

Abstract
Traumatic brain injury (TBI) in children <4 years of age leads to long-term deficits in cognitive and learning abilities that can persist or even worsen as these children age into adolescence. In this study, the role of glucocorticoid receptor (GR) function in the dorsal hippocampus (DH) in hippocampal-dependent cognitive function and synaptic plasticity were assessed following injury to the 11-day-old rat. Brain injury produced significant impairments in spatial learning and memory in the Morris water maze in male and female rats at 1-month post-injury (adolescence), which was accompanied by impairments in induction and maintenance of long-term potentiation (LTP) in the CA1 region of the DH. Brain injury resulted in a significant decrease in the expression of the glucocorticoid-inducible gene, serum- and glucocorticoid-kinase 1 (sgk1), suggestive of an impairment in GR transcriptional activity within the hippocampus. Lentiviral transfection of the human GR (hGR) in the DH improved spatial learning and memory in the Morris water maze and attenuated LTP deficits following TBI. GR overexpression in the DH was also associated with a significant increase in the mRNA expression levels of sgk1, and the glutamate receptor subunits GluA1 and GluA2 within the hippocampus. Overall, these findings support an important role for dorsal hippocampal GR function in learning and memory deficits following pediatric TBI and suggest that these effects may be related to the regulation of glutamate receptor subunit expression in the DH.
Keywords: cognition, glucocorticoid receptors, hippocampus, long-term potentiation, pediatric TBI
Significance Statement
Traumatic brain injury (TBI) is the leading cause of injury-induced mortality in children <4 years of age; survivors are faced with life-long cognitive impairments that manifest in early adolescence. In this study, we explored the role of glucocorticoid receptor (GR) dysfunction in the dorsal hippocampus (DH) on the effects of pediatric TBI on hippocampal-dependent learning and synaptic plasticity during adolescence. The results indicate that viral transfection of the human GR in the DH following pediatric TBI improves spatial learning and facilitates long-term potentiation (LTP), potentially through upregulating the expression of glutamate receptor subunits GluA1 and GluA2. These findings provide novel support for the GR system as a target for improving long-term cognitive outcomes in pediatric TBI patients.
Introduction
Children <4 years of age are at the highest risk for TBI, with ∼250,000 injuries in this age group reported each year in the United States.1–3 Impairments in learning and memory abilities are among the most prevalent outcomes after childhood TBI,4,5 often leading to poor academic performance and requiring placement in special education classrooms.6 These impairments resulting from TBI during early childhood can become more apparent over time as children reach adolescence.6–9 Although improvements in post-traumatic neurocritical supportive care over the past two decades have significantly reduced mortality, there remains a lack of therapeutic treatments to limit these chronic deficits in survivors of childhood TBI.10–12
Stressful or traumatic events that occur during early critical periods of development can lead to long-term changes in the expression of GRs within limbic regions including the hippocampus, amygdala, and hypothalamus.13–15 Few previous studies have investigated changes in GR expression as an outcome of pediatric TBI, although a significant decrease in GR expression in the cortex was reported up to 72 h following focal cerebral ischemia in neonate rats.16 We and others have reported impairments in hippocampal-dependent spatial learning and memory following pediatric TBI.17–21 Although the importance of GR-mediated actions of glucocorticoids in learning and memory is well established, whether impaired function of GRs may contribute to long-term deficits in cognitive function following pediatric TBI has yet to be explored.
GRs and mineralocorticoid receptors (MRs) are members of the steroid hormone receptor family, and therefore are ligand-inducible transcription factors.22 During baseline conditions of low circulating glucocorticoids, GRs are predominantly found in the cytoplasm,23 whereas MRs are typically almost fully occupied by corticosterone during baseline conditions and are thus found predominantly in the nucleus. As corticosterone levels rise, the GR complex dissociates from the GR co-chaperone FK506-binding protein 51 (FKBP5), allowing it to bind corticosterone and recruit FK506-binding protein 52 (FKBP4), enabling it to translocate into the nucleus and regulate the genome.23
The genomic actions of glucocorticoids within the dorsal hippocampal formation strongly influence learning and memory processes. GRs are abundantly expressed in the hippocampus and are critical for mediating cellular and transcriptional effects, which facilitate the consolidation of information, particularly during the context of a stressful learning paradigm such as the Morris water maze (MWM).24–26 The removal of endogenous glucocorticoids by adrenalectomy, genetic disruption of GR function, and pharmacological inhibition of GRs all profoundly impair the performance of animals in spatial learning tasks.25–28 There is also evidence that GRs are necessary for the expression of long-term potentiation (LTP) in the hippocampus,29 an important cellular mechanism involved in learning. In contrast, glucocorticoids were reported to enhance object recognition memory only when the training conditions were modified to increase emotional arousal, highlighting the selective role of glucocorticoids in memory consolidation for emotionally arousing and/or stressful experiences.30,31
The effects of GR on spatial learning and LTP may be mediated through serum- and glucocorticoid-inducible kinase 1 (sgk1), a primary glucocorticoid-induced gene 32 and downstream target of the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinases (ERK) signaling pathway.33 Knockout of the sgk1 gene similarly impairs both spatial learning34 and LTP35 in mice. Glucocorticoids, acting through sgk-1, affect learning ability and synaptic plasticity via regulating the expression of glutamatergic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-D-aspartate (NMDA) receptors.36–39 We hypothesized that impairments in GR function may underlie deficits in both hippocampal-dependent spatial learning/memory and synaptic plasticity following pediatric TBI, and that overexpression of GRs in the DH would reverse these deficits through upregulating the hippocampal expression of sgk1 and regulating glutamatergic AMPA and NMDA receptor subunit expression
Methods
Experimental design
Animals in each litter were randomly assigned to receive a closed head brain injury (n = 78) or were treated as sham-injured controls (n = 74). Sham- and brain-injured animals from a subset of litters were randomly assigned to receive a microinjection into the DH containing a GR-expressing lentivirus (n = 48) or control lentivirus (n = 44). Animals were tested beginning at 1 month after injury in the MWM (spatial learning/memory) and spatial object recognition test (spatial memory). Behavioral testing was performed in adolescent rats (38–50 days old) because of our previous findings demonstrating sustained cognitive deficits at 1 month following TBI in neonate rats.17,40 Moreover, cognitive and behavioral symptoms following TBI in infants and toddlers can become more noticeable as they age into adolescence,41,42 validating that this is a clinically relevant time point for assessing post-traumatic cognitive impairments. At the end of behavioral assessments, subgroups of animals were randomly assigned to generate tissue for histological analysis, long-term potentiation, quantitative real-time polymerase chain reaction (qRT-PCR), or Western blots.
TBI
Surgical procedures were performed in accordance with the rules and regulations of the Institutional Animal Care and Use Committee at Drexel University and the National Institutes of Health (NIH) guide for the care and use of Laboratory animals (NIH Publications No. 8023, revised 1978). Raghupathi and Huh17 originally characterized the pediatric injury model used in this study, which has been used in multiple subsequent studies.40,43,44 Timed-pregnant (E20) dams were ordered from Charles River Laboratories (Wilmington MA) and housed under 12 h light:12 h dark conditions with access to food and water ad libitum. On post-natal day 11 (the neurological equivalent of a child <4 years of age,45–47 male and female Sprague Dawley rat pups were anesthetized using isoflurane (Patterson Veterinary, Greeley CO, 5% induction, 2–3% maintenance), and an incision of 2 cm was made to expose the skull. Animals were then transferred to a plastic rodent restrainer (Braintree Scientific, Braintree MA, USA) and moved onto the stage of an electronic controlled cortical impact device (eCCI, Custom Design and Fabrication, Richmond VA, USA). The impactor tip (5 mm diameter) was positioned over the left parietal cortex midway between the bregma and lambda sutures and was driven into the intact skull at a velocity of 5 m/sec (3 mm distance from zero point, 100 ms dwell time). After receiving the impact, animals were placed on their backs and the time to right themselves (righting reflex) and time to return to normal breathing (apnea) were recorded. Animals were then re-anaesthetized, and the presence of a skull fracture and hematoma were recorded prior to the scalp being sutured closed. The severity of the hematoma was determined based on the degree and extent of discoloration at or around the site of impact, and was characterized as mild, moderate, or severe, as previously described.48 Sham-injured animals were surgically prepared but were not injured. Animals were allowed to recover in a separate cage before being placed back with the dam. Surgical procedures and recovery were performed on heating pads at 37°C to maintain the body temperature.
Lentivirus injections
The lentivirus plasmid encoding the green fluorescent protein (GFP)-glucocorticoid receptor was subcloned into a modified pCSC-SP-PW-GFP (Addgene plasmid # 12337, a gift from Inder Verma).49 The modified plasmid, pBOB-mcs, was created by removal of the GFP by restriction digestion using XbaI/PmeI sites and subcloning of a multiple cloning site 5′ TCTAGAGGATCCACCGGTGGCCGCCTGGGCCCGTTAACGTCTCGAGTTTAAAC 3′ into the XbaI/PmeI sites. The GFP-glucocorticoid receptor was then cut from pEGFP-GR (Addgene plasmid # 47504, a gift from Alice Wong) using the restriction sites NheI/BamHI and subcloned into the XbaI/BamHI sites of pBOB-mcs. Successful clones were isolated and verified by DNA sequencing (GENEWIZ, South Plainfield, NJ) and Western blot analyses. Viral stocks were generated by calcium-phosphate transfection of 293T cells with plasmid encoding either pBOB-EGFP-GR or pBOB-GFP plus Mb1, Rev and vesicular stomatitis virus G (VSV G)-plasmid encoding envelope glycoprotein. The supernatants were collected 72 h after transfection and concentrated by sucrose gradient ultracentrifugation. Purified virus was suspended in Tris buffer containing rat albumin and mannitol, aliquoted, and stored at −80°C until further use. Lentiviral titers were estimated using a commercially available p24 ELISA kit (HIV-1 p24 antigen ELISA, Aalto, Ireland). The lentiviral titers for the GFP-GR lentivirus and GFP-control lentiviruses were 1.81 × 107 viral particles/μL and 3.19 × 107 viral particles/μL, respectively. Both viruses were prepared by the Dr. George Smith Lab (Temple University, Philadelphia, PA). The GFP-GR lentivirus or GFP-control lentivirus was unilaterally microinjected into the left DH (1 μL delivered at 0.2 μL/min) on post-natal day 18, 1 week following TBI or sham surgery, to allow the animals time to recover. The coordinates used for the injection were (in mm) -2.5 A/P, +2.75 M/L, and -3.25 D/V.
MWM
Spatial learning was assessed in the MWM between 5 and 6 weeks post-injury as previously described.48 Testing was conducted between 8:00 and 10:00 AM on each day. The maze consisted of a 1-m diameter circular pool containing 18°C water made opaque with white non-toxic paint. The length of time taken (latency) and the distance traveled to reach a submerged, hidden platform were recorded. The platform was submerged 2 cm below the surface of the water. Rats were initially placed in the center of the pool and guided to fixed start points located around the periphery of the pool. Visual cues were placed around the pool to help animals locate the hidden platform. Each animal was exposed to four trials/day over four consecutive training blocks (16 total trials). If an animal did not reach the platform within 120 sec, a latency of 120 sec was recorded for that trial. The average latency to reach the platform was averaged among the four trials on each training day, and data are presented as a function of training day to demonstrate the “learning curve.”
