

Abstract
Over the past 20 years, diagnostic testing for genetic diseases has evolved, leading to variable diagnostic certainty for individuals included in long-term natural history studies. Using genotype and phenotype data from an ongoing natural history study of CLN3 disease, we developed a hierarchical diagnostic confidence scheme with three major classes: Definite, Probable, or Possible CLN3 disease. An additional level, CLN3 Disease PLUS, includes individuals with CLN3 disease plus an additional disorder with a separate etiology that substantially affects the phenotype. Within the Definite and Probable CLN3 disease classes, we further divided individuals into subclasses based on phenotype. After assigning participants to classes, we performed a blinded reclassification to assess the reliability of this scheme. A total of 134 individuals with suspected CLN3 disease were classified: 100 as Definite, 21 as Probable, and 7 as Possible. Six individuals were classified as CLN3-PLUS. Phenotypes included the classical juvenile-onset syndromic phenotype, a “vision loss only” phenotype, and an atypical syndromic phenotype. Some individuals were too young to fully classify phenotype. Test-retest reliability showed 96% agreement. We created a reliable diagnostic confidence scheme for CLN3 disease that has excellent face validity. This scheme has implications for clinical research in CLN3 and other rare genetic neurodegenerative disorders.
Keywords: Batten disease, diagnosis, lysosomal disorders, natural history, neurodegenerative disease, neuronal ceroid lipofuscinosis
1 |. INTRODUCTION
The types, precision, and availability of molecular diagnostic testing for genetic diseases have evolved substantially over the last several years.1 With increasing use of newer methods, including next-generation sequencing (NGS), grouping individuals diagnosed with older methods together with those diagnosed genetically may be confounded by differing levels of diagnostic accuracy or precision. This is a particular challenge for research studies on individual rare diseases where evaluation of small and potentially heterogeneous cohorts may lead to inaccurate conclusions. To maximize sample sizes while limiting confounds, a reasonable degree of diagnostic certainty is required. Diagnostic certainty can be a challenge when there is substantial phenotypic heterogeneity, especially when NGS yields variants of uncertain significance (VUS), although this can be mitigated by following best practices in genomic analysis.2
The neuronal ceroid lipofuscinoses (NCLs) are a group of rare genetic neurodegenerative lysosomal diseases, most of which are inherited in an autosomal recessive manner. NCLs are characterized by accumulation of autofluorescent lipopigment in the lysosomes. NCLs typically result in vision loss, seizures, movement disorders, cognitive decline, and premature death. Many individuals also develop behavior and mood problems, sleep problems, and swallowing dysfunction. Historically, NCLs were grouped based on age at symptom onset into infantile, late infantile, juvenile, and adult-onset forms. At least 12 genetic etiologies are now recognized; the current convention is to use a gene-based nomenclature for grouping.3 Many individuals have had their specific diagnosis revised due to the change from age-based to gene-based nomenclature.4 Currently in the United States, the most prevalent form is CLN3 disease (CLN3; OMIM# 204200), classic juvenile NCL (JNCL; Batten disease), which typically begins in the middle of the first decade of life and progresses until eventual death in the third decade.
Three major phenotypes have been described in association with pathogenic CLN3 variants. The classical (syndromic) JNCL phenotype has onset between the ages of 4 and 6 years with rapid vision loss followed by behavior problems and cognitive decline. Epilepsy begins around age 10 years followed by sleep problems and motor decline.5 Most individuals lose independent mobility and self-care in the late teens at which time cardiac dysrhythmia and feeding problems often develop. The classical JNCL phenotype is relatively homogeneous and is the most common of the phenotypes.6,7 Two less common phenotypes have also been described. One is “vision loss only,” but with no other symptoms well into adulthood.8 The other is a “protracted course” with typical childhood onset of initial symptoms, but with slower accumulation of additional symptoms, slower progression of overall disease severity, or both.8,9 This phenotype has not been described quantitatively.
In CLN3 disease, the most common variant is a ~1 kb deletion of exons 7 and 8 of CLN3.10 This variant accounts for about 80% of disease alleles; 70% of individuals with genetically confirmed CLN3 disease are homozygous for the common 1 kb deletion in the University of Rochester Batten Center (URBC) database, consistent with previous reports.11 While the exact function of the CLN3 protein is unknown, the common deletion likely results in loss of function.12
In the pregene era, diagnosis was based on electron microscopy demonstrating “fingerprint body” inclusions or vacuolated lymphocytes.13,14 Since identification of the CLN3 gene, diagnostic testing has evolved substantially. Initial genetic testing for CLN3 disease was limited to identification of the common 1 kb deletion soon after the gene was cloned in 199510,15; the presence of a different pathogenic variant in trans with the common deletion was often inferred based on clinical and microscopic evidence. Although other pathogenic variants were described by 1997,16 routine clinical sequencing of CLN3 was not performed until well after 2000. Currently, clinical sequencing of CLN3 is widely available and CLN3 disease is included in many commercial genetic testing panels. Our center has been studying the natural history of NCLs since 2002 and thus our database includes individuals with a variety of diagnostic evidence.
