

Abstract
Cervical cancer is one of the most common types of gynecological tumors. Lysophosphatidic acid (LPA), as a bioactive lipid medium, plays an important role in numerous physiological and pathophysiological processes, including the stimulation of cell migration and tumor cell invasion. LPA is increased in the plasma of patients with cervical cancer. Doxorubicin hydrochloride (DOX) is used as a first-line drug in the treatment of cervical cancer in clinics, however, the effect and molecular mechanism of LPA on DOX-induced apoptosis in cervical cancer cells remain unclear. Therefore, the present study aimed to explore the effect and underlying molecular mechanism of LPA on DOX-induced apoptosis in cervical cancer cells. HeLa cells were treated as a control group or with LPA (10 µmol/l), DOX (4 µmol/l) or LPA (10 µmol/l) + DOX (4 µmol/l) for 24 h. Using transmission electron microscopy the results demonstrated that LPA reduced cell death and the degree of chromatin aggregation in DOX-induced HeLa cells. Reverse transcription-quantitative PCR demonstrated that LPA significantly downregulated caspase-3 mRNA expression levels in DOX-induced HeLa cells. Moreover, western blotting demonstrated that LPA significantly reduced caspase-3 and cleaved caspase-3 protein expression levels in DOX-induced HeLa, C33A and SiHa cells. Furthermore, flow cytometry demonstrated that LPA may prevent apoptosis in DOX-induced HeLa cells (P<0.05). Using dichloro-dihydro-fluorescein diacetate assay, it was demonstrated that LPA significantly reduced the intracellular ROS levels induced by DOX. In summary, the present study indicated that LPA may protect HeLa cells from apoptosis induced by DOX. These findings have provided experimental evidence that LPA may be a potential therapeutic target for the treatment of cervical cancer.
Keywords: lysophosphatidic acid, doxorubicin hydrochloride, cervical cancer, apoptosis, reactive oxygen species
Introduction
Lysophosphatidic acid (LPA) is a simple glycerol phospholipid that serves an important role in numerous physiological and pathophysiological processes, including the stimulation of cell migration, tumor cell invasion, neurite retraction, as well as proliferation stimulation of a variety of normal and tumorigenic cells (1). LPA is also increased in the plasma of patients with cervical cancer (2). Elevated plasma LPA levels represent a potential biomarker for certain types of gynecological cancer (3,4). As a bioactive lipid medium, LPA is an endogenous lysophospholipid with signaling properties outside of the cell. Moreover, LPA signals via specific G-protein-coupled receptors, known as LPA1-6 (5,6).
Cervical cancer is one of the most common types of gynecological tumors (7). An estimated 500,000 new cases of cervical cancer are diagnosed each year worldwide, which leads to 280,000 cancer-related deaths annually (8). Recurrence and metastasis events are often associated with a poor prognosis and the five-year survival rate of patients with locally advanced cervical cancer is as high as 75–85% following treatment via surgical resection, radiotherapy and chemotherapy (9).
Doxorubicin hydrochloride (DOX) induces the apoptosis of cancer cells via numerous mechanisms and serves an anticancer role as a first-line drug in cancer treatment (10–12). However, the traditional DOX-based chemotherapy regimen has received numerous negative evaluations, which includes the development of drug resistance and the development of the epithelial-mesenchymal transformation progression (13). DOX induces oxidative stress, which is characterized by the accumulation of reactive oxygen species (ROS) and a reduction in antioxidant defenses. Oxidative stress injury is closely related to apoptosis, an important antitumor mechanism, in which ROS are involved (14). Other studies have reported that ROS have an important role in tumorigenesis. The high level of ROS in cancer cells is the product of a high metabolism, which can maintain cancer cell proliferation and also stimulates cell invasion and metastasis (15,16).
The molecular mechanism of LPA on DOX-induced apoptosis in cervical cancer cells remains unclear. The hypothesis of the present study, is that as a bioactive lipid medium, LPA may protect cervical cancer cells from apoptosis via inhibiting caspase-3 expression and reducing oxidative stress injury in cancer cells. Therefore, the aim of the present study was to investigate this interaction to potentially identify a novel therapeutic target and approach for the treatment of cervical cancer.
Materials and methods
Reagents
The human cervical cancer HeLa (cat. no. CL0134), C33A (cat. no. CL0385) and SiHa (cat. no. CL0280) cell lines were purchased from Hunan Fenghui Biotechnology Co., Ltd. RPMI-1640 culture medium was purchased from Zhejiang Senrui Biotechnology Co., Ltd. FBS was purchased from Omnimabs. Penicillin-streptomycin mixed solution, the ROS detection kit (cat. no. CA1410) and DOX were purchased from Solarbio Science & Technology Co., Ltd. The TRIzol® RNA isolation kit was purchased from Jiangsu Cowin Biotech Co., Ltd. LPA was purchased from Sigma-Aldrich (Merck KGaA). The reverse transcription (RT) kit and SYBR Green® Real-Time PCR Master Mix were purchased from Toyobo Life Science. Primers were synthesized by Shanghai Personal Biotechnology Co., Ltd. The BCA kit was purchased from Beyotime Institute of Biotechnology. The rabbit polyclonal anti-β-actin primary antibody was purchased from Biosharp Life Sciences (cat. no. BL005B). The rabbit polyclonal caspase-3 primary antibody (cat. no. ab13847) and the donkey anti-rabbit IgG heavy chain and light chain Alexa Fluor 680-conjugated secondary antibody (cat. no. ab175772) were purchased from Abcam. The Annexin V-FITC Apoptosis Detection Kit (cat. no. 556547) was purchased from Becton Dickinson and Company. Superoxide dismutase (SOD; cat. no. A001-3) and malondialdehyde (MDA; cat. no. A003-2) detection kits were purchased from Nanjing Jiancheng Bioengineering Institute. ImageJ software (V1.48) for densitometry reading of bands of the western blot analysis was provided by the National Institutes of Health.