At 24 h following the final training day, spatial memory was tested in two probe trials during which the platform was removed from the maze. Animals went through two probe trials that consisted of 120 sec each, with an inter-trial interval of ∼15 min. Animals were placed in the pool in the same manner as they were during training. Probe trials were recorded with an overhead camera and the time spent in various zones surrounding either the platform location or the perimeter of the maze was computed (AccuScan, San Diego Instruments, San Diego CA, USA). The scores received in the two probe trials were averaged for each animal and presented as time spent in the platform or periphery zone.
Tissue preparation for extracellular recordings
LTP was measured in the DH after the conclusion of behavioral testing (5–7 weeks post-injury; post-natal day 48–60). Rats were anesthetized with an intraperitoneal injection of Euthasol (100 mg/kg) and transcardially perfused with 60 mL of carboxygenated (5% CO2, 95% H20) slicing artificial cerebrospinal fluid (ACSF) consisting of the following (in mM): 126 NaCl, 10 glucose, 26 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 5 MgCl2, and 1 CaCl2, pH of 7.4. Brains were then rapidly removed and glued to the slicing stage of a vibrating microtome (Leica Microsystems, Buffalo Grove, IL), and 400 μM horizontal slices containing the DH were obtained between 3 and 4.5 mm from the dorsal surface. Slices were incubated at 37°C for 1 h in oxygenated artificial cerebrospinal fluid (aCSF) containing the following (in mM): 126 NaCl, 10 glucose, 26 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 1 MgCl2, and 2 CaCl2, pH 7.4. Brain slices were then kept at room temperature for at least 1 h prior to recording.
Field potential recording and LTP quantification
Brain slices were individually transferred to a recording chamber and continually perfused with oxygenated aCSF maintained at 34°C. The Schaffer Collateral pathway was stimulated with a bipolar electrode and excitatory field post-synaptic potentials (fEPSPs) were recorded in the stratum radiatum (SR) of area CA1 using borosilicate glass pipettes filled with aCSF (resistance 1–2 MΩ). Signals were acquired using an axon MultiClamp 700A amplifier and pClamp 9.2 data acquisition software (Molecular Devices), digitized using a DigiData 1332A digitizer (Molecular Devices) at 10 kHz, and low pass filtered at 1kHz. Input/output (I/O) curves were conducted using stimulation intensities from 10 to 80 V (duration = 0.2 ms), and the stimulation level that produced the half maximal response was used. Baseline field potentials were recorded every 30 sec for 20 min. If a stable baseline was obtained, LTP was induced by theta-burst stimulation, consisting of 5 × 100 Hz bursts (5 pulses per burst, 200 ms inter-burst interval) at the test pulse intensity, which was repeated four times (10 sec interval in between each series of bursts). Field potentials were subsequently recorded every 30 sec for 60 min. The amplitude of the field potentials was measured in Clampfit 10.5 (Molecular Devices, San Jose, CA), and reported as the percent change from baseline measurements (average of the last 10 responses prior to Theta Burst Stimulation [TBS]). Potentiation was assessed from fEPSPs evoked 10 and 60 min after high frequency stimulation (HFS) (average of 10 responses taken at each time point).
Histological confirmation of lentivirus expression
A subset of animals was transcardially perfused at 4 weeks following injury using 0.9% saline containing heparin (1000 units/L, Sagent Pharmaceuticals, Schaumburg, IL, USA) followed by 10% formalin (Fisher Scientific, Pittsburgh, PA). Brains were post-fixed in formalin for 24 h before being transferred to a cryoprotective solution containing 30% sucrose. Brains were frozen between -40 and -50°C and 40 μM coronal sections were obtained between 2 mm and 8 mm posterior to bregma using a freezing sliding microtome. Sections were incubated in anti-GFP (1:500; Invitrogen, Waltham, MA, USA) at 4°C for 16–24 h, and were incubated in an anti-rabbit secondary antibody conjugated to Alexa Fluor 488 (1:500, ThermoFisher Scientific, Waltham, MA, USA) for 2 h at room temperature. Sections were then mounted on gelatin-coated glass slides and cover-slipped using Vectashield (Vector Laboratories, Burlingame, CA, USA) mounting medium. GFP expression was visualized in three non-adjacent dorsal hippocampal sections per animal using a fluorescent microscope.
qRT-PCR
A subset of animals was used to harvest tissue from the hippocampus for qRT-PCR analysis immediately following the 1st day of training in the MWM. The DH was micro-dissected from coronal sections taken between 2 mm and 4 mm posterior to bregma and stored in RNA-Later (Sigma-Aldrich, St. Louis MO, USA) until processed. The ventral hippocampus (VH) was taken between 5 mm and 7 mm posterior to bregma. As described previously,40,50 the RNA from the tissue was extracted using a RNeasy Mini Kit (Qiagen Inc.) along with DNase 1 (Qiagen Inc.). RNA yields were measured with a NanoDrop Lite spectrophotometer (Thermo Electron North America LLC, Madison WI, USA) and resulted in A260/A280 ratios of 2.0–2.1, indicating high purity. RNA was converted to cDNA using SuperScript® VILO™ Master Mix (Invitrogen, Grand Island, NY, USA) in a SimpliAmp™ Thermal Cycler (Applied Biosystems, Waltham, MA, USA). Triplicate samples of cDNA, SYBR Green PCR reagent (Applied Biosystems, Grand Island, NY, USA), and target primer or cyclophilin primer were run on a 96-Well MicroAmp® Fast Optical Reaction Plates (Applied Biosystems). The protocol was set to be 2 min at 50°C, 10 min at 95°C, 40 cycles of 15 sec at 95°C, and 1 min at 60°C. Primers were designed using the NCBI primer design tool (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) and purchased from ThermoFisher Scientific. The primers were validated by testing primer dilutions against a series of cDNA dilutions and determining the efficiency of the standard curve [10^(-1/slope)], as well as running a melting curve analysis to confirm primer specificity. The concentration and sequences of the primers used are listed in Table 1.
Table 1.
Primers Used for Analysis of mRNA Expression in the Dorsal Hippocampus of Sham- and Brain-Injured Rats
| Gene | Accession number | Concentration (nM) | Forward | Reverse |
|---|---|---|---|---|
| Cyc | NM_017101.1 | 200 | GTGTTCTTCGACATCACGGCT | CTGTCTTTGGAACTTTGTCTGCA |
| GR (rat) | NM_053859.2 | 200 | AACATGTTAGGTGGGCGTCA | AGTTTCTGAAGCCTGGTATCGC |
| MR (rat) (rat) | NM_053427.1 | 200 | AGAAAGGTGCTCACGACGTT | GCCAGTCACACCATTGGAGA |
| Sgk1 | NM_001193568.1 | 50 | TCAGGAGCCCGAACTTATGAA | GGACCCAGGTTGATTTGTTGA |
| GR (human) (human) | NM_000176.3 | 50 | GGACCACCTCCCAAACTCTG | GCTGTCCTTCCACTGCTCTT |
| GluA1 | NM_031608.1 | 200 | GAAATGTGCAGTTCAACGAGAAAG | TTTCGGATTCCATCATGTTTCAT |
| GluA2 | NM_017261.2 | 200 | CTACCAATGGGACAAGTTCG | CAGGATTACACGCCGTT |
| GluN1 | NM_001270602.1 | 100 | CACCAGACTAAAGATAGTGACAATCCA | CCTCTTTGCATGTCCCATCA |
| GluN2A | NM_012573.4 | 100 | AGGACAGCAAGAGGAGCAAG | ACCTCAAGGATGACCGAAGA |
| GluN2B | NM_012574.1 | 200 | TGAGTGAGGGAAGAGAGAGAGG | ATGGAAACAGGAATGGTGGA |
All sequences are listed as 5′-3′.
Cyc, cyclophilin-A; GR, glucocorticoid receptor; MR, mineralocorticoid receptor; Sgk1; serum-and-glucocorticoid inducible kinase 1; GluA1, glutamate ionotropic receptor α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) type subunit 1; GluA2, glutamate ionotropic receptor AMPA type subunit 2; GluN1, glutamate ionotropic receptor NMDA type subunit 1; GluN2A, glutamate ionotropic receptor N-methyl-d-aspartate (NMDA) type subunit 2A; GluN2B, glutamate ionotropic receptor NMDA type subunit 2B.
Corticosterone analysis
Trunk blood was collected from animals immediately after the fourth trial on the 1st day of MWM training. Blood samples were centrifuged at 4°C for 10 min at 2000g, after which the blood serum was collected into a separate tube for analysis. Corticosterone was measured by liquid chromatography-high resolution mass spectrometry modified from a previously reporter steroid assay.51 Briefly, 5-20 μL of sample was spiked with 50 μL of 100 pg/ μL d3-corticosterone in methanol. Samples were extracted by liquid-liquid extraction by addition of 250 μL of water, 150 μL of saturated sodium chloride in water, 10 μL of HCl, and 1.3 mL of methyl-tertbutyl-ether and vortexed for 10 min. Phases were separated with 5 min of centrifugation at 2000g, the lower layer was frozen at -80°C and the supernatant transferred to a new tube and then evaporated to dryness under nitrogen gas. Extracts were derivatized using Girard P reagent by adding 200 μL of 10% (v/v) acetic acid in water, 20 μL of Girard P reagent at 1 mg/mL in water, vortex mixing for 10 sec, incubation for 10 min at 60°C, evaporation to dryness under nitrogen gas, and resuspension in 100 μL of 80:20 (v/v) methanol:water. Ten microliters of each sample were analyzed on an Ultimate 3000 HPLC coupled to a Q Exactive Plus mass spectrometer. The double charged doubly derivatized form of corticosterone was predominantly observed and was used for peak integration. Calibration curves were prepared in an identical manner to the samples, spanning 3.9–62.5 pg/μL and 15–5000 pg/μL to provide linear ranges for interpolation of sample concentration.
