

During the decoding of a subset of mRNAs, a proportion of ribosomes productively shift to the −1 reading frame at specific slippage-prone sites in a phenomenon known as programmed −1 ribosomal frameshifting (−1 PRF)1 to generate a frameshifted, C-terminally unique protein. The first experimentally verified occurrence of functionally utilized non-retroelement derived −1 PRF in humans has been reported in the mRNA encoding the immune-functioning C-C chemokine receptor 5 (CCR5)2. Here, we show that frameshifting does not occur during CCR5 decoding. Apart from its importance in understanding expression of a gene relevant to cancer, an HIV-1 receptor (and the associated claimed rationale for generating the first humans derived from genetically modified embryos), the findings imply that caution is appropriate in assessing suggestions of widespread regulated mRNA decay by advantageous frameshifting in cellular gene expression3.
PRF is often measured with fused dual-luciferase reporter systems (Figure 1A). The upstream reporter (often Renilla luciferase; R-luc) monitors zero-frame translation and the downstream reporter (Firefly luciferase; F-luc) monitors an alternative reading frame. Ribosomes that frameshift synthesize an R-luc-F-luc fusion protein, whereas those that do not yield R-luc alone4. Frameshifting is measured as the ratio of F-luc/R-luc activities, normalized to the ratio expressed from an in-frame control (IFC) reporter expressing identical protein sequence as the product generated by frameshifting (Figure 1B).
Figure 1: Evaluation of the original evidence for CCR5 frameshifting:
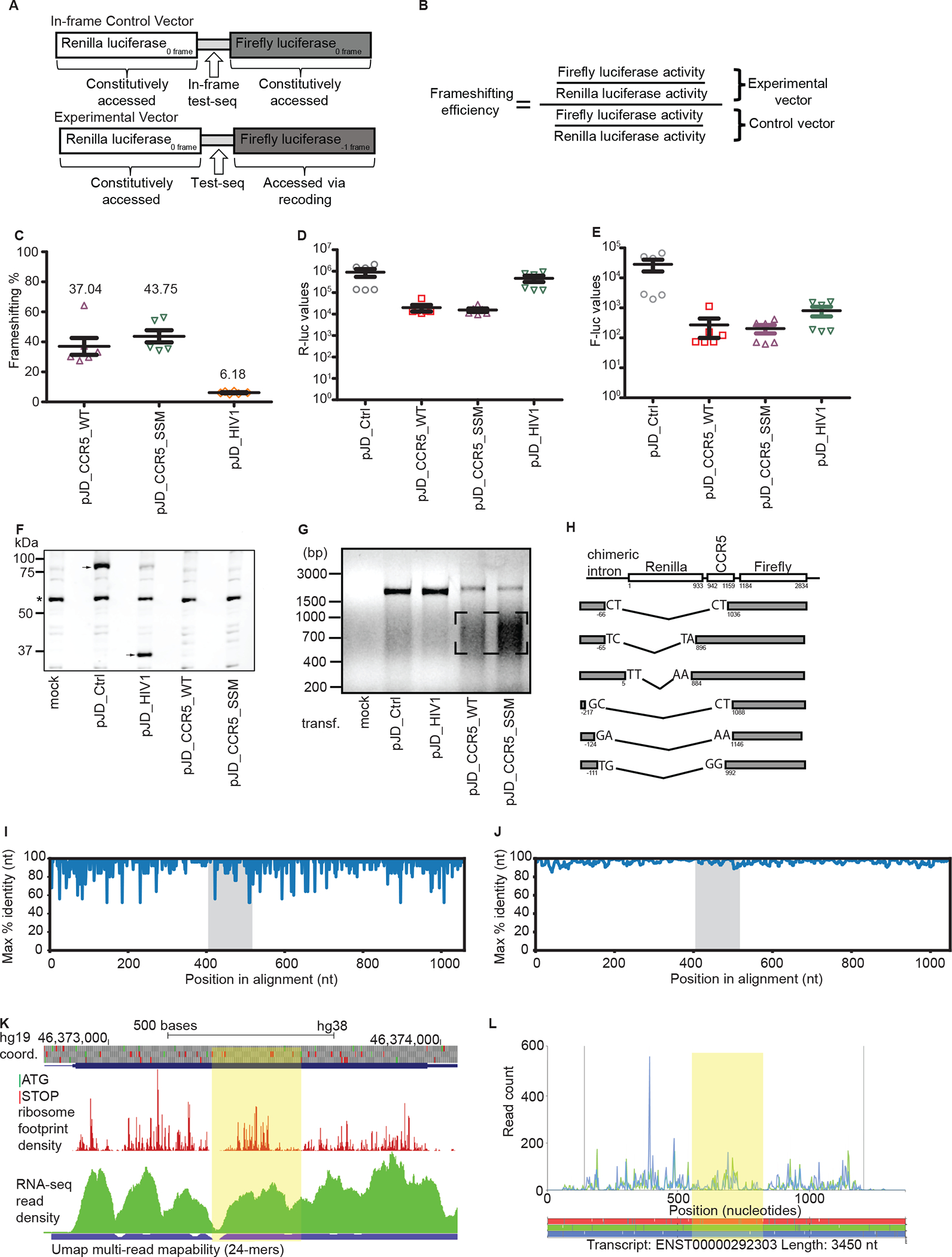
a. Graphical representation of the test sequence and in-frame control. b. Formula for calculating the −1 PRF efficiency. c. Relative firefly to Renilla luciferase activities, normalized to an in-frame control (pJD_Ctrl) in HeLa cells. d. Renilla luciferase activities. e. Firefly luciferase activities. c-e Each data point represents an independently transfected well of cells; error bars represent SEM; n = 2 (three separate wells of cells per sample were transfected on two different days). f. Representative anti-R-Luc immunoblot of lysates from HEK293T cells transfected with dual luciferase reporter constructs used in c-e. An asterisk indicates detection of a non-specific product that serves as a loading control. Arrow(s) indicate the presence of the reference Renilla luciferase that is always fused to firefly luciferase (pJD_Ctrl) or that is only Renilla luciferase (pJD_HIV-1). No Renilla luciferase is detected in pJD_CCR5_WT or pJD_CCR5_SSM. g. RT-PCR of dual luciferase reporter constructs used in c-e. cDNA was synthesized using gene-specific primers and analyzed by gel electrophoresis. h. Schematic representation (not to scale) of the RNA rearrangements identified by sequencing of the lower molecular mass cDNA products (see box in g). Numbers under the boxes represent location in sequence relative to the Renilla luciferase start codon. i. Single nucleotide resolution conservation analysis (max % identity) of the CCR5 CDS for primates only, excluding Lemur catta. j. 10-nucleotide sliding window analysis of conservation in the CCR5 CDS for primates only, excluding Lemur catta. The area containing