

Abstract
The combination of ion/ion chemistry with commercially available ion mobility/mass spectrometry systems has allowed rich structural information to be obtained for gaseous protein ions. Recently, the simple modification of such an instrument with an electrospray reagent source has allowed three-dimensional gas-phase interrogation of protein structures through covalent and non-covalent interactions coupled with collision cross section measurements. However, the energetics of these processes have not yet been studied quantitatively. In this work, previously developed Monte Carlo simulations of ion temperatures inside traveling wave ion guides are used to characterize the energetics of the transition state of activated ubiquitin cation/sulfo-benzoyl-HOAt reagent anion long-lived complexes formed via ion/ion reactions. The ΔH‡ and ΔS‡ of major processes observed from collisional activation of long-lived gas phase ion/ion complexes, namely collision induced unfolding (CIU), covalent bond formation, or neutral loss of the anionic reagent via intramolecular proton transfer, were determined. Covalent bond formation via ion/ion complexes was found to be significantly lower energy compared to unfolding and bond cleavage. ΔG‡ of activation of all three processes lie between 55 and 75 kJ/mol, easily accessible with moderate collisional activation. Bond formation is favored over reagent loss at lower activation energies, whereas reagent loss becomes competitive at higher collision energies. Though ΔG‡ between CIU of a precursor ion and covalent bond formation of its ion/ion product complex are comparable, our data suggest covalent bond formation does not require extensive isomerization.
Keywords: Native mass spectrometry, Ion/Ion reactions, Ion mobility, CIU, Activation Energy
Graphical Abstract
Introduction
The production of multiply charged gas phase ions from biomolecules by electrospray (ESI)1 allows mass spectrometers (MS) to be used as reaction vessels for ion/ion2–3 and ion/electron reactions4. Importantly, product ions of these reactions retain charge, necessary for mass spectral detection. The analytical applications of these reactions are numerous5–6, including spectral deconvolution and charge manipulation, mixture analysis, structural determination, and gas phase sequencing. Ion/ion reactions have also been used to synthesize long-lived complexes in the gas phase. By using bifunctional reagents that form a long-lived complex (due to a relatively high energy dissociation barrier via, e.g., gas phase salt bridge formation, that allows for significant collisional cooling7) and contain moieties that form covalent bonds, such as aldehydes8 or activated esters9–10, new covalent bonds can be efficiently formed. More recently, covalent bond formation via ion/ion reactions has been used to characterize gas-phase tertiary structures of protein ions, via covalent chemical crosslinking11, covalent labeling12, and salt bridge sensitive covalent reactions13.
Careful measurements using ion mobility-mass spectrometry (IM-MS) approaches have shown that a strong correlation exists between gaseous and solution protein structures14–23, especially when unintentional ion heating via energetic collisions or other mechanisms is minimized, and this is supported by molecular dynamics simulations of proteins during and after the electrospray process24–25. These observations make gas phase measurements of protein structures attractive due to the sensitivity, speed, and specificity of MS26. Since the timescale of MS experiments dictates that chemistry of gaseous ions is typically under kinetic control, relevant protein structural information can be obtained if isomerization to extended conformations (via, e.g., activating collisions) is minimized or takes place only after structurally sensitive studies are conducted. Therefore, gas-phase covalent chemistry can be effectively used for protein structural determination, but only if proteins do not lose their overall structure under the timescales and energies used for covalent bond formation.
Previously, the McLuckey group used a combination of quantum mechanical calculations and ion/ion reactions coupled with ion collisional activation through dipolar DC in a collision quadrupole to study the energetics and kinetics involved in covalent bond formation via gas phase long-lived complexes10, 27. They determined that if the electrostatic group (e.g., a sulfonate) necessary for long-lived complex formation remained on the analyte ion after covalent reaction, formation of the covalent bond becomes the rate limiting step. By varying the timescale and magnitude of collisional activation, conditions in which the main product is covalent bond formation can be implemented. Intramolecular proton transfer to and neutral loss of the entire reagent becomes favorable at higher collision energies, which the authors attributed to a “looser” transition state (i.e., a more positive activation entropy) for reagent loss (bond dissociation) than covalent bond formation. Additionally, they determined the effects of using a more reactive reagent ion, sulfo-benzoyl-1-hydroxy-7-azabenzonitrazole (sulfo-benzoyl-HOAt) than sulfo-benzoyl-N-hydroxysuccinimide (sulfo-benzoyl-NHS), finding that sulfo-benzoyl-HOAt formed a higher ratio of covalent products to reagent loss than sulfo-benzoyl-NHS under similar activation conditions. Density functional theory calculations showed that, using a model system of methylamine and acetyl-HOAt versus acetyl-NHS, HOAt gas phase reactions had a 12.6 kJ/mol lower activation barrier than NHS reactions.
