

ABSTRACT
Iminosugar compounds are monosaccharide mimetics with broad but generally weak antiviral activities related to inhibition of enzymes involved in glycobiology. Miglustat (N-butyl-1-deoxynojirimycin), which is approved for the treatment of lipid storage diseases in humans, and UV-4 [N-(9-methoxynonyl)-1-deoxynojirimycin] inhibit the replication of hepatitis A virus (HAV) in cell culture (50% inhibitory concentrations [IC50s] of 32.13 μM and 8.05 μM, respectively) by blocking the synthesis of gangliosides essential for HAV cell entry. We used a murine model of hepatitis A and targeted mass spectrometry to assess the capacity of these compounds to deplete hepatic gangliosides and modify the course of HAV infection in vivo. Miglustat, given by gavage to Ifnar1−/− mice (4,800 mg/kg of body weight/day) depleted hepatic gangliosides by 69 to 75% but caused substantial gastrointestinal toxicity and failed to prevent viral infection. UV-4, similarly administered in high doses (400 mg/kg/day), was well tolerated but depleted hepatic gangliosides by only 20% after 14 days. UV-4 depletion of gangliosides varied by class. Several GM2 species were paradoxically increased, likely due to inhibition of β-glucosidases that degrade gangliosides. Both compounds enhanced, rather than reduced, virus replication. Nonetheless, both iminosugars had surprising anti-inflammatory effects, blocking the accumulation of inflammatory cells within the liver. UV-4 treatment also resulted in a decrease in serum alanine aminotransferase (ALT) elevations associated with acute hepatitis A. These anti-inflammatory effects may result from iminosugar inhibition of cellular α-glucosidases, leading to impaired maturation of glycan moieties of chemokine and cytokine receptors, and point to the potential importance of paracrine signaling in the pathogenesis of acute hepatitis A.
IMPORTANCE Hepatitis A virus (HAV) is a common cause of viral hepatitis. Iminosugar compounds block its replication in cultured cells by inhibiting the synthesis of gangliosides required for HAV cell entry but have not been tested for their ability to prevent or treat hepatitis A in vivo. We show that high doses of the iminosugars miglustat and UV-4 fail to deplete gangliosides sufficiently to block HAV infection in mice lacking a key interferon receptor. These compounds nonetheless have striking anti-inflammatory effects on the HAV-infected liver, reducing the severity of hepatitis despite enhancing chemokine and cytokine expression resulting from hepatocyte-intrinsic antiviral responses. We propose that iminosugar inhibition of cellular α-glucosidases impairs the maturation of glycan moieties of chemokine and cytokine receptors required for effective signaling. These data highlight the potential importance of paracrine signaling pathways in the inflammatory response to HAV and add to our understanding of HAV pathogenesis in mice.
KEYWORDS: antiviral agents, ganglioside, hepatitis A virus, host response, iminosugar, liver immunology, miglustat, picornavirus
INTRODUCTION
Hepatitis A virus (HAV) remains a threat to human health in many regions of the world despite the availability of highly effective, inactivated vaccines developed decades ago. The virus circulates freely in sub-Saharan Africa, where most children are infected before the age of 5 years (1). Large outbreaks of hepatitis A also continue to occur in well-developed countries. In recent years, these outbreaks have included a multinational epidemic in western Europe involving over 1,400 individuals, mostly men who have sex with men (2), and community outbreaks of hepatitis A associated with homelessness and injection drug use in the United States (3). While causing substantial morbidity, most HAV infections are self-limited. Acute liver failure is unusual in the setting of HAV infection, typically occurring in ∼0.5% of infected persons. However, the risk of fatal, fulminant hepatitis increases significantly with advancing age or underlying chronic liver disease (4). An outbreak in San Diego, CA, from 2017 to 2018 involved over 590 confirmed cases and resulted in 20 deaths, a surprisingly high mortality rate (3.4%) (5).
There are no approved antiviral therapies for acute HAV infection, and only limited efforts have been made to identify antivirals that target the virus (6). This reflects both the absence of persistent HAV infections and the ability of vaccines and passively transferred immunoglobulins to protect against acute hepatitis A. Moreover, given that the liver injury in acute hepatitis A appears to be immune mediated (7), it is not clear that effective antiviral suppression would shorten the course of the disease or lessen the mortality of fulminant hepatitis A. Although HAV is classified within the Picornaviridae family (genus Hepatovirus), it differs substantially from the enteroviruses for which effective antivirals have long been sought. The structure of the HAV capsid lacks the hydrophobic pocket into which compounds with potent antienterovirus activity bind (8), rendering the virus relatively resistant to compounds such as pleconaril (9). Similarly, both enviroxime and rupintrivir, antivirals that target nonstructural proteins of enteroviruses, have little if any activity against HAV in cell culture (9). A peptidyl monofluoromethyl ketone inhibitor of 3Cpro, the only protease expressed by hepatoviruses, has been identified and shown to have low-micromolar activity against HAV in cell culture (10), but it was never further developed. Sofosbuvir has been suggested recently to be active against the HAV polymerase (11). However, it has antiviral activity in cell culture only at concentrations far above that required to inhibit the replication of hepaciviruses and even then only in some cell lines.
HAV is an unusual hepatotropic picornavirus that circulates in the blood in a membrane-cloaked quasienveloped form and is shed in feces as naked, nonenveloped virions (12). We recently identified 39 candidate host factors for HAV using a genome-wide CRISPR-based screening strategy (13). Among these factors were multiple enzymes and transporters required for the synthesis of gangliosides, complex glycosphingolipids with head groups containing one or more sialic acid moieties. The lead hit in two independent screens was UGCG, which encodes glucosylceramide synthase (GCS), an enzyme catalyzing the first step in ganglioside synthesis (13). Subsequent studies demonstrated that a crucial late step in the cellular entry of both quasienveloped and naked HAV virions is mediated by endosomal gangliosides, particularly the disialoganglioside GD1a (13). Accordingly, Huh-7.5 human hepatoma cells became nearly completely nonpermissive for HAV infection when treated with an iminosugar inhibitor of GCS, miglustat (N-butyl-1-deoxynojirimycin). In cultured Huh-7.5 cells treated with concentrations of miglustat far below that causing cellular toxicity, hepatovirus virions taken up by endocytosis became stalled during entry in endolysosomes and failed to uncoat (13).
Iminosugar compounds such as miglustat are monosaccharide mimetics that inhibit a wide range of enzymes involved in glycobiology (14). Marketed as Zavesca, miglustat is approved for the treatment of Gaucher disease and Niemann-Pick type C disease, both autosomal recessive lipid storage diseases. Iminosugars impede the replication of a broad spectrum of enveloped viruses, including influenza virus, flaviviruses, and filoviruses, by inhibiting host α-glucosidases required for oligosaccharide trimming of immature N-linked glycans within the endoplasmic reticulum (ER) (15–18). In contrast, the inhibition of HAV replication by miglustat in cultured cells results from its capacity to inhibit GCS and thus deplete gangliosides (13). Whether this in vitro antiviral effect would be replicated in vivo is uncertain. In addition to blocking ganglioside synthesis by inhibiting GCS, iminosugars may interfere with the degradation of gangliosides by inhibiting β-glucosidases that mediate the degradation of gangliosides (19, 20). Importantly, the 50% inhibitory concentration (IC50) of miglustat for nonlysosomal glucosylceramidase (GBA2) has been reported to be considerably lower than that for GCS (0.15 μM versus 20.5 to 22.9 μM), suggesting that miglustat may have the potential to enhance ganglioside abundances at lower doses (21, 22).
