

Abstract
Background: The alternative transcriptional isoform of Bruton’s tyrosine kinase, BTK-C, is expressed in a wide variety of epithelial tumor types where it impacts apoptosis resistance, therapeutic escape, and glucose uptake. The initial exon in BTK-C encodes a 34 amino acid extension of the amino terminus of the canonical BTK-A isoform. Its function is unknown.
Materials and Methods: Site-directed mutagenesis, acylation assays and expression studies in cancer cell lines were used to determine the effects that the BTK-C first exon sequence has on kinase activity, subcellular localization and cell physiology. Analysis of BTK-C expression in tumors was conducted using genomic databases.
Results: BTK-C is palmitoylated on two cysteine residues. BTK-C localization at the plasma membrane is dependent upon phosphatidylinositol 3,4,5-triphosphate (PIP3) levels as well as palmitoylation. In epithelial cancer cells, both BTK-A and BTK-C isoforms are recruited to the plasma membrane; however, BTK-A also localizes to the nucleus whereas BTK-C has a primarily perinuclear distribution. Transcription of the BTK-C isoform is inversely correlated with expression of commonly activated breast cancer signaling receptors in breast tumors. In MDA-MB-231 cells, BTK-C expression confers modest increases in proliferation and glucose uptake rates compared to BTK-A.
Conclusion: Palmitoylation affects localization and regulation of BTK-C in epithelial tumor cells where it functions as an important survival factor. Expression of either palmitoylated or non-palmitoylated kinase isoforms that function in PI3K signaling may be a common regulatory feature as nine other soluble kinases in the human genome possess similarly encoded alternative N-termini (ANT).
Keywords: Epithelial cancers, BTK, palmitoylation, PI3K, alternative N-terminus
The phosphatidylinositol 3-kinase (PI3K) pathway is commonly activated in a variety of cancers (1). Increased activity of upstream receptors and mutations in PI3K components lead to production of phosphatidylinositol 3,4,5-triphosphate, PIP3, on the inner leaflet of the plasma membrane. PIP3 levels recruit and activate effectors through interaction with pleckstrin homology (PH) domains in a variety of effector proteins. PIP3 levels are also controlled by the phosphatase and tensin homologue (PTEN) (2). Genomic alterations affect several points in the pathway and are frequently found in most solid tumor types (3-5). Loss of PTEN is the most common alteration, however; between a quarter and a half of tumors of various epithelial origins carry mutations in genes in the pathway (1).
We previously identified an isoform of the PH domain-containing kinase, Bruton’s tyrosine kinase (BTK), in an RNAi kinome screen as a critical survival factor for breast cancer cells (6). ShRNA-mediated knockdown of this BTK isoform (BTK-C; UniProt: Q06187-2, Ensembl: ENST00000621635.4) had a larger effect on reducing survival and proliferation than other targeted kinases with established roles in breast cancer including HER2/neu, EGFR, Src, and others. The BTK-C isoform identified in breast and prostate cancer cells (7) is similar to the original BTK isoform, which we refer to as BTK-A for clarity, but contains an alternative first exon encoding parts of a 34 amino acid amino-terminal extension (6). The BTK-C protein provides cell survival activities in breast and prostate cancer cells (8) whereas BTK-A is expressed in hematopoietic cells (9) during maturation of B-cells (10-12). In hematopoietic cells, B-cell receptor engagement by antigen stimulates PI3K and the resulting PIP3 accumulation at the plasma membrane recruits and activates BTK-A. The importance of BTK-A to B cell survival is underscored by the development BTK inhibitors such as ibrutinib, acalabrutinib and others (13). These BTK inhibitors also target BTK-C decreasing tumor cell proliferation by reducing resistance to apoptosis which ultimately may be due to effects on glucose transport (6). Decreased survival signaling caused by BTK-C inhibition also results in decreased therapeutic escape in breast cancer cells suggesting that it may be useful as an adjuvant therapy (6,7). Importantly, inhibition of BTK-C prevents activation of the AKT signaling pathway by NRG or EGF that has been shown to promote growth factor-driven lapatinib resistance in HER2+ breast cancer cells (14-17). BTK-C signaling is involved in the appearance of ligand-dependent lapatinib resistance in treated HER2-positive breast cancer cell populations.
In this study, we show that the N-terminal extension of the BTK-C isoform contains a palmitoylation sequence which alters its subcellular localization. The palmitoylated form of BTK is expressed in 10-25% of a wide variety of cancers (7). In breast tumors that do not express BTK-C, increased levels of other signaling inputs are commonly found. In this sense, the BTK-C isoform may function in cancer cells by providing increased effector function in the PI3K pathway. The unique structure of the BTK gene allows for both palmitoylated and non-palmitoylated forms of the kinase to be produced. This appears to be a common feature of mammals as we find similar exon arrangements in other tyrosine kinases and pleckstrin homology-containing kinase genes.
Materials and Methods
Tissue culture. Cell lines MD-AMB-231, BT549, SUM149, SKBr3 and ΦNX-AMPHO were obtained from the American Type Culture Collection (ATCC). MD-AMB-231, SUM149, SKBr3, and Phoenix AMPHO cells were maintained in DMEM (Hyclone, Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS) (Hyclone). BT549 and LNCaP C4-2b cells were cultured in RPMI (Hyclone) with 10% FBS (Hyclone). All culture media contained 100 U/μl penicillin-streptomycin. All cell lines were authenticated in March 2016 by the SUNY-Albany Center for Functional Genomics Molecular Core Facility using a short tandem repeat method (Promega GenePrint 10 system, Madison, WI, USA). To minimize the influence of components contributed by bovine serum, all stimulation experiments were performed using 1% FBS unless otherwise indicated (18,19). Cells were imaged as described. As localization was the key attribute being tested, images were normalized with respect to brightness and contrast. In growth and uptake experiments, differences between control and treatment conditions were evaluated using Student’s t-test for pairwise comparisons. Growth and glucose experiments were repeated 3 times. The average of at least three biological replicates were normalized to control and presented as mean±standard deviation.
Reporter construction and viral transgenesis. GFP fusion constructs were generated using a BTK pleckstrin homology domain fusion protein vector, PH-Btk-GFP (Addgene Plasmid #51463). BTK constructs (BTK-A R28C; BTK-C; BTK-C R28C; BTK-C C13A, C16A; BTK-C C13A, C16A, R28C) were generated in this plasmid using QuikChange site directed mutagenesis. The exon1C sequence was subcloned from the original cDNA isolate (20). To prevent internal translational initiation at the BTK-A ATG (6), this codon was changed to Ala in all BTK-C constructs. Production of either the 77 kDa BTK-A isoform or the 65 kDa p65BTK (21) was not observed for BTK-C constructs. GFP reporters were subcloned into MARXIVpuro REV plasmid (22). Full-length mutant proteins were produced by subcloning into a BTK gene with a C-terminal Flag tag expressed from a MARXIVpuro REV vector. DNA sequence was confirmed by DNA sequence analysis for all constructs. ΦNX-Ampho cells were transfected with Flag-tagged versions of BTK as described (23). Empty vector was used as a control. A lentivirus-born shRNA targeting the chromosomal BTK 3’UTR TRCN0000000358 (Sigma-Aldrich, Saint Louis, MO, USA) was used to silence endogenous BTK isoform expression such that each line predominantly expresses the wild-type or mutant version of the intended isoform.
