

Abstract
Cardiorenal syndrome type 1 (CRS-1) is acute kidney injury (AKI) due to acute worsening of cardiac function. More than 20% of patients with acute heart failure develop AKI, and AKI predicts poor outcome. Although a number of potential pathways have been suggested as heart-kidney connectors which might drive the syndrome, there are significant barriers to investigation, such as a paucity of animal models, a lack of specific biomarkers, and an inconsistent temporal and causal relationship between changes in cardiac flow and development of renal dysfunction. Thus, mechanisms of heart-kidney interaction are still unclear, and there is no specific or effective therapy for CRS-1. This review therefore focuses on mitigating these challenges in the investigation of CRS-1. We review the available in vitro and in vivo models and focus on mechanistic insights gained from those models. In particular, we focus on non-flow and endocrine mediators of CRS-1 such as heart-derived messengers which alter renal function and which may represent targetable pathways in this syndrome. As precise connectors of heart-kidney interaction remain unclear, the establishment of animal and relevant cell-culture models and further investigation are required.
Keywords: Cardiorenal syndrome, Acute kidney injury, Cardiac arrest and cardiopulmonary resuscitation, Cysteine-glycine-rich protein-3 (CSRP3)
Introduction
Acute cardiorenal syndrome (CRS-1) is a common and morbid complication of acute cardiovascular disease. It is characterized as acute kidney injury (AKI) following rapid worsening of cardiac function, such as acute myocardial infarction, acute decompensated heart failure, and cardiac surgery [1] (Fig. 1A). Frequency of CRS-1 in cardiovascular diseases is well reported. For example, 43% of patients recovered from cardiac arrest suffer from CRS-1 [2]. Heart failure patients with AKI suffer much greater mortality than those without AKI, and CRS-1 also associates with longer hospital stay and higher risk of readmission. Thus, CRS-1 is a driver of mortality and morbidity in patients with cardiovascular disease. Furthermore, AKI patients, including those with CRS-1, suffer elevated risk of chronic kidney disease (CKD). Therefore, understanding CRS-1 is critically important.
Figure 1.
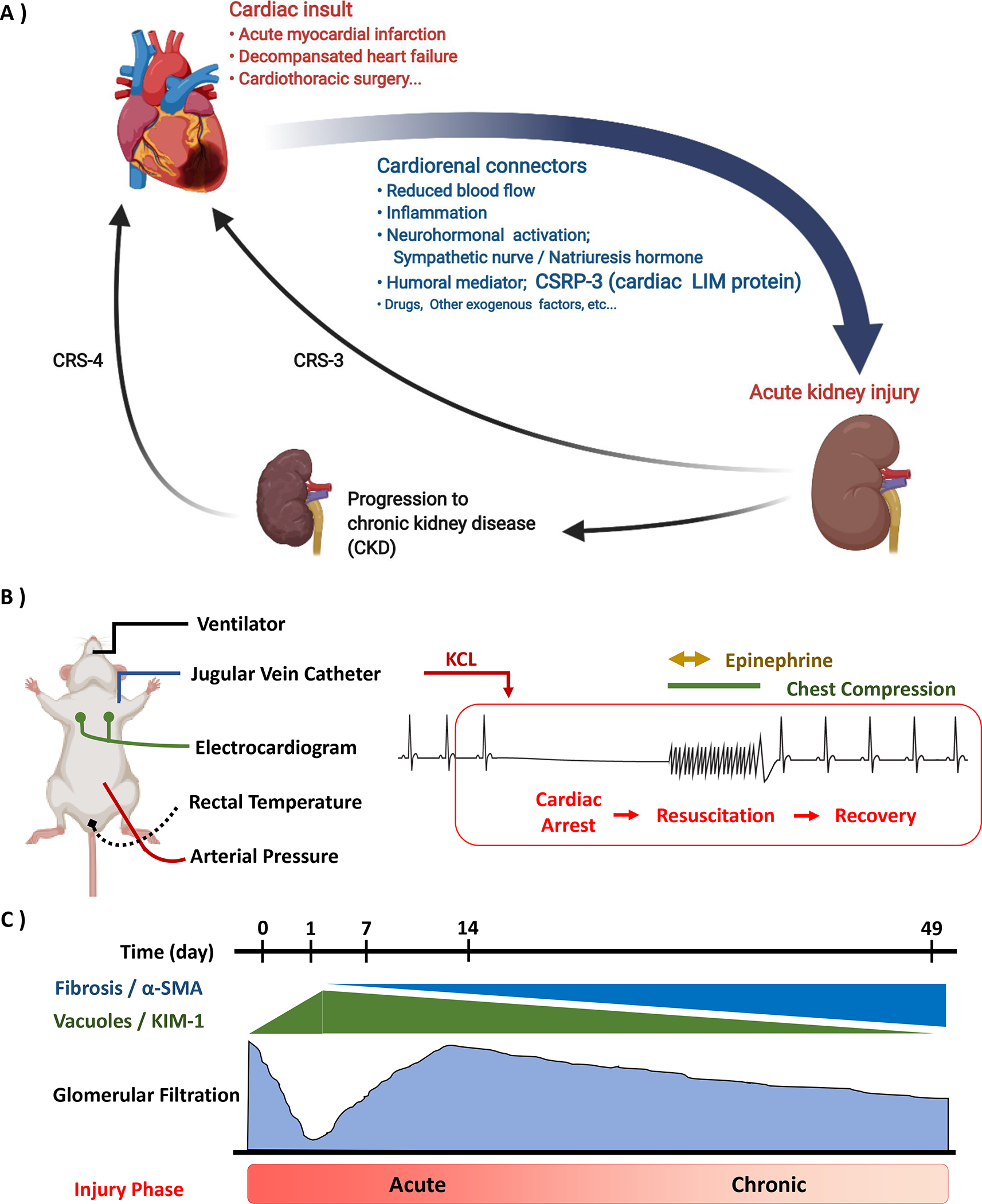
Critical concepts in modeling cardiorenal syndrome, type 1 (CRS-1). A: Schema of cardiorenal syndrome. Acute kidney injury (AKI) following acute cardiovascular disease defines CRS-1. CRS-1 is mediated by various cardiorenal connectors, including loss of blood flow, immune mediators, and endocrine mediators such as cardiac LIM protein (Cysteine-glycine-rich protein-3; CSRP3). CRS-1 may induce or lead to progression of chronic kidney disease (CKD). Both AKI and CKD worsen the risk of further cardiovascular disease (CRS-3 and −4). B: Cardiac arrest and cardiopulmonary resuscitation (CA/CPR) is a translational model of acute cardiorenal syndrome. Under isoflurane anesthesia, mice are carefully monitored by orotracheal ventilation, electrