-low and Hoechst red-low corner regions in scatter plots of fluorescence-activated cell sorting and therefore termed as the side population [SP]. The SP was found to be enriched for hematopoietic stem cells (Goodell et al., 1996). Afterwards, many laboratories have observed the SP in various tissues, including endometrium and myometrium, from adults and, in general, this population shows properties of somatic stem cells (Ono et al., 2007; Masuda et al., 2010; Cervello et al., 2011). Along these lines, several studies demonstrate a SP in LM with characteristics of stem-like cells that are considered crucial for LM growth (Mas et al., 2012; Ono et al., 2012, 2013). Furthermore, distinct cell populations within LM tissue at different differentiation stages were identified based on the cell surface markers, CD34 and CD49b. The authors evaluated that cells with CD34+/CD49b+ had stem cell-like characteristics and were highly enriched in the side population [LSC], CD34+/CD49b– cells were intermediate [LIC] and CD34–/CD49b– cells were fully differentiated [LDC] (Yin et al., 2015). DNA methylation seems to play an important role in LM, but to elucidate its role in stem cell regulation, Liu et al. (2020) performed Methyl Cap sequencing across all three populations of cells. Their data show that when compared to LIC and LDC, LSC harbored several thousands of differentially methylated regions [DMRs]. Most of the differential regions in LSC were hypermethylated with no drastic difference between the LIC and LDC populations. Concurrently, there was the lowest expression of two DNA demethylases in LSC, TET methylcytosine dioxygenase 1 and 3, but no significant changes in DNMTs. Key here is that the LSC population of cells, as opposed to LIC and LDC populations, harbored extensive DNA methylation in genes required for differentiation, such as estrogen receptor 1 [ESR1], TIMP metallopeptidase inhibitor 3 [TIMP3], receptor tyrosine kinase like orphan receptor 2 [ROR2] and myosin heavy chain 11 [MYH11], which suppressed their expression and the ability to develop into differentiated LM cells (Liu et al., 2020). A more recent study by this group found that within the LSC population, the progesterone receptor [PR] gene contained significant hypermethylation at several intronic regions. Treatment with 5-Azacytidine [5-Aza], a drug that inhibits DNMT, activated PR signaling and stimulated LSC differentiation. The authors suggested that the increased level of DNA methylation within PR and its target cistrome interfered with PR signaling and inhibited the expression of genes critical for progesterone-induced LSC differentiation (Liu et al., 2021a). Interestingly, since LSC cells are deficient in steroid hormone receptors, they are possibly resistant to the hormonal therapies used to treat LM. After treatment, these cells may proliferate and differentiate, leading to tumor growth, as has been observed in breast cancer (Liu and Wicha, 2010). Expectedly, in the Liu et al. (2020) study, the application of 5-Aza promoted differentiation of the LSC population, raising the possibility that it may sensitize them toward hormone therapy (Liu et al., 2020). When testing this drug’s effect on hormonal therapy, Liu et al. (2021a) observed that 5-Aza treatment synergized with an antiprogestin to reduce tumor size in a xenograft mouse model, highlighting the feedback loop between progesterone signaling and DNA methylation as a potential therapeutic target that may decrease LSC stemness and tumor growth (Liu et al., 2021a). However, owing to potential toxicity associated with compounds interfering with DNA methylation globally, further studies should be performed to determine success revolving around the treatment designed to target LSC and avert the formation of novel and unwanted tumors.
Impact of enhancer methylation on RANKL gene expression
With the finding of the LM stem cell population and its important function in this benign tumor growth (Yin et al., 2015), researchers are now attempting to target this population specifically. Breast cancer research has identified a paracrine pathway where progesterone signals expansion and differentiation of the PR-negative mammary stem cells (Joshi et al., 2010). This pathway involves the secretion of receptor activator of nuclear factor κ-Β ligand [RANKL] from PR-positive mammary epithelial cells in response to progesterone. RANKL then acts on its receptor, receptor activator of nuclear factor κ-Β [RANK], which is present on stem cells (Joshi et al., 2010; Sigl et al., 2016). As LMs are characterized as hormone-sensitive tumors, Ikhena et al. (2018) therefore speculated a similar phenomenon in LM stem cells and demonstrated LM growth suppression in a mouse xenograft model in the presence of RANK-Fc (a RANK/RANKL pathway inhibitor). Accordingly, compared to myometrium, the LM intermediate population [LIC] showed the highest expression of RANKL, while stem cells [LSC] exhibited high levels of RANK (Ikhena et al., 2018). Moreover, RANKL transcription responds to progesterone signaling with increased sensitivity in LM cells (Liu et al., 2019). To shed some light on the interplay between progesterone and the RANK/RANKL pathway in LM, Liu et al. (2019) found a distal progesterone receptor binding site [PRBS] located 87 kb upstream of the RANKL transcription start site (Liu et al., 2019). This region was enriched for H3K27ac, which is an active enhancer mark indicating it as a potential enhancer region. Interestingly, Methyl Cap-Seq for robust genome-wide DNA methylation profiling revealed a DMR in the vicinity of this binding site (Brinkman et al., 2010; Liu et al., 2019). The hypothesis that methylation at this site prevents binding of PR and thus decreases RANKL expression was tested by 5-Aza treatment of LM cells. Furthermore, this site was found to be demethylated in LM tissues as compared to normal myometrium (Liu et al., 2019). The authors proposed that both the DMR and the distal PRBS compose a novel RANKL distal regulatory element that functions as an enhancer to regulate RANKL expression. Overall, DNA hypomethylation was suggested to be involved in recruiting and enhancing the binding of PR to the enhancer and strengthening the looping interaction between the distal PRBS and the RANKL promoter. A positive correlation between a mutation in MED12 and increased binding of PR to PRBS and RANKL expression was observed, with concomitant higher affinity binding of PR to mutant MED12 (Liu et al., 2019). Altogether, this study suggests a possible relation between TF binding, histone modification and DNA methylation to the enhancer and its integration into LM phenotype. Although this comprehensive study has identified a new regulatory element for RANKL, the factors that determine methylation changes at this element still need to be determined as it is plausible that those factors are crucial in development of this disease. While distal PR targeting enhancers are possible regulators, clustered regularly interspaced short palindromic repeat [CRISPR]-mediated ablation of the sites will conclusively establish the role of this enhancer in PR/progestin-mediated regulation of RANKL. We further emphasize that causal experiments are necessary to follow-up on the regulatory roles of DNA methylation uncovered by gene-specific and global DNA methylation studies in regulating LM subtype-specific gene expression and LM genesis.
Role of 5-hydroxy-methylcytosine
Various studies in recent years have focused on understanding 5-hmC in the context of solid and hematological tumors. The loss of 5-hmC levels has been associated with higher grade and increased metastasis in various tumors. At the same time, the reduced expression and altered function of TET enzymes were observed in these tumors (Kudo et al., 2012; Yang et al., 2013). In contrast to malignant tumors, benign tumors such as LM have higher levels of 5-hmC as well as upregulated TET1 and TET3 levels compared to normal myometrium. Knockdown of TET1 or TET3 coordinated with decreased 5-hmC levels and reduced cellular proliferation (Navarro et al., 2014). Interestingly, increased expression of TET3 is found to be linked with aberrant expression of H19 long non-coding RNA [H19 lncRNA] in LM. H19 lncRNA, by acting as a molecular sponge for miRNA let-7, regulates TET3 levels in LM, possibly explaining the increase in 5-hmC levels (Cao et al., 2019). The global levels of this mark in LM are in stark contrast to malignant cancer, raising the need for further investigations to understand the role of 5-hmC in benign tumors. The ease of measuring 5-hmC in cell-free DNA and the availability of cost-effective high throughput methods for its quantitation has made this mark conducive for diagnosis and prognosis purposes (Xu and Gao, 2020). The possibility of using 5-hydroxymethylation as an epigenomic signature in detecting LMs or subtypes shall be explored in future studies.
Although 5-hmC is an intermediate in the 5mC to C conversion pathways, its role in transcriptional regulation has also been scrutinized. Previous studies have found that 5-hmC is enriched at gene bodies, promoters and TF binding sites (Nestor et al., 2012). However, correlation between the presence of 5-hmC and gene expression has not yet been established and it is speculated that it is cell-type specific (Nestor et al., 2012; Tan et al., 2013). Researchers have shown that not only are there changes in the levels of 5-hmC in cancer cells compared to healthy cells, but also there is a change in the distribution of this modification in stem cells from a tet2-mutant AML murine model (Han et al., 2016; Shi et al., 2017). It would be interesting to evaluate the redistribution of 5-hmC in LM and its association with gene expression, as the abundance of this modification increases in this diseased state.
Histone modifications in uterine leiomyoma
Background
In addition to genomic DNA methylation, post-translational modifications of histones hold importance in the epigenomic regulation of gene expression owing to their role in altering chromatin structure. Histones are defined as nucleoproteins because of their functions associated with DNA. Eukaryotes possess five canonical histone proteins: H1, H2A, H2B, H3 and H4. To create a more organized and compacted DNA, ∼ 147 base pairs of DNA are wrapped around a histone core (consisting of dimers of H2A, H2B, H3 and H4) to form a nucleosome (Zhou et al., 2019). H1 functions to add stability to the nucleosome by binding to inter-nucleosomal space (Li and Zhu, 2015). Histones interact well with the negatively charged DNA and are themselves considered basic as they are composed largely of the positively charged amino acids—lysine and arginine. The histone proteins and DNA can form two categories of chromatin: heterochromatin and euchromatin. Heterochromatin has a condensed structure. This tight coiling, in general, prohibits access of the transcriptional machinery to DNA, thus rendering it transcriptionally silent. On the other hand, euchromatin is loosely packed, therefore it is accessible to TFs and generally transcriptionally active (Morrison and Thakur, 2021).
One mechanism capable of remodeling chromatin structure and regulating gene expression consists of the covalent post-translational modifications of histones. These modifications include methylation, acetylation, phosphorylation, ubiquitination and sumoylation among others (Castillo et al., 2017). When a TF binds to DNA, it can recruit coactivator enzymes such as histone acetyltransferases [HATs]. HATs catalyze the addition of acetyl groups to lysine residues found on the N-terminal tails of histones. This event neutralizes the histone’s positive charge and reduces the affinity to the adjacent nucleosome, resulting in destabilization of the nucleosome’s higher-order helix (Luger et al., 1997; Castillo et al., 2017). This modification can be regulated via histone deacetylases [HDACs], which remove the acetyl group from histone proteins and impose a repressive effect (Wang et al., 2020). Similar to histone acetylation, histone phosphorylation is generally associated with transcriptional activation. The histone tails contain serine, threonine and tyrosine residues that can be phosphorylated or dephosphorylated. Protein kinases mediate phosphorylation by transferring phosphates to these acceptor sites, while protein phosphatases mediate dephosphorylation. Phosphorylation of histones creates a repulsive force between the added negatively charged phosphate group and the negatively charged phosphates of the DNA backbone, leading to a more open chromatin structure (Banerjee and Chakravarti, 2011; Rossetto et al., 2012). Other epigenomic enzymes include histone methyltransferases [HMTs], which catalyze the transfer of a methyl group from SAM to locations of the histone tails containing arginine or lysine residues, and demethylase enzymes, which work to remove the methyl groups (D'Oto et al., 2016). The likely outcome of methylation varies. It has a transcriptional silencing (tri-methylation at the ninth lysine residue of the histone H3 protein [H3K9me3], tri-methylation at the 27th lysine residue of the histone H3 protein [H3K27me3]) effect, similar to the outcome of DNA methylation, but activation of gene transcription (tri-methylation at the fourth lysine residue of the histone H3 protein [H3K4me3]) has also been observed (D'Oto et al., 2016; Gong and Miller, 2019). In addition, TFs, cofactors and non-histone proteins, such as β-catenin, are also targeted by HAT and HDAC enzymes.
The epigenomic studies pertaining to LM pathogenesis have mainly focused on DNA methylation; however, as histone modifications also have the potential to play an equally important function in chromatin alterations, researchers are now exploring the effect of histone modifications in this disease.
Genome-wide histone epigenomic alterations in uterine leiomyoma
Considering that the uterus is a highly steroid-responsive tissue, it is not surprising that LM growth and development is influenced by sex steroid hormones (Marsh and Bulun, 2006). To determine the effect of environmental estrogens on LM development, various studies have been pursued. Recent work on a commonly consumed phytoestrogen, genistein, has revealed that it has both inhibitory and stimulatory effects in LM cells depending on its concentration (Di et al., 2008; Di et al., 2012; Castro et al., 2016). At a low concentration of 1 μg/ml, genistein stimulated cell proliferation by activating the mitogen-activated protein kinase p44/42 signaling pathway [MAPKp44/42] via ERα (Di et al., 2008). Yu et al. (2016) showed that genistein-induced phosphorylation of MAPKp44/42 in immortalized human uterine LM [ht-UtLM] cells increased the activation of its downstream effector mitogen- and stress-activated protein kinase 1 [MSK1], which in turn modified histone H3 to histone H3 phosphorylated at serine 10 [H3S10ph]. Colocalization of phospho-MSK1 and H3S10ph in the nuclei of ht-UtLM cells was also observed yet was abolished in the presence of MEK1 inhibitor PD98059. Genes related to cell proliferation and altered by genistein, such as inhibitor of DNA binding 1 [ID1] and c-MYB, showed enrichment of H3S10ph at their promoter regions by ChIP-qPCR assay (Yu et al., 2016). This serves to indicate the possibility that genistein’s induction of the MAPKp44/42/MSK1/H3S10ph axis leads to a more open chromatin structure allowing for the transcriptional upregulation of genes involved in promoting cell proliferation in hormonally driven LM growth. Interestingly, histone phosphorylation has also been shown to play a regulatory role in androgen responsiveness of prostate cancer cells (Banerjee and Chakravarti, 2011; Kim et al., 2016). In LM, steroid hormones and growth receptors crosstalk to drive its development (Borahay et al., 2015). Nevertheless, it is unknown if the above-mentioned mechanism holds true for steroid hormones in vivo or if it is a specific effect of this phytoestrogen. Further studies using antibodies to H3S10ph are warranted to establish a genome-wide role of H3S10ph in LM biology.
TET3 knockdown diminishes chromatin accessibility
As mentioned above, TET3 expression is elevated in LM versus matched myometrium and is regulated by the H19/let-7 axis (Navarro et al., 2014; Cao et al., 2019). Likewise, downregulation of either H19 or TET3 leads to decreased protein levels of several LM-promoting genes: TGF-β receptor 2 [TGFBR2], thrombospondin 1 [THBS1], Rho GTPase activating protein 26 [ARHGAP26], secreted protein acidic and cysteine rich [SPARC], collagen type V alpha 2 chain [COL5A2], collagen type IV alpha 1 chain [COL4A1] and collagen type III alpha 1 chain [COL3A1] (Cao et al., 2019). In addition, this study explored the interaction of TET3 with promoter regions of two such LM-promoting genes: the binding of TET3 at the promoter of TGFBR2 and THBS1 was decreased in the TET3 knockdown cells compared to the control with concurrently increased DNA methylation. Previous findings have shown that within the promoter regions of actively transcribed genes, there is an enrichment of H3K4me3, while enrichment of H3K27me3 is associated with inactive promoters (Barski et al., 2007). Consistent with this idea, ChIP analysis revealed a significant reduction in H3K4me3 and an increase in H3K27me3 at these promoters owing to TET3 knockdown (Cao et al., 2019). Thus, it may be inferred that the knockdown of TET3 promotes a heterochromatin conformation yielding reduced DNA accessibility at the promoters of genes that enhance fibrosis and, thereby, repressed transcription. On the other hand, it may be plausible to assume that overexpression of TET3 can create a more accessible chromatin structure, promoting transcription of the ECM component genes (ARHGAP26, SPARC, COL5A2, COL4A1 and COL3A1) and TGF-β pathway genes (TGFBR2 and THBS1) in LM. Nevertheless, experimental data to substantiate this hypothesis and to emphasize the role of TET proteins in this disease is required.
Hierarchical clustering and disease prediction based on DNA methylation and histone modifications
A recent critical study found differential transcriptomic profiles of LM subtypes were based on their mutation status (Mehine et al., 2016). Can differential epigenomic profiles similarly segregate the LM subtypes? To answer this question in the context of DNA methylation, Sato et al. (2016) focused on 12 of the 120 aberrantly methylated genes by utilizing combined bisulfite restriction analysis and criteria of similar alteration in at least 70% of the LM cases. The purpose of this study was to construct a hierarchical clustering system for clinical application. A combination of 10 genes (ALX homeobox 1 [ALX1], cerebellin 1 precursor [CBLN1], corin, serine peptidase [CORIN], forkhead box P1 [FOXP1], GATA2, IgLON family member 5 [IGLON5], neuronal pentraxin 2 [NPTX2], neurotrophic receptor tyrosine kinase 2 [NTRK2], prolactin [PRL], plus STEAP4 metalloreductase [STEAP4]) was established to differentiate normal myometrium from LM. Furthermore, LM showed sub-clusters, but this substructure was independent of the MED12 mutation [MED12mt] status. Here, the authors mainly relied on altered methylation at 10 genes for separating the different conditions. This approach could differentiate normal from the diseased state but could not identify LM subtypes (Sato et al., 2016). Nevertheless, it showed that an altered epigenomic profile in addition to transcriptomic changes has the potential to segregate LM from normal myometrium. As an additional possibility, utilization of a hierarchical clustering system by a comprehensive epigenomic analysis can provide an opportunity to reveal LM subtypes. In this regard, in a key study, George et al. (2019) performed unsupervised clustering of 10 normal myometrium and 24 LM samples based on highly variable 1% DNA methylome identified in an Infinium Methylation EPIC array [EPIC] that includes enhancer sites. They observed the segregation of normal myometrium and LM, and further clustering of LM into MED12mt, overexpressed HMGA2 [HMGA2hi] and overexpressed HMGA1 [HMGA1hi] subtypes. Compared to normal myometrial tissues, homeobox A13 [HOXA13] was also identified to be hypomethylated and upregulated in LM. This overexpression correlated with homeotic transformation in the myometrium to a more cervical stroma phenotype. This observation identified a potential event that leads to the development of LM since cervical stroma and LM are both characterized by a significant amount of ECM. Additionally, it established global DNA methylation as a powerful experimental and clinical test to classify LM (George et al., 2019).