BTK acylation. HEK293 cells plated on 6 wells plates were transfected using X-tremeGENE HP transfection reagent (Sigma-Aldrich) with 1 μg of BTK-C, BTK-A, or BTK-C C13A, C16A with 2 μg of zDHHC5 plasmid. Twenty-four hours after transfection, cells were incubated with 100 μM of palmitic acid alkyne (in DMEM with 1mg/ml defatted BSA) for 4h at 37˚C. Cells were washed twice in PBS, then lysed on ice in 50 mM Tris HCl, pH 7.4 with 150 mM NaCl, 1 mM EDTA, 1% TRITON X-100 and protein inhibitors. Cell lysate supernatants were used for immunoprecipitation with Anti-Flag M2 affinity gel (Sigma-Aldrich) at 4˚C overnight. The affinity gel was washed with 0.5 ml of TBS three times and incubated with click chemistry reaction mixture (Click-&-Go Click Chemistry Reaction Buffer Kit, Click Chemistry Tools, Scottsdale, AZ, USA) containing 20 μM biotin picolyl azide for 30 min at room temperature, separated by SDS PAGE and blotted.
Protein methods. Subcellular fractionation was performed using standard methods as described (24). Immunoblots were completed following standard protocols. Primary antibodies used were: P-Btk (Y223) (Cell Signaling Inc., Danvers, MA; #5082S), Flag Tag DYKDDDDK (D6W58) (Cell Signaling Inc., #14793S), Purified mouse anti-Btk (pY551)/Itk (pY511) (BD Biosciences, San Jose, CA, USA; #558034), Histone H3 (Cell Signaling Inc., #9715S), Na/K ATPase (Epitomics, Burlingame, CA, USA; #2047-X). Secondary antibodies: Anti-rabbit IgG, HRP-linked Antibody (Cell Signaling Inc., #7074S), Anti-mouse IgG, HRP-linked Antibody (Cell Signaling Inc., #7076S).
Results
The cancer cell survival BTK-C isoform is phylogenetically conserved. Our previous studies have shown that the BTK-C isoform, which has a transcriptional initiation site approximately 10 kb upstream of the BTK-A isoform (Figure 1A), is the predominant form expressed in solid tumor cells in humans (6-8). Analysis of recent RNAseq data from the Genotype-Tissue Expression (GTEx) project confirms previous results indicating that the BTK-C isoform is expressed in relatively few tissues and at low levels under normal conditions (Supplemental Figure 1). GTEx data show that the greatest expression of BTK-C is in testis, with lower levels of expression in both spleen and EBV-transformed lymphocytes. There is essentially no expression in other tissues.
Figure 1. Overview of the BTK locus in humans. (A) BTK-C transcription is initiated at an alternative transcriptional start site. Encoded sequence in this exon is phylogenetically conserved in mammals. (B) The key feature encoded in this sequence is one or more predicted palmitoylation sites. (C) The BTK-C isoform is expressed in approximately 15% of breast cancer tumors. BTK-C expression is not correlated with sample type. (D) Volcano plot of reverse-phase protein array data of BTK-C -expressing and -non-expressing tumors from the TCGA Firehose Legacy dataset. Only those protein species which have statistically significant (p<0.05 and q<0.05) differences in abundance between the two groups are labeled. Human breast tumors that do not express BTK-C are enriched in common breast cancer signaling inputs including ERBB2, the estrogen receptor, androgen receptor and the PIP3-dependent serine threonine kinase, PDPK1.

An analysis of several species’ genomes indicates that the exon 1C sequence is conserved in mammals [Figure 1B and (7)]. The 20 organisms in both primate and vertebrate list of the UCSC Genome Browser (25) all display sequence conservation within this region. In each case, this includes encoded amino acid sequences of exon 1C as well as encoded amino acid sequences in exon 2 that are upstream of the start codon of the BTK-A isoform. In each organism, transcription initiation occurs divergently within a few hundred bases of the RPL36 ribosomal protein gene. The range of additional protein sequence is between 32 and 34 amino acids and is predicted to be unstructured. Importantly, analysis of the protein motifs found in these conserved sequences indicates that each contains potential palmitoylation sites (Figure 1B). One or two cysteine residues are found in each exon 1C encoded amino acid sequence which score highly in the CSS-Palm palmitoylation motif prediction algorithm (26). Of note, some rodent sequences including mouse and rat share conserved sequences in the appropriate positions, however, a stop codon is found at the corresponding fourth amino acid of BTK-C and transcripts that map to this area are not annotated in mouse genome sequences. We have isolated the mouse sequence by PCR and confirmed the presence of the stop codon at this position.
BTK-C expression in breast tumors. Analysis of expression of the BTK splice isoforms in tumor tissues in the TCGA database using the Tumor Splice Variant Database (TSV DB) (27) indicates that virtually all tumor samples show expression of some BTK isoform within the tumor (Figure 1C). BTK-A is widely expressed in the samples, although it is difficult to assess whether this is due to the presence of B-cell infiltrates in these tumors as BTK-A is expressed in hematopoietic cells at levels approximately 1,000 times higher than those in solid tumor cells [(6,7) and not shown]. BTK-C expression is observed in 5-25% of tumors in a wide variety of cancers. This includes 14.4% (158/1,100) of breast tumors, 12.5% (51/408) of bladder tumors, 23.5% (118/501) of lung squamous cell carcinomas and 5.8% (29/495) of prostate tumors.
Tumor samples from The Cancer Genome Atlas (TCGA) Firehose Legacy breast cancer database were parsed into two groups: tumors that expressed BTK-C (RNAseq reads mapped to exon1C >0) and those that do not express BTK-C (RNAseq reads mapped to exon1C=0). Analysis of attributes of the two groups indicated that tumors expressing BTK-C were more likely to be either ER or PR negative (Supplementary Figure 2). There was, however, no statistically significant difference in overall survival between the groups. Analysis of transcripts differentially expressed in BTK-C expressing tumors are enriched in function-appropriate gene ontology biological process groups of ER sequestered calcium release, regulation of Toll-like receptor 2 signaling pathway and leukocyte aggregation. When compared to non-expressers, tumors expressing BTK-C show a statistically significant increase in a single protein species as measured by reverse-phase protein array: increased phosphorylation on threonine 346 of the hypoxia-induced, lipid metabolism gene, NDRG1(28-30) (Figure 1D). Tumors that do not express BTK-C share clinical attributes having greater ER positivity or PR positivity which is also reflected in both transcriptomic and proteomic analysis. The top scoring gene ontology class of messages enriched in tumors not expressing BTK-C was “breast development and signaling” which includes transcripts for the estrogen receptor (ESR1), androgen receptor (AR), progesterone receptor (PGR) and the EGF family member ERBB4, among others. Many of these proteins were also found to exhibit statistically significant higher levels of expression in proteomic analysis of tumors not expressing BTK-C including the estrogen receptor, the androgen receptor, and ERBB2 among others (Figure 1D). Although expression of BTK-C does not track with a specific breast cancer subtype, taken together, the results indicate that tumors expressing BTK-C are likely to have reduced levels of canonical breast cancer signaling receptor proteins.