To maximize valid inclusion of data from individuals with absent or incomplete genetic testing and to maximize the likelihood of identifying discrete phenotypes in our natural history research studies, we sought to develop a hierarchical diagnostic confidence scheme. Similar schemes have been used to support diagnostic confidence in disorders such as multiple sclerosis and Parkinson disease that predominantly rely on clinical diagnosis.17,18 Clinical diagnostic criteria are commonly used for syndromes such as Neurofibromatosis type 1 (NF1; OMIM# 162200) or Dravet syndrome (OMIM# 607208) that have a genetic basis, but these are not hierarchical confidence schemes.19,20 Valid, reliable, and utile hierarchical confidence schemes have potential value for both clinical and research applications in many rare genetic disorders.
2 |. PATIENTS AND METHODS
We created a diagnostic confidence scheme using data from individuals enrolled in the URBC ongoing longitudinal natural history study (NCT01873924). Phenotype information is collected using the Unified Batten Disease Rating Scale (UBDRS), a disease-specific instrument that assesses severity in four domains and records caregiver-reported age-at-onset for seven core features of the disease.7 We also collected results of diagnostic testing such as genetic testing, electron microscopy, and presence of lymphocytic vacuoles. We have been performing genetic testing for most participants at our center since 2004.21 Individuals with suspected NCL of any form were recruited between 2002 and 2020 for participation in the natural history study. All individuals suspected to have CLN3 disease were included in this analysis. Individuals were excluded from this analysis if they had another form of NCL, no diagnosis of an NCL, or if there was insufficient information to accurately assess their phenotype.
We devised a hierarchical scheme with three classes based on diagnostic certainty (Table 1).
TABLE 1.
Diagnostic confidence classes. Each subclassification requires both diagnostic and phenotype Information
| Definite CLN3 disease | Probable CLN3 disease | Possible CLN3 disease | CLN3 disease PLUS | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 1A | 1B | 1C | 1D | 2A | 2B | 2C | 2D | 3 | CLN3-PLUS |
| Classical Syndromic JNCL | Vision Loss Only | Too Young to Determine | Atypical Syndromic | Classical Syndromic JNCL | Vision Loss Only | Too young to determine | Atypical syndromic | Classical syndromic JNCL | |
| Diagnostic information | |||||||||
| Disease-causing variant on both alleles OR Homozygous for common deletion without features of another disorder |
Disease-causing variant on both alleles | Disease-causing variant on both alleles AND NOT Deletion homozygote |
Disease-causing variant on both alleles | Disease-causing variant on one allele only AND/OR Fingerprint bodies/lymphocytic vacuoles OR Sibling with genetically confirmed CLN3 |
Disease-causing variant on one allele only AND/OR Fingerprint bodies/lymphocytic vacuoles OR Sibling with genetically confirmed CLN3 |
Disease-causing variant on one allele only AND/OR Fingerprint bodies/lymphocytic vacuoles OR Sibling with genetically confirmed CLN3 |
Disease-causing variant on one allele only AND/OR Fingerprint bodies/lymphocytic vacuoles OR Sibling with genetically confirmed CLN3 |
Genetic testing not performed AND Microscopy not performed |
Genetic or laboratory evidence for CLN3 disease |
| Phenotype information | |||||||||
| Characteristic CLN3 disease phenotype | Vision loss only at age 12 years or older | Vision loss only or presymptomatic at age < 12 years | Atypical age-at-onset or rate-of-progression | Characteristic CLN3 disease phenotype | Vision loss only at age 12 Years or older | Vision loss only or presymptomatic at age <12 years | Atypical age-at-onset or rate-of-progression | Characteristic CLN3 disease phenotype | CLN3 disease phenotype plus non-NCL neurologic features |
Class 1—
Definite CLN3 Disease: Individuals in class 1 have pathogenic variants identified on both alleles of CLN3. Pathogenicity is here defined by the American College of Medical Genetics, the Association for Molecular Pathology,22 as employed in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/docs/clinsig/).
Class 2—
Probable CLN3 Disease: Individuals in class 2 have some diagnostic evidence for CLN3 disease in addition to a phenotype consistent with CLN3 disease. Such evidence could include an identified pathogenic variant on only one allele, fingerprint bodies demonstrated with electron microscopy, lymphocytic vacuoles, or a combination.13,14 Class 2 also includes individuals without genetic or microscopy testing results, but who have symptoms of CLN3 disease and have one or more siblings with genetically confirmed CLN3 disease.
Class 3—
Possible CLN3 Disease. Individuals in class 3 have a clinical diagnosis of CLN3 disease, with a classical JNCL phenotype, but no available laboratory testing results.
We defined another group of individuals with definite or probable CLN3 disease who had an additional medical condition of a separate etiology that substantially impacted the neurobehavioral phenotype. We termed this group CLN3 Disease PLUS (CLN3-PLUS).
We created subclasses based on quantitative phenotypic data obtained using the UBDRS. These subclasses were defined in reference to the classical JNCL phenotype, using data from individuals who were homozygous for the common 1 kb deletion. Individuals in this reference group have a relatively homogeneous phenotype.6 For classification purposes, we evaluated phenotypic features quantitatively using data on age-at-onset of core symptoms and UBDRS physical subscale severity scores in relationship to age.