To identify if there are subtype-specific histone modifications, Leistico et al. (2021) utilized chromatin immunoprecipitation followed by NGS [ChIP-seq] against ‘active’ histone modifications, namely H3K4me3 (promoter mark), H3K27ac (present at active promoter and enhancer) and H3K4me1 (enhancer mark) in LM and matched myometrium from 21 patients. Owing to computational challenges in analyzing heterogenous ChIP-seq datasets, the authors employed a tensor decomposition method called decomposition and classification of epigenomic tensors [DeCET]. Using DeCET, epigenomic features could distinguish myometrium from LM subtypes and, interestingly, could also classify them in > 95% of the cases analyzed (Leistico et al., 2021). This model has the capacity to categorize diseased state based on a small number of datasets. Thus, it could serve as an effective diagnostic tool when combined with small-scale ChIP-seq procedures, such as CUT&RUN or ChIL-seq. Overall, this suggests that, in addition to distinct DNA methylation profiles, there are LM- and subtype-specific histone modifications (Fig. 1). The identification of HOXA13 as an important dysregulated factor by independently performed DNA methylation and histone modifications studies suggests a convergence of these two epigenomic features in LM pathogenesis (George et al., 2019; Leistico et al., 2021). Furthermore, enrichment of FOS, JUN at hypomethylated regions, especially in MED12 mutant LM, corroborates the finding that activator protein-1 (AP-1) is one of the crucial factors in modifying chromatin architecture in this disease (Moyo et al., 2020). Collectively, these studies established a critical role of epigenome regulation of LM. Various studies highlight the coordinated effects of DNA methylation and histone modifications in several important processes such as embryonic development, X-chromosome inactivation, etc. There is evidence suggesting the dependence of each arm on the other in different conditions (Cedar and Bergman, 2009). Consequently, a comprehensive integrative analysis with DNA methylation and histone modifications needs to be performed to understand the interplay between these two arms of epigenomic regulation in the context of this disease. In some fibrotic diseases, methyl-CpG-binding protein 2 [MeCP2] has been studied as a bridge between these two epigenomic events (discussed later). Perhaps, its role may be explored in LM.
Figure 1.
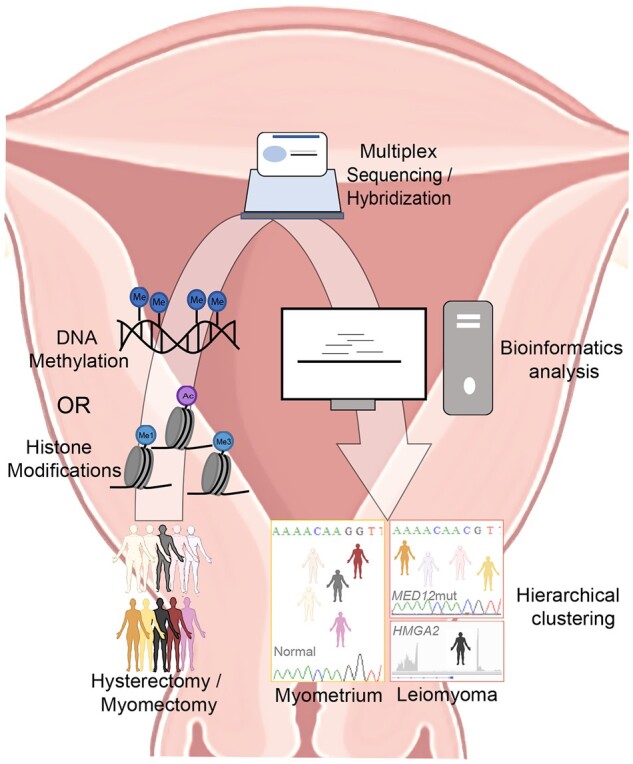
Schematic illustration of workflow employed to identify histone modification/DNA methylation patterns that discriminate uterine leiomyoma from normal myometrium. Tissue samples from patients undergoing hysterectomy or myomectomy were frozen until use. Following appropriate processing, samples were prepared either for ChIP-sequencing (histone modifications, Leistico et al., 2021) or hybridization (DNA methylation, George et al., 2019). Data were analyzed using various bioinformatic tools and unsupervised clustering was performed. Epigenomic alterations could cluster myometrium separately from uterine leiomyoma [LM] and, furthermore, LM into subtypes (MED12mut indicates mediator complex subunit 12 mutated LM subtype; HMGA2, overexpressed high mobility group AT-hook 2 LM subtype).
Role of enhancers in uterine leiomyoma
Background
TFs can recognize specific response elements outside of the promoter proximal regions. An enhancer is a collection of several response elements which regulates gene expression by a variety of signals and acts to amplify gene transcription levels. Like promoters, enhancers are considered cis-regulatory regions, but unlike promoters, they are not necessarily adjacent to the target gene TSS. In humans, these regions can be located as far as 2 million base pairs away, either upstream or downstream, from the affected gene (van Heyningen and Bickmore, 2013). Owing to the large distance between the promoter and enhancer regions, DNA must often be bent into a hairpin loop to form enhancer–promoter contact (Fig. 2). This is believed to occur through lineage-specific DNA-binding TFs—bound at promoters and enhancers—interacting together or recruiting the looping factors necessary to form the long-range contact. Binding proteins, such as CCCTC-binding factor [CTCF] and cohesins, may also play a role in this interaction (Pennacchio et al., 2013).
Figure 2.

Altered H3K27Ac and enhancer architecture in MED12 mutant [G44D/S] leiomyoma. Schematic of the proposed (Moyo et al., 2020) differential activator protein 1 [AP-1] and cyclin-dependent kinase 8 [CDK8]/mediator complex subunit 12 [MED12] occupancy, altered enhancer architecture and altered acetylation of the 27th lysine of histone H3 [H3K27Ac] involved in gene dysregulation of AP-1 and extracellular matrix [ECM] target genes in the pathogenesis of G44D/S-mutated MED12 uterine leiomyoma [LM]. Font size represents the strength of recruitment of factors (MED12, CDK8, Cyclin C, AP-1) and arrow thickness represents transcriptional output. Compared to normal myometrium (A), in LM, a subset of genes (ADAM19 indicates ADAM metallopeptidase domain 19; FN1, fibronectin 1) shows higher CDK8/MED12 binding, increased H3K27ac signals found at the enhancer region and enriched AP-1 binding activity. The resulting stronger enhancer–promoter contact found in MED12 mutant [G44D/S] LM leads to the activation of LM-specific gene program (B). Another subset of genes (EFEMP1 indicates EGF containing fibulin extracellular matrix protein 1; ETS2, ETS proto-oncogene 2, transcription factor) in LM shows lower CDK8/MED12 binding, decreased H3K27ac signals found at the enhancer region and depleted AP-1 binding activity. The resulting weaker enhancer–promoter contact found in MED12 mutant [G44D/S] LM leads to the repression of LM-specific gene program (C). Adapted from ‘Eukaryotic Gene Regulation—Transcriptional Initiation’, by BioRender.com (2020). Retrieved from https://app.biorender.com/biorender-templates
Ample studies have recognized how alterations in promoter regions affect gene expression, but little has been explored in the context of altered enhancer landscape and resulting gene dysregulation. Recent data suggests that changes in enhancer landscape plays a key role in disease pathogenesis (Krijger and de Laat, 2016). The aberrant enhancer epigenome and changes in TF occupancy have been proposed to lead to alterations in enhancer 3D architecture, bringing about dysregulated RNA polymerase II gene transcription. Recently, this postulation has been translated into research on LM formation.
DNA methylation landscape at enhancers in uterine leiomyoma
As discussed earlier, George et al. (2019) utilized EPIC to analyze genome-wide DNA methylation in LM and normal myometrium. One of their observations was a small shift in overall distribution of methylation at enhancer regions, from partially methylated domains to highly methylated domains in LM. While analyzing TF binding sites at differentially methylated loci located distally, they found that both MED12mt and HMGA2hi LM were enriched for enhancer of zeste 2 polycomb repressive complex 2 subunit [EZH2] and SUZ12 polycomb repressive complex 2 subunit [SUZ12] (components of Polycomb repressive complex 2 [PRC2]) binding sites in hypermethylated cytosines. Furthermore, ERα binding sites were distributed at hypomethylated sites. Interestingly, they noticed hypomethylation in a segment of the HMGA2 gene body in HMGA2 overexpressing LM compared to normal myometrial and other subtypes (George et al., 2019). In HMGA2hi LM, 12q14-15 chromosomal rearrangements are generally considered a reason for the higher expression of this gene (Ashar et al., 1995; Schoenmakers et al., 1995). Two of the cases in the study did not show the translocation at these loci using fluorescence in situ hybridization probes but still overexpressed HMGA2. The authors considered the observed hypomethylation to be responsible for this phenomenon (George et al., 2019). They located a CTCF binding site near the 3′ end distal hypomethylation locus of the HMGA2 gene to further explore it. CTCF is a TF that can bind at enhancer sites to form chromatin loops and/or attract transcriptional coactivators, repressors and RNA Polymerase II (Holwerda and de Laat, 2013). Using FANTOM5 and Chromatin Interaction Analysis by Paired-End Tag Sequencing interaction data, a CTCF looping region was observed with the HMGA2 gene within this region. DNA methylation profiles to infer accessibility of chromatin showed that this region possesses more accessibility or a more open chromatin structure in HMGA2hi LM (George et al., 2019). Although further analysis needs to be performed to confirm this observation, this alteration in chromatin structure caused by hypomethylation gives rise to the possibility of a more influential CTCF-mediated looping in HMGA2hi LM that facilitates interaction between the distal enhancer and the HMGA2 gene promoter, initiating increased gene transcription.
Enhancer/histone epigenome dysregulation in uterine leiomyoma
The genome-wide screening of 21 patients for histone modifications via DeCET revealed that the majority (70%) of the changes in LM occur at distal enhancers (Leistico et al., 2021). It is similar to the observation in MED12mt LM published previously in a separate report (discussed below) (Moyo et al., 2020). The enhancer sites with increased activity in LM were located further away from promoters than those with decreased activity. Overall, these observations advocate the role of long-range interactions in the pathogenesis of LM. Leistico et al. (2021) further found that the alterations in histone modifications were mainly confined within the chromatin contact domains (annotated in HeLa cells). These contact domains may perhaps restrict alterations in the epigenome as the tumors evolve. The genes that were upregulated and within 10 kb of the sites that gained histone modification signals were enriched with gene ontology [GO] terms related to collagen and ECM, in accordance with the LM etiology. Integration of assay for transposase-accessible chromatin using sequencing [ATAC-seq] peaks with DeCET regions showed enrichment of HOXA (most notably HOXA9 and HOXA10) and serum response factor motifs in the diseased state. At the same time, compared to the diseased state, the normal condition had more nuclear receptor subfamily 3 group C member 1 [NR3C1] and E26 transformation-specific [ETS] family TF binding sites. Consistent with ATAC data, interestingly Yin et al. (2013) found that NR3C1, also known as glucocorticoid receptor, was a key downregulated nuclear receptor in LM. Furthermore, the posterior HOXA cluster showed transcriptional as well as epigenomic changes, especially in HOXA13 (Leistico et al., 2021). HOX proteins play an important role in reproductive system development and function, and are dysregulated in various tumors (Cillo et al., 2001; Daftary and Taylor, 2006). To explore the role of HOXA13 in the context of LM, Leistico et al. (2021) knocked down and overexpressed HOXA13 in primary LM and myometrial cells, respectively. The key GO terms related to genes modulated when HOXA13 expression was modified were related to ECM. As stated earlier, different arms of epigenomic regulation (i.e. DNA methylation and histone modifications) can converge, as seen in the case of HOXA13, to regulate gene expression in LM pathology (George et al., 2019; Leistico et al., 2021). Previous publications suggest that DNA methylation brings about chromatin changes by altering histone modifications (Kondo, 2009). Further knowledge of the interaction between these two mechanisms in this disease may offer better integrative mechanistic details.
AP-1-driven aberrant enhancer regulation in uterine leiomyoma
Mutations in MED12 have been observed in roughly 70% of patients diagnosed with LM, with the most prevalent occurring in exon 2 of the gene (Makinen et al., 2011). MED12 is a subunit of the mediator of the RNA polymerase II transcription complex and is normally involved in regulating RNA polymerase II-dependent transcription (Baranov et al., 2019).
To identify genome-wide epigenomic changes in MED12mt LM, Moyo et al. (2020) pursued NGS on frozen tissues. To keep variables to a minimum for this study, only cases with missense mutations of glycine, at the 44th codon in exon 2 of MED12, to either aspartate [G44D] or serine residue [G44S] were followed-up. The transcriptomic data revealed a clear distinction between G44D/S LM and adjacent matched myometrium (Moyo et al., 2020). Notably, H3K27ac ChIP-seq yielded features that could distinguish the diseased state from normal. As observed above, most of the differential H3K27ac signal was located distal to promoters in intergenic and intronic regions, suggesting the role of enhancers. Moreover, ∼ 46% of the deregulated genes did not show a significant change in H3K27ac at their promoters, which included genes such as fibronectin 1 [FN1], a disintegrin, ADAM metallopeptidase domain 19 [ADAM19] and collagen Type XII alpha 1 chain [COL12A1]. In order to define the relation between distal H3K27ac signal and transcriptomic changes, Moyo et al. (2020) performed promoter capture Hi-C (high throughout chromosome conformation capture). While capture Hi-C identifies all genomic interactions, promoter-capture HiC is more specific to determining altered enhancer interactions within promoters, thereby analyzing ‘productive interactions’. About 1835 of 2715 differentially acetylated H3K27 enhancer regions, associated with differentially expressed genes, had an insignificant level of change in H3K27 acetylation in their corresponding promoters. Enhancer regions with altered contacts which showed increased acetylation status were positively correlated with increased promoter contact strength, whereas depleted acetylation had varying contact strength. Some genes involved in ECM formation that demonstrated differential gene expression related to changes in promoter contact strength include FN1, ADAM19, ETS proto-oncogene 2 transcription factor [ETS2], EFEMP1, COL6A3 and COL12A1. Furthermore, capture Hi-C showed that in LM, 672 genes displayed differential enhancer usage, wherein decreased H3K27ac levels in one enhancer for a specific gene occurred simultaneously with an increase in H3K27ac levels in another enhancer associated with the same gene (Moyo et al., 2020). A possible implication of this may allow for the alteration of strong enhancers with weak ones, and vice versa, resulting in changes in gene expression.
The mediator complex, comprising a core mediator and a CDK8 submodule, forms a stable complex that regulates transcription. Specifically, MED12 is one of the subunits of the CDK8 submodule that consists of CDK8, MED12, MED13 and cyclin C (Allen and Taatjes, 2015; Soutourina, 2018). A previous study proposed that one possible mechanism leading to the dysregulation of transcription in LM by mutant MED12 is the decreased binding affinity of cyclin C and CDK8 at active transcription sites (Turunen et al., 2014). However, Moyo et al. (2020) demonstrated that it is not a global loss of cyclin C/CDK8 binding but rather altered CDK8/MED12 binding that leads to LM-specific enhancer features. This postulation was complemented by the positive correlation observed between altered H3K27ac and changes in the CDK8 submodule chromatin occupancy in the enhancer region (Moyo et al., 2020). This modified enhancer architecture, caused by changes in chromatin from histone acetylation and altered enhancer–promoter contact, provides a potential mechanism for the gene dysregulation that occurs outside the promoter-proximal area in MED12mt LM. Moreover, understanding how the interaction of mutated MED12 with other members of the CDK8 submodule brings about changes in their DNA-binding, as observed in LM, may ultimately provide a mechanism for initiation of this disease. Again, while compelling correlative data emerges linking epigenomic changes to LM gene expression and possibly disease development and progression, in a majority of the cases, causal experiments are lacking. With the use of CRISPR-based gene editing, correlative altered enhancer and epigenome function in LM gene regulation can now be causally studied.
Additionally, it was observed that the motif of TF AP-1 was enriched at differentially acetylated enhancers (Moyo et al., 2020). The AP-1 term is used for dimeric TFs composed of subunits of JUN, FOS, activating transcription factor or musculoaponeurotic fibrosarcoma that recognize a common motif (Bejjani et al., 2019). ChIP-seq analysis of FOS and JUN uncovered a reduction in AP-1 occupancy in chromatin in LM. While it has already been reported that LM show down-regulation of JUN and FOS mRNA, the Moyo et al. (2020) study demonstrated that a loss of JUN or FOS gene expression led to an accompanying decrease in the binding of AP-1 at enhancer sites that contained reduced H3K27ac and therefore resulted in the dysregulation of previously identified AP-1 target genes and ECM-associated genes, among others (Lessl et al., 1997; Skubitz and Skubitz, 2003; Moyo et al., 2020). Strikingly, sites where AP-1 gained or lost occupancy in LM were also bound by CDK8 and MED12, with changes in their occupancy in the same direction. The simultaneous CRISPR/Cas-9-mediated depletion of JUN and FOS proteins in primary human uterine smooth muscle cells [HUtSMC] revealed differential expression of 1894 genes compared to the negative control, including those involved in ECM organization. Furthermore, changes in H3K27 acetylation patterns in AP-1-depleted cells were similar to those observed in LM, with the majority of changes occurring at promoter-distal regions (Moyo et al., 2020). This resemblance in both gene expression and H3K27ac levels between LM and the AP-1-depleted cells implies that the loss of AP-1 gene expression is linked to the aberrant epigenome and dysregulation of ECM genes observed in LM. Overall, the findings propose that aberrant enhancer regulation in LM related to differential AP-1 and CDK8/MED12 occupancy, altered enhancer–promoter contact and altered H3K27 acetylation are important mechanisms in LM pathogenesis (Fig. 2). The subunits of AP-1 did not show differential H3K27ac signal in their promoters in the diseased state; differential enhancer usage was proposed to be a mechanism leading to decreased expression of these genes (Moyo et al., 2020). Identification of the factors that cause the use of alternate enhancers may ultimately be beneficial in understanding the development of this disease.