Here, we describe experiments aimed at determining whether miglustat or a more potent, chemically related iminosugar inhibitor of GCS, N-(9-methoxynonyl)-1-deoxynojirimycin (UV-4), can alter the course of acute HAV infection in a murine model of acute hepatitis A. We show that high doses of these compounds fail to inhibit HAV replication in vivo but nonetheless mitigate hepatic inflammation associated with infection, thus providing insight into pathogenesis in this murine model of hepatitis A.
RESULTS
Antihepatovirus activities of iminosugar compounds in cell culture.
We assessed the in vitro antiviral activities of miglustat and UV-4 in Huh-7.5 human hepatoma cells using a nanoluciferase (NLuc)-expressing reporter virus derived from the HM175 strain of human HAV, 18f-NLuc (23) (Fig. 1A). Cells were pretreated for 72 h with a range of drug concentrations prior to infection with naked, nonenveloped virus and harvested at 12 h postinfection (p.i.) for nanoluciferase assays. Previous studies suggest that the IC50 of miglustat for HAV under these conditions is about 44 μM and that the abundance of individual ganglioside classes is reduced by 72 to 97% in cells treated with 200 μM miglustat (13). These experiments defined a somewhat lower IC50 for miglustat, 32.13 μM (Fig. 1B). UV-4 provided significantly more potent suppression of HAV infection, with an IC50 of 8.05 μM.
FIG 1.
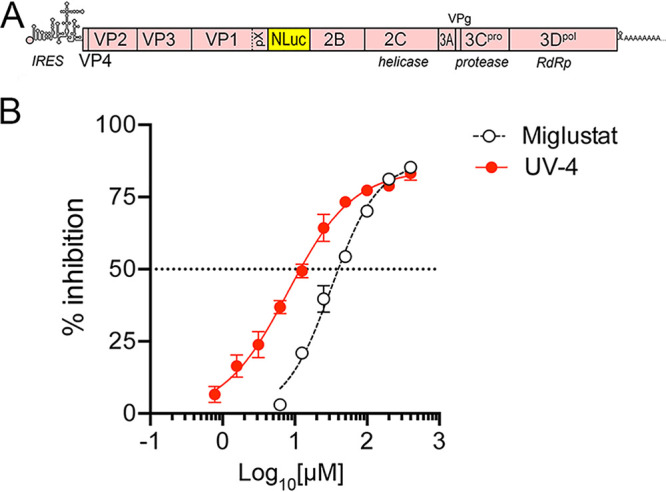
Antiviral activity of miglustat (N-butyl-1-deoxynojirimycin) and UV-4 [N-(9-methoxynonyl)-1-deoxynojirimycin] against HM175/18f-NLuc virus in Huh-7.5 cells. (A) Organization of the reporter virus genome showing the insertion of the nanoluciferase (NLuc) sequence between VP1pX and 2B, where it is flanked by 3Cpro cleavage sites. IRES, internal ribosome entry site; RdRp, RNA-dependent RNA polymerase. (B) Percent inhibition of NLuc activity expressed by HM175/18f-NLuc virus in cells treated with increasing concentrations of iminosugar compounds. Cells were treated for 72 h prior to infection with naked, nonenveloped virus and harvested for NLuc assays at 12 h p.i. Data shown are means ± standard deviations (SD) from 3 technical replicates fit to a variable-slope (four-parameter) nonlinear regression model. The estimated IC50s were 32.13 μM (95% confidence interval, 27.75 to 37.21 μM) for miglustat and 8.045 μM (95% confidence interval, 6.410 to 10.10 μM) for UV-4.
HAV infection in Ifnar1−/− mice pretreated with miglustat.
We used Ifnar1−/− mice lacking the expression of the type I interferon (IFN) receptor to assess the potential of miglustat to prevent HAV infection in vivo. As with any animal model of infectious disease, there may be important differences from infections in human. However, Ifnar1−/− mice are highly permissive for infection with wild-type HAV and develop an acute inflammatory disease of the liver similar to that in human hepatitis A, marked by hepatocellular apoptosis, mixed mononuclear intrahepatic cell infiltrates, and elevated serum levels of liver enzymes (alanine aminotransferase [ALT]) (24, 25). In an effort to deplete gangliosides required for cell entry of HAV, 9-week-old female Ifnar1−/− mice (n = 4) were treated with miglustat, given by gavage as a single daily 100-mg dose (∼4,800 mg/kg of body weight/day), beginning 4 days prior to virus challenge (Fig. 2A). A matching group of sham-treated mice (n = 4) received an equal volume of water by gavage. Previous studies suggest that mice receiving this dose of miglustat should achieve serum miglustat concentrations in excess of 20 μM, sufficient to inhibit GCS (22), and that bone marrow gangliosides should be reduced by about 70% after 6 days of treatment (26). On the day of virus challenge (day 0) (Fig. 2A), 3 mice in each group were inoculated intravenously (i.v.) with 2 × 109 genome equivalents (GE) of 6th-mouse-passage HM175 virus (“mp6” inoculum) (Fig. 2A). Beginning 4 to 5 days after the initiation of treatment (days 0 to 1 postinfection), some animals receiving miglustat developed diarrhea and displayed ruffled fur and evidence of dehydration. This was mitigated by subcutaneous fluid replacement and switching to a regimen of 50 mg miglustat twice daily (Fig. 2A). However, increasing gastrointestinal toxicity necessitated early necropsy and led to the premature death of one infected animal on day 4 postinfection.
FIG 2.

HAV infection in miglustat-treated Ifnar1−/− mice. (A) Experimental scheme showing two groups of 3 mice receiving miglustat (100 mg per day) or an equal volume of the water control by gavage for 4 days prior to i.v. challenge with mouse-passage 6 (mp6) HAV. Due to diarrhea in the treated animals, the miglustat dosage was modified as shown beginning on the day of infection. Miglustat-treated animals 8 and 7 were found moribund on days 4 and 5 p.i., respectively. (B) HAV RNA quantified by RT-PCR in feces from mice at days 4 and 5 p.i. (fecal samples were not available from mice 7 and 8 on day 5). (C) HAV RNA quantified by RT-PCR in liver tissues at necropsy on day 5 (day 4 for mouse 8). (D) Serum ALT activities at necropsy on day 5 (day 4 for mouse 8; no serum available from mouse 7). The dashed horizontal line represents the upper limit of normal. (E) Liver sections from HAV-infected animal 3 (no drug group), showing numerous apoptotic hepatocytes and diffuse parenchymal inflammatory cell infiltrates (top), and animal 6 (miglustat group), showing normal hepatic architecture, no apoptosis, and no inflammatory infiltrates (bottom). (F) Histopathology scores for hepatic inflammation from reading of 10 randomly selected 40× microscopic fields in a blind manner. Liver from mouse 8 was a poor-quality sample and not included. (G) Immunohistochemical staining for cleaved caspase 3 in liver sections from animals 3 and 6 (left to right). (H) Mean numbers of hepatocytes staining for cleaved caspase 3 per 20× microscopic field in 10 randomly selected fields of view. (I) H&E-stained sections of spleens from animals 1, 3, 5, and 6 (left to right).
Despite the early termination of this experiment, some conclusions could be drawn concerning the impact of miglustat on the course of infection. No reductions were observed in fecal shedding of virus at days 4 and 5 p.i. or in the abundance of HAV RNA in the liver at necropsy (Fig. 2B and C). On the contrary, there was a trend toward increased replication and shedding of virus in the treated animals, although neither achieved statistical significance due to the small number of animals. Despite this, serum ALT levels were lower in the miglustat-treated mice (Fig. 2D). Histological examination of liver tissues from two treated animals showed a surprising absence of inflammatory cell infiltrates (Fig. 2E and F), given that both samples contained more HAV RNA than livers from untreated animals with prominent hepatitis (Fig. 2C). Immunohistochemical staining for cleaved caspase 3 also suggested a reduced frequency of hepatocellular apoptosis in the HAV-infected animals treated with miglustat (Fig. 2G and H). There was marked splenic atrophy with a prominent loss of white pulp in miglustat-treated mice (Fig. 2I).