Age-at-onset phenotype was assessed using five core features of the NCLs: vision loss, cognitive decline, behavior problems, seizures, and motor difficulties. Sleep problems were not used in this analysis due to highly variable age-at-onset. Feeding dysfunction was not used because of the late age-at-onset and resulting paucity of data. We calculated the average age-at-onset and SD of each the five symptoms in the reference group. Individuals not in the reference group who had symptom age-at-onset more than two standard deviations from the mean were flagged as potential outliers. Those who had three or more symptoms with an age-at-onset outside 2 SDs from the mean were considered to have an atypical age-at-onset phenotype.
UBDRS physical subscale severity reflects abnormalities in vision, speech, tone, strength, gait, coordination, and presence of involuntary movements, with higher scores reflecting greater severity. The physical subscale positively correlates with age and disease progression.6,23 We assessed age-related severity in two ways: (a) cross-sectionally using the most recent data point and (b) longitudinally (for individuals with two or more serial assessments) based on the individual’ slope. For the reference group, we performed a simple linear regression of physical severity score vs age-at-evaluation using the most recently obtained data point from all individuals in that group. We calculated the 95% confidence interval of the slope and the 95% prediction interval. Individuals who had a slope of longitudinally collected data outside of the 95% confidence interval or who had a data point outside of the 95% prediction interval were considered to have atypical physical subscale severity.
Compared to the reference group, we defined several specific phenotypic subclasses. One subclass of individuals had the classical JNCL phenotype. Classical CLN3 disease is a progressive disease with different symptoms presenting at different ages, with most individuals having vision loss as the initial symptom.23,24 A second subclass had vision loss only with no other symptoms.8,25 Individuals with classical JNCL have multiple symptoms by age 12 years.6,7,23,26 Therefore, we further subdivided those with vision loss only based on age (less than 12 years or 12 years and older). A third subclass had multiple symptoms, but with atypical age-at-onset, atypical age-related severity, or both. Using this framework, we divided class 1 (Definite CLN3 disease) and class 2 (Probable CLN3 disease) into four subclasses: A—Classical Syndromic JNCL; B—Vision Loss Only (beyond age 12 years); C—Too Young to Determine, these individuals were presymptomatic or had vision loss only but were less than 12 years old; and D—Atypical Syndromic. Class 3 was not subdivided because it included only those with a classical JNCL phenotype. Individuals homozygous for the common deletion were assigned to Class 1A regardless of age, given the homogenous phenotype.6 Sub-grouping was based on the status of the affected individual at the time of the most recent evaluation.
All individuals were classified systematically by two of the authors (M.C.M. and J.W.M.) using a decision tree designed according to the criteria above (Figure 1). To determine the reliability of our scheme, all individuals not known to be homozygous for the 1 kb common deletion were re-evaluated by the same authors with J.W.M. blinded to the initial classification. Percent agreement between the first and second classifications was calculated.
FIGURE 1.
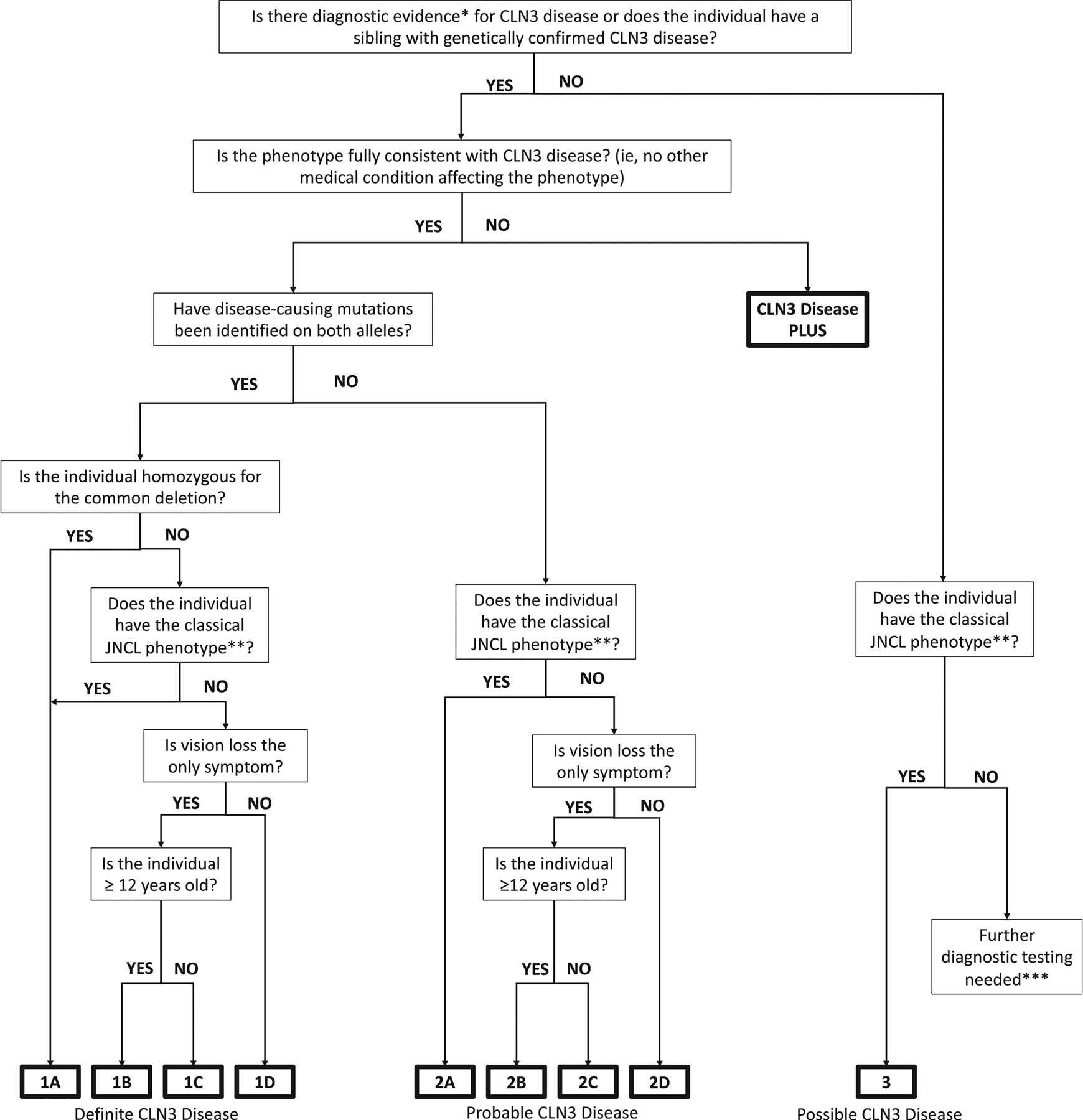