Overall, it is becoming clear that enhancer dysfunction seems to be an important phenomenon in the etiology of these uterine benign tumors. Of note is that the genome-wide DNA methylation (George et al., 2019), histone modifications (Leistico et al., 2021) and promoter capture Hi-C (Moyo et al., 2020) studies were performed in tissues, thereby enhancing the physiological relevance of these findings. Therefore, closely mapping the genes that are deregulated in LM, their distal regulatory regions and in turn regulation of those regions may offer potential therapeutic targets. More studies dissecting primary events from secondary effects may be helpful in identifying driver events leading to LM formation. Furthermore, loci-specific DNA methylation or histone modifications could provide information about deregulated genes; however, genome-wide studies give a better picture of the chromatin architecture and regulation by enhancers. Compounds currently under clinical trials for other diseases targeting the epigenome and chromatin-modifying enzymes may be proven useful in non-surgical therapeutic intervention in LM.
Histone variants, chromatin remodelers and a new subtype of uterine leiomyoma
While altered DNA methylation and histone modifications have now been linked to LM, the role of histone variants and chromatin remodelers is not well established. Additionally, the previously known exclusive mutations in LM do not cover genetic mutational status of all LMs, thereby necessitating more extensive studies using a larger number of human samples. A tour de force comprehensive study, using a large cohort of patient samples and state of the art genomic, epigenomic, chromatin-state and molecular analyses, now sheds new light on LM genesis, identification of the potential role of the SRCAP chromatin complex and the role of histone variant H2A.Z in LM (Berta et al., 2021). This comprehensive study on 2263 fibroids from 728 women helped identify a new molecular subtype of LM, thereby advancing our knowledge of additional exclusive mutations in genes defining LM subtypes (Berta et al., 2021). This new subgroup of patient samples, which represented ∼ 1.8% of the LM cases analyzed, harbored mutations in six out of nine genes encoding the chromatin remodeler SRCAP complex subunits, with YEATS domain containing 4 [YEATS4] being most frequently mutated. Epigenomic silencing of the normal allele by DNA methylation of YEATS4 was also observed in tumors carrying heterozygous YEATS4 mutations. As validation for their finding, the authors found that protein truncating germline mutations in YEATS4 and another gene of this complex, zinc finger HIT-type containing 1 [ZNHIT1], are risk factors for the development of LM (Berta et al., 2021). SRCAP is a chromatin remodeler complex that carries out histone, specifically H2A.Z, exchange/deposition (Giaimo et al., 2019). H2A.Z is found at the nucleosomes flanking the promoter region, centromeres and boundaries of chromatin (Gerhold and Gasser, 2014). The functional roles of H2A.Z include DNA repair, maintaining genome integrity and controlling gene transcription (Giaimo et al., 2019).
To determine if SRCAP alteration affected protein levels of H2A.Z, Berta et al. (2021) performed immunohistochemistry and found that SRCAP-altered LM had very weak to no staining, suggesting that the level of H2A.Z is reduced in this subtype. Myometrium, MED12mt LM and LM with loss of FH had the strongest expression, while HMGA2 overexpressing LM had moderate expression. Moving forward, the ChIP-seq of H2A.Z revealed significant loss of H2A.Z binding not only in SRCAP-altered LM, as expected, but also surprisingly in MED12mt LM that displayed strong H2A.Z staining. These results suggest that additional regulatory mechanisms may be involved in H2A.Z deposition in MED12mt LMs. These results also suggest that dysregulated H2A.Z deposition may be a common phenomenon in all LM subtypes, which may also involve different mechanisms based on the disease subtypes. Considering that histone/nucleosome occupancy dictates open/closed chromatin states and dysregulated chromatin occupancy of H2A.Z is observed in ChIP-seq analysis, the authors performed ATAC-seq to identify open/closed chromatin states in a genome-wide manner. Their ATAC-seq analysis showed that YEATS4-mutated LM have a more open chromatin structure at the TSS of active or bivalent genes and these regions showed reduced H2A.Z binding (Berta et al., 2021). This finding is consistent with previous observations made in other model systems (Giaimo et al., 2019). Of note, bivalent genes house both activating (H3K4me3 or H3K4me1) and repressing (H3K27me3) histone marks within promoter or enhancer regions. These associated genes are expressed at low levels but poised for rapid gene activation following cellular cues (Bernhart et al., 2016).
The authors then addressed the key question regarding dysregulated H2A.Z deposition and gene expression changes associated with SRCAP-altered LMs. Using RNA sequencing analysis, the authors found that YEATS4-mutated LM displays upregulation of differentiation genes driven by H2A.Z sensitive bivalent promoters. These results suggest that, in SRCAP-altered LM, these gene sets are primed to be regulated based on their promoter bivalency and that rapid dysregulated differentiation may be a contributing factor in LM genesis. The authors then examined expression of genes whose promoters showed differential H2A.Z occupancy in all disease subtypes. Interestingly, the results show that for HMGA2, HMGA1 and FH LM subtypes, decreased H2A.Z binding correlated with downregulation, while increased H2A.Z promoter occupancy correlated with upregulation of target gene expression. However, in YEATS4- and MED12mt LM subtypes, this direct correlation between H2A.Z and gene expression was not apparent. For example, the authors found that in YEATS4-mutated LM with H2A.Z lost in TSS, 204 genes were overexpressed while 296 genes were under expressed. GO of overexpressed genes identified morphogenetic pathways while under expressed genes failed to identify any specific biological pathways. These results suggest that the gene regulation linking H2A.Z deposition and SRCAP complex mutation is complicated and may also involve non-promoter bound H2A.Z-mediated regulation as well as H2A.Z deposition-independent functions of SRCAP complex mutations in this LM subtype. Consistent with this thought, the authors found, using H3K27ac-HiChIP-based chromatin state capture assays, that decreased H2A.Z binding outside of TSS (promoter distal/potential enhancer sites) correlates with decreased gene expression of these enhancer connected promoters (Berta et al., 2021).
Additionally, Berta et al. (2021) found upregulation in components of canonical PRC-1 [cPRC1], which were Chromobox 2, 4 and 8 [CBX2, 4 and 8]. Consistent with the findings presented in Berta et al., two independent studies also found these genes to be upregulated in LMs (Mehine et al., 2016; Berta et al., 2021; Leistico et al., 2021). cPRC1 is involved in development by bringing about epigenetic silencing of genes (Geng and Gao, 2020). Furthermore, genes coding for TFs involved in development (HOXA13 and SATB2) along with steroid 5 alpha-reductase 2 [SRD5A2] and hydroxysteroid 17-beta dehydrogenase 6 [HSD17B6], genes that encode dihydrotestosterone synthesis, were upregulated in all subtypes of LM (Berta et al., 2021). Leistico et al. (2021) had a similar observation in their RNA-seq screen (Leistico et al., 2021). Again, global gene expression analysis coupled with genome-wide DNA methylation and histone modification studies identify developmental TF HOXA13 as a key dysregulated factor in LM (George et al., 2019; Berta et al., 2021; Leistico et al., 2021). These independent studies emphasize the robustness and usefulness of genome-wide studies using tissue preparations in molecularly characterizing LMs (George et al., 2019; Berta et al., 2021; Leistico et al., 2021).
Although researchers in the Berta et al. (2021) study have used many of the latest multi-omics techniques to characterize the molecular landscape of LM and found a new subtype of LM (Berta et al., 2021), there are still a few questions that can open up new research areas in the future. MED12mt LM show high protein expression of H2A.Z but have reduced chromatin binding. What causes this alteration needs to be understood. H2A.Z occupancy in LM showed an inverse relationship with DNA methylation at binding sites and a positive correlation with chromatin accessibility; however, these associations were weak in the tumors bearing SRCAP complex mutations (Berta et al., 2021). It remains elusive as to how changes in H2A.Z binding cause formation of LM in SRCAP tumors. Berta et al. (2021) independently reported how the epigenetic landscape of LM changes upon H2A.Z loading deficiency, and how this epigenetic instability may be involved in LM genesis (Berta et al., 2021). Similarly, altered chromatin binding and resulting changes in 3D-chromatin structure have been observed by other groups (George et al., 2019; Moyo et al., 2020; Leistico et al., 2021). Collectively, these independent studies highlight the possibility of altered enhancer architecture being, at least partially, responsible for the pathogenesis of this disease, but causal and functional studies are largely lacking and need to be performed to move the field forward. However, it is important to note that the proneness to LM development in women together with germline mutations within SRCAP complex genes may unveil clinical implications that are of potential use therapeutically and in genetic counseling settings when advising women who are affected by LM or at risk of LM development (Ordulu, 2021).
Epigenomics in other fibrotic diseases
DNA methylation
Like LM, many loci-specific DNA methylation events have been identified in various other fibrotic diseases. Genes that are methylated and downregulated include RAS protein activator like 1 [RASAL1] (tumor suppressor gene found in kidney fibrosis), peroxisome proliferator activated receptor gamma [PPARG or PPARγ] (antifibrotic gene in liver fibrosis), Fli-1 proto-oncogene, ETS TF [FLI1] [suppressor of collagen expression, systemic sclerosis] and Thy-1 cell surface antigen [THY1] (TGFβ inhibitor, idiopathic pulmonary fibrosis [IPF]), among others (Yao and Li, 2015; O'Reilly, 2017). Hypoxia in cardiac fibrosis has been understood as one of the major drivers for increased global methylation by deregulating DNMT1 and DNMT3b. The pro-fibrotic effects of hypoxia could be counteracted by a general DNMT inhibitor, 5-aza-dC. In addition, treatment with 5-azacytidine, another DNMT inhibitor and ribose analog of 5-aza-dC, attenuates folic acid-induced renal fibrosis (Yao and Li, 2015). Thus, it seems that modulators of DNA methylation can provide relief in some fibrotic diseases. However, specific DNMT inhibitors may be more beneficial by reducing the potential side effects of global DNA methylation inhibitors. In the liver, MeCP2 is considered an important regulator of fibrosis. MeCP2 binds to methylated DNA and, by interacting with HDACs, causes gene repression. MeCP2 is found to regulate PPARγ expression in liver fibrosis, thus leading to increased pro-fibrotic molecules in this disease. It also decreases the expression of RASAL1 in kidney fibrosis and thereby increases Ras-GTP signaling. RASAL1 is hypermethylated in fibrotic hearts. This hypermethylation could be reversed not only by non-specific broad-spectrum demethylating agents but also, interestingly, by an endogenous antifibrotic agent, BMP7 (O'Reilly, 2017). This treatment concept may be explored even in LM to target specific DNA methylation loci to minimize side effects. As we now know from LM, enhancers play an important role in the manifestation of this disease. Potentially, similar phenomena also occur in other fibrotic diseases and, thus, more comprehensive studies should be performed exploring enhancer malfunction in these and other fibrotic diseases.
Histone modifications
General HDAC inhibitors [HDACi] have been associated with decreasing fibrotic burden in various tissues through distinct signaling pathways, for example, in renal fibrosis via TGFβ and EGF receptor signaling, in bleomycin-induced lung fibrosis by apoptosis, and in liver fibrosis by inhibiting TGFβ and vascular endothelial growth factor pathway (Yao and Li, 2015; O'Reilly, 2017). The success of HDACi may be attributed to their dysregulated HDAC activity in fibrosis, but it seems to be highly dependent on tissue type. To develop successful treatment, the specific HDAC enzymes deregulated in each condition should be determined and treated appropriately. Not only HDACs but also HATs can also contribute to fibrosis; for example, EP300 is increased in systemic sclerosis and is regulated by TGFβ1 (O'Reilly, 2017). Therefore, a good understanding of which HDACs and HATs are aberrantly expressed during fibrosis is required for specific treatment with minimum toxic effects. In addition to acetylation, histone methylation is an important modification in regulating pro-fibrotic gene expression. Several loci-specific occurrences have been reported by different groups. These include increased H3K9me2 near the PPARγ promoter in liver fibrosis that leads to reduced PPARγ expression; lung fibroblasts from IPF patients show reduced H3K9me3, H3K27me3 and DNA methylation at the prostaglandin-endoperoxide synthase 2 (also referred to as COX2) promoter (Yao and Li, 2015; O'Reilly, 2017). In liver fibrosis, Jumonji domain containing 1A (an H3K9 demethylase) is involved in regulating fibrosis by regulating PPARγ; in systemic sclerosis, treatment with DZNep (a methyltransferase EZH2 inhibitor) causes more ECM formation by upregulation of Fra2 (AP-1 family member) and increases fibrotic phenotype (O'Reilly, 2017). At the COX2 promoter in lung fibroblasts, the reduced accumulation of histone methyltransferases G9a, EZH2 and DNMTs as well as binding protein heterochromatin protein 1, Polycomb protein complex 1 and MeCP2 have been implicated in alteration of histone modifications at this site. EZH2 and ASH1 (an H3K4 methyltransferase) have also been implicated in liver fibrosis. ASH1 seems to be regulated by MeCP2, thus inducing pro-fibrotic genes (O'Reilly, 2017). This and other examples, as discussed above, link DNA methylation with histone modifications via MeCP2. The role of MeCP2 has not yet been established in LM; it may provide a good opportunity to scrutinize these two important arms of epigenetic modifications in this disease. Also, the above histone modifications studied in various fibrotic diseases suggest a diverse string of events, all leading to the fibrotic state. Thus, it is important to perform genome-wide histone modification studies to identify common and tissue-specific epigenomic events in various tissue fibrosis pathologies. In such a study, the tensor decomposition method employed for LM may be beneficial to link specific epigenome alterations of the disease.
Racial disparities of uterine leiomyoma
LM has been found to disproportionately affect Black women, with the prevalence being two to three times greater as compared to White women (Catherino et al., 2013; Eltoukhi et al., 2014). Black women are particularly affected with an increased incidence, earlier age of onset, larger and faster growing fibroids and greater severity of symptoms (Marshall et al., 1997; Moorman et al., 2013; Bray et al., 2017). Furthermore, an online study surveying Black and White women was conducted to evaluate symptoms and impairments, concerns regarding treatment and the effect on employment and social life. It reported that Black women suffered from more severe symptoms and anemia; were more likely to miss work owing to symptoms; expressed more interference in daily and social activities; had a higher degree of impaired relationships with family, friends and significant others; and were significantly more concerned with the effects of treatment relating to fertility and a successful pregnancy. It is also noteworthy that although Black women expressed higher interest than White women in uterine-sparing procedures (Stewart et al., 2013), they are still 2.4 times more likely to undergo hysterectomy, possibly owing to the increased severity (Wechter et al., 2011; Richard-Davis, 2013). Overall, the increased incidence and severity of the disease in Black women has attracted many to identify the influence that ethnicity has in LM development and growth.
There are a wide range of risk factors and exposures that are posited that place Black women at an increased risk for fibroids over White women. The field of epigenomics includes gene–environment interactions that can potentially include social environments. Specifically, the idea behind social and behavioral epigenomics is that socio-cultural factors can generate trauma-associated epigenetic changes, which impact the expression of certain genes (Mulligan, 2021). It is well known that Black women experience racism that promulgates negative social and economic conditions (the social and economic determinants of health), which in turn negatively impact health and thus promote health inequities. The experience of racism and the social determinants of health and their influences on LM have been studied (Zota and VanNoy, 2021). For example, one study observed the association between increased major life discrimination events and altered DNA methylation at nine CpG sites within disease-associated genes (Barcelona de Mendoza et al., 2018). Additionally, the disproportionate suffering of Black women with LM might arise from other environmental factors that induce chronic stress, such as the experiences of adverse childhood experiences [ACEs] and economic hardships, both of which are known to contribute to negative health outcomes (Hughes et al., 2017; Aroke et al., 2019). It has already been shown that exposure to early childhood abuse is associated with the incidence of clinically detectable LM (Boynton-Jarrett et al., 2011). Not only do Black children experience a higher quantity of ACE compared to children from other races, but they are also more likely to grow up in lower socio-economic status [SES] neighborhoods (Aroke et al., 2019). Needham et al. (2015) found that low childhood SES was associated with changes in global DNA methylation, leading to the differential expression of stress- and inflammation-related genes (Needham et al., 2015). While studying the relation between psychological stress and LM, a study found that chronic stress, which Black women were determined to be more exposed to, correlates with increased hypothalamic–pituitary axis activity. This eventually leads to the increased release of sex steroid hormones that drive LM development (Qin et al., 2019). Interestingly, it has also been reported that upwards mobility in Black women results in a significantly increased likelihood of racial discrimination (Colen et al., 2018). While upwards mobility is thought to serve a health protective function in White women, it is thought that these same benefits are likely suppressed in racial minorities owing to the continuance of race-based unfair treatment (Colen et al., 2018). As such, this suggests that the disparity faced by racially oppressed Black women can possibly be related to socio-cultural factors, such as segregation, systemic racism, discrimination, economic hardship and other stressors, which influence the expression of LM-specific genes through imposing changes on the epigenome.