Miglustat depletion of hepatic gangliosides.
To determine the extent to which the ganglioside abundance was reduced by miglustat, we quantified 56 individual ganglioside species in liver tissue collected at necropsy from both treated and untreated animals using targeted mass spectrometry. Interestingly, the proportional abundances of different ganglioside classes differed remarkably from what we determined previously in Huh-7.5 human hepatoma cells (13) (Fig. 3A and B; see also Table S1 in the supplemental material). Whereas GM3 was dominant in the livers of uninfected mice, comprising >80% of gangliosides, we previously found that GM2 was dominant in hepatoma cells, in which it comprised over 70% of gangliosides. GD1a species, which appear most active in mediating HAV cell entry (13), comprised up to 10% of gangliosides in mouse liver but only ∼0.6% of gangliosides in Huh-7.5 cells (Fig. 3B). Gangliosides were quantified by different mass spectrometry methods in these two studies (labeled versus label free). This could lead to some variation in the reported percentages of each ganglioside, but methodological differences are unlikely to result in such large differences. Based on summed ion intensities, miglustat treatment reduced the total ganglioside content in liver tissue by about 70% in both infected and uninfected animals (Fig. 3C). With the possible exception of GD3, which was present only in very low abundance, miglustat-mediated reductions were similar for each ganglioside class, including GD1a. Thus, only minor changes in the relative amounts of different ganglioside classes accompanied miglustat treatment (Fig. 3D). Each of the 56 gangliosides measured in the assay was reduced by at least 50% in the treated, HAV-infected animals (Fig. 3E). However, this level of ganglioside depletion was insufficient to inhibit HAV infection (Fig. 2).
FIG 3.

Hepatic ganglioside abundance in miglustat-treated mice. (A) Ganglioside synthetic pathways. Each step in the pathway is catalyzed by a specific enzyme (39). GCS, glucosylceramide synthase, encoded by the gene UGCG. (B) Class composition of gangliosides in untreated Ifnar1−/− mouse livers (n = 8) (left) versus Huh-7.5 cells (n = 3 technical replicates) (right). Huh-7.5 cell data are from a study by Das et al. (13). (C) Ion intensities of different ganglioside classes in liver tissues from miglustat-treated and untreated naive mice (n = 1 each) (left) and HAV-infected mice (n = 2 and 3, respectively) (right) in the experiment shown in Fig. 2. Based on summed intensities, miglustat reduced the overall ganglioside abundance in HAV-infected livers by 74.9%. GM3 ganglioside was reduced by 73.7%, GM2 was reduced by 82.7%, GM1 was reduced by 78.8%, and GD1a was reduced by 78.3% compared to untreated mice. GD3 comprised less than 0.01% of gangliosides in both treated and untreated animals. (D) Class composition of gangliosides in livers from untreated and miglustat-treated mice. Data shown are the mean percentages that each ganglioside class represented of all gangliosides, ± SD, based on summed intensities. Each symbol represents the liver from an individual animal. (E) Volcano plot showing differences (fold changes) in the abundances of 56 individual ganglioside species in a repeat analysis of the liver tissues from treated versus untreated HAV-infected mice (see Table S1 in the supplemental material). Probability was determined by an unpaired t test with Šidák-Holm correction for multiple comparisons. Solid symbols indicate a false discovery rate (FDR) of <0.01%; the horizontal line indicates a P value of 0.05.
Miglustat treatment of established HAV infection in Ifnar1−/− mice.
To better understand the reduced hepatic inflammation observed with miglustat in the above-described experiment, we administered miglustat, or a sham water control, to groups of male (n = 4) or female (n = 5) mice beginning 5 days after intravenous inoculation of HAV (Fig. 4A). This is near the peak of infection, when ALT levels are maximal with this inoculum. As described above, miglustat was given by gavage at a total dose of ∼4,500 mg/kg/day. Animals were necropsied approximately 84 h (3.5 days) after the initiation of therapy. Consistent with previous observations of greater liver injury in HAV-infected male than in female Ifnar1−/− mice (I. Misumi, S. M. Lemon, and J. K. Whitmire, unpublished data), serum ALT levels were significantly higher in male than in female mice at the start of treatment (Fig. 4B). ALT levels fell subsequently in both treated and untreated mice, without any apparent miglustat effect, resulting in ALT levels in male mice being only slightly higher than and no longer significantly different from those in female mice at necropsy (Fig. 4B). Miglustat treatment resulted in significant increases in intrahepatic viral RNA abundances in both males and females (Fig. 4C). HAV RNA levels were generally higher in female than in male livers, but this difference achieved statistical significance only in treated animals (Fig. 4C). Histological examination of liver sections, with scoring for apoptotic hepatocytes and inflammatory cell infiltrates in a blind manner by an experienced veterinary hepatic pathologist, revealed no difference in the frequencies of apoptotic hepatocytes despite a marked decrease in the number of inflammatory foci in miglustat-treated mice (Fig. 4D). Hepatocytes from 4 of 5 treated mice (but no control mice) demonstrated diffuse cytoplasmic microvesicular changes (Fig. 4E). Similar numbers of hepatocytes stained positively for cleaved caspase 3 in livers from miglustat- versus sham-treated animals, confirming that there was no reduction in apoptosis, but there was a striking absence of inflammatory cells surrounding apoptotic hepatocytes in the treated animals (Fig. 4F and G). Collectively, these data confirm that treatment with high doses of miglustat reduces hepatic inflammation while enhancing viral replication or reducing viral clearance from the liver of Ifnar1−/− mice.
FIG 4.

Miglustat treatment of Ifnar1−/− mice with established HAV infection. (A) Experimental design. Groups of female (F) (n = 5 per group) or male (M) (n = 4) mice, infected 5 days previously with mouse-passaged mp6-HM175 virus, were treated with miglustat (50 mg twice daily by gavage) or the sham water control for 3.5 days prior to necropsy. q.d, once a day. (B) Serum ALT on day 5 and at necropsy on day 8. Symbols representing results from mice treated with miglustat are shaded. (C) Intrahepatic HAV RNA at necropsy on day 8. (D) Mean numbers of apoptotic hepatocytes (left) and inflammatory foci (right) per 40× field of view in H&E-stained liver sections. (E) Liver of a miglustat-treated animal showing hepatocytes with multiple small vesicles in the cytoplasm (arrows). (F) Mean number of liver cells staining for cleaved caspase 3 per 20× field of view. (G) Apoptotic hepatocytes staining positively for cleaved caspase 3 in sham (left)- and miglustat (right)-treated mice. (H) Spleen weight at necropsy on day 8. (I) Representative images of H&E-stained splenic sections. (J) Representative images of splenic sections stained immunohistochemically for cleaved caspase 3. (K) Infection-induced changes in intrahepatic cytokine and chemokine mRNA abundances in miglustat- and sham-treated animals. Data shown represent the ΔCT values between cytokine and actin transcripts, with baseline values for each derived from the means of results from 3 to 4 individual uninfected, untreated Ifnar1−/− mice (baseline shown only for Ifng). Statistical testing was done by two-way analysis of variance (ANOVA) with Tukey’s multiple-comparison test (C) or a Mann-Whitney test (D, H, and K).
As observed in animals treated prior to infection (Fig. 2J), miglustat treatment led to a sharp reduction in splenic mass (Fig. 4H). Spleens of treated mice showed a minimal to moderate loss of lymphocytes in the periarteriolar lymphoid sheath (Fig. 4I). Disruption of the interface with the marginal zone was concurrent with a relative expansion of the red pulp. In normal mice, the white pulp constituted approximately 50% of the area of the spleen. In more markedly affected spleens from treated animals, this area was reduced to approximately one-third that in tissue from untreated animals with overall shrinkage of the spleen. These changes were associated with a marked increase in the number of cleaved caspase 3-positive cells (lymphocytes as well dendritic cells or macrophages) within the red pulp (Fig. 4J).
Although the liver injury associated with HAV is incompletely understood, previous studies suggest that hepatocellular apoptosis and hepatic inflammation result from interferon regulatory factor 3 (IRF3)-mediated transcriptional responses in HAV-infected Ifnar1−/− mice (24). Unlike Ifnar1−/− mice, Mavs−/− mice, which fail to transcriptionally activate IRF3, and Irf3−/− Irf7−/− mice, which lack IRF3 expression, do not develop inflammatory changes in the liver when infected with HAV despite being equally if not more permissive to infection (24). Antibody depletion of CD8+ or CD4+ T cells or NK/NK-T cells or elimination of macrophages by treatment with clodronate liposomes prior to infection also has no impact on the development of hepatitis in Ifnar1−/− mice (24). Despite the reductions in inflammation in miglustat-treated animals, HAV-induced increases in intrahepatic Isg15, Ccl5 (RANTES), Ccl2 (monocyte chemoattractant protein 1 [MCP-1]), and Cxcl10 transcripts were greater in the treated than in the untreated animals (Fig. 4K). These cytokines are directly induced by activated IRF3 (24). These data thus indicate that IRF3 signaling was not impaired in these mice and that the reductions in inflammation were not due to decreased chemokine expression. Unlike these genes that are under direct IRF3 transcriptional control, increases in Tnfa (tumor necrosis factor alpha [TNF-α]) and Ifng (interferon gamma [IFN-γ]) expression occur later and do not correlate temporally with viral load in the liver of Ifnar1−/− mice (24). Except for two treated animals with markedly increased expression, Tnfa transcript levels were similar in treated and untreated animals. There was no increase in Ifng expression in the infected mice, but Ifng transcripts were markedly reduced from baseline levels in the miglustat-treated animals (Fig. 4K).
We also examined the impact of miglustat treatment on leukocytes in the liver and spleen. HAV infection normally results in a significant accumulation of leukocytes in the liver of Ifnar1−/− mice (24). This was reduced by about 30% by miglustat treatment (Fig. 5A). CD8+ and CD4+ T cell recruitment were both reduced by miglustat, although treatment led to no differences in the proportions of cells expressing the activation marker CD44 (Fig. 5B and C). Miglustat treatment resulted in only a small decrease in macrophage accumulation and no defect in infection-related increases in monocytes, neutrophils, or Kupffer cells. The impact of miglustat on cell numbers was more severe in the spleen, where all cell types were reduced in number by 82 to 89% (Fig. 5D and E). This included a large reduction in CD8+ T lymphocytes staining positively for CD44, which was not observed in the liver (Fig. 5C and F).
FIG 5.

Cells present in the liver and spleen of naive mice and mice with established HAV infection following miglustat or sham treatment as shown in Fig. 4A. (A) Total number of leukocytes present in the liver. (B) Cell types present in the liver. cDC, conventional dendritic cells. (C) Proportions of intrahepatic CD4+ and CD8+ lymphocytes staining positively for the activation marker CD44. (D) Total number of leukocytes in the spleen. (E) Cell types present in the spleen. (F) Proportions of CD4+ and CD8+ lymphocytes in splenic tissue staining positively for the activation marker CD44 (n = 4 in each group). P values are shown only for significant pairwise differences from untreated, infected mice, determined by 2-way ANOVA with Šidák’s multiple-comparison test (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
Taken collectively, these results are consistent with a model in which enhanced replication of the virus secondary to miglustat treatment boosts IRF3-dependent gene expression and leads to continued hepatocellular apoptosis. Despite exuberant chemokine induction, inflammatory cells fail to accumulate within the liver due to either aberrant maturation of N-linked glycans on key cellular receptor molecules or drug-related cell depletion (see Discussion).
HAV infection in Ifnar1−/− mice treated with UV-4.
The above-described experiments were carried out using maximal doses of miglustat that engendered clear gastrointestinal toxicity. To achieve a clearer view of the impact of iminosugar inhibitors on HAV infection, we pretreated mice with UV-4, which has been shown to be active against flaviviruses in mice and is about 4-fold more active than miglustat in inhibiting HAV replication in Huh-7.5 cells (Fig. 1B). Beginning 7 days prior to infection (day −7), groups of 8-week-old female Ifnar1−/− mice (n = 6) were given 4 mg UV-4 in water, or an equal volume of the water control, by gavage twice daily (total dose of ∼400 mg/kg/day) (Fig. 6A). This UV-4 dosage is over 10-fold higher than that shown previously to have significant antiviral activity against dengue virus in mice (17). Five animals in the no-drug group were inoculated i.v. with 2 × 109 GE of 10th-mouse-passage HAV (HM175-mp10), an inoculum that is somewhat less robust in inducing hepatitis than the HM175-mp6 inoculum used in the experiments described above. Twice-daily gavage with UV-4 (or the water control) continued until necropsy at 7 days p.i. (Fig. 6A). The drug was well tolerated, and the UV-4-treated animals remained healthy, with no evidence of gastrointestinal toxicity. Surprisingly, the UV-4-treated mice shed significantly more virus in feces at both 4 and 7 days p.i. (Fig. 6B). The abundance of HAV RNA was increased about 3-fold in the livers of treated versus untreated animals at necropsy, but this did not reach statistical significance (P = 0.0733 by a two-tailed t test) (Fig. 6C).
FIG 6.

HAV infection in UV-4-treated Ifnar1−/− mice. (A) Infection scheme with 3 groups of mice. Groups of mice received 4 mg UV-4 [N-(9-methoxynonyl)-1-deoxynojirimycin] in 200 μl water (n = 6) or an equal volume of the water control (n = 5) twice daily (b.i.d.) by gavage for 7 days prior to and 6 days following i.v. challenge with HM175-mp10 mouse-passaged HAV. A single mouse (mouse 1) was monitored in parallel with no drug and no virus challenge as a contemporary control. Mice were necropsied at 7 days p.i. (B) HAV RNA quantified by RT-qPCR in feces from mice at days 4 and 7 p.i. (C) HAV RNA quantified by RT-PCR in liver tissues at necropsy. (D) Serum ALT activities at necropsy. The dashed horizontal line represents the upper limit of normal. (E) Histopathology and cleaved caspase 3-positive cell scores for liver tissues from reading of 10 randomly selected microscopic fields performed in a blind manner. (F) Representative microscopic fields of hematoxylin- and eosin-stained liver tissue from mouse 5 (no drug; pathology score = 15) (top) and mouse 7 (UV-4 treated; pathology score = 5) (bottom). Arrows indicate apoptotic hepatocytes, some with a surrounding inflammatory infiltrate. (G) RT-qPCR quantitation of cytokine transcripts in liver tissue collected at 7 days p.i. Data are presented as fold changes from the uninfected, untreated control animal (mouse 1), relative to actin mRNA. (H) Splenic histopathology scores in UV-4-treated and untreated mice. Spleens were scored in a blind manner according to the following scale: 1 for normal, 2 for a minimal (approximately <30%) decrease in white pulp, 3 for a moderate decrease in the overall size and disrupted organization of the periarteriolar lymphoid sheath, and 4 for a complete loss of lymphoid cells. In all panels, pairwise comparisons were performed by a two-tailed Mann-Whitney test; significant (P < 0.05) P values are noted.
Serum ALT levels, which are typically linked closely to HAV replication in this murine model (24, 25), were significantly lower in treated than in untreated mice (Fig. 6D). Histological examination of liver tissues performed in a blind manner also revealed significantly fewer foci of inflammation and lower numbers of cleaved caspase 3-positive apoptotic hepatocytes in the UV-4-treated animals (Fig. 6E and F). Both TNF-α and IFN-γ expression levels were significantly reduced in the livers of UV-4-treated mice, whereas Isg15 and Ccl5 transcripts were slightly increased in abundance (Fig. 6G). There was a moderate loss of white pulp in the spleen of one animal in each treatment group but no difference overall in splenic histology in the UV-4-treated versus untreated animals (Fig. 6H). Taken collectively, these data show that UV-4 has no significant antiviral activity against HAV in mice at this dosage but nonetheless mitigates inflammation resulting from viral replication in the liver.
Hepatic ganglioside abundance in UV-4-treated animals.
Targeted mass spectrometry analyses indicated that UV-4 treatment reduced the total hepatic ganglioside abundance by only 20% (Fig. 7A; Table S2). Not all ganglioside classes were equally affected. Analysis of raw ion intensities revealed an increase in the relative abundance of GM2, from 4.4% of all gangliosides in untreated mice to 7.9% in UV-4-treated mice (Fig. 7B). Whereas most ganglioside species were reduced in abundance, several GM2 species (especially those with long acyl tails, GM2 d18:1/22:0 and GM2 d18:0/24:0) were increased in UV-4-treated animals (Fig. 7C and D). These increases likely reflect variable inhibition of different ganglioside-specific β-glucosidases mediating ganglioside degradation (27).
FIG 7.

Hepatic ganglioside abundance in UV-4-treated mice. (A) Summed mean ion intensities of ganglioside classes in liver tissue from untreated (n = 4) versus UV-4-treated (n = 5) mice. Based on summed ion intensities, UV-4 reduced the overall hepatic ganglioside abundance by 19.9%. The GD1a ganglioside abundance was reduced by 10.5%, GM1 was reduced by 23.8%, and GM3 was reduced by 32.6%, whereas the GM2 abundance was increased by 55.8%. (B) Class composition of gangliosides in livers from untreated and UV-4-treated mice. Data shown are the mean percentages that each ganglioside class represents of all gangliosides, ± SD. GD3 comprised less than 0.1% of gangliosides in both treated and untreated animals. Statistical significance was determined by a t test with Holm-Šidák correction for multiple comparisons. (C) A volcano plot shows the differences detected in the abundances of individual ganglioside species in liver tissues from treated versus untreated HAV-infected mice. Probability was determined by an unpaired t test with Holm-Šidák correction for multiple comparisons. Solid symbols indicate an FDR of <0.01%; the horizontal line indicates a P value of 0.05. (D) Mean raw ion intensity values ± SD for ganglioside GM2 species in liver tissues from untreated (n = 4) or UV-4-treated (n = 5) mice. Statistical significance was determined by two-way ANOVA.
DISCUSSION
Following its endocytosis, HAV must interact with gangliosides expressed within the endolysosomal membrane for uncoating to occur and for the viral genome to be delivered to ribosomes to initiate the translation of viral proteins (13). The antiviral activity of miglustat stems from its ability to block the de novo synthesis of gangliosides by inhibiting glucosylceramide synthase (GCS), an enzyme that catalyzes the conjugation of UDP-glucose to ceramide to produce lactosylceramide as the initial step in the synthesis of all gangliosides (Fig. 3A) (27). Hepatovirus replication is thus blocked in cell culture by the depletion of cellular gangliosides (13). This mechanism of action is distinct from that underlying the antiviral activity of iminosugar compounds against enveloped viruses such as dengue virus, which is thought to involve disruption of the trimming of glycans on glycoproteins, a process normally mediated by host α-glucosidases (16, 17).
Very high doses of miglustat (4,800 mg/kg/day) resulted in a 70% reduction in ganglioside abundance within the livers of Ifnar1−/− mice (Fig. 3C and E). This degree of depletion was without antiviral effects and may even have been associated with increased replication of virus (Fig. 2C). UV-4, also used at a relatively high dose (400 mg/kg/day), reduced the abundance of most ganglioside species by less than 2-fold but paradoxically increased the abundance of many GM2 species (Fig. 7B to D). This was associated with a highly significant 30-fold increase in fecal virus shedding in treated animals (Fig. 6B) and a 3-fold increase in the intrahepatic viral load that did not achieve statistical significance (Fig. 6C). Gangliosides are enzymatically degraded within lysosomes by β-glucosidases that, like GCS, are inhibited by miglustat (27). Any contrary increases in ganglioside abundance may have been greatest within lysosomes, where gangliosides function in HAV entry (13). An additional factor that may have contributed to the lack of antihepatovirus activity in vivo is the 10-fold-higher relative abundance of GD1a in mouse liver than in Huh-7.5 cells (13) (Fig. 3B). Exogenous GD1a is ∼10-fold more active than other gangliosides in blocking HAV infection of Huh-7.5 cells (13), suggesting that it is more active in mediating viral entry. Residual levels of GD1a in particular may have been sufficient for continued HAV cell entry in both miglustat- and UV-4-treated animals.
Impaired immune control may have also contributed to greater HAV replication in the treated animals. Miglustat and UV-4 treatment resulted in reduced inflammatory cell populations and IFN-γ expression in the livers of treated animals (Fig. 2E and F, Fig. 4D and G, and Fig. 6E and F). Inflammatory cell infiltrates were sharply reduced in miglustat-treated animals despite greater induction of chemokine expression in the liver (Fig. 2I and Fig. 4K). Increases in chemokine expression were consistent with increases in viral RNA in the treated animals (Fig. 2C and Fig. 4C) and likely resulted directly from MAVS-dependent transcriptional activation of IRF3, as shown previously (24). Despite robust replication of the virus, there is no inflammatory response to HAV infection in Mavs−/− mice or in Irf3−/−/Irf7−/− mice lacking IRF3 expression (24).
Chao et al. (28) recently demonstrated that proinflammatory MAVS-dependent NF-κB responses in murine astrocytes are boosted by lactosylceramide, which they found to promote the interaction of MAVS with cytosolic phospholipase A (cPLA2). By inhibiting the synthesis of lactosylceramide, the first intermediate in ganglioside synthesis (Fig. 3A), these investigators found that miglustat reduced the inflammatory response in experimental murine autoimmune encephalitis (28). While possible, a similar suppression of MAVS signaling within hepatocytes seems an unlikely explanation for the reduced inflammation in miglustat- or UV-4-treated mice, as the transcription of Isg15 and Ccl5, both of which are directly regulated transcriptionally by IRF3 (29, 30), remained strongly induced, and in some cases even increased, in treated mice (Fig. 4K and Fig. 6G).