the putative frameshift region is highlighted in grey. The accession numbers of the sequences used are indicated in bold in Supplementary Table 1B. k-l. GWIPS-viz (k) and Trips-Viz (l) visualizations of ribosome profiling data obtained in monocytes. The region downstream of the putative frameshift site up to the next −1 frame stop codon is highlighted in yellow. GWIPS-viz screenshot displaying ribosome footprints aligning to the genomic locus corresponding to the protein coding exon of CCR5. Reduced density is observed at the frameshift site and immediately downstream due to low mapability (blue bar; ambiguous mapping is disallowed). Trips-Viz screenshot displaying ribosome footprints aligning to the CCR5 ENSEMBL transcript; only footprints supporting translation in the zero (blue) and −1 (green) frames are shown. Ambiguous read mapping is allowed.
We obtained and sequenced the exact constructs from the original report that claimed 10% efficient CCR5 frameshifting and noticed that their IFC reporter did not contain any CCR5 sequence. Whereas a control construct containing the −1 PRF site from HIV-1 showed frameshifting activities at the expected level (~6%), apparent −1 PRF rates from the CCR5 −1 PRF construct were variable and generally much higher than previously reported (Figure 1C)2. Furthermore, a slippery site mutant (SSM) of the CCR5 −1 PRF site in which frameshifting should be abolished showed equally high apparent −1 PRF. Absolute luciferase activities were reduced in cells transfected with reporters containing CCR5 sequence compared to the HIV-1 control and IFC. A greater decrease in R-luc relative to F-luc led to the increase in F-luc/R-luc ratios and thus inflated the estimated −1 PRF (Figure 1C–F). Since the reporters were introduced as plasmids, the observed effect could be due to cryptic splicing of the reporter RNA. RT-PCR of reporter RNAs showed that for cells transfected with the CCR5 reporters, there was significantly less full-length cDNA and a simultaneous appearance of smaller molecular mass products (Figure 1G). Sequencing of the smaller cDNAs revealed many different rearrangements within the reporter mRNA, mostly involving an upstream chimeric intron in the reporter plasmid 5′ leader and the CCR5 cassette. These findings suggest that sequences within the CCR5 cassette stimulate variable rearrangements within the reporter RNA, possibly by activation of cryptic splice sites (Figure 1H).
We then reanalyzed the evolutionary conservation of the predicted CCR5 −1 PRF site, which was reported as highly conserved in higher primates2. Primate CCR5 coding DNA sequences (CDSs) were aligned and conservation was measured at single-nucleotide resolution (Figure 1I) and in a 10-nucleotide sliding window (Figure 1J). This analysis showed that the entire CDS, not just the frameshift region, is highly conserved among primates. Thus, the claim that the frameshift site is highly conserved among the higher primates does not indicate that the (putative) frameshift signal is subject to purifying selection. In mammalian orthologues of human CCR5 identified by BLAST5 in the RefSeq database6, we observed substitutions in many species that produce sequences inconsistent with known −1 PRF slippery sites1 (Extended Figure 1).