Western blot analysis
Brain tissue was homogenized in lysis buffer A (10mM Hepes, 1.5mM MgCl2, 10mM KCl, 0.5mM DTT, 0.05% NP40, 0.05M sodium vanadate) adding one tablet of Complete Protease Inhibitor (ThermoScientific, Pierce Protease Inhibitor Mini Tablets, A32953) per 10 mL lysis buffer A. The homogenate was then centrifuged at 3000g for 10 min at 4°C. After centrifugation, the supernatant was collected and kept as the cytoplasmic fraction (CF). The resulting pellet was re-suspended with equal volume of lysis buffer B (5mM Hepes, 1.5 mM MgCl2, 0.2mM EDTA, 0.5mM DTT, 26% glycerol, 300 mM NaCl) and sat on ice for 30 min. After centrifugation (20,000g, 20 min, 4°C) the supernatant was collected and kept as the nuclear fraction (NF). Protein concentration was measured by Bio-Rad DC protein assay using TCAN Plate Reader, after which the supernatant of both the NFs and CFs were denatured in 80% sample buffer (125mM Tris, 4% sodium dodecyl sulfate [SDS], 0.005% bromophenol blue, 20% glycerol), 20% β-mercaptoethanol (BME) and boiled for 5 min. Samples were separated by SDS electrophoresis (Mini-PROTEAN 11 Cell chamber, Bio-Rad Laboratories, USA) in 4–20% polyacrylamide gel (GenScript SurePAGE, Bis-Tris, 10 × 8 cm). Electrophoresis was performed in a Tris-MOPS-SDS (50mM Tris Base, 50mM MOPS, 0.1% SDS, 1 mM EDTA, pH 8.5) electrophoretic running buffer (GenScript, Cat.No.:M00138) at a voltage of 100 V. After electrophoresis, the proteins were transferred to a 0.2 μm polyvinylidene difluoride (PVDF) membrane (BioRad Trans-Blot Turbo Transfer Pack, Cat#1704156) using Bio-Rad Trans-Blot Turbo Transferring System at a current of 1.3 A, 25 V, for 7 min.
Membranes were incubated overnight at 4°C on a rotary platform with the following antibodies: HMGCR (rabbit monoclonal, dilution 1:1000, ab183127; Abcam, Cambridge MA, USA) and secondary (goat anti-rabbit IgG antibody, dilution 1:30,000 Licor IRDye 800CW). The membrane containing CF samples were then incubated, as detailed, with anti-actin, clone C4 (mouse monoclonal, dilution 1:5000 Chemicon MAB1501R) and secondary (goat anti-mouse IgG antibody, dilution 1:30,000, Licor IRDye 680CW). The membrane containing NF samples were then incubated, as detailed, with Lamin A/C (mouse monoclonal, dilution 1:1000 Active Motif AB_2793218) and secondary (goat anti-mouse immunoglobulin [Ig]G antibody, dilution 1:30,000, Licor IRDye 680CW). Fluorescent detection was visualized using the Li-Cor system. The staining intensity of the bands corresponding to the analyzed proteins was assessed by using Image Studio 5.2.5 (Li-Cor).
Statistical analysis
All data are presented as mean ± standard error of the mean (SEM). All statistical tests were performed using Statistica 7 (StatSoft, Tulsa, OK, USA). All data sets were confirmed to contain a normal distribution and homogenous variances, as indicated by a Shapiro–Wilk and Levene's test, respectively. An independent samples t test was used for comparisons between two means. For comparisons between more than two means, an analysis of variance (ANOVA) was used. For comparisons between more than two means across multiple time points or conditions, a repeated measures ANOVA (RMANOVA) was used (e.g., I/O responses in the CA1 hippocampus). Correlation analysis was used to quantify the strength of the relationship between two independent variables (e.g., hippocampal sgk1 mRNA vs. latency to platform). Post-hoc tests, when necessary, were conducted using the Neuman–Keuls correction. In all cases, a p value <0.05 was considered significant.
Results
Acute response to injury
Closed-head injury in 11-day-old rats resulted in a skull fracture and hematoma. All animals were included in the study and evaluated in the various outcomes regardless of hematoma severity.
In animals from cohort 1 (no virus), there was an increase in the righting reflex latency in animals that received a brain injury compared with those that received a sham injury (status; F[1,63] = 152, p < 0.0001, ANOVA, Table 2), which did not differ between males and females (sex; F[1,63] = 0.98, p = 0.32, ANOVA). Brain injury also resulted in brief period of apnea, which did not differ between male and female animals (T[31] = 1.2, p = 0.23, t test). The number of animals that were classified as having either a mild, moderate, or severe hematoma was not significantly different between male and female animals; χ2(1, n=31) = 0.07, p = 0.5, χ2 test.
Table 2.
Acute Neurological Status of Rats in the Study
| Group |
n (Male, Female) |
Righting reflex (sec) |
Apnea (sec) |
Hematoma |
||
|---|---|---|---|---|---|---|
| Mild |
Moderate |
Severe |
||||
| Group 1 (No Virus-MWM) | ||||||
| Sham | 29 (14, 15) | 57.6 ± 3.9 | - | - | - | - |
| Injured | 31 (16, 15) | 189.4 ± 10.1* | 16.1 ± 0.8 | 11 | 20 | 0 |
| Group 2 (LV-GR-MWM) | ||||||
|---|---|---|---|---|---|---|
| Sham+Control |
16 (7, 9) |
52.9 ± 5.4 |
- |
- |
- |
- |
| Sham+GR |
17 (8, 9) |
50.9 ± 3.4 |
- |
- |
- |
- |
| Injured+Control |
16 (8, 8) |
221.9 ± 9.8* |
20.5 ± 0.9 |
3 |
11 |
2 |
| Injured+GR | 19 (10, 9) | 217.5 ± 14.3* | 21.8 ± 1.3 | 4 | 13 | 2 |
| Group 3 (1 Day MWM) | ||||||
|---|---|---|---|---|---|---|
| Sham+Control |
8 (4, 4) |
50.9 ± 6.7 |
- |
- |
- |
- |
| Sham+GR |
10 (6, 4) |
58.9 ± 7.8 |
- |
- |
- |
- |
| Injured+Control |
8 (4, 4) |
206.1 ± 29.5* |
18.4 ± 2.1 |
1 |
6 |
1 |
| Injured+GR | 10 (5, 5) | 245.9 ± 16.1 | 18.4 ± 1.1 | 3 | 5 | 2 |
Three separate cohorts of 11-day-old male and female rat pups were randomly assigned to either sham- or brain-injured groups. Sham- and brain-injured rats in the first cohort which did not receive virus injections were used for behavioral testing and were subsequently used for long-term potentiation recordings in hippocampal slices. The second and third animal cohorts were randomly assigned to receive an injection of lentivirus (LV) containing glucocorticoid receptor (GR) or control lentivirus. Subsets of the animals tested in the behavioral assays were randomly assigned to be used for histology or long-term potentiation recordings in hippocampal slices. Animals tested in the Morris water maze (MWM) for 1 day were subsequently used for and mRNA or protein measurements using quantitative real-time polymerase chain reaction (qRT-PCR) and Western blot, respectively. Latency to regain righting reflex and times of apnea were recorded as described in the Methods.
p < 0.05 compared with sham-injured rats.
In animals that were injected with either GFP-control lentivirus (control) or GFP-GR-lentivirus (LV-GR) and tested in the MWM at 4 weeks post-injury (cohort 2), there was an increase in righting reflex latency in animals that received a brain injury compared with those that received a sham injury (status; F[59] = 333.8, p < 0.0001, ANOVA, Table 2), which did not differ between males and females (sex; F[1,59] = 3.2, ANOVA, p = 0.07), or between animals designated to receive an injection of control virus or LV-GR (virus; F[1,59] = 0.02, p = 0.89, ANOVA). The apnea time did not differ between male and female animals (sex; F[1,31] = 0.69, p = 0.41, ANOVA), or between animals designated to receive an injection of either control virus or LV-GR (virus; F[1,31] = 0.56, p = 0.46, ANOVA). Moreover, the number of animals that were classified as having either a mild, moderate, or severe hematoma was not significantly different between animals designated to receive an injection of either control virus or LV-GR (χ2[2, n=35] = 0.05, p = 0.97, χ2 test), confirming the randomization of animals to virus groups.
A third cohort of animals was used for the molecular characterization of corticosterone, GR/MR, skg1, and glutamate receptor expression following 1 day of MWM training. There was an increase in righting reflex latency in animals that received a brain injury compared with those that received a sham injury (status; F[1, 28] = 118.9, p < 0.0001, Table 2, ANOVA), which did not differ between males and females (sex; F[1,28] = 0.06, p = 0.8, ANOVA), or between animals designated to receive an injection of control virus or LV-GR (virus; F[1,28] = 2.1, p = 0.16, ANOVA). The apnea time did not differ between male and female animals (sex; F[1,13[ = 0.1, p = 0.76, ANOVA), or between animals designated to receive an injection of either control virus or LV-GR (virus; F[1,13] = 0.006, p = 0.93, ANOVA). Moreover, the number of animals that were classified as having either a mild, moderate, or severe hematoma was not significantly different between animals designated to receive an injection of either control virus or LV-GR (χ2[2, n=18) = 1.2, p = 0.54, χ2 test).
Pediatric TBI impairs spatial learning during adolescence
We have previously reported that TBI in 11-day-old rats impairs spatial learning in the MWM at 4 weeks post-injury.17 In the current study, we sought to determine whether spatial learning deficits persist at a later time point, and to examine whether these deficits may differ between male and female animals. Therefore, we evaluated spatial learning and memory in the MWM at 6 weeks following TBI or sham injury in male and female rats (Fig 1A). The latency to locate the platform during training in the MWM was significantly increased in brain-injured animals compared with their sham-injured counterparts (Fig 1B); status; F[1,45] = 15.7, p = 0.0002, RMANOVA. The distance traveled to reach the platform across training days was also higher in brain-injured animals (Fig 1C); status; F[1,38] = 11.1, p = 0.002, RMANOVA. The swim speed during the four training blocks did not differ between sham and injured animals (data not shown). During the probe trials, brain-injured animals spent significantly more time in the peripheral zone (status; F[1,45] = 5, p = 0.03, ANOVA) and less time in the platform zone (status; F[1,45] = 7.3, p = 0.009, ANOVA) compared with sham-injured animals (Fig 1E). TBI did not affect the total distance traveled during the probe trial (Fig 1D); status; F[1,45] = 1.8, p = 0.18, ANOVA. Although the effect of injury on the performance of animals during the training or probe trials did not differ between male and female rats, females exhibited a significant increase in latency to reach the platform during training (sex; F[1,45] = 4, p = 0.05, ANOVA) and in the time spent in the peripheral zone during the probe trial (sex; F[1,45] = 4.9, p = 0.03, ANOVA) compared with males, regardless of injury status. Overall, these data confirm that pediatric TBI results in deficits in spatial learning and spatial memory that were sustained up to 6 weeks post-injury in male and female rats.
FIG. 1.

Hippocampal-based cognitive deficits persist for at least 6 weeks following pediatric traumatic brain injury (TBI). Spatial learning and memory were assessed in the Morris water maze at 6 weeks following TBI (n = 26) or sham injury (n = 23) in male and female rats. (A) Experimental timeline, (B) Latency (in sec) to locate the hidden platform during four consecutive training days in sham and brain-injured animals, (C) distance (in cm) to reach the hidden platform during training days, (D) total distance (in cm) traveled during the probe trial, (E) total time (in sec) spent in the platform zone or the peripheral zone during the probe trial in sham and brain-injured animals. Bars represent group mean values and error bars represent standard error of the mean. *p < 0.05.