An alternative explanation for the lack of inflammatory cells accumulating within the liver in treated mice is a failure of chemokines such as RANTES (Ccl5) and MCP-1 (Ccl2) to recruit cells to the liver. This could result from a lack of proper glycan maturation and potential misfolding of essential cellular glycoprotein receptors secondary to iminosugar inhibition of α-glucosidases in the endoplasmic reticulum. The antiviral action of UV-4 against dengue virus results from such activity (31). Moreover, dampened signaling responses to IFN-γ and TNF-α receptor engagement have been reported in cells treated with either miglustat or UV-4 (32). This scenario is consistent with the absence of inflammatory cells normally found to surround cleaved caspase 3-positive hepatocytes in miglustat-treated mice (Fig. 4G). A similar phenomenon may explain the splenic atrophy observed in these mice, as homeostatic lymphotoxin-α1β2 signaling is essential for the maintenance of normal MAdCAM-1+ (mucosal addressin cell adhesion molecule 1) endothelial cell architecture in the spleen (33, 34). In addition, the generalized lymphopenia evident in spleens of miglustat-treated animals (Fig. 4I and J and Fig. 5E) may have contributed to the reduction in cells infiltrating the infected liver as well as the decreases observed in intrahepatic Ifng expression (Fig. 4K and Fig. 6G). However, the loss of IFN-γ expression probably played no role in the anti-inflammatory effects of the iminosugar compounds, as Ifng was not induced above basal levels in untreated, infected Ifnar1−/− mice with hepatitis (Fig. 4K and Fig. 6G). In addition, previous studies show that hepatic inflammation is just as severe in double-knockout Ifngr1−/− Ifnar1−/− mice lacking both type I and type II IFN receptors (24).
In summary, neither miglustat nor UV-4, both used at relatively high doses, provides any useful suppression of HAV replication in Ifnar1−/− mice. However, both iminosugar compounds negatively impact the accumulation of inflammatory cells in the livers of HAV-infected mice, likely by disrupting chemokine and cytokine signaling. How such host-targeted effects of iminosugar compounds influence the immune responses to other virus infections should be considered when these compounds are employed for their antiviral effect.
MATERIALS AND METHODS
Virus and cells.
Huh-7.5 human hepatoma cells and HM175/p16 virus have been described previously (13, 35, 36). Viral inocula for mouse infections were prepared from lysates of liver from Mavs−/− mice infected with 6th-mouse-passage (HM175-mp6) or 10th-mouse-passage (HM175-mp10) virus, as described previously (24).
Iminosugar compounds.
Miglustat (N-butyl-1-deoxynojirimycin), also known as UV-1, and UV-4 [N-(9-methoxynonyl)-1-deoxynojirimycin] were provided by Emergent Biosolutions, Inc., Gaithersburg, MD. In both in vitro and in vivo experiments, UV-4 was administered as the hydrochloride salt, also known as UV-4B.
RT-qPCR for HAV RNA and cytokine mRNA.
RNA was extracted from liver tissue using TRIzol reagent (Invitrogen Life Technologies) according to the manufacturer’s suggested protocol. Fecal RNA was isolated using the QIAamp viral RNA isolation kit (Qiagen). cDNA was synthesized using a SuperScript III first-strand synthesis supermix kit (Invitrogen). HAV RNA was quantified by reverse transcription-quantitative PCR (RT-qPCR) using iTaq SYBR green (Bio-Rad) with a real-time PCR detection system (Bio-Rad), as described previously (13). HAV RNA was determined by reference to a standard curve generated with synthetic HAV RNA. For the quantification of liver cytokines, primers for Isg15, Ccl5, Ccl2, Tnfa, Cxcl10, Ifng, and β-actin were purchased from Sigma; qPCR was carried out with iTaq universal SYBR green supermix (Bio-Rad).
Cell-based antiviral activity assays.
Huh-7.5 cells (104) were plated in wells of a 96-well plate. The cells were refed 24 h later with Dulbecco’s modified Eagle’s medium (DMEM) containing increasing concentrations of miglustat or UV-4 and 10% fetal bovine serum. Cells were inoculated 72 h later with 18f-NLuc virus (9 × 106 GE); virus was allowed to adsorb for 2 h at 37°C and then washed off and replaced with fresh medium containing iminosugar compounds. Cells were harvested for NLuc assays at 12 h p.i. NLuc assays were carried out as described previously (13).
HAV infection in Ifnar1−/− mice.
Female Ifnar1−/− mice were intravenously inoculated with virus at 6 to 8 weeks of age. Mice were housed in individual cages for the collection of fecal pellets and serum samples. Livers and spleens were harvested at necropsy and stored in RNAlater (Thermo Fisher Scientific), snap-frozen on dry ice and kept at −80°C, or fixed in 10% formalin for 48 h and stored in 70% ethanol for histology. All experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill.
Alanine aminotransferase.
Mouse sera (2.5 μl) were diluted 1:2 in phosphate-buffered saline (PBS) and assayed for alanine aminotransferase (ALT) activity using the alanine aminotransferase activity assay kit (Reitman-Frankel method; Elabscience) according to the manufacturer’s suggested protocol.
Histopathological examination of mouse liver and spleen.
Sections (4-μm thickness) cut from formalin-fixed, paraffin-embedded liver and spleen were stained with hematoxylin and eosin (H&E) and examined for histological changes by light microscopy. Liver sections were scored in a blind manner for inflammation by a veterinary hepatic pathologist by enumerating foci of inflammation in 10 randomly selected 10× fields of view.
Hepatic and splenic leukocyte analysis.
Single-cell suspensions were prepared from the spleen and liver for analysis of leukocytes. Perfused livers were cut into small pieces and digested at 37°C for 30 min with 100 U/ml type IV collagenase (Gibco/Thermo Scientific, Waltham, MA) and 10 mg/ml DNase I (Sigma-Aldrich) in DMEM containing 1 mM CaCl2 and 1 mM MgCl2. After passing through a 70-μm nylon cell strainer, hepatocytes were spun down briefly by centrifugation at 50 × g for 2 min and discarded. Washed and pelleted cells were resuspended with a 40% isotonic Percoll solution (GE Healthcare) and overlaid on a cushion of an 80% Percoll solution. Intrahepatic leukocytes were carefully removed from the Percoll solution after centrifugation at 800 × g for 20 min at room temperature (RT). Spleens were physically disrupted over a 70-μm nylon cell strainer (Corning, NY). Single-cell preparations from spleens and livers were rinsed and resuspended in 10% RPMI 1640 medium and then surface stained with combinations of fluorescently labeled monoclonal antibodies (mAbs) procured from BioLegend (see Table S3 in the supplemental material). Fluorescently labeled cells were quantified by flow cytometry using a FACSCalibur cytometer (BD Biosciences), and the data were analyzed with FlowJo software (TreeStar). All mAbs were purchased from BioLegend.
Extraction of gangliosides from mouse liver.
Mouse liver was washed to remove blood using PBS and blot dried, and 200 mg of tissue was then weighed, added to 750 μl liquid chromatography-mass spectrometry (LC-MS)-grade water (Fisher Scientific), and homogenized using a Precellys Evolution homogenizer (Bertin Technologies) at 6,800 rpm for 30 s in 2 cycles. The homogenate was mixed with 2 ml LC-MS-grade methanol (Fisher Scientific) and 1 ml high-performance liquid chromatography (HPLC)-grade chloroform (Fisher Scientific) and then centrifuged at 2,500 rpm at room temperature for 15 min. The supernatant was collected and mixed with 650 μl of LC-MS-grade water and then recentrifuged at 2,500 rpm at room temperature for 15 min. The upper layer thus formed was collected and dried under a stream of nitrogen gas. The dried ganglioside extract was reconstituted using chloroform-methanol-water (2:1:0.1, vol/vol/vol) to 1 mg/μl (tissue wt/vol) before liquid chromatography–multiple-reaction monitoring (LC-MRM) analysis.
RPLC-MRM analysis of label-free gangliosides.