Abnormalities in vitamin D levels have already been observed in various estrogen-dependent gynecological disease pathologies, such as in endometriosis, polycystic ovary syndrome and ovarian cancer (Colonese et al., 2015). Vitamin D may be obtained through the diet, but the majority of natural vitamin D is produced by the conversion of 7-dehydrocholesterol to vitamin D3 in the skin by means of sun exposure. To form the biologically active metabolite, vitamin D3 is first hydroxylated in the liver to produce 25-hydroxyvitamin D3 (calcidiol), the major form of circulating vitamin D, which is then further hydroxylated in the kidney and other extra-renal tissues to form the active metabolite 1, 25 dihydroxyvitamin D3 (calcitriol) (Bikle, 2014). Vitamin D deficiency, or hypovitaminosis D, has also been identified as a potential LM risk factor with several studies finding an association between low serum levels of vitamin D3 and increased incidence of LM (Baird et al., 2013; Sabry et al., 2013; Ciebiera et al., 2016; Halder and Al-Hendy, 2016; Ciebiera et al., 2020; Mohammadi et al., 2020). Furthermore, Othman et al. (2018) reported that the tissue concentration of calcitriol was lower in LM compared to its adjacent myometrium; however, they did not find a significant correlation between the level of calcitriol within LM and tumor volume (Othman et al., 2018). Physiologically, vitamin D is known to play a role in regulating calcium and phosphorus metabolism, but in vitro studies of LM have suggested its role in inducing apoptosis, suppressing the action of matrix metalloproteinases, and downregulating ER and PR levels (Sharan et al., 2011; Halder et al., 2013; Al-Hendy et al., 2015). Calcitriol has also been implicated in serving to inhibit cell proliferation and deposition of ECM by reducing TGF-β3 effects in human LM cell lines (Halder et al., 2011). The authors also found that women undergoing hysterectomy for symptomatic LM had significantly higher expression of CYP24A1, a gene encoding 24-hydroxylase enzyme, compared to normal myometrium from women undergoing the same procedure for non-LM indications. Since this enzyme catalyzes the degradation of calcidiol and calcitriol, the authors indicated that the overexpression of 24-hydroxylase may be implicated in the mechanism that sustains a hypovitaminosis D state in LM (Othman et al., 2018). Interestingly, vitamin D insufficiency is roughly 10-fold more prevalent among Black women compared with White women, likely because of their darker skin pigmentation, which reduces vitamin D production from sunlight exposure (Nesby-O'Dell et al., 2002; Harris, 2006). This predisposition can provide a partial explanation for the ethnic disparity observed with regards to LM. One recent study also proposed that low vitamin D receptor [VDR] expression may be associated with the development and growth of LM (Lima et al., 2021). In this recent study, 67% of the evaluated women were of Black descent, thus it would be interesting to examine if the expression of VDR varies among races. On the contrary, in an expression profiling study of 48 nuclear receptors from 22 patients that included 36% Black women, Yin et al. (2013) reported that VDR was a hyperexpressed receptor in LM when compared with myometrium (Yin et al., 2013). Similarly, Leistico et al. (2021) found increased expression of VDR in their RNA-seq analysis from LM compared to matched myometrium that included 30% Black women. This discrepancy may be explained by the analysis of the different molecular entities that are protein (Lima et al., 2021) and RNA (Yin et al., 2013; Leistico et al., 2021). On the other hand, it may not be wrong to assume that the inconsistencies are race-related as there is differential representation of Black women in the studies. Nevertheless, many studies have further found that Vitamin D supplementation is a potentially effective, non-surgical treatment in reducing LM-related symptoms and the volume of LM (Hajhashemi et al., 2019; Davari Tanha et al., 2021; Halder et al., 2012; Suneja et al., 2021). In one such preliminary study which aimed to identify the role of vitamin D supplementation in women with concurrent hypovitaminosis D and LM, it was demonstrated that supplementation therapy with calcidiol restored normal vitamin D serum levels and stabilized the growth of LM, preventing progression of the disease and thus reducing the need for traditional surgical or other management options (Table III) (Ciavattini et al., 2016). As such, the antifibrotic effects of vitamin D should be further investigated owing to the potential use as a novel therapeutic and an explanation for the ethnic disparity seen within this disease. Similarly, studies employing 24-hydroxylase inhibitors or 24-hydroxylase-resistant calcidiol analogs may be of use to better understand the effect of augmenting vitamin D levels on LM incidence and as possible therapeutics (Othman et al., 2018). Furthermore, clinician and public health educator interventions, such as employing 25-hydroxy vitamin D tests and supplementation to those with LM or at risk of developing the disease, can lessen the burden of the disease and LM growth.
Table III.
Current medical, radiological and surgical management options for uterine leiomyoma.
| Name | Function | Limitations | Source | |
|---|---|---|---|---|
| Medical management | Non-steroidal anti-inflammatory agents [NSAIDs] | Improves dysmenorrhea and menorrhagia; works by inhibiting cyclooxygenase enzyme involved in the inflammatory process | May cause gastrointestinal [GI] bleeding and ulcers | (Vane and Botting, 1998) |
| Selective progesterone receptor modulators [SPRMs] | Reduces bleeding, pain, fibroid volume; not associated with the adverse symptoms of a hypoestrogenic state; works by inhibiting cell proliferation and inducing apoptosis | May cause liver damage; may cause progesterone receptor modulator-associated endometrial changes | (Donnez and Dolmans, 2016) | |
| Gonadotropin releasing hormone [GnRH] agonist | Reduces heavy menstrual bleeding, uterine volume and fibroid volume; works by desensitizing the GnRH receptor producing a hypogonadotropic state and thereby reducing secretion of estradiol and progesterone | Short-term use may cause adverse symptoms associated with a hypoestrogenic state; long-term use may result in reduced bone mineral density and cause histological changes in uterine leiomyoma | (Lethaby et al., 2001; Donnez and Dolmans, 2016) | |
| GnRH antagonist | Provides faster symptom relief than GnRH agonists, reduces tumor volume and uterine size; works by competitively binding to GnRH receptor | Compared to GnRH agonists, GnRH antagonists are more expensive and require daily injections due to their short half life | (Lethaby et al., 2001; Giuliani et al., 2020) | |
| Combined estrogen-progesterone contraceptives | Reduces menorrhagia | Has no effect on decreasing tumor volume | (Marret et al., 2012) | |
| Tranexamic acid | Improves abnormal uterine bleeding and symptoms by acting as an antifibrinolytic agent | May produce GI and musculoskeletal symptoms | (De La Cruz and Buchanan, 2017) | |
| Progestogen-only | Progestogen: a synthetic progesterone hormone that may be administered orally or through injections; reduces menorrhagia | Progestogen-associated histopathological changes may lead to misdiagnosis of leiomyosarcoma or smooth muscle tumor of uncertain malignant potential; risk of IUS expulsion; overall lack of high-quality evidence assessing its efficacy; mixed study results regarding the influence on uterine and tumor volume | (Senol et al., 2015; Sohn et al., 2018; Donnez, 2020; Sangkomkamhang et al., 2020) | |
| Progestogen-releasing intrauterine system [IUS]: reduces menorrhagia; the device is placed inside the uterus and works by secreting progestogen to suppress endometrial lining and therefore reduce blood flow | ||||
| Interventional Radiology | Uterine Artery Embolization [UAE] | Provides symptom relief and reduces tumor volume; works by limiting blood supply to the tumor and inducing ischemic necrosis | Risk of infection; may compromise blood supply to ovaries or other organs; increased likelihood of reintervention within 2 to 5 years after initial procedure; may impact fertility; post embolization syndrome | (Gupta et al., 2014; Zupi et al., 2016) |
| Magnetic resonance-guided high-intensity focused ultrasound [MRgHIFU] | Significant symptom relief and tumor shrinkage for at least 12 months; works via using a magnetic resonance imaging [MRI] device to monitor thermal ablation using ultrasonic energy to cause coagulative necrosis within fibroid tissue | May cause abdominal pain, abdominal edema, vaginal discharge, skin burn, and damage to surrounding tissue; inclusion criteria is met by only a portion of patients; can compromise fertility; higher cost due to reinterventions and the need of an MRI device; procedure time is longer compared to USgHIFU | (Gorny et al., 2011; Zupi et al., 2016; Wang et al., 2018) | |
| Ultrasound guided-high intensity focused ultrasound [USgHIFU] | Significant symptom relief and tumor shrinkage; works by using a diagnostic ultrasound device to locate the target region to induce thermal ablation using ultrasonic energy and result in coagulative necrosis within the tissue |
|
(Wang et al., 2018; Liu et al., 2021b; Marinova et al., 2021) | |
| Cryomyolysis | Reduces tumor volume, bleeding, pelvic pain, and urinary frequency; works by limiting primary blood supply to the tumor via cryoablation | May impact fertility; exclusion criteria limits women who can undergo this treatment; lack of histological examination of fibroids | (Zupi et al., 2016) | |
| Radiofrequency thermal ablation (Acessa) | Reduces tumor volume and provides symptom relief; works by using a thin needle to target the tumors via radiofrequency heat, causing coagulative necrosis and reabsorption of the tumor by surrounding myometrium | May result in skin burns; Possibility of reoccurrence and reintervention; limited long term data for pregnancy and pregnancy outcomes | (Lee and Yu, 2016) | |
| Surgical Management | Myomectomy |
|
Risk of hemorrhage; possibility of being converted to hysterectomy; risk of uterine rupture; possible scarring which can affect fertility; likelihood of reintervention within 5 to 10 years due to fibroid reoccurrence | (De La Cruz and Buchanan, 2017; Flyckt et al., 2017; Piecak and Milart, 2017) |
| Hysterectomy | Definitive cure for fibroids; surgical procedure to remove the uterus via open or minimally invasive approach | Fertility is not preserved; vaginal cuff cellulitis; risk of infection; infected pelvic hematoma or abscess; venous thromboembolic complications | (Clarke-Pearson and Geller, 2013; De La Cruz and Buchanan, 2017) |
Pan et al. (2007) performed proteomic and genomic analysis on samples from Black and White women, which revealed a similar molecular environment in the two groups. Still, there was a difference in the expression level of genes and proteins involved in regulating the inflammatory response, apoptosis, cell cycle and ECM turnover between the two cohorts, potentially giving rise to the difference in LM incidence between the two ethnic groups (Pan et al., 2007).
In an effort to identify risk alleles that differ in frequency among Black and White women, Wise et al. (2012) performed an admixture-based genome-wide scan. However, they were unable to find a specific highly differentiated locus that is responsible for the increased risk of LM in Black women (Wise et al., 2012). In a more recent study, which conducted a multi-stage genome-wide association study [GWAS] in Black women, Hellwege et al. (2017) identified several single nucleotide polymorphisms [SNPs] near cytohesin 4 [CYTH4], ER degradation enhancing alpha-mannosidase like protein 1 [EDEM1], junctophilin 1 [JPH1] and CUB and sushi multiple domains 1 [CSMD1] genes as potential loci to be associated with LM risk in Black women (Hellwege et al., 2017). However, another study was not able to validate these findings (Bray et al., 2018). In another GWAS, Valimaki et al. (2018) reported 22 genome-wide susceptibility loci among genes involved in genitourinary development and genetic stability, which when compiled into a polygenic risk score for LM association, revealed that Black women entailed the highest population-specific risk score compared to ‘Other white background’, ‘Indian’, ‘Irish’ and ‘Northern Finland Birth Cohort’ ethnicities (Valimaki et al., 2018). Various other factors, such as SNPs within genes involved in estrogen synthesis, genes related to the retinoic-acid pathway and aberrant miRNA expression levels, have since been identified as potential molecular mechanisms explaining the difference in LM development among ethnic groups but have had conflicting results when reproduced (Commandeur et al., 2015). For example, Wang et al. (2007) performed a global miRNA profile analysis and found that a subset of miRNAs, including miR-23a/b, let-7s, miR-145, miR-197, miR-411 and miR-412, were significantly different in LM of Black women compared to White women (Wang et al., 2007).
As discussed in above sections, recent multi-omics studies such as RNA sequencing (Moyo et al., 2020; Berta et al., 2021; Leistico et al., 2021), histone modifications (Berta et al., 2021; Leistico et al., 2021) and DNA methylation (George et al., 2019; Berta et al., 2021) have found differential features between LM subtypes based on the mutation status. These studies included patients with different racial backgrounds; however, the genomic and epigenomic changes found in LM do not identify any distinctive signatures that could justify the observed ethnic disparities. As noted by Pan et al. (2007), possibly it is the intensity of the response to the mutational driver that leads to greater severity of this condition in Black women compared to their White counterparts (Pan et al., 2007). Overall, it seems that the greater incidence and severity of LM related to the impact of ancestry of African American, African and Black descent remains unclear and may be attributable to a combination of genetic, socio-economic and environmental factors. Furthermore, more intensified studies involving a larger number of patients are clearly needed to identify and confirm the proposed molecular mechanisms and events that may explain the difference in LM incidence and LM size between ethnic groups.
Translational values and future therapy
The available treatment options for LM form three main categories: medical management, interventional radiology and surgical management (summarized in Table III) (Vilos et al., 2015; Sohn et al., 2018; Giuliani et al., 2020). When it comes to choosing which treatment route to undergo, various factors are considered, including the size and location of LM, the patient’s age, the severity of symptoms and the desire to preserve fertility or the uterus (De La Cruz and Buchanan, 2017). Additionally, since the shrinkage of tumors and relief of symptoms is correlated with menopause, asymptomatic women or those approaching menopause may choose expectant management, or clinical surveillance, before discussing further treatment options (Vilos et al., 2015).
Currently, gonadotropin-releasing hormone [GnRH] agonists and selective PR modulators [SPRMs] show the most potential as medical therapies owing to the abundance of evidence supporting their ability to shrink LM and improve symptoms (Sohn et al., 2018; Lethaby et al., 2001). GnRH agonists work by desensitizing GnRH receptors, resulting in a hypogonadotropic hypogonadal state (or pseudo menopause) (Ortmann et al., 2002). Since LMs are steroid-responsive tissues, this hypoestrogenic state is thought to restrict the growth of the tumor (Sohn et al., 2018). Unfortunately, this compound is limited to short-term therapy owing to its association with reducing bone mineral density and imposing histological changes in LM during long-term use (Palomba et al., 1999). Alternatively, SPRMs are synthetic steroids that can act as agonists or antagonists on PR in progesterone-sensitive tissues, such as LM (Wagenfeld et al., 2016). Some of the current SPRMs that have shown to be effective in the treatment of LM include mifepristone and ulipristal acetate [UPA]. Vilaprisan is still being investigated; meanwhile, Asoprisnil and Telapristone have been discontinued because of safety concerns (Islam et al., 2020). Several clinical trials confirmed UPA’s efficacy in treating LM in various races, such as the PEARL I–IV studies completed on White women in Europe (Donnez et al., 2012a, 2012b, 2014, 2016), VENUS I and II studies completed on Black women in the USA (Liu et al., 2018; Simon et al., 2018), and a phase III randomized control trial on Japanese women in Asia (Osuga et al., 2021). Although UPA had been approved in Canada and Europe, in March 2020, the Europe Medicines Agency suspended the use of UPA in treating LM until its safety is reevaluated owing to its serious risk of causing liver damage in women taking the drug (Ekanem and Talaulikar, 2021). Critics have suggested that the clinical promise of UPA has been overlooked because of its association with liver injury (Liu, 2021). They have pointed out that the risk of drug-induced liver injury is 10 out of 100 000, while the risk of death as a result of laparoscopic hysterectomy and abdominal hysterectomy of LM in the USA is 12 out of 100 000 and 32 out of 100 000, respectively (Siedhoff et al., 2015; Liu, 2021). Nevertheless, there are ongoing trials that compare UPA with other drugs. The results of these trials will be of great value in determining their clinical relevance by adding to the evidence regarding their safety and undesirable effects.
Despite the possibility of reduced tumor volume and symptomatic relief provided by medical management (Sohn et al., 2018), women who have severe symptoms and pain or suffer from adverse side effects often rely on surgical or radiological interventions. Unfortunately, LM removal [via methods such as minimally invasive techniques or uterine artery embolization] is not always a long-term solution as there is a risk for reoccurrence, which can require future reinterventions (De La Cruz and Buchanan, 2017). Furthermore, since many women are now delaying childbearing past the age of 35 years, uterine-sparing treatment options are highly sought (Balasch and Gratacos, 2012; Sohn et al., 2018). But how can LM be treated without resorting to invasive surgical procedures that have the possibility of affecting a woman’s fertility? One possibility revolves around a better understanding of the epigenome. Epigenomic alterations are unlike mutations in the sense of being potentially reversible and having great plasticity (Miranda Furtado et al., 2019). Thus, understanding these events can have substantial significance when it comes to developing future mono or combinatorial non-surgical therapies.
‘Epi-drugs’, or drugs that target epigenetic marks by acting on enzymes responsible for epigenomic alterations, are already an emerging area of therapy for cancer (Bennett and Licht, 2018). In the context of LM, there are various enzymes that can serve as targets for these epi-drugs. For instance, hypomethylating agents that inhibit DNMT [DNMTi] can reactivate silenced genes such as those which serve tumor suppressor functions. In another example, Carbajo-Garcia et al. (2021) performed DNA methylation reversion in human LM primary cells using 5-aza-dC, a DNMTi. They suggested this DNMTi as an effective treatment option owing to its ability to inhibit cell proliferation, ECM formation and the WNT/β-catenin pathway (Carbajo-Garcia et al., 2021).
Alternatively, HAT and HDAC enzymes can serve as targets as well. Consistently, Moyo et al. (2020) and Leistico et al. (2021) observed extensive alterations in enhancer H3K27 acetylation, a modification catalyzed by EP300 (Moyo et al., 2020; Leistico et al., 2021). The enzyme EP300 serves as a coactivator of gene transcription by directly acetylating promoter-binding histones via its catalytic HAT domain. It is also the key cofactor involved in TGF-β-induced signaling associated with driving fibrogenesis (HWang et al., 2020). The high expression and signaling associated with its transcriptional regulation have been observed in various fibrotic diseases such as liver (Yao et al., 2018) and cardiac (Bugyei-Twum et al., 2014) fibrosis. Therefore, it would be of potential use to generate potent small molecular inhibitors that target EP300’s HAT domain in the hope of blocking its catalytic activity.
Furthermore, acetylated lysine residues—such as those carried out by EP300—are recognized as binding sites for bromodomain [BRD] proteins such as BRD2, BRD3 and BRD4. These proteins are members of the bromodomain and extra terminal [BET] family of proteins, which act as epigenomic readers that play a critical role in regulating transcription by recruiting additional TFs and transcriptional machinery (Borck et al., 2020). BET inhibitors have been pursued in cardiovascular clinical trials (Schooling and Zhao, 2019) and clinical development for treating cancer (Pervaiz et al., 2018). For this reason, BET inhibitors may serve as potential antifibrotic agents in the case of already acetylated lysine residues in the context of LM. On another note, enhancers play a key role in regulating the tissue-specific expression patterns of genes, so it is conceivable that changes in enhancer activity result in the enhancer malfunction that constitutes the dysregulation of LM-specific genes. Rai et al. recently reported that the acetyltransferase EP300 inhibitor prevented cardiac fibrosis in a murine model (Rai et al., 2017, 2019). It would be interesting to translate this finding into LM-based animal models to see if there is a similar antifibrotic effect. BRD4 is also found to be enriched in enhancer regions (Lee et al., 2017), so it may be plausible that BET inhibitor-induced repression of BRD4 binding may displace the mediator complex from the enhancer, preventing its interaction with transcriptional machinery (Loven et al., 2013; Shorstova et al., 2021).
On the contrary, several HDACi (vorinostat, romidepsin, belinostat and panobinostat) have also been approved by the US Food and Drug Administration to serve as treatments for various cancers by exerting antitumor effects such as cell cycle arrest, mitotic cell death, inducing apoptosis and autophagy, inhibiting angiogenesis and accumulating reactive oxygen species (Singh et al., 2018). Similarly, in human LM tissue samples, and ht-UtLM and HUtSMC cell lines, treatment using HDACi (HDACi VIII and apicidin) showed an antiproliferative effect and induced cell cycle arrest and apoptosis (Ali et al., 2020).