Pediatric TBI impairs LTP in the DH
To investigate the cellular mechanisms underlying spatial learning deficits, we measured LTP in the hippocampal CA1 region. There was no effect of injury on fEPSP amplitude in response to varying stimulus intensities (Fig. 2B), suggesting that TBI did not impair basal synaptic transmission. The fEPSP amplitude (normalized to baseline) following HFS of the Schaffer Collaterals was significantly lower in hippocampal slices from brain-injured animals than in hippocampal slices from sham animals (Fig. 2C, D); status; F[1,16] = 11.8, p = 0.003, RMANOVA. When we compared the average fEPSP amplitude at 10 min or 60 min following HFS to evaluate LTP induction and maintenance, respectively, we determined that amplitudes were significantly lower in injured slices at both time points than in sham slices (Fig. 2E); status; F[1,34] = 20, p < 0.001, ANOVA. Together, these data demonstrate that pediatric TBI results in sustained deficits in LTP expression in the dorsal hippocampus.
FIG. 2.

Pediatric traumatic brain injury (TBI) impairs long-term potentiation (LTP) in the dorsal hippocampus. LTP was measured in the CA1 region of the dorsal hippocampus of sham and brain-injured animals (n = 11 sham slices, 10 injured slices). (A) Representative excitatory field post-synaptic potentials (EPSP) traces demonstrating input/output responses in the CA1 region. (B) The amplitude of field EPSPs (fEPSPs) in response to varying levels of stimulation in sham and brain-injured slices. (C) Representative fEPSP traces demonstrating amplitudes at baseline, 10 min, and 60 min following theta-burst stimulation. (D) The average fEPSP amplitude (displayed as percent of baseline values) during the 20-min baseline recording and 60-min post-stimulation recording period. (E) The fEPSP amplitude averaged at 10 min or 60 min following theta-burst stimulation in sham and brain-injured animals. Bars represent group mean values and error bars represent standard error of the mean. HFS, high frequency stimulation. *p < 0.05.
Sgk1 mRNA expression in the DH is decreased after pediatric TBI
The presence of a learning/memory deficit in the MWM, a stressful learning paradigm, but not in object location memory (Supplementary Methods and Results; Supplementary Fig. S1), prompted us to consider whether the spatial learning impairment may be related to an altered stress response in brain-injured animals, which might impede the consolidation of information during stressful learning. Thus, we measured the effect of bath application of moderate/high concentrations of corticosterone on hippocampal LTP, with the expectation that the effects of corticosterone on LTP would be blocked in brain-injured hippocampal slices. Interestingly, we found that corticosterone bath application in brain-injured hippocampal slices was in fact able to facilitate LTP induction but failed to restore maintenance (Supplementary Methods and Results; Supplementary Fig. S2), which was suggestive of an impairment in GR activity.39 Because both GRs and the GR target gene sgk1 have been implicated in LTP maintenance,35,39 we sought to determine whether altered expression of GR or sgk1 in the DH may influence deficits in spatial learning following pediatric TBI.
To address this question, we harvested tissue from the DH immediately following 1 day of MWM training for qRT-PCR analysis of GR, MR, and sgk1 mRNA. The objective of this experiment was to characterize the effects of TBI on molecular changes induced by stressful learning. We selected this time point of 1 day based on prior evidence that the MWM induces a stress response that is gradually attenuated with repeated exposures.24 Additionally, the expression of specific genes that are induced by stress such as pERK1/2, which is also regulated by the GR target gene sgk1,52,53 peaks in the CA1 region of the hippocampus after the 1st day of training and then gradually declines.54 Because we were primarily interested in identifying stress-dependent changes in signaling pathways important for learning acquisition during MWM training, we determined that we were most likely to capture relevant changes in the expression of GR-dependent genes (e.g., sgk1) during these early training days.
We first measured serum corticosterone levels to determine whether the hypothalamic–pituitary–adrenal (HPA)-axis stress response to training may differ between sham and brain-injured animals. The baseline corticosterone concentration in animals that did not undergo MWM training was 17.1 ± 6.5 pg/μL in sham animals and 16.5 ± 7.9 pg/μL in brain-injured animals (data not shown). In animals that had undergone 1 day of training, we observed much higher corticosterone concentrations in the 200–300 pg/μL range (Fig. 3A), which did not differ between sham and brain-injured animals (status; F[1,9] = 1.9, p = 0.2, ANOVA), confirming that training induced a stress response that was similar in sham and brain-injured animals.
FIG. 3.
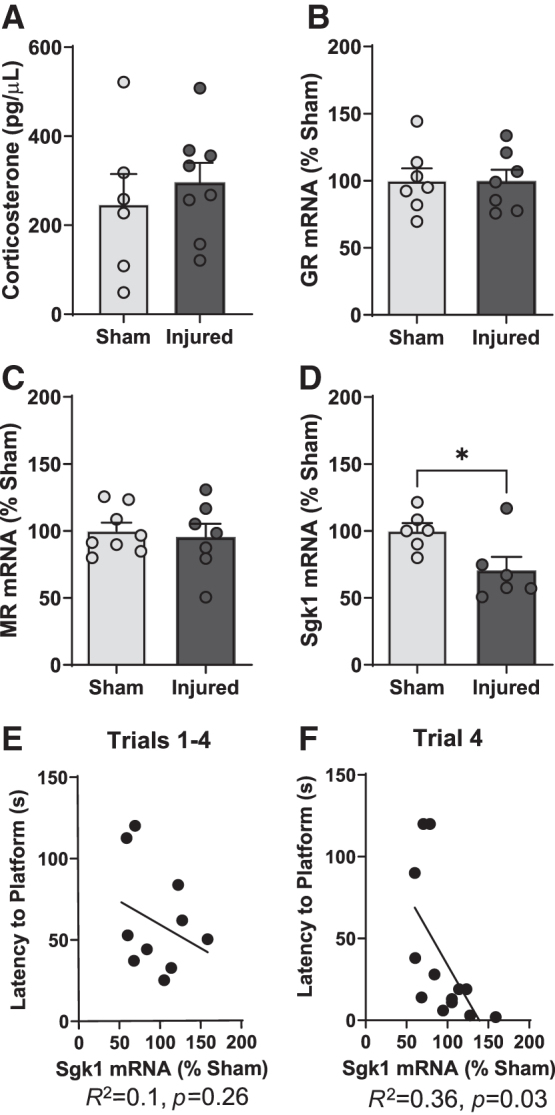
Decreased hippocampal sgk1 mRNA expression is associated with spatial learning impairments in brain-injured animals. In a subset of animals (n = 8 sham and 7 brain-injured animals), the mRNA expression of glucocorticoid-associated genes was measured in the dorsal hippocampus immediately following day 1 of water maze training. (A) Serum corticosterone levels (displayed as pg/μL) in sham and brain-injured animals. (B,C) The mRNA expression levels (displayed as percent of sham expression) of glucocorticoid receptors (GR) (B) and mineralocorticoid receptors (MR) (C) in the dorsal hippocampus. (D) Serum- and glucocorticoid-kinase 1 (Sgk1) mRNA expression (displayed as percent of sham expression) in the dorsal hippocampus. (E,F) Scatter plots demonstrating the relationship between Sgk1 mRNA expression level in the dorsal hippocampus with latency to reach the platform across the four trials during the 1st day of water maze training (E), and latency to reach the platform during the fourth trial of water maze training (F). Bars represent group averages and error bars represent standard error of the mean. *p < 0.05.
Our qRT-PCR results indicated that there was no difference in the expression of hippocampal GR mRNA between sham and injured animals (status; F[1,10] = 0.06, p = 0.8, ANOVA, Fig 3B). Similarly, the mRNA levels of the MR in the hippocampus did not differ between sham and injured animals (Fig 3C). However, there was a significant decrease in hippocampal sgk1 mRNA in brain-injured animals compared with sham animals (status; F[1,8] = 5.4, p = 0.04, ANOVA, Fig. 3D). A correlation analysis was calculated to determine the strength of the association between latency to the platform during water maze training and sgk1 mRNA expression in the DH. The correlation coefficient was significant for both latency (R2 = 0.36, p = 0.03, Fig. 3F) and distance to the platform (R2 = 0.38, p = 0.026, data not shown) during the fourth learning trial, but not for the latency to reach the platform averaged across four trials (R2 = 0.1, p = 0.26, Fig. 3E). These results indicate that pediatric TBI does not result in altered expression of hippocampal GR or MR mRNA but does lead to a decrease in the expression of the GR target gene (sgk1). Overall, these data suggest an impairment in GR transcriptional activity in brain-injured animals that is associated with impairment in learning acquisition in the MWM.
Confirmation of lentivirus expression in the DH
In the next set of experiments, we sought to determine whether viral transfection of functional GRs in the DH, as a mechanism to replace dysfunctional GRs, would attenuate deficits in spatial learning and hippocampal LTP in brain-injured animals. Therefore, we used a lentivirus containing GFP to transfect human GR (hGR) in the DH. Robust GFP immunoreactivity was observed predominantly in the CA1 and dentate gyrus regions of the DH at 3 weeks following injection (Fig. 4B and D) but not in the VH (Fig 4C and E). Moreover, qRT-PCR results determined that the expression of hGR was significantly elevated in the DH of LV-GR animals compared with control virus animals (virus; F[1,25] = 14.4, p = 0.04, ANOVA, Fig. 4F). The expression of hGR was not altered in the VH (virus; F[1,21] = 2.8, p = 0.4, ANOVA, Fig. 4G). These data confirm that LV-GR injections resulted in an increase in GR expression that was restricted to the DH.
FIG. 4.

Confirmation of lentivirus (LV) expression in the dorsal hippocampus. Green fluorescent protein (GFP) immunoreactivity, human glucocorticoid receptor (hGR), and serum- and glucocorticoid-kinase 1 (Sgk1) mRNA expression were evaluated in the dorsal hippocampus to confirm LV-mediated GR overexpression (n = 6–8/group). (A) Illustration of the timing of LV-GR or control virus injection into the dorsal hippocampus. (B,C) Representative images of GFP expression in the CA1 of the dorsal (B) and ventral (C) hippocampus. (D,E) Representative images of GFP expression in the dentate gyrus (DG) of the dorsal (D) ventral (E) hippocampus. (F,G) The mRNA expression levels (displayed as percent of sham control expression) of Sgk1 in the dorsal hippocampus (F) and ventral hippocampus (G). Bars represent group averages and error bars represent standard error of the mean. *p < 0.05. Color image is available online.