A binary Vanquish ultrahigh-performance liquid chromatography (UHPLC) system (Thermo Scientific) coupled with a Quantiva triple-quadrupole mass spectrometer (Thermo Fisher Scientific, Haverhill, MA) was used for LC-MRM analysis. For chromatographic separation, a Cortecs C18 column (2.6-mm internal diameter [i.d.] by 100 mm, 1.6 μm; Waters) and the following mobile phases were used: mobile phase A contained 60:40 acetonitrile-water with 10 mM ammonium formate and 0.1% formic acid, and mobile phase B contained 90:10 isopropanol-acetonitrile with 10 mM ammonium formate and 0.1% formic acid. The gradient was 30% mobile phase B (0 min), 50% mobile phase B (1 min), 70% mobile phase B (7 min), 99% mobile phase B (13 min), 30% mobile phase B (13.1 min), and column equilibration for an additional 2 min at 30% mobile phase B. The column was maintained at 40°C at a flow rate of 350 μl/min. The injection volume was 10.0 μl. The following parameters were set for the mass spectrometer: 20 Arb (arbitrary units), 7 Arb, 1 Arb, 300°C, and 300°C for sheath gas, auxiliary gas, sweep gas, ion transfer tube temperature, and vaporizer temperature, respectively. The ion source was operated using heated electrospray ionization (ESI) with the ion spray voltage set at 3,000 V in negative-ion mode. The collision cell gas pressure was 1.5 mtorr, and the collision energy for each ganglioside class was optimized using standards. Scheduled MRM mode was employed for analysis of all gangliosides based on a previously reported method (37), with some modifications. First, a list of esterified fatty acids in ganglioside extracts of mouse liver was determined by fatty acid analysis; next, the theoretical mass and transition list of all potential gangliosides were generated based on the fatty acid list and theoretical fragment ions. Fragment ions were then verified using PRM (parallel-reaction monitoring) mode in a Q Exactive HF mass spectrometer (Thermo Scientific). The retention time was verified against known ganglioside standards. Raw data files were acquired in XCalibur 2.2 (Thermo Scientific) and processed in Skyline (20.1.0.28) (38).
Statistical analysis.
Statistical calculations were carried out using Prism 6.0 (GraphPad).
ACKNOWLEDGMENTS
We thank Kevin Spurgers and Anthony Treston of Emergent Biosolutions, Inc., Gaithersburg, MD, for generously providing the iminosugar compounds and for helpful discussions and reviews of the manuscript.
This work was supported in part by grants from the U.S. National Institutes of Health: R01-AI103083, R01-AI131685, and R01-AI150095 (S.M.L.) and R01-DK123499 (Q.Z.).
Footnotes
Supplemental material is available online only.
Contributor Information
Stanley M. Lemon, Email: smlemon@med.unc.edu.
J.-H. James Ou, University of Southern California.
REFERENCES
- 1.Jacobsen KH. 2018. Globalization and the changing epidemiology of hepatitis A virus. Cold Spring Harb Perspect Med 8:a031716. 10.1101/cshperspect.a031716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ndumbi P, Freidl GS, Williams CJ, Mårdh O, Varela C, Avellón A, Friesema I, Vennema H, Beebeejaun K, Ngui SL, Edelstein M, Smith-Palmer A, Murphy N, Dean J, Faber M, Wenzel J, Kontio M, Müller L, Midgley SE, Sundqvist L, Ederth JL, Roque-Afonso A-M, Couturier E, Klamer S, Rebolledo J, Suin V, Aberle SW, Schmid D, De Sousa R, Augusto GF, Alfonsi V, Del Manso M, Ciccaglione AR, Mellou K, Hadjichristodoulou C, Donachie A, Borg M-L, Sočan M, Poljak M, Severi E, Members of the European Hepatitis A Outbreak Investigation Team . 2018. Hepatitis A outbreak disproportionately affecting men who have sex with men (MSM) in the European Union and European Economic Area, June 2016 to May 2017. Euro Surveill 23:1700641. 10.2807/1560-7917.ES.2018.23.33.1700641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Foster MA, Hofmeister MG, Kupronis BA, Lin Y, Xia GL, Yin S, Teshale E. 2019. Increase in hepatitis A virus infections—United States, 2013–2018. MMWR Morb Mortal Wkly Rep 68:413–415. 10.15585/mmwr.mm6818a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shin EC, Jeong SH. 2018. Natural history, clinical manifestations, and pathogenesis of hepatitis A. Cold Spring Harb Perspect Med 8:a031708. 10.1101/cshperspect.a031708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wooten DA. 2019. Forgotten but not gone: learning from the hepatitis A outbreak and public health response in San Diego. Top Antivir Med 26:117–121. [PMC free article] [PubMed] [Google Scholar]
- 6.Debing Y, Neyts J, Thibaut HJ. 2014. Molecular biology and inhibitors of hepatitis A virus. Med Res Rev 34:895–917. 10.1002/med.21292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lemon SM, Ott JJ, Van Damme P, Shouval D. 2018. Type A viral hepatitis: a summary and update on the molecular virology, epidemiology, pathogenesis and prevention. J Hepatol 68:167–184. 10.1016/j.jhep.2017.08.034. [DOI] [PubMed] [Google Scholar]
- 8.Wang X, Ren J, Gao Q, Hu Z, Sun Y, Li X, Rowlands DJ, Yin W, Wang J, Stuart DI, Rao Z, Fry EE. 2015. Hepatitis A virus and the origins of picornaviruses. Nature 517:85–88. 10.1038/nature13806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Debing Y, Kaplan GG, Neyts J, Jochmans D. 2013. Rapid and convenient assays to assess potential inhibitory activity on in vitro hepatitis A replication. Antiviral Res 98:325–331. 10.1016/j.antiviral.2013.03.016. [DOI] [PubMed] [Google Scholar]
- 10.Morris TS, Frormann S, Shechosky S, Lowe C, Lall MS, Gauss-Muller V, Purcell RH, Emerson SU, Vederas JC, Malcolm BA. 1997. In vitro and ex vivo inhibition of hepatitis A virus 3C proteinase by a peptidyl monofluoromethyl ketone. Bioorg Med Chem 5:797–807. 10.1016/s0968-0896(97)88649-x. [DOI] [PubMed] [Google Scholar]
- 11.Jiang W, Muhammad F, Ma P, Liu X, Long G. 2018. Sofosbuvir inhibits hepatitis A virus replication in vitro assessed by a cell-based fluorescent reporter system. Antiviral Res 154:51–57. 10.1016/j.antiviral.2018.04.007. [DOI] [PubMed] [Google Scholar]
- 12.Feng Z, Hensley L, McKnight KL, Hu F, Madden V, Ping L, Jeong SH, Walker C, Lanford RE, Lemon SM. 2013. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 496:367–371. 10.1038/nature12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Das A, Barrientos RC, Shiota T, Madigan V, Misumi I, McKnight KL, Sun L, Li Z, Meganck RM, Li Y, Kaluzna E, Asokan A, Whitmire JK, Kapustina M, Zhang Q, Lemon SM. 2020. Gangliosides are essential endosomal receptors for quasi-enveloped and naked hepatitis A virus. Nat Microbiol 5:1069–1078. 10.1038/s41564-020-0727-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horne G, Wilson FX, Tinsley J, Williams DH, Storer R. 2011. Iminosugars past, present and future: medicines for tomorrow. Drug Discov Today 16:107–118. 10.1016/j.drudis.2010.08.017. [DOI] [PubMed] [Google Scholar]
- 15.Tyrrell BE, Sayce AC, Warfield KL, Miller JL, Zitzmann N. 2017. Iminosugars: promising therapeutics for influenza infection. Crit Rev Microbiol 43:521–545. 10.1080/1040841X.2016.1242868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Warfield KL, Alonzi DS, Hill JC, Caputo AT, Roversi P, Kiappes JL, Sheets N, Duchars M, Dwek RA, Biggins J, Barnard D, Shresta S, Treston AM, Zitzmann N. 2020. Targeting endoplasmic reticulum α-glucosidase I with a single-dose iminosugar treatment protects against lethal influenza and dengue virus infections. J Med Chem 63:4205–4214. 10.1021/acs.jmedchem.0c00067. [DOI] [PubMed] [Google Scholar]
- 17.Warfield KL, Plummer EM, Sayce AC, Alonzi DS, Tang W, Tyrrell BE, Hill ML, Caputo AT, Killingbeck SS, Beatty PR, Harris E, Iwaki R, Kinami K, Ide D, Kiappes JL, Kato A, Buck MD, King K, Eddy W, Khaliq M, Sampath A, Treston AM, Dwek RA, Enterlein SG, Miller JL, Zitzmann N, Ramstedt U, Shresta S. 2016. Inhibition of endoplasmic reticulum glucosidases is required for in vitro and in vivo dengue antiviral activity by the iminosugar UV-4. Antiviral Res 129:93–98. 10.1016/j.antiviral.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller JL, Tyrrell BE, Zitzmann N. 2018. Mechanisms of antiviral activity of iminosugars against dengue virus. Adv Exp Med Biol 1062:277–301. 10.1007/978-981-10-8727-1_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kolter T, Sandhoff K. 2010. Lysosomal degradation of membrane lipids. FEBS Lett 584:1700–1712. 10.1016/j.febslet.2009.10.021. [DOI] [PubMed] [Google Scholar]
- 20.van der Spoel AC, Mott R, Platt FM. 2008. Differential sensitivity of mouse strains to an N-alkylated imino sugar: glycosphingolipid metabolism and acrosome formation. Pharmacogenomics 9:717–731. 10.2217/14622416.9.6.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang P, Dwek RA, Pollock S, Zitzmann N, Butters TD, Alonzi D, Kiappes J, Ramstedt U. 6 November 2014. Glycolipid inhibition using iminosugars. WIPO patent WO 2014/1794338 A2.