Aside from their potential to serve as monotherapies, the combination of various epi-drugs with each other or with other antifibrotic therapies may be advantageous as well. In cancer cells, this combination has been hypothesized to serve as a practical approach toward sensitizing cells to given therapies and reduce the likelihood of acquired resistance to specific treatments (Morel et al., 2020). For example, in EZH2-aberrant solid tumors, BET inhibitors have been shown to re-sensitize cancer cells to EZH2 inhibitors via disrupting H3K27ac-mediated signaling (Huang et al., 2018). Therefore, compounds that target the epigenome, such as bromodomain inhibitors and inhibitors of HATs, HDACs, HMTs, HDMs, DNMTs and TET enzymes, either alone or in combination, may serve as a promising therapeutic strategy and should be investigated in the context of LM (Fig. 3). Nevertheless, the limitations of these proposed therapies, such as adverse systemic effects and limited tolerability, are a significant challenge. Can it be that low doses are sufficient to affect the hypersensitive LM cells without causing harm to distant healthy cells? Can sequential scheduling of combination therapies be more effective than concurrent administration? Will the targeted delivery of these drugs utilizing antibody–drug conjugates or liposomal particles reduce the inadvertent systemic effects (Morel et al., 2020)? These are all questions to take into consideration while seeking to develop novel epigenome-targeting drugs.
Figure 3.

Epigenomic dysregulation in uterine leiomyoma and potential therapeutic targets. Histone modification (1), aberrant promoter methylation (2) and altered enhancer architecture (3) are three categories of epigenomic changes that are implicated in the pathogenesis of uterine leiomyomas. Epi-drugs (HDACi indicates histone deacetylase inhibitor; HATi, histone acetyltransferase inhibitor; BETi, bromodomain and extra-terminal motif protein inhibitor; HDMi, histone demethylase inhibitor; HMTi, histone methyltransferase inhibitor; TETi, ten-eleven translocation protein inhibitor; DNMTi, DNA methyltransferase inhibitor), which target the enzymes involved in modulating epigenomic marks (HDAC indicates histone deacetylase; HAT, histone acetyltransferase; BRD, bromodomain protein; HDM, histone demethylase; HMT, histone methyltransferase; TET, ten-eleven translocation protein; DNMT, DNA methyltransferase), may play a critical role in managing tumor development. Adapted from ‘Regulation of Transcription in Eukaryotic Cells’, by BioRender.com (2020). Retrieved from https://app.biorender.com/biorender-templates
With future studies focused on histone and DNA modifying enzymes and on epigenome reader proteins, the innovative treatment options can be utilized in various in vitro assays and translated into murine/animal-based studies, serving as models for a preliminary screen, to test the efficacy and effectiveness in targeting the epigenome in LM. Additionally, organoid and spheroid tumor models may hold an advantage over traditional cell lines in these pre-clinical assays as they are 3D units with a similar composition to the primary tissue, making them more representative of in vivo conditions (Gunti et al., 2021). It is important to note that to aid in the usefulness of these systems for future mechanistic studies, it will be important to do a systemic analysis of the epigenome in established cell lines, primary LM and myometrium cells, spheroids and organoid models, and human tissues.
We also note that with the advent of genomics, and our ability to characterize subtypes of LM, there is great hope to develop disease subtype-specific treatments. Additionally, common dysregulated pathways in all subtypes of LM can also be targeted for future therapeutics. For example, all LM subtypes demonstrate an increase in expression of the cPRC1 that epigenetically silences genes. Therefore, the use of small molecular inhibitors targeting this epigenome regulatory complex may provide therapeutic benefits for treatment of LM. Similarly, inhibiting the enzyme HSD17B6 involved in the biosynthetic pathway for the synthesis of dihydrotestosterone, found overexpressed in all LM subtypes, might be a future therapeutic choice for LM treatment (Berta et al., 2021; Ordulu, 2021). Of course, extensive functional cell- and animal-based studies need to be conducted prior to any potential human clinical trials and therapeutic use of all epigenomic drugs discussed in this review.
Overall, the ability to change the cell landscape can provide a promising non-surgical treatment of LM that affects roughly 70% of all women globally. Along these lines, continued investigation on the epigenomic dysregulation in LM is critically needed to better grasp this still poorly understood disease and for future therapeutic interventions.
Knowledge gaps, emerging areas and concluding remarks
Aberrant epigenomic alterations are common molecular markers in tumor cells. While the field of epigenomics has advanced in other disorders and tumor-bearing diseases, its role in the context of LM etiology is still evolving. Recent studies discussed in this review have, to some extent, expanded our understanding of the role of epigenomics in LM development. This review largely focuses on how DNA methylation and histone modifications in the promoter or enhancer regions can dysregulate gene expression, which aids in developing the LM phenotype. Aside from pathways revolving around ECM production, genome-wide DNA methylation, histone modification and 3D chromatin analysis are being used to identify other biological pathways, such as those involving the HOX family of TFs or tumor suppressor proteins, that will be used in future studies to better understand this disease.
Within the context of promoter methylation status, the trend observed conveyed that hypermethylation and hypomethylation within the promoter are associated with gene silencing and activation, respectively. Thus, the genome-wide DNA methylation patterns identify LM-specific signatures that can serve as biomarkers for the identification of the disease and clinical outcomes. Furthermore, the utilization of a hierarchical clustering system can possibly provide an opportunity to reveal information on the subtypes of LM and differentiate them from rare types of uterine smooth muscle tumors [USMT], such as LMS, LM with bizarre nuclei [LM-BN] and smooth muscle tumors of uncertain malignant potential [STUMP]. LMS are malignant USMT with unfavorable 5-year survival rates (Giuntoli et al., 2003). LM-BN, previously named as atypical leiomyoma [ALM], is an intermediate grade USMT with an overall benign clinical course and occasional recurrence (Bell et al., 1994). STUMP are tumors that cannot, with certainty, be histologically determined as benign or malignant (Lu and Chen, 2014; Rizzo et al., 2020). Owing to some overlapping histologic and molecular features between STUMP, ALM and LMS, it is difficult to diagnose and manage these tumors (Bell et al., 1994; Layfield et al., 2000). Therefore, there is a need to identify genomic and/or epigenomic features that may differentiate between these tumor types. In this regard, recently, Gao et al. (2021) conducted whole genome copy number analysis by NGS in LMS and LM-BN (Gao et al., 2021). In this study, they applied a GISTIC analysis to identify the high-resolution genomic copy number alteration [CNA] specific to LMS and LM-BN. This study demonstrates both LM-BN and LMS as being genomic unstable tumors and sharing many CNA, including loss of genomic regions of Retinoblastoma [RB] and tumor protein p53 [TP53], suggesting a genetic association for these two tumor types. Furthermore, a frequent loss of TET2 and DNMT1 genomic regions in LMS was observed. Conconi et al. (2021) performed comparative genomic hybridization array and DNA methylation studies on LMS, STUMP and LM. They identified a few genes, such as protein kinase, DNA-activated, catalytic subunit [PRKDC] and pumilio RNA-binding family member 2 [PUM2], that may have prognostic value for STUMP. Although their analysis for DNA methylation was restricted to only four samples, they found varying methylation features in the STUMP that relapsed as LMS (Conconi et al., 2021). These results seem promising for the epigenomic field, and perhaps a more detailed analysis of DNA methylation as well as histone modifications can be performed on these samples to identify distinctive patterns with diagnostic/prognostic value. To overcome the limitation of sample recruitment owing to rare presentations of these tumors, formalin-fixed paraffin-embedded [FFPE] tissues may be utilized for various epigenomic modalities. Importantly, it was found that the stem cell-like population within LM is insensitive to hormonal treatments and can proliferate, leading to benign tumor growth (Conconi et al., 2021). Further research is needed to establish an understanding of abnormal epigenomic regulation within these populations of cells to develop efficient therapeutic approaches.
The regulation of histone modifications in LM has historically been less understood as compared to DNA methylation, but recent publications, including those from our laboratory, have shed light on the significance this plays in disease development. The interplay of cell signaling pathways and histone modification can explain the altered 3D chromatin structure, which aids in developing the LM-specific gene dysregulation resulting in its pathogenesis. Understanding pathways or trans-regulatory elements that induce histone modification will allow for target options for suppressing the fibroid phenotype.
The application of an altered enhancer landscape and its effect on gene regulation is a new approach to a broader understanding of what drives LM development and requires more attention. The interplay of epigenomic marks, TF occupancy and enhancer chromatin structure plays a key role in transmitting information from millions of base pairs away for the regulation of gene expression. Interestingly, in mutant MED12 LM, modified enhancer architecture provides a novel potential mechanism of gene dysregulation that occurs outside the promoter proximal area. Whether enhancer malfunction also plays critical roles in non-MED12 mutant LM will be a key future question to be addressed. The next challenge will be to use animal-based experiments with drugs targeting the epigenome, in combination with current drugs or alone, which will lead to better therapeutic outcomes.
With the advent of new technologies extracting pathologically relevant data at the single cell level, there are now opportunities to understand various diseases at an unparalleled depth. These high-resolution multi-omics tools have been utilized to study various fibrotic organs such as lung, liver and kidney (Henderson et al., 2020). Single-cell RNA sequencing [scRNA-seq] has revealed the presence of a subpopulation of macrophages that are pro-fibrotic and are involved in progression of lung (Misharin et al., 2017; Reyfman et al., 2019) and liver fibrosis (Ramachandran et al., 2019). In kidney fibrosis, scRNA-seq along with other modalities, namely ATAC-seq and spatial transcriptomics, led to the identification of NKD inhibitor of WNT signaling pathway 2 [NKD2], a gene that is differentially expressed in myofibroblasts and can be targeted in kidney fibrosis (Kuppe et al., 2021). The compatibility of these latest technologies with the use of preserved patient tissues, and the availability of FFPE tissues for even the rarest forms of LM, provide new avenues for research in the uterine LM field. In this context, single-cell transcriptomic analysis, as well as spatial gene expression studies, may be useful in dissecting the heterogeneity of various uterine fibroids based on genetic status and/or size. These studies may provide specific targets for the therapeutic treatment of this benign tumor with little to no effect on the normal tissues.
To reiterate, despite this condition affecting a significant number of women worldwide, LM has been an overlooked public health issue. In addition, research revolving around this disease has remained understudied compared to other non-malignant conditions (Ciavattini et al., 2013), although the National Institutes of Health and private funding agencies are now investing financial and scientific efforts to better understand this disease, which is a key for future therapies. An integrative knowledge linking genetic mutations, altered signaling and epigenomic events in the context of patients will be vital to better understand and treat this disease. In addition, using a larger number of patient samples will also be critical to better understand how the social–environmental context influences LM in Black women: particularly because environmental factors, such as experiences of childhood adverse effects, the experience of racial discrimination/racism, economic hardship and experience of redlining and residential segregation, are thought to influence the epigenome and gene expression. Now, bioinformatic programs exist that can interpret these events and analyze extensive patient data sets. The implementation of artificial intelligence-based predictions may guide us toward a better understanding of how epigenomic and enhancer dysregulation result in LM development and LM health disparity.
Data availability
No new data were generated or analyzed in support of this research.
Authors’ roles
O.W.M. and P.S. are the co-first authors. O.W.M wrote the initial draft of the manuscript. O.W.M., P.S. and D.C. were responsible for literature search, manuscript drafting, figures, tables and critical discussions. J.B.P., J.-J.W., S.E.B. and M.A.S provided discussion and comments on the manuscript. D.C. conceived and oversaw the review. All authors read and commented on the manuscript.
Funding
This review is supported, in part, by the National Institutes of Health (USA) grants R01HD089552 and P50HD098580 and the Anna Lapham endowment of Northwestern University.
Conflict of interest
The authors declare no competing interests.
Contributor Information
Oliwia W Mlodawska, Division of Reproductive Science in Medicine, Department of Obstetrics and Gynecology, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA.
Priyanka Saini, Division of Reproductive Science in Medicine, Department of Obstetrics and Gynecology, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA.
J Brandon Parker, Division of Reproductive Science in Medicine, Department of Obstetrics and Gynecology, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA.
Jian-Jun Wei, Department of Pathology, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA; Robert H. Lurie Comprehensive Cancer Center of Northwestern University, Chicago, IL 60611, USA.
Serdar E Bulun, Division of Reproductive Science in Medicine, Department of Obstetrics and Gynecology, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA.
Melissa A Simon, Department of Obstetrics and Gynecology, Center for Health Equity Transformation, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA.
Debabrata Chakravarti, Division of Reproductive Science in Medicine, Department of Obstetrics and Gynecology, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA; Robert H. Lurie Comprehensive Cancer Center of Northwestern University, Chicago, IL 60611, USA; Department of Pharmacology, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA.