Belew et al2 used ribosome profiling data obtained in HeLa cells7 as supporting evidence for frameshifting. However, due to negligible levels of CCR5 expression in HeLa, the coverage of ribosome profiling data for CCR5 is indistinguishable from background noise and no reliable conclusion can be made from these data. Using GWIPS-viz8 and Trips-Viz9 to visualize ribosome footprint density in monocytes10, where CCR5 is expressed, (Figure 1K–L), we were unable to detect transitions in reading frames or changes in footprint density exceeding the expected heterogeneity of ribosome footprint density profiles.
Next we inserted the same CCR5 frameshift and negative control sequences tested in the original article into the most recent iteration of the dual luciferase reporter, pSGDluc11. Here, test sequence is flanked by tandem StopGo sequences that prevent with high efficiency peptide bond formation at a specific site, resulting in expression of reporters as separate proteins. This arrangement avoids potential artefacts that can arise when the test sequence product fusion alters the individual reporter activities or their stability. Furthermore, cryptic splice sites within R-luc were removed (see Methods). Importantly, we also generated IFC reporters expressing the same protein sequence as the putative frameshift product (Figure 2A).
Figure 2: Additional approaches to CCR5 −1 PRF detection:
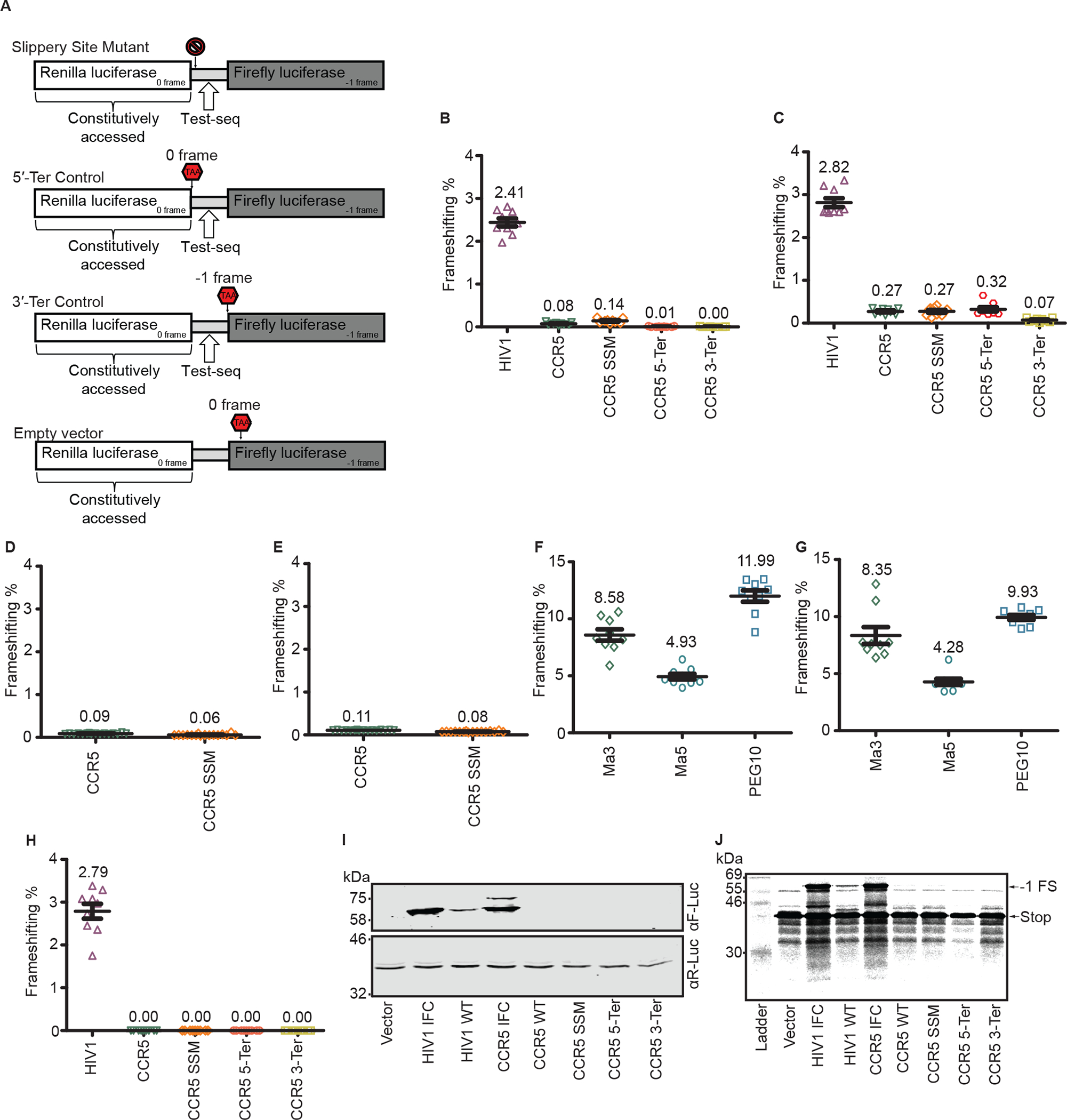
a. Graphical representation of negative controls. The circular cross represents a mutated slippery site, and the red stop signs represent constructs with a stop codon in the appropriately labeled frame. All four of these constructs are meant to establish a background baseline for F-luc luminescence. b. Frameshifting efficiencies for experiments performed in HeLa cells, normalized to the HIV-1 or CCR5 in-frame control vector. c. Frameshifting efficiencies for experiments performed in HEK293T cells, normalized to the HIV-1 or CCR5 in-frame control vector. d. Frameshifting efficiencies for experiments performed in HeLa cells, normalized to the CCR5 in-frame control vector. e. Frameshifting efficiencies for experiments performed in HEK293T cells, normalized to the HIV-1 or CCR5 in-frame control vector. f. Frameshifting efficiencies for experiments performed in HeLa cells, normalized to the respective in-frame controls PNMA3, PNMA5 or PEG10. g. Frameshifting efficiencies for experiments performed in HEK293T cells, normalized to the respective in-frame controls Ma3, Ma5 or PEG10. Each data point represents an independently transfected well of cells; error bars represent standard error of measurement (SEM); n = 3 (three separate wells of cells (b,c,f,g) or four separate wells of cells (d,e) per sample were transfected on three different days). The slip site mutants used were the following: U_UUA_AAA → G_CGC_GCG for b-c, U_UUA_AAA → A_UUC_AAA for d-e. h. Frameshifting efficiency for RNA transfection experiments performed in HeLa cells, normalized to the CCR5 or HIV-1 