Diagnostic confidence flow chart (* Diagnostic evidence includes disease-causing variant on one or both alleles, fingerprint bodies, or lymphocytic vacuoles. ** Classic JNCL phenotype as defined by age-at-onset of core features of the disease as well as progression of the disease. *** Without diagnostic evidence and with an atypical phenotype, further investigation is necessary to determine if the individual has CLN3 disease, a different NCL, or a different diagnosis entirely)
3 |. RESULTS
Data from 136 individuals with clinically suspected CLN3 disease were identified from the URBC database. Two individuals were excluded because of insufficient information on symptom age-at-onset or the physical subscale was not completed. We included 134 individuals (63 M, 71 F; median age 15.3 years [range 0.2–31.1 years]) for classification. Of these, 100 individuals met the criteria for class 1, 22 individuals met the criteria for class 2, 6 individuals met the criteria for class 3, and 6 individuals were classified as CLN3-PLUS.
Of the 100 individuals in class 1, 93 were classified as 1A, 2 were classified as 1B, 2 were classified as 1C, and 3 were classified as 1D. All three individuals classified as 1D had slower disease progression or later onset compared to the classical phenotype.
Of the 22 individuals in class 2, 13 had the common deletion on one allele but sequencing was not performed, 1 had a sibling with genetically confirmed CLN3 disease, 1 had two deletions in exon 12 of CLN3, but it was unknown whether they were in trans, and 7 had electron microscopy showing fingerprint inclusions. Twenty individuals were classified as 2A and two individuals were classified as 2D. Both individuals classified as 2D had slower disease progression or later onset compared to the classical phenotype. No individuals met the criteria for subclasses 2B or 2C.
All six individuals in class 3 had previous diagnostic testing, as reported by a caregiver, but the results were not available to us.
Individuals classified as CLN3-PLUS had additional diagnoses of autism associated with known autism genetic variants (n = 2), failure to thrive as an infant with multifocal brain lesions on MRI (n = 1), a novel quadruplication in CLN3 with early language delay (n = 1), or early-onset developmental delay (n = 2). Of the six children classified as CLN3-PLUS, four met the criteria for a definite CLN3 diagnosis and two met the criteria for a probable CLN3 diagnosis (one had a known pathogenic variant on one allele and one had a sibling with genetic confirmation but did not have confirmation themselves). Four had an identified etiology for their additional diagnosis.
In the blinded reliability analysis, we re-evaluated 27 individuals from class 1 who were not homozygous for the common deletion and all 22 individuals in class 2. This analysis resulted in changing the classification for 2 individuals from class 1: one individual was re-classified from 1D to 1A because of initial evaluation error and one from 1D to CLN3-PLUS due to subsequent confirmation of early global developmental delay and early-onset epilepsy. We did not re-classify anyone from class 2. No individual changed classification due to ambiguity in the classification scheme. The percent agreement between the first and second classifications was 96%.
4 |. DISCUSSION
We created a hierarchal diagnostic confidence scheme that can be applied to individuals with clinically suspected CLN3 disease. As diagnostic testing has evolved over the past 20 years, the type and amount of available diagnostic information has changed. In a rare disease where the amount of data is already limited, we developed this scheme to facilitate inclusion of data from individuals with different degrees of diagnostic certainty but with the potential to weight or exclude based on that certainty.