Cell culture and treatment
HeLa, C33A and SiHa cells were cultured in RPMI-1640 medium containing 10% FBS and 1% penicillin-streptomycin in an incubator with 5% CO2 at 37°C. Cells were inoculated into 6-well plates, according to the required number of cells for the experiment, at 37°C with 5% CO2 for 24 h. Cells were subsequently incubated with LPA (10 µmol/l), DOX (4 µmol/l) or LPA (10 µmol/l) + DOX (4 µmol/l) at 37°C with 5% CO2 for 24 h.
Cell morphology evaluation
To detect cell morphology changes of HeLa cells after LPA and DOX treatment, HeLa cells (5×105 cells/well) were treated at 37°C for 24 h with LPA (10 µmol/l), DOX (4 µmol/l) or LPA (10 µmol/l) + DOX (4 µmol/l). Following centrifugation at 200 × g for 10 min at room temperature, the supernatant was discarded and the cells were washed once with PBS. The morphology of HeLa cells in the different treatment groups was observed and the cells were imaged using an optical microscope (magnification ×100; BX53 and DP80 models; Olympus Corporation).
Detection of apoptosis via transmission electron microscopy
To detect ultrastructural morphology of HeLa cells after LPA and DOX treatment by transmission electron microscope, HeLa cells in each treatment group were washed with PBS, digested into a single cell suspension using trypsin and centrifuged at 200 × g for 10 min at room temperature. The supernatant was discarded following the termination of digestion. A total of 3×106 cells were fixed using 3% glutaraldehyde for 30 min at room temperature and the supernatant was discarded. Subsequently the cells were incubated with a fresh fixation solution at 4°C for 24 h. The cells were then washed three times with precooled PBS and incubated with 1% osmic acid for further fixation at 4°C for 2 h. Following gradient dehydration, immersion and embedding, 70-nm ultra-thin sections of the treated HeLa cells were produced. The sections were collected using a copper net and were stained using 2% uranyl acetate for 30 min at room temperature and subsequently lead citrate (0.665 g lead nitrate and 0.88 g sodium citrate was dissolved in 15 ml distilled water. Next, 4 ml 1N sodium hydroxide was added and a final volume of 25 ml was created with distilled water) for 15 min at room temperature. After drying, images of the cells were obtained using a transmission electron microscope (magnification, ×2,000; HT7700 model; Hitachi. Ltd.).
RT-quantitative PCR (RT-qPCR)
To determine caspase-3 and caspase-8 at the mRNA level as a result of apoptosis in HeLa cells treated with LPA and DOX by qPCR, total cellular RNA was extracted from HeLa cells using the TRIzol method according to the manufacturer's protocol. Subsequently RNA was reverse transcribed into complementary DNA using a RT kit according to the manufacturer's protocol and the thermocycling conditions were as follows: 37°C for 15 min and 98°C for 5 min. SYBR Green® Real-Time PCR Master Mix was used for qPCR. The following qPCR primers were used: GAPDH forward (F), 5′-AGAAGGCTGGGGCTCATTTG-3′ and reverse (R), 5′-AGGGGCCATCCACAGTCTTC-3′; caspase-3 F, 5′-GTGGAGGCCGACTTCTTGTATGC-3′ and R, 5′-TGGCACAAAGCGACTGGATGAAC-3′; and caspase-8 F, 5′-CGGATGAGGCTGACTTTCTGCTG-3′ and R, 5′-GGCTCTGGCAAAGTGACTGGATG-3′. The reaction system was prepared according to the manufacturer's protocol. The thermocycling conditions for qPCR were as follows: Initial denaturation at 95°C for 30 sec; and 40 cycles of denaturation at 95°C for 5 sec, annealing at 55°C for 10 sec and elongation at 72°C for 15 sec. GAPDH was used as the internal reference gene and the relative mRNA expression levels were determined using the 2−ΔΔCq method (17).
Western blotting
To determine caspase-3 and cleaved caspase-3 at the protein level as a result of apoptosis in HeLa cells treated with LPA and DOX by western blotting, HeLa, C33A and SiHa cells were collected following treatment. Total protein was extracted using RIPA buffer purchased from Beijing Solarbio Science & Technology Co., Ltd., (cat. no. R0020; RIPA:PMSF, 100:1) and the protein concentration was determined using a BCA kit. Total protein (20 µg per lane) was separated on a 12% gel using SDS-PAGE. Separated proteins were electrically transferred onto a PVDF membrane, which was then blocked using 5% skimmed milk at room temperature for 1 h. Subsequently the membranes were incubated with primary antibodies, rabbit polyclonal to caspase-3 (1:1,000; cat. no. ab13847; Abcam) for 12 h at 4°C and the membranes were then washed with TBS with 0.05% Tween-20 three times. Following the primary incubation the membranes were incubated with the HRP-conjugated secondary antibody, donkey anti-rabbit IgG H&L Alexa Fluor® 680 (1:10,000 dilution; cat. no. ab175772; Abcam) at room temperature for 1 h. Protein bands were visualized using ECL reagent and images were acquired using the Amersham Imager 600 (Cytiva).
