

Abstract
Ribonucleotide reductases (RNRs) catalyze the reduction of ribonucleotides to deoxyribonucleotides, thereby playing a key role in DNA replication and repair. Escherichia coli class Ia RNR is an α2β2 enzyme complex that uses a reversible multistep radical transfer (RT) over 32 Å across its two subunits, α and β, to initiate, using its metallo-cofactor in β2, nucleotide reduction in α2. Each step is proposed to involve a distinct proton-coupled electron-transfer (PCET) process. An unresolved step is the RT involving Y356(β) and Y731(α) across the α/β interface. Using 2,3,5-F3Y122-β2 with 3,5-F2Y731-α2, GDP (substrate) and TTP (allosteric effector), a Y356• intermediate was trapped and its identity was verified by 263 GHz electron paramagnetic resonance (EPR) and 34 GHz pulse electron–electron double resonance spectroscopies. 94 GHz 19F electron-nuclear double resonance spectroscopy allowed measuring the interspin distances between Y356• and the 19F nuclei of 3,5-F2Y731 in this RNR mutant. Similar experiments with the double mutant E52Q/F3Y122-β2 were carried out for comparison to the recently published cryo-EM structure of a holo RNR complex. For both mutant combinations, the distance measurements reveal two conformations of 3,5-F2Y731. Remarkably, one conformation is consistent with 3,5-F2Y731 within the H-bond distance to Y356•, whereas the second one is consistent with the conformation observed in the cryo-EM structure. The observations unexpectedly suggest the possibility of a colinear PCET, in which electron and proton are transferred from the same donor to the same acceptor between Y356 and Y731. The results highlight the important role of state-of-the-art EPR spectroscopy to decipher this mechanism.
1. Introduction
Ribonucleotide reductases (RNRs) catalyze the conversion of four nucleoside di- or triphosphates (ND(T)Ps) to deoxyribonucleoside di- or triphosphates (dND(T)Ps) in all organisms (Figure 1).1−3 RNRs are highly regulated enzymes playing an important role in controlling the ratio and relative amounts of dNTPs essential for the fidelity of DNA replication and repair. Imbalance in dNTP pools results in genomic instability and leads to disease states.4−6 RNRs’ essential role has made them targets for cancer and, more recently, antibiotic therapeutics.6−12
Figure 1.
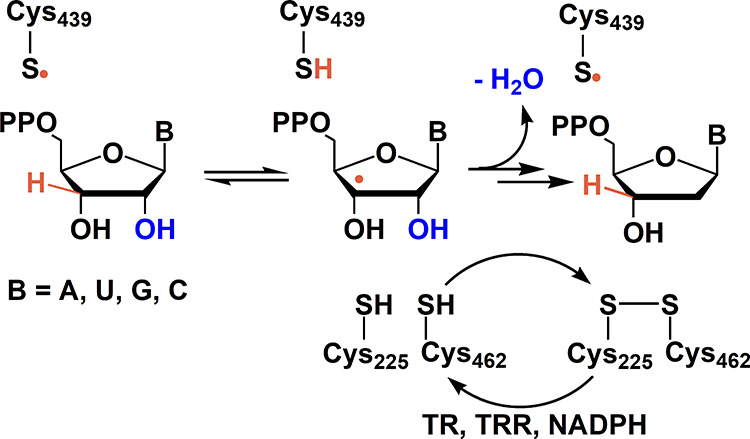
Reduction of NDPs to dNDPs catalyzed by Escherichia coli class Ia RNR. The reduction is initiated by a thiyl radical (C439•), and the reducing equivalents are provided by the oxidation of C225 and C462 to a disulfide. Multiple turnovers require a redoxin reducing system such as thioredoxin (TR), thioredoxin reductase (TRR), and nicotinamide adenine dinucleotide phosphate (NADPH).
The E. coli class Ia RNR, a prototype model system for human RNR,6 is composed of two subunits, α13 and β,14 both required for activity. Based on their α2 and β2 structures, Uhlin and Eklund proposed a symmetrical α2β2 docking model (Figure 2A) for active RNR, which has played a central role in the experimental design.13 The model for substrate activation and chemistry requires that the diferric tyrosyl radical (Y122•) cofactor located in β2 oxidizes C439 to a thiyl radical in the active site of α2, which, in turn, initiates NDP reduction (Figures 1 and 2C). Thiyl radical formation is proposed to occur by a radical transfer (RT) pathway, which involves five or six radical intermediates (Figure 2C),15 each generated by proton-coupled electron-transfer (PCET) steps.16−19
Figure 2.

Docking model13 (A) and cryo-EM structure38 (B) of the α2β2 complex of E. coli class Ia RNR and the proposed RT pathway, (C) and (D), respectively. (A) The docking model based on the shape complementarity of subunits α213 and β2.14 (B) Cryo-EM structure of an α2β2 complex of RNR generated when E52Q/F3Y122-β2, wt-α2 GDP (substrate) and TTP (effector) were quenched at 50 s (pdb code: 6W4X).38 Asymmetry of the complex is indicated by α′β′ (disordered pair) and αβ (ordered pair). (C) The proposed forward RT pathway based on many experiments.20−27,30−33 W48 is shown in parentheses as there currently is no direct evidence for its involvement. The red and blue double arrows describe electron and proton transfers, respectively. Evidence for the bold water molecules has been reported recently.27,28 (D) An intact RT pathway within αβ including Y356 and its position relative to Y731 is visible for the first time in the cryo-EM structure.38 Distances between RT residues are indicated; the 19F atoms of 2,3,5-F3Y122 present in the cryo-EM structure have been omitted. Interfacial residue Q52 (E52 in wt-RNR) is included as it was important for stabilizing the α2β2 complex in the cryo-EM experiment.
Central for developing this model has been the ability to replace pathway Ys site-selectively with unnatural amino acids (UAAs) that have allowed the generation and thermodynamic trapping of pathway radical intermediates. The tyrosyl radicals (Y•s) were studied by a suite of multifrequency electron paramagnetic resonance (EPR)20−30 methods as well as by transient absorption spectroscopic methods using photo-β2 RNRs.30−34
Despite much insight into nature’s design for radical initiation in RNRs, elucidating the molecular basis for the RT across the α/β subunit interface has been hampered by the lack of structural information about the C-terminal tail of all βs (residues 341–375 in E. coli RNR), essential for α/β subunit interaction.35−37 The location of Y356 in the RT pathway within this tail was thus unknown. Recently, a near-atomic resolution cryo-EM structure of a trapped α2β2E. coli complex was obtained (Figure 2B).38 It was generated from the incubation of a double mutant of β2, E52Q/F3Y122-β2, with wt-α2, substrate (GDP), and allosteric effector (TTP) with freeze-quenching at 50 s. The 2,3,5-F3Y122 substitution allowed the generation of one dGDP product and accumulation of one pathway radical at Y356•. The E52Q mutation was important for successfully trapping the α2β2 complex. The E52 residue resides at the α/β-interface and is essential for activity, enabling proton release during Y356 oxidation in the RT.33,39
The cryo-EM structure (Figure 2B) revealed an asymmetric α2β2 complex, consistent with earlier results.37,40 It also revealed the residues in the C-terminal tail of β (341–375) in an ordered αβ pair, the intact RT pathway including the location of Y356 and its location relative to Y731(α) (Figure 2D) for the first time. The entire C-terminal tail in α′/β′, where chemistry has occurred and Y356• is supposedly trapped, remains disordered.
The importance of Y356 during RT has been established by many different methods that often led to the detection of the Y356• intermediate. Recent studies to identify the proton acceptor during its oxidation in forward RT revealed that the most reasonable candidates, E52(β) and E350(β), both conserved and essential,36,39,41 are unlikely to be the ultimate acceptors.33,34,42 These residues are located at ∼7 Å (E52) and ∼14 Å (E350) distances from the phenol-oxygen atom of Y356 in the ordered αβ pair of the cryo-EM structure,38 too far for direct proton or H atom transfer with Y356.43 A variety of 1H and 17O high-frequency electron-nuclear double resonance (ENDOR) experiments on Y356•,27,28 kinetic studies using RNRs with FnY35633 and a photo-oxidant appended to the C355 mutant of β, and pH studies of Y356• formation using F2Y35642 all support the interaction of Y356• with water (Figure 2C).
Efforts to understand the residues involved in managing the proton to support the PCET between Y356 and Y731 across the α/β interface have been less successful. The cryo-EM structure shows an O–O distance between Y356 and Y731 of ∼8 Å in ordered αβ, with Y731 in its unusual stacked conformation with Y730 as in previous X-ray structures of α2 alone.13 While a number of pulsed electron double resonance (PELDOR) experiments6 revealed sharp distance distributions consistent with little Y356• flexibility, several different experiments reported the mobility of Y731. In a crystal structure of NH2Y730-α2 alone, Y731 was found in a conformation where it is flipped away from the stacked conformation with NH2Y730.44 PELDOR studies on a double mutant R411A-NH2Y731-α2 under turnover conditions revealed a conformational change of 3 Å in trapped NH2Y731•, consistent with a flipping toward the α/β interface.26 Subsequent studies using photo-β2 with the same α2 mutations revealed dynamic/rapid conformational changes of Y731.30 Another EPR study by Yokoyama et al. suggested the flipping of F2Y731•,23 which was trapped as a minority radical species in NO2Y122-β2/F2Y731-α2. Molecular dynamics (MD) simulations using the cryo-EM structure and the α/β interface in water also support the flexibility of Y731,45 with movement away from the stacked conformation with Y730. The studies together support a model for PCET between Y356• and Y731 across the α/β interface that could involve a movement of Y731 toward the interface (Figure 2C), with consequences for their PCET chemistry. However, structural or spectroscopic evidence for interaction between Y356• and Y731 has never been observed.
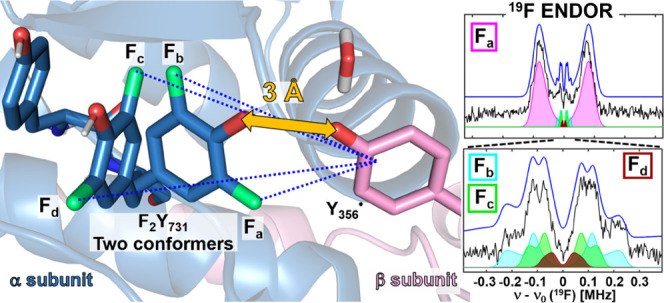
In this article, we use 19F–Y analogues introduced site-specifically into E. coli RNR, F3Y122-β2 (or the double mutant E52Q/F3Y122-β2), incubated with 3,5-F2Y731-α2, GDP, and TTP to generate and trap Y356•. F2Y731 was chosen for its symmetric 19F substitution pattern and minimally perturbed reduction potential relative to Y.46,47 The Y356• location and identity are established using 34 GHz PELDOR and 263 GHz EPR spectroscopies, respectively. 19F ENDOR spectroscopy48,49 at 94 GHz is used in an effort to determine the distances across the subunit interface between the trapped Y356•(β) and the 19F nuclei of F2Y731(α). The ENDOR spectra give unambiguous evidence for two conformations of F2Y731. One conformation is consistent with the structure observed by cryo-EM (ordered αβ pair). The second conformation indicates a flipping of F2Y731 toward Y356•. The results have important implications for the PCET mechanism across the α/β interface.
2. Materials and Methods
2.1. Preparation of RNR Mutants and Activity Assays
The RNR mutants F3Y122-β2, E52Q/F3Y122-β2, F2Y731-α2, and 17O–Y-wt-α2 were expressed and purified, as previously described.39,44,50 Activities of (E52Q)F3Y122-β2/F2Y731-α2 and wt-β2/17O–Y-α2 were determined using the spectrophotometric assay (Supporting Information (SI) 1, Table S1).51
2.2. EPR Sample Preparation
The Y356• intermediate was trapped by incubating a solution of F2Y731-α2, GDP, and TTP in assay buffer (50 mM HEPES, 15 mM MgSO4, 1 mM EDTA, pH 7.6) with F3Y122-β2 or E52Q/F3Y122-β2 in assay buffer. Glycerol concentrations were optimized (Figure S1) and typically added to ∼20% of the final volume to prolong phase memory times TM for PELDOR and ENDOR measurements. The final concentrations were ∼80 μM α2β2, ∼1 mM GDP, and ∼200 μM TTP. The reaction mixture was transferred to either 34 GHz EPR tubes (Q-band) (12 μL, 1.5 mm inner diameter (ID) Suprasil tube, Wilmad) or 94 GHz (W-band) tubes (4.4 μL, 0.7 mm ID clear fused quartz tubes) and quenched by freezing in liquid nitrogen at reaction times (TQ) of 40–80 s (Q-band) or 35–55 s (W-band). A second set of samples were prepared with TQ > 100 s. Two hundred and sixty-three GHz EPR samples were prepared in Suprasil capillaries (ID 0.2 mm, Vitrocom) without glycerol and quenched at TQ = 15–20 s. All samples are summarized in SI 2, Table S2.
2.3. 263 GHz EPR Spectroscopy
High-frequency (HF) 263 GHz echo-detected EPR spectra were recorded with a commercial spectrometer, as previously reported.52 Details on the spectral acquisition are given in SI 3.
2.4. 34 GHz PELDOR Spectroscopy
Four-pulse PELDOR experiments53,54 were performed at 34 GHz (Q-band) on a commercial Bruker ELEXSYS E580 EPR spectrometer, as previously reported.27 An optimized temperature of 50 K was selected, where high sensitivity is achieved and unreacted F3Y122• does not contribute to the spin echo under conditions used for data collection (SI 4.1–4.3). MW pulses were amplified by a pulsed 170 W TWT amplifier (Model 187Ka, Applied Systems Engineering) with typical pulse lengths of 14–16 ns for the pump π-pulse at the center of the overcoupled resonator. The observer frequency was set to −105 MHz from the dip center, leading to observer π-pulse lengths of 24–28 ns. The τ1 value was 250 ns, and τ2 values were optimized based on TM measurements (SI 4.2). Shot repetition times were 4–6 ms. Time traces were recorded at three different observer positions (Figure S5) and their intensities were summed, reflecting their respective EPR signal strengths at that excitation position. Traces were analyzed with DeerAnalysis 2019,55 using Tikhonov regularization (L-curve criterion for α parameter) and checked for consistency using neural network analysis.56,57
2.5. 94 GHz ENDOR Spectroscopy
Pulsed EPR and ENDOR experiments at 94 GHz (W-band) were performed on a commercial Bruker ELEXSYS E680 EPR spectrometer, as previously described.25 Using a 2 W MW amplifier, typical π/2 pulse lengths of 10–12 ns were achieved. EPR (echo-detected) spectra and signal contributions are illustrated in SI 5.1. Shot repetition times were optimized to 2–4 ms based on T1 measurements (SI 5.2).
19F Mims ENDOR spectra of the Y356• were recorded using radio frequency (RF) pulses amplified by a 250 W RF amplifier (250A250A Amplifier Research). RF pulse lengths of 22 μs were used for 19F nuclei with ∼1.6 MHz couplings or 44 μs for couplings ≤∼250 kHz. RF pulse lengths were optimized using Rabi nutation experiments. Stochastic RF acquisition58−60 with 20 shots per point was used. To observe 19F couplings of different sizes, the adjustment of the interpulse delay τ in the Mims sequence was crucial. For couplings on the order of 1.6 MHz, two measurements with τ values of 236 and 266 ns were performed and summed subsequently (normalized to the number of scans) to attenuate the proton background. For smaller couplings, ≤∼250 kHz, τ was optimized to 620–622 ns (SI 5.3). ENDOR spectra were recorded at three different observer positions (Figure S8) and summed up with intensities reflecting their respective EPR signal strengths at that excitation position.
Data were collected at two temperatures. At 50 K, ENDOR sensitivity was higher than that at 80 K, where usually the signal of unreacted F3Y122• disappears due to faster relaxation.27 As a downside, at 50 K, the unreacted F3Y122• contributed to the echo intensity of the Mims sequence at short interpulse delays τ. The contribution of F3Y122• led to 19F ENDOR background signals, which had to be removed during data processing (SI 5.4). As a control for the background correction procedure, we repeated representative 19F ENDOR measurements at 80 K (SI 5.5–5.6) where no background of F3Y122• was present. The results obtained at 50 and 80 K are fully consistent. In addition to the 19F background, broad, overlapping 1H resonances associated with the 3,5-H atoms of Y35627 were identified by their changes observed with τ value changes and they were subtracted from the 19F spectra, as illustrated in SI 5.4.
17O ENDOR control experiments were performed using similar parameters described in our recent 17O ENDOR study28 and are reported in SI 6.
2.6. Simulations of ENDOR Data
Mims ENDOR simulations of the Y356• were performed using EasySpin’s saffron routine.61 The g tensor was gx = 2.0062, gy = 2.0044, and gz = 2.0022.27 In the molecular frame, gx is aligned along the C–O• bond of Y356•, while gy is perpendicular to this direction and in the plane of the aromatic ring. The strongly coupled β-proton of Y356• was included using previously reported hyperfine coupling (HFC) parameters.27 For simulating the 19F ENDOR spectra with τ = 620–622 ns, the C3 and C5 protons27 of Y356• were included. The 19F ENDOR line width parameter was simulated as 25 kHz for couplings below 0.5 MHz.49 For larger couplings, a line width of 250 kHz was used. Chemical shift anisotropies were not resolved in the 94 GHz 19F ENDOR spectra.62
2.7. Structural Models for ENDOR Analysis
Due to the large parameter space associated with the two Fs of F2Y731 and, as will become clear, their multiple side-chain conformations, a fitting routine that generates the most likely set of HFC parameters by minimizing residuals (rmsd) is not possible. We therefore used an approach similar to that described previously to analyze the PCET steps within α2 using NH2Y731 and the X-ray structure of α2 to position Y730 and C439.25 In the present case, the small models were constructed starting from pdb 6W4X, the recent cryo-EM structure (resolution 3.3–5.5 Å).38 Y356 from β and Y731 and Y730 from α were extracted from the ordered α/β pair (Figure 2B,D). 19F atoms at C3 and C5 of Y731 were introduced using PyMOL.63 The peptide bonds connecting each tyrosine to their protein backbone were replaced by NHR and −CRO (Figure 3) groups, and their xyz coordinates were not changed compared to the cryo-EM structure. Density functional theory (DFT)-based, constrained geometry optimization using ORCA64 resulted in the model structure S1 of the triad Y356–F2Y731–Y730. Further representative conformations of the triad were obtained by rotating around Cα/Cβ and Cβ-phenol bonds displacing the phenol side chains of Y356 and F2Y731, as illustrated in Figure 3. Resulting models to fit the spectroscopic data are designated SX (X =1, 2, 3,...5) and are summarized in Tables S6 and S7 in SI 8. A water molecule binding to Y356• was also introduced into each model, with a binding geometry based on our previous studies (H-bond length ca. 1.8 Å, angle C4–O•···H ca. 120°, C3–C4–O•···H dihedral ca. 20°).27,28 The effect of H-bonds on the spin density distribution,65,66 further technical details on the DFT calculations, and the adaptation of the DFT-predicted parameters to the ENDOR simulations are described in the results section and summarized in SI 7. Contributions of the different conformations were assessed by rmsd analysis. Orientation-selective 19F spectra were then simulated using one set of parameters for all spectra.
Figure 3.
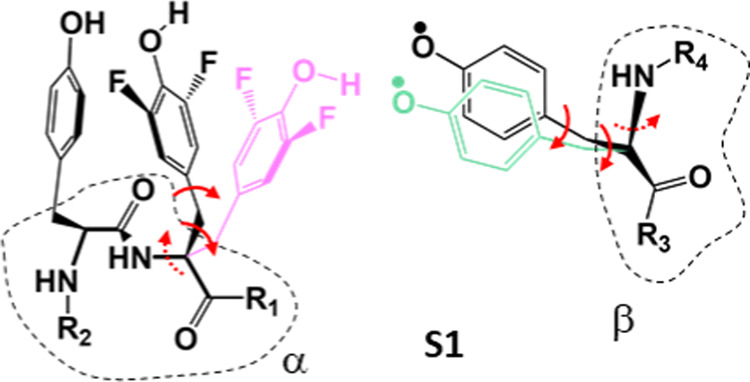
Models for the Y-triad. The black conformation corresponds to S1 but without the water molecule. The pink orientation of F2Y731 illustrates a flipped conformation, and the green orientation of Y356• represents a repositioning of the radical toward F2Y731, used in models S2–S5. Atom positions of the backbone are from the cryo-EM structure within ≤∼0.5 Å. R1–R4 peptide chains have been replaced by H atoms in S1–S5. Red arrows indicate a rotation around a bond, and dashed arrows indicate small rotations (Table S6).
3. Results
3.1. Characterization of RNR Constructs Using Activity Measurements, High-Field EPR, and PELDOR
The first part of the investigation required examination of the new RNR constructs that contain the 19F labels in F2Y731. Steady-state activities are reported in Table S1. Spectrophotometric assays revealed a specific activity of 560 nmol/(mg·min) (ca. 7% of wt) for F3Y122-β2/F2Y731-α2, defined with respect to the mass of β2 in the assay. In contrast, an activity of only 6 nmol/(mg·min), that is, the lower limit of detection, was measured for E52Q/F3Y122-β2/F2Y731-α2. The latter finding was expected, as the E52Q mutation disrupts steady-state activity.39
Nevertheless, both constructs are capable of one turnover and allowed trapping of the intermediate Y356• for EPR samples during back-radical transfer.67 Moreover, glycerol is required in the sample preparation to prolong spin relaxation in the EPR experiments. Thus, the glycerol content (v%) was also optimized based on its effect on RNR activity (SI 1) and a value of 20 v% was selected for almost all samples (SI 2, Table S2). We characterized the structure of the trapped radical in F3Y122-β2/F2Y731-α2 and E52Q/F3Y122-β2/F2Y731-α2 by 263 GHz EPR (SI 3). In all quenched reaction mixtures, two radical species were observed (Figure S2). One contribution arose from the unreacted F3Y122• and was readily identified by its large gx value (2.0082) and its characteristic 19F HFC structure. After subtracting a reference spectrum of F3Y122•, the spectrum of the intermediate became visible (Figure S3). This radical was identified as Y356• due to the characteristic low gx value of 2.0062 (reference spectrum of Y356• is shown in Figures S2 and S3), as reported with F3Y122-β2/wt-α2.27 The analysis of the HF-EPR spectra also revealed no other radical species.
PELDOR spectroscopy (34 GHz) was then used to measure the diagonal distance between Y356• in one αβ pair and F3Y122• in the second one (Figure 4). The orientation-averaged time traces exhibit clear oscillations. Indistinguishable results were obtained for various sample preparation conditions (SI 4). For comparison, a time trace of F3Y122-β2/wt-α2 was also measured (Figure 4, green). Distance distributions with a single peak centered at 3.03 ± 0.02 nm (Figure 4) and a width (full width at half-maximum (FWHM); Table S4) of 0.09–0.14 nm were obtained for all samples. The observed distance is typical for F3Y122•–Y356• pairs.6,27 From PELDOR and HF-EPR, we conclude that Y356• is the observed radical, as previously characterized using wt-α2 for incubation.27
Figure 4.

Orientation-averaged 34 GHz PELDOR time traces of F3Y122-β2/F2Y731-α2 (∼80 μM, TQ = 77 s, blue line), E52Q/F3Y122-β2/F2Y731-α2 (∼80 μM, TQ = 44 s, red), and F3Y122-β2/wt-α2 (green) along with fits (dotted lines). Distance distributions are shown as the inset. A cartoon illustrates the assignment of distance peaks to radical pairs. A symmetric representation was chosen as the experiments reported herein do not inform about the asymmetry in the protein complex.
It is interesting to consider the observed distance within the framework of the new cryo-EM structure.38 The detected radical intermediate (Y356•) is thought to be produced during reverse RT in the first turnover.67 If the first turnover was occurring for instance in the α′β′ pair, see the notation from the cryo-EM structure (Figure 2B), then the observed PELDOR distance should be between Y356•(β′) and F3Y122•(β). However, in the cryo-EM structure, the C-terminal β′ tail is disordered at the interface, indicating that the trapped state might be different under the conditions of the EPR experiments. Because of the disorder, the distance between F3Y122•(β) and Y356•(β′) cannot be measured in the cryo-EM structure. If we consider the opposite diagonal distance, i.e., between the centroids68 of the Tyr-O, C1, C3, and C5 atoms of F3Y122• in β′ and Y356• in β, then the PELDOR distance of 3.0 nm is in agreement with this structure. We note that many such distances have been measured with other constructs.6 All give a sharp 3 nm distance feature, suggesting that the Y356• conformation is constrained. Our model for half-site RNR reactivity15 requires that the complex interconverts to allow for alternating PCET in αβ and α′β′. When the Y356• is trapped, the interconversion is slow. The kinetics of this structural interconversion and the mechanism of switching remain to be established but are likely to be critical for comparing results from different experimental setups.
3.2. Distance Measurements across the RNR α/β Interface Using 94 GHz 19F ENDOR
3.2.1. 19F ENDOR Detects Y356•–19F2Y731 Distances
19F ENDOR spectra of Y356• in F3Y122-β2/F2Y731-α2 (black) and E52Q/F3Y122-β2/F2Y731-α2 (red) were obtained after summing three background-corrected, orientation-selective spectra in the range of ±4 MHz around the 19F Larmor frequency ν0(19F) (Figure 5A). When using short τ values (236 and 266 ns), prominent resonances are observed at ±∼0.8 MHz in both samples. These resonances are attributed to one 19F nucleus, Fa, with a peak separation of ∼1.6 ± 0.1 MHz (purple, dashed lines). Additionally, sharp features are observed in a ±250 kHz region around ν0(19F). These resonances were investigated using a larger τ value of 620 ns, which enhances the sensitivity for smaller couplings (Figure 5B).49 For both samples, the spectra in Figure 5B can be interpreted as a superposition of two Pake patterns contributed by two 19F nuclei, designated as Fb and Fc. Pake patterns result from purely dipolar coupling and allow assignment of the corresponding dipolar HFC T by reading off the splitting between the sharp, central peaks: Tb = 250 ± 15 kHz (cyan, dashed lines) and Tc = 150 ± 15 kHz (green, dashed lines). These peaks are contributed by molecules in which the 19F-radical interspin vector is perpendicular to the external magnetic field B0. Using the point-dipole approximation (eq 1)49
| 1 |
we can estimate interspin distances of Rb = 6.7 ± 0.2 Å and Rc = 7.9 ± 0.3 Å, with the centroid of the O, C1, C3, and C5 atoms of Y356• as a point of reference.68 Aside from the central peaks, Pake patterns are also characterized by shoulders appearing at twice the coupling strength (2·T = T∥). These features are contributed by molecules with interspin vectors parallel to B0. The dipolar approximation does not apply for the stronger coupling Ta due to the shorter distance, <5 Å.49
Figure 5.
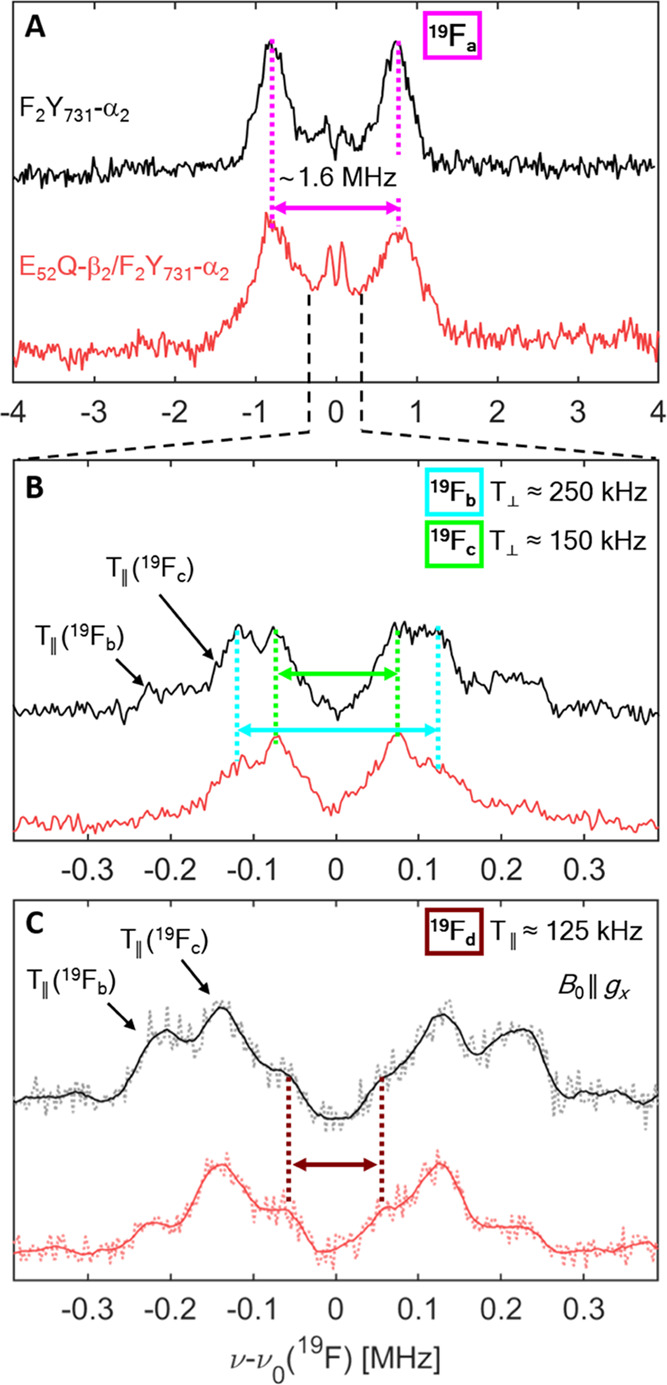
94 GHz 19F Mims ENDOR spectra of F3Y122-β2/F2Y731-α2 (80 μM, TQ = 50 s, black lines) and E52Q/F3Y122-β2/F2Y731-α2 (80 μM, TQ = 35 s, red lines) at T = 50 K. Spectra in panels (A) and (B) were obtained by adding three orientation-selective spectra. (A) Measurement with short τ values (∼250 ns). (B) Measurement with larger τ values (∼620 ns). (C) Orientation-selective spectra with B0 a∥ gx and τ = 620 ns after data point smoothing with the Savitzky–Golay filter (full lines). Original data are shown as dotted lines. Measurement time per spectrum is 30–40 h (A) and 50–60 h (B). Analysis of the spectra in panels (A)–(C) requires consideration of four nuclei 19Fa–19Fd, as marked by arrows and colored dashed lines.
The observation of three distinct 19F resonances in Figure 5A,B requires at least two conformations of F2Y731. Since each conformation contributes two 19F–Y356• spin pairs, a fourth set of resonances (Fd) is expected but not clearly resolved in the spectra obtained by summing up three orientation-selective measurements. An indication for coupling to a fourth nucleus Fd was provided by the orientation-selective measurements with B0 aligned along gx (Figure 5C). Here, strong selectivity for the parallel components of Fb and Fc was observed. In addition, shoulders on the inside of the two most prominent features are observed, which suggest the parallel coupling of the fourth atom Fd. Further analysis of the orientation-selective spectra is discussed below and will confirm this assignment.
Interestingly, the size of the observed HFCs (peak positions) is conserved in both F3Y122-β2/F2Y731-α2 and E52Q/F3Y122-β2/F2Y731-α2 mutants, but the spectrum of E52Q/F3Y122-β2/F2Y731-α2 in Figure 5A appears broader, suggesting more heterogeneity in this mutant.
3.2.2. Examination of Structural Models of the Triad Y730–F2Y731–Y356•
To rationalize the 19F ENDOR spectra, structural models of the tyrosine triad were built (Section 2.7 and Figure 3) and the DFT-predicted 19F HFCs were compared with the experimental values in Figure 5. The starting point for modeling is the cryo-EM structure.38 Model S1 (Figure 3, black) is identical to this structure, with two 19F nuclei replacing the 3,5-H atoms in Y731. This structure results in HFCs of 65 kHz and 114 kHz (see also SI 8, Table S8), the latter approaching but not quite matching the 150 kHz indicated for Fc in Figure 5B given DFT uncertainties up to 20%. The 65 kHz coupling could potentially be attributed to the fourth 19F nucleus, Fd.
To increase the coupling strength in S1, either the position of F2Y731 or of Y356• had to be readjusted for the spin centers to come closer. An increase of Tc from 114 to ∼150 kHz for Fc would require reducing the interspin distance by roughly 1 Å based on eq 1. To maintain the stacked arrangement of F2Y731 and Y730, observed in almost all available structures, we adjusted the position of O–Y356• by ca. 1 Å, which is still well within the resolution of the cryo-EM structure, as indicated in green color in Figure 3 (Table S6). This resulted in model S2, illustrated in Figure 6. We note that in model S2, as well as in all other models, a water molecule was introduced in the vicinity of Y356• (Section 2.7), the presence of which was reported earlier.27,28 The H-bonding water molecule affects Y356•’s spin density distribution and, consequently, also the effective 19F-radical HFCs. As detailed in SI 7, the resulting geometrical changes are minor and amount to ca. 0.1–0.2 Å.
Figure 6.
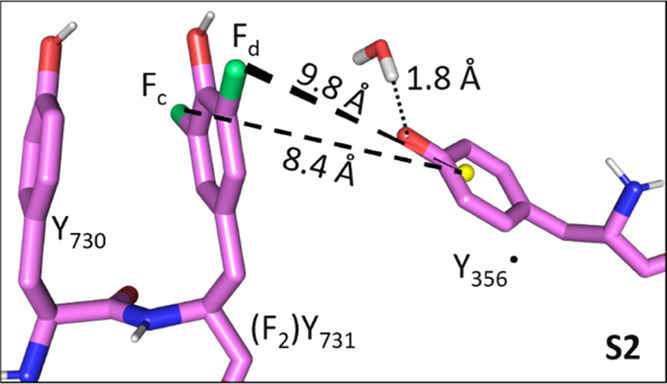
Model S2, 19F–Y356• distances are indicated by dashed lines (centroid of Y356• as a point of reference). Fluorine, oxygen, and nitrogen atoms are in green, red, and blue, respectively. H2O was included based on our previous results.27,28
In S2, the 19F–Y356• distances are 9.8 and 8.4 Å, the latter consistent with the estimate for Rc based on the dipolar approximation (eq 1). DFT analysis of S2 predicts coupling constants of 85 and 153 kHz, reproducing the coupling of Fc in Figure 5B within the estimated uncertainty. The 85 kHz coupling could be attributed to Fd. When the triad shown in S2 is incorporated back into the cryo-EM structure, the position of Y356• was found to fulfill the PELDOR diagonal distance of 3.0 nm (Figure 4 and Table S7).
Nevertheless, it is clear that neither model S2 nor reorienting the ring plane of F2Y731 (model S3, Figure S16) is able to reproduce the observed strong HFCs of Fa.
We therefore examined the possibility that a second conformation between the interfacial Ys might result in a second pair of stronger 19F HFCs. This proposal is reasonable based on previous evidence from different types of experiments that Y731 can flip.23,26,30,44,45 A small model based on the flipped Y-dyad taken from the X-ray structure of NH2Y730-α244 (without β2) could not be placed into the cryo-EM structure using pair fitting (in PyMOL) of the ring atoms to superimpose the Y730 side chains since clashes resulted (SI 8, Figure S17). This is in principle expected because this structure is missing the β subunit, which provides structural constraints. We thus focused on αβ and returned to model S2, adjusted the dihedral angles around Cα–Cβ and N–Cα of Y731 (Table S6), until the DFT-predicted HFC couplings reached the range of the experimental values for Fa and Fb. Representative structures that fulfilled the 19F HFCs are shown as models S4 and S5 (Figure 7), in which the fluorophenol groups are flipped by about 50–70° toward the subunit interface.
Figure 7.
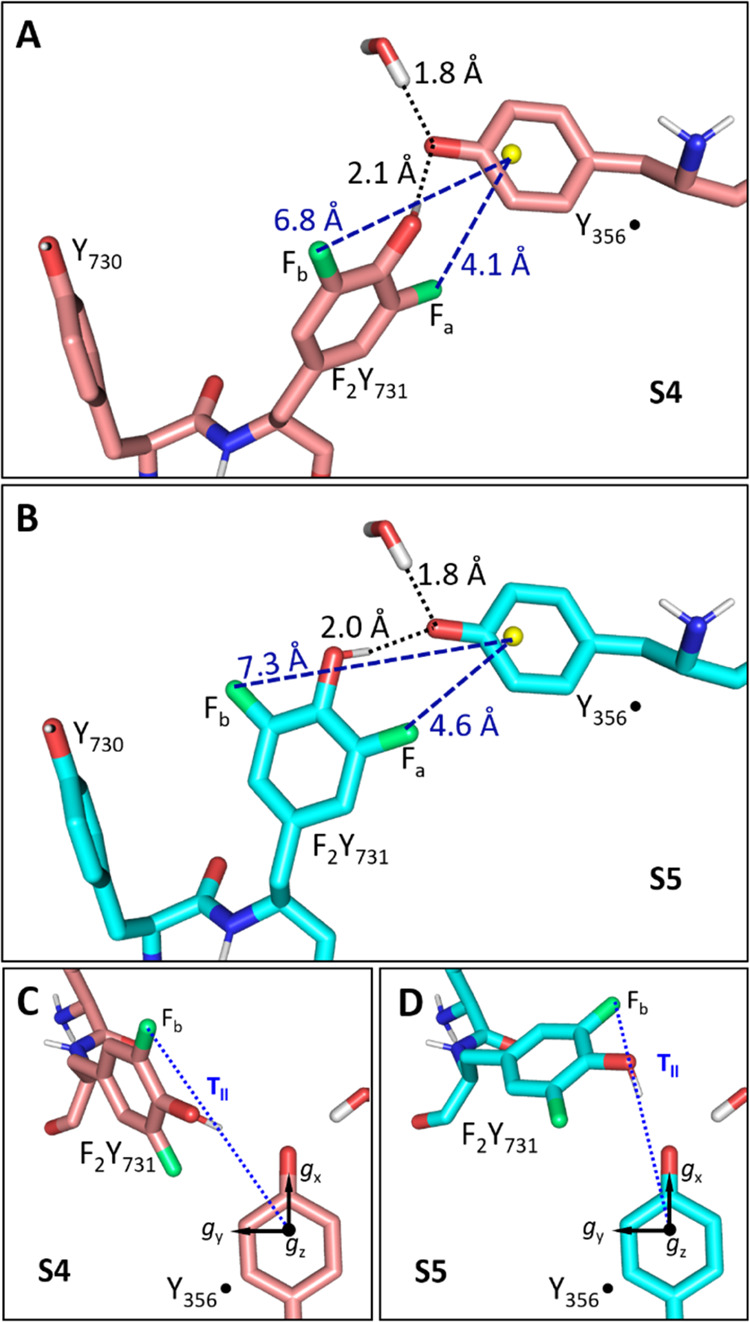
Models S4 (A) and S5 (B). (A) Model S4 (fluorine, oxygen, and nitrogen atoms in green, red, and blue, respectively). H-bond lengths and the 19F-centroid (Y356•, yellow sphere) distances are indicated. (B) Model S5 (cyan sticks, colors as in panel A). (C and D) Top view of the models shown in panels (A) and (B). In panels (C) and (D), the g tensor of Y356• is indicated along with the parallel component of the dipolar HFC tensor of the distal 19F nucleus Fb.
In S4 (Figure 7A,C), the 19F nuclei reside at distances of 4.1 and 6.8 Å from the centroid of Y356•. For the proximal 19F atom (Fa), DFT predicts a dipolar coupling constant Ta of ∼1.0 MHz and a negative, isotropic coupling constant aiso,a of −0.8 MHz. This combination leads to a splitting of ∼1.8 MHz for S4, similar to the ∼1.6 MHz observed experimentally for Fa (Figure 5A). The larger of the two 19F-radical distances in S4 agrees well with the estimate for Rb, yielding a coupling constant Tb of 254 kHz, in agreement with the resonances of Fb (Figure 5B).
In a second model with a flipped Y731 (S5, Figure 7B,D), a distinct orientation of Y731 and Y356 was considered to account for orientation selection (see also next section). In S5, the 19F–Y356• distances are 4.6 and 7.3 Å. The interspin vector from the distal Fb to the centroid of Y356• is nearly parallel to the direction of gx (Figure 7D) and distinct from S4 (Figure 7C). It has a DFT-derived HFC of Tb = 246 kHz. For the proximal 19F nucleus Fa, a dipolar coupling constant of Ta ≈ 0.8 MHz with a negative isotropic coupling constants aiso,a of ca −1.0 MHz is predicted and leads to an expected peak separation of ∼1.8 MHz as in S4.
A comparison of DFT-predicted HFCs from all models, S1–S5, and the experimental values is shown in Figure 8. More details on geometrical parameters of the five models are summarized in Table S7. We note that the combination of S2 with either S4 or S5 could satisfy the experimentally observed peak separations in Figure 5.
Figure 8.
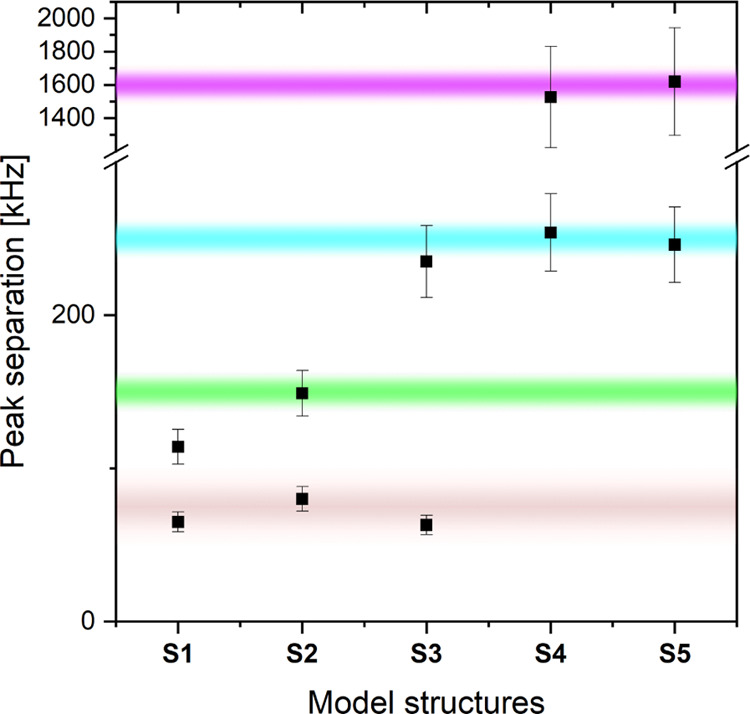
Comparison of experimentally observed peak separation from Figures 5 and 9 (purple (Fa), cyan (Fb), green (Fc), and brown (Fd) shadings indicate the range of uncertainty) with DFT-predicted peak positions (black squares) for models S1–S5. For the DFT values, an error of ±20% (Fa, this nucleus exhibits isotropic and anisotropic coupling) or ±10% (Fb–Fd, these nuclei show purely dipolar coupling) is estimated.
Finally, both S4 and S5, when integrated back into the framework of the cryo-EM structure,38 give centroid–centroid distances between F2Y731 in αβ and F3Y122• in α′β′ of 35.0 and 35.5 Å, respectively, both very similar to the constraints measured in our previous PELDOR experiments.26
3.2.3. Spectral Simulations Including a Superposition of Stacked and Flipped Y731 Conformations
The DFT analysis indicated that it is possible to find mutual conformations of F2Y731 and Y356, which individually satisfy some observed 19F–Y356• distances. To examine whether a superposition of these conformations can reproduce the ENDOR spectra, we also considered the orientation-selected ENDOR spectra, which pose additional constraints with respect to the sum spectra of Figure 5.
Representative orientation-selected spectra, corresponding to the black sum spectra of Figure 5, are displayed in Figure 9. In the small coupling region (Figure 9B), we observe that T∥(Fb) appears enhanced at gx, suggesting an orientation of the Fb dipolar tensor parallel to gx. Therefore, a structure similar to S5 likely describes the data better than S4, as illustrated in Figure 7C,D, where the orientation of the dipolar vector with respect to gx is displayed.
Figure 9.

94 GHz 19F Mims ENDOR spectra on F3Y122-β2/F2Y731-α2 (80 μM, TQ = 50 s, black lines) at T = 50 K. (A) Measurement with short τ values (∼250 ns). (B) Measurement with larger τ values (∼620 ns). Simulations including four different 19F atoms (Fa–Fd) are shown as blue lines and are based on S2 and S5 (Tables 1 and 2). Contributions of individual 19F atoms are shown as shaded areas: purple (Fa), cyan, (Fb), green (Fc), and brown (Fd).
Using these orientational constraints, global simulations of the orientation-selective ENDOR spectra based on models S2 and S5 were carried out with the DFT-predicted parameters listed in Table 1 and the ratio (i.e., the relative contribution of S2 and S5) varied until a minimum of residual could be found (SI 9). rmsd from these simulations for all samples amount to ca. 0.1 or 10% at the optimized ratios (Figure S18). We observed that the simulation of the large coupling Fa (Figure 9A) is not very sensitive to the weighting of S2 and S5. This is expected as, under those experimental conditions, the resonances of Fb–Fd are suppressed by the Mims blind spot in the center of the spectrum. Instead, the ratio Fb/Fc affects the simulations of the small coupling region, as can be seen in Figure 9B by the decomposition of the simulation into the individual contributions. We note that the obtained weighting of the flipped conformation slightly varies between samples from 18 to 33% within an error of 5% for each sample (Table 2). Therefore, we estimate that the flipped conformation represents on average 25 ± 10% of the molecular ensemble.
Table 1. Parameters Used for the ENDOR Simulations.
| atom (model) | F–Y356•a [Å] | Ax, Ay, Azb [kHz] | aiso [kHz] |
|---|---|---|---|
| Fa (S5) | 4.6 | 580, −1668, −1952 | –1013 |
| Fb (S5) | 7.3 | –246, −246, 492 | 0 |
| Fc (S2) | 8.4 | –159, −159, 318 | 0 |
| Fd (S2) | 10.0 | –83, −83, 166 | 0 |
Coupling constants Ai consider the anisotropic and the isotropic coupling constants (Ti and aiso, respectively): Ai = Ti + aiso. Euler angles for relating the A to g tensors are reported in Table S8. An error of ±15 kHz was estimated for couplings <500 kHz, while an error of ±125 kHz is estimated for the 1.6 MHz coupling (ca. 50% of the ENDOR line width parameter in both cases).
Table 2. Ratios of the Stacked Model S2 and the Flipped Model S5 from ENDOR Simulations.
| RNR mutant | TQ [s] | contribution of flipped (S5)a |
|---|---|---|
| F3Y122-β2/F2Y731-α2 | 50 | 33% |
| F3Y122-β2/F2Y731-α2 | 143 | 22% |
| E52Q/F3Y122-β2/F2Y731-α2 | 35 | 18% |
| E52Q/F3Y122-β2/F2Y731-α2 | 153 | 25% |
Estimated error: ±5%; see Figure S18.
The representative best simulation for one sample F3Y122-β2/F2Y731-α2 is superimposed on the experimental data in Figure 9. Remarkably, the simulation of the orientation-selective spectrum at B0 ∥ gx captures the selectivity for T∥(Fb) and T∥(Fc) and also reproduces the shoulders on the inner side, which were tentatively assigned to Fd in the discussion of Figure 5C. Given the challenges of the simulation procedure, we find that the obtained simulation reproduces the experimental data very satisfactorily.
3.3. 17O ENDOR with (E52Q)-F3Y122-β2/17O–Y-wt-α2
An independent effort was made to obtain experimental evidence for a flipped Y731 conformation in the trapped complex. We investigated whether a 17O ENDOR signal might be observable with a sample prepared using uniformly labeled 17O–Y-wt-α2 (17O in the phenol groups). This experiment was motivated by our recent successful observation of a 17O ENDOR signal from water H-bonded to Y356•.28 DFT calculations predicted a 17O–Y731–Y356• coupling of ∼0.5 MHz for the flipped structure S5, slightly smaller than observed for H-bonded 17OH2 (0.7 MHz) (Table S9). We further considered issues that might make detection of this interaction more challenging. 17O has a lower gyromagnetic ratio than 19F (γ(19F)/γ(17O) ≈ 6.95) and its quadrupolar coupling may lead to signal broadening. In addition, the 17O–Y731-α2 is only 35–40%-labeled based on the available 17O–Y used during expression (SI 6). A reference ENDOR signal, with a comparable concentration of predicted 17O spins in close proximity to Y356• (i.e., ca 10–20 μM), is shown in Figure S15. Despite potential unexpected issues, we proceeded with the experiment as 17O should be a sensitive nucleus at short distances (≲3 Å) and the 17O–Y731 coupling for the stacked conformation should not be detectable, allowing us to test the flipped Y731 model. As shown in SI 6.2, we were not able to observe any 17O couplings in three independently prepared samples. We have considered several possible explanations for these observations that may be related either to the experiment or to the use of FnY probes: (1) the 17O coupling might be smaller than the DFT prediction and not detectable; (2) F2Y731 could experience a different flipping ratio or rate of flipping relative to Y731; (3) the F3Y122• used to initiate radical transfer in the experiment is likely reduced to its phenolate, not phenol as with Y122•, and could play a role for the subunit interaction. These scenarios will be further discussed in the next section.
4. Discussion
In this paper, we report the use of 94 GHz 19F ENDOR spectroscopy, which has provided new insight into the chemistry of RT between Y356(β) and Y731(α) of E. coli RNR located at the subunit interface (Figure 2C,D). Success was possible using enzymes with site-specifically incorporated FnYs: F3Y122-β2 (or E52Q/F3Y122-β2) and F2Y731-α2, which, when incubated with substrate (GDP) and effector (TTP), allowed trapping of the Y356• pathway radical in an “active” α2β2 complex during the reverse RT pathway process. PELDOR and HF-EPR analysis established the location of the trapped radical, and the double mutant provided a direct link to the recent cryo-EM structure.38 The studies allowed measurement of the 19F–Y731 hyperfine couplings to Y356•, which report on their interspin distances and provide interesting mechanistic implications.
Analysis of 94 GHz 19F ENDOR spectra of the Y356• required careful evaluation and subtraction of 19F signals associated with unreduced F3Y122• and 1H backgrounds. Nevertheless, comparison of the spectra acquired at 50 and 80 K allowed unambiguous assignment of three distinct couplings between F2Y731 and Y356•.
Construction of small models of the three Ys and their DFT-predicted 19F HFC couplings, ENDOR orientation selection, and spectral simulations indicated that the 19F spectra are consistent with a mixture of flipped and stacked conformations of F2Y731 with respect to Y730, with flipped contributions of 25 ± 10% among the samples. While the flexibility of Y731 has been reported previously, the present results provide the first evidence for a conformation, in which the two pathway residues are located at an O–O distance of ∼3 Å, with potentially important consequences for understanding the interfacial PCET step. The presence of both conformations simultaneously suggests that they are energetically similar and may exist in equilibrium.
A number of different types of experiments have previously reported multiple Y731 conformations.26,30 In one study, in which CDP/ATP was incubated with wt-β2/R411A-NH2Y731-α2, an NH2Y731• intermediate trapped in the forward RT was observed.26 The flipping was detected by PELDOR spectroscopy by its unusual Y356•/NH2Y731• distance. This distance, however, was only observed in conjunction with an additional mutation at α-R411A. This residue sits in the α/β interface. In addition, transient absorption experiments in solution using the same α-R411A mutation and a photo-oxidant indicated a kPCET between Y356F-photoβ2 and Y731 much faster than dNDP formation, ∼104 s–1 versus 1–10 s–1.30
On the other hand, neither in the cryo-EM structure with E52Q/F3Y122-β2 nor in the 17O ENDOR experiments, which both employed F3Y122• and wt-α2, was the flipped conformation of Y731 observed. Thus, while the role of F2Y731 in potentiating flipping is still unclear, the F3-phenolate generated at residue 122 during RT may not be the basis for a flipped Y731 conformation. In addition, the conditions for freeze-quenching the cryo-EM and ENDOR samples are very distinct in terms of protein concentration and glycerol content. A protein concentration of ∼80 μM had to be used for EPR samples, exceeding physiological RNR concentrations (ca. 1 μM). At elevated protein concentrations, the formation of α4β4 complexes has been reported.69,70 However, these complexes are incapable of producing Y356• and should not affect the analysis of EPR experiments, in which Y356• was observed selectively.
Overall, the complex interplay between Y356(β), Y731(α), R411(α), and other residues at the subunit interface is likely to be crucial for regulating the communication between the two redox-active Ys across the α/β interface.
Inspecting the predicted HFC parameters of the phenolic proton of F2Y731 with respect to Y356• is another interesting source of information. The DFT calculations predicted HFCs of ∼6 MHz in models S4 and S5. It is important to rationalize this finding in the context of previous 1H ENDOR studies on H-bond interactions to Y356•.27 In those studies, a 1H coupling in the range of 6 MHz was observed and assigned to one (or 2 equiv) H-bonded water molecule(s). The presence of the second water molecule was postulated to explain the unprecedented low gx value of Y356•, i.e., 2.0062.27 The sharp peaks observed in our recent 263 GHz 17O ENDOR experiments support the presence of only a single water molecule.28 Given the similarity of coupling constants for the H-bonded protons for Y731 from either model S4 or S5, the flipped conformation provides an explanation for the 1H coupling consistent with these previous 1H ENDOR data. To date, however, no ENDOR study has provided information on the interplay between stacked/flipped Y731 and the water binding at Y356•, which may be a key feature to control PCET across the interface. Interestingly, no distribution of gx values at Y356• is observed, indicating that the electrostatic environment is well defined and similar in both Y731 conformations. A mechanism, by which Y731 replaces a water molecule as a H-bond donor to Y356• upon flipping, could explain this finding.
4.1. Implication of Flipped Y731 in PCET across α/β
Observation of flipped F2Y731 in close distance to Y356•, trapped in an active RNR complex, enables the examination of a mechanism for the PCET step between Y356• and Y731 for the first time.
The current hypothesis for interfacial PCET involving water, as noted above, was based on the ENDOR studies and the H-bond to Y356• assigned to water.27,28 Recent MD simulations45 based on the cryo-EM structure supported the role of water first suggested by Nick et al.27 The simulations additionally showed that water molecules can be present at the α/β interface including between Y356 and Y731, between Y356 and β-E52 (an interface residue), and support a pathway for water to escape to the bulk solvent.38,45 Interestingly, MD also revealed an equilibrium between flipped and stacked conformation for Y731, both populated at room temperature.45 Nevertheless, the reported flipped Y731 structure from the MD study still shows a long O–O distance to Y356 (∼8 Å on average), precluding a direct interaction between the two Ys.45
Thus, the mechanism of PCET between Y356 and Y731 (i.e., during reverse and forward RTs) remained to be resolved due to the long Y356–Y731 distance (∼8 Å) observed in the cryo-EM structure.38 We note that the published cryo-EM structure and ENDOR data have distinct problems. The resolution of the cryo-EM structure was insufficient to resolve waters. The ENDOR studies only detected water in the first coordination sphere of Y356•, i.e., in a distance range of ∼3 Å.27,28
The 19F ENDOR data presented here, despite the issues raised, provide evidence for close interaction between the two Ys across the subunit interface in an active RNR construct. In our ENDOR-derived model S5, the O–O distance between Y356•–Y731 amounts to 3.0 ± 0.2 Å, with a similar value in the related model S4. This distance is within the range of the distances reported for the pathway pair C439–Y730 (O–S: 3.7 Å in the X-ray structure of α2 versus 3.4 Å in α-NH2Y730)13,44 as well as for the pair Y730–Y731 (O–O: 3.3 Å in α2 versus 2.7 Å in α-NH2Y730).13,44 For these pairs, independent quantum chemical calculations predicted a colinear PCET mechanism,24,71,72 in which the electron and proton are transferred individually in one step from the same donor to the same acceptor, although a water-assisted PCET has been proposed and discussed for the C439–Y730 pair.73 Recently, also an alternative, glutamate (E623)-mediated proton transfer for the RT between Y731 and Y730, has been proposed based on MD simulations and QM/MM analysis.74 A key conclusion from the latter study based on the analysis of E623 was that forward and reverse RTs are different. Interestingly, our earlier large-scale DFT calculation on the pathway triad C439–Y730–Y731 predicted that the coordination of a water molecule to Y730• can stabilize this radical intermediate and the transition states to the next pathway intermediates, Y731• and C439•.24 Therefore, the calculation pointed to a functional role of water in PCET without its direct involvement as a proton donor or acceptor. Based on these considerations, we propose that our current results are consistent with a model of colinear PCET mechanism for the RT Y356•(β) – Y731(α) ⇄ Y356(β) – Y731•(α). This mechanism requires a conformational change of Y731 during the long-range RT, as the next step (Y731•(α) – Y730(α) ⇄ Y731(α) – Y730•(α)) occurs in the stacked conformation of the Y731/Y730 pair.
5. Conclusions
Use of site-specifically incorporated unnatural amino acids and kinetic trapping in conjunction with high-field ENDOR, PELDOR, and EPR spectroscopies has given new insight into the PCET involving Y356•(β) and Y731(α) across the RNR subunit interface. 19F ENDOR revealed two sets of hyperfine coupling constants for F2Y731 caused by the occurrence of two distinct conformations. One set of hyperfine couplings is consistent with a stacked Y731 conformation at an ∼8 Å distance (O–O) to Y356•, as observed by cryo-EM. However, much larger 19F couplings revealed a second conformation, in which F2Y731 is flipped toward Y356• at a much shorter O–O distance of ∼3 Å. This distance is similar to distances between other Y pairs on the RT pathway in α, for which colinear PCET has been established.
These results reveal again the ability and importance of EPR spectroscopic methods and new experimental designs for the detection of multiple conformations in a biological machinery.
Acknowledgments
The authors thank Dr. M. Hiller for the initial recording of 263 GHz and Anna Doppler for assistance in EPR sample preparation and initial PELDOR measurements. J.S. would like to thank Dan Nocera for the continuous discussions about PCET and his comments on this manuscript. Sabine König is acknowledged for assistance with the mass spectrometry experiments. Dr. Dalvin Méndez-Hernández and Prof. Dr. Ana L. Moore are gratefully acknowledged for providing their structural models of water-binding phenoxyl radicals.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c02906.
Measurement of RNR activity, EPR sample preparation, EPR experiments (263, 34, and 94 GHz spectroscopies including relaxation measurements, radical yield determination, and background corrections), 17O ENDOR data, modeling of the tyrosine triad, and ENDOR simulations (PDF)
Author Present Address
¶ Frankfurt Cancer Institute, Goethe University Frankfurt, 60596 Frankfurt, Germany
Author Present Address
◆ Department of Medicine II, Hematology/Oncology, University Hospital Frankfurt, 60590 Frankfurt, Germany
Author Present Address
▼ Department of Chemistry, Texas A&M University, College Station, Texas 77843, United States.
The authors acknowledge funding from the Max Planck Society, the German DFG priority program SPP 1601, the ERC-BIO-enMR grant (contract 101020262), a fellowship of the graduate school GGNB (A.K., program Physics of Biological and Complex Systems), and the NIH grants GM047274 (C.C.) and GM-29595 (J.S. and C.C.). Open access funded by Max Planck Society.
The authors declare no competing financial interest.
Notes
Original spectroscopic data associated with Figures 5 and 9 as well as all xyz coordinates of the model structures can be accessed via the open database Göttingen Research Online (https://doi.org/10.25625/YXHC63).
This paper published ASAP on June 2, 2022 with a missing author (Manas K. Ghosh). The paper was corrected and the revised manuscript reposted on June 17, 2022.
Supplementary Material
References
- Hofer A.; Crona M.; Logan D. T.; Sjöberg B.-M. DNA Building Blocks: Keeping Control of Manufacture. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 50–63. 10.3109/10409238.2011.630372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordlund P.; Reichard P. Ribonucleotide Reductases. Annu. Rev. Biochem. 2006, 75, 681–706. 10.1146/annurev.biochem.75.103004.142443. [DOI] [PubMed] [Google Scholar]
- Stubbe J.; Nocera D. G. Radicals in Biology: Your Life Is in Their Hands. J. Am. Chem. Soc. 2021, 143, 13463–13472. 10.1021/jacs.1c05952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aye Y.; Li M.; Long M.; Weiss R. Ribonucleotide Reductase and Cancer: Biological Mechanisms and Targeted Therapies. Oncogene 2015, 34, 2011–2021. 10.1038/onc.2014.155. [DOI] [PubMed] [Google Scholar]
- Zimanyi C. M.; Chen P. Y.-T.; Kang G.; Funk M. A.; Drennan C. L. Molecular Basis for Allosteric Specificity Regulation in Class Ia Ribonucleotide Reductase from Escherichia Coli. eLife 2016, 5, e07141 10.7554/eLife.07141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene B. L.; Kang G.; Cui C.; Bennati M.; Nocera D. G.; Drennan C. L.; Stubbe J. Ribonucleotide Reductases: Structure, Chemistry, and Metabolism Suggest New Therapeutic Targets. Annu. Rev. Biochem. 2020, 89, 45–75. 10.1146/annurev-biochem-013118-111843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker C. H.; Banzon J.; Bollinger J.; Stubbe J.; Samano V.; Robins M.; Lippert B.; Jarvi E.; Resvick R. 2′-Deoxy-2′-Methylenecytidine and 2′-Deoxy-2′, 2′-Difluorocytidine 5′-Diphosphates: Potent Mechanism-Based Inhibitors of Ribonucleotide Reductase. J. Med. Chem. 1991, 34, 1879–1884. 10.1021/jm00110a019. [DOI] [PubMed] [Google Scholar]
- Hertel L. W.; Boder G. B.; Kroin J. S.; Rinzel S. M.; Poore G. A.; Todd G. C.; Grindey G. B. Evaluation of the Antitumor Activity of Gemcitabine (2′, 2′-Difluoro-2′-Deoxycytidine). Cancer Res. 1990, 50, 4417–4422. [PubMed] [Google Scholar]
- Narasimhan J.; Letinski S.; Jung S. P.; Gerasyuto A.; Wang J.; Arnold M.; Chen G.; Hedrick J.; Dumble M.; Ravichandran K.; Levitz T.; Cui C.; Drennan C. L.; Stubbe J.; Karp G.; Branstrom A. Ribonucleotide Reductase, a Novel Drug Target for Gonorrhea. eLife 2022, 11, e67447 10.7554/eLife.67447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutia B. M.; Frame M. C.; Subak-Sharpe J. H.; Clark W. N.; Marsden H. S. Specific Inhibition of Herpesvirus Ribonucleotide Reductase by Synthetic Peptides. Nature 1986, 321, 439–441. 10.1038/321439a0. [DOI] [PubMed] [Google Scholar]
- Shao J.; Zhou B.; Chu B.; Yen Y. Ribonucleotide Reductase Inhibitors and Future Drug Design. Curr. Cancer Drug Targets 2006, 6, 409–431. 10.2174/156800906777723949. [DOI] [PubMed] [Google Scholar]
- Aye Y.; Stubbe J. Clofarabine 5′-Di and-Triphosphates Inhibit Human Ribonucleotide Reductase by Altering the Quaternary Structure of Its Large Subunit. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 9815–9820. 10.1073/pnas.1013274108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlin U.; Eklund H. Structure of Ribonucleotide Reductase Protein R1. Nature 1994, 370, 533–539. 10.1038/370533a0. [DOI] [PubMed] [Google Scholar]
- Nordlund P.; Eklund H. Structure and Function of the Escherichia Coli Ribonucleotide Reductase Protein R2. J. Mol. Biol. 1993, 232, 123–164. 10.1006/jmbi.1993.1374. [DOI] [PubMed] [Google Scholar]
- Minnihan E. C.; Nocera D. G.; Stubbe J. Reversible, Long-Range Radical Transfer in E. Coli Class Ia Ribonucleotide Reductase. Acc. Chem. Res. 2013, 46, 2524–2535. 10.1021/ar4000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes-Schiffer S. Proton-Coupled Electron Transfer: Moving Together and Charging Forward. J. Am. Chem. Soc. 2015, 137, 8860–8871. 10.1021/jacs.5b04087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukier R. I.; Nocera D. G. Proton-Coupled Electron Transfer. Annu. Rev. Phys. Chem. 1998, 49, 337–369. 10.1146/annurev.physchem.49.1.337. [DOI] [PubMed] [Google Scholar]
- Reece S. Y.; Nocera D. G. Proton-Coupled Electron Transfer in Biology: Results from Synergistic Studies in Natural and Model Systems. Annu. Rev. Biochem. 2009, 78, 673–699. 10.1146/annurev.biochem.78.080207.092132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbe J.; Nocera D. G.; Yee C. S.; Chang M. C. Radical Initiation in the Class I Ribonucleotide Reductase: Long-Range Proton-Coupled Electron Transfer?. Chem. Rev. 2003, 103, 2167–2202. 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- Bennati M.; Weber A.; Antonic J.; Perlstein D. L.; Robblee J.; Stubbe J. Pulsed ELDOR Spectroscopy Measures the Distance between the Two Tyrosyl Radicals in the R2 Subunit of the E. Coli Ribonucleotide Reductase. J. Am. Chem. Soc. 2003, 125, 14988–14989. 10.1021/ja0362095. [DOI] [PubMed] [Google Scholar]
- Bennati M.; Robblee J. H.; Mugnaini V.; Stubbe J.; Freed J. H.; Borbat P. EPR Distance Measurements Support a Model for Long-Range Radical Initiation in E. Coli Ribonucleotide Reductase. J. Am. Chem. Soc. 2005, 127, 15014–15015. 10.1021/ja054991y. [DOI] [PubMed] [Google Scholar]
- Seyedsayamdost M. R.; Chan C. T.; Mugnaini V.; Stubbe J.; Bennati M. PELDOR Spectroscopy with DOPA-β2 and NH2Y-α2s: Distance Measurements between Residues Involved in the Radical Propagation Pathway of E. Coli Ribonucleotide Reductase. J. Am. Chem. Soc. 2007, 129, 15748–15749. 10.1021/ja076459b. [DOI] [PubMed] [Google Scholar]
- Yokoyama K.; Smith A. A.; Corzilius B.; Griffin R. G.; Stubbe J. Equilibration of Tyrosyl Radicals (Y356•, Y731•, Y730•) in the Radical Propagation Pathway of the Escherichia Coli Class Ia Ribonucleotide Reductase. J. Am. Chem. Soc. 2011, 133, 18420–18432. 10.1021/ja207455k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argirevic T.; Riplinger C.; Stubbe J.; Neese F.; Bennati M.; ENDOR; Spectroscopy; Calculations DFT. Evidence for the Hydrogen-Bond Network within α2 in the PCET of E. Coli Ribonucleotide Reductase. J. Am. Chem. Soc. 2012, 134, 17661–17670. 10.1021/ja3071682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick T. U.; Lee W.; Koßmann S.; Neese F.; Stubbe J.; Bennati M. Hydrogen Bond Network between Amino Acid Radical Intermediates on the Proton-Coupled Electron Transfer Pathway of E. Coli α2 Ribonucleotide Reductase. J. Am. Chem. Soc. 2015, 137, 289–298. 10.1021/ja510513z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasanmascheff M.; Lee W.; Nick T. U.; Stubbe J.; Bennati M. Radical Transfer in E. Coli Ribonucleotide Reductase: A NH2Y731/R411A-α Mutant Unmasks a New Conformation of the Pathway Residue 731. Chem. Sci. 2016, 7, 2170–2178. 10.1039/C5SC03460D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick T. U.; Ravichandran K. R.; Stubbe J.; Kasanmascheff M.; Bennati M. Spectroscopic Evidence for a H Bond Network at Y356 Located at the Subunit Interface of Active E. Coli Ribonucleotide Reductase. Biochemistry 2017, 56, 3647–3656. 10.1021/acs.biochem.7b00462. [DOI] [PubMed] [Google Scholar]
- Hecker F.; Stubbe J.; Bennati M. Detection of Water Molecules on the Radical Transfer Pathway of Ribonucleotide Reductase by 17O Electron–Nuclear Double Resonance Spectroscopy. J. Am. Chem. Soc. 2021, 143, 7237–7241. 10.1021/jacs.1c01359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokern Y.; Eltzner B.; Huckemann S. F.; Beeken C.; Stubbe J.; Tkach I.; Bennati M.; Hiller M. Statistical Analysis of ENDOR Spectra. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2023615118 10.1073/pnas.2023615118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene B. L.; Taguchi A. T.; Stubbe J.; Nocera D. G. Conformationally Dynamic Radical Transfer within Ribonucleotide Reductase. J. Am. Chem. Soc. 2017, 139, 16657–16665. 10.1021/jacs.7b08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holder P. G.; Pizano A. A.; Anderson B. L.; Stubbe J.; Nocera D. G. Deciphering Radical Transport in the Large Subunit of Class I Ribonucleotide Reductase. J. Am. Chem. Soc. 2012, 134, 1172–1180. 10.1021/ja209016j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song D. Y.; Pizano A. A.; Holder P. G.; Stubbe J.; Nocera D. G. Direct Interfacial Y731 Oxidation in α2 by a Photoβ2 Subunit of E. Coli Class Ia Ribonucleotide Reductase. Chem. Sci. 2015, 6, 4519–4524. 10.1039/C5SC01125F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui C.; Greene B. L.; Kang G.; Drennan C. L.; Stubbe J.; Nocera D. G. Gated Proton Release during Radical Transfer at the Subunit Interface of Ribonucleotide Reductase. J. Am. Chem. Soc. 2021, 143, 176–183. 10.1021/jacs.0c07879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene B. L.; Stubbe J.; Nocera D. G. Photochemical Rescue of a Conformationally Inactivated Ribonucleotide Reductase. J. Am. Chem. Soc. 2018, 140, 15744–15752. 10.1021/jacs.8b07902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Climent I.; Sjoeberg B. M.; Huang C. Y. Carboxyl-Terminal Peptides as Probes for Escherichia Coli Ribonucleotide Reductase Subunit Interaction: Kinetic Analysis of Inhibition Studies. Biochemistry 1991, 30, 5164–5171. 10.1021/bi00235a008. [DOI] [PubMed] [Google Scholar]
- Climent I.; Sjoeberg B. M.; Huang C. Y. Site-Directed Mutagenesis and Deletion of the Carboxyl Terminus of Escherichia Coli Ribonucleotide Reductase Protein R2. Effects on Catalytic Activity and Subunit Interaction. Biochemistry 1992, 31, 4801–4807. 10.1021/bi00135a009. [DOI] [PubMed] [Google Scholar]
- Sjöberg B. M.; Karlsson M.; Jörnvall H. Half-Site Reactivity of the Tyrosyl Radical of Ribonucleotide Reductase from Escherichia Coli. J. Biol. Chem. 1987, 262, 9736–9743. 10.1016/S0021-9258(18)47996-3. [DOI] [PubMed] [Google Scholar]
- Kang G.; Taguchi A. T.; Stubbe J.; Drennan C. L. Structure of a Trapped Radical Transfer Pathway within a Ribonucleotide Reductase Holocomplex. Science 2020, 368, 424–427. 10.1126/science.aba6794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q.; Parker M. J.; Taguchi A. T.; Ravichandran K.; Kim A.; Kang G.; Shao J.; Drennan C. L.; Stubbe J. Glutamate 52-β at the α/β Subunit Interface of Escherichia Coli Class Ia Ribonucleotide Reductase is Essential for Conformational Gating of Radical Transfer. J. Biol. Chem. 2017, 292, 9229–9239. 10.1074/jbc.M117.783092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy B.; Decout J.-L.; Béguin C.; Fontecave M.; Allard P.; Kuprin S.; Ehrenberg A. NMR Studies of Binding of 5-FdUDP and DCDP to Ribonucleoside-Diphosphate Reductase from Escherichia Coli. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1995, 1247, 284–292. 10.1016/0167-4838(94)00241-8. [DOI] [PubMed] [Google Scholar]
- Ravichandran K.; Minnihan E. C.; Lin Q.; Yokoyama K.; Taguchi A. T.; Shao J.; Nocera D. G.; Stubbe J. Glutamate 350 Plays an Essential Role in Conformational Gating of Long-Range Radical Transport in Escherichia Coli Class Ia Ribonucleotide Reductase. Biochemistry 2017, 56, 856–868. 10.1021/acs.biochem.6b01145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravichandran K. R.; Taguchi A. T.; Wei Y.; Tommos C.; Nocera D. G.; Stubbe J. A >200 meV Uphill Thermodynamic Landscape for Radical Transport in Escherichia Coli Ribonucleotide Reductase Determined Using Fluorotyrosine-Substituted Enzymes. J. Am. Chem. Soc. 2016, 138, 13706–13716. 10.1021/jacs.6b08200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover S. D.; Parada G. A.; Markle T. F.; Ott S.; Hammarström L. Isolating the Effects of the Proton Tunneling Distance on Proton-Coupled Electron Transfer in a Series of Homologous Tyrosine-Base Model Compounds. J. Am. Chem. Soc. 2017, 139, 2090–2101. 10.1021/jacs.6b12531. [DOI] [PubMed] [Google Scholar]
- Minnihan E. C.; Seyedsayamdost M. R.; Uhlin U.; Stubbe J. Kinetics of Radical Intermediate Formation and Deoxynucleotide Production in 3-Aminotyrosine-Substituted Escherichia Coli Ribonucleotide Reductases. J. Am. Chem. Soc. 2011, 133, 9430–9440. 10.1021/ja201640n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt C. R.; Li P.; Kang G.; Stubbe J.; Drennan C. L.; Hammes-Schiffer S. Conformational Motions and Water Networks at the α/β Interface in E. Coli Ribonucleotide Reductase. J. Am. Chem. Soc. 2020, 142, 13768–13778. 10.1021/jacs.0c04325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravichandran K. R.; Zong A. B.; Taguchi A. T.; Nocera D. G.; Stubbe J.; Tommos C. Formal Reduction Potentials of Difluorotyrosine and Trifluorotyrosine Protein Residues: Defining the Thermodynamics of Multistep Radical Transfer. J. Am. Chem. Soc. 2017, 139, 2994–3004. 10.1021/jacs.6b11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt C. R.; Sequeira R.; Tommos C.; Hammes-Schiffer S. Computing Proton-Coupled Redox Potentials of Fluorotyrosines in a Protein Environment. J. Phys. Chem. B 2021, 125, 128–136. 10.1021/acs.jpcb.0c09974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells G. B.; Makinen M. W. ENDOR-Determined Molecular Geometries of Spin-Labeled Fluoroanilides in Frozen Solution. J. Am. Chem. Soc. 1988, 110, 6343–6352. 10.1021/ja00227a012. [DOI] [Google Scholar]
- Meyer A.; Dechert S.; Dey S.; Höbartner C.; Bennati M. Measurement of Angstrom to Nanometer Molecular Distances with 19F Nuclear Spins by EPR/ENDOR Spectroscopy. Angew. Chem., Int. Ed. 2020, 59, 373–379. 10.1002/anie.201908584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minnihan E. C.; Young D. D.; Schultz P. G.; Stubbe J. Incorporation of Fluorotyrosines into Ribonucleotide Reductase Using an Evolved, Polyspecific Aminoacyl-tRNA Synthetase. J. Am. Chem. Soc. 2011, 133, 15942–15945. 10.1021/ja207719f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thelander L.; Sjöberg B.-M.; Eriksson S.. Ribonucleoside Diphosphate Reductase (Escherichia Coli). In Methods in Enzymology; Academic Press, 1978; Vol. 51, pp 227–237. [DOI] [PubMed] [Google Scholar]
- Tkach I.; Bejenke I.; Hecker F.; Kehl A.; Kasanmascheff M.; Gromov I.; Prisecaru I.; Höfer P.; Hiller M.; Bennati M. 1H High Field Electron-Nuclear Double Resonance Spectroscopy at 263 GHz/9.4 T. J. Magn. Reson. 2019, 303, 17–27. 10.1016/j.jmr.2019.04.001. [DOI] [PubMed] [Google Scholar]
- Martin R. E.; Pannier M.; Diederich F.; Gramlich V.; Hubrich M.; Spiess H. W. Determination of End-to-end Distances in a Series of TEMPO Diradicals of up to 2.8 Nm Length with a New Four-pulse Double Electron Electron Resonance Experiment. Angew. Chem., Int. Ed. 1998, 37, 2833–2837. . [DOI] [PubMed] [Google Scholar]
- Pannier M.; Veit S.; Godt A.; Jeschke G.; Spiess H. W. Dead-Time Free Measurement of Dipole–Dipole Interactions between Electron Spins. J. Magn. Reson. 2011, 213, 316–325. 10.1016/j.jmr.2011.08.035. [DOI] [PubMed] [Google Scholar]
- Jeschke G.; Chechik V.; Ionita P.; Godt A.; Zimmermann H.; Banham J.; Timmel C.; Hilger D.; Jung H. DeerAnalysis2006—a Comprehensive Software Package for Analyzing Pulsed ELDOR Data. Appl. Magn. Reson. 2006, 30, 473–498. 10.1007/BF03166213. [DOI] [Google Scholar]
- Chiang Y.-W.; Borbat P. P.; Freed J. H. The Determination of Pair Distance Distributions by Pulsed ESR Using Tikhonov Regularization. J. Magn. Reson. 2005, 172, 279–295. 10.1016/j.jmr.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Worswick S. G.; Spencer J. A.; Jeschke G.; Kuprov I. Deep Neural Network Processing of DEER Data. Sci. Adv. 2018, 4, eaat5218 10.1126/sciadv.aat5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epel B.; Arieli D.; Baute D.; Goldfarb D. Improving W-Band Pulsed ENDOR Sensitivity—Random Acquisition and Pulsed Special TRIPLE. J. Magn. Reson. 2003, 164, 78–83. 10.1016/S1090-7807(03)00191-5. [DOI] [PubMed] [Google Scholar]
- Bruggemann W.; Niklas J. R. Stochastic ENDOR. J. Magn. Reson., Ser. A 1994, 108, 25–29. 10.1006/jmra.1994.1084. [DOI] [Google Scholar]
- Rizzato R.; Kaminker I.; Vega S.; Bennati M. Cross-Polarisation Edited ENDOR. Mol. Phys. 2013, 111, 2809–2823. 10.1080/00268976.2013.816795. [DOI] [Google Scholar]
- Stoll S.; Schweiger A. EasySpin, a Comprehensive Software Package for Spectral Simulation and Analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Kehl A.; Hiller M.; Hecker F.; Tkach I.; Dechert S.; Bennati M.; Meyer A. Resolution of Chemical Shift Anisotropy in 19F ENDOR Spectroscopy at 263 GHz/9.4 Tesla. J. Magn. Reson. 2021, 107091 10.1016/j.jmr.2021.107091. [DOI] [PubMed] [Google Scholar]
- DeLano W. L. Pymol: An Open-Source Molecular Graphics Tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Neese F. The ORCA Program System. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 73–78. 10.1002/wcms.81. [DOI] [Google Scholar]
- Lucarini M.; Mugnaini V.; Pedulli G. F.; Guerra M. Hydrogen-Bonding Effects on the Properties of Phenoxyl Radicals. An EPR, Kinetic, and Computational Study. J. Am. Chem. Soc. 2003, 125, 8318–8329. 10.1021/ja034963k. [DOI] [PubMed] [Google Scholar]
- Méndez-Hernández D. D.; Baldansuren A.; Kalendra V.; Charles P.; Mark B.; Marshall W.; Molnar B.; Moore T. A.; Lakshmi K.; Moore A. L. HYSCORE and DFT Studies of Proton-Coupled Electron Transfer in a Bioinspired Artificial Photosynthetic Reaction Center. iScience 2020, 23, 101366 10.1016/j.isci.2020.101366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravichandran K. R.; Minnihan E. C.; Wei Y.; Nocera D. G.; Stubbe J. Reverse Electron Transfer Completes the Catalytic Cycle in a 2,3,5-Trifluorotyrosine-Substituted Ribonucleotide Reductase. J. Am. Chem. Soc. 2015, 137, 14387–14395. 10.1021/jacs.5b09189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riplinger C.; Kao J. P.; Rosen G. M.; Kathirvelu V.; Eaton G. R.; Eaton S. S.; Kutateladze A.; Neese F. Interaction of Radical Pairs Through-Bond and through-Space: Scope and Limitations of the Point– Dipole Approximation in Electron Paramagnetic Resonance Spectroscopy. J. Am. Chem. Soc. 2009, 131, 10092–10106. 10.1021/ja901150j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando N.; Brignole E. J.; Zimanyi C. M.; Funk M. A.; Yokoyama K.; Asturias F. J.; Stubbe J.; Drennan C. L. Structural Interconversions Modulate Activity of Escherichia Coli Ribonucleotide Reductase. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 21046–21051. 10.1073/pnas.1112715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimanyi C. M.; Ando N.; Brignole E. J.; Asturias F. J.; Stubbe J.; Drennan C. L. Tangled up in Knots: Structures of Inactivated Forms of E. Coli Class Ia Ribonucleotide Reductase. Structure 2012, 20, 1374–1383. 10.1016/j.str.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegbahn P. E. M.; Eriksson L.; Himo F.; Pavlov M. Hydrogen Atom Transfer in Ribonucleotide Reductase (RNR). J. Phys. Chem. B 1998, 102, 10622–10629. 10.1021/jp9827835. [DOI] [Google Scholar]
- Kaila V. R. I.; Hummer G. Energetics of Direct and Water-Mediated Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2011, 133, 19040–19043. 10.1021/ja2082262. [DOI] [PubMed] [Google Scholar]
- Chen X.; Ma G.; Sun W.; Dai H.; Xiao D.; Zhang Y.; Qin X.; Liu Y.; Bu Y. Water Promoting Electron Hole Transport between Tyrosine and Cysteine in Proteins via a Special Mechanism: Double Proton Coupled Electron Transfer. J. Am. Chem. Soc. 2014, 136, 4515–4524. 10.1021/ja406340z. [DOI] [PubMed] [Google Scholar]
- Reinhardt C. R.; Sayfutyarova E. R.; Zhong J.; Hammes-Schiffer S. Glutamate Mediates Proton-Coupled Electron Transfer Between Tyrosines 730 and 731 in Escherichia Coli Ribonucleotide Reductase. J. Am. Chem. Soc. 2021, 143, 6054–6059. 10.1021/jacs.1c02152. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.