We further subdivided classes 1 and 2 based on phenotype but did not further subdivide class 3 due to the lower level of diagnostic confidence. Although CLN3 disease has a relatively homogenous phenotype in relation to other NCLs, the “vision loss only” and “protracted” phenotypes can be distinguished from the classical JNCL phenotype. Individuals homozygous for the common deletion who were less than 12 years of age (n = 7) and had vision loss as their only symptom were automatically classified as 1A rather than 1C based on the homogeneity of phenotype among deletion homozygotes. To our knowledge, there are no data reporting an individual homozygous for the common deletion with a phenotype other than the classical JNCL phenotype unless they have a co-occurring disorder.6 The one individual who was homozygous for the common deletion but who had an atypical (autistic) phenotype had an additional variant in a gene that has been associated with autism.27 This individual was therefore classified as CLN3-PLUS.
Diagnostic confidence based on precision of laboratory testing is important, but knowledge of the phenotype is also essential. Because rare disease cohorts are limited in size, a single outlier could have a disproportionate impact on results and could bias conclusions. Detailed knowledge of the CLN3 disease phenotypes has allowed us to suspect the presence of an additional disorder in some individuals with atypical phenotypes leading to subsequent inclusion in the CLN3-PLUS class. The two children with early developmental delay had unknown etiologies for the developmental disorder, but their CLN3 disease phenotype was clearly modified by the coexisting, nonprogressive neurodevelopmental disorder. Four percent of our sample was classified as CLN3-PLUS, which is near the expected prevalence of intellectual and developmental disability (IDD) in the population at large which was 7% in 2016.28 IDDs represent just one example of disorders prevalent in the population at large that may co-occur at a similar frequency in individuals with a rare disease. Therefore, identifying individuals with dual diagnoses and excluding them is integral to having a complete picture of the population.
Having a rare disease does not preclude an individual from also having a more common disorder. Thus, other children in the cohort may have additional but less severe neurobehavioral disorders that do not rise to the level of detection because of the severity of the CLN3 disease phenotype. These may include ADHD, learning disability, common childhood challenging behaviors (difficult toilet training, temper tantrums, and sleep difficulty), developmental coordination disorder, or others. Despite this likelihood, the classical CLN3 disease phenotype is quite homogeneous across individuals as reflected in the large preponderance of individuals in classes 1A and 2A (84%). All five individuals (4%) with an atypical course (classes 1D and 2D) had a milder than expected disease course, supporting our inference that the CLN3 syndromic phenotype is substantially more severe than these common neurobehavioral conditions. Additionally, the evolution of NCL symptoms early in the CLN3 disease course occurs at an age when many milder neurodevelopmental symptoms first manifest and therefore the impact may not be easily distinguished.
Our diagnostic confidence scheme is not static. Individuals with incomplete information can be re-classified at higher confidence levels based on new diagnostic information. Younger individuals with an indeterminate phenotype can be re-classified as additional symptoms develop or as time progresses without the evolution of new symptoms. For example, individuals who are initially classified as 1C but for whom additional follow-up information is obtained beyond the age of 12 years would always be reclassified based on that information. Similarly, an individual classified as 1B who subsequently sustains a significant traumatic brain injury would be reclassified as CLN3-PLUS for any future evaluations. These reclassifications maintain the integrity of the scheme but might influence which natural history data might be included in specific studies.
This diagnostic confidence scheme has implications for stratification within research studies including natural history studies, clinical trials, and biological studies based on tissue derived from individuals with different degrees of diagnostic confidence. For natural history studies, segregation of those with vision loss only or a protracted course phenotype from those with the classic syndromic CLN3 disease phenotype will likely decrease the variability of the sample. Individuals in classes 2 and 3 can then be compared those in classes 1A to inform decisions regarding inclusion or exclusion in the larger sample based on phenotype. For clinical trials, it would likely be advantageous to stratify based on phenotype in order to maximize the ability to compare to existing natural history data and would enhance the quality of matching beyond age- and sex-based approaches. Furthermore, exclusion of those with milder or more slowly progressing phenotypes will increase the likelihood of detecting a change from expected disease progression during the limited time duration that would be practical for clinical trial design. Alternatively, detecting and excluding outliers with atypically aggressive phenotypes would be helpful to avoid biasing a trial against efficacy. Because most future clinical trials will likely require genetic confirmation, the genotype-phenotype association is expected to be more important than the method of laboratory diagnosis.
For clinical care, the use of the diagnostic confidence scheme has more limited utility in prognostication of disease progression. This is especially true for individuals under the age of 12 years who are not homozygous for the common deletion. Although phenotypes have been reported for some genetic variants, much of the available data on genotype-phenotype associations are incomplete or nonspecific (https://www.ncbi.nlm.nih.gov/clinvar/?term=CLN3%5Bgene%5D, December 21, 2020; https://www.ucl.ac.uk/drupal/site_ncl-disease/mutation-and-patient-database, December 21, 2020).