References
- Al-Hendy A, Diamond MP, El-Sohemy A, Halder SK.. 1,25-dihydroxyvitamin D3 regulates expression of sex steroid receptors in human uterine fibroid cells. J Clin Endocrinol Metab 2015;100:E572–E582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali M, Shahin SM, Sabri NA, Al-Hendy A, Yang Q.. Activation of beta-catenin signaling and its crosstalk with estrogen and histone deacetylases in human uterine fibroids. J Clin Endocrinol Metab 2020;105: e1517–e1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen BL, Taatjes DJ.. The Mediator complex: a central integrator of transcription. Nat Rev Mol Cell Biol 2015;16:155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoto S, Fushimi M, Yura K, Okamura K.. Diversification of CpG-island promoters revealed by comparative analysis between human and rhesus monkey genomes. Mamm Genome 2020;31:240–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong MJ, Jin Y, Allen EG, Jin P.. Diverse and dynamic DNA modifications in brain and diseases. Hum Mol Genet 2019;28:R241–R253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroke EN, Joseph PV, Roy A, Overstreet DS, Tollefsbol TO, Vance DE, Goodin BR.. Could epigenetics help explain racial disparities in chronic pain? J Pain Res 2019;12:701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashar HR, Fejzo MS, Tkachenko A, Zhou X, Fletcher JA, Weremowicz S, Morton CC, Chada K.. Disruption of the architectural factor HMGI-C: DNA-binding AT hook motifs fused in lipomas to distinct transcriptional regulatory domains. Cell 1995;82:57–65. [DOI] [PubMed] [Google Scholar]
- Baird DD, Hill MC, Schectman JM, Hollis BW.. Vitamin d and the risk of uterine fibroids. Epidemiology 2013;24:447–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasch J, Gratacos E.. Delayed childbearing: effects on fertility and the outcome of pregnancy. Curr Opin Obstet Gynecol 2012;24:187–193. [DOI] [PubMed] [Google Scholar]
- Banerjee T, Chakravarti D.. A peek into the complex realm of histone phosphorylation. Mol Cell Biol 2011;31:4858–4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranov VS, Osinovskaya NS, Yarmolinskaya MI.. Pathogenomics of uterine fibroids development. Int J Mol Sci 2019;20:6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcelona de Mendoza V, Huang Y, Crusto CA, Sun YV, Taylor JY.. Perceived racial discrimination and DNA methylation among African American women in the InterGEN study. Biol Res Nurs 2018;20:145–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K.. High-resolution profiling of histone methylations in the human genome. Cell 2007;129:823–837. [DOI] [PubMed] [Google Scholar]
- Bejjani F, Evanno E, Zibara K, Piechaczyk M, Jariel-Encontre I.. The AP-1 transcriptional complex: local switch or remote command? Biochim Biophys Acta Rev Cancer 2019;1872:11–23. [DOI] [PubMed] [Google Scholar]
- Bell SW, Kempson RL, Hendrickson MR.. Problematic uterine smooth muscle neoplasms. A clinicopathologic study of 213 cases. Am J Surg Pathol 1994;18:535–558. [PubMed] [Google Scholar]
- Bennett RL, Licht JD.. Targeting epigenetics in cancer. Annu Rev Pharmacol Toxicol 2018;58:187–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhart SH, Kretzmer H, Holdt LM, Juhling F, Ammerpohl O, Bergmann AK, Northoff BH, Doose G, Siebert R, Stadler PF. et al. Changes of bivalent chromatin coincide with increased expression of developmental genes in cancer. Sci Rep 2016;6:37393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berta DG, Kuisma H, Valimaki N, Raisanen M, Jantti M, Pasanen A, Karhu A, Kaukomaa J, Taira A, Cajuso T. et al. Deficient H2A.Z deposition is associated with genesis of uterine leiomyoma. Nature 2021;596:398–403. [DOI] [PubMed] [Google Scholar]
- Bikle DD. Vitamin D metabolism, mechanism of action, and clinical applications. Chem Biol 2014;21:319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borah BJ, Nicholson WK, Bradley L, Stewart EA.. The impact of uterine leiomyomas: a national survey of affected women. Am J Obstet Gynecol 2013;209:319.e311–319.e320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borahay MA, Al-Hendy A, Kilic GS, Boehning D.. Signaling pathways in leiomyoma: understanding pathobiology and implications for therapy. Mol Med 2015;21:242–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borck PC, Guo LW, Plutzky J.. BET epigenetic reader proteins in cardiovascular transcriptional programs. Circ Res 2020;126:1190–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borggrefe T, Yue X.. Interactions between subunits of the Mediator complex with gene-specific transcription factors. Semin Cell Dev Biol 2011;22:759–768. [DOI] [PubMed] [Google Scholar]
- Boynton-Jarrett R, Rich-Edwards JW, Jun HJ, Hibert EN, Wright RJ.. Abuse in childhood and risk of uterine leiomyoma: the role of emotional support in biologic resilience. Epidemiology 2011;22:6–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray MJ, Edwards TL, Wellons MF, Jones SH, Hartmann KE, Velez Edwards DR.. Admixture mapping of uterine fibroid size and number in African American women. Fertil Steril 2017;108:1034–1042.e1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray MJ, Wellons MF, Jones SH, Torstenson ES, Edwards TL, Velez Edwards DR.. Transethnic and race-stratified genome-wide association study of fibroid characteristics in African American and European American women. Fertil Steril 2018;110:737–745.e734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman AB, Simmer F, Ma K, Kaan A, Zhu J, Stunnenberg HG.. Whole-genome DNA methylation profiling using MethylCap-seq. Methods 2010;52:232–236. [DOI] [PubMed] [Google Scholar]
- Bugyei-Twum A, Advani A, Advani SL, Zhang Y, Thai K, Kelly DJ, Connelly KA.. High glucose induces Smad activation via the transcriptional coregulator p300 and contributes to cardiac fibrosis and hypertrophy. Cardiovasc Diabetol 2014;13:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukulmez O, Doody KJ.. Clinical features of myomas. Obstet Gynecol Clin North Am 2006;33:69–84. [DOI] [PubMed] [Google Scholar]
- Cao T, Jiang Y, Wang Z, Zhang N, Al-Hendy A, Mamillapalli R, Kallen AN, Kodaman P, Taylor HS, Li D. et al. H19 lncRNA identified as a master regulator of genes that drive uterine leiomyomas. Oncogene 2019;38:5356–5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbajo-Garcia MC, Corachan A, Segura-Benitez M, Monleon J, Escrig J, Faus A, Pellicer A, Cervello I, Ferrero H.. 5-aza-2'-deoxycitidine inhibits cell proliferation, extracellular matrix formation and Wnt/beta-catenin pathway in human uterine leiomyomas. Reprod Biol Endocrinol 2021;19:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardozo ER, Clark AD, Banks NK, Henne MB, Stegmann BJ, Segars JH.. The estimated annual cost of uterine leiomyomata in the United States. Am J Obstet Gynecol 2012;206:211.e211–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carranza-Mamane B, Havelock J, Hemmings R, Cheung A, Sierra S, Carranza-Mamane B, Case A, Cathie D, Graham J, Havelock J. et al. The management of uterine fibroids in women with otherwise unexplained infertility. J Obstet Gynaecol Can 2015;37:277–285. [DOI] [PubMed] [Google Scholar]
- Castillo J, Lopez-Rodas G, Franco L.. Histone post-translational modifications and nucleosome organisation in transcriptional regulation: some open questions. Adv Exp Med Biol 2017;966:65–92. [DOI] [PubMed] [Google Scholar]
- Castro L, Gao X, Moore AB, Yu L, Di X, Kissling GE, Dixon D.. A high concentration of genistein induces cell death in human uterine leiomyoma cells by autophagy. Expert Opin Environ Biol 2016,5;S1–003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catherino WH, Eltoukhi HM, Al-Hendy A.. Racial and ethnic differences in the pathogenesis and clinical manifestations of uterine leiomyoma. Semin Reprod Med 2013;31:370–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedar H, Bergman Y.. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 2009;10:295–304. [DOI] [PubMed] [Google Scholar]
- Cervello I, Mas A, Gil-Sanchis C, Peris L, Faus A, Saunders PT, Critchley HO, Simon C.. Reconstruction of endometrium from human endometrial side population cell lines. PLoS One 2011;6:e21221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Li E.. Structure and function of eukaryotic DNA methyltransferases. Curr Top Dev Biol 2004;60:55–89. [DOI] [PubMed] [Google Scholar]
- Ciavattini A, Delli Carpini G, Serri M, Vignini A, Sabbatinelli J, Tozzi A, Aggiusti A, Clemente N.. Hypovitaminosis D and “small burden” uterine fibroids: opportunity for a vitamin D supplementation. Medicine (Baltimore) 2016;95:e5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciavattini A, Di Giuseppe J, Stortoni P, Montik N, Giannubilo SR, Litta P, Islam MS, Tranquilli AL, Reis FM, Ciarmela P.. Uterine fibroids: pathogenesis and interactions with endometrium and endomyometrial junction. Obstet Gynecol Int 2013;2013:173184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciebiera M, Ali M, Zgliczynska M, Skrzypczak M, Al-Hendy A.. Vitamins and uterine fibroids: current data on pathophysiology and possible clinical relevance. Int J Mol Sci 2020;21:5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciebiera M, Włodarczyk M, Słabuszewska-Jóźwiak A, Nowicka G, Jakiel G.. Influence of vitamin D and transforming growth factor beta3 serum concentrations, obesity, and family history on the risk for uterine fibroids. Fertil Steril 2016;106:1787–1792. [DOI] [PubMed] [Google Scholar]
- Ciebiera M, Wlodarczyk M, Wrzosek M, Meczekalski B, Nowicka G, Lukaszuk K, Ciebiera M, Slabuszewska-Jozwiak A, Jakiel G.. Role of transforming growth factor beta in uterine fibroid biology. Int J Mol Sci 2017;18:2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke-Pearson DL, Geller EJ.. Complications of hysterectomy. Obstet Gynecol 2013;121:654–673. [DOI] [PubMed] [Google Scholar]
- Cillo C, Cantile M, Faiella A, Boncinelli E.. Homeobox genes in normal and malignant cells. J Cell Physiol 2001;188:161–169. [DOI] [PubMed] [Google Scholar]
- Colen CG, Ramey DM, Cooksey EC, Williams DR.. Racial disparities in health among nonpoor African Americans and Hispanics: the role of acute and chronic discrimination. Soc Sci Med 2018;199:167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonese F, Lagana AS, Colonese E, Sofo V, Salmeri FM, Granese R, Triolo O.. The pleiotropic effects of vitamin D in gynaecological and obstetric diseases: an overview on a hot topic. Biomed Res Int 2015;2015:986281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commandeur AE, Styer AK, Teixeira JM.. Epidemiological and genetic clues for molecular mechanisms involved in uterine leiomyoma development and growth. Hum Reprod Update 2015;21:593–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conconi D, Redaelli S, Lissoni AA, Cilibrasi C, Perego P, Gautiero E, Sala E, Paderno M, Dalpra L, Landoni F.. Genomic and epigenomic profile of uterine smooth muscle tumors of uncertain malignant potential (STUMPs) revealed similarities and differences with leiomyomas and leiomyosarcomas. Int J Mol Sci 2021;221580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daftary GS, Taylor HS.. Endocrine regulation of HOX genes. Endocr Rev 2006;27:331–355. [DOI] [PubMed] [Google Scholar]
- Dagur G, Suh Y, Warren K, Singh N, Fitzgerald J, Khan SA.. Urological complications of uterine leiomyoma: a review of literature. Int Urol Nephrol 2016;48:941–948. [DOI] [PubMed] [Google Scholar]
- Davari Tanha F, Feizabad E, Vasheghani Farahani M, Amuzegar H, Moradi B, Sadeh SS.. The effect of vitamin D deficiency on overgrowth of uterine fibroids: a blinded randomized clinical trial. Int J Fertil Steril 2021;15:95–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Cruz MS, Buchanan EM.. Uterine fibroids: diagnosis and treatment. Am Fam Physician 2017;95:100–107. [PubMed] [Google Scholar]
- de Mello VD, Pulkkinen L, Lalli M, Kolehmainen M, Pihlajamaki J, Uusitupa M.. DNA methylation in obesity and type 2 diabetes. Ann Med 2014;46:103–113. [DOI] [PubMed] [Google Scholar]
- Di X, Andrews DM, Tucker CJ, Yu L, Moore AB, Zheng X, Castro L, Hermon T, Xiao H, Dixon D.. A high concentration of genistein down-regulates activin A, Smad3 and other TGF-beta pathway genes in human uterine leiomyoma cells. Exp Mol Med 2012;44:281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di X, Yu L, Moore AB, Castro L, Zheng X, Hermon T, Dixon D.. A low concentration of genistein induces estrogen receptor-alpha and insulin-like growth factor-I receptor interactions and proliferation in uterine leiomyoma cells. Hum Reprod 2008;23:1873–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnez J. Uterine fibroids and progestogen treatment: lack of evidence of its efficacy: a review. J Clin Med 2020;9:3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnez J, Dolmans MM.. Uterine fibroid management: from the present to the future. Hum Reprod Update 2016;22:665–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnez J, Donnez O, Matule D, Ahrendt HJ, Hudecek R, Zatik J, Kasilovskiene Z, Dumitrascu MC, Fernandez H, Barlow DH. et al. Long-term medical management of uterine fibroids with ulipristal acetate. Fertil Steril 2016;105:165–173.e164. [DOI] [PubMed] [Google Scholar]
- Donnez J, Tatarchuk TF, Bouchard P, Puscasiu L, Zakharenko NF, Ivanova T, Ugocsai G, Mara M, Jilla MP, Bestel E. et al. Ulipristal acetate versus placebo for fibroid treatment before surgery. N Engl J Med 2012a;366:409–420. [DOI] [PubMed] [Google Scholar]
- Donnez J, Tomaszewski J, Vazquez F, Bouchard P, Lemieszczuk B, Baro F, Nouri K, Selvaggi L, Sodowski K, Bestel E. et al. Ulipristal acetate versus leuprolide acetate for uterine fibroids. N Engl J Med 2012b;366:421–432. [DOI] [PubMed] [Google Scholar]
- Donnez J, Vazquez F, Tomaszewski J, Nouri K, Bouchard P, Fauser BC, Barlow DH, Palacios S, Donnez O, Bestel E. et al. PEARL III and PEARL III Extension Study Group. Long-term treatment of uterine fibroids with ulipristal acetate. Fertil Steril 2014;101:1565–1573.e1561–e1518. [DOI] [PubMed] [Google Scholar]
- D'Oto A, Tian QW, Davidoff AM, Yang J.. Histone demethylases and their roles in cancer epigenetics. J Med Oncol Ther 2016;1:34–40. [PMC free article] [PubMed] [Google Scholar]
- Ehrlich KC, Paterson HL, Lacey M, Ehrlich M.. DNA hypomethylation in intragenic and intergenic enhancer chromatin of muscle-specific genes usually correlates with their expression. Yale J Biol Med 2016;89:441–455. [PMC free article] [PubMed] [Google Scholar]
- Ehrlich M. DNA hypermethylation in disease: mechanisms and clinical relevance. Epigenetics 2019;14:1141–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekanem E, Talaulikar V.. Medical therapy for fibroids: what next for ulipristal acetate? Adv Ther 2021;38:137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elhamamsy AR. Role of DNA methylation in imprinting disorders: an updated review. J Assist Reprod Genet 2017;34:549–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltoukhi HM, Modi MN, Weston M, Armstrong AY, Stewart EA.. The health disparities of uterine fibroid tumors for African American women: a public health issue. Am J Obstet Gynecol 2014;210:194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falahati Z, Mohseni-Dargah M, Mirfakhraie R.. Emerging roles of long non-coding RNAs in uterine leiomyoma pathogenesis: a review. Reprod Sci 2021. https://link.springer.com/article/10.1007/s43032-021-00571-w. [DOI] [PubMed] [Google Scholar]
- Fleischer AC, James AE Jr, Millis JB, Julian C.. Differential diagnosis of pelvic masses by gray scale sonography. AJR Am J Roentgenol 1978;131:469–476. [DOI] [PubMed] [Google Scholar]
- Florence AM, Fatehi M.. Leiomyoma. Treasure Island, FL: StatPearls, 2021. [Google Scholar]
- Flyckt R, Coyne K, Falcone T.. Minimally invasive myomectomy. Clin Obstet Gynecol 2017;60:252–272. [DOI] [PubMed] [Google Scholar]
- Fusco A, Fedele M.. Roles of HMGA proteins in cancer. Nat Rev Cancer 2007;7:899–910. [DOI] [PubMed] [Google Scholar]
- Gallagher WM, Currid CA, Whelan LC.. Fibulins and cancer: friend or foe? Trends Mol Med 2005;11:336–340. [DOI] [PubMed] [Google Scholar]
- Gao T, Finkelman BS, Ban Y, Li Y, Yin P, Bulun SE, Lu X, Ha C, Wei JJ.. Integrated histologic and molecular analysis of uterine leiomyosarcoma and 2 benign variants with nuclear atypia. Cancer Sci 2021;112:2046–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Z, Gao Z.. Mammalian PRC1 complexes: compositional complexity and diverse molecular mechanisms. Int J Mol Sci 2020;21:8594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George JW, Fan H, Johnson B, Carpenter TJ, Foy KK, Chatterjee A, Patterson AL, Koeman J, Adams M, Madaj ZB. et al. Integrated epigenome, exome, and transcriptome analyses reveal molecular subtypes and homeotic transformation in uterine fibroids. Cell Rep 2019;29:4069–4085.e4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhold CB, Gasser SM.. INO80 and SWR complexes: relating structure to function in chromatin remodeling. Trends Cell Biol 2014;24:619–631. [DOI] [PubMed] [Google Scholar]
- Giaimo BD, Ferrante F, Herchenrother A, Hake SB, Borggrefe T.. The histone variant H2A.Z in gene regulation. Epigenetics Chromatin 2019;12:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuliani E, As-Sanie S, Marsh EE.. Epidemiology and management of uterine fibroids. Int J Gynaecol Obstet 2020;149:3–9. [DOI] [PubMed] [Google Scholar]
- Giuntoli RL 2nd, Metzinger DS, DiMarco CS, Cha SS, Sloan JA, Keeney GL, Gostout BS.. Retrospective review of 208 patients with leiomyosarcoma of the uterus: prognostic indicators, surgical management, and adjuvant therapy. Gynecol Oncol 2003;89:460–469. [DOI] [PubMed] [Google Scholar]
- Gong F, Miller KM.. Histone methylation and the DNA damage response. Mutat Res Rev Mutat Res 2019;780:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC.. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med 1996;183:1797–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorny KR, Woodrum DA, Brown DL, Henrichsen TL, Weaver AL, Amrami KK, Hangiandreou NJ, Edmonson HA, Bouwsma EV, Stewart EA. et al. Magnetic resonance-guided focused ultrasound of uterine leiomyomas: review of a 12-month outcome of 130 clinical patients. J Vasc Interv Radiol 2011;22:857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunti S, Hoke ATK, Vu KP, London NR Jr.. Organoid and spheroid tumor models: techniques and applications. Cancers (Basel) 2021;13:874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta JK, Sinha A, Lumsden MA, Hickey M.. Uterine artery embolization for symptomatic uterine fibroids. Cochrane Database Syst Rev 2014:issue 12, Art # CD005073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajhashemi M, Ansari M, Haghollahi F, Eslami B.. The effect of vitamin D supplementation on the size of uterine leiomyoma in women with vitamin D deficiency. Caspian J Intern Med 2019;10:125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder S, Al-Hendy A.. Hypovitaminosis D and high serum transforming growth factor beta-3: important biomarkers for uterine fibroids risk. Fertil Steril 2016;106:1648–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder SK, Goodwin JS, Al-Hendy A.. 1,25-dihydroxyvitamin D3 reduces TGF-beta3-induced fibrosis-related gene expression in human uterine leiomyoma cells. J Clin Endocrinol Metab 2011;96:E754–E762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder SK, Osteen KG, Al-Hendy A.. Vitamin D3 inhibits expression and activities of matrix metalloproteinase-2 and -9 in human uterine fibroid cells. Hum Reprod 2013;28:2407–2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder SK, Sharan C, Al-Hendy A.. 1,25-dihydroxyvitamin D3 treatment shrinks uterine leiomyoma tumors in the Eker rat model. Biol Reprod 2012;86:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D, Lu X, Shih AH, Nie J, You Q, Xu MM, Melnick AM, Levine RL, He C.. A highly sensitive and robust method for genome-wide 5hmC profiling of rare cell populations. Mol Cell 2016;63:711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris SS. Vitamin D and African Americans. J Nutr 2006;136:1126–1129. [DOI] [PubMed] [Google Scholar]