- 22.Walden CM, Sandhoff R, Chuang CC, Yildiz Y, Butters TD, Dwek RA, Platt FM, van der Spoel AC. 2007. Accumulation of glucosylceramide in murine testis, caused by inhibition of beta-glucosidase 2: implications for spermatogenesis. J Biol Chem 282:32655–32664. 10.1074/jbc.M702387200. [DOI] [PubMed] [Google Scholar]
- 23.Das A, Hirai-Yuki A, Gonzalez-Lopez O, Rhein B, Moller-Tank S, Brouillette R, Hensley L, Misumi I, Lovell W, Cullen JM, Whitmire JK, Maury W, Lemon SM. 2017. TIM1 (HAVCR1) is not essential for cellular entry of either quasi-enveloped or naked hepatitis A virions. mBio 8:e00969-17. 10.1128/mBio.00969-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirai-Yuki A, Hensley L, McGivern DR, Gonzalez-Lopez O, Das A, Feng H, Sun L, Wilson JE, Hu F, Feng Z, Lovell W, Misumi I, Ting JP, Montgomery S, Cullen J, Whitmire JK, Lemon SM. 2016. MAVS-dependent host species range and pathogenicity of human hepatitis A virus. Science 353:1541–1545. 10.1126/science.aaf8325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirai-Yuki A, Whitmire JK, Joyce M, Tyrrell DL, Lemon SM. 2019. Murine models of hepatitis A virus infection. Cold Spring Harb Perspect Med 9:a031674. 10.1101/cshperspect.a031674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ersek A, Xu K, Antonopoulos A, Butters TD, Santo AE, Vattakuzhi Y, Williams LM, Goudevenou K, Danks L, Freidin A, Spanoudakis E, Parry S, Papaioannou M, Hatjiharissi E, Chaidos A, Alonzi DS, Twigg G, Hu M, Dwek RA, Haslam SM, Roberts I, Dell A, Rahemtulla A, Horwood NJ, Karadimitris A. 2015. Glycosphingolipid synthesis inhibition limits osteoclast activation and myeloma bone disease. J Clin Invest 125:2279–2292. 10.1172/JCI59987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kolter T. 2012. Ganglioside biochemistry. ISRN Biochem 2012:506160. 10.5402/2012/506160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chao CC, Gutiérrez-Vázquez C, Rothhammer V, Mayo L, Wheeler MA, Tjon EC, Zandee SEJ, Blain M, de Lima KA, Takenaka MC, Avila-Pacheco J, Hewson P, Liu L, Sanmarco LM, Borucki DM, Lipof GZ, Trauger SA, Clish CB, Antel JP, Prat A, Quintana FJ. 2019. Metabolic control of astrocyte pathogenic activity via cPLA2-MAVS. Cell 179:1483–1498.e22. 10.1016/j.cell.2019.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grandvaux N, Servant MJ, tenOever B, Sen GC, Balachandran S, Barber GN, Lin R, Hiscott J. 2002. Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J Virol 76:5532–5539. 10.1128/jvi.76.11.5532-5539.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Genin P, Algarte M, Roof P, Lin R, Hiscott J. 2000. Regulation of RANTES chemokine gene expression requires cooperativity between NF-kappa B and IFN-regulatory factor transcription factors. J Immunol 164:5352–5361. 10.4049/jimmunol.164.10.5352. [DOI] [PubMed] [Google Scholar]
- 31.Sayce AC, Alonzi DS, Killingbeck SS, Tyrrell BE, Hill ML, Caputo AT, Iwaki R, Kinami K, Ide D, Kiappes JL, Beatty PR, Kato A, Harris E, Dwek RA, Miller JL, Zitzmann N. 2016. Iminosugars inhibit dengue virus production via inhibition of ER alpha-glucosidases—not glycolipid processing enzymes. PLoS Negl Trop Dis 10:e0004524. 10.1371/journal.pntd.0004524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller JL, Hill ML, Brun J, Pountain A, Sayce AC, Zitzmann N. 2019. Iminosugars counteract the downregulation of the interferon γ receptor by dengue virus. Antiviral Res 170:104551. 10.1016/j.antiviral.2019.104551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zindl CL, Kim TH, Zeng M, Archambault AS, Grayson MH, Choi K, Schreiber RD, Chaplin DD. 2009. The lymphotoxin LTalpha(1)beta(2) controls postnatal and adult spleen marginal sinus vascular structure and function. Immunity 30:408–420. 10.1016/j.immuni.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tan JKH, Watanabe T. 2018. Determinants of postnatal spleen tissue regeneration and organogenesis. NPJ Regen Med 3:1. 10.1038/s41536-018-0039-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jansen RW, Newbold JE, Lemon SM. 1988. Complete nucleotide sequence of a cell culture-adapted variant of hepatitis A virus: comparison with wild-type virus with restricted capacity for in vitro replication. Virology 163:299–307. 10.1016/0042-6822(88)90270-x. [DOI] [PubMed] [Google Scholar]
- 36.Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol 76:13001–13014. 10.1128/jvi.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ikeda K, Shimizu T, Taguchi R. 2008. Targeted analysis of ganglioside and sulfatide molecular species by LC/ESI-MS/MS with theoretically expanded multiple reaction monitoring. J Lipid Res 49:2678–2689. 10.1194/jlr.D800038-JLR200. [DOI] [PubMed] [Google Scholar]
- 38.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. 2010. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26:966–968. 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Groux-Degroote S, Guerardel Y, Delannoy P. 2017. Gangliosides: structures, biosynthesis, analysis, and roles in cancer. Chembiochem 18:1146–1154. 10.1002/cbic.201600705. [DOI] [PubMed] [Google Scholar]