

Abstract
Background.
Second hand smoke (SHS) exposure is associated with pediatric asthma, and oxidative stress is believed to play a role in mediating this association. The nuclear factor (erythroid-derived 2)-like 2 (NFE2L2) is important for the defense against oxidative stress.
Objective.
To explore interactions between NFE2L2 genotype and SHS exposure in pediatric asthma risk.
Methods.
We used a genotyped subset of patients of European ancestry (N=669, median age at enrollment=6.8 years) enrolled in the clinical cohort Greater Cincinnati Pediatric Clinic Repository as the study population, and a population-based pediatric cohort (N=791) to replicate our findings. History of asthma diagnosis was obtained from medical records, and SHS exposure was obtained from questionnaires. Four NFE2L2 tagging SNPs were included in the analysis, and interactions between SHS and NFE2L2 genotype were evaluated using logistic regression.
Results.
Three of the analyzed SNPs, rs10183914, rs1806649, and rs2886161, interacted significantly with SHS exposure to increase asthma risk (p≤0.02). The interaction was replicated in an independent cohort for rs10183914 (p=0.04). Interactions between SHS exposure and NFE2L2 genotype were also associated with an increased risk of hospitalization (p=0.016). In stratified analyses, NFE2L2 genotype was associated with daily asthma symptoms in children with SHS exposure (OR=3.1; p=0.048). No association was found in children without SHS exposure.
Examination of publicly available chromatin immunoprecipitation followed by sequencing (ChIP-seq) datasets confirmed the presence of active histone marks and binding sites for particular transcription factors overlapping the coordinates for the significantly associated SNPs.
Conclusions & Clinical Relevance.
Our study provides evidence that NFE2L2 genotype interacts with SHS exposure to affect both asthma risk and severity in children and identifies a population of children at increased risk of asthma development.
Keywords: asthma, NFE2L2, second hand smoke, gene-environment interaction, children
Introduction
Exposure to second hand smoke (SHS) has repeatedly been shown to be associated with asthma development in both children and adults, and in the Surgeon General’s 2006 report it was concluded that there is sufficient evidence for a causal relationship between exposure to SHS and asthma prevalence in school-aged children[1]. In addition, SHS exposure has been shown to be associated with asthma exacerbation in children, especially in preschool-age children[2-4].
While there is a clear causal relationship between SHS and childhood asthma, not all children exposed to SHS develop asthma. Genetic variation may modulate the response to SHS, which may explain why some individuals are more susceptible to SHS exposure than others[5-7]. An example of this type of effect is the association of asthma development with the interaction between SHS, as well as in utero smoke exposure, and the antioxidant gene glutathione S-transferase M1 (GSTM1)[8-10]. Other candidate genes that have been shown to modify the association between SHS exposure and asthma include glutathione S-transferase P1 (GSTP1)[11, 12], the immune response gene ICAM1[13], NAT1[14], and the ciliary gene DNAH9[15].
The nuclear factor (erythroid-derived 2)-like 2 (NFE2L2) is a basic leucine zipper transcription factor that regulates the expression of antioxidant genes by binding to antioxidant response elements (AREs). Often called the master switch in the antioxidant defense system, NFE2L2 is activated by low levels of oxidative stress and reactive oxygen species (ROS), leading to the transcription of a large number of protective antioxidant genes. A role for NFE2L2 in asthma development is supported by studies on animal models[16]. NFE2L2-deficient mice have increased susceptibility to ovalbumin-driven airway inflammation and hyperresponsiveness[17], and allergic airway inflammatory responses induced by diesel exhaust particles in mice are increased in NFE2L2(−/−) mice[18].
The role of NFE2L2 in asthma development in humans has not yet been fully established, and only a few studies have explored the way NFE2L2 modifies the impact of oxidative stressors on lung function. The association between annual forced expiratory volume (FEV1) decline and smoking behavior in adults has been shown to be modified by NFE2L2 genotype[19], and a study of Hungarian children demonstrated that traffic-related air pollution, as assessed by nitrogen dioxide concentrations, modifies the association between NFE2L2 genotype and infection-induced asthma prevalence[20]. However, there have been no studies of interactions between exposure to SHS and NFE2L2 genotype in pediatric asthma.
The objective of this study was to test the hypothesis that NFE2L2 genotype and SHS exposure interact to increase the risk of childhood asthma. Given the association between smoke exposure and asthma severity, including hospitalization[21], we also evaluated associations between asthma phenotype and interactions between SHS and NFE2L2 genotype. We used a clinical cohort from the Greater Cincinnati area as the discovery population and the Cincinnati Genomic Control Cohort to confirm our findings.
Methods
Study populations
The study population used for this study was a genotyped subset of patients of European ancestry enrolled in the Greater Cincinnati Pediatric Clinic Repository (GCPCR), a clinical biorepository of over 7000 children with and without allergic diseases. Subjects were recruited from various clinics at Cincinnati Children’s Hospital Medical Center (CCHMC). To replicate our findings, we used white participants from the Genomic Control Cohort (GCC), a population cohort of 1,020 children age 3-18 years old from the Greater Cincinnati region[22, 23]. Recruitment, as well as the demographic and clinical characteristics of each of these cohorts has been described in detail previously[22-24]. Briefly, children with asthma and other allergic conditions were recruited to the GCPCR from the allergy/immunology, pulmonary, and dermatology outpatient specialty clinics and from the Emergency Department at CCHMC. Following written informed consent, a study-specific questionnaire was administered, and participants were asked to provide a buccal or saliva sample for DNA isolation. Genotyped GCPCR participants had at least 250ng of DNA with an OD of 1.6 to 2.2. Age in the GCPCR is defined as the age at the time of enrollment. The GCC participants were recruited to be representative of the Greater Cincinnati area. All GCC participants completed a questionnaire that included asthma, allergy and skin questions similar to those included in GCPCR and provided a blood sample for genetic analysis. This study was approved by the CCHMC Institutional Review Board under protocols 2010-0438 (GCPCR) and 2008-0089 (GCC).
Asthma diagnosis
Diagnosis of asthma was based on standard GCPCR definitions[22]. Briefly, a GCPCR participant with asthma diagnosed according to American Thoracic Society[25] criteria from the pulmonary or allergy clinics at CCHMC was considered asthmatic. Children with asthma diagnoses from non-specialty clinics or primary care physicians were excluded from the genotyping analyses. Asthma in the GCC cohort was defined as parental reports of physician diagnosis.
Information about symptom scores was available for 301 of the 444 participants in GCPCR who had a history of asthma. Respiratory symptom frequency in children diagnosed with asthma in the GCPCR was based our validated score of the frequency of cough, wheezing, shortness of breath, and/or chest tightness[22]. Scores ranges from 0-4, with 4 being daily symptoms. Subjects were defined as having daily symptoms if they scored a 4 for any of the four respiratory symptoms. Information about ever having been hospitalized was available for 436 participants with a history of asthma.
Comparable information about asthma severity was not available for participants in GCC.
Environmental exposure
Exposure to second-hand smoke (SHS) was assessed through detailed questionnaires administered at enrollment and at follow-up visits. SHS exposure was defined as having reported the child spending time in the same area as someone who smokes, including time spent at home, in daycare, in the car, or at someone else’s house.
DNA isolation and genotyping
For both study populations, DNA was isolated from buccal swabs as described previously [26]. Study participants genotyped for this study were white and aged between 5 and 17 years. Four NFE2L2 tagging SNPs were selected using HapMap for European ancestry (CEU)[27]. Coordinates of the analyzed SNPs can be found in Supplemental Table 1. Included SNPs had pairwise tagging SNP r2 values of at least 0.8. For both study populations, the four analyzed SNPs were in Hardy-Weinberg equilibrium (p > 0.05 in GCPCR; p > 0.001 in GCC), missing call rates were < 5%, and MAF (minor allele frequency) were > 10%. The four selected SNPs were in strong linkage disequilibrium, as shown in Figure 1. High-throughput genotyping was performed by the CCHMC Genetic Variation and Gene Discovery Core using the Illumina Golden Gate assay (http://www.illumina.com). Genotypes were assigned using GenomeStudio V2010.1 Software (Illumina, San Diego, CA). We used existing information in GCC on genotyping previously performed as described above for the replication analysis. Available SNP genotypes included rs10183914, rs6726395, and rs1806649, but not rs2886161.
Figure 1:

LD plot showing degree of linkage disequilibrium for each pair of NFE2L2 SNPs analyzed in this study calculated as D’ (the difference between the observed and the expected frequency of a given haplotype).
Genetic variant functional interpretation
In order to identify gene regulatory mechanisms potentially impacted by the genetic variants examined in this study, we collected lung cell functional genomics datasets from a variety of sources, including ChIP-seq datasets from the Gene Expression Omnibus[28], Roadmap Epigenomics[29], and ReMap-ChIP[30], and super enhancers from Hnisz et al.[31]. For datasets collected from GEO, we uniformly processed the sequencing reads to call peaks using the MARIO pipeline[32]. The genomic coordinates of each of the genetic variants were then intersected with the coordinates of these functional genomics dataset “peaks” using bedtools [33] and custom scripts.
Statistics
Population characteristics of children with versus without asthma in genotyped participants were compared using the Wilcoxon rank-sum test and Chi-square test for GCPCR and GCC cohort separately. The genotyped study population in GCPCR was also compared to all eligible white participants in GCPCR not included in the study. For each cohort, logistic regression models were performed to examine the associations of NFE2L2 SNPs with asthma in the genotyped participants, and also stratified by SHS exposure. Logistic regression models of asthma were also performed to formally test the interaction between SHS and NFE2L2. For the regression models of asthma risk, SNPs were coded as copy numbers of minor allele. Because of the smaller sample size and relatively small number of participants with a history of hospitalization in the models of hospitalization, SNP genotypes were modeled as binary variables to maximize power, with the minor allele homozygous genotype vs. the heterozygous and major allele homozygous genotypes. Age and Sex were included as covariates in all the logistic regression models. P values of Wald tests were reported. The correlations among our four SNPs of interest are high, with a mean correlation of 0.59. The LD adjusted Bonferroni correction for the four tests in the GCPCR cohort was calculated using the freely available Simple Interactive Statistical Analyses Software (http://www.quantitativeskills.com/sisa/), and the resulting cutoff for significance was p < 0.028. In all other analyses, a p-value < 0.05 was considered statistically significant. All the analyses were performed in R software, version 3.22 (https://www.r-project.org).
Results
Demographics
The number of genotyped white participants (participants of European ancestry) in GCPCR was 941. Of the genotyped participants, 246 were missing information about SHS exposure and 26 lacked conclusive information about asthma status. The remaining 669 participants included in this study had information about both variables. When compared with the larger group of white participants who had information about sex, age of enrollment, and asthma history (N=2914), the study population was representative with regard to sex and household income (Supplemental Table 2). Age (6.8 vs. 5.9, p=0.005) and the proportion of participants with a history of asthma was higher in the analyzed subgroup compared with the larger cohort (66.3%vs 40.4%, p<0.0001), which is expected as asthmatic cases were selected for genotyping and the larger GCPCR includes participants 0-18 but the genotyped subset restricted to ages 5-17. A current or past diagnosis of asthma was on record for 444 of the study participants, whereas 225 participants had no history of asthma (Table 1). Forty-four point six percent of asthmatic children had been exposed to SHS on a regular basis, which was significantly higher than the 26.7% exposed non-asthmatic children (p<0.001). In an adjusted regression analysis adjusted for age and sex, SHS remained significantly associated with asthma (OR=2.05; p<0.001). Fifty-seven point four percent of the asthmatics were male, compared with 60.4% of the non-asthmatics. The median enrollment age of asthmatic children was 7.9 years. Non-asthmatic children were significantly younger (4.1 years, p<0.001). Although household incomes were slightly higher for non-asthmatic children than for asthmatic children, this difference was not statistically significant (p=0.67). Among the asthmatic, 20.2% had daily asthma symptoms and 23.8% had been hospitalized in the past for asthma exacerbation.
Table 1.
Demographic characteristics of the genotyped subset of the GCPCR population, stratified for past or present asthma diagnosis.
| Variable1 | All participants (669) |
Asthma (n=444) |
no asthma (n=225) |
P-value |
|---|---|---|---|---|
| Male sex (%) | 58.4 | 57.4 | 60.4 | 0.50 |
| Age at enrollment (median years, IQR) | 6.8 (3.4, 11.0) | 7.9 (5.4, 12.0) | 4.1 (1.9, 8.1) | <.0001 |
| SHS exposure (%) | 38.6 | 44.6 | 26.7 | <0.001 |
| Household income2 | 0.67 | |||
| < $30K (%) | 22.4 | 23.1 | 21.3 | |
| $30K - $70K (%) | 28.2 | 29.0 | 26.8 | |
| > $70K (%) | 49.3 | 47.9 | 51.9 | |
| Daily asthma symptoms (%)3 | NA | 20.2 | NA | |
| Ever hospitalized (%)4 | NA | 23.8 | NA |
Categorical variables were presented as %, and p values were obtained by Chi-squared test. Continuous variable Age of enrollment was treated as a continuous variable and was presented as median (IQR). P value for age of enrollment was obtained by Wilcoxon rank sum test.
Missing for 115 participants.
Information about asthma symptom score was available for 301 of the participants with a history of asthma. Daily asthma symptoms were defined as having ever reported having cough, wheezing, shortness of breath, and/or chest tightness with a daily frequency.
Information about hospitalization was available for 436 of the participants with a history of asthma.
The lifetime history of asthma in the GCC control population was 12.3% (97 out of 791 participants). Neither sex nor SHS exposure was associated with asthma (Table 2). Age- and sex-adjusted logistic regression confirmed that SHS was not associated with asthma in GCC (OR=1.06; p=0.83). Similar to the GCPCR study population, asthma was associated with age of enrollment in the GCC, with asthmatic participants being older than non-asthmatics (12.6 vs 11.1, respectively; p=0.01). Asthma was significantly associated with income in GCC, and a higher proportion of asthmatics resided in households with income levels < < $30K compared with non-asthmatics (p=0.001).
Table 2.
Demographics for the genotyped GCC population, stratified for past or present asthma diagnosis.
| Variable1 | All participants (791) | Asthma (n=97) | No asthma (694) | P-value |
|---|---|---|---|---|
| Male sex (%) | 49.4 | 49.5 | 49.4 | 0.92 |
| Age at enrollment (median years, IQR) | 11.4 (7.6 - 14.6) | 12.6 (9.1 - 15.4) | 11.1 (7.4 - 14.5) | 0.01 |
| SHS exposure (%) | 19.8 | 20.6 | 19.7 | 0.95 |
| Household income | 0.001 | |||
| < $30K (%) | 10.1 | 20.6 | 8.7 | |
| $30K - $75K (%) | 39.8 | 33.0 | 40.7 | |
| > $75K (%) | 50.1 | 46.4 | 50.6 |
Categorical variables were presented as %, and p values were obtained by Chi-squared test. Age of enrollment was treated as a continuous variable and was presented as median years (IQR). P value for age of enrollment was obtained by Wilcoxon rank sum test.
NFE2L2 genotype interacts with SHS to increase the risk of asthma
We first analyzed associations of NFE2L2 with asthma in the genotyped GCPCR study cohort. Four SNPs were analyzed, and as shown in Figure 1 these SNPs were in strong linkage disequilibrium. None of the analyzed SNPs were significantly associated with asthma (Table 3A). Stratifying for SHS, however, revealed that the association between NFE2L2 and asthma differed by SHS exposure. Two of the SNPs, rs1806649, and rs2886161 were significantly associated with asthma in SHS-exposed children (p = 0.02, and 0.02, respectively), whereas rs10183914 was nominally associated with asthma in exposed children (p = 0.04). No NFE2L2 SNPs were associated with asthma in children without exposure to SHS. Based on regression modelling, there was a linear increase in the predicted percentage of children with asthma as a function of the number of rs10183914 minor alleles (the A allele) in the exposed, but not unexposed, group (Figure 2). Formal interaction analysis confirmed that the association between SHS and asthma was significantly modified by NFE2L2 genotype (p = 0.02, 0.01, and 0.02 for the interaction terms between SHS and rs10183914, rs1806649, and rs2886161, respectively).
Table 3.
Interaction analyses of NFE2L2 SNPs*SHS in asthma risk.
| A. GCPCR study population. | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Minor/ Major allele |
MAF | All (n=669) | SHS (n=258) | No SHS (n=411) | SNP*SHS Interaction (n=669) |
||||
| OR1 (95% CI) |
P | OR (95% CI) |
P | OR (95% CI) |
P | OR (95% CI) |
P | |||
| rs10183914 | A/G | 0.360 | 1.02 (0.79,1.32) |
0.89 | 1.70 (1.03,2.90) |
0.04 | 0.84 (0.62,1.14) |
0.25 | 1.99 (1.11,3.65) |
0.02 |
| rs6726395 | A/G | 0.456 | 0.96 (0.75,1.22) |
0.74 | 1.35 (0.87,2.12) |
0.18 | 0.82 (0.61,1.10) |
0.19 | 1.63 (0.97, 2.77) |
0.07 |
| rs1806649 | A/G | 0.239 | 1.02 (0.77,1.37) |
0.88 | 2.09 (1.14,4.10) |
0.02 | 0.80 (0.57,1.12) |
0.20 | 2.55 (1.28, 5.34) |
0.01 |
| rs2886161 | G/A | 0.338 | 0.91 (0.69,1.18) |
0.46 | 0.58 (0.37,0.91) |
0.02 | 1.14 (0.82,1.59) |
0.44 | 0.51 (0.29,0.90) |
0.02 |
| B. GCC replication cohort. | ||||||||||
| SNP | Minor/ Major allele |
MAF | All (n=791) | SHS (n=157) | No SHS (n=634) | SNP*SHS interaction (791) |
||||
| OR (95% CI) |
P | OR (95% CI) |
P | OR (95% CI) |
P | OR (95% CI) |
P | |||
| rs10183914 | A/G | 0.36 | 0.88 (0.64,1.20) |
0.42 | 1.48 (0.74,2.96) |
0.27 | 0.73 (0.50,1.05) |
0.1 | 2.20 (1.02,4.76) |
0.04 |
| rs6726395 | A/G | 0.456 | 0.94 (0.69,1.27) |
0.68 | 1.22 (0.62,2.40) |
0.56 | 0.85 (0.60,1.20) |
0.36 | 1.55 (0.75,3.25) |
0.24 |
| rs1806649 | A/G | 0.239 | 1.06 (0.75,1.46) |
0.75 | 1.35 (0.67,2.63) |
0.39 | 0.96 (0.64,1.40) |
0.83 | 1.52 (0.69,3.27) |
0.29 |
Odds ratios (ORs) for life time asthma risk were obtained using logistic regression analysis. The models were adjusted for age and sex. ORs refer to increase in risk for each additional minor allele. Genotypes were coded as copy numbers of minor allele (additive/continuous).
Figure 2:

Predicted probability of asthma as a function of the number of rs10183914 minor alleles (the A allele). Predicted probabilities were calculated using logistic regression. (A) GCPCR. (B) GCC.
Genotyping results for rs10183914, rs6726395, and rs1806649 were available in GCC, which enabled us to test our results for these three SNPs in an independent control cohort representative of the general pediatric population in the Greater Cincinnati region. As in the GCPCR, the predicted percentage of children with asthma increased linearly as a function of the number of rs10183914 minor A alleles in the exposed, but not unexposed, group (Figure 2). We observed a significant interaction between rs10183914 genotype and SHS that was associated with lifetime asthma prevalence (p=0.04; Table 3B). As in the GCPCR, no main-effect associations between NFE2L2 genotype and asthma were found. As mentioned above, household income was significantly associated with asthma in GCC. Adjusting for income in GCC did not, however, alter the results, and the interaction term was still significantly associated with asthma risk (p=0.03)
The interaction of rs10183914 and SHS exposure is associated with risk for increased asthma severity.
Since we observed that the interaction between NFE2L2 and SHS exposure is associated with the risk of asthma, we next examined associations with asthma severity defined by history of symptoms in children diagnosed with asthma as well as history of hospitalization. In the unstratified analysis including both exposed and unexposed children in GCPCR, rs10183914 genotype was not significantly associated with history of asthma symptom frequency, although there was a trend towards a greater percentage of daily symptoms for participants with the AA genotype (Figure 3, all). When the analysis was stratified for SHS exposure, the number of SHS-exposed participants with the AA genotype who had ever experienced daily symptoms was increased at more than double that observed for SHS-exposed participants with the AG or GG genotype (OR=3.1; p = 0.048). By contrast, there was no association between rs10183914 genotype and symptom frequency in unexposed participants. We also found that an interaction between rs6726395 and SHS exposure was significantly associated with ever having been hospitalized for asthma exacerbation in children with a history of asthma (Table 4). These results showed that interactions between NFE2L2 genotype and SHS exposure is associated with both increased asthma prevalence and a more severe phenotype, and that the rs10183914 AA allele is a risk allele for both asthma prevalence and severity.
Figure 3:
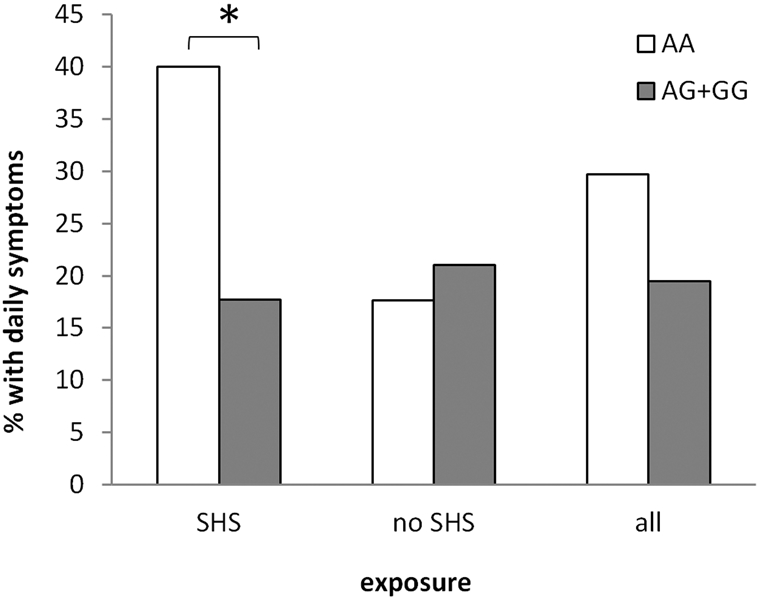
Percentage of asthmatic GCPCR participants who had ever reported daily asthma symptoms as a function of SHS exposure, stratified for rs10183914 genotype. SNPs were coded as binary (AA: the minor allele homozygous genotype; AG+GG: the heterozygous and major allele homozygous genotypes). Daily asthma symptoms was defined as having ever reported cough, wheezing, shortness of breath, and/or chest tightness with an average daily frequency. N = 301. Asterix indicates significance at a significance level of 0.05.
Table 4.
Interaction between SHS and NFE2L2 genotype in GCPCR in the regression of ever having been hospitalized for participants with a history of asthma.
| SNP1 | OR2 (95% CI) |
SNP*SHS interaction p value |
|---|---|---|
| rs10183914 | 0.55 (0.18, 1.55) |
0.27 |
| rs6726395 |
0.21
(0.05, 0.70) |
0.016 |
| rs1806649 | 0.59 (0.22, 1.58) |
0.3 |
| rs2886161 | 1.59 (0.60, 4.28) |
0.35 |
SNPs were coded as binary (0: the minor allele homozygous genotype; 1: the heterozygous and major allele homozygous genotypes).
Odds ratios and p values of the SNP by SHS interaction term were calculated from logistic regression models of ever having been hospitalized, adjusted for SHS (second-hand smoke), age and sex. Total sample size N=436. Number of participants ever having been hospitalized N=104.
Potential functional mechanisms impacted by the NFE2L2 SNPs
All three genetic variants that interacted significantly with SHS exposure to increase the risk asthma are located within non-coding regions of the genome. It is therefore likely that their functional impact, if any, involves alteration to gene regulatory mechanisms such as the binding of transcription factors. Functional genomics experiments such as ChIP-seq provide information on the presence of particular histone marks, which can indicate likely enhancer regions in a given cell population, as well as the binding of particular transcription factors. Such datasets are therefore useful for identifying genetic variants that are located in likely enhancers in relevant cell types, and even for identifying the particular transcription factors whose binding might be altered by the variant. We therefore used in silico analyses to explore the potential functional impact of each of the genetic variants in the lung using publicly available data. To this end, we collected lung cell functional genomics datasets including ChIP-seq from a variety of sources (see Methods). We then intersected the genomic coordinates of each of the three variants with these lung cell functional genomic peak coordinates.
This analysis revealed that all three of the variants found to significantly interact with SHS (rs10183914, rs1806649, and rs2886161, p = 0.02, 0.01, and 0.02, respectively) intersect at least seven lung functional genomics datasets (Supplemental Table 3), indicating that they might act by altering gene regulatory mechanisms in the lung. For example, the genomic coordinates of the replicated SNP, rs10183914, intersected the H3K36me3 and H3K79me2 histone marks in multiple lung-derived experimental data sets. Another of the analyzed SNPs, rs1806649, is located within a predicted super enhancer in lung primary cells and normal human lung fibroblasts[31]. This region also contains high levels of activating histone marks in the A-549 lung cancer cell line, such as H3K4me1, H3K4me3, and H3K27ac (Supplemental Table 3). Further, rs1806649 falls within ChIP-seq peaks for the CEBPB transcription factor in three different lung datasets (two in A-549 cells and one in IMR-90 cells) (Supplemental Table 3), suggesting that this variant might functionally act by altering the binding of CEBPB in lung cells.
Discussion
In this study, we found that NFE2L2 genotype modified the interaction between SHS exposure and asthma in a clinical cohort as well as in a population-based cohort. Moreover, the NFE2L2 SNP rs10183914 also interacted with SHS exposure to increase the risk of severe asthma defined by daily asthma symptoms and hospitalization. To our knowledge, this is the first study demonstrating an interaction between second-hand smoke exposure and NFE2L2 genotype in the risk, as well as severity, of childhood asthma. NFE2L2 genotype was only a risk factor for asthma in smoke-exposed children; we found no association between NFE2L2 genotype and asthma in the analysis unstratified for SHS exposure. These results agree with the lack of association between childhood asthma and NFE2L2 genotype in a study by Cordova et al.[34], and illustrates the importance of evaluating genotype associations in the context of environmental exposures. It has long been acknowledged that asthma-associated SNPs found in GWA studies account for only part of the heritability of asthma, which has been estimated to be between 35% and 95%[35]. Gene-environment interactions have been suggested to be a factor partly explaining this “missing” heritability[36]. This study has helped to fill this gap by identifying a gene-environment interaction that explains a component of asthma heritability specific to SHS-exposed children.
One significant difference between GCPCR and GCC was in the association between SHS exposure and asthma. We found an association in the GCPCR, with an OR of 2.05 (p<0.001), whereas in GCC, SHS exposure was not associated with asthma. While smoke exposure in childhood is clearly associated with asthma and lung function, we did not find any such relationship in GCC, however, rates of SHS exposure and asthma were lower in GCC, which lowered the power to detect an association. It is also possible that the asthmatic participants in GCPCR, who were recruited from hospital clinics, had more severe asthma compared with the asthmatics in the community-based GCC. An interaction between genotype and environmental exposure may, however, exist even in the absence of a main effect of environmental exposure on asthma if the association for individuals with the risk genotype is obscured by the lack of an association in individuals without the risk genotype.
Numerous studies have shown that SHS negatively impacts lung functioning and increases asthma risk in children[37]. Tobacco smoke is an extremely complex mixture of over 7000 chemicals[38], including compounds associated with oxidative stress and inflammation in biological systems[39, 40]. Although an association between asthma and oxidative stress has been established, the causal relationship and mechanisms involved are not well understood. Endogenous reactive oxygen species (ROS) are generated by many types of inflammatory cells, including neutrophils and eosinophils, whereas SHS and traffic-related air pollution are examples of external sources of ROS. Low levels of oxidative stress play important roles in cell signaling, including NF-κB-driven inflammatory pathways. High levels of ROS, however, can overwhelm the antioxidant response system through glutathione depletion and cause worsening of inflammation and cellular damage through the oxidation of biological macromolecules. NFE2L2 regulates a large number of oxidative stress related genes by binding to AREs[41]. Among the target genes of NFE2L2 are members of the gene family glutathione S-transferases (GSTs)[42], which are important in the detoxification of electrophilic compounds and products of oxidative stress. Transfection of a human bronchial epithelial cell line with NFE2L2-specific siRNA has been shown to reduce steroid-induced enhancement of airway epithelial barrier integrity in cultured human bronchial epithelial cells[43]. Furthermore, children with severe asthma have been found to have greater oxidation and lower concentrations of glutathione, which was associated with posttranslational modifications of NFE2L2[44].
Not only postnatal but also prenatal exposure to tobacco smoke can affect lung function and asthma risk[45]. Several GSTs, notably GSTM1 and GSTP1, have been shown to modify the association between intrauterine smoke exposure and asthma risk, further demonstrating the importance of oxidative stress-related pathways in the development of asthma[8]. In a study of interactions between maternal smoking and maternal genotype of important antioxidant genes, no evidence was found for modification by maternal NFE2L2 genotype of the association between prenatal smoke exposure and later asthma prevalence and lung function[46]. It is plausible that altered maternal detoxification capacity could expose the developing fetus to increased levels of oxidants and toxic metabolites. We did not, however, have information about prenatal smoke exposure or maternal genotype, and were therefore not able to address this.
NFE2L2-linked SNPs previously shown to modify the association between airway pollution and asthma include rs6721961[20], which is located in the promoter region of NFE2l2, and rs2588882[42], which is located downstream of NFE2L2 in the untranslated 3’-end of HNRNPA3. To the best of our knowledge, the replicated NFE2L2 SNP rs10183914 has not previously been associated with disease, whereas rs2886161 and rs1806649 have been linked to age of onset of Parkinson's disease[47], a disease known to involve oxidative stress. Rs1806649 has also been associated with an increased risk of COPD mortality[48].
The three SNPs that were significant in our interaction models were all located in intronic regions of NFE2L2, and all were highly linked in our cohort. Although our study was not designed to address functionality of the SNPs, examination of functional genomics datasets showed overlap between the SNP sequences and several regulatory DNA elements in the lung, suggesting that all might be capable of functionally altering gene regulatory mechanisms. The SNP with the most evidence of functionality, rs1806649, overlaps with ChIP-seq peaks for CCAAT enhancer binding protein beta (CEBPB), a transcription factor important in regulating the expression of genes involved in immune and inflammatory responses[49-51]. Members of the CEBP gene family are associated with airway inflammation and hyper-responsiveness[52], and human airway smooth muscle cells from asthmatic individuals have increased CXCL8 production due to binding of CEBPB to the CXCL8 promoter[50]. Intriguingly, expression of CEBPB has been shown to be significantly down-regulated in smokers compared to never smokers[53, 54]. Furthermore, CCAAT enhancer binding protein alpha (CEBPA), which is closely related to CEBPB and shares the same DNA binding motif, has been found to mediate the suppression of airway smooth muscle cell proliferation by cigarette smoke[55].
Our study has some limitations. SHS was only available for a subset of the GCPCR participants, which limited the statistical power of the analyses. Furthermore, the GCPCR is a clinical cohort, and may not be representative of the general population. Nevertheless, the findings for one of the SNPs, rs10183914, was replicated in the population-based GCC. NFE2L2 allele frequencies were very different in white and African-American children, and we were not able to analyze African-American children separately because of low sample sizes. Therefore, our study was limited to white children.
In conclusion, this study has shown that the interaction between NFE2L2 genotype and SHS exposure in early life is associated with an increased risk for asthma development and asthma severity. Furthermore, an examination of functional genomics datasets suggested that all analyzed SNPs that interacted significantly with SHS might be capable of functionally altering gene regulatory mechanisms in lung cells. Additional studies using in vitro and animal models are needed to further elucidate the causal relationship and mechanisms behind the interaction between NFE2L2 and SHS.
Supplementary Material
Funding Source:
This work was supported by NIH grant U19AI070235.
Abbreviations:
- ARE
antioxidant response element
- ChIP
chromatin immunoprecipitation
- DNAH9
Dynein Axonemal Heavy Chain 9
- GCC
Genomic Control Cohort
- GCPCR
Greater Cincinnati Pediatric Clinic Repository
- ICAM1
Intercellular Adhesion Molecule 1
- NAT1
N-acetyltransferase 1
- NFE2L2
nuclear factor (erythroid-derived 2)-like 2
- ROS
reactive oxygen species
- SHS
second hand smoke
- SNP
single nucleotide polymorphism
Footnotes
COI Statement: GKKH has acted as a paid consultant to Hoth Therapeutics and received research grants from Adare and the NIH.
References
- 1.Department of Health and Human Services, Centers for Disease Control and Prevention,, Coordinating Center for Health Promotion,, National Center for Chronic Disease Prevention and Health Promotion,, Office on Smoking and Health,, The Health Consequences of Involuntary Exposure to Tobacco Smoke: A Report of the Surgeon General. In: Department of Health and Human Services; ed. Atlanta, GA. , 2006. [PubMed] [Google Scholar]
- 2.Kanchongkittiphon W, Mendell MJ, Gaffin JM, Wang G, Phipatanakul W, Indoor environmental exposures and exacerbation of asthma: an update to the 2000 review by the Institute of Medicine. Environ Health Perspect 2015;123: 6–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perzanowski MS, Divjan A, Mellins RB, Canfield SM, Rosa MJ, Chew GL, Rundle A, Goldstein IF, Jacobson JS, Exhaled NO among inner-city children in New York City. J Asthma 2010;47: 1015–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang HC, McGeady SJ, Yousef E, Patient, home residence, and neighborhood characteristics in pediatric emergency department visits for asthma. J Asthma 2007;44: 95–8. [DOI] [PubMed] [Google Scholar]
- 5.London SJ, Romieu I, Gene by environment interaction in asthma. Annu Rev Public Health 2009;30: 55–80. [DOI] [PubMed] [Google Scholar]
- 6.Hussein YM, Shalaby SM, Zidan HE, Sabbah NA, Karam NA, Alzahrani SS, CD14 tobacco gene-environment interaction in atopic children. Cell Immunol 2013;285: 31–7. [DOI] [PubMed] [Google Scholar]
- 7.Rava M, Smit LA, Nadif R, Gene-environment interactions in the study of asthma in the postgenomewide association studies era. Curr Opin Allergy Clin Immunol 2015;15: 70–8. [DOI] [PubMed] [Google Scholar]
- 8.Gilliland FD, Li YF, Dubeau L, Berhane K, Avol E, McConnell R, Gauderman WJ, Peters JM, Effects of glutathione S-transferase M1, maternal smoking during pregnancy, and environmental tobacco smoke on asthma and wheezing in children. Am J Respir Crit Care Med 2002;166: 457–63. [DOI] [PubMed] [Google Scholar]
- 9.Kabesch M, Hoefler C, Carr D, Leupold W, Weiland SK, von Mutius E, Glutathione S transferase deficiency and passive smoking increase childhood asthma. Thorax 2004;59: 569–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rogers AJ, Brasch-Andersen C, Ionita-Laza I, Murphy A, Sharma S, Klanderman BJ, Raby BA, The interaction of glutathione S-transferase M1-null variants with tobacco smoke exposure and the development of childhood asthma. Clin Exp Allergy 2009;39: 1721–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee YL, Lee YC, Guo YL, Associations of glutathione S-transferase P1, M1, and environmental tobacco smoke with wheezing illness in school children. Allergy 2007;62: 641–7. [DOI] [PubMed] [Google Scholar]
- 12.Li YF, Gauderman WJ, Conti DV, Lin PC, Avol E, Gilliland FD, Glutathione S-transferase P1, maternal smoking, and asthma in children: a haplotype-based analysis. Environ Health Perspect 2008;116: 409–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li YF, Lin CC, Tai CK, Interaction of intercellular adhesion molecule 1 (ICAM1) polymorphisms and environmental tobacco smoke on childhood asthma. Int J Environ Res Public Health 2014;11: 6504–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brooks CC, Martin LJ, Pilipenko V, He H, LeMasters GK, Lockey JE, Bernstein DI, Ryan PH, Khurana Hershey GK, Biagini Myers JM, NAT1 genetic variation increases asthma risk in children with secondhand smoke exposure. J Asthma 2019: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dizier MH, Nadif R, Margaritte-Jeannin P, Barton SJ, Sarnowski C, Gagne-Ouellet V, Brossard M, Lavielle N, Just J, Lathrop M, Holloway JW, Laprise C, Bouzigon E, Demenais F, Interaction between the DNAH9 gene and early smoke exposure in bronchial hyperresponsiveness. Eur Respir J 2016;47: 1072–81. [DOI] [PubMed] [Google Scholar]
- 16.Cho HY, Kleeberger SR, Association of Nrf2 with airway pathogenesis: lessons learned from genetic mouse models. Arch Toxicol 2015;89: 1931–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, Yamamoto M, Kensler TW, Tuder RM, Georas SN, Biswal S, Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med 2005;202: 47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li YJ, Takizawa H, Azuma A, Kohyama T, Yamauchi Y, Takahashi S, Yamamoto M, Kawada T, Kudoh S, Sugawara I, Nrf2 is closely related to allergic airway inflammatory responses induced by low-dose diesel exhaust particles in mice. Clin Immunol 2010;137: 234–41. [DOI] [PubMed] [Google Scholar]