Detection of apoptosis via flow cytometry
Flow cytometry was used to detect apoptosis of HeLa cells after treatment using Annexin V staining. Following treatment with LPA (10 µmol/l), DOX (4 µmol/l) or LPA (10 µmol/l) + DOX (4 µmol/l), HeLa cells were collected, washed with precooled PBS and centrifuged at 200 × g for 10 min at 4°C. Subsequently, the supernatant was discarded and the cells were resuspended in 100 µl 1X buffer solution with 5 µl FITC-Annexin V and 5 µl PI. The cells were incubated at room temperature for 15 min in the dark and subsequently 400 µl 1X buffer solution was added. After passing the cells through a 400-mesh filter, apoptotic cells were detected via flow cytometry (Beckman Coulter, Inc.). The plotted cells in Q1, Q2 and Q4 of the graph were used to calculate the apoptosis rate.
Detection of active oxygen levels
To detect the level of ROS of HeLa cells treated with LPA and DOX using Rosup detection kit, HeLa cells were inoculated into 96-well plates at a density of 2×104 cells/well and were treated with the different combinations of drugs at 37°C for 24 h. Subsequently, a ROS detection kit was used according to the manufacturer's protocol. The positive control cell group was stimulated using the ROS positive control Rosup (1:1,000) for 30 min at room temperature and the supernatant was discarded. Rosup was provided as a component of the ROS detection kit. To each group dichloro-dihydro-fluorescein diacetate (1:1,000) was added and the cells were incubated at 37°C for 20 min. Then the cells were washed with PBS three times. The fluorescence intensity was directly measured without image capture using a multifunctional spectrophotometer (SpectraMax M5; Molecular Devices, LLC) at an excitation wavelength of 488 nm and an emission wavelength of 525 nm.
SOD detection
As SOD balances ROS by scavenging free radicals and prevents cell damage caused by superoxide anion radicals (18), SOD levels in HeLa cells were detected using a SOD detection kit according to the manufacturer's instructions. The cells were inoculated at 5×105 cells/ml in 6-well plates and were cultured at 37°C with 5% CO2 for 24 h. Following centrifugation at 200 × g for 10 min at room temperature, the supernatant was discarded and the cells were treated as the control group, or with LPA, DOX or LPA + DOX for 24 h. The cells were digested with trypsin, collected and centrifuged at 200 × g for 10 min at room temperature. The supernatant was discarded and the cells were washed using 1 ml PBS. Subsequently the cells were collected into 1.5 ml Eppendorf tubes. Following centrifugation at 200 × g for 10 min at room temperature, the supernatant was discarded and 300 µl 0.5% Triton X-100 lysis buffer was added to each tube. The cells were incubated for 30 min on ice and were then centrifuged at 1,200 × g for 8 min at 4°C. The supernatant was collected and the cellular protein concentration was detected using a BCA kit. The extracted protein was added to distilled water, enzyme working solution and substrate application solution according to the SOD kit manufacturer's protocol. Following the reaction, the cells were incubated at 37°C for 20 min. SOD levels were quantified using an ELISA microplate reader to detect the absorbance value at 450 nm. SOD activity was calculated according to the formula provided by the manufacturer (SOD detection kit; Nanjing Jiancheng Bioengineering Institute.
MDA detection
As MDA is a widely detected marker of oxidative damage and its elevated level is the result of lipid peroxidation (19), MDA levels in HeLa cells were determined using the thiobarbituric acid method. Cells were inoculated at a density of 5×105 cells/ml in 6-well plates and were cultured in an incubator at 37°C with 5% CO2 for 24 h. Following centrifugation at 200 × g for 10 min at room temperature, the supernatant was discarded and the cells were treated as the control group or with LPA, DOX or LPA + DOX for 24 h at 37°C. Subsequently, cells were digested with trypsin, collected and centrifuged at 200 × g for 10 min at room temperature. The supernatant was discarded and the cells were washed with 1 ml PBS prior to being collected in a 1.5 ml Eppendorf tube. Following centrifugation at 200 × g for 5 min at 4°C, the supernatant was discarded and 300 µl 0.5% Triton X-100 lysis buffer was added to each tube. The cells were incubated for 30 min on ice and then centrifuged at 1,200 × g for 8 min at 4°C. The supernatant was collected and the cell protein concentration was determined using a BCA kit. The extracted protein was used for the measurement of MDA level using an MDA detection kit (cat. no. A003-2) according to the manufacturer's protocol. The absorbance value of MDA was determined using a UV spectrophotometer at 532 nm. MDA levels were quantified according to the formula provided by the manufacturer (MDA detection kit; Nanjing Jiancheng Bioengineering Institute).
Statistical analysis
Data are presented as the mean ± SD. All data were statistically analyzed using GraphPad Prism 5.0 (GraphPad Software, Inc.). One-way ANOVA was used to statistically compare more than two groups and Tukey's post hoc test was used to compare the different groups following ANOVA. P<0.05 was considered to indicate a statistically significant difference.