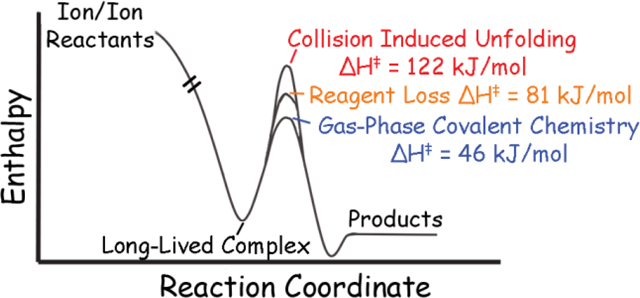
In this work, we applied the recently developed Monte Carlo simulation approach to determining ion temperatures of polypeptides in traveling wave ion guides28–29 to determine the activation thermochemistry for covalent reactions of long-lived complexes formed by ion/ion reactions of native like ubiquitin ions with sulfo-benzoyl-HOAt anions. The choice of ubiquitin ions was made due to the relatively low number of degrees of freedom30 in ubiquitin ions versus most proteins as well as the lack of disulfide bonds, making ubiquitin more susceptible to collision induced unfolding (CIU)31 processes, and thus, a more sensitive “thermometer” ion. Sulfo-benzoyl-HOAt anions were used due to their inherent reactivity (relatively low activation energies) and facile formation by negative ion electrospray. Herein, we have made the first quantitative measurements of activation enthalpy, entropy, and free energy for covalent bond formation, reagent loss, and CIU via ion/ion reactions with sulfo-benzoyl-HOAt anions and collisional activation (Scheme 1). By measuring these activation energies, a much clearer picture of fates of ion/ion and collisional activation products has emerged.
Scheme 1.

A summary of the possible reactions and products that are formed from ion/ion reactions followed by collisional activation. Covalently modified (red circle) and charge reduced species of the protein cation are formed, which can either remain in their native-like state or be unfolded/refolded (twisted star) by CIU processes. Additionally, unreacted protein is still subject to CIU during the activation step. The activation energies for the products labeled I, II, and III were measured in this study.
Experimental Section
Materials
Ubiquitin from bovine erythrocytes, myoglobin from horse heart, and ammonium acetate were purchased from Sigma-Aldrich. Acetonitrile, formic acid, and dimethylformamide (DMF) were purchased from Fisher Scientific. 1-Hydroxy-7-azabenzonitrazole (HOAt) was purchased from TCI America (Portland, OR). 1-Ethyl-3-(3-(dimethylamino)-propyl) carbodiimide hydrochloride (EDC) was purchased from Thermo Scientific (Rockford, IL). 3-Sulfobenzoic acid monosodium salt was purchased from Alfa Aesar (Ward Hill, MA). Water was obtained from a Milli-Q Millipore A10 water purification system at a resistivity of 18 MΩ•cm or greater. Sulfo-benzoyl HOAt was synthesized by combining EDC, sulfobenzoic acid, and HOAt at 100 mM in DMF and diluting 100-fold into acetonitrile. Ubiquitin was dissolved into 10 mM aqueous ammonium acetate. Myoglobin was dissolved into 50/50/0.1 vol/vol/vol water/acetonitrile/formic acid to produce apo-myoglobin ions. Samples were used without further purification.
Ion Mobility-Mass Spectrometry
All experiments were performed on a Synapt G2-Si quadrupole/ion mobility/time-of-flight mass spectrometer (Waters Corp.) with a nanolockspray source modified for positive electrospray/negative electrospray ion/ion reactions (Figure 1) 32. Ubiquitin was loaded into borosilicate capillaries pulled into nanoelectrospray tips by a Flaming-Brown micropipette puller (Sutter Instruments) and ionized by the application of ~1 kV by a platinum wire inserted into the capillary in contact with the electrospray solution.
Figure 1.

Illustration of the two major experiment types, ion/ion reactions with collisional activation to form covalent products (A) and CIU (B). Collisional activation occurs in the transfer cell for ion/ion reactions and collisional activation occurs in the trap cell for CIU.
For ion/ion reactions, anions were infused at 2 ml/min into the nanolockspray reference emitter and ionized at ~−1.5 kV. ETD mode was used to inject anions and cations sequentially into the helium-filled trap cell, where one of the switching trap dc potentials was used to trigger an external switching electrospray power supply (GAA Custom Electronics) to synchronize ionization of each polarity with suitable lens voltages to transmit the ions to the trap cell (10–20 ml/min helium flow rates). ETD refill times and refill intervals were each 1 second. Products of the ion/ion reactions were either collisionally activated entering the helium and mobility cells or by transfer collision energy (transfer cell argon flow rate of 1 ml/min, 300 m/s, 2 V traveling wave) for determining activation energies. The helium cell helium flow rate was 100 ml/min or greater, and the mobility cell nitrogen flow rate was 50 ml/min with a 40 V traveling wave with the wave velocity ramped from 3400 to 600 m/s over the course of the mobility separation.
CIU was performed in CID mode (argon in the trap cell, 6 ml/min flow rate, 300 m/s, 2 V traveling wave) by increasing the trap CE by increments of 0.5 V. The helium cell flow rate was set to the maximum value of 200 ml/min for CIU experiments since the unreacted 6+ ubiquitin ion was observed to unfold to a partially unfolded state at flow rates below 180 ml/min. This is likely due to less thermalization with helium in the helium cell prior to the nitrogen-containing drift cell at lower helium flow rates as the ions pass through the potentially activating pressure gradient (~0.04 mbar to 3 mbar). The mobility cell nitrogen flow rate was again 50 ml/min with a 40 V traveling wave. Under these conditions, with lower trap CE voltages, unintentional CIU was minimized (vide infra). All data was taken in triplicate measurements.
A previously established procedure for traveling wave experimental collision cross section (CCS) calibration was used33, with reported CCS values as the average of triplicate measurements. Ubiquitin ion mobility spectra were calibrated against denatured apo-myoglobin nitrogen cross sections from the literature34.