- Hellwege JN, Jeff JM, Wise LA, Gallagher CS, Wellons M, Hartmann KE, Jones SF, Torstenson ES, Dickinson S, Ruiz-Narvaez EA. et al. A multi-stage genome-wide association study of uterine fibroids in African Americans. Hum Genet 2017;136:1363–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson NC, Rieder F, Wynn TA.. Fibrosis: from mechanisms to medicines. Nature 2020;587:555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holwerda SJ, de Laat W.. CTCF: the protein, the binding partners, the binding sites and their chromatin loops. Philos Trans R Soc Lond B Biol Sci 2013;368:20120369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Yan J, Zhang M, Wang Y, Chen Y, Fu X, Wei R, Zheng XL, Liu Z, Zhang X. et al. Targeting epigenetic crosstalk as a therapeutic strategy for EZH2-aberrant solid tumors. Cell 2018;175:186–199.e119. [DOI] [PubMed] [Google Scholar]
- Hughes K, Bellis MA, Hardcastle KA, Sethi D, Butchart A, Mikton C, Jones L, Dunne MP.. The effect of multiple adverse childhood experiences on health: a systematic review and meta-analysis. Lancet Public Health 2017;2:e356–e366. [DOI] [PubMed] [Google Scholar]
- Hwang CF, Chien CY, Huang SC, Yin YF, Huang CC, Fang FM, Tsai HT, Su LJ, Chen CH.. Fibulin-3 is associated with tumour progression and a poor prognosis in nasopharyngeal carcinomas and inhibits cell migration and invasion via suppressed AKT activity. J Pathol 2010;222:367–379. [DOI] [PubMed] [Google Scholar]
- Hwang SY, Park SY, Hong JY, Lee SY, Shin JH, Na Y, Sohn MH, Yoon HG, Kwon Y.. Field-based rational design of p300 histone acetyltransferase inhibitor and systematic evaluation as an anti-fibrotic agent. Chem Commun (Camb) 2020;56:9795–9798. [DOI] [PubMed] [Google Scholar]
- Ikhena DE, Liu S, Kujawa S, Esencan E, Coon JSt, Robins J, Bulun SE, Yin P.. RANKL/RANK pathway and its inhibitor RANK-Fc in uterine leiomyoma growth. J Clin Endocrinol Metab 2018;103:1842–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam MS, Afrin S, Jones SI, Segars J.. Selective progesterone receptor modulators-mechanisms and therapeutic utility. Endocr Rev 2020;41:bnaa012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam MS, Protic O, Stortoni P, Grechi G, Lamanna P, Petraglia F, Castellucci M, Ciarmela P.. Complex networks of multiple factors in the pathogenesis of uterine leiomyoma. Fertil Steril 2013;100:178–193. [DOI] [PubMed] [Google Scholar]
- Jabbari K, Bernardi G.. Cytosine methylation and CpG, TpG (CpA) and TpA frequencies. Gene 2004;333:143–149. [DOI] [PubMed] [Google Scholar]
- Joshi PA, Jackson HW, Beristain AG, Di Grappa MA, Mote PA, Clarke CL, Stingl J, Waterhouse PD, Khokha R.. Progesterone induces adult mammary stem cell expansion. Nature 2010;465:803–807. [DOI] [PubMed] [Google Scholar]
- Karmon AE, Cardozo ER, Rueda BR, Styer AK.. MicroRNAs in the development and pathobiology of uterine leiomyomata: does evidence support future strategies for clinical intervention? Hum Reprod Update 2014;20:670–687. [DOI] [PubMed] [Google Scholar]
- Katerndahl CDS, Rogers ORS, Day RB, Cai MA, Rooney TP, Helton NM, Hoock M, Ramakrishnan SM, Nonavinkere Srivatsan S, Wartman LD. et al. Tumor suppressor function of Gata2 in acute promyelocytic leukemia. Blood 2021;138:1148–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y, Kaneda M, Hata K, Kumaki K, Hisano M, Kohara Y, Okano M, Li E, Nozaki M, Sasaki H.. Role of the Dnmt3 family in de novo methylation of imprinted and repetitive sequences during male germ cell development in the mouse. Hum Mol Genet 2007;16:2272–2280. [DOI] [PubMed] [Google Scholar]
- Kaur G, Dufour JM.. Cell lines: valuable tools or useless artifacts. Spermatogenesis 2012;2:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazanets A, Shorstova T, Hilmi K, Marques M, Witcher M.. Epigenetic silencing of tumor suppressor genes: paradigms, puzzles, and potential. Biochim Biophys Acta 2016;1865:275–288. [DOI] [PubMed] [Google Scholar]
- Kim EJ, Lee SY, Woo MK, Choi SI, Kim TR, Kim MJ, Kim KC, Cho EW, Kim IG.. Fibulin-3 promoter methylation alters the invasive behavior of non-small cell lung cancer cell lines via MMP-7 and MMP-2 regulation. Int J Oncol 2012;40:402–408. [DOI] [PubMed] [Google Scholar]
- Kim JY, Yu J, Abdulkadir SA, Chakravarti D.. KAT8 regulates androgen signaling in prostate cancer cells. Mol Endocrinol 2016;30:925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Yoon HY, Kim SK, Kim YW, Kim EJ, Kim IY, Kim WJ.. EFEMP1 as a novel DNA methylation marker for prostate cancer: array-based DNA methylation and expression profiling. Clin Cancer Res 2011;17:4523–4530. [DOI] [PubMed] [Google Scholar]
- Klenotic PA, Munier FL, Marmorstein LY, Anand-Apte B.. Tissue inhibitor of metalloproteinases-3 (TIMP-3) is a binding partner of epithelial growth factor-containing fibulin-like extracellular matrix protein 1 (EFEMP1). Implications for macular degenerations. J Biol Chem 2004;279:30469–30473. [DOI] [PubMed] [Google Scholar]
- Kondo Y. Epigenetic cross-talk between DNA methylation and histone modifications in human cancers. Yonsei Med J 2009;50:455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijger PH, de Laat W.. Regulation of disease-associated gene expression in the 3D genome. Nat Rev Mol Cell Biol 2016;17:771–782. [DOI] [PubMed] [Google Scholar]
- Kudo Y, Tateishi K, Yamamoto K, Yamamoto S, Asaoka Y, Ijichi H, Nagae G, Yoshida H, Aburatani H, Koike K.. Loss of 5-hydroxymethylcytosine is accompanied with malignant cellular transformation. Cancer Sci 2012;103:670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundaje A, , MeulemanW, , ErnstJ, , BilenkyM, , YenA, , Heravi-MoussaviA, , KheradpourP, , ZhangZ, , WangJ, , Ziller MJet al. ; Roadmap Epigenomics Consortium. . Integrative analysis of 111 reference human epigenomes. Nature 2015;518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuppe C, Ibrahim MM, Kranz J, Zhang X, Ziegler S, Perales-Paton J, Jansen J, Reimer KC, Smith JR, Dobie R. et al. Decoding myofibroblast origins in human kidney fibrosis. Nature 2021;589:281–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagana AS, Scioscia M.. Endometrial cancer in women with adenomyosis: an underestimated risk? Int J Fertil Steril 2020;14:260–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagana AS, Vergara D, Favilli A, La Rosa VL, Tinelli A, Gerli S, Noventa M, Vitagliano A, Triolo O, Rapisarda AMC. et al. Epigenetic and genetic landscape of uterine leiomyomas: a current view over a common gynecological disease. Arch Gynecol Obstet 2017;296:855–867. [DOI] [PubMed] [Google Scholar]
- Launonen V, Vierimaa O, Kiuru M, Isola J, Roth S, Pukkala E, Sistonen P, Herva R, Aaltonen LA.. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci U S A 2001;98:3387–3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, Low HM, Kin Sung KW, Rigoutsos I, Loring J. et al. Dynamic changes in the human methylome during differentiation. Genome Res 2010;20:320–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layfield LJ, Liu K, Dodge R, Barsky SH.. Uterine smooth muscle tumors: utility of classification by proliferation, ploidy, and prognostic markers versus traditional histopathology. Arch Pathol Lab Med 2000;124:221–227. [DOI] [PubMed] [Google Scholar]
- Lee BB, Yu SP.. Radiofrequency ablation of uterine fibroids: a review. Curr Obstet Gynecol Rep 2016;5:318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Park YK, Park S, Jang Y, Waring N, Dey A, Ozato K, Lai B, Peng W, Ge K.. Brd4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nat Commun 2017;8:2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtonen R, Kiuru M, Vanharanta S, Sjoberg J, Aaltonen LM, Aittomaki K, Arola J, Butzow R, Eng C, Husgafvel-Pursiainen K. et al. Biallelic inactivation of fumarate hydratase (FH) occurs in nonsyndromic uterine leiomyomas but is rare in other tumors. Am J Pathol 2004;164:17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leistico JR, Saini P, Futtner CR, Hejna M, Omura Y, Soni PN, Sandlesh P, Milad M, Wei JJ, Bulun S. et al. Epigenomic tensor predicts disease subtypes and reveals constrained tumor evolution. Cell Rep 2021;34:108927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessl M, Klotzbuecher M, Schoen S, Reles A, Stockemann K, Fuhrmann U.. Comparative messenger ribonucleic acid analysis of immediate early genes and sex steroid receptors in human leiomyoma and healthy myometrium. J Clin Endocrinol Metab 1997;82:2596–2600. [DOI] [PubMed] [Google Scholar]
- Lethaby A, Vollenhoven B, Sowter M.. Pre-operative GnRH analogue therapy before hysterectomy or myomectomy for uterine fibroids. Cochrane Database Syst Rev 2001:CD000547. [DOI] [PubMed] [Google Scholar]
- Li G, Zhu P.. Structure and organization of chromatin fiber in the nucleus. FEBS Lett 2015;589:2893–2904. [DOI] [PubMed] [Google Scholar]
- Lima MSO, da Silva BB, de Medeiros ML, Dos Santos AR, do Nascimento Brazil ED, Filho W, Ibiapina JO, Brito AGA, Costa PVL.. Evaluation of vitamin D receptor expression in uterine leiomyoma and nonneoplastic myometrial tissue: a cross-sectional controlled study. Reprod Biol Endocrinol 2021;19:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JH. Medical treatment with ulipristal acetate for uterine fibroids: will it be resurrected? Fertil Steril 2021;116:80. [DOI] [PubMed] [Google Scholar]
- Liu JH, Soper D, Lukes A, Gee P, Kimble T, Kroll R, Mallick M, Chan A, Gillard P, Harrington A. et al. Ulipristal acetate for treatment of uterine leiomyomas: a randomized controlled trial. Obstet Gynecol 2018;132:1241–1251. [DOI] [PubMed] [Google Scholar]
- Liu S, Wicha MS.. Targeting breast cancer stem cells. J Clin Oncol 2010;28:4006–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Yin P, Kujawa SA, Coon JS, Okeigwe I, Bulun SE.. Progesterone receptor integrates the effects of mutated MED12 and altered DNA methylation to stimulate RANKL expression and stem cell proliferation in uterine leiomyoma. Oncogene 2019;38:2722–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Yin P, Xu J, Dotts AJ, Kujawa SA, Coon VJ, Zhao H, Dai Y, Bulun SE.. Progesterone receptor-DNA methylation crosstalk regulates depletion of uterine leiomyoma stem cells: a potential therapeutic target. Stem Cell Reports 2021a;16:2099–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Yin P, Xu J, Dotts AJ, Kujawa SA, Coon VJ, Zhao H, Shilatifard A, Dai Y, Bulun SE.. Targeting DNA methylation depletes uterine leiomyoma stem cell-enriched population by stimulating their differentiation. Endocrinology 2020;161:bqaa143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wu X, Wu A, Gong C, Wang Z, Zhang L.. Ultrasound-guided high intensity focused ultrasound ablation for uterine fibroids: long-term outcomes and factors affecting local recurrence. Int J Hyperthermia 2021b;38:1341–1348. [DOI] [PubMed] [Google Scholar]
- Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA.. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013;153:320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Chen J.. [Introduction of WHO classification of tumours of female reproductive organs, fourth edition]. Zhonghua Bing Li Xue Za Zhi 2014;43:649–650. [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ.. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997;389:251–260. [DOI] [PubMed] [Google Scholar]
- Maekawa R, Sato S, Yamagata Y, Asada H, Tamura I, Lee L, Okada M, Tamura H, Takaki E, Nakai A. et al. Genome-wide DNA methylation analysis reveals a potential mechanism for the pathogenesis and development of uterine leiomyomas. PLoS One 2013;8:e66632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinen N, Mehine M, Tolvanen J, Kaasinen E, Li Y, Lehtonen HJ, Gentile M, Yan J, Enge M, Taipale M. et al. MED12, the mediator complex subunit 12 gene, is mutated at high frequency in uterine leiomyomas. Science 2011;334:252–255. [DOI] [PubMed] [Google Scholar]
- Marinova M, Ghaei S, Recker F, Tonguc T, Kaverina O, Savchenko O, Kravchenko D, Thudium M, Pieper CC, Egger EK. et al. Efficacy of ultrasound-guided high-intensity focused ultrasound (USgHIFU) for uterine fibroids: an observational single-center study. Int J Hyperthermia 2021;38:30–38. [DOI] [PubMed] [Google Scholar]
- Marret H, Fritel X, Ouldamer L, Bendifallah S, Brun JL, De Jesus I, Derrien J, Giraudet G, Kahn V, Koskas M. et al. CNGOF (French College of Gynecology and Obstetrics). Therapeutic management of uterine fibroid tumors: updated French guidelines. Eur J Obstet Gynecol Reprod Biol 2012;165:156–164. [DOI] [PubMed] [Google Scholar]
- Marsh EE, Al-Hendy A, Kappus D, Galitsky A, Stewart EA, Kerolous M.. Burden, prevalence, and treatment of uterine fibroids: a survey of U.S. women. J Womens Health (Larchmt) 2018;27:1359–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh EE, Bulun SE.. Steroid hormones and leiomyomas. Obstet Gynecol Clin North Am 2006;33:59–67. [DOI] [PubMed] [Google Scholar]
- Marsh EE, Chibber S, Wu J, Siegersma K, Kim J, Bulun S.. Epidermal growth factor-containing fibulin-like extracellular matrix protein 1 expression and regulation in uterine leiomyoma. Fertil Steril 2016;105:1070–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall LM, Spiegelman D, Barbieri RL, Goldman MB, Manson JE, Colditz GA, Willett WC, Hunter DJ.. Variation in the incidence of uterine leiomyoma among premenopausal women by age and race. Obstet Gynecol 1997;90:967–973. [DOI] [PubMed] [Google Scholar]
- Mas A, Cervello I, Gil-Sanchis C, Faus A, Ferro J, Pellicer A, Simon C.. Identification and characterization of the human leiomyoma side population as putative tumor-initiating cells. Fertil Steril 2012;98:741–751.e746. [DOI] [PubMed] [Google Scholar]
- Masuda H, Matsuzaki Y, Hiratsu E, Ono M, Nagashima T, Kajitani T, Arase T, Oda H, Uchida H, Asada H. et al. Stem cell-like properties of the endometrial side population: implication in endometrial regeneration. PLoS One 2010;5:e10387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehine M, Kaasinen E, Heinonen HR, Makinen N, Kampjarvi K, Sarvilinna N, Aavikko M, Vaharautio A, Pasanen A, Butzow R. et al. Integrated data analysis reveals uterine leiomyoma subtypes with distinct driver pathways and biomarkers. Proc Natl Acad Sci USA 2016;113:1315–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehine M, Kaasinen E, Makinen N, Katainen R, Kampjarvi K, Pitkanen E, Heinonen HR, Butzow R, Kilpivaara O, Kuosmanen A. et al. Characterization of uterine leiomyomas by whole-genome sequencing. N Engl J Med 2013;369:43–53. [DOI] [PubMed] [Google Scholar]
- Mehine M, Makinen N, Heinonen HR, Aaltonen LA, Vahteristo P.. Genomics of uterine leiomyomas: insights from high-throughput sequencing. Fertil Steril 2014;102:621–629. [DOI] [PubMed] [Google Scholar]
- Mesquita FS, Dyer SN, Heinrich DA, Bulun SE, Marsh EE, Nowak RA.. Reactive oxygen species mediate mitogenic growth factor signaling pathways in human leiomyoma smooth muscle cells. Biol Reprod 2010;82:341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda Furtado CL, Dos Santos Luciano MC, Silva Santos RD, Furtado GP, Moraes MO, Pessoa C.. Epidrugs: targeting epigenetic marks in cancer treatment. Epigenetics 2019;14:1164–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, Chen CI, Anekalla KR, Joshi N, Williams KJN. et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med 2017;214:2387–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi R, Tabrizi R, Hessami K, Ashari H, Nowrouzi-Sohrabi P, Hosseini-Bensenjan M, Asadi N.. Correlation of low serum vitamin-D with uterine leiomyoma: a systematic review and meta-analysis. Reprod Biol Endocrinol 2020;18:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn F, Weber M, Rebhan M, Roloff TC, Richter J, Stadler MB, Bibel M, Schubeler D.. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol Cell 2008;30:755–766. [DOI] [PubMed] [Google Scholar]
- Moore LD, Le T, Fan G.. DNA methylation and its basic function. Neuropsychopharmacology 2013;38:23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorman PG, Leppert P, Myers ER, Wang F.. Comparison of characteristics of fibroids in African American and white women undergoing premenopausal hysterectomy. Fertil Steril 2013;99:768–776.e761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moosavi A, Ardekani AM.. Role of epigenetics in biology and human diseases. Iran Biomed J 2016;20:246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel D, Jeffery D, Aspeslagh S, Almouzni G, Postel-Vinay S.. Combining epigenetic drugs with other therapies for solid tumours – past lessons and future promise. Nat Rev Clin Oncol 2020;17:91–107. [DOI] [PubMed] [Google Scholar]
- Morrison O, Thakur J.. Molecular complexes at euchromatin, heterochromatin and centromeric chromatin. Int J Mol Sci 2021;22:6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyo MB, Parker JB, Chakravarti D.. Altered chromatin landscape and enhancer engagement underlie transcriptional dysregulation in MED12 mutant uterine leiomyomas. Nat Commun 2020;11:1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan CJ. Systemic racism can get under our skin and into our genes. Am J Phys Anthropol 2021;175:399–405. [DOI] [PubMed] [Google Scholar]
- Navarro A, Yin P, Monsivais D, Lin SM, Du P, Wei JJ, Bulun SE.. Genome-wide DNA methylation indicates silencing of tumor suppressor genes in uterine leiomyoma. PLoS One 2012;7:e33284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro A, Yin P, Ono M, Monsivais D, Moravek MB, Coon JS, Dyson MT, Wei J-J, Bulun SE.. 5-Hydroxymethylcytosine promotes proliferation of human uterine leiomyoma: a biological link to a new epigenetic modification in benign tumors. J Clin Endocrinol Metab 2014;99:E2437–E2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needham BL, Smith JA, Zhao W, Wang X, Mukherjee B, Kardia SL, Shively CA, Seeman TE, Liu Y, Diez Roux AV.. Life course socioeconomic status and DNA methylation in genes related to stress reactivity and inflammation: the multi-ethnic study of atherosclerosis. Epigenetics 2015;10:958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesby-O'Dell S, Scanlon KS, Cogswell ME, Gillespie C, Hollis BW, Looker AC, Allen C, Doughertly C, Gunter EW, Bowman BA.. Hypovitaminosis D prevalence and determinants among African American and white women of reproductive age: third National Health and Nutrition Examination Survey, 1988–1994. Am J Clin Nutr 2002;76:187–192. [DOI] [PubMed] [Google Scholar]