- 19.Masuko H, Sakamoto T, Kaneko Y, Iijima H, Naito T, Noguchi E, Hirota T, Tamari M, Hizawa N, An interaction between Nrf2 polymorphisms and smoking status affects annual decline in FEV1: a longitudinal retrospective cohort study. BMC Med Genet 2011;12: 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ungvari I, Hadadi E, Virag V, Nagy A, Kiss A, Kalmar A, Zsigmond G, Semsei AF, Falus A, Szalai C, Relationship between air pollution, NFE2L2 gene polymorphisms and childhood asthma in a Hungarian population. J Community Genet 2012;3: 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Z, May SM, Charoenlap S, Pyle R, Ott NL, Mohammed K, Joshi AY, Effects of secondhand smoke exposure on asthma morbidity and health care utilization in children: a systematic review and meta-analysis. Ann Allergy Asthma Immunol 2015;115: 396–401 e2. [DOI] [PubMed] [Google Scholar]
- 22.Butsch Kovacic M, Biagini Myers JM, Lindsey M, Patterson T, Sauter S, Ericksen MB, Ryan P, Assa'ad A, Lierl M, Fischer T, Kercsmar C, McDowell K, Lucky AW, Sheth AP, Hershey AD, Ruddy RM, Rothenberg ME, Khurana Hershey GK, The Greater Cincinnati Pediatric Clinic Repository: A Novel Framework for Childhood Asthma and Allergy Research. Pediatr Allergy Immunol Pulmonol 2012;25: 104–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baye TM, Butsch Kovacic M, Biagini Myers JM, Martin LJ, Lindsey M, Patterson TP, He H, Ericksen MB, Gupta J, Tsoras AM, Lindsley A, Rothenberg ME, Wills-Karp M, Eissa NT, Borish L, Khurana Hershey GK, Differences in candidate gene association between European ancestry and African American asthmatic children. PLoS One 2011;6: e16522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biagini Myers JM, Martin LJ, Kovacic MB, Mersha TB, He H, Pilipenko V, Lindsey MA, Ericksen MB, Bernstein DI, LeMasters GK, Lockey JE, Khurana Hershey GK, Epistasis between serine protease inhibitor Kazal-type 5 (SPINK5) and thymic stromal lymphopoietin (TSLP) genes contributes to childhood asthma. J Allergy Clin Immunol 2014;134: 891–99 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Standardization of Spirometry, 1994 Update. American Thoracic Society. Am J Respir Crit Care Med 1995;152: 1107–36. [DOI] [PubMed] [Google Scholar]
- 26.Kovacic MB, Myers JM, Wang N, Martin LJ, Lindsey M, Ericksen MB, He H, Patterson TL, Baye TM, Torgerson D, Roth LA, Gupta J, Sivaprasad U, Gibson AM, Tsoras AM, Hu D, Eng C, Chapela R, Rodriguez-Santana JR, Rodriguez-Cintron W, Avila PC, Beckman K, Seibold MA, Gignoux C, Musaad SM, Chen W, Burchard EG, Hershey GK, Identification of KIF3A as a novel candidate gene for childhood asthma using RNA expression and population allelic frequencies differences. PLoS One 2011;6: e23714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.International HapMap C, Altshuler DM, Gibbs RA, Peltonen L, Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E, Schaffner SF, Yu F, Peltonen L, Dermitzakis E, Bonnen PE, Altshuler DM, Gibbs RA, de Bakker PI, Deloukas P, Gabriel SB, Gwilliam R, Hunt S, Inouye M, Jia X, Palotie A, Parkin M, Whittaker P, Yu F, Chang K, Hawes A, Lewis LR, Ren Y, Wheeler D, Gibbs RA, Muzny DM, Barnes C, Darvishi K, Hurles M, Korn JM, Kristiansson K, Lee C, McCarrol SA, Nemesh J, Dermitzakis E, Keinan A, Montgomery SB, Pollack S, Price AL, Soranzo N, Bonnen PE, Gibbs RA, Gonzaga-Jauregui C, Keinan A, Price AL, Yu F, Anttila V, Brodeur W, Daly MJ, Leslie S, McVean G, Moutsianas L, Nguyen H, Schaffner SF, Zhang Q, Ghori MJ, McGinnis R, McLaren W, Pollack S, Price AL, Schaffner SF, Takeuchi F, Grossman SR, Shlyakhter I, Hostetter EB, Sabeti PC, Adebamowo CA, Foster MW, Gordon DR, Licinio J, Manca MC, Marshall PA, Matsuda I, Ngare D, Wang VO, Reddy D, Rotimi CN, Royal CD, Sharp RR, Zeng C, Brooks LD, McEwen JE, Integrating common and rare genetic variation in diverse human populations. Nature 2010;467: 52–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, Yefanov A, Lee H, Zhang N, Robertson CL, Serova N, Davis S, Soboleva A, NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res 2013;41: D991–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, Amin V, Whitaker JW, Schultz MD, Ward LD, Sarkar A, Quon G, Sandstrom RS, Eaton ML, Wu YC, Pfenning AR, Wang X, Claussnitzer M, Liu Y, Coarfa C, Harris RA, Shoresh N, Epstein CB, Gjoneska E, Leung D, Xie W, Hawkins RD, Lister R, Hong C, Gascard P, Mungall AJ, Moore R, Chuah E, Tam A, Canfield TK, Hansen RS, Kaul R, Sabo PJ, Bansal MS, Carles A, Dixon JR, Farh KH, Feizi S, Karlic R, Kim AR, Kulkarni A, Li D, Lowdon R, Elliott G, Mercer TR, Neph SJ, Onuchic V, Polak P, Rajagopal N, Ray P, Sallari RC, Siebenthall KT, Sinnott-Armstrong NA, Stevens M, Thurman RE, Wu J, Zhang B, Zhou X, Beaudet AE, Boyer LA, De Jager PL, Farnham PJ, Fisher SJ, Haussler D, Jones SJ, Li W, Marra MA, McManus MT, Sunyaev S, Thomson JA, Tlsty TD, Tsai LH, Wang W, Waterland RA, Zhang MQ, Chadwick LH, Bernstein BE, Costello JF, Ecker JR, Hirst M, Meissner A, Milosavljevic A, Ren B, Stamatoyannopoulos JA, Wang T, Kellis M, Integrative analysis of 111 reference human epigenomes. Nature 2015;518: 317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheneby J, Gheorghe M, Artufel M, Mathelier A, Ballester B, ReMap 2018: an updated atlas of regulatory regions from an integrative analysis of DNA-binding ChIP-seq experiments. Nucleic Acids Res 2018;46: D267–D75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, Young RA, Super-enhancers in the control of cell identity and disease. Cell 2013;155: 934–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harley JB, Chen X, Pujato M, Miller D, Maddox A, Forney C, Magnusen AF, Lynch A, Chetal K, Yukawa M, Barski A, Salomonis N, Kaufman KM, Kottyan LC, Weirauch MT, Transcription factors operate across disease loci, with EBNA2 implicated in autoimmunity. Nat Genet 2018;50: 699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quinlan AR, Hall IM, BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26: 841–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cordova EJ, Jimenez-Morales S, Centeno F, Martinez-Hernandez A, Martinez-Aguilar N, Del-Rio-Navarro BE, Gomez-Vera J, Orozco L, NFE2L2 gene variants and susceptibility to childhood-onset asthma. Rev Invest Clin 2011;63: 407–11. [PubMed] [Google Scholar]