Monte Carlo Simulations
The methodology behind the Monte Carlo simulations has been previously described28–29. Briefly, the probability of ion/neutral collisions at each time point was calculated according to ion mean free paths as a function of ion CCS and gas pressures in the trap and transfer cells. The calculations of potentials in these cells were based on calculations from Mortensen et al35. Timepoints were sampled twenty times during each collision interval for the entire length of the trap or transfer cell. The fraction of available kinetic energy converted into internal energy, x, was determined considering the Maxwell-Boltzmann distribution of argon velocities at 298 K, the reduced mass of the ion-neutral collision pair, and the velocity of the ion as a function of mass and DC and traveling wave potentials. Effective temperatures of ions were determined by the following relationship:
| (1) |
Teff is the effective ion temperature, T0 is 298 K, z is the ion charge state, e is the elementary charge in coulombs, V is the collision voltage, Natoms is the number of atoms in the protein ion, and kB is the Boltzmann constant. Temperatures and total activation times were extracted from the simulations and used for calculating activation energies.
Data Analysis
Ion/ion reaction data were analyzed directly in MassLynx 4.2 (Waters Corp.) with mass spectral peak intensity ratios of the ion/ion complex to the sum of ion/ion complex, covalent product, and proton transfer peak intensities extracted for Eyring analyses. CIU data were analyzed with CIUSuite 236, using default values for smoothing and gaussian fitting. Peak areas from the different gaussians were used to determine the ratio of the intensity of the peak representing the preactivated distribution to the total intensity of the ion mobility distribution (i.e., the relative abundance of the most compact peak). The natural logarithms of the ratio of the negative natural logarithm of the intensity ratios to effective temperature were plotted against the reciprocal of the product of the effective temperature and the Boltzmann constant. The activation enthalpy is determined from the slope of the plot, and the activation entropy is extracted from the intercept from an algebraic rearrangement of the Eyring equation:
| (2) |
[R] represents the intensity of the reactant (the long-lived ion/ion complex or most compact peak for CIU), [P] represents the intensities of ion/ion or CIU products, ΔH‡ is the activation enthalpy, ΔS‡ is the activation entropy, Teff is effective ion temperature, t is the reaction time (collisional heating time), kB is the Boltzmann constant, and h is Planck’s constant. T and t are estimated by Monte Carlo simulations28–29 that consider the mass of the reactant, the pressure of background gas, the DC and traveling wave voltages, and the lengths of the trap, helium, mobility, and transfer cells.
Results and Discussion
Activation Energies of the Transition States of Covalent and Proton Transfer Reactions
Covalent reactions and proton transfer gas phase ion/ion chemistries between multiply charged cations and “sticky” anionic reagents are known to proceed through the formation of long-lived intermediates stabilized by strong electrostatic interactions5, 8, 10, 27, 37. Therefore, reaction rates of the covalent modification reactions, observed via neutral loss of the leaving group and proton transfer from the polypeptide to the reagent, can be readily probed by slow-heating methods10, 27. The reagent used for this study was sulfo-benzoyl HOAt, shown to be favored on kinetic grounds to proton transfer with only a small van der Waals barrier between the nascent covalent product complex and loss of the HOAt leaving group10. We have recently used this reagent to probe gas phase structures of ubiquitin cations formed from aqueous and low pH, methanolic solutions12, 38. Since it is of interest to differentiate between gas phase structures resulting directly from dehydration of the ions from droplets in the electrospray process and structures produced by activation and refolding of proteins into additional structures (i.e., CIU), the activation enthalpies, entropies, and free energies for the covalent and proton transfer reactions are valuable for characterizing the kinetics and mechanisms of these reactions versus CIU processes.
The ion/ion reaction products of ubiquitin 6+ ions and sulfo-benzoyl HOAt 1− ions formed in the trap cell with the application of various collision energies in the transfer cell are shown in Figure 2. Lower collision energies, as exemplified in Fig. 2 A, (20 V transfer collision energy) result in minimal formation of covalent reaction products. However, when the collision energy is increased to 50 V (Fig. 2 B), substantial covalent product [M+6H+SB]5+ nominal m/z 1777 is observed for both single and double additions of the reagent with the neutral loss of the HOAt leaving group from the ion/ion product complex [M+6H+SB-HOAT]5+ at nominal m/z 1751. Finally, at 65 V transfer collision energy (Fig. 2 C), the main product from a single addition of the reagent is neutral loss of the entire reagent (nominally proton transfer), while for the double addition peak, up to two covalent reactions are observed. It is clear from these experiments and previous studies10, 27 that the covalent product is favored under kinetic grounds, while the proton transfer product is entropically favored, as is likely from a bond-forming reaction versus a bond-breaking reaction.
Figure 2.

Reaction of ubiquitin (star) 6+ ions with sulfo-benzoyl (red circle) HOAt (green circle) 1− ions with a transfer collision energy of (A) 20 V, (B) 50 V, and (C) 65 V.