Figure 2. The BTK-C isoform is palmitoylated. (A) Diagram of BTK isoform C-terminal Flag-tagged constructs. (B) Expression of Flag-tagged constructs in MDA-MB-231 cells. (C) Detection scheme for acylated BTK isoforms. Cells were incubated with 17-ODYA. Flag immunoprecipitates were reacted with azide-activated biotinylation reagent and detected through reaction with streptavidin-linked horseradish peroxidase. (D) HEK293 cells were transfected with 1μg of BTK-A, BTK-C, or BTK-C C13A,C16A and 2 μg of a plasmid expressing zDHHC5. 24h after transfection cells were incubated for 4 h with 100 μM palmitic acid alkyne (17-ODYA). Cell lysate supernatants were immunoprecipitated with Anti-Flag M2 affinity gel (Sigma) at 4˚C overnight, subjected to click chemistry with 20 μM biotin picolyl azide for 30 m at room temperature before electrophoresis and blotting.

The BTK-C isoform but not BTK-A is palmitoylated. Analysis of the predicted BTK-C sequence with motif identification algorithms indicated that cysteine residues 13 and 16 in the BTK-C 34 amino acid N-terminal extension are highly scoring predicted Type II palmitoylation residues (26) (Figure 1B). Conserved motifs for N-glycosylation or N-myristoylation were not found. Palmitoylation is a dynamic post-translational modification of proteins in which palmitate is covalently bonded to cysteine residues of the mature protein. The linkage occurs through a reversible thioester bond catalyzed by any of a large family of protein acyl transferases the substrate specificities of which are at present poorly understood (31). Palmitoylation was originally thought to be a mechanism for targeting to lipid rafts in the plasma membrane. It is now more generally considered to play an important role in subcellular trafficking of substrates many of which function in signal transduction (32-34). So that the functional significance of the predicted palmitoylation sites at Cys13 and Cys16 within the N terminal domain of human BTK-C could be determined, these sites were mutated to alanine residues using in vitro mutagenesis (Figure 2A).
To determine whether the predicted palmitoylation sites of BTK-C are acylated, Flag-tagged versions of BTK-C wt, BTK-A wt, or BTK-C:C13A,C16A were transfected (1 μg each) and expressed in HEK293 cells (Figure 2B) with 2 μg of a plasmid expressing the plasma membrane-localized palmitoyltransferase, zDHHC5 (35). The cells were subjected to acyl incorporation assays (Figure 2C) in which modification with an acyl analogue is evidenced using click chemistry modification after FLAG based immunoprecipitation (36). Analogues of palmitic acid, 17-octadecynoic acid (17-ODYA, 13266 Cayman Chemical, Ann Arbor, MI, USA), palmitoleic acid (25362 Cayman Chemical), oleic acid (9002078 Cayman Chemical) and arachidonic acid (10538 Cayman Chemical) were tested for incorporation. As shown in Figure 2D, label is increased in BTK-C but not BTK-A, or the BTK-C C13A, C16A double mutant. We failed to observe incorporation of palmitoleic acid, oleic acid and arachidonic acid (not shown). We were also unable to see incorporation without co-expression of the palmitoyl transferase; nevertheless, these experiments confirm that the N terminal domain of BTK-C is a substrate for palmitoylation.
Palmitoylation affects BTK-C cellular localization. BTK is established as soluble kinase that is recruited to the membrane by PI3K activity (9,37-39). Fusions of the pleckstrin homology domain of BTK (amino acids 1-177) with GFP are widely used to monitor PIP3 levels in cells (20). We modified this construct to incorporate the exon 1C sequence of BTK-C and several site-directed mutations (Figure 3A). Each construct was subcloned into a PuroMaRX IV-based retrovirus and transduced into a variety of cell lines. Mutation of either cysteine residue by itself in the exon 1C sequence is largely without effect on subcellular localization of BTK-C (Supplementary Figure 3). Mutation of both cysteine 13 and cysteine 16, however, caused the reporter construct to localize in the cell interior (Figure 3B).
Figure 3. Subcellular distribution of the BTK isoforms in breast cancer cells. (A) Diagram of BTK pleckstrin homology domain with GFP reporters. Expression of each reporter was driven by a CMV promoter in a retroviral vector. (B) Localization of wild type and mutant isoform reporters in breast cancer cell lines MD-AMB-231 (triple negative; PTEN+), BT549 (triple negative; PTEN–), SUM149 (triple negative; PTEN–) and SKBr3 (luminal HER2 enriched; PTEN+). BTK-A isoform is found in the plasma membrane (red arrows) and to a greater degree in the nuclei of cancer cells (white arrows). BTK-C is also found on the plasma membrane but exhibits a greater degree of perinuclear localization (gray arrows). Mutation of the two palmitoylation sites reduces membrane localization of BTK-C. Mutation of arginine 28 of BTK (arginine 62 and BTK-C sequence) also decreases membrane staining. (C) Subcellular fractionation showing increased levels of BTK-A in the nucleus and greater BTK-C in membrane fractions. (D) BTK reporters localize with actin in cancer cells. Scale bars: 100 μm.

Both BTK-A GFP and BTK-C GFP fusions exhibit significant plasma membrane localization; however, images focused on the mid-depth of cancer cells show that the BTK-A isoform is largely localized within the nucleus whereas the BTK-C isoform has a primarily perinuclear distribution (Figure 3B). The nuclear localization of BTK-A in solid tumor cells is not surprising. The BTK-A isoform has been shown to shuttle between the cytoplasm and nucleus in B cells. During this process, export from the nucleus requires association with the LIAR protein (40). This interaction occurs within the SH3 domain which is missing in these pleckstrin homology domain reporters but is retained in the full-length constructs. Analysis of BTK-GFP fusion signal in the cells with CellProfiler image quantification analysis (41) showed that approximately 60% more BTK-A signal is localized to the nucleus compared to BTK-C. Fractionation of cells expressing Flag-tagged, full-length versions of BTK-A and BTK-C and subsequent immunoblotting of nuclear and cellular membranes shows that BTK-C is more abundant in cellular membranes compared to BTK-A which is found in higher amounts in the nucleus (Figure 3C).
Mutations in the palmitoylation sequences at the cysteine residues in BTK-C (C13A, C16A) substantially decreased plasma membrane localization of BTK-C-GFP. Additionally, mutation of the amino acid corresponding to arginine 28 of the BTK-A sequence to cysteine as occurs in mice (42) (37,43) also abolished membrane localization. This position in the pleckstrin homology domain interacts with PIP3 during activation. The same R28C mutation in BTK-C causes it to fail to localize to the membrane indicating that BTK-C requires both palmitoylation and PI3K signaling to associate with the membrane. A common downstream effect of BTK activation due to B cell receptor engagement is actin polymerization. We observed that considerable BTK-A and BTK-C reporter localization is juxtaposed to actin filaments in the breast cells (Figure 3D).