- Nestor CE, Ottaviano R, Reddington J, Sproul D, Reinhardt D, Dunican D, Katz E, Dixon JM, Harrison DJ, Meehan RR.. Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res 2012;22:467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomoto S, Kanda M, Okamura Y, Nishikawa Y, Qiyong L, Fujii T, Sugimoto H, Takeda S, Nakao A.. Epidermal growth factor-containing fibulin-like extracellular matrix protein 1, EFEMP1, a novel tumor-suppressor gene detected in hepatocellular carcinoma using double combination array analysis. Ann Surg Oncol 2010;17:923–932. [DOI] [PubMed] [Google Scholar]
- O'Reilly S. Epigenetics in fibrosis. Mol Aspects Med 2017;54:89–102. [DOI] [PubMed] [Google Scholar]
- Ono M, Maruyama T, Masuda H, Kajitani T, Nagashima T, Arase T, Ito M, Ohta K, Uchida H, Asada H. et al. Side population in human uterine myometrium displays phenotypic and functional characteristics of myometrial stem cells. Proc Natl Acad Sci USA 2007;104:18700–18705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono M, Qiang W, Serna VA, Yin P, Coon JS, Navarro A, Monsivais D, Kakinuma T, Dyson M, Druschitz S. et al. Role of stem cells in human uterine leiomyoma growth. PLoS One 2012;7:e36935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono M, Yin P, Navarro A, Moravek MB, Coon JS, Druschitz SA, Serna VA, Qiang W, Brooks DC, Malpani SS. et al. Paracrine activation of WNT/beta-catenin pathway in uterine leiomyoma stem cells promotes tumor growth. Proc Natl Acad Sci USA 2013;110:17053–17058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordulu Z. Deranged chromatin drives uterine fibroid tumours. Nature 2021;596:349–351. [DOI] [PubMed] [Google Scholar]
- Ortmann O, Weiss JM, Diedrich K.. Gonadotrophin-releasing hormone (GnRH) and GnRH agonists: mechanisms of action. Reprod Biomed Online 2002;5 Suppl 1:1–7. [DOI] [PubMed] [Google Scholar]
- Osuga Y, Nakano Y, Yamauchi Y, Takanashi M.. Ulipristal acetate compared with leuprorelin acetate for Japanese women with symptomatic uterine fibroids: a phase III randomized controlled trial. Fertil Steril 2021;116:189–197. [DOI] [PubMed] [Google Scholar]
- Othman ER, Ahmed E, Sayed AA, Hussein M, Abdelaal II, Fetih AN, Abou-Taleb HA, Yousef AA.. Human uterine leiomyoma contains low levels of 1, 25 dihdroxyvitamin D3, and shows dysregulated expression of vitamin D metabolizing enzymes. Eur J Obstet Gynecol Reprod Biol 2018;229:117–122. [DOI] [PubMed] [Google Scholar]
- Palomba S, Affinito P, Di Carlo C, Bifulco G, Nappi C.. Long-term administration of tibolone plus gonadotropin-releasing hormone agonist for the treatment of uterine leiomyomas: effectiveness and effects on vasomotor symptoms, bone mass, and lipid profiles. Fertil Steril 1999;72:889–895. [DOI] [PubMed] [Google Scholar]
- Pan Q, Luo X, Chegini N.. Genomic and proteomic profiling I: leiomyomas in African Americans and Caucasians. Reprod Biol Endocrinol 2007;5:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennacchio LA, Bickmore W, Dean A, Nobrega MA, Bejerano G.. Enhancers: five essential questions. Nat Rev Genet 2013;14:288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervaiz M, Mishra P, Gunther S.. Bromodomain drug discovery – the past, the present, and the future. Chem Rec 2018;18:1808–1817. [DOI] [PubMed] [Google Scholar]
- Piecak K, Milart P.. Hysteroscopic myomectomy. Prz Menopauzalny 2017;16:126–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard PJ, Briere JJ, Alam NA, Barwell J, Barclay E, Wortham NC, Hunt T, Mitchell M, Olpin S, Moat SJ. et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet 2005;14:2231–2239. [DOI] [PubMed] [Google Scholar]
- Popp B, Erber R, Kraus C, Vasileiou G, Hoyer J, Burghaus S, Hartmann A, Beckmann MW, Reis A, Agaimy A.. Targeted sequencing of FH-deficient uterine leiomyomas reveals biallelic inactivating somatic fumarase variants and allows characterization of missense variants. Mod Pathol 2020;33:2341–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H, Lin Z, Vasquez E, Xu L.. The association between chronic psychological stress and uterine fibroids risk: a meta-analysis of observational studies. Stress Health 2019;35:585–594. [DOI] [PubMed] [Google Scholar]
- Quade BJ, Weremowicz S, Neskey DM, Vanni R, Ladd C, Dal Cin P, Morton CC.. Fusion transcripts involving HMGA2 are not a common molecular mechanism in uterine leiomyomata with rearrangements in 12q15. Cancer Res 2003;63:1351–1358. [PubMed] [Google Scholar]
- Rai R, Sun T, Ramirez V, Lux E, Eren M, Vaughan DE, Ghosh AK.. Acetyltransferase p300 inhibitor reverses hypertension-induced cardiac fibrosis. J Cell Mol Med 2019;23:3026–3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai R, Verma SK, Kim D, Ramirez V, Lux E, Li C, Sahoo S, Wilsbacher LD, Vaughan DE, Quaggin SE. et al. A novel acetyltransferase p300 inhibitor ameliorates hypertension-associated cardio-renal fibrosis. Epigenetics 2017;12:1004–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, Portman JR, Matchett KP, Brice M, Marwick JA. et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019;575:512–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen KD, Helin K.. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev 2016;30:733–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray SK. The transcription regulator Kruppel-like factor 4 and its dual roles of oncogene in glioblastoma and tumor suppressor in neuroblastoma. For Immunopathol Dis Therap 2016;7:127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren W, Gao L, Song J.. Structural basis of DNMT1 and DNMT3A-mediated DNA methylation. Genes (Basel) 2018;9:620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, Fernandez R, Akbarpour M, Chen CI, Ren Z. et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med 2019;199:1517–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard-Davis G. Uterine fibroid: the burden borne by African American women. J Womens Health (Larchmt) 2013;22:793–794. [DOI] [PubMed] [Google Scholar]
- Rizzo A, Ricci AD, Saponara M, DE Leo A, Perrone AM, DE Iaco P, Pantaleo MA, Nannini M.. Recurrent uterine smooth-muscle tumors of uncertain malignant potential (STUMP): state of the art. Anticancer Res 2020;40:1229–1238. [DOI] [PubMed] [Google Scholar]
- Rossetto D, Avvakumov N, Cote J.. Histone phosphorylation: a chromatin modification involved in diverse nuclear events. Epigenetics 2012;7:1098–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabry M, Halder SK, Allah AS, Roshdy E, Rajaratnam V, Al-Hendy A.. Serum vitamin D3 level inversely correlates with uterine fibroid volume in different ethnic groups: a cross-sectional observational study. Int J Womens Health 2013;5:93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadr-Nabavi A, Ramser J, Volkmann J, Naehrig J, Wiesmann F, Betz B, Hellebrand H, Engert S, Seitz S, Kreutzfeld R. et al. Decreased expression of angiogenesis antagonist EFEMP1 in sporadic breast cancer is caused by aberrant promoter methylation and points to an impact of EFEMP1 as molecular biomarker. Int J Cancer 2009;124:1727–1735. [DOI] [PubMed] [Google Scholar]
- Sam M, Matthew RP, Patel V, Manolea F, Low G.. Limitations in US diagnosis of adenomysosis. Radiographics 2019;39:303–304. [DOI] [PubMed] [Google Scholar]
- Sangkomkamhang US, Lumbiganon P, Pattanittum P.. Progestogens or progestogen-releasing intrauterine systems for uterine fibroids (other than preoperative medical therapy). Cochrane Database Syst Rev 2020;11:CD008994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Maekawa R, Tamura I, Shirafuta Y, Shinagawa M, Asada H, Taketani T, Tamura H, Sugino N.. SATB2 and NGR1: potential upstream regulatory factors in uterine leiomyomas. J Assist Reprod Genet 2019;36:2385–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Maekawa R, Yamagata Y, Tamura I, Lee L, Okada M, Jozaki K, Asada H, Tamura H, Sugino N.. Identification of uterine leiomyoma-specific marker genes based on DNA methylation and their clinical application. Sci Rep 2016;6:30652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenmakers EF, Wanschura S, Mols R, Bullerdiek J, Van den Berghe H, Van de Ven WJ.. Recurrent rearrangements in the high mobility group protein gene, HMGI-C, in benign mesenchymal tumours. Nat Genet 1995;10:436–444. [DOI] [PubMed] [Google Scholar]
- Schooling CM, Zhao JV.. How might Bromodomain and extra-terminal (BET) inhibitors operate in cardiovascular disease? Am J Cardiovasc Drugs 2019;19:107–111. [DOI] [PubMed] [Google Scholar]
- Senol T, Kahramanoglu I, Dogan Y, Baktiroglu M, Karateke A, Suer N.. Levonorgestrel-releasing intrauterine device use as an alternative to surgical therapy for uterine leiomyoma. Clin Exp Obstet Gynecol 2015;42:224–227. [PubMed] [Google Scholar]
- Sharan C, Halder SK, Thota C, Jaleel T, Nair S, Al-Hendy A.. Vitamin D inhibits proliferation of human uterine leiomyoma cells via catechol-O-methyltransferase. Fertil Steril 2011;95:247–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi DQ, Ali I, Tang J, Yang WC.. New insights into 5hmC DNA modification: generation, distribution and function. Front Genet 2017;8:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorstova T, Foulkes WD, Witcher M.. Achieving clinical success with BET inhibitors as anti-cancer agents. Br J Cancer 2021;124:1478–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siedhoff MT, Wheeler SB, Rutstein SE, Geller EJ, Doll KM, Wu JM, Dl C-P.. Laparoscopic hysterectomy with morcellation vs abdominal hysterectomy for presumed fibroid tumors in premenopausal women: a decision analysis. Am J Obstet Gynecol 2015;212:591.e591–e598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigl V, Jones LP, Penninger JM.. RANKL/RANK: from bone loss to the prevention of breast cancer. Open Biol 2016;6:160230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JA, Catherino W, Segars JH, Blakesley RE, Chan A, Sniukiene V, Al-Hendy A.. Ulipristal acetate for treatment of symptomatic uterine leiomyomas: a randomized controlled trial. Obstet Gynecol 2018;131:431–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AK, Bishayee A, Pandey AK.. Targeting histone deacetylases with natural and synthetic agents: an emerging anticancer strategy. Nutrients 2018;10:731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skubitz KM, Skubitz AP.. Differential gene expression in uterine leiomyoma. J Lab Clin Med 2003;141:297–308. [DOI] [PubMed] [Google Scholar]
- Smith J, Sen S, Weeks RJ, Eccles MR, Chatterjee A.. Promoter DNA hypermethylation and paradoxical gene activation. Trends Cancer 2020;6:392–406. [DOI] [PubMed] [Google Scholar]
- Sohn GS, Cho S, Kim YM, Cho CH, Kim MR, Lee SR; Working Group of Society of Uterine Leiomyoma. Current medical treatment of uterine fibroids. Obstet Gynecol Sci 2018;61:192–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soutourina J. Transcription regulation by the Mediator complex. Nat Rev Mol Cell Biol 2018;19:262–274. [DOI] [PubMed] [Google Scholar]
- Steinmann S, Kunze P, Hampel C, Eckstein M, Bertram Bramsen J, Muenzner JK, Carle B, Ndreshkjana B, Kemenes S, Gasparini P. et al. DAPK1 loss triggers tumor invasion in colorectal tumor cells. Cell Death Dis 2019;10:895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart EA. Uterine fibroids. Lancet 2001;357:293–298. [DOI] [PubMed] [Google Scholar]
- Stewart EA, Cookson CL, Gandolfo RA, Schulze-Rath R.. Epidemiology of uterine fibroids: a systematic review. BJOG 2017;124:1501–1512. [DOI] [PubMed] [Google Scholar]
- Stewart EA, Nicholson WK, Bradley L, Borah BJ.. The burden of uterine fibroids for African-American women: results of a national survey. J Womens Health (Larchmt) 2013;22:807–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Styer AK, Rueda BR.. The epidemiology and genetics of uterine leiomyoma. Best Pract Res Clin Obstet Gynaecol 2016;34:3–12. [DOI] [PubMed] [Google Scholar]
- Sun S, , BonaffiniPA, , NougaretS, , FournierL, , DohanA, , ChongJ, , SmithJ, , AddleyH, , Reinhold C.. How to differentiate uterine leiomyosarcoma from leiomyoma with imaging. Diagn Interv Imaging 2019;100:619–634. [DOI] [PubMed] [Google Scholar]
- Suneja A, Faridi F, Bhatt S, Guleria K, Mehndiratta M, Sharma R.. Effect of vitamin D3 supplementation on symptomatic uterine leiomyoma in women with hypovitaminosis D. J Mid-Life Health 2021;12:53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Aoki M, Miyagawa C, Murakami K, Takaya H, Kotani Y, Nakai H, Matsumura N.. Differential diagnosis of uterine leiomyoma and uterine sarcoma using magnetic resonance images: a literature review. Healthcare (Basel) 2019;7:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L. et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009;324:930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan L, Xiong L, Xu W, Wu F, Huang N, Xu Y, Kong L, Zheng L, Schwartz L, Shi Y. et al. Genome-wide comparison of DNA hydroxymethylation in mouse embryonic stem cells and neural progenitor cells by a new comparative hMeDIP-seq method. Nucleic Acids Res 2013;41:e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, Leigh I, Gorman P, Lamlum H, Rahman S. et al.; Multiple Leiomyoma Consortium. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 2002;30:406–410. [DOI] [PubMed] [Google Scholar]
- Turunen M, Spaeth JM, Keskitalo S, Park MJ, Kivioja T, Clark AD, Makinen N, Gao F, Palin K, Nurkkala H. et al. Uterine leiomyoma-linked MED12 mutations disrupt mediator-associated CDK activity. Cell Rep 2014;7:654–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valimaki N, Kuisma H, Pasanen A, Heikinheimo O, Sjoberg J, Butzow R, Sarvilinna N, Heinonen HR, Tolvanen J, Bramante S. et al. Genetic predisposition to uterine leiomyoma is determined by loci for genitourinary development and genome stability. Elife 2018;7:e37110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heyningen V, Bickmore W.. Regulation from a distance: long-range control of gene expression in development and disease. Philos Trans R Soc Lond B Biol Sci 2013;368:20120372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vane JR, Botting RM.. Anti-inflammatory drugs and their mechanism of action. Inflamm Res 1998;47 Suppl 2:S78–S87. [DOI] [PubMed] [Google Scholar]
- Vilos GA, Allaire C, Laberge P-Y, Leyland N; SPECIAL CONTRIBUTORS. The management of uterine leiomyomas. J Obstet Gynaecol Can 2015;37:157–178. [DOI] [PubMed] [Google Scholar]
- Wagenfeld A, Saunders PT, Whitaker L, Critchley HO.. Selective progesterone receptor modulators (SPRMs): progesterone receptor action, mode of action on the endometrium and treatment options in gynecological therapies. Expert Opin Ther Targets 2016;20:1045–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LH, Wu CF, Rajasekaran N, Shin YK.. Loss of tumor suppressor gene function in human cancer: an overview. Cell Physiol Biochem 2018;51:2647–2693. [DOI] [PubMed] [Google Scholar]
- Wang P, Wang Z, Liu J.. Role of HDACs in normal and malignant hematopoiesis. Mol Cancer 2020;19:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Zhang X, Obijuru L, Laser J, Aris V, Lee P, Mittal K, Soteropoulos P, Wei JJ.. A micro-RNA signature associated with race, tumor size, and target gene activity in human uterine leiomyomas. Genes Chromosom Cancer 2007;46:336–347. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wang ZB, Xu YH.. Efficacy, efficiency, and safety of magnetic resonance-guided high-intensity focused ultrasound for ablation of uterine fibroids: comparison with ultrasound-guided method. Korean J Radiol 2018;19:724–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechter ME, Stewart EA, Myers ER, Kho RM, Wu JM.. Leiomyoma-related hospitalization and surgery: prevalence and predicted growth based on population trends. Am J Obstet Gynecol 2011;205:492.e491–e495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams ARW. Uterine fibroids – what's new? F1000Res 2017;6:2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise LA, Ruiz-Narvaez EA, Palmer JR, Cozier YC, Tandon A, Patterson N, Radin RG, Rosenberg L, Reich D.. African ancestry and genetic risk for uterine leiomyomata. Am J Epidemiol 2012;176:1159–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak A, Wozniak S.. Ultrasonography of uterine leiomyomas. Prz Menopauzalny 2017;16:113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Gao H.. Hydroxymethylation and tumors: can 5-hydroxymethylation be used as a marker for tumor diagnosis and treatment? Hum Genomics 2020;14:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata Y, Maekawa R, Asada H, Taketani T, Tamura I, Tamura H, Ogane J, Hattori N, Shiota K, Sugino N.. Aberrant DNA methylation status in human uterine leiomyoma. Mol Hum Reprod 2009;15:259–267. [DOI] [PubMed] [Google Scholar]
- Yang H, Liu Y, Bai F, Zhang JY, Ma SH, Liu J, Xu ZD, Zhu HG, Ling ZQ, Ye D. et al. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene 2013;32:663–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Mas A, Diamond MP, Al-Hendy A.. The mechanism and function of epigenetics in uterine leiomyoma development. Reprod Sci 2016;23:163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao HW, Li J.. Epigenetic modifications in fibrotic diseases: implications for pathogenesis and pharmacological targets. J Pharmacol Exp Ther 2015;352:2–13. [DOI] [PubMed] [Google Scholar]
- Yao W, Wang T, Huang F.. p300/CBP as a key nutritional sensor for hepatic energy homeostasis and liver fibrosis. Biomed Res Int 2018;2018:8168791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Lo JH, Kim JY, Marsh EE, Kim JJ, Ghosh AK, Bulun S, Chakravarti D.. Expression profiling of nuclear receptors identifies key roles of NR4A subfamily in uterine fibroids. Mol Endocrinol 2013;27:726–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin P, Lin Z, Reierstad S, Wu J, Ishikawa H, Marsh EE, Innes J, Cheng Y, Pearson K, Coon JS. et al. Transcription factor KLF11 integrates progesterone receptor signaling and proliferation in uterine leiomyoma cells. Cancer Res 2010;70:1722–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin P, Ono M, Moravek MB, Coon JS, Navarro A, Monsivais D, Dyson MT, Druschitz SA, Malpani SS, Serna VA. et al. Human uterine leiomyoma stem/progenitor cells expressing CD34 and CD49b initiate tumors in vivo. J Clin Endocrinol Metab 2015;100:E601–E606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Ham K, Gao X, Castro L, Yan Y, Kissling GE, Tucker CJ, Flagler N, Dong R, Archer TK. et al. Epigenetic regulation of transcription factor promoter regions by low-dose genistein through mitogen-activated protein kinase and mitogen-and-stress activated kinase 1 nongenomic signaling. Cell Commun Signal 2016;14:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou K, Gaullier G, Luger K.. Nucleosome structure and dynamics are coming of age. Nat Struct Mol Biol 2019;26:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zota AR, VanNoy BN.. Integrating intersectionality into the exposome paradigm: a novel approach to racial inequities in uterine fibroids. Am J Public Health 2021;111:104–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zupi E, Centini G, Sabbioni L, Lazzeri L, Argay IM, Petraglia F.. Nonsurgical alternatives for uterine fibroids. Best Pract Res Clin Obstet Gynaecol 2016;34:122–131. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data were generated or analyzed in support of this research.