Quantifying the changes in enthalpy and entropy from the electrostatically bound complex to the transition states for the two reactions was enabled by the previously described Monte Carlo simulation approach to determining ion temperatures and reaction times for protein ions traversing traveling wave ion guides28–29. Under the pressures and voltages used for the ion/ion studies, the reactant ion populations rapidly reach a steady-state temperature near which they remain until detection. For ubiquitin 5+ ions, the fraction of kinetic energy converted into internal energy is 0.22, and the activation time in the transfer cell is 0.34 ms, and for the 6+ ions, the fraction of kinetic energy converted into internal energy is 0.11, and the activation time is 0.46 ms. The differences in efficiencies and times are due to the m/z of the ions as well as the transfer cell pressures measured on the days on which measurements were made. The resulting Eyring plots (ln(k/T) vs 1/kBT) for products formed from the 5+ and 6+ ion/ion electrostatic complexes are shown in Figure 3. For the complex formed from the 5+ ubiquitin ion (Fig. 3 A), there is a linear region defined by a single slope, indicating that for the collision energies used, the major observed reaction was covalent bond formation without significant competition from the proton transfer pathways. The ion/ion complex formed from reaction with the 6+ ion (Fig. 3 B) shows two linear regions, one with a slope of smaller magnitude (black), and one at higher temperatures with a slope of larger magnitude (red), indicating that at higher collision energies, proton transfer becomes a dominant process. The extracted activation energies are given in Table 1. ΔH‡ of covalent bond formation for both charge states are much less than the ΔH‡ of proton transfer to the reagent and its subsequent neutral loss, consistent with the observation in Figure 2 that proton transfer only becomes competitive at higher activation energies. At the highest energies (e.g., Fig. 2 C), proton transfer becomes favorable as the transition state is more disordered than the transition state for covalent bond formation, evidenced by a positive activation entropy for proton transfer versus a negative activation entropy for covalent bond formation. This reflects the ability in previous experiments to tune the reaction conditions in the trap traveling wave cell appropriately12, 32 such that covalent bond formation was the only observed ion/ion product, ensuring that intramolecular proton transfer to sulfo-benzoyl HOAt does not measurably occur, in agreement with the measurements here that covalent bond formation is the kinetically favored product. To ensure that our results were significant (i.e., that proton transfer is significantly higher in activation enthalpy and entropy than covalent bond formation), we explored the sensitivity of activation energies on changes in the efficiency of conversion of collision energies into internal energies (x, vide supra) and overall activation time. Previous estimates of the maximum possible uncertainty in activation times were t +/− 25% and estimates of the maximum possible uncertainty in energy conversion efficiency are x +/− 0.0228–29. According to the Eyring equation (Equation 2), the activation enthalpy (and effective temperature) does not depend on the heating time, but the activation entropy and free energy do. All three activation energies are dependent on the efficiency of the conversion of kinetic energy to internal energy. The uncertainties in Table 1 and 2 are given as the square root of the sum of the square of +/− half of the ranges obtained for each ΔH‡ and ΔS‡ and the square of the standard error for each ΔH‡ and ΔS‡ determined from triplicate measurements on three separate days. The effective temperature uncertainty is given by +/− half of the average effective temperature range across the various x values. The uncertainty in ΔG‡ is the square root of the sum of the squared ΔH‡ and TΔS‡ uncertainties.
Figure 3.

Eyring plots for collisional activation of electrostatic ion/ion complexes formed by reaction with (A)5+and (B)6+charge states of ubiquitin. The black regression line is for covalent bond formation and the red line is for proton transfer.
Table 1.
Activation Energies Covalent Bond Formation and Proton Transfer
| Covalent Bond Formation | Proton Transfer | |||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| Ion | ΔH‡, kJ/mol | ΔS‡, J/mol•K | ΔG‡, kJ/mol | ΔH‡, kJ/mol | ΔS‡, J/mol•K | ΔG‡, kJ/mol |
|
| ||||||
| [M+5H+SB-HOAT]4+ | 29.0 ± 1.9 | −105 ± 10 | 65.8 ± 3.5 | - | - | - |
| [M+6H+SB-HOAT]5+ | 46.0 ± 6.5 | −65 ± 25 | 68.8 ± 10.3 | 81.1 ± 9.5 | 27 ± 33 | 70.3 ± 16.8 |
Table 2.
Activation Energies CIU
| Ion | ΔH‡, kJ/mol | ΔS‡, J/mol-K | ΔG‡, kJ/mol |
|---|---|---|---|
|
| |||
| [M+5H]5+ | 114 ± 14 | 181 ± 48 | 57.3 ± 20.5 |
| [M+6H]6+ | 122 ± 18 | 192 ± 60 | 59.7 ± 26.9 |
Interestingly, the two charge states’ activation enthalpies are different, despite the fact that both ions have similar overall gas phase structures (as determined by their CCS, vide infra) and both result in the same residues, lysine 29 and arginine 54, being labeled12. Additionally, the significantly higher activation energy of the neutral loss of the reagent channel (proton transfer) than covalent bond formation likely precludes the necessity of the fragmentation of the sulfonate ionic bond to the protein prior to the formation of the covalent product. A possible explanation is that the extra coulombic repulsion from the sixth charge increases the energetic barrier for the reaction. The exploration of this difference in activation energies warrants further experimental and computational investigation.
Collision Induced Unfolding Energies of Native-like Ubiquitin Cations
To provide further insight into the mechanism of covalent reactions and proton transfer facilitated by long-lived, electrostatically bound complexes generated via ion/ion reactions, activation energies of CIU transitions for 5+ and 6+ ions of ubiquitin electrosprayed from native-like conditions were measured. CIU “fingerprints” for the ubiquitin 5+ and 6+ charge states are shown in Figure 4 A and B, respectively, detailing structural transitions as a function of the trap collision energy. The transitions are labeled with calibrated nitrogen collision cross sections (nm2). CIU were obtained using collision energies up to 25 V; however, the CCS did not significantly change between 12 V and 25 V, so the plots in Figure 4 are truncated at 12 V for clarity of the transition regions. The 5+ charge state (Fig. 4 A) is resistant to refolding into a more elongated structure upon activation. The higher energy conformer family is only 3% larger than the CCS measured at lower energies and is 2% smaller than the lowest energy conformer family measured for the 6+ ion (Fig. 4 B). However, the higher energy conformer family for the 5+ charge state is present even at the lowest activation energies, evidence of multiple compact conformers that reflect individual solution conformations that slightly compact upon lifting into the gas phase. This has been previously observed using ion mobility studies of ubiquitin electrosprayed from conditions that are known to stabilize the native state14, 20, 39. Therefore, the higher energy conformer family likely also reflects a slightly compacted solution state. The robustness of the lowest charge states of proteins in the gas-phase to unfolding23, 40–42 makes them particularly good candidates to probe via our ion/ion covalent chemistry, and native MS methods in general.