Regulation of BTK-C localization and activity by PIP3 levels. Activation of BTK-A in B cells has been extensively studied. Adsorption of BTK-A to the B cell plasma membrane is driven by binding to PIP3 which opens the BTK monomer conformation allowing dimerization, auto-activation (Y223 phosphorylation) and transactivation (Y551 phosphorylation) (9,37-39). Results from several lines of experimentation indicate that the additional sequence at the BTK-C amino terminus does not alter the regulation of the kinase by PIP3. The positively charged R28 residue in the PH domain has been shown to contact the 5’-PO4 of PIP3 (37,43) and mutation of this residue makes the kinase insensitive to PI3K signaling (42). Mutation of the analogous arginine in BTK-C (R62 in BTK-C) causes it to fail to localize to the membrane (Supplementary Figure 3). Both BTK-A and BTK-C isoforms exhibit significant localization in the plasma membrane of cells that harbor mutations in PTEN, the phosphatase that removes the D3 phosphate and reduces PIP3 levels (Figure 4A and B). For example, MDA-MB-231 cells which are PTEN+ display less plasma membrane staining when compared to SUM149 cells which are PTEN–. In these cells, BTK-A also shows increased plasma membrane localization but to a lesser degree than BTK-C. Since there are a limited number of PTEN– cell lines, we also used pharmacological inhibition with IPI-145 (Selleckchem, Houston, TX, USA) (44), Duvelisib, a highly selective PI3K δ/γ inhibitor that decreases the membrane signal of both BTK-A and BTK-C GFP fusions. In breast cells, this is accompanied by significant toxicity (not shown). However, there is less toxicity in the PTEN– LNCaP C42b prostate cancer cell line that expresses BTK-C, where IPI-145 inhibition dramatically decreases the degree of membrane localization observed in controls (Figure 4B). Similarly, expressing wild-type PTEN in the PTEN– LNCaP C42b prostate cancer cell line decreases BTK-C activation whereas expressing dominant negative mutants (45) does not (Figure 4D). This PIP3-dependent activation may occur through BTK-C dimer formation through juxtaposed membrane-bound PH domains as occurs with the BTK-A (39). Using an assay developed by Chung et al. (39), co-transfecting BTK-C with the BTK-C-GFP construct which contains the PH domain but is missing the kinase domain reduces Y551 phosphorylation compared with vector or BTK-C only (Figure 4D). This result is consistent with its interfering with dimer formation as has been shown for similar BTK kinase domain constructs in other transfection-based studies (39).
Figure 4. BTK isoform localization and activation is responsive to PI3K pathway signaling in solid tumor cells. (A) BTK isoform localization in MDA-MB-231 (PTEN+) and SUM149 (PTEN–) cells. (B) Pharmacological inhibition of PI3K decreases association of BTK-A and BTK-C with the plasma membrane in LNCaP C4-2b (PTEN–) cells. Scale bar: 100 μm. (C) Activating tyrosine phosphorylation of the BTK isoforms in transiently transfected HEK293 cells. Y551 phosphorylation is dependent on other kinases, Y223 autophosphorylation results from BTK activation. Values represent fold tyrosine-phosphorylated signal normalized to total transfected BTK control (anti-Flag) determined by densitometry. (D) PIP3 dependence of BTK-C activation in transfected LNCaP C4-2b (PTEN–) cells. PTEN activity by expression of PTEN dominant negative mutants increases BTK-C activation. Additionally, a BTK-C non-kinase domain construct (PH-EGFP) capable of dimerizing with full length BTK reduces activating phosphorylation. In each case, 24 hours after transfection cell lysates were collected for immunoblotting.
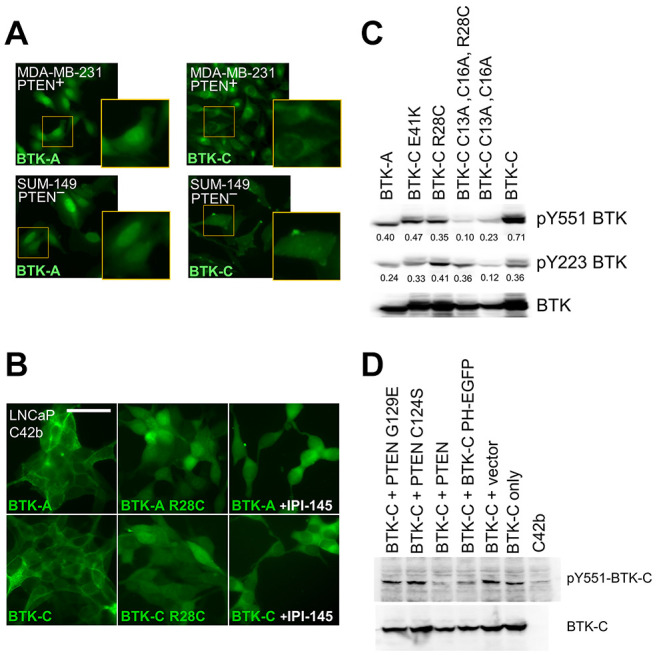
We assessed the in vivo activity of the BTK isoforms by co-transfecting wild-type and mutant constructs into HEK293 cells (Figure 2 and Supplementary Figure 4). BTK-A and BTK-C are both activated in the cells as judged by tyrosine phosphorylation at Y223 and Y551. As shown in Figure 4C, BTK-C: R28C (50% BTK-C wt), BTK-C: C13A, C16A (33% BTK-C wt) and BTK-C: C13A, C16A, R28C (14% BTK-C wt) proteins exhibit lower Y551 phosphorylation levels suggesting that BTK-C activation by other activating kinases may depend on its correct localization which requires both palmitoylation and PIP3 binding ability (9). BTK transactivation was less impacted by these mutations with only the BTK-C: C13A, C16A (33% BTK-C wt). Expression of BTK-C with a mutation analogous to BTK-A E41K (E75K in BTK-C numbering) which increases the positive charge in the PH domain and has been shown to increase membrane recruitment and activation (42), does not exhibit appreciably increased activation in these cells. PLCγ2, an established BTK effector in hematopoietic cells, is expressed in a variety of epithelial cancer cells (46). As shown in Figure 5B, overexpression of either isoform increases phosphorylation of PLCγ2 on tyrosine 759. Interestingly, mutation of only the palmitoylation residues does not impact PLCγ2 phosphorylation appreciably, although mutation of both palmitoylation and PIPs binding site does.
Figure 5. (A) Activating tyrosine phosphorylation of the BTK isoforms. BTK-A and BTK-C constructs were transfected into HEK293 cell and after 24 hours stimulated with insulin. Values represent fold tyrosine-phosphorylated signal normalized to total BTK-A or BTK-C (anti-Flag) determined by densitometry. (B) Phosphorylation of the BTK target tyrosine 759 of PLCγ2 in transfected HEK293 cells. (C) BTK-C expression increases proliferation rate of MDA-MB-231 (PTEN+) but not SUM149 (PTEN-) cells. Cells were seeded in 96 well plates and fixed with 4% formaldehyde and counted at 72 hours. Data were normalized to control and are presented as mean±SD. *p<0.05 using Student’s t-test, n=3. (D) BTK-C increases glucose uptake in MDA-MB-231 (PTEN+) but not SUM149 (PTEN-) cells. Cells were seeded and treated with 100 μM 2-NBDG for 15 min. Fluorescence images were acquired with an InCell 2200 and overall fluorescence intensity quantified. Data were normalized to control and are presented as mean±SD. *p<0.05 using Student’s t-test, n=3.

BTK-C activity in cancer cells in vitro. Previous studies have shown that both BTK-A and BTK-C are active in proliferating breast and prostate cancer lines under normal conditions in vitro (6,8). To determine whether the two isoforms of BTK responded differently to stimulation, we examined activation in response to insulin stimulation under reduced serum conditions. Insulin is a common activator of PI3K in cancer cells (47) and has been shown to cause activation of BTK in other cell types (48). Insulin stimulation increases Y551 phosphorylation, reflecting activation by other kinases, two-fold in BTK-A but considerably less (~24%) in BTK-C. Transactivation, reflecting phosphorylation of BTK by its dimerized partner, is largely unchanged in BTK-A and BTK-C under these conditions (Figure 5A). Other ligands including EGF, NRG and RANKL have a similar impact (not shown) increasing activating phosphorylation levels above unstimulated levels only modestly. These results are in accord with the original observations that the kinase is active in proliferating cancer cells (6,8) and suggests that further work is needed to identify the pathway or pathways responsible for activation in a given cell type. Nevertheless, even without exogenous stimulation, the baseline BTK activity has functional consequences that correlate with the palmitoylation status of BTK in breast cancer cells. We observe a statistically significant increase in proliferation, which was reproduced in multiple experiments, of 17.6% in PTEN+ MDA-MB-231 cells expressing BTK-C (+ endogenous BTK-targeting shRNA) compared to BTK-A (+ endogenous BTK-targeting shRNA) (Figure 5C). Effects of the expression of the BTK-C C13A, C16A double mutant does not differ appreciably from the BTK-A isoform (data not shown). Cell proliferation rates of PTEN– SUM149 cells exhibited similar rates of growth irrespective of the isoform expressed. Since our previous work has shown that BTK-C activity is correlated with glucose uptake in breast cancer cells (6), both lines expressing full length BTK-A and BTK-C wild-type constructs were tested for effects on glucose uptake by assaying 2-NBDG fluorescence. The results show that in MDA-MB-231 cells, there is a reproducible 25% increase in glucose uptake in those cells expressing BTK-C when compared to BTK-A (Figure 5D). Similar to the impacts on proliferation, there are no significant differences in glucose uptake activity between BTK-A and BTK-C constructs in PTEN– SUM149 cells. We surmise that the lack of significant differences between the two isoforms when expressed in the PTEN– cell is due to constitutive activation of the non-palmitoylated form.
PI3K effector kinase isoform generation through alternative start site selection. Results of these experiments on BTK-C indicate that an alternative amino terminus can impact kinase localization and function. BTK is one of 5 TEC family kinases encoded in the human genome (49). Although the TXK kinase does not contain a bona fide PH domain, it is the most highly related TEC kinase to BTK (50). Previous studies have established that the 5’ end of the TXK mRNA possesses two alternative translational start sites, one of which encodes a run of cysteine residues that is palmitoylated and produces a predominantly perinuclear localized product (50). Each of the other TEC kinases has a single 5’ end version and none are predicted to be palmitoylated.
To extend the observations on the TEC kinases, we determined how many kinases in the human genome transducing signals at the plasma membrane, have the potential to encode either palmitoylated or non-palmitoylated versions. An analysis of all soluble, non-receptor tyrosine kinases and all PH domain-containing serine/threonine kinases (Supplementary Figure 5) showed that 4 others have alternative first exons: FRK, ABL1, ABL2 and PDPK1 (Figure 6A); these encode either palmitoylated or non-palmitoylated amino termini analogous to BTK-A and BTK-C (Figure 6B). Three other kinases (JAK1, JAK2 and UFO) are similar to TXK (50), having the potential for either palmitoylated or non-palmitoylated amino termini determined by translational initiation site choice (Supplementary Figure 5). Additionally, HCK, whose expression involves both alternative transcriptional and translational start sites and myristoylation, has previously been demonstrated to have both palmitoylated and non-palmitoylated versions (Figure 6B) (51,52). Analysis of coding regions for these 10 kinases shows that those isoforms with confirmed or predicted palmitoylation sites are enriched in cysteine residues near the amino terminus. Those with the potential for alternative translational start sites are also enriched in methionine, with most having at least one methionine occurring after the palmitoylation site, as occurs in BTK and TXK to produce a non-palmitoylated version. The predicted and established non-palmitoylated isoform sequences of these kinases are depleted of both these amino acids over the first 50 amino acids (Figure 6B and Supplementary Figure 5). In this way, amino terminus choice appears to be a binary switch that impacts the localization and activity of these kinases. These kinases, which we classify as alternative N-terminal (ANT) kinases, include several tyrosine kinases as well as PDPK1, a major PI3K-effector serine threonine kinase (53). Determining the function of these altered isoforms will require considerable additional investigation. However, it underscores the notion that alternative amino termini, as occurs with BTK, is a common feature of kinases with important roles in cancer cell signaling.
Figure 6. Alternative N-terminal kinases in the human genome. (A) Exon maps of kinases with alternative first exons that either do or do not contain palmitoylation sites, as occurs with BTK-A and BTK-C. (B) Alternate N terminal sequences of tyrosine kinases TXK, FRK, HCK, ABL1, ABL2, and the serine/threonine kinase PDPK1 resemble BTK. Isoforms containing palmitoylation sequences in the amino terminal region are enriched in cysteine and methionine residues in predicted protein products. The encoded alternative isoforms represent a binary switch as non-palmitoylated forms are depleted of cysteine and non-start codon methionine residues near the N terminus.

Discussion
Our previous findings showed that the BTK-C isoform is expressed in human breast and prostate cancer cells and that it plays a crucial role in epithelial cancer cell survival. In this study, we find that the 34 amino acid extension of BTK-C contains a phylogenetically conserved palmitoylation site that localizes the kinase to membranes and has context-dependent functional consequences. Although originally identified as the predominant isoform expressed in solid tumor cells, this work shows that BTK-C is not expressed at significant levels in most normal tissues or cells. Its expression profile and its role as a critical survival factor for breast and other solid tumor cells makes BTK-C a specific, druggable target for treating a wide range of epithelial cancers.
We observed that the BTK-C isoform is palmitoylated but only when the plasma membrane-localized palmitoyltransferase zDHHC5 was also overexpressed. This reaction appears to be inefficient in these experiments (Figure 2) which might reflect another, non-tested acyl group as the primary possible modification. Nevertheless, mutation of the acylated cysteines C13 and C16 had obvious effects on the subcellular localization of the BTK kinase in cells. BTK-C displays significant perinuclear localization as occurs with several palmitoylated family kinases, such as SRC, LYN and YES (54), as well as the TEC family TXK. Although palmitoylation is important to the activity of these kinases, the impact that palmitoylation has on the dynamics of signaling is poorly understood in these instances and complicated somewhat by the fact that these enzymes are also myristoylated. Association of BTK-C with the membrane required either C13 or C16 and a functional PH domain for membrane association and impacted kinase activation (Figure 5). These results may indicate that BTK-C has a somewhat different mechanism of activation from BTK-A which is activated at the plasma membrane through PIP3 interaction alone (37,43).
Both the palmitoylated and non-palmitoylated isoforms responded to PI3K signaling in increased plasma membrane association. However, molecular mechanisms responsible for phosphorylation-based activation of the kinases (Figure 4C and Figure 5A) in solid tumor cells may be less straightforward than those involved in activation by B cell receptor signaling. Expression of full-length BTK-C improved glucose uptake and proliferation of triple negative MDA-MB-231 cells but did not have a significant effect on PTEN– SUM149 cells. We surmise that increased localization of BTK-A in the PTEN– SUM149 cells reduces the difference in activity of the isoforms. In these cells and in general, the lack of dramatic differences between the isoforms may reflect the fact that the palmitoylated isoform exists primarily as a targeting mechanism in cells that have larger surface areas, receptor repertoires, and cytoplasmic volumes as well as more developed endomembrane systems. B cells, which express the BTK-A isoform, are significantly different from cells of epithelial origin that express BTK-C in these respects (55-57).
Finally, the arrangement of the BTK gene with alternative 5’ exons encoding alternative amino termini, appears to be an underappreciated paradigm in soluble kinases in mammals. 10 kinases possess encoded, phylogenetically conserved, alternative N termini with potential impacts on tumor biology and progression. For example, BTK-C expression in breast tumors is correlated with lower protein expression of common breast cancer signaling receptors, consistent with the notion that expression of BTK-C or other palmitoylated kinases in solid tumors increases the level of PI3K effector activity irrespective of signaling inputs. There are other possible interpretations and although additional experimentation is required, this explanation would fit with the observed effects of BTK-C expression in solid tumors including increased survival, apoptosis resistance, and chemotherapeutic escape (6-8).
Supplementary Material
Supplementary Figures and additional information are available at the following website: http://www.albany.edu/cancergenomics/faculty/dconklin/datasets.html
Conflicts of Interest
Some of the work in this manuscript has been used to support one or more patent applications owned by the Research Foundation for the State University of New York and is related to the current interests of Cancer Molecular DesignWorks, LLC in which DC and XW have financial interests.
Authors’ Contributions
MK and XW performed tissue culture, imaging and molecular signaling experiments. DSC performed the bioinformatics data mining. EV and FK engineered mutant BTK constructs. EA and LK performed additional tissue culture based experiments. SD confirmed the sequence of the mouse BTK-C exon 1. DSC, MK and XW conceived and designed the study, interpreted the data and drafted the manuscript.
Acknowledgements
We thank members of the Conklin lab for help with experimentation and critical reading of the manuscript and Dr. Maurine Linder for helpful discussions. The plasmid expressing zDHHC5 was a gift of Dr Masaki Fukata, National Institutes of Natural Sciences, Aichi, Japan. The results shown here are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. This work was supported by NCI R01CA136658 and a Technology Accelerator Fund Award 69344 from The Research Foundation for the State University of New York to DSC and a grant from the Capital Region Medical Research Institute to XW.
References
- 1.Millis SZ, Ikeda S, Reddy S, Gatalica Z, Kurzrock R. Landscape of phosphatidylinositol-3-kinase pathway alterations across 19 784 diverse solid tumors. JAMA Oncol. 2016;2(12):1565–1573. doi: 10.1001/jamaoncol.2016.0891. [DOI] [PubMed] [Google Scholar]
- 2.Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer. 2011;11(4):289–301. doi: 10.1038/nrc3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levine DA, Bogomolniy F, Yee CJ, Lash A, Barakat RR, Borgen PI, Boyd J. Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clin Cancer Res. 2005;11(8):2875–2878. doi: 10.1158/1078-0432.CCR-04-2142. [DOI] [PubMed] [Google Scholar]
- 4.Lee JW, Soung YH, Kim SY, Lee HW, Park WS, Nam SW, Kim SH, Lee JY, Yoo NJ, Lee SH. PIK3CA gene is frequently mutated in breast carcinomas and hepatocellular carcinomas. Oncogene. 2005;24(8):1477–1480. doi: 10.1038/sj.onc.1208304. [DOI] [PubMed] [Google Scholar]
- 5.Wu G, Xing M, Mambo E, Huang X, Liu J, Guo Z, Chatterjee A, Goldenberg D, Gollin SM, Sukumar S, Trink B, Sidransky D. Somatic mutation and gain of copy number of PIK3CA in human breast cancer. Breast Cancer Res. 2005;7(5):R609–R616. doi: 10.1186/bcr1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eifert C, Wang X, Kokabee L, Kourtidis A, Jain R, Gerdes MJ, Conklin DS. A novel isoform of the B cell tyrosine kinase BTK protects breast cancer cells from apoptosis. Genes Chromosomes Cancer. 2013;52(10):961–975. doi: 10.1002/gcc.22091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang X, Kokabee L, Kokabee M, Conklin DS. Bruton’s tyrosine kinase and its isoforms in cancer. Front Cell Dev Biol. 2021;9:668996. doi: 10.3389/fcell.2021.668996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kokabee L, Wang X, Sevinsky CJ, Wang WL, Cheu L, Chittur SV, Karimipoor M, Tenniswood M, Conklin DS. Bruton’s tyrosine kinase is a potential therapeutic target in prostate cancer. Cancer Biol Ther. 2015;16(11):1604–1615. doi: 10.1080/15384047.2015.1078023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mohamed AJ, Yu L, Bäckesjö CM, Vargas L, Faryal R, Aints A, Christensson B, Berglöf A, Vihinen M, Nore BF, Smith CI. Bruton’s tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol Rev. 2009;228(1):58–73. doi: 10.1111/j.1600-065X.2008.00741.x. [DOI] [PubMed] [Google Scholar]
- 10.Aoki Y, Isselbacher KJ, Pillai S. Bruton tyrosine kinase is tyrosine phosphorylated and activated in pre-B lymphocytes and receptor-ligated B cells. Proc Natl Acad Sci USA. 1994;91(22):10606–10609. doi: 10.1073/pnas.91.22.10606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawakami Y, Kitaura J, Hata D, Yao L, Kawakami T. Functions of Bruton’s tyrosine kinase in mast and B cells. J Leukoc Biol. 1999;65(3):286–290. doi: 10.1002/jlb.65.3.286. [DOI] [PubMed] [Google Scholar]
- 12.Khan WN, Alt FW, Gerstein RM, Malynn BA, Larsson I, Rathbun G, Davidson L, Müller S, Kantor AB, Herzenberg LA. Defective B cell development and function in Btk-deficient mice. Immunity. 1995;3(3):283–299. doi: 10.1016/1074-7613(95)90114-0. [DOI] [PubMed] [Google Scholar]
- 13.Uckun FM, Venkatachalam T. Targeting solid tumors with BTK inhibitors. Front Cell Dev Biol. 2021;9:650414. doi: 10.3389/fcell.2021.650414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lovitt CJ, Shelper TB, Avery VM. Evaluation of chemotherapeutics in a three-dimensional breast cancer model. J Cancer Res Clin Oncol. 2015;141(5):951–959. doi: 10.1007/s00432-015-1950-1. [DOI] [PubMed] [Google Scholar]
- 15.Weigelt B, Lo AT, Park CC, Gray JW, Bissell MJ. HER2 signaling pathway activation and response of breast cancer cells to HER2-targeting agents is dependent strongly on the 3D microenvironment. Breast Cancer Res Treat. 2010;122(1):35–43. doi: 10.1007/s10549-009-0502-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Konecny GE, Pegram MD, Venkatesan N, Finn R, Yang G, Rahmeh M, Untch M, Rusnak DW, Spehar G, Mullin RJ, Keith BR, Gilmer TM, Berger M, Podratz KC, Slamon DJ. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006;66(3):1630–1639. doi: 10.1158/0008-5472.CAN-05-1182. [DOI] [PubMed] [Google Scholar]
- 17.Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, Peng J, Lin E, Wang Y, Sosman J, Ribas A, Li J, Moffat J, Sutherlin DP, Koeppen H, Merchant M, Neve R, Settleman J. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487(7408):505–509. doi: 10.1038/nature11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng X, Baker H, Hancock WS, Fawaz F, McCaman M, Pungor E Jr. Proteomic analysis for the assessment of different lots of fetal bovine serum as a raw material for cell culture. Part IV. Application of proteomics to the manufacture of biological drugs. Biotechnol Prog. 2006;22(5):1294–1300. doi: 10.1021/bp060121o. [DOI] [PubMed] [Google Scholar]
- 19.Vande Voorde J, Ackermann T, Pfetzer N, Sumpton D, Mackay G, Kalna G, Nixon C, Blyth K, Gottlieb E, Tardito S. Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Sci Adv. 2019;5(1):eaau7314. doi: 10.1126/sciadv.aau7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Várnai P, Balla T. Visualization of phosphoinositides that bind pleckstrin homology domains: calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools. J Cell Biol. 1998;143(2):501–510. doi: 10.1083/jcb.143.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grassilli E, Pisano F, Cialdella A, Bonomo S, Missaglia C, Cerrito MG, Masiero L, Ianzano L, Giordano F, Cicirelli V, Narloch R, D’Amato F, Noli B, Ferri GL, Leone BE, Stanta G, Bonin S, Helin K, Giovannoni R, Lavitrano M. A novel oncogenic BTK isoform is overexpressed in colon cancers and required for RAS-mediated transformation. Oncogene. 2016;35(33):4368–4378. doi: 10.1038/onc.2015.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hannon GJ, Sun P, Carnero A, Xie LY, Maestro R, Conklin DS, Beach D. MaRX: an approach to genetics in mammalian cells. Science. 1999;283(5405):1129–1130. doi: 10.1126/science.283.5405.1129. [DOI] [PubMed] [Google Scholar]
- 23.Sevinsky CJ, Khan F, Kokabee L, Darehshouri A, Maddipati KR, Conklin DS. NDRG1 regulates neutral lipid metabolism in breast cancer cells. Breast Cancer Res. 2018;20(1):55. doi: 10.1186/s13058-018-0980-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holden P, Horton WA. Crude subcellular fractionation of cultured mammalian cell lines. BMC Res Notes. 2009;2:243. doi: 10.1186/1756-0500-2-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12(6):996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ren J, Wen L, Gao X, Jin C, Xue Y, Yao X. CSS-Palm 2.0: an updated software for palmitoylation sites prediction. Protein Eng Des Sel. 2008;21(11):639–644. doi: 10.1093/protein/gzn039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun W, Duan T, Ye P, Chen K, Zhang G, Lai M, Zhang H. TSVdb: a web-tool for TCGA splicing variants analysis. BMC Genomics. 2018;19(1):405. doi: 10.1186/s12864-018-4775-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bartlett JM, Thomas J, Ross DT, Seitz RS, Ring BZ, Beck RA, Pedersen HC, Munro A, Kunkler IH, Campbell FM, Jack W, Kerr GR, Johnstone L, Cameron DA, Chetty U. Mammostrat as a tool to stratify breast cancer patients at risk of recurrence during endocrine therapy. Breast Cancer Res. 2010;12(4):R47. doi: 10.1186/bcr2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bartlett JM, Bloom KJ, Piper T, Lawton TJ, van de Velde CJ, Ross DT, Ring BZ, Seitz RS, Beck RA, Hasenburg A, Kieback D, Putter H, Markopoulos C, Dirix L, Seynaeve C, Rea D. Mammostrat as an immunohistochemical multigene assay for prediction of early relapse risk in the tamoxifen versus exemestane adjuvant multicenter trial pathology study. J Clin Oncol. 2012;30(36):4477–4484. doi: 10.1200/JCO.2012.42.8896. [DOI] [PubMed] [Google Scholar]
- 30.Vieira AF, Schmitt F. An update on breast cancer multigene prognostic tests-emergent clinical biomarkers. Front Med (Lausanne) 2018;5:248. doi: 10.3389/fmed.2018.00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aicart-Ramos C, Valero RA, Rodriguez-Crespo I. Protein palmitoylation and subcellular trafficking. Biochim Biophys Acta. 2011;1808(12):2981–2994. doi: 10.1016/j.bbamem.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 32.Linder ME, Deschenes RJ. New insights into the mechanisms of protein palmitoylation. Biochemistry. 2003;42(15):4311–4320. doi: 10.1021/bi034159a. [DOI] [PubMed] [Google Scholar]
- 33.Resh MD. Palmitoylation of ligands, receptors, and intracellular signaling molecules. Sci STKE. 2006;2006(359):re14. doi: 10.1126/stke.3592006re14. [DOI] [PubMed] [Google Scholar]
- 34.Resh MD. Trafficking and signaling by fatty-acylated and prenylated proteins. Nat Chem Biol. 2006;2(11):584–590. doi: 10.1038/nchembio834. [DOI] [PubMed] [Google Scholar]
- 35.Howie J, Reilly L, Fraser NJ, Vlachaki Walker JM, Wypijewski KJ, Ashford ML, Calaghan SC, McClafferty H, Tian L, Shipston MJ, Boguslavskyi A, Shattock MJ, Fuller W. Substrate recognition by the cell surface palmitoyl transferase DHHC5. Proc Natl Acad Sci USA. 2014;111(49):17534–17539. doi: 10.1073/pnas.1413627111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin BR, Cravatt BF. Large-scale profiling of protein palmitoylation in mammalian cells. Nat Methods. 2009;6(2):135–138. doi: 10.1038/nmeth.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baraldi E, Djinovic Carugo K, Hyvönen M, Surdo PL, Riley AM, Potter BV, O’Brien R, Ladbury JE, Saraste M. Structure of the PH domain from Bruton’s tyrosine kinase in complex with inositol 1,3,4,5-tetrakisphosphate. Structure. 1999;7(4):449–460. doi: 10.1016/s0969-2126(99)80057-4. [DOI] [PubMed] [Google Scholar]
- 38.Wang Q, Pechersky Y, Sagawa S, Pan AC, Shaw DE. Structural mechanism for Bruton’s tyrosine kinase activation at the cell membrane. Proc Natl Acad Sci USA. 2019;116(19):9390–9399. doi: 10.1073/pnas.1819301116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chung JK, Nocka LM, Decker A, Wang Q, Kadlecek TA, Weiss A, Kuriyan J, Groves JT. Switch-like activation of Bruton’s tyrosine kinase by membrane-mediated dimerization. Proc Natl Acad Sci USA. 2019;116(22):10798–10803. doi: 10.1073/pnas.1819309116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gustafsson MO, Hussain A, Mohammad DK, Mohamed AJ, Nguyen V, Metalnikov P, Colwill K, Pawson T, Smith CI, Nore BF. Regulation of nucleocytoplasmic shuttling of Bruton’s tyrosine kinase (Btk) through a novel SH3-dependent interaction with ankyrin repeat domain 54 (ANKRD54) Mol Cell Biol. 2012;32(13):2440–2453. doi: 10.1128/MCB.06620-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McQuin C, Goodman A, Chernyshev V, Kamentsky L, Cimini BA, Karhohs KW, Doan M, Ding L, Rafelski SM, Thirstrup D, Wiegraebe W, Singh S, Becker T, Caicedo JC, Carpenter AE. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 2018;16(7):e2005970. doi: 10.1371/journal.pbio.2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mohamed AJ, Nore BF, Christensson B, Smith CI. Signalling of Bruton’s tyrosine kinase, Btk. Scand J Immunol. 1999;49(2):113–118. doi: 10.1046/j.1365-3083.1999.00504.x. [DOI] [PubMed] [Google Scholar]
- 43.Hyvönen M, Saraste M. Structure of the PH domain and Btk motif from Bruton’s tyrosine kinase: molecular explanations for X-linked agammaglobulinaemia. EMBO J. 1997;16(12):3396–3404. doi: 10.1093/emboj/16.12.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Winkler DG, Faia KL, DiNitto JP, Ali JA, White KF, Brophy EE, Pink MM, Proctor JL, Lussier J, Martin CM, Hoyt JG, Tillotson B, Murphy EL, Lim AR, Thomas BD, Macdougall JR, Ren P, Liu Y, Li LS, Jessen KA, Fritz CC, Dunbar JL, Porter JR, Rommel C, Palombella VJ, Changelian PS, Kutok JL. PI3K-δ and PI3K-γ inhibition by IPI-145 abrogates immune responses and suppresses activity in autoimmune and inflammatory disease models. Chem Biol. 2013;20(11):1364–1374. doi: 10.1016/j.chembiol.2013.09.017. [DOI] [PubMed] [Google Scholar]
- 45.Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. 1998;280(5369):1614–1617. doi: 10.1126/science.280.5369.1614. [DOI] [PubMed] [Google Scholar]
- 46.Jackson JT, Mulazzani E, Nutt SL, Masters SL. The role of PLCγ2 in immunological disorders, cancer, and neurodegeneration. J Biol Chem. 2021;297(2):100905. doi: 10.1016/j.jbc.2021.100905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hopkins BD, Goncalves MD, Cantley LC. Insulin-PI3K signalling: an evolutionarily insulated metabolic driver of cancer. Nat Rev Endocrinol. 2020;16(5):276–283. doi: 10.1038/s41574-020-0329-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nore BF, Vargas L, Mohamed AJ, Brandén LJ, Bäckesjö CM, Islam TC, Mattsson PT, Hultenby K, Christensson B, Smith CI. Redistribution of Bruton’s tyrosine kinase by activation of phosphatidylinositol 3-kinase and Rho-family GTPases. Eur J Immunol. 2000;30(1):145–154. doi: 10.1002/1521-4141(200001)30:1<145::AID-IMMU145>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 49.Smith CI, Islam TC, Mattsson PT, Mohamed AJ, Nore BF, Vihinen M. The Tec family of cytoplasmic tyrosine kinases: mammalian Btk, Bmx, Itk, Tec, Txk and homologs in other species. Bioessays. 2001;23(5):436–446. doi: 10.1002/bies.1062. [DOI] [PubMed] [Google Scholar]
- 50.Debnath J, Chamorro M, Czar MJ, Schaeffer EM, Lenardo MJ, Varmus HE, Schwartzberg PL. rlk/TXK encodes two forms of a novel cysteine string tyrosine kinase activated by Src family kinases. Mol Cell Biol. 1999;19(2):1498–1507. doi: 10.1128/MCB.19.2.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robbins SM, Quintrell NA, Bishop JM. Myristoylation and differential palmitoylation of the HCK protein-tyrosine kinases govern their attachment to membranes and association with caveolae. Mol Cell Biol. 1995;15(7):3507–3515. doi: 10.1128/MCB.15.7.3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carréno S, Gouze ME, Schaak S, Emorine LJ, Maridonneau-Parini I. Lack of palmitoylation redirects p59Hck from the plasma membrane to p61Hck-positive lysosomes. J Biol Chem. 2000;275(46):36223–36229. doi: 10.1074/jbc.M003901200. [DOI] [PubMed] [Google Scholar]
- 53.Gagliardi PA, Puliafito A, Primo L. PDK1: At the crossroad of cancer signaling pathways. Semin Cancer Biol. 2018;48:27–35. doi: 10.1016/j.semcancer.2017.04.014. [DOI] [PubMed] [Google Scholar]
- 54.Sato I, Obata Y, Kasahara K, Nakayama Y, Fukumoto Y, Yamasaki T, Yokoyama KK, Saito T, Yamaguchi N. Differential trafficking of Src, Lyn, Yes and Fyn is specified by the state of palmitoylation in the SH4 domain. J Cell Sci. 2009;122(Pt 7):965–975. doi: 10.1242/jcs.034843. [DOI] [PubMed] [Google Scholar]
- 55.Fletcher CDM. Elsevier Limited. 2013. Diagnostic histopathology of tumors. 4th edn. [Google Scholar]
- 56.Li J, Yin W, Jing Y, Kang D, Yang L, Cheng J, Yu Z, Peng Z, Li X, Wen Y, Sun X, Ren B, Liu C. The coordination between B cell receptor signaling and the actin cytoskeleton during B cell activation. Front Immunol. 2019;9:3096. doi: 10.3389/fimmu.2018.03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tanaka S, Baba Y. B cell receptor signaling. Adv Exp Med Biol. 2020;1254:23–36. doi: 10.1007/978-981-15-3532-1_2. [DOI] [PubMed] [Google Scholar]