GR overexpression in the DH improves spatial learning and memory in the MWM
We then evaluated spatial learning and memory in the MWM in LV-GR and control animals during adolescence (4 weeks post-injury) (Fig. 5A). There was a significant main effect of both status (status; F[1,51] = 31.4, p < 0.001, RMANOVA) and the type of virus (virus; F[1,51] = 4.2, p = 0.04, RMANOVA) on the latency to reach the platform during water maze training (Fig. 5B). Overall, brain-injured animals exhibited a longer latency to locate the platform compared with their sham-injured counterparts, and LV-GR animals exhibited a significantly shorter latency to reach the platform compared with control animals (Fig. 5B). There was also a significant interaction between the type of virus and training day on the latency to reach the platform during training (virus × days; F[3,153] = 3.3, p = 0.02, RMANOVA). A post-hoc analysis indicated that the latency to reach the platform was significantly shorter in LV-GR animals on day 2 of training than in control animals (p = 0.007) (Fig. 5B). The distance to reach the platform during training remained significantly higher in brain-injured animals than in sham animals (status; F[1,41] = 4.3, p = 0.04, RMANOVA) and was not affected by the type of virus (virus; F[1,41] = 1.7, p = 0.2, RMANOVA) (Fig. 5C). GR overexpression did not affect swim speed during the training blocks (Fig. 5D).
FIG. 5.

Viral transfection of human glucocorticoid receptor (hGR) in the dorsal hippocampus improves spatial learning and memory. Spatial learning and memory were assessed in the Morris water maze at 4 weeks post-injury following control or lentivirus (LV)-GR injection into the dorsal hippocampus in sham (n = 14 control and 14 GR) and brain-injured (n = 13 control and 13 GR) animals. (A) Experimental timeline, (B) the latency (displayed in sec) to locate the platform, (C) the distance (displayed in cm) traveled to reach the platform, (D) the swim speed (displayed as cm/sec) during training days, (E) the total time (displayed in sec) spent in the platform zone, (F) the total time spent in the peripheral zone, (G) the total distance traveled during the probe trial. Bar graphs represent group averages and error bars represent standard error of the mean. *p < 0.05, #p < 0.05 relative to control.
During the probe trials, there was a main effect of injury status on time spent in the platform zone (status; F[1,56] = 8.1, p = 0.006, ANOVA) and peripheral zone (status; F[1,56] = 4.4, p = 0.04, ANOVA). There were also significant interaction effects between injury status and type of virus on time spent in the platform (status × virus; F[1,56] = 4.4, p = 0.04, ANOVA) and peripheral zones (status × virus; F[1,56] = 4.1, p = 0.04, ANOVA) of the pool. Post-hoc analyses revealed that compared with sham control animals, brain-injured control animals spent significantly less time in the platform zone (p = 0.005, Fig. 5E) and more time in the peripheral zone (p = 0.02, Fig. 5F). However, brain-injured animals did not differ from the sham control (p = 0.2) or sham GR (p = 0.6) groups in time spent in the platform zone (Fig. 5E). Brain-injured GR animals similarly did not differ from the sham control (p = 0.7) or sham GR (p = 0.9) groups in time spent in the peripheral zone (Fig. 5F). GR overexpression did not affect the total distance traveled during the probe trial (Fig. 5G); virus; F[1,46] = 0.002, p = 0.9, ANOVA. Together, these data demonstrate that GR transfection in the DH improved both spatial learning and spatial memory in the MWM following TBI. In contrast, we did not find a significant effect of GR overexpression on object location memory in male or female animals (Supplementary Methods and Results, Supplementary Fig. S3).
GR overexpression attenuates LTP deficits in the DH after pediatric TBI
We then measured LTP in the hippocampus of LV-GR and control animals to determine whether improved learning acquisition was mediated by enhanced hippocampal synaptic plasticity. GR overexpression did not affect the amplitude of evoked fEPSPs in response to various levels of stimulation (virus; F[1,29] = 0.33, p = 0.56, RMANOVA, Fig. 6A). GR overexpression significantly increased LTP expression in the brain-injured hippocampus relative to control brain-injured levels (virus; F[1,13] = 10.43, p = 0.006, RMANOVA, Fig. 6C). Analysis of the average fEPSP amplitude at 10 min following HFS revealed a significant interaction between status and virus (status × virus; F[1,22] = 5.3, p = 0.03, ANOVA, Fig. 6D). Post-hoc tests indicated that the fEPSP amplitude was significantly decreased in control brain-injured slices compared with both control sham-injured slices (p = 0.002) and LV-GR brain-injured slices (p = 0.01). GR overexpression did not significantly affect LTP expression in the sham-injured hippocampus relative to control sham-injured levels (virus; F[1,9] = 0.13, p = 0.72, RMANOVA, Fig. 6B). However, analysis of the average fEPSP amplitude at 60 min post-stimulation revealed a significant effect of virus (virus; F[1,22] = 4.7, p = 0.04, ANOVA, Fig. 6E), with an increase in fEPSP amplitude in LV-GR animals compared with control animals. Overall, these data indicate that GR overexpression in the DH attenuates deficits in the induction of LTP in the hippocampus following brain injury, and leads to an overall enhancement of LTP maintenance in the hippocampus.
FIG. 6.

Viral transfection of human glucocorticoid receptor (hGR) attenuates long-term potentiation (LTP) deficits in the dorsal hippocampus following pediatric traumatic brain injury (TBI). LTP was measured in the CA1 region of the dorsal hippocampus in control or GR-overexpressing sham- and brain-injured animals. (A) The amplitude of baseline input/output responses in the CA1 hippocampus in response to varying stimulus intensity. (B) The effect of hippocampal hGR transfection on excitatory field post-synaptic potentials (fEPSP) amplitude (normalized to baseline values during the 20-min baseline recording and following theta-burst stimulation in sham-injured hippocampal slices (n = 4 control slices, 7 GR slices). (C) The effect of hippocampal hGR transfection on fEPSP amplitude (normalized to baseline values) during the 20-min baseline recording and following theta-burst stimulation in brain-injured hippocampal slices (n = 7 control slices, 8 GR slices). (D) The average fEPSP amplitude at 10 min following theta-burst stimulation in hippocampal slices from control and lentivirus (LV)-GR sham and brain-injured animals. (E) The average fEPSP amplitude at 60 min following theta-burst stimulation in hippocampal slices from control and LV-GR sham and brain-injured animals. Bars represent group averages and error bars represent standard error of the mean. *p < 0.05; **p<0.01.
GR overexpression enhances GR function and regulates glutamatergic AMPA receptor (AMPAR) mRNA expression in the DH
We next wanted to characterize molecular changes that may underlie the improvement in learning acquisition that we observed in LV-GR animals. It was apparent that performance of LV-GR mice was significantly better than that of control mice by the 2nd training day, after animals had only undergone 1 day of training (Fig. 5B). Therefore, we sought to determine which molecular mechanisms may be involved in this effect during these early training days. Corticosterone and/or stress have been shown to regulate the expression and synaptic trafficking of AMPAR subunits,38,39,55 which play a prominent role in mediating changes in synaptic strength that accompany memory consolidation. A previous study found that mice trained in more stressful conditions in the MWM exhibit an increase in the synaptic expression of the AMPAR subunit GluA2 in the hippocampus relative to mice trained under less stressful conditions after just 1 day of training.55 Therefore, we collected hippocampal tissue from LV-GR and control animals after 1 day of training in the MWM for molecular analyses of GR, sgk1, and glutamate receptor subunit expression.
We first determined whether GR overexpression may have influenced corticosterone levels. Although brain injury did not affect corticosterone levels (status; F[1,24] = 0.17, p = 0.68, ANOVA, Fig. 7B), corticosterone levels were significantly lower in LV-GR animals than in control animals (virus; F[1,24] = 6.8, p = 0.01, ANOVA, Fig. 7B). To confirm that GR-overexpression enhanced GR transcriptional activity, we measured sgk1 mRNA using qRT-PCR. There was a significant increase in sgk1 mRNA in LV-GR animals relative to control animals in the DH (virus; F[1,23] = 18.8, p = 0.02, ANOVA, Fig. 7C), but no difference in sgk1 mRNA expression in the VH (virus; F[1,22] = 4.8, p = 0.3, ANOVA, data not shown). These data confirmed that LV-GR animals exhibited an increase in GR activity, as evidenced by higher expression of sgk1 mRNA. LV-GR animals also exhibited lower corticosterone levels, likely because of enhanced GR-mediated negative feedback. When we examined the relationship between sgk1 mRNA in the DH and performance in the MWM in control or LV-GR animals, we again found that the correlation coefficient was significant for both latency (R2 = 0.21, p = 0.02, Fig. 7H) and distance to the platform (R2 = 0.22, p = 0.02, data not shown) during the fourth learning trial, but not for the latency to reach the platform averaged across four trials (R2 = 0.01, p = 0.65, Fig 7G). Thus, our data suggests that the increase in sgk1 mRNA expression in LV-GR animals is associated with improved learning acquisition during MWM training.
FIG. 7.

Glucocorticoid receptors (GRs) regulate glutamate receptor subunit mRNA expression in the dorsal hippocampus. The effects of viral transfection of human GR (hGR) on corticosterone, serum- and glucocorticoid-kinase 1 (Sgk1), and glutamate receptor subunit mRNA expression levels were measured in the dorsal hippocampus immediately following the 1st day of water maze training in sham and brain-injured lentivirus (LV)-GR and control animals (n = 5–8/group). (A) Experimental timeline; (B) corticosterone levels (displayed as pg/μL); (C) Sgk1 mRNA expression (displayed as percent of sham control expression); (D–F) the mRNA expression levels (displayed as percent of sham control expression) of GluA1 (D), GluA2 (E), and GluN1 (F); (G) the relationship between sgk1 mRNA and latency to reach the platform averaged among four trials, (H) the relationship between sgk1 mRNA and latency to reach the platform during the fourth trial; (I–K) the relationship between latency to reach the platform during the fourth trial and (expressed as percent of sham control expression) GluA1 mRNA (I), GluA2 mRNA (J), and GluN1 mRNA (K). Error bars represent standard error of the mean. *p < 0.05.
Our analysis of glutamate receptor subunit expression revealed that there was a significant effect of injury status on the mRNA levels of GluA1 (status; F[1,22] = 34.4, p < 0.0001, ANOVA), GluA2 (status; F[1,21] = 6.8, p = 0.01, ANOVA), and GluN1 (status; F[1,22] = 11, p = 0.003, ANOVA), with lower mRNA levels observed in injured animals relative to sham animals after the 1st day of water maze training (Fig. 7D–F). There was also a decrease in GluN2A mRNA following brain injury that approached significance (status; F[1,26] = 3.4, p = 0.056, ANOVA, data not shown). There was no effect of injury on the expression of GluN2B mRNA (status; F[1,26] = 0.02, p = 0.87, ANOVA, data not shown). GR overexpression resulted in a significant increase in the expression of GluA1 mRNA (virus; F[1,22] = 9.2, p = 0.006, Fig. 7D) and GluA2 mRNA (virus; F[1,21] = 5.3, p = 0.03, Fig. 7E) in the DH, and a slight increase in the expression of GluN1 mRNA that did not reach significance (virus; F[1,22] = 3.6, p = 0.07, Fig. 7F). GR overexpression did not affect GluN2A mRNA (virus; F[1,26] = 0.15, p = 0.8, ANOVA, data not shown), or GluN2B mRNA (virus; F[1,26] = 0.57, p = 0.4, ANOVA, data not shown). Additionally, a correlation analysis was calculated to determine the strength of the association between latency to the platform during water maze training and GluA1, GluA2, and GluN1 subunit mRNA expression in the DH. The correlation coefficient was significant for GluA1 (R2 = 0.36, p = 0.002, Fig. 7I), GluA2 (R2 = 0.31, p = 0.007, Fig. 7J), and GluN1 (R2 = 0.2, p = 0.035, Fig. 7K). Overall, LV-GR animals exhibited shorter latencies to reach the platform and higher GluA1 and GluA2 mRNA expression than their control counterparts. These results indicate that post-training expression of several glutamate receptor subunits is decreased in the DH of brain-injured animals, and that GR overexpression in the DH resulted in an increase in the mRNA levels of specific glutamate AMPAR subunits following water maze training.