Figure 4.

CIU fingerprints of ubiquitin (A) 5+and (B) 6+charge states. CCS values determined by calibration with nitrogen values from denatured myoglobin.
In contrast, the 6+ charge state unfolds from a compact state into a slightly unfolded state upon collisional activation (Fig. 4 B). For the 6+ charge state, a partially unfolded state emerges at higher collision energies. Recently, 1 ms, 300 K MD simulations for the ubiquitin 6+ charge state undertaken by the Konermann group revealed that a partially unfolded state can be populated which then refolds back into the native-like state25. For example, one of the trajectories of their simulation illustrated that the main differences between the partially unfolded state and native-like state is that the two N-terminal beta strands (before proline 19) are rotated slightly away from the main alpha helix, and that the beta turn (including alanine 46 and glycine 47) is rotated away from the core of the protein. However, the overall fold of the protein remains intact. Thus, for the 5+ and 6+ charge states, neither CIU transition results in full loss of the main solution structure. If CIU of the charge states were to precede covalent bond formation, these “unfolded” structures probed by ion/ion chemistry maintain almost all the solution structure.
Since reduction in overall charge stabilizes the compact conformers of gas-phase proteins40, 42–43, and the presence of the bound sulfonate anion likely changes the proton transfer potential energy surface of the protein, the CIU activation energies of the bare protein ions cannot be directly compared to the unfolding energetics of long-lived ion/ion complexes. The instrument geometry prohibits the determination of activation energies from CIU studies of ion/ion reaction covalent products. However, key mechanistic differences in these processes can be inferred by comparing their energies and entropies qualitatively. The effective temperatures at various trap collision energies were again determined by simulation of the efficiency of energy deposition and reaction times and used to construct Eyring plots for CIU of the 5+ and 6+ charge states to determine activation enthalpies and entropies.
The first major difference between activation thermochemistry of CIU (Table 2) and covalent bond formation via ion/ion reactions is the positive activation entropy for CIU. This is consistent with previous measurements of activation entropies during CIU29, and indicates that the transition state between the folded and partially folded structures is more disordered than the more compact, native-like state. Thus, the transition states for covalent reactions from the ion/ion long-lived complex are very different than for collision-induced unfolding and dissociation (i.e., proton transfer), reflecting bond forming versus bond breaking processes.
Though, as expected the activation enthalpy for CIU, a non-covalent bond breaking reaction, is much higher than the enthalpy required for covalent bond formation, the mechanism of reaction is very different, indicated, again, by the large differences in activation entropy. The differences in energies between Gibbs free energies of activation (calculated using the average activation effective temperature for each study) for CIU and covalent bond formation are not significant. This suggests that on the timescale of a mass spectrometric experiment, CIU of the unreacted precursors is competitive with covalent bond formation from the product ion/ion complexes. However, the activation enthalpy and entropy measurements alone cannot inform on whether these processes are competing or consecutive. For example, covalent modification may only proceed following an intermediate formed by CIU or not require any unfolding at all. Therefore, the ion/ion reactions were studied without the application of activation energy (i.e., under conditions where Teff ≈ 300 K).
CIU, Covalent Bond Formation, and Proton Transfer without Collisional Energy
Figure 5 A shows the ion/ion reaction product spectrum using the gentlest settings that allow for transmission of the ubiquitin 6+ ion in ETD mode. The helium cell helium flow rate was 200 ml/min, the mobility cell nitrogen flow rate was 50 ml/min, and the transmission cell argon flow rate was 0.5 ml/min. The voltage gradient from the trap cell to the mobility cell, where activation is known to occur44, was minimized. The voltage difference between the trap exit and helium cell entrance was only 7 V, and the voltage difference between the helium cell exit and the mobility cell bias was only 10 V. Under these conditions, the ion/ion reaction is not as efficient, since there is less cation/anion overlap with smaller voltage gradients, as the anions can more easily escape the trap cell with a less positive potential between the trap and helium cell (in other words, a smaller DC barrier to anions). However, upon careful inspection, the covalent reaction product is still formed (Fig. 5 B) without significant CIU. As the ion/ion reaction occurs prior to injection into the helium and mobility cells, the extent of activation will be less than for the precursor unreacted 6+ charge state, since the reduction of charge reduces the lab frame collision energy at the same potentials applied to the electrodes. The mobility spectrum for the unreacted 6+ ion (Fig. 5 C) is very similar to the spectrum obtained from a nitrogen drift tube ion mobility/mass spectrometer34, suggesting CIU is not occurring under these conditions or that the ions have cooled back into a similar conformation to the starting conformation. Figure 5 D shows the ion/ion product complex mobility spectrum, with the inset showing the spectrum from covalent product formation. The CCS of the products are slightly smaller (~2%) than the 6+ precursor, and in between the CCS of the two major conformer families observed for the 5+ unreacted ions, clearly illustrating that no major structural change is required to generate the covalent product. Thus, the observation of the covalent product without significant activation and the differences in reaction mechanisms between the ion/ion processes and CIU implied by the very different activation enthalpies and entropies suggest that covalent bond formation via gas phase ion/ion reactions are a useful probe of gas phase protein structures after electrospray from aqueous conditions.