- 35.Ober C, Yao TC, The genetics of asthma and allergic disease: a 21st century perspective. Immunological Reviews 2011;242: 10–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee JU, Kim JD, Park CS, Gene-Environment Interactions in Asthma: Genetic and Epigenetic Effects. Yonsei Med J 2015;56: 877–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vanker A, Gie RP, Zar HJ, The association between environmental tobacco smoke exposure and childhood respiratory disease: a review. Expert Rev Resp Med 2017;11: 661–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rodgman A, Perfetti TA, The chemical components of tobacco and tobacco smoke. 2nd Edn. Boca Raton: CRC Press, 2013. [Google Scholar]
- 39.Milnerowicz H, Sciskalska M, Dul M, Molecular mechanisms of the impact of smoke-oxidants. Exp Toxicol Pathol 2015;67: 377–82. [DOI] [PubMed] [Google Scholar]
- 40.de Carvalho FO, Felipe FA, de Melo Costa AC, Teixeira LG, Silva ER, Nunes PS, Shanmugam S, de Lucca Junior W, Quintans JS, de Souza Araujo AA, Inflammatory Mediators and Oxidative Stress in Animals Subjected to Smoke Inhalation: A Systematic Review. Lung 2016;194: 487–99. [DOI] [PubMed] [Google Scholar]
- 41.Liu Q, Gao Y, Ci X, Role of Nrf2 and Its Activators in Respiratory Diseases. Oxid Med Cell Longev 2019;2019: 7090534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klaassen CD, Reisman SA, Nrf2 the rescue: Effects of the antioxidative/electrophilic response on the liver. Toxicology and applied pharmacology 2010;244: 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shintani Y, Maruoka S, Gon Y, Koyama D, Yoshida A, Kozu Y, Kuroda K, Takeshita I, Tsuboi E, Soda K, Hashimoto S, Nuclear factor erythroid 2-related factor 2 (Nrf2) regulates airway epithelial barrier integrity. Allergology International 2015;64: S54–S63. [DOI] [PubMed] [Google Scholar]
- 44.Fitzpatrick AM, Stephenson ST, Hadley GR, Burwell L, Penugonda M, Simon DM, Hansen J, Jones DP, Brown LAS, Thiol redox disturbances in children with severe asthma are associated with posttranslational modification of the transcription factor nuclear factor (erythroid-derived 2)-like 2. J Allergy Clin Immun 2011;127: 1604–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.He Z, Wu H, Zhang S, Lin Y, Li R, Xie L, Li Z, Sun W, Huang X, Zhang CJP, Ming WK, The association between secondhand smoke and childhood asthma: A systematic review and meta-analysis. Pediatr Pulmonol 2020. [DOI] [PubMed] [Google Scholar]
- 46.Henderson AJ, Newson RB, Rose-Zerilli M, Ring SM, Holloway JW, Shaheen SO, Maternal Nrf2 and gluthathione-S-transferase polymorphisms do not modify associations of prenatal tobacco smoke exposure with asthma and lung function in school-aged children. Thorax 2010;65: 897–902. [DOI] [PubMed] [Google Scholar]
- 47.von Otter M, Landgren S, Nilsson S, Zetterberg M, Celojevic D, Bergstrom P, Minthon L, Bogdanovic N, Andreasen N, Gustafson DR, Skoog I, Wallin A, Tasa G, Blennow K, Nilsson M, Hammarsten O, Zetterberg H, Nrf2-encoding NFE2L2 haplotypes influence disease progression but not risk in Alzheimer's disease and age-related cataract. Mech Ageing Dev 2010;131: 105–10. [DOI] [PubMed] [Google Scholar]
- 48.Figarska SM, Vonk JM, Boezen HM, NFE2L2 polymorphisms, mortality, and metabolism in the general population. Physiological Genomics 2014;46: 411–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huber R, Pietsch D, Panterodt T, Brand K, Regulation of C/EBP beta and resulting functions in cells of the monocytic lineage. Cell Signal 2012;24: 1287–96. [DOI] [PubMed] [Google Scholar]
- 50.John AE, Zhu YM, Brightling CE, Pang LH, Knox AJ, Human Airway Smooth Muscle Cells from Asthmatic Individuals Have CXCL8 Hypersecretion Due to Increased NF-kappa B p65, C/EBP beta, and RNA Polymerase II Binding to the CXCL8 Promoter. Journal of Immunology 2009;183: 4682–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsukada J, Yoshida Y, Kominato Y, Auron PE, The CCAAT/enhancer (C/EBP) family of basic-leucine zipper (bZIP) transcription factors is a multifaceted highly-regulated system for gene regulation. Cytokine 2011;54: 6–19. [DOI] [PubMed] [Google Scholar]
- 52.Miglino N, Roth M, Tamm M, Borger P, Asthma and COPD - The C/EBP Connection. Open Respir Med J 2012;6: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mori M, Bjermer L, Erjefalt JS, Stampfli MR, Roos AB, Small airway epithelial-C/EBPbeta is increased in patients with advanced COPD. Respir Res 2015;16: 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Didon L, Barton JL, Roos AB, Gaschler GJ, Bauer CMT, Berg T, Stampfli MR, Nord M, Lung Epithelial CCAAT/Enhancer-binding Protein-beta Is Necessary for the Integrity of Inflammatory Responses to Cigarette Smoke. Am J Resp Crit Care 2011;184: 233–42. [DOI] [PubMed] [Google Scholar]
- 55.Guan P, Cai WT, Yu HP, Wu ZY, Li W, Wu J, Chen J, Feng GQ, Cigarette smoke extract promotes proliferation of airway smooth muscle cells through suppressing C/EBP- expression. Exp Ther Med 2017;13: 1408–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.