Clinical criteria for diagnosis and diagnostic confidence schemes have been used successfully in other neurologic disorders. The specific schemes have varied based on the uses for which they were designed. Some schemes were designed prior to discovery of the gene responsible for the condition, often motivated by a desire to define more homogeneous clinical phenotypes.19,29 For other conditions, such as Dravet syndrome, clinical diagnostic criteria define a syndrome which may be caused by different types of genetic variants.20 Still others were designed to increase certainty of diagnosis based on clinical criteria in diseases that are not primarily genetic.17,18 These schemes bear some similarities to each other but differ by specific criteria and purposes. Of course, diagnostic confidence schemes are not limited to neurologic disorders. A scheme that has some similarities to ours is the one developed for Alport syndrome (OMIM# 301050; 203780).30
Hierarchical diagnostic confidence schemes similar to ours may have utility for other genetic diseases but must be tailored to the specific disease. Many of these disorders present challenges for natural history studies, clinical trial design, and providing accurate anticipatory guidance based on newborn screening results. Newborn screening presents unique clinical challenges in some diseases for which positive newborn screen result does not provide the basis for accurate prediction of disease onset, disease course, or other phenotypic features.31 Specific relevant examples include Krabbe disease, (GALC; OMIM# 245200), metachromatic leukodystrophy (ARSA; OMIM# 250100), X-linked adrenoleukodystrophy (ABCD1; OMIM# 300100), and CLN2 disease (TPP1; OMIM# 204500).
Krabbe disease (KD) is an autosomal recessive disorder with multiple phenotypes.32 Diagnosis and confirmation are based on enzymatic or biochemical assay, genetic testing, or a combination. Natural history studies have shown that there is no simple genotype-phenotype correlation in KD, and there can be multiple phenotypes within a kindred.33,34 With inclusion of KD in newborn screening programs, diagnostic confidence is increasingly important.
MLD has been categorized into late-infantile, juvenile (early and late), and adult-onset forms.35 In general, earlier onset form progress more rapidly than later onset form, but type of initial symptom also predicts rate of disease progression.35 Age at symptoms onset of rate of progression are also generally related to the degree of lysosomal arylsulfatase A deficiency.36 Categorization and diagnostic confidence are important considerations for testing and developing disease-modifying treatments.
Specific genotype-phenotype correlations have not been described in X-ALD.37,38 Indeed, even monozygotic twins with X-ALD may have different phenotypes.39 Development of a valid hierarchical scheme could allow researchers to categorize individuals at risk based on susceptibility to develop disease and the expected phenotype while segregating them from individuals who are presymptomatic or who will never develop disease. Given current knowledge, such a scheme may not help predict the exact phenotype an individual will display given their genotype but may have prognostic potential in the future.
Diagnostic confidence for CLN2 disease, the classic late-infantile NCL, has increasing importance due to a recently approved enzyme replacement therapy and work toward inclusion in newborn screening.40 The most common presentation of CLN2 disease is in the late-infantile period. However, many individuals with CLN2 disease have a later onset disease.41,42 Stratification into these two groups based on a valid diagnostic confidence scheme has implications for surveillance, stratification in clinical trials, and timing of therapy initiation.41
For the many other genetic neurologic disorders, even with widespread use of chromosomal microarray analysis and NGS, diagnostic criteria and diagnostic confidence schemes have high potential importance. This is especially true when genetic testing reveals VUSs.
There are several limitations of our scheme. Although we actively recruited participants, most were members of a convenience sample ascertained at annual meetings of the Batten Disease Support and Research Association Family Conference and diagnostic information for many individuals was obtained from medical records. There was likely an ascertainment bias with under-sampling of more severely affected individuals because greater disease severity creates a higher burden of travel to study sites. Additionally, those with milder than expected phenotypes may not be identified, may not have participation in research as a priority, or may not attend family conferences. Although the collection of data over a long period of time has allowed us to accrue information on a relatively large sample of individuals with CLN3 disease, the amount and quality of diagnostic information available have varied over the years. Early in this work, genetic testing was largely limited to detection of the common deletion and several individuals had been diagnosed without the use of genetic testing. For the past 15 years, sequencing has been employed more routinely, which has provided more diagnostic precision. Our sample includes individuals with different types of diagnostic information. Even within specific diagnostic test classes, the quality and quantity of information available are variable.
Our data demonstrate the utility of a diagnostic confidence scheme for stratification of data obtained with different diagnostic tests. Increased recognition of genetic heterogeneity and phenotypic pleiotropy has shown that the combined use of genotype and phenotype information remains essential for complete disease characterization. This diagnostic confidence scheme will aid CLN3 disease researchers in navigating these challenges and ensuring that data represent the true phenotypic spectrum in CLN3 disease.
Synopsis.
A diagnostic confidence scheme for CLN3 disease (juvenile neuronal ceroid lipofuscinosis; Batten disease).
ACKNOWLEDGMENTS
The authors thank the participants and their families and the Batten Disease Support and Research Association for ongoing collaboration and support.