Figure 5.

Ion/ion reaction of ubiquitin 6+with sulfo-benzoyl HOAt anions under low energy conditions. (A) Mass spectrum of the reaction. (B) 10x zoom of the product of a single ion/ion reaction showing the formation of a covalent product. (C) Mobility spectrum of the unreacted 6+ions. (D) Mobility spectra of the ion/ion product complex and (inset) the covalent reaction product.
A source of internal energy from the protein cations that could possibly lead to rearrangement, fragmentation, or other reactions is the charge recombination energy from the sulfonate and proton. The proton affinity of hydrogen sulfate, a surrogate for sulfonate, is 1338 kJ/mol45, so the ion/ion reaction is expected to be very exothermic. Acid/base chemistry performed by ion/ion reactions with protein cations and stable anions do not show evidence for bond dissociation, even for fragmentation of non-covalent bonds, such as heme loss from myoglobin46, which has been shown by blackbody infrared radiative dissociation47 and collisional activation in a traveling ion guide with temperatures modeled by Monte Carlo simulations29 to be approximately 90 kJ/mol. The enthalpy of reaction for proton transfer from myoglobin through a hopping mechanism to a reagent anion is ~400 – 800 kJ/mol (the enthalpy difference between the proton affinity of the anion and the protein). The authors’ rationale for not observing heme loss was the high helium collision rate with the bath gas (~.001 mbar), where there are roughly 1 million thermalizing collisions per second that remove the energy released from the ion/ion reaction faster than dissociation occurs. Another explanation is that most of the proton transfer energy is partitioned into the neutral reagent instead of into the protein43. As the reactions investigated here proceed through the condensation of the protein cations and anion reagents into a single product, all of the energy released from the highly exothermic ion/ion reaction will be deposited into the product ion7. Again, the observed ion/ion product complex does not show unfolding. Thus, the energy produced by the exothermic ion/ion reaction is rapidly removed, likely via collisions with helium background gas (roughly 0.04 mbar) or other processes, before unfolding is observed. Therefore, it is unlikely that the energy released from ion/ion recombination is available for the covalent reaction, proton transfer, CIU, or other chemical processes.
Conclusions
Observations that gas phase, three-dimensional structural data from ion/ion reactions with proteins electrosprayed from native-like and other solution conditions strongly correlate with X-ray crystal structures, molecular modeling, and CCS measurements have been experimentally grounded by the measurement of transition state thermodynamics. We have described three possible outcomes for protein ions participating in ion/ion reactions with reagents that can form covalent bonds: bond formation, collision induced unfolding, and neutral loss of the reagent. The results from measurements of the free energies of activation show that CIU of the unreacted precursor ion and covalent bond formation via the ion/ion product long-lived complex are competitive under gentle activation conditions. At higher energies, neutral loss of the reagent via intramolecular proton transfer becomes competitive. The transition states for covalent bond formation and CIU/proton transfer are markedly different, illustrated by the relatively low activation enthalpy and negative activation entropy for covalent bond formation versus the relatively high enthalpies (>> 60 kJ/mol) and positive entropies for CIU/proton transfer. This coupled with the evidence of small amounts of covalent bond formation without the application of activation energy much beyond thermal energies suggest that CIU is not a rate-determining step for covalent bond formation, in agreement with the fact that data derived from gas phase covalent labeling of compact protein ions correlates most strongly with compact structures. In sum, our results suggest that gas phase ion/ion chemistry will be a useful probe of protein three-dimensional structure. The approach that has been developed will also have high utility in the future for studying transition state energetics of all kinds of chemical reactions that proceed via long-lived complex formation.
Supplementary Material
Acknowledgement
This work was supported by the National Institutes of Health (NIH) under Grant R21GM134408–01 (IKW), the Indiana University-Purdue University Indianapolis School of Science (IKW), and an American Society for Mass Spectrometry Research Award (JSP).
Footnotes
Associated Content
• Plots of covalent bond formation versus reagent loss, Eyring plots for CIU, and a table of parameters and results of the Monte Carlo simulations.
References
- 1.Fenn JB, Electrospray Wings for Molecular Elephants (Nobel Lecture). Angew. Chem. Int. Ed 2003, 42 (33), 3871–3894. [DOI] [PubMed] [Google Scholar]
- 2.McLuckey SA; Stephenson JL, Ion ion chemistry of high-mass multiply charged ions. Mass Spectrom. Rev 1998, 17 (6), 369–407. [DOI] [PubMed] [Google Scholar]
- 3.Syka JE; Coon JJ; Schroeder MJ; Shabanowitz J; Hunt DF, Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. U. S. A 2004, 101 (26), 9528–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zubarev RA; Kelleher NL; McLafferty FW, Electron Capture Dissociation of Multiply Charged Protein Cations. A Nonergodic Process. J. Am. Chem. Soc 1998, 120 (13), 3265–3266. [Google Scholar]
- 5.Foreman DJ; McLuckey SA, Recent Developments in Gas-Phase Ion/Ion Reactions for Analytical Mass Spectrometry. Anal. Chem 2020, 92 (1), 252–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zubarev RA, Electron-capture dissociation tandem mass spectrometry. Curr. Opin. Biotechnol 2004, 15 (1), 12–6. [DOI] [PubMed] [Google Scholar]
- 7.McLuckey SA; Herron WJ; Stephenson JL Jr; Goeringer DE, Cation Attachment to Multiply Charged Anions of Oxidized Bovine Insulin A-chain. J. Mass Spectrom 1996, 31 (10), 1093–1100. [DOI] [PubMed] [Google Scholar]
- 8.Han H; McLuckey SA, Selective covalent bond formation in polypeptide ions via gas-phase ion/ion reaction chemistry. J. Am. Chem. Soc 2009, 131 (36), 12884–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mentinova M; McLuckey SA, Covalent Modification of Gaseous Peptide Ions with N-Hydroxysuccinimide Ester Reagent Ions. J. Am. Chem. Soc 2010, 132 (51), 18248–18257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bu J; Peng Z; Zhao F; McLuckey SA, Enhanced Reactivity in Nucleophilic Acyl Substitution Ion/Ion Reactions Using Triazole-Ester Reagents. J. Am. Soc. Mass. Spectrom 2017, 28 (7), 1254–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Webb IK; Mentinova M; McGee WM; McLuckey SA, Gas-phase intramolecular protein crosslinking via ion/ion reactions: ubiquitin and a homobifunctional sulfo-NHS ester. J. Am. Soc. Mass. Spectrom 2013, 24 (5), 733–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carvalho VV; See Kit MC; Webb IK, Ion Mobility and Gas-Phase Covalent Labeling Study of the Structure and Reactivity of Gaseous Ubiquitin Ions Electrosprayed from Aqueous and Denaturing Solutions. J. Am. Soc. Mass. Spectrom 2020, 31 (5), 1037–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pitts-McCoy AM; Harrilal CP; McLuckey SA, Gas-Phase Ion/Ion Chemistry as a Probe for the Presence of Carboxylate Groups in Polypeptide Cations. J. Am. Soc. Mass. Spectrom 2019, 30 (2), 329–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wyttenbach T; Bowers MT, Structural Stability from Solution to the Gas Phase: Native Solution Structure of Ubiquitin Survives Analysis in a Solvent-Free Ion Mobility–Mass Spectrometry Environment. The Journal of Physical Chemistry B 2011, 115 (42), 12266–12275. [DOI] [PubMed] [Google Scholar]
- 15.Seo J; Hoffmann W; Warnke S; Bowers MT; Pagel K; von Helden G, Retention of Native Protein Structures in the Absence of Solvent: A Coupled Ion Mobility and Spectroscopic Study. Angew. Chem. Int. Ed. Engl 2016, 55 (45), 14173–14176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li J; Taraszka JA; Counterman AE; Clemmer DE, Influence of solvent composition and capillary temperature on the conformations of electrosprayed ions: unfolding of compact ubiquitin conformers from pseudonative and denatured solutions11Dedicated to Professor Michael T. Bowers on the occasion of his 60th birthday. Int. J. Mass spectrom 1999, 185–187, 37–47. [Google Scholar]
- 17.Shi H; Pierson NA; Valentine SJ; Clemmer DE, Conformation Types of Ubiquitin [M+8H]8+ Ions from Water:Methanol Solutions: Evidence for the N and A States in Aqueous Solution. The Journal of Physical Chemistry B 2012, 116 (10), 3344–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Silveira JA; Fort KL; Kim D; Servage KA; Pierson NA; Clemmer DE; Russell DH, From solution to the gas phase: stepwise dehydration and kinetic trapping of substance P reveals the origin of peptide conformations. J. Am. Chem. Soc 2013, 135 (51), 19147–53. [DOI] [PubMed] [Google Scholar]
- 19.Shi H; Atlasevich N; Merenbloom SI; Clemmer DE, Solution Dependence of the Collisional Activation of Ubiquitin [M + 7H]7+ Ions. J. Am. Soc. Mass. Spectrom 2014, 25 (12), 2000–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi H; Clemmer DE, Evidence for two new solution states of ubiquitin by IMS-MS analysis. The journal of physical chemistry. B 2014, 118 (13), 3498–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.El-Baba TJ; Woodall DW; Raab SA; Fuller DR; Laganowsky A; Russell DH; Clemmer DE, Melting Proteins: Evidence for Multiple Stable Structures upon Thermal Denaturation of Native Ubiquitin from Ion Mobility Spectrometry-Mass Spectrometry Measurements. J. Am. Chem. Soc 2017, 139 (18), 6306–6309. [DOI] [PubMed] [Google Scholar]
- 22.Ruotolo BT; Robinson CV, Aspects of native proteins are retained in vacuum. Curr. Opin. Chem. Biol 2006, 10 (5), 402–408. [DOI] [PubMed] [Google Scholar]
- 23.Zhong Y; Han L; Ruotolo BT, Collisional and Coulombic unfolding of gas-phase proteins: high correlation to their domain structures in solution. Angew. Chem. Int. Ed. Engl 2014, 53 (35), 9209–12. [DOI] [PubMed] [Google Scholar]