Results
LPA protects HeLa cells from DOX-induced apoptosis
The morphology of HeLa cells in the different treatment groups was determined using a light microscope. The results demonstrated that there was no significant change in cell morphology in the control and LPA-treated groups. However, cell shrinkage and abscission in the DOX-treated group were observed and the degree of cell abscission and shrinkage in the LPA + DOX treated group was reduced compared with the DOX group (Fig. 1A). Using transmission electron microscopy it was determined that the cell morphology and nuclear condensation of the control and LPA groups remained unchanged. However, in the DOX group, chromatin gathered at the edge of the nuclear membrane and broken nuclear membrane and cells were observed. Moreover, in the LPA + DOX group, nuclear chromatin gathered at the edge of the nuclear membrane and an increased vacuole was observed; however, the percentage of apoptotic cells was less compared with the DOX group (Fig. 1B). Moreover, the results indicated that LPA may have protected HeLa cells from DOX-induced apoptosis. Subsequently, apoptosis was detected via flow cytometry. The results demonstrated that there was no significant change in the apoptotic rate in the control and LPA groups. However, the apoptotic rate of the DOX group increased significantly, whereas the apoptotic rate of the LPA + DOX group decreased by 23.24% compared with the DOX group (P<0.05) (Fig. 2A and B). These results indicated that LPA potentially protected HeLa cells from DOX-induced apoptosis.
Figure 1.

Morphology of HeLa cells treated with DOX and LPA. (A) Images were captured using an optical microscope. Magnification, ×100. Scale bar, 100 µm. (B) Images were captured using a transmission electron microscope. Magnification, ×2,000. Scale bar, 20 µm. Arrows indicate membrane broken, nuclear fragmentation, and chromatin condensation. DOX, doxorubicin hydrochloride; LPA, lysophosphatidic acid; MB, membrane broken, NF, nuclear fragmentation; CC, chromatin condensation.
Figure 2.

Determination of the apoptotic rate using flow cytometry. (A) Flow cytometry was used to detect apoptosis in the control, LPA, DOX and LPA + DOX HeLa cell groups. The percentage of apoptotic cells in Q1, Q2 and Q4 are as indicated. (B) Quantification of the apoptotic rate was determined using the flow cytometric data. Data are presented as the mean ± SD (n=3). *P<0.05 and **P<0.01 vs. the control. DOX, doxorubicin hydrochloride; LPA, lysophosphatidic acid; n.s., no significance.
LPA downregulates the mRNA expression levels of caspase-3 and caspase-8 in DOX-induced HeLa cells
RT-qPCR was used to determine the mRNA expression levels of caspase-3 and caspase-8 in HeLa cells in the different treatment groups. The results demonstrated that there were no significant changes in the mRNA expression levels of caspase-3 and caspase-8 in the control and LPA-treated groups. However, the caspase-3 mRNA expression levels significantly increased in the DOX group (P<0.01), whereas they decreased in the LPA + DOX group by 35.61% compared with the DOX group (P<0.05) (Fig. 3A). The mRNA expression levels of caspase-8 in the DOX group significantly increased (P<0.01), whereas they significantly decreased in the LPA + DOX by 41.38% compared with the DOX group (P<0.05) (Fig. 3B). These results indicated that LPA potentially downregulated the mRNA expression levels of caspase-3 and caspase-8, which suggested that LPA may protect HeLa cells from DOX-induced apoptosis via the downregulation of these proapoptotic genes.
Figure 3.

LPA decreases the mRNA expression of caspase-3 and caspase-8 in DOX-induced HeLa cells. (A) Caspase-3 mRNA expression levels in the control, LPA, DOX and LPA + DOX HeLa cell groups. (B) Caspase-8 mRNA expression levels in the control, LPA, DOX and LPA + DOX HeLa cell groups. Data are presented as the mean ± SD (n=3). *P<0.05 and **P<0.01 vs. the control. DOX, doxorubicin hydrochloride; LPA, lysophosphatidic acid; n.s., no significance.
LPA downregulates the protein expression levels of caspase-3 in DOX-induced cervical cancer cells
Western blotting demonstrated that there were no significant changes in the protein expression levels of caspase-3 and cleaved caspase-3 in the control and LPA groups. However, the protein expression levels of caspase-3 and cleaved caspase-3 in the DOX group were significantly increased in HeLa, C33A and SiHa cells (P<0.01). Moreover, the protein expression levels of caspase-3 and cleaved caspase-3 in the LPA + DOX group decreased compared with the DOX group in HeLa (Fig. 4A), C33A (Fig. 4D) and SiHa cells (Fig. 4G). Densitometric analysis of the western blotting results demonstrated that caspase-3 protein expression levels in HeLa cells significantly decreased by 33.13% (P<0.05; Fig. 4B) and cleaved caspase-3 protein expression levels significantly decreased by 33.88% (P<0.05; Fig. 4C) in the LPA + DOX group compared with the DOX group. Moreover, caspase-3 protein expression levels in C33A cells significantly decreased by 39.38% (P<0.05; Fig. 4E) and cleaved caspase-3 protein expression levels significantly decreased by 36.89% (P<0.05; Fig. 4F) in the LPA + DOX group compared with the DOX group. Furthermore, caspase-3 protein expression levels in SiHa cells significantly decreased by 44.73% (P<0.01; Fig. 4H) and cleaved caspase-3 protein expression levels significantly decreased by 31.78% (P<0.05; Fig. 4I) in the LPA + DOX group compared with the DOX group. These results indicated that LPA potentially downregulated the protein expression levels of apoptosis-related proteins in DOX-induced cervical cancer HeLa, C33A and SiHa cells.