GR overexpression facilitates GR nuclear translocation in the DH after brain injury
The data from this study establish a sustained cognitive deficit following TBI that is associated with reduced transcriptional activity of GRs in the hippocampus and that is attenuated by overexpression of GR in the DH. Although we did not observe a difference in GR mRNA expression following injury, we sought to determine whether TBI may affect GR protein levels or nuclear translocation, which could explain the impairment in GR function. Therefore, we measured the expression of GR protein in the nuclear and CF following water maze training in sham and brain-injured animals using Western blot. The bands corresponding to GR in the CF are illustrated in Figure 8A, and bands corresponding to GR in the NF are illustrated in Figure 8B. There was a significant interaction effect on GR expression in the CF (status × virus; F[1,24] = 9, p = 0.006, ANOVA, Fig. 8C). Post-hoc tests indicated that GR expression was significantly higher in DH samples from injured control animals than in those from sham-injured control animals (p < 0.05), or from their brain-injured GR counterparts (p < 0.05). Moreover, there was a main effect of virus type on GR expression in the NF (virus; F[1,22] = 4.6, p = 0.04, ANOVA Fig. 8D), with higher GR protein levels in DH samples from sham and brain-injured GR groups than in those from sham and brain-injured control groups. Finally, we sought to determine whether GR overexpression may affect the NF to CF ratio of GR expression. There was a significant interaction effect on the GR NF/CF ratio (status × virus; F[1,21] = 8.9, p = 0.007, ANOVA, Fig. 8E). Post-hoc tests indicated that the ratio of GR protein in the NF relative to the CF was significantly decreased in brain-injured control animals compared with both sham-injured control animals (p < 0.05) and brain-injured GR animals (p < 0.05). Overall, these findings suggest that GR overexpression in the DH resulted in an increase in GR nuclear translocation following water maze training, particularly in brain-injured animals.
FIG. 8.

Glucocorticoid receptor (GR) overexpression in the dorsal hippocampus facilitates GR nuclear translocation in brain-injured animals. The effect of viral transfection of human GR (hGR) on GR protein levels in the dorsal hippocampus was measured using Western blot in dorsal hippocampal samples harvested immediately following the 1st day of water maze training. Representative blots demonstrating GR and actin bands in the cytoplasmic fraction (n = 6–8/group) are shown in A; representative blots demonstrating GR and lamin bands in the nuclear fraction (n = 6–8/group) are shown in B. (C) The signal of the GR band (normalized to actin signal and displayed as percent of sham control expression) in the cytoplasmic fraction. (D) The signal of the GR band (normalized to lamin signal and displayed as percent of sham control expression) in the nuclear fraction. (E) The ratio of normalized GR signal in the nuclear fraction relative to the cytoplasmic fraction (displayed as percent of sham control expression). Error bars represent standard error of the mean. *p < 0.05. NF, nuclear fraction; CF, cytoplasmic fraction. Color image is available online.
Discussion
The purpose of this study was to investigate the role of the GR activity in the hippocampus in cognitive and behavioral outcomes following brain trauma in the immature rat. Through the utilization of a viral approach to manipulate GR expression in the hippocampus, these experiments emphasize the significant role that stress response systems play in the chronic outcomes of pediatric brain trauma. Our findings indicate that moderate TBI in neonate rats results in deficits in spatial learning and hippocampal LTP, which are associated with impaired function of GRs in the DH during adolescence, as evidenced by a reduction in the expression of the GR target gene sgk1 in the DH following water maze training. The lentiviral overexpression of hGRs in the DH attenuated deficits in spatial learning and memory, and hippocampal LTP induction and maintenance. Moreover, GR overexpression also led to an increase in the expression of GluA1 and GluA2 mRNA in the DH following water maze training, suggesting that these beneficial effects of GR overexpression may be mediated in part by the regulation of glutamate receptor subunit expression.
Early life trauma is known to increase vulnerability to cognitive, social, and behavioral impairments later in life, although specific mechanisms underlying this association are not fully understood. Interestingly, these results suggest that the sequelae of brain injury may differ from other forms of early life stress (e.g., neglect, abuse). Whereas early life adversity results in exaggerated responses to stress, both clinical data56 and data from these experiments suggest that early life TBI may dampen stress responsivity, suggestive of differences between the two models. Moreover, although both models share some overlap in behavioral outcomes, there may be important differences in underlying mechanisms, suggesting that the same treatment may not be similarly effective for both types of early life trauma.
Pediatric TBI resulted in chronic impairments in the induction and expression of LTP in the DH, without affecting CA1 I/O responses to Schaffer collateral stimulation. These findings differ from previous evidence of reductions in the amplitude of evoked responses in the CA1 hippocampus following lateral fluid percussion (LFP) injury in adult animals.57,58 However, a previous study reported that baseline I/O responses in the CA1 region of the hippocampus were not altered after a mild weight drop injury, a model of closed head injury, in juvenile animals,59 suggesting that the effects of TBI on baseline I/O function in pediatric animals may differ from that in adult animals.
Our findings that bath application of both moderate and high concentrations of corticosterone during HFS increase the magnitude of LTP induction following pediatric TBI corroborate previous evidence that corticosterone facilitates hippocampal LTP when it is temporally associated with the timing of HFS.60 In contrast to this study, we did not observe an effect of corticosterone on LTP in sham hippocampal slices. This discrepancy may be the result of differences in the LTP protocols implemented, as the prior study used a stimulus frequency of 10 Hz to induce LTP whereas in the current study we used 100 Hz stimulation. Therefore, it is possible that the LTP protocol we used resulted in a saturation that occluded any further plasticity. Importantly, neither of the two concentration of corticosterone was effective in increasing the expression of LTP at 1 h following stimulation in brain-injured hippocampal slices. Previous studies have demonstrated impairments in the expression but not the induction of LTP following downregulation of GRs29 and sgk1,35 and pharmacological inhibition of GRs was shown to selectively attenuate the long-term but not the immediate effects of corticosterone on the surface expression of the AMPAR GluA2 subunit in response to a chemical LTP stimulus.39 In contrast, inhibition of MRs resulted in an attenuation of the effects of corticosterone GluA2 trafficking immediately following a chemical LTP stimulus but did not alter the long-term effects of corticosterone on surface AMPAR subunit expression.39 Together, these data suggest that the facilitation of LTP induction by corticosterone in brain-injured hippocampal slices may have been mediated by hippocampal MRs, which are left intact following TBI, whereas the inability of corticosterone to facilitate the expression of LTP may be a consequence of impairments in GR function.
A novel finding of this study is that overexpression of hGR in the DH led to an overall improvement in spatial learning and memory. Although the effects of GR overexpression in the hippocampus on cognitive function have not previously been studied, GR overexpression in the forebrain resulted in mild cognitive dysfunction in mice,61 suggesting that forebrain and hippocampal GRs exert distinct effects on learning processes. Although a number of previous studies have established that a loss of GR function significantly impairs spatial learning,25–28 this is the first study to our knowledge to provide evidence for impairment in hippocampal GR function as a mechanism underlying deficits in cognitive function following pediatric TBI, and for targeting GRs in the hippocampus as an effective strategy to attenuate learning deficits in brain-injured animals. Moreover, our findings suggest that long-term deficits in spatial learning following pediatric TBI may be a consequence of an impaired cellular stress response, which will have important clinical implications for the development of targeted therapies to treat learning and memory deficits in pediatric TBI patients.
Notably, our results indicate that GR overexpression in the DH not only attenuated deficits in the expression of LTP, but also facilitated LTP induction in brain-injured hippocampal slices. These results were somewhat surprising because mice lacking either a functional GR29 or sgk135 gene exhibit deficits in the expression but not induction of LTP. Our results therefore suggest that although GRs are not necessary for LTP induction, GR overexpression is sufficient to facilitate LTP induction following pediatric TBI. These effects are likely related to the ability of GR overexpression to increase the expression of the AMPAR GluA1 and GluA2 subunits in the DH, thus possibly enhancing the excitatory effects of glutamate and, in turn, the amount of CA2+ influx in response to stimulation as well the trafficking of AMPARs to the cell surface. The findings from this study support previous evidence that acute stress enhances hippocampal LTP and increases AMPAR GluA1/GluA2 phosphorylation and surface expression in a mouse model of Alzheimer's disease, and that these effects are mediated through GRs.38 Further, these results are in line with evidence that the GluA1 AMPAR subunit is required for hippocampal LTP but does not significantly regulate basal synaptic transmission.62 Overall, these findings support an important role in the GR-mediated actions of glucocorticoids in hippocampal synaptic plasticity and impairments in plasticity that arise following TBI, which are potentially mediated through their actions on glutamate receptor subunit expression. Future studies will be necessary to determine the precise mechanisms by which GR activation regulates these processes.
Interestingly, although male and female rats were similarly impaired in spatial learning and hippocampal LTP, only female animals exhibited an impairment in spatial object location memory. Further, GR overexpression in the DH did not have a significant effect on the discrimination ratio in this task, which is in line with previous evidence that glucocorticoids do not influence memory in object recognition tasks under baseline conditions.30,31 Further, these data suggest that the impairment in object location memory in female rats following TBI is likely independent from deficits in GR function and may instead be mediated by a different cognitive mechanism. Therefore, the mechanisms underlying these sex-specific impairments in object location memory following pediatric TBI will be an important topic of future investigation.
The early post-natal period in rats is significantly shaped by developmental processes that are underway during the first few post-natal weeks, with full adult maturation occurring at approximately post-natal day 60.63 Namely, the glucocorticoid system, and GRs in particular, undergo a rapid increase in expression during the first post-natal weeks, reaching adult levels by 1 post-natal month.64,65 It is important to note that glucocorticoids are also important for normal brain development.66 Pre-natal exposure to glucocorticoid treatments has been associated with decreased cortical thickness, altered neuronal proliferation, and disrupted myelination of white matter tracts,66–68 all of which are processes that are similarly disrupted by pediatric TBI. Although beyond the scope of this project, it is possible that GR overexpression following pediatric TBI may have affected some these other developmental processes.