in-frame control vector. Each data point represents an independently transfected well of cells; error bars represent SEM; n = 3 (three separate wells of cells per sample were transfected on three different days). i. Representative immunoblots of HeLa cells transfected with dual luciferase constructs. The experiment was repeated three times. Note that the more slowly migrating bands in the R-luc westerns and for CCR5 IFC in the F-luc western likely correspond to incomplete polypeptide separation at the StopGo sequences16. j. In vitro translation of capped RNAs in rabbit reticulocyte lysates. The experiment was repeated twice. Image exposure was adjusted uniformly to detect potential low levels of frameshifting.
The reference luciferase (Renilla) displayed similar activity for all constructs (Extended Figure 2A). The F-luc activity for the WT HIV-1 frameshift construct was >40 fold higher than that of WT CCR5 and its negative controls. Furthermore, the F-luc activity for WT CCR5 was similar to its negative controls (Extended Figure 2B). We found the HIV-1 −1 PRF efficiency to be comparable to previously reported findings in HeLa cells12. However, CCR5 WT −1 PRF efficiency was ~0.08%, indistinguishable from its negative controls, indicating that −1 PRF on the CCR5 sequence is not above background (Figure 2B). These same experiments in HEK293T cells gave near-identical results to those obtained in HeLa cells (Extended Figure 2C, 2D, and Figure 2C).
The cloning and assaying of CCR5 frameshifting were also completed independently by a different laboratory (see Extended Methods). For both HeLa and HEK293T cells, we again found that the efficiency of −1 PRF at the CCR5 signal is similar to background (Extended Figure 2E, 2F, 2G, 2H, and Figure 2D, 2E).
As a further control, we cloned into pSGDluc three validated human retrotransposon-derived −1 PRF signals1 (from PNMA3, PNMA5 and PEG10). These sequences were again assayed in both HeLa and HEK293T cells (Extended Figure 2I, 2J, 2K, 2L and Figure 2F, 2G). All three signals stimulated −1 PRF far above background.
To control for possible mRNA-level effects such as cryptic splicing, 5′-capped reporter RNAs were generated in vitro using T7 RNA polymerase. Again, no −1 PRF was observed for CCR5 or its negative controls, whereas the −1 PRF efficiency for HIV-1 was similar to that found from the DNA transfections (Extended Figure 2M, 2N, Figure 2H).
We transfected HeLa cells and probed cell lysates with antibodies against F-luc and R-luc (Figure 2I). As expected, we observed similar R-luc abundance for all constructs. We also detected F-luc for the HIV-1 and CCR5 IFCs, and for the WT HIV-1 construct. However, no F-luc expression was observed for CCR5 WT. Finally, a rabbit reticulocyte lysate in vitro translation system was programmed with capped RNAs (Figure 2J). A −1 PRF product was detectable for HIV-1 WT, co-migrating with the equivalent products produced from the HIV-1 and CCR5 IFCs. Again, no −1 PRF product was detectable for CCR5 WT.