Figure 4.

Effect of LPA on the protein expression levels of caspase-3 and cleaved caspase-3 in DOX-induced HeLa, C33A and SiHa cells. After HeLa, C33A and SiHa cells were treated as the control or with LPA, DOX or LPA + DOX for 24 h, the cells were harvested and lysed for western blotting. (A) Western blotting was performed to determine caspase-3 and cleaved caspase-3 protein expression levels in HeLa cells. (B) Semi-quantification of caspase-3 protein expression levels in HeLa cells was performed using ImageJ software. (C) Semi-quantification of cleaved caspase-3 protein expression levels in HeLa cells was performed using ImageJ software. (D) Western blotting was performed to determine caspase-3 and cleaved caspase-3 protein expression levels in C33A cells. (E) Semi-quantification of caspase-3 protein expression levels in C33A was performed using ImageJ software. (F) Semi-quantification of cleaved caspase-3 protein expression levels in C33A cells was performed using ImageJ software. (G) Western blotting was performed to determine caspase-3 and cleaved caspase-3 protein expression levels in SiHa cells. (H) Semi-quantification of caspase-3 protein expression levels in SiHa cells was performed using ImageJ software. (I) Semi-quantification of cleaved caspase-3 protein expression levels in SiHa cells was performed using ImageJ software. Data are presented as the mean ± SD (n=3). *P<0.05 and **P<0.01 vs. the control. DOX, doxorubicin hydrochloride; LPA, lysophosphatidic acid; n.s., no significance.
LPA reduces oxidative stress injury in DOX-induced HeLa cells
The results of the ROS detection assay demonstrated that there was no significant change in ROS levels in the control and LPA groups. However, ROS levels in the DOX group significantly increased (P<0.01), whereas ROS levels in the LPA + DOX group significantly decreased by 24.71% (P<0.05) compared with the DOX group (Fig. 5A). The SOD detection assay demonstrated that there was no significant change in SOD levels in the control and LPA groups. However, the SOD level in the DOX group significantly decreased (P<0.01), whereas the SOD level in the LPA + DOX group significantly increased by 24.94% (P<0.05) compared with the DOX group (Fig. 5B). The MDA detection assay demonstrated that there was no significant change in MDA levels in the control and LPA groups. However, MDA levels in the DOX group significantly increased (P<0.01), whereas MDA levels in the LPA + DOX group significantly decreased by 28.81% (P<0.01) compared with the DOX group (Fig. 5C). These results indicated that LPA potentially reduced the oxidative stress injury of DOX-induced HeLa cells.
Figure 5.

LPA reduces oxidative stress injury of DOX-induced HeLa cells. (A) Reactive oxygen species fluorescence intensity levels of the control, LPA, DOX and LPA + DOX HeLa cell groups. (B) SOD levels of the control, LPA, DOX and LPA + DOX HeLa cell groups. (C) MDA levels of the control, LPA, DOX and LPA + DOX HeLa cell groups. Data are presented as the mean ± SD (n=3). *P<0.05 and **P<0.01 vs. the control. DOX, doxorubicin hydrochloride; LPA, lysophosphatidic acid; SOD, superoxide dismutase; MDA, malondialdehyde; n.s., no significance.
Discussion
A loss of apoptosis is considered to be a characteristic of cancer cells. Apoptosis can quickly and selectively remove harmful, defective or damaged cells from the body (20). Therefore, molecules or mechanisms that are proapoptotic may be potential therapeutic targets in cancer treatment. In the present study, the effect of LPA on DOX-induced apoptosis in cervical cancer cells was investigated. The results demonstrated that LPA was an anti-apoptotic factor that potentially protected cervical cancer HeLa cells from DOX-induced apoptosis by reducing oxygen stress injury and inhibiting caspase-3 expression. The associated mechanisms by which LPA potentially protects HeLa cells from DOX-induced apoptosis are presented in Fig. 6.
Figure 6.
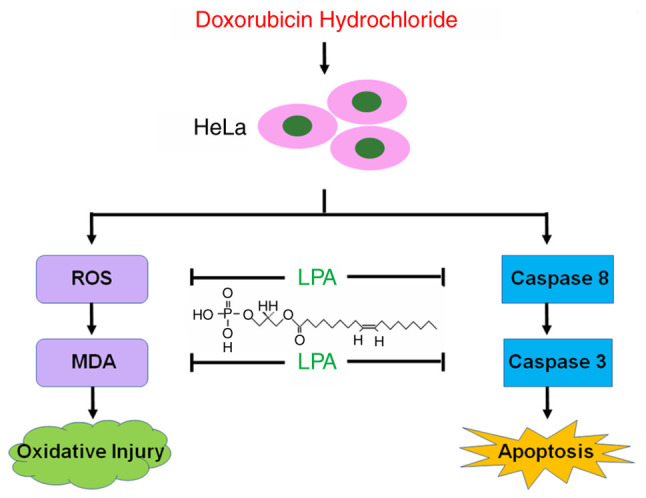
The diagram illustrates that LPA potentially protects HeLa cells from DOX-induced apoptosis via inhibiting caspase-3 expression and LPA reduces intracellular ROS level induced by DOX to prevent HeLa cells from oxygen stress injury. LPA, lysophosphatidic acid; DOX, doxorubicin; ROS, reactive oxygen species.
Previous studies have reported that cell pyknosis is the result of chromatin agglutination in the nucleus and is the most significant feature of apoptosis (21). Furthermore, the decrease or disappearance of cell surface villi, contraction of the nuclear membrane, reduction of nuclear volume, chromatin aggregation and vacuoles in the cytoplasm, are also typical ultrastructural changes of apoptotic cells (22). The present study demonstrated that the control and LPA groups displayed normal cellular morphology. However, DOX treatment of the cells resulted in significant changes in cell morphology, including cell shrinkage, roundness, a reduced volume and severe shedding. Compared with the DOX group, the LPA + DOX group exhibited a significant improvement in cell morphology and cell availability. Transmission electron microscopy demonstrated that the nucleus, nuclear membrane and chromatin state remained unchanged in the control and LPA groups. However, chromatin aggregation and cell fragmentation were observed in the cells of the DOX group, which suggested that these cells were undergoing apoptosis. The flow cytometric analysis confirmed the aforementioned result. The degree of apoptosis in the LPA + DOX group was significantly lower compared with the DOX group, which suggested that LPA potentially reduced the DOX-induced apoptosis of cervical cancer cells.
Caspase-3 is an important regulatory factor of cancer cells during apoptosis (23). Both endogenous and exogenous apoptotic signaling pathways can lead to the activation of caspase-3 (24). In order to further understand the mechanism by which LPA protected cervical cancer cells from DOX-induced apoptosis, the expression levels of caspase-3 were analyzed. The results demonstrated that DOX treatment of HeLa cells resulted in a significant increase in the caspase-3 mRNA expression levels. Compared with the DOX group, the mRNA expression levels of caspase-3 gene in the LPA + DOX group were significantly decreased. The activated form of caspase-3 is cleaved caspase-3 and once activated it will cleave a large number of cytoskeleton-related proteins, cell cycle regulatory proteins and DNA repair and DNA degradation-related molecules (25,26), which results in cell death. Therefore, the effect of DOX on caspase-3 and cleaved caspase-3 protein expression levels was further investigated in cervical cancer cells treated with LPA. The results demonstrated that the protein expression levels of caspase-3 and cleaved caspase-3 protein significantly increased and the degree of apoptosis was severe in the DOX group. However, the protein expression levels of caspase-3 and cleaved caspase-3 significantly decreased in the LPA + DOX group compared with the DOX group. Furthermore, the results of the present study demonstrated that at both the mRNA and protein expression levels, DOX-induced apoptosis was inhibited by LPA via decreasing caspase-3 expression. It was also previously reported that LPA inhibits cisplatin-induced apoptosis of cervical cancer cells (8,16), which is consistent with the results of the present study. Therefore, further investigation into the role of LPA in DOX-induced cell death and the downstream signaling pathway of LPA will potentially provide novel insights into the clinical treatment of cervical cancer.
Numerous studies have reported that oxidative stress is strongly associated with arthritis, cancer, autoimmune diseases, aging and cardiovascular and neurodegenerative diseases (15,27,28). High ROS levels can disrupt the structure of the mitochondrial membrane, which leads to a shift in membrane permeability and ultimately to apoptosis (29). ROS regulates the tumor suppressor p53 signal, which releases cytochrome c that activates caspase-3 (30). Therefore, as DOX can induce oxidative stress, several oxidative stress indicators were detected in the present study. This was to determine if LPA affected DOX-induced apoptosis in cervical cancer cells via the inhibition of oxidative stress. The results demonstrated that LPA inhibited DOX-induced apoptosis by significantly decreasing ROS levels in cervical cancer cells. ROS-mediated oxidative stress is a complex reaction between free radical production and the antioxidant defense of the body (31). SOD balances ROS by scavenging free radicals and prevents cell damage caused by superoxide anion radicals (18). Moreover, MDA is a widely detected oxidative damage marker and elevated MDA levels are the result of lipid peroxidation (19). SOD levels are negatively correlated with MDA levels and the quantification of SOD and MDA levels can infer the metabolism of free radicals, which therefore reflects the degree of oxidative stress in the cells (32). Subsequently, SOD and MDA levels were determined in HeLa cells in the present study. The results demonstrated that there was a significant decrease in SOD levels in the DOX group compared with an increase of 24.94% in the LPA + DOX group. Moreover, MDA levels were significantly increased in the DOX group, whereas there was a significant decrease of 28.81% in the LPA + DOX group. These results suggested that LPA potentially reduced DOX-induced oxidative stress damage in HeLa cells.
In conclusion, the present study indicated that LPA potentially protected HeLa cells from DOX-induced apoptosis via a reduction in caspase-3 expression levels. Furthermore, LPA was demonstrated to reduce intracellular ROS levels, which were induced by DOX, to prevent HeLa cells from oxygen stress damage. These results indicated that LPA could be a novel therapeutic target in cervical cancer treatment. Based on the finding that LPA potentially protects HeLa cells from DOX-induced apoptosis, further research may be extended to the directions such as discovering an antagonist to LPA receptor, using an animal model to investigate if combined use of an antagonist to LPA receptor and DOX could improve the effect of anticancer drugs, developing LPA receptor inhibitors and targeting LPA as a new strategy for cervical cancer therapy.