A limitation of the current study is the evaluation of GR levels at only one delayed post-injury time point. Both clinical and pre-clinical literature have pointed to a significant need to understand mechanisms underlying pediatric TBI-induced functional changes that often do not become apparent until adolescence/adulthood, and to develop treatment strategies to mitigate these symptoms as they arise.12 Although we determined that reduced GR-mediated transcription was associated with learning impairments at the adolescent time point, we cannot rule out the possibility that abnormal GR function during critical developmental periods may affect various processes such as learning and synaptic plasticity and may have contributed to these sustained impairments. Therefore, assessment of GR levels at different time points, including more acutely post-injury, will be an important avenue of future pediatric TBI research.
Overall, these findings demonstrate a novel role for impairments in hippocampal GR function in spatial learning and LTP deficits following pediatric TBI. To the best of our knowledge, this is the first study to demonstrate that overexpression of GRs in the DH improves spatial learning, memory, and synaptic plasticity in the hippocampus following TBI in pediatric animals. Moreover, we have identified the regulation of the GR target gene sgk1 and glutamate receptor subunits GluA1 and GluA2 in the DH as a mechanism underlying these effects of GR overexpression on hippocampal-dependent learning. Our findings provide support for the GR system as a potential therapeutic target for mitigating learning and memory impairments following pediatric TBI.
Supplementary Material
Authors' Contributions
D.L., J.W.H., and R.R. designed the experiments; D.L. and Z.L.R. ran the experiments; A.B. and N.W.S. prepared and ran the samples for measuring serum corticosterone and analyzed the data thereof; G.M.S. designed the experiments with the GR virus and provided the lentiviral construct; and D.L. and R.R. prepared the first draft of the manuscript. All authors edited the manuscript.
Funding Information
These studies were supported, in part, by grants from the National Institutes of Health R01 NS110898 (J.W.H. and R.R.) and Commonwealth Universal Research Enhancement from the Pennsylvania Department of Health SAP 410-007-9710 (R.R.) and SAP 410-007-7079 (R.R.). Additional support was provided by a grant from the Brain Injury Association of America's Brain Injury Research Fund to DL. Visit www.biausa.org/research.
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
References
- 1. Faul, M., and Coronado, V. (2015). Epidemiology of traumatic brain injury. Handb. Clin. Neurol. 127, 3–13. [DOI] [PubMed] [Google Scholar]
- 2. Araki, T., Yokota, H., and Morita, A. (2017). Pediatric traumatic brain injury: characteristic features, diagnosis, and management. Neurol. Med. Chir. (Tokyo) 57, 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Coronado, V.G., Xu, L., Basavaraju, S.V., McGuire, L.C., Wald, M.M., Faul, M.D., Guzman, B.R., Hemphill, J.D., and Centers for Disease Control and Prevention (2011). Surveillance for traumatic brain injury-related deaths—United States, 1997–2007. MMWR Surveill. Summ. 60, 1–32. [PubMed] [Google Scholar]
- 4. DeMaster, D., Johnson, C., Juranek, J., and Ewing-Cobbs, L. (2017). Memory and the hippocampal formation following pediatric traumatic brain injury. Brain. Behav 7, e00832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Anderson, V., Catroppa, C., Morse, S., Haritou, F., and Rosenfeld, J. (2000). Recovery of intellectual ability following traumatic brain injury in childhood: impact of injury severity and age at injury. Pediatr. Neurosurg. 32, 282–290. [DOI] [PubMed] [Google Scholar]
- 6. Ewing-Cobbs, L., Prasad, M.R., Kramer, L., Cox, C.S.Jr., Baumgartner, J., Fletcher, S., Mendez, D., Barnes, M., Zhang, X., and Swank, P. (2006). Late intellectual and academic outcomes following traumatic brain injury sustained during early childhood. J. Neurosurg. 105, 287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Treble-Barna, A., Schultz, H., Minich, N., Taylor, H.G., Yeates, K.O., Stancin, T., and Wade, S.L. (2017). Long-term classroom functioning and its association with neuropsychological and academic performance following traumatic brain injury during early childhood. Neuropsychology 31, 486–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Catroppa, C., Anderson, V.A., Morse, S.A., Haritou, F., and Rosenfeld, J.V. (2008). Outcome and predictors of functional recovery 5 years following pediatric traumatic brain injury (TBI). J. Pediatr. Psychol. 33, 707–718. [DOI] [PubMed] [Google Scholar]
- 9. Catroppa, C., Godfrey, C., Rosenfeld, J.V., Hearps, S.S., and Anderson, V.A. (2012). Functional recovery ten years after pediatric traumatic brain injury: outcomes and predictors. J. Neurotrauma 29, 2539–2547. [DOI] [PubMed] [Google Scholar]
- 10. Kochanek, P.M., Tasker, R.C., Carney, N., Totten, A.M., Adelson, P.D., Selden, N.R., Davis-O'Reilly, C., Hart, E.L., Bell, M.J., Bratton, S.L., Grant, G.A., Kissoon, N., Reuter-Rice, K.E., Vavilala, M.S., and Wainwright, M.S. (2019). Guidelines for the Management of Pediatric Severe Traumatic Brain Injury, Third Edition: Update of the Brain Trauma Foundation Guidelines, Executive Summary. Neurosurgery 84, 1169–1178. [DOI] [PubMed] [Google Scholar]
- 11. Thurman, D.J. (2016). The epidemiology of traumatic brain injury in children and youths: a review of research since 1990. J. Child. Neurol. 31, 20–27. [DOI] [PubMed] [Google Scholar]
- 12. Huh, J.W., and Raghupathi, R. (2019). Therapeutic strategies to target acute and long-term sequelae of pediatric traumatic brain injury. Neuropharmacology 145, 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Krugers, H.J., Arp, J.M., Xiong, H., Kanatsou, S., Lesuis, S.L., Korosi, A., Joels, M., and Lucassen, P.J. (2017). Early life adversity: lasting consequences for emotional learning. Neurobiol Stress 6, 14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Arnett, M.G., Pan, M.S., Doak, W., Cyr, P.E., Muglia, L.M., and Muglia, L.J. (2015). The role of glucocorticoid receptor-dependent activity in the amygdala central nucleus and reversibility of early-life stress programmed behavior. Transl. Psychiatry 5, e542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Felszeghy, K., Gaspar, E., and Nyakas, C. (1996). Long-term selective down-regulation of brain glucocorticoid receptors after neonatal dexamethasone treatment in rats. J. Neuroendocrinol. 8, 493–499. [DOI] [PubMed] [Google Scholar]
- 16. Lee, B.H., Wen, T.C., Rogido, M., and Sola, A. (2007). Glucocorticoid receptor expression in the cortex of the neonatal rat brain with and without focal cerebral ischemia. Neonatology 91, 12–19. [DOI] [PubMed] [Google Scholar]
- 17. Raghupathi, R., and Huh, J.W. (2007). Diffuse brain injury in the immature rat: evidence for an age-at-injury effect on cognitive function and histopathologic damage. J. Neurotrauma 24, 1596–1608. [DOI] [PubMed] [Google Scholar]
- 18. Geddes, R.I., Peterson, B.L., Stein, D.G., and Sayeed, I. (2016). Progesterone treatment shows benefit in female rats in a pediatric model of controlled cortical impact injury. PLoS One 11, e0146419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Geddes, R.I., Sribnick, E.A., Sayeed, I., and Stein, D.G. (2014). Progesterone treatment shows benefit in a pediatric model of moderate to severe bilateral brain injury. PLoS One 9, e87252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Newell, E.A., Todd, B.P., Luo, Z., Evans, L.P., Ferguson, P.J., and Bassuk, A.G. (2020). A Mouse model for juvenile, lateral fluid percussion brain injury reveals sex-dependent differences in neuroinflammation and functional recovery. J Neurotrauma 37, 635–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Uysal, N., Baykara, B., Kiray, M., Cetin, F., Aksu, I., Dayi, A., Gurpinar, T., Ozdemir, D., and Arda, M.N. (2013). Combined treatment with progesterone and magnesium sulfate positively affects traumatic brain injury in immature rats. Turk. Neurosurg. 23, 129–137. [DOI] [PubMed] [Google Scholar]
- 22. Stahn, C. and Buttgereit, F. (2008). Genomic and nongenomic effects of glucocorticoids. Nat. Clin. Pract. Rheumatol. 4, 525–533. [DOI] [PubMed] [Google Scholar]
- 23. Mifsud, K.R., and Reul, J. (2018). Mineralocorticoid and glucocorticoid receptor-mediated control of genomic responses to stress in the brain. Stress 21, 389–402. [DOI] [PubMed] [Google Scholar]
- 24. Aguilar-Valles, A., Sanchez, E., de Gortari, P., Balderas, I., Ramirez-Amaya, V., Bermudez-Rattoni, F., and Joseph-Bravo, P. (2005). Analysis of the stress response in rats trained in the water-maze: differential expression of corticotropin-releasing hormone, CRH-R1, glucocorticoid receptors and brain-derived neurotrophic factor in limbic regions. Neuroendocrinology 82, 306–319. [DOI] [PubMed] [Google Scholar]
- 25. Oitzl, M.S., de Kloet, E.R., Joels, M., Schmid, W., and Cole, T.J. (1997). Spatial learning deficits in mice with a targeted glucocorticoid receptor gene disruption. Eur. J. Neurosci. 9, 2284–2296. [DOI] [PubMed] [Google Scholar]
- 26. Oitzl, M.S., Reichardt, H.M., Joels, M. and de Kloet, E.R. (2001). Point mutation in the mouse glucocorticoid receptor preventing DNA binding impairs spatial memory. Proc. Natl. Acad. Sci. U. S. A. 98, 12,790–12,795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Conrad, C.D., Lupien, S.J., and McEwen, B.S. (1999). Support for a bimodal role for type II adrenal steroid receptors in spatial memory. Neurobiol. Learn. Mem. 72, 39–46. [DOI] [PubMed] [Google Scholar]
- 28. Rousse, I., Beaulieu, S., Rowe, W., Meaney, M.J., Barden, N., and Rochford, J. (1997). Spatial memory in transgenic mice with impaired glucocorticoid receptor function. Neuroreport 8, 841–845. [DOI] [PubMed] [Google Scholar]
- 29. Steckler, T., Rammes, G., Sauvage, M., van Gaalen, M.M., Weis, C., Zieglgansberger, W., and Holsboer, F. (2001). Effects of the monoamine oxidase A inhibitor moclobemide on hippocampal plasticity in GR-impaired transgenic mice. J. Psychiatr. Res. 35, 29–42. [DOI] [PubMed] [Google Scholar]