Apart from the mutant state of several neurodegenerative disease genes, we deduce that there are currently no known validated non-retroelement derived cellular genes in humans that functionally utilize efficient −1 PRF. Knowledge of the extent to which −1 PRF is used in human gene expression is of paramount importance for ongoing studies of compounds targeting viral −1 frameshifting13,14.
Extended Methods
Alignment of CCR5 slippery sites
The NCBI RefSeq database was searched with tblastn using the human CCR5 amino acid sequence derived from mRNA accession NM_000579.3 as query on December 4th, 2018 using default parameters, with the number of hits expanded from 100 to 500. Only sequences with >70% query coverage were used, and the best sequence match for each organism was selected based on ‘Max Score’. These nucleotide sequences were aligned with MUSCLE15. The alignment was trimmed to only include the putative frameshift region of CCR5. The alignment image was generated using the box-shade server (https://embnet.vital-it.ch/software/BOX_form.html).
CCR5 primate CDS analysis
Of the sequences obtained in the ortholog search, only those belonging to the taxonomic descriptor of ‘primate’ (taxid 9443) were used. Lemur catta sequences were excluded since the original manuscript states that the CCR5 frameshift signal is not conserved in Lemur catta. Full length CDS nucleotide sequences were aligned with MUSCLE15 and then the nucleotide identity percentage (as compared to the consensus sequence) either at single nucleotide resolution or a smoothed 10 nucleotide window was plotted. If a gap in the alignment position was the highest identity, it was ignored.
Vector construction
Vectors pJD_Ctrl, pJD_HIV-1, pJD_CCR5_WT and pJD_CCR5_SSM were provided by the Dinman lab. The vectors used in Figure 1C–H are: pJD_Ctrl – dual luciferase reporter with both luciferases in 0 frame; pJD_HIV-1 – dual luciferase reporter with HIV-1 −1 PRF site between Renilla luciferase (0 frame) and firefly luciferase (−1 frame); pJD_CCR5_WT – dual luciferase reporter with CCR5 −1 PRF site between Renilla luciferase (0 frame) and firefly luciferase (−1 frame); and pJD_CCR5_SSM – as pJD_CCR5_WT but with the slippery site mutation (TTTAAAA → CGCGCGC). pSGDlucV3.0 (Addgene 119760) is a version of pSGDLuc generated by introducing silent mutations into the Renilla coding sequence to disrupt two potential donor splice sites (tgggtaagt).
For Extended Figures 2A, 2B, 2C, 2D, 2I, 2J, 2K, 2L, 2M, 2N and Figures 2B, 2C, 2F, and 2G, pSGDlucV3.0 was digested with PspXI and BglII. Overlapping oligonucleotides for insert sequences were obtained from Sigma Aldrich (Supplementary Table 1), resuspended at 100 μM, diluted to 10 μM, and then mixed together at a 1:100 dilution (100 nM). This 1:100 dilution of the forward and reverse oligonucleotide was heated to 95 °C. 2 μL of this mix was ligated into 40 ng of linearized backbone vector using NEB T4 ligase.
For Extended Figures 2E, 2F, 2G, 2H and Figures 2D and 2E, pSGDlucV3.0 was digested with PspXI and BglII. Using forward and reverse PCR primers (Supplementary Table 1), the CCR5 WT, SSM and IFC were all amplified from genomic DNA with enzyme overhangs. These PCR products were subsequently digested and ligated into pSGDlucV3.0.
Transfections and dual luciferase assays
For Figure 1C–E, HeLa cells were seeded in a 96-well format at a density of 1×104 cells per well and transfected 24 h post seeding with 50 ng reporter plasmid using Lipofectamine LTX with Plus reagent (Thermo Fisher Scientific). 24 h after transfection cells were lysed in 30 μl 1x passive lysis buffer (Promega) and luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega) by injection of 100 μl of each reagent.
For Extended Figures 2A, 2B, 2C, 2D, 2I, 2J, 2K, 2L, 2M, 2N and Figures 2B, 2C, 2F, and 2G, HeLa and HEK293T cells were reverse transfected with 100 ng of plasmid using 0.2 μL Lipofectamine 2000 in a 96-well format. Transfection mix was added to 30,000 cells and incubated for 1–2 min at room temperature, before adding fetal calf serum.
The next day, media was removed from cells and 100 μL of fresh 1X passive lysis buffer (Promega) was added to each well. Samples were frozen and thawed before luciferase activity was measured by adding 20 μL of lysate and 20 μL of each reagent, as per the Promega protocol.