- 24.McAllister RG; Metwally H; Sun Y; Konermann L, Release of Native-like Gaseous Proteins from Electrospray Droplets via the Charged Residue Mechanism: Insights from Molecular Dynamics Simulations. J. Am. Chem. Soc 2015, 137 (39), 12667–12676. [DOI] [PubMed] [Google Scholar]
- 25.Bakhtiari M; Konermann L, Protein Ions Generated by Native Electrospray Ionization: Comparison of Gas Phase, Solution, and Crystal Structures. The Journal of Physical Chemistry B 2019, 123 (8), 1784–1796. [DOI] [PubMed] [Google Scholar]
- 26.McLafferty FW, Mass spectrometry across the sciences. Proc. Natl. Acad. Sci. U. S. A 2008, 105 (47), 18088–18089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bu J; Fisher CM; Gilbert JD; Prentice BM; McLuckey SA, Selective Covalent Chemistry via Gas-Phase Ion/ion Reactions: An Exploration of the Energy Surfaces Associated with N-Hydroxysuccinimide Ester Reagents and Primary Amines and Guanidine Groups. J. Am. Soc. Mass. Spectrom 2016, 27 (6), 1089–1098. [DOI] [PubMed] [Google Scholar]
- 28.Donor MT; Mroz Austin M.; Prell JS, Experimental and theoretical investigation of overall energy deposition in surface-induced unfolding of protein ions. Chemical Science 2019, 10 (14), 4097–4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Donor MT; Shepherd SO; Prell JS, Rapid Determination of Activation Energies for Gas-Phase Protein Unfolding and Dissociation in a Q-IM-ToF Mass Spectrometer. J. Am. Soc. Mass. Spectrom 2020, 31 (3), 602–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lifshitz C, Recent developments in applications of RRKM-QET. Int. J. Mass Spectrom. Ion Processes 1992, 118–119, 315–337. [Google Scholar]
- 31.Dixit SM; Polasky DA; Ruotolo BT, Collision induced unfolding of isolated proteins in the gas phase: past, present, and future. Curr. Opin. Chem. Biol 2018, 42, 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Webb IK; Morrison LJ; Brown J, Dueling electrospray implemented on a traveling-wave ion mobility/time-of-flight mass spectrometer: Towards a gas-phase workbench for structural biology. Int. J. Mass spectrom 2019, 444, 116177. [Google Scholar]
- 33.Sun Y; Vahidi S; Sowole MA; Konermann L, Protein Structural Studies by Traveling Wave Ion Mobility Spectrometry: A Critical Look at Electrospray Sources and Calibration Issues. J. Am. Soc. Mass. Spectrom 2016, 27 (1), 31–40. [DOI] [PubMed] [Google Scholar]
- 34.May JC; Jurneczko E; Stow SM; Kratochvil I; Kalkhof S; McLean JA, Conformational landscapes of ubiquitin, cytochrome c, and myoglobin: Uniform field ion mobility measurements in helium and nitrogen drift gas. Int. J. Mass spectrom 2018, 427, 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mortensen DN; Susa AC; Williams ER, Collisional Cross-Sections with T-Wave Ion Mobility Spectrometry without Experimental Calibration. J. Am. Soc. Mass. Spectrom 2017, 28 (7), 1282–1292. [DOI] [PubMed] [Google Scholar]
- 36.Polasky DA; Dixit SM; Fantin SM; Ruotolo BT, CIUSuite 2: Next-Generation Software for the Analysis of Gas-Phase Protein Unfolding Data. Anal. Chem 2019, 91 (4), 3147–3155. [DOI] [PubMed] [Google Scholar]
- 37.Prentice BM; McLuckey SA, Gas-phase ion/ion reactions of peptides and proteins: acid/base, redox, and covalent chemistries. Chem. Commun 2013, 49 (10), 947–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheung See Kit M; Carvalho VV; Vilseck JZ; Webb IK, Gas-phase ion/ion chemistry for structurally sensitive probes of gaseous protein ion structure: Electrostatic and electrostatic to covalent cross-linking. Int. J. Mass spectrom 2021, 463, 116549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koeniger SL; Merenbloom SI; Clemmer DE, Evidence for Many Resolvable Structures within Conformation Types of Electrosprayed Ubiquitin Ions. The Journal of Physical Chemistry B 2006, 110 (13), 7017–7021. [DOI] [PubMed] [Google Scholar]
- 40.Bornschein RE; Ruotolo BT, Ion mobility-mass spectrometry of charge-reduced protein complexes reveals general trends in the collisional ejection of compact subunits. Analyst 2015, 140 (20), 7020–7029. [DOI] [PubMed] [Google Scholar]
- 41.Zhou M; Liu W; Shaw JB, Charge Movement and Structural Changes in the Gas-Phase Unfolding of Multimeric Protein Complexes Captured by Native Top-Down Mass Spectrometry. Anal. Chem 2020, 92 (2), 1788–1795. [DOI] [PubMed] [Google Scholar]
- 42.Stiving AQ; Jones BJ; Ujma J; Giles K; Wysocki VH, Collision Cross Sections of Charge-Reduced Proteins and Protein Complexes: A Database for Collision Cross Section Calibration. Anal. Chem 2020, 92 (6), 4475–4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laszlo KJ; Bush MF, Analysis of Native-Like Proteins and Protein Complexes Using Cation to Anion Proton Transfer Reactions (CAPTR). J. Am. Soc. Mass. Spectrom 2015, 26 (12), 2152–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Merenbloom SI; Flick TG; Williams ER, How hot are your ions in TWAVE ion mobility spectrometry? J. Am. Soc. Mass. Spectrom 2012, 23 (3), 553–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.House JE; Kemper KA, Proton affinities of sulfate and bisulfate ions. J. Therm. Anal 1987, 32 (6), 1855–1858. [Google Scholar]
- 46.Stephenson JL; Van Berkel GJ; McLuckey SA, Ion-ion proton transfer reactions of bio-ions involving noncovalent interactions: Holomyoglobin. J. Am. Soc. Mass. Spectrom 1997, 8 (6), 637–644. [Google Scholar]
- 47.Gross DS; Zhao Y; William ER, Dissociation of heme-globin complexes by blackbody infrared radiative dissociation: Molecular specificity in the gas phase? J. Am. Soc. Mass. Spectrom 1997, 8 (5), 519–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.