Acknowledgements
Not applicable.
Funding Statement
The present study was supported by the National Key Technology R&D Program of the Ministry of Science and Technology (grant no. 2013GA740103), and the project of Shandong Province Higher Educational Science and Technology Program (grant no. J14LK15), the Shandong Medical and Health Science and Technology Development Project (grant no. 2018WS063), the Shandong Provincial Natural Science Foundation of China (grant no. ZR2020KC016) and the Weifang Science and Technology Bureau (grant No. 2020YQFK013).
Availability of data and materials
The datasets used during the present study are available from the corresponding authors upon reasonable request.
Authors' contributions
XW, HW and XM performed the majority of the experiments. YX, WH, AH, YL and HJ performed the cell culture, western blotting and analyzed the data. XW, XY and ZH designed, supervised the study, wrote and finalized the manuscript. XW, XM and XY confirm the authenticity of all the raw data. All authors read and approved the final manuscript and agree to be accountable for all aspects of the research in ensuring that the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Murph MM, Scaccia LA, Volpicelli LA, Radhakrishna H. Agonist-induced endocytosis of lysophosphatidic acid-coupled LPA1/EDG-2 receptors via a dynamin2- and Rab5-dependent pathway. J Cell Sci. 2003;116:1969–1980. doi: 10.1242/jcs.00397. [DOI] [PubMed] [Google Scholar]
- 2.Minis E, Holcomb K, Sisti G, Nasioudis D, Kanninen TT, Athanasiou A, Frey MK, Chapman-Davis E, Caputo TA, Witkin SS. Evaluation of lysophosphatidic acid in vaginal fluid as a biomarker for ovarian cancer: A pilot study. Eur J Obstet Gynecol Reprod Biol X. 2019;2:100012. doi: 10.1016/j.eurox.2019.100012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramachandran S, Ramaswamy S, Cho CH, Parthasarathy S. Lysophosphatidic acid induces glycodelin gene expression in cancer cells. Cancer Lett. 2002;177:197–202. doi: 10.1016/S0304-3835(01)00807-2. [DOI] [PubMed] [Google Scholar]
- 4.Kim NH, Sadra A, Park HY, Oh SM, Chun J, Yoon JK, Huh SO. HeLa E-Box Binding protein, HEB, inhibits promoter activity of the lysophosphatidic acid receptor gene Lpar1 in neocortical neuroblast cells. Mol Cells. 2019;42:123–134. doi: 10.14348/molcells.2018.0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu S, Jiang H, Min L, Ning T, Xu J, Wang T, Wang X, Zhang Q, Cao R, Zhang S, Zhu S. Lysophosphatidic acid mediated PI3K/AKT activation contributed to esophageal squamous cell cancer progression. Carcinogenesis. 2021;42:611–620. doi: 10.1093/carcin/bgaa143. [DOI] [PubMed] [Google Scholar]
- 6.Feng Y, Xiao M, Zhang Z, Cui R, Jiang X, Wang S, Bai H, Liu C, Zhang Z. Potential interaction between lysophosphatidic acid and tumor-associated macrophages in ovarian carcinoma. J Inflamm (Lond) 2020;17:23. doi: 10.1186/s12950-020-00254-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Obol JH, Lin S, Obwolo MJ, Harrison R, Richmond R. Knowledge, attitudes, and practice of cervical cancer prevention among health workers in rural health centres of Northern Uganda. BMC Cancer. 2021;21:110. doi: 10.1186/s12885-021-07847-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sui Y, Yang Y, Wang J, Li Y, Ma H, Cai H, Liu X, Zhang Y, Wang S, Li Z, et al. Lysophosphatidic acid inhibits apoptosis induced by cisplatin in cervical cancer cells. Biomed Res Int. 2015;2015:598386. doi: 10.1155/2015/598386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nie H, Bu F, Xu J, Li T, Huang J. 29 immune-related genes pairs signature predict the prognosis of cervical cancer patients. Sci Rep. 2020;10:14152. doi: 10.1038/s41598-020-70500-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abdoul-Azize S, Buquet C, Li H, Picquenot JM, Vannier JP. Integration of Ca2+ signaling regulates the breast tumor cell response to simvastatin and doxorubicin. Oncogene. 2018;37:4979–4993. doi: 10.1038/s41388-018-0329-6. [DOI] [PubMed] [Google Scholar]
- 11.Wei T, Xiaojun X, Peilong C. Magnoflorine improves sensitivity to doxorubicin (DOX) of breast cancer cells via inducing apoptosis and autophagy through AKT/mTOR and p38 signaling pathways. Biomed Pharmacother. 2020;121:109139. doi: 10.1016/j.biopha.2019.109139. [DOI] [PubMed] [Google Scholar]
- 12.Jawad B, Poudel L, Podgornik R, Steinmetz NF, Ching WY. Molecular mechanism and binding free energy of doxorubicin intercalation in DNA. Phys Chem Chem Phys. 2019;21:3877–3893. doi: 10.1039/C8CP06776G. [DOI] [PubMed] [Google Scholar]