For Figure 2H, HeLa cells were reverse transfected with 100 ng of capped RNA using 0.5 μL Lipofectamine 2000 in a 96-well format. Transfection mix was added to 50,000 cells and incubated for 1–2 min at room temperature, before adding fetal calf serum. Six hours later, media was removed from cells and 100 μL of fresh 1X passive lysis buffer (Promega) was added to each well. Samples were frozen and thawed before luciferase activity was measured by adding 20 μL of lysate and 20 μL of each reagent, as per the Promega protocol.
For Extended Figures 2E, 2F, 2G, 2H and Figures 2D and 2E, HeLa and HEK293T cells were reverse transfected with 25 ng of plasmid using 0.2 μL Lipofectamine 2000 in a half-area 96-well format. Transfection mix was added to 40,000 cells. The next day, transfected cells were lysed in 12.6 μL of 1X passive lysis buffer (Promega) and luciferase activity was measured by injection of 25 μL of each reagent.
Background level readings (empty vector for firefly luciferase for Figures 2B, 2C, 2F, 2G, and 2H and water transfected cells for Figure 2D and 2E, and water transfected cells for Renilla luciferase) were subtracted from all firefly and Renilla luciferase readings. Frameshift efficiencies were calculated by dividing firefly luciferase values by Renilla luciferase values and then dividing the relative ratios by the average F-Luc/R-Luc ratio of the in-frame control.
T7 RNA transcription
Circular DNA vectors used for dual luciferase assays were all linearized with ApaLI. The Invitrogen mMessage mMachine T7 kit (AM1344) was used to generate capped transcripts with the 2X NTP/CAP solution provided. After DNase I treatment, mRNAs were concentrated with the Zymo Research RNA Clean and Concentration™-5 (R1016) kit. The integrity of these mRNAs was verified on a 1% non-denaturing agarose gel.
Western blot
For Figure 1F, 0.75 × 106 cells were reverse transfected in a 6-well plate with 1.0 μg of plasmid with 7.5 μL of Lipofectamine 2000 for 24 h. Cells were lysed in 100 μL of 1× PLB. Proteins were resolved by SDS-PAGE and transferred to nitrocellulose membranes (Protran), which were incubated at 4°C overnight with anti-R-Luc (MBLPM047). Immunoreactive bands were detected on membranes after incubation with appropriate fluorescently labeled secondary antibodies using a LI-COR Odyssey® Infrared Imaging Scanner.
For Figure 2I, 0.75 × 106 cells were reverse transfected in a 6-well plate with 2500 ng of plasmid with 7.5 μL of Lipofectamine 2000 overnight. The following day, cells were scraped and lysed in 100 μL of 1x PLB. Lysates were equalized by Renilla luciferase activity. Membranes were blocked with LICOR blocking buffer at room temperature for 1 h. Firefly luciferase (Promega G745A) and Renilla luciferase (MBL PM047) antibodies were incubated at a 1:1000 dilution in LICOR blocking buffer overnight and the secondary LICOR antibodies (LICOR IRDye 926–32214 Donkey anti Goat 800 for F-Luc and LICOR IRDye 926–68023 Donkey anti Rabbit 680 for R-Luc), were incubated at a 1:20,000 dilution for 1 h at room temperature. Membranes were imaged with a LICOR Odyssey CLx. Membranes were also stained with Ponceau as an additional loading control.
In vitro translation
RNAs were translated in 9.5 μL of RRL (lacking methionine, 20 μM amino acids), 0.5 μL of 0.2 MBq [35S]-Met, and 200 ng of RNA. Reactions were incubated for 60–90 min. Proteins were resolved on 5%–12% SDS-PAGE gels and dried gels were exposed to a Fujifilm Imaging Plate Phosphor Screen (BAS-IP SR 2025). The screen was scanned using a Typhoon FLA 7000 (GE Healthcare). The image was analyzed using GelQuantNET.
cDNA synthesis and cloning of putative splicing products
HeLa cells were seeded into 6-well plates at a density of 2.5 × 105 cells per well, and transfected the next day with 1.5 μg reporter plasmid using Lipofectamine LTX with Plus reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. Cells were harvested 24 h after transfection using the E.Z.N.A. Total RNA kit (Omega Bio-tek) according to manufacturer’s instructions. 1 μg total RNA was reverse transcribed to cDNA using the iScript cDNA synthesis kit (Bio-rad). 2 μl of the cDNA was used as template in a 50 μl PCR using Phusion polymerase and reporter specific primers (Fwd primer: 5′-GCG ATC GAA TTC GAC ACA ACA GTC TCG AAC TTA AGC TG-3′; Rev primer: 5′-CCA GAG GAA TTC ATT ATC AGT GC-3′; bold letters depict restriction sites within the primers). The PCR products were analyzed by Agarose gel electrophoresis and visualized by ethidium bromide staining.
The smaller molecular mass PCR products were excised from the agarose gel and the DNA was extracted using the Wizard SV gel and PCR clean-up system (Promega). PCR products were digested with EcoRI (NEB) and BamHI (NEB) and cloned into a EcoRI/BamHI-linearized pUC19 vector, and transformed into E. coli DH5α. Six individual clones were picked, the DNA extracted and analyzed by Sanger sequencing.
Data availability statement
The data used to support our findings are either publicly available or were generated in this study and are included in the source data.
Extended Data
Extended Figure 1: Alignment of mammalian CCR5 sequences for the putative −1 PRF region in human:

Sequence alignment of CCR5 CDS regions obtained from tblastn hits to human CCR5, trimmed to only include the frameshift region. The red box indicates the putative slippery site. See Supplementary Table 1B for a list of the accession numbers used.
Extended Figure 2: Luciferase values for each experiment:
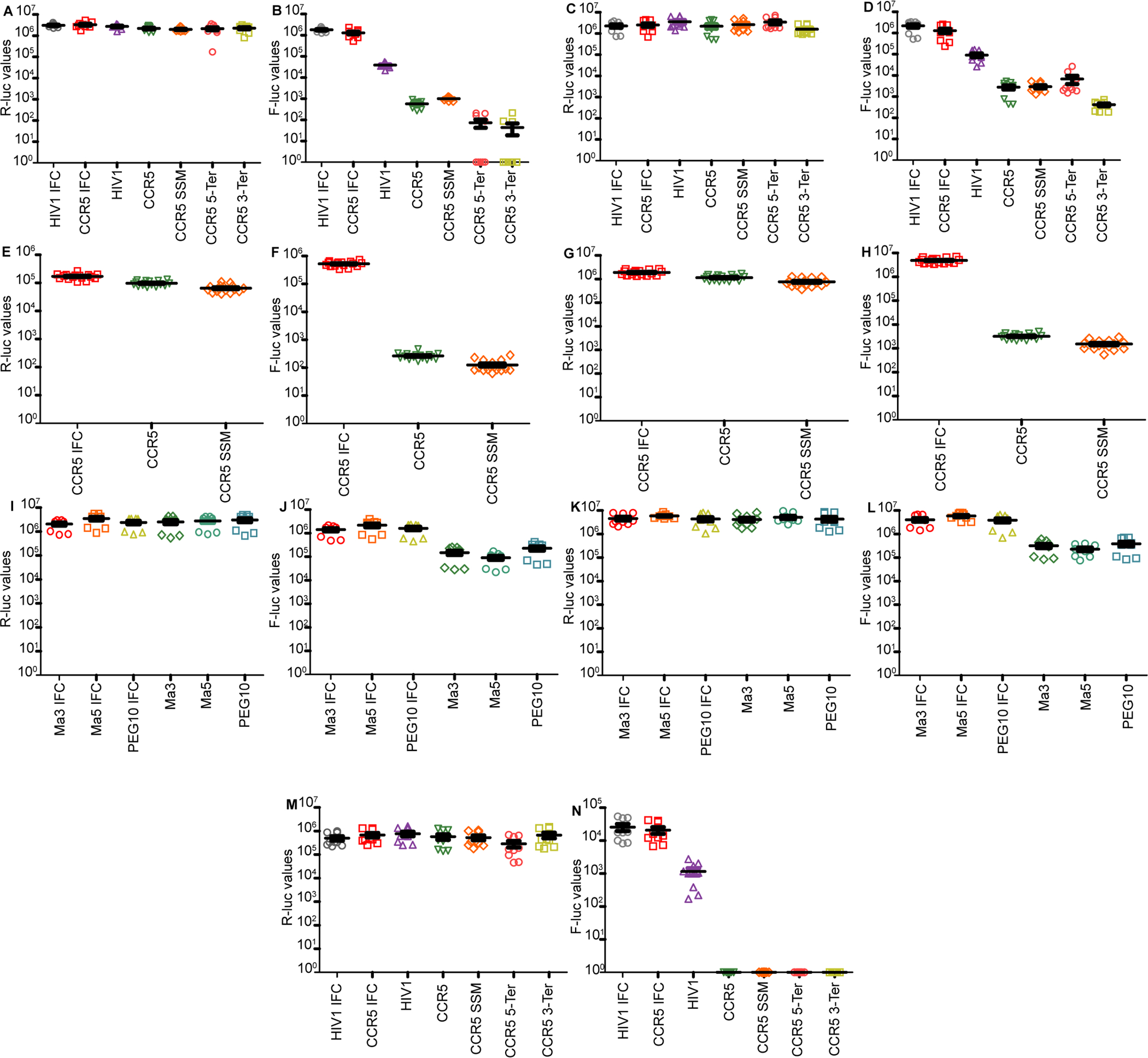
a. Absolute Renilla luciferase values for CCR5 DNA transfection experiments performed in HeLa cells. b. Absolute firefly luciferase values for CCR5 DNA transfection experiments performed in HeLa cells. c. Absolute Renilla luciferase values for CCR5 DNA transfection experiments performed in HEK293T cells. d. Absolute firefly luciferase values for CCR5 DNA transfection experiments performed in HEK293T cells. e. Absolute Renilla luciferase values for CCR5 DNA transfection experiments performed in HeLa cells f. Absolute firefly luciferase values for CCR5 DNA transfection experiments performed in HeLa cells g. Absolute Renilla luciferase values for CCR5 DNA transfection experiments performed in HEK293T cells h. Absolute firefly luciferase values for CCR5 DNA transfection experiments performed in HEK293T cells i. Absolute Renilla luciferase values for retrotransposon transfection experiments performed in HeLa cells. j. Absolute firefly luciferase values for retrotransposon transfection experiments performed in HeLa cells. k. Absolute Renilla luciferase values for retrotransposon transfection experiments performed in HEK293T cells. l. Absolute firefly luciferase values for retrotransposon transfection experiments performed in HEK293T cells. m. Absolute Renilla luciferase values for CCR5 RNA transfection experiments performed in HeLa cells. n. Absolute firefly luciferase values for CCR5 RNA transfection experiments performed in HeLa cells.