- 30. Roozendaal, B., Okuda, S., Van der Zee, E.A., and McGaugh, J.L. (2006). Glucocorticoid enhancement of memory requires arousal-induced noradrenergic activation in the basolateral amygdala. Proc. Natl. Acad. Sci. U. S. A. 103, 6741–6746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Okuda, S., Roozendaal, B., and McGaugh, J.L. (2004). Glucocorticoid effects on object recognition memory require training-associated emotional arousal. Proc. Natl. Acad. Sci. U. S. A. 101, 853–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Anacker, C., Cattaneo, A., Musaelyan, K., Zunszain, P.A., Horowitz, M., Molteni, R., Luoni, A., Calabrese, F., Tansey, K., Gennarelli, M., Thuret, S., Price, J., Uher, R., Riva, M.A., and Pariante, C.M. (2013). Role for the kinase SGK1 in stress, depression, and glucocorticoid effects on hippocampal neurogenesis. Proc. Natl. Acad. Sci. U. S. A. 110, 8708–8713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee, C.T., Tyan, S.W., Ma, Y.L., Tsai, M.C., Yang, Y.C., and Lee, E.H. (2006). Serum- and glucocorticoid-inducible kinase (SGK) is a target of the MAPK/ERK signaling pathway that mediates memory formation in rats. Eur. J. Neurosci. 23, 1311–1320. [DOI] [PubMed] [Google Scholar]
- 34. Tsai, K.J., Chen, S.K., Ma, Y.L., Hsu, W.L., and Lee, E.H. (2002). sgk, a primary glucocorticoid-induced gene, facilitates memory consolidation of spatial learning in rats. Proc. Natl. Acad. Sci. U. S. A. 99, 3990–3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ma, Y.L., Tsai, M.C., Hsu, W.L., and Lee, E.H. (2006). SGK protein kinase facilitates the expression of long-term potentiation in hippocampal neurons. Learn. Mem. 13, 114–118. [DOI] [PubMed] [Google Scholar]
- 36. Tai, D.J., Su, C.C., Ma, Y.L. and Lee, E.H. (2009). SGK1 phosphorylation of IkappaB Kinase alpha and p300 Up-regulates NF-kappaB activity and increases N-Methyl-D-aspartate receptor NR2A and NR2B expression. J. Biol. Chem. 284, 4073–4089. [DOI] [PubMed] [Google Scholar]
- 37. Lang, F., Bohmer, C., Palmada, M., Seebohm, G., Strutz-Seebohm, N., and Vallon, V. (2006). (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol. Rev. 86, 1151–1178. [DOI] [PubMed] [Google Scholar]
- 38. Wang, M., Ramasamy, V.S., Samidurai, M., and Jo, J. (2019). Acute restraint stress reverses impaired LTP in the hippocampal CA1 region in mouse models of Alzheimer's disease. Sci. Rep. 9, 10,955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Groc, L., Choquet, D., and Chaouloff, F. (2008). The stress hormone corticosterone conditions AMPAR surface trafficking and synaptic potentiation. Nat. Neurosci. 11, 868–870. [DOI] [PubMed] [Google Scholar]
- 40. Lengel, D., Huh, J.W., Barson, J.R., and Raghupathi, R. (2020). Progesterone treatment following traumatic brain injury in the 11-day-old rat attenuates cognitive deficits and neuronal hyperexcitability in adolescence. Exp. Neurol. 330, 113,329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ryan, N.P., Catroppa, C., Cooper, J.M., Beare, R., Ditchfield, M., Coleman, L., Silk, T., Crossley, L., Beauchamp, M.H., and Anderson, V.A. (2015). The emergence of age-dependent social cognitive deficits after generalized insult to the developing brain: a longitudinal prospective analysis using susceptibility-weighted imaging. Hum. Brain Mapp. 36, 1677–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Choe, M.C., Valino, H., Fischer, J., Zeiger, M., Breault, J., McArthur, D.L., Leung, M., Madikians, A., Yudovin, S., Lerner, J.T., and Giza, C.C. (2016). Targeting the epidemic: interventions and follow-up are necessary in the pediatric traumatic brain injury clinic. J. Child. Neurol. 31, 109–115. [DOI] [PubMed] [Google Scholar]
- 43. Hanlon, L.A., Raghupathi, R., and Huh, J.W. (2017). Differential effects of minocycline on microglial activation and neurodegeneration following closed head injury in the neonate rat. Exp. Neurol. 290, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hanlon, L.A., Raghupathi, R., and Huh, J.W. (2019). Depletion of microglia immediately following traumatic brain injury in the pediatric rat: Implications for cellular and behavioral pathology. Exp. Neurol. 316, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Porterfield, S.P. (1994). Vulnerability of the developing brain to thyroid abnormalities: environmental insults to the thyroid system. Environ. Health Perspect. 102, Suppl. 2, 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rice, D., and Barone, S.Jr. (2000). Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ. Health Perspect. 108, Suppl. 3, 511–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yager, J.Y., and Thornhill, J.A. (1997). The effect of age on susceptibility to hypoxic-ischemic brain damage. Neurosci. Biobehav. Rev. 21, 167–174. [DOI] [PubMed] [Google Scholar]
- 48. Huh, J.W., Widing, A.G., and Raghupathi, R. (2008). Midline brain injury in the immature rat induces sustained cognitive deficits, bihemispheric axonal injury and neurodegeneration. Exp. Neurol. 213, 84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Marr, R.A., Guan, H., Rockenstein, E., Kindy, M., Gage, F.H., Verma, I., Masliah, E., and Hersh, L.B. (2004). Neprilysin regulates amyloid Beta peptide levels. J. Mol. Neurosci. 22, 5–11. [DOI] [PubMed] [Google Scholar]
- 50. Pandey, S., Badve, P.S., Curtis, G.R., Leibowitz, S.F., and Barson, J.R. (2019). Neurotensin in the posterior thalamic paraventricular nucleus: inhibitor of pharmacologically relevant ethanol drinking. Addict. Biol. 24, 3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Frey, A.J., Wang, Q., Busch, C., Feldman, D., Bottalico, L., Mesaros, C.A., Blair, I.A., Vachani, A., and Snyder, N.W. (2016). Validation of highly sensitive simultaneous targeted and untargeted analysis of keto-steroids by Girard P derivatization and stable isotope dilution-liquid chromatography-high resolution mass spectrometry. Steroids 116, 60–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Won, M., Park, K.A., Byun, H.S., Kim, Y.R., Choi, B.L., Hong, J.H., Park, J., Seok, J.H., Lee, Y.H., Cho, C.H., Song, I.S., Kim, Y.K., Shen, H.M., and Hur, G.M. (2009). Protein kinase SGK1 enhances MEK/ERK complex formation through the phosphorylation of ERK2: implication for the positive regulatory role of SGK1 on the ERK function during liver regeneration. J. Hepatol. 51, 67–76. [DOI] [PubMed] [Google Scholar]
- 53. Liu, H., Yu, J., Xia, T., Xiao, Y., Zhang, Q., Liu, B., Guo, Y., Deng, J., Deng, Y., Chen, S., Naray-Fejes-Toth, A., Fejes-Toth, G., and Guo, F. (2014). Hepatic serum- and glucocorticoid-regulated protein kinase 1 (SGK1) regulates insulin sensitivity in mice via extracellular-signal-regulated kinase 1/2 (ERK1/2). Biochem. J. 464, 281–289. [DOI] [PubMed] [Google Scholar]
- 54. Carter, S.D., Mifsud, K.R. and Reul, J.M. (2015). Distinct epigenetic and gene expression changes in rat hippocampal neurons after Morris water maze training. Front. Behav. Neurosci. 9, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Conboy, L., and Sandi, C. (2010). Stress at learning facilitates memory formation by regulating AMPA receptor trafficking through a glucocorticoid action. Neuropsychopharmacology 35, 674–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ewing-Cobbs, L., Prasad, M.R., Cox, C.S.Jr., Granger, D.A., Duque, G., and Swank, P.R. (2017). Altered stress system reactivity after pediatric injury: Relation with post-traumatic stress symptoms. Psychoneuroendocrinology 84, 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Miyazaki, S., Katayama, Y., Lyeth, B.G., Jenkins, L.W., DeWitt, D.S., Goldberg, S.J., Newlon, P.G., and Hayes, R.L. (1992). Enduring suppression of hippocampal long-term potentiation following traumatic brain injury in rat. Brain Res. 585, 335–339. [DOI] [PubMed] [Google Scholar]
- 58. Witgen, B.M., Lifshitz, J., Smith, M.L., Schwarzbach, E., Liang, S.L., Grady, M.S., and Cohen, A.S. (2005). Regional hippocampal alteration associated with cognitive deficit following experimental brain injury: a systems, network and cellular evaluation. Neuroscience 133, 1–15. [DOI] [PubMed] [Google Scholar]
- 59. White, E.R., Pinar, C., Bostrom, C.A., Meconi, A., and Christie, B.R. (2017). Mild traumatic brain injury produces long-lasting deficits in synaptic plasticity in the female juvenile hippocampus. J. Neurotrauma 34, 1111–1123. [DOI] [PubMed] [Google Scholar]
- 60. Wiegert, O., Joels, M., and Krugers, H. (2006). Timing is essential for rapid effects of corticosterone on synaptic potentiation in the mouse hippocampus. Learn. Mem. 13, 110–113. [DOI] [PubMed] [Google Scholar]
- 61. Wei, Q., Hebda-Bauer, E.K., Pletsch, A., Luo, J., Hoversten, M.T., Osetek, A.J., Evans, S.J., Watson, S.J., Seasholtz, A.F., and Akil, H. (2007). Overexpressing the glucocorticoid receptor in forebrain causes an aging-like neuroendocrine phenotype and mild cognitive dysfunction. J. Neurosci. 27, 8836–8844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Terashima, A., Suh, Y.H., and Isaac, J.T.R. (2019). The AMPA Receptor Subunit GluA1 is Required for CA1 Hippocampal long-term potentiation but is not essential for synaptic transmission. Neurochem. Res. 44, 549–561. [DOI] [PubMed] [Google Scholar]
- 63. Semple, B.D., Blomgren, K., Gimlin, K., Ferriero, D.M., and Noble-Haeusslein, L.J. (2013). Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 106–107, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rosenfeld, P., Sutanto, W., Levine, S., and De Kloet, E.R. (1988). Ontogeny of type I and type II corticosteroid receptors in the rat hippocampus. Brain Res. 470, 113–118. [DOI] [PubMed] [Google Scholar]
- 65. Rosenfeld, P., van Eekelen, J.A., Levine, S., and de Kloet, E.R. (1993). Ontogeny of corticosteroid receptors in the brain. Cell Mol. Neurobiol. 13, 295–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Davis, E.P., Sandman, C.A., Buss, C., Wing, D.A., and Head, K. (2013). Fetal glucocorticoid exposure is associated with preadolescent brain development. Biol. Psychiatry 74, 647–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Antonow-Schlorke, I., Helgert, A., Gey, C., Coksaygan, T., Schubert, H., Nathanielsz, P.W., Witte, O.W., and Schwab, M. (2009). Adverse effects of antenatal glucocorticoids on cerebral myelination in sheep. Obstet. Gynecol. 113, 142–151. [DOI] [PubMed] [Google Scholar]
- 68. Scheepens, A., van de Waarenburg, M., van den Hove, D., and Blanco, C.E. (2003). A single course of prenatal betamethasone in the rat alters postnatal brain cell proliferation but not apoptosis. J. Physiol. 552, 163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.