Funding information
Batten Disease Support and Research Association; Beyond Batten Disease Foundation; Eunice Kennedy Shriver National Institute of Child Health and Human Development, Grant/Award Number: P50HD103536; National Institute of Neurological Disorders and Stroke, Grant/Award Numbers: U01NS101946, K12NS066098, U54NS065768, R01NS060022; Noah’s Hope/Hope4Bridget; Our Promise to Nicholas Foundation; School of Medicine and Dentistry, University of Rochester, Grant/Award Number: Wilbur Smith Pediatric Neurology Fund
CONFLICT OF INTEREST
Ms. Masten declares no conflict of interest. Ms. Corre has received research support from the Child Neurology Foundation and the American Academy of Neurology. Dr. Paciorkowski has been funded by the NIH. Dr. Vierhile declares no conflict of interest. Dr. Adams has been funded by the NIH. She has received compensation as a consultant for Neurogene Inc, and Amicus Therapeutics, and as a faculty member and Delphi panel member for activities organized by National MPS Society. She has received research support through the University of Rochester from Beyond Batten Disease Foundation, Amicus Therapeutics, Neurogene, Inc., and Abeona Therapeutics. Dr. Vermilion has received research support from the American Academy of Neurology and the Tourette Association of America. Ms. Zimmerman declares no conflict of interest. Dr. Augustine has been funded by the NIH. She has received compensation as a consultant for Signant Health, BioMarin Pharmaceutical, Neurogene Inc, Taysha Gene Therapies, and Amicus Therapeutics. She serves on DSMBs for PTC Therapeutics. She has received research support through the University of Rochester from Beyond Batten Disease Foundation, Abeona Therapeutics, Amicus Therapeutics, and Neurogene, Inc. Dr. Mink has been funded by the NIH. He has received compensation as a consultant for Biogen, Applied Therapeutics, Neurogene, Taysha Gene Therapies, and Amicus Therapeutics. He serves on DSMBs for PTC Therapeutics. He has received research support through the University of Rochester from Beyond Batten Disease Foundation, Abeona Therapeutics, Amicus Therapeutics, and Neurogene Inc. None of the above entities were involved in study design, data collection, data analysis, decision to publish, or writing this manuscript.
Footnotes
ETHICS STATEMENT
This study was approved by the University of Rochester Research Subjects Review Board. Informed consent was obtained from a parent/legal guardian for each participant. This study was conducted in accordance with the Declaration of Helsinki.
Data Availability Statement
De-identified data will be provided upon request.
REFERENCES
- 1.Claussnitzer M, Cho JH, Collins R, et al. A brief history of human disease genetics. Nature. 2020;577(7789):179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marshall CR, Chowdhury S, Taft RJ, et al. Best practices for the analytical validation of clinical whole-genome sequencing intended for the diagnosis of germline disease. NPJ Genom Med. 2020;5:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Williams RE, Mole SE. New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses. Neurology. 2012; 79(2):183–191. [DOI] [PubMed] [Google Scholar]
- 4.Zhong N, Moroziewicz DN, Ju W, et al. Heterogeneity of late-infantile neuronal ceroid lipofuscinosis. Genet Med. 2000;2(6): 312–318. [DOI] [PubMed] [Google Scholar]
- 5.Augustine EF, Adams HR, Beck CA, et al. Standardized assessment of seizures in patients with juvenile neuronal ceroid lipofuscinosis. Dev Med Child Neurol. 2015;57(4):366–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kwon JM, Adams H, Rothberg PG, et al. Quantifying physical decline in juvenile neuronal ceroid lipofuscinosis (Batten disease). Neurology. 2011;77(20):1801–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marshall FJ, de Blieck EA, Mink JW, et al. A clinical rating scale for batten disease: reliable and relevant for clinical trials. Neurology. 2005;65(2):275–279. [DOI] [PubMed] [Google Scholar]
- 8.Ku CA, Hull S, Arno G, et al. Detailed clinical phenotype and molecular genetic findings in CLN3-associated isolated retinal degeneration. JAMA Ophthalmol. 2017;135(7):749–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kousi M, Lehesjoki AE, Mole SE. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum Mutat. 2012;33(1):42–63. [DOI] [PubMed] [Google Scholar]
- 10.The International Batten Disease Consortium. Isolation of a novel gene underlying batten disease, CLN3. Cell. 1995;82(6): 949–957. [DOI] [PubMed] [Google Scholar]
- 11.Haskell RE, Carr CJ, Pearce DA, Bennett MJ, Davidson BL. Batten disease: evaluation of CLN3 mutations on protein localization and function. Hum Mol Genet. 2000;9(5):735–744. [DOI] [PubMed] [Google Scholar]
- 12.Mirza M, Vainshtein A, DiRonza A, et al. The CLN3 gene and protein: what we know. Mol Genet Genomic Med. 2019;7(12):e859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anderson GW, Goebel HH, Simonati A. Human pathology in NCL. Biochim Biophys Acta. 2013;1832(11):1807–1826. [DOI] [PubMed] [Google Scholar]
- 14.Aula P, Rapola J, Andersson LC. Distribution of cytoplasmic vacuoles in blood T and B lymphocytes in two lysosomal disorders. Virchows Arch B Cell Pathol. 1975;18(4):263–271. [DOI] [PubMed] [Google Scholar]