Supplementary Material
Acknowledgements
We thank Ashton T. Belew for his courage in alerting key outside individuals in May 2017 to problems with his work and recently in providing us with crucial information about the original plasmids. This work was supported by grants from NIH (R35GM118070 and R01AI133348) to J.S.K., the Wellcome Trust (106207) to A.E.F., SFI-HRB-Wellcome Trust Investigator in Science award [210692/Z/18] to P.V.B. and Science Foundation Ireland and the Irish Research Council (13/IA/1853 and IRCLA/2019/74n) to J.F.A.. A.-L.S. was supported by DFG STE 2509/2–1, S.J.K. was supported by an Irish Research Council studentship and Y.A.K. by an award of the Winston Churchill Foundation of the USA, a Graduate Research Fellowship from the National Science Foundation and the Knight-Hennessy Scholarship. Y.A.K. acknowledges the support of his father A.S. Khan.
Footnotes
Competing Interests Statement
All authors declare no competing interests.
References
- 1.Atkins JF, Loughran G, Bhatt PR, Firth AE & Baranov PV Ribosomal frameshifting and transcriptional slippage: From genetic steganography and cryptography to adventitious use. Nucleic Acids Res. 44, 7007–7078 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belew AT et al. Ribosomal frameshifting in the CCR5 mRNA is regulated by miRNAs and the NMD pathway. Nature 512, 265–269 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Belew AT, Dinman JD Cell cycle control (and more) by programmed −1 ribosomal frameshifting: implications for disease and therapeutics.Cell Cycle 14, 172–178 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grentzmann G, Ingram JA, Kelly PJ, Gesteland RF & Atkins JF A dual-luciferase reporter system for studying recoding signals. RNA 4, 479–86 (1998). [PMC free article] [PubMed] [Google Scholar]
- 5.Altschul SF, Gish W, Miller W, Myers EW & Lipman DJ Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990). [DOI] [PubMed] [Google Scholar]
- 6.O’Leary NA et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo H, Ingolia NT, Weissman JS & Bartel DP Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466, 835–840 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Michel AM, Kiniry SJ, O’Connor PBF, Mullan JP & Baranov PV GWIPS-viz: 2018 update. Nucleic Acids Res. 46, D823–D830 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kiniry SJ, O’Connor PBF, Michel AM & Baranov PV Trips-Viz: A transcriptome browser for exploring Ribo-Seq data. Nucleic Acids Res. 47, D847–D852 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Su X et al. Interferon-γ regulates cellular metabolism and mRNA translation to potentiate macrophage activation. Nat. Immunol. 16, 838–849 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loughran G, Howard MT, Firth AE & Atkins JF Avoidance of reporter assay distortions from fused dual reporters. Rna 23, 1285–1289 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biswas P, Jiang X, Pacchia AL, Dougherty JP & Peltz SW The Human Immunodeficiency Virus Type 1 Ribosomal Frameshifting Site Is an Invariant Sequence Determinant and an Important Target for Antiviral Therapy. J. Virol. 78, 2082–2087 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun Y, Abriola L, Surovtseva YV, Lindenbach BD, Guo JU Restriction of SARS-CoV-2 replication by targeting programmed −1 ribosomal frameshifting in vitro. bioRxiv 10.21.349225 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhatt PR et al. Structural basis of ribosomal frameshifting during translation of the SARS-CoV-2 RNA genome. Science (80-. ) (2021). doi: 10.1126/science.abf3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edgar RC MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kendra JA et al. Functional and structural characterization of the chikungunya virus translational recoding signals. J. Biol. Chem. 293, 17536–17545 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data used to support our findings are either publicly available or were generated in this study and are included in the source data.