- 13.Xu J, Liu D, Niu H, Zhu G, Xu Y, Ye D, Li J, Zhang Q. Resveratrol reverses Doxorubicin resistance by inhibiting epithelial-mesenchymal transition (EMT) through modulating PTEN/Akt signaling pathway in gastric cancer. J Exp Clin Cancer Res. 2017;36:19. doi: 10.1186/s13046-016-0487-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pilco-Ferreto N, Calaf GM. Influence of doxorubicin on apoptosis and oxidative stress in breast cancer cell lines. Int J Oncol. 2016;49:753–762. doi: 10.3892/ijo.2016.3558. [DOI] [PubMed] [Google Scholar]
- 15.Islam MT. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol Res. 2017;39:73–82. doi: 10.1080/01616412.2016.1251711. [DOI] [PubMed] [Google Scholar]
- 16.Prasad S, Gupta SC, Tyagi AK. Reactive oxygen species (ROS) and cancer: Role of antioxidative nutraceuticals. Cancer Lett. 2017;387:95–105. doi: 10.1016/j.canlet.2016.03.042. [DOI] [PubMed] [Google Scholar]
- 17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 18.Liu R, Gang L, Shen X, Xu H, Wu F, Sheng L. Binding characteristics and superimposed antioxidant properties of caffeine combined with superoxide dismutase. ACS Omega. 2019;4:17417–17424. doi: 10.1021/acsomega.9b02205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saribal D, Hocaoglu-Emre FS, Karaman F, Mırsal H, Akyolcu MC. Trace element levels and oxidant/antioxidant status in patients with alcohol abuse. Biol Trace Elem Res. 2020;193:7–13. doi: 10.1007/s12011-019-01681-y. [DOI] [PubMed] [Google Scholar]
- 20.Xu X, Lai Y, Hua ZC. Apoptosis and apoptotic body: Disease message and therapeutic target potentials. Biosci Rep. 2019;39:BSR20180992. doi: 10.1042/BSR20180992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elmore S. Apoptosis: A review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X, Li Y, Tang X, Shang X, Zhao Z, Jiang Y, Li Y. Stenotrophomonas maltophilia outer membrane protein A induces epithelial cell apoptosis via mitochondrial pathways. J Microbiol. 2020;58:868–877. doi: 10.1007/s12275-020-0235-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang M, Qi L, Li L, Li Y. The caspase-3/GSDME signal pathway as a switch between apoptosis and pyroptosis in cancer. Cell Death Discov. 2020;6:112. doi: 10.1038/s41420-020-00349-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma M, Wang X, Liu N, Shan F, Feng Y. Low-dose naltrexone inhibits colorectal cancer progression and promotes apoptosis by increasing M1-type macrophages and activating the Bax/Bcl-2/caspase-3/PARP pathway. Int Immunopharmacol. 2020;83:106388. doi: 10.1016/j.intimp.2020.106388. [DOI] [PubMed] [Google Scholar]
- 25.Kim JM, Ghosh SR, Weil AC, Zirkin BR. Caspase-3 and caspase-activated deoxyribonuclease are associated with testicular germ cell apoptosis resulting from reduced intratesticular testosterone. Endocrinology. 2001;142:3809–3816. doi: 10.1210/endo.142.9.8375. [DOI] [PubMed] [Google Scholar]
- 26.Zhang C, Feng X, He L, Zhang Y, Shao L. The interrupted effect of autophagic flux and lysosomal function induced by graphene oxide in p62-dependent apoptosis of F98 cells. J Nanobiotechnology. 2020;18:52. doi: 10.1186/s12951-020-00605-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wan T, Wang Z, Luo Y, Zhang Y, He W, Mei Y, Xue J, Li M, Pan H, Li W, et al. FA-97, a New synthetic caffeic acid phenethyl ester derivative, protects against oxidative stress-mediated neuronal cell apoptosis and scopolamine-induced cognitive impairment by activating Nrf2/HO-1 signaling. Oxid Med Cell Longev. 2019;2019:8239642. doi: 10.1155/2019/8239642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nogueira V, Hay N. Molecular pathways: Reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin Cancer Res. 2013;19:4309–4314. doi: 10.1158/1078-0432.CCR-12-1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peng N, Jin L, He A, Deng C, Wang X. Effect of sulphoraphane on newborn mouse cardiomyocytes undergoing ischaemia/reperfusion injury. Pharm Biol. 2019;57:753–759. doi: 10.1080/13880209.2019.1680705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song S, Chu L, Liang H, Chen J, Liang J, Huang Z, Zhang B, Chen X. Protective effects of dioscin against doxorubicin-induced hepatotoxicity via regulation of Sirt1/FOXO1/NF-κb Signal. Front Pharmacol. 2019;10:1030. doi: 10.3389/fphar.2019.01030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rasool M, Malik A, Basit Ashraf MA, Parveen G, Iqbal S, Ali I, Qazi MH, Asif M, Kamran K, Iqbal A, et al. Evaluation of matrix metalloproteinases, cytokines and their potential role in the development of ovarian cancer. PLoS One. 2016;11:e0167149. doi: 10.1371/journal.pone.0167149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wei LF, Zhang HM, Wang SS, Jing JJ, Zheng ZC, Gao JX, Liu Z, Tian J. Changes of MDA and SOD in brain tissue after secondary brain injury with seawater immersion in rats. Turk Neurosurg. 2016;26:384–288. doi: 10.5137/1019-5149.JTN.8265-13.1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used during the present study are available from the corresponding authors upon reasonable request.