- 15.Jarvela I, Mitchison HM, Munroe PB, O’Rawe AM, Mole SE, Syvanen AC. Rapid diagnostic test for the major mutation underlying batten disease. J Med Genet. 1996;33(12):1041–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Munroe PB, Mitchison HM, O’Rawe AM, et al. Spectrum of mutations in the batten disease gene, CLN3. Am J Hum Genet. 1997;61(2):310–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162–173. [DOI] [PubMed] [Google Scholar]
- 18.Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord. 2015;30(12):1591–1601. [DOI] [PubMed] [Google Scholar]
- 19.Ferner RE, Huson SM, Thomas N, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44(2):81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu YW, Sullivan J, McDaniel SS, et al. Incidence of Dravet syndrome in a US population. Pediatrics. 2015;136(5):e1310–e1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rothberg PG, Ramirez-Montealegre D, Frazier SD, Pearce DA. Homogeneous polymerase chain reaction nucleobase quenching assay to detect the 1-kbp deletion in CLN3 that causes batten disease. J Mol Diagn. 2004;6(3):260–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masten MC, Williams JD, Vermilion J, et al. The CLN3 disease staging system: a new tool for clinical research in batten disease. Neurology. 2020;94(23):e2436–e2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cialone J, Adams H, Augustine EF, et al. Females experience a more severe disease course in batten disease. J Inherit Metab Dis. 2012;35(3):549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuper WFE, van Alfen C, Rigterink RH, Fuchs SA, van Genderen MM, van Hasselt PM. Timing of cognitive decline in CLN3 disease. J Inherit Metab Dis. 2018;41(2):257–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mink JW, Augustine EF, Adams HR, Marshall FJ, Kwon JM. Classification and natural history of the neuronal ceroid lipofuscinoses. J Child Neurol. 2013;28(9):1101–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Filges I, Sparagana S, Sargent M, et al. Brain MRI abnormalities and spectrum of neurological and clinical findings in three patients with proximal 16p11.2 microduplication. Am J Med Genet A. 2014;164a(8):2003–2012. [DOI] [PubMed] [Google Scholar]
- 28.Zablotsky B, Black LI, Blumberg SJ. Estimated prevalence of children with diagnosed developmental disabilities in the United States, 2014–2016. NCHS Data Brief. 2017;291: 1–8. [PubMed] [Google Scholar]
- 29.Bruno MK, Hallett M, Gwinn-Hardy K, et al. Clinical evaluation of idiopathic paroxysmal kinesigenic dyskinesia: new diagnostic criteria. Neurology. 2004;63(12):2280–2287. [DOI] [PubMed] [Google Scholar]
- 30.Kashtan CE. Alport syndrome: achieving early diagnosis and treatment. Am J Kidney Dis. 2021;77(2):272–279. [DOI] [PubMed] [Google Scholar]
- 31.Kwon JM, Steiner RD. I’m fine; I’m just waiting for my disease: the new and growing class of presymptomatic patients. Neurology. 2011;77(6):522–523. [DOI] [PubMed] [Google Scholar]
- 32.Orsini JJ EM, Wasserstein MP, Caggana M. Krabbe Disease. GeneReviews® [Internet] 2000. Jun 19 [Updated 2018 Oct 11]; https://www.ncbi.nlm.nih.gov/books/NBK1238/. [Google Scholar]
- 33.Duffner PK, Barczykowski A, Kay DM, et al. Later onset phenotypes of Krabbe disease: results of the world-wide registry. Pediatr Neurol. 2012;46(5):298–306. [DOI] [PubMed] [Google Scholar]
- 34.Madsen AMH, Wibrand F, Lund AM, Ek J, Duno M, Ostergaard E. Genotype and phenotype classification of 29 patients affected by Krabbe disease. JIMD Rep. 2019;46(1):35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kehrer C, Elgün S, Raabe C, et al. Association of age at onset and first symptoms with disease progression in patients with metachromatic Leukodystrophy. Neurology. 2021;96(2):e255–e266. [DOI] [PubMed] [Google Scholar]
- 36.Shaimardanova AA, Chulpanova DS, Solovyeva VV, et al. Metachromatic leukodystrophy: diagnosis, modeling, and treatment approaches. Front Med (Lausanne). 2020;7:576221. 10.3389/fmed.2020.576221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith KD, Kemp S, Braiterman LT, et al. X-linked adrenoleukodystrophy: genes, mutations, and phenotypes. Neurochem Res. 1999;24(4):521–535. [DOI] [PubMed] [Google Scholar]
- 38.Takano H, Koike R, Onodera O, Sasaki R, Tsuji S. Mutational analysis and genotype-phenotype correlation of 29 unrelated Japanese patients with X-linked adrenoleukodystrophy. Arch Neurol. 1999;56(3):295–300. [DOI] [PubMed] [Google Scholar]
- 39.Di Rocco M, Doria-Lamba L, Caruso U. Monozygotic twins with X-linked adrenoleukodystrophy and different phenotypes. Ann Neurol. 2001;50(3):424. [DOI] [PubMed] [Google Scholar]
- 40.Fietz M, AlSayed M, Burke D, et al. Diagnosis of neuronal ceroid lipofuscinosis type 2 (CLN2 disease): expert recommendations for early detection and laboratory diagnosis. Mol Genet Metab. 2016;119(1–2):160–167. [DOI] [PubMed] [Google Scholar]
- 41.Kohan R, Carabelos MN, Xin W, et al. Neuronal ceroid lipofuscinosis type CLN2: a new rationale for the construction of phenotypic subgroups based on a survey of 25 cases in South America. Gene. 2013;516(1):114–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lourenco CM, Pessoa A, Mendes CC, et al. Revealing the clinical phenotype of atypical neuronal ceroid lipofuscinosis type 2 disease: insights from the largest cohort in the world. J Paediatr Child Health. 2021;57(4):519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
De-identified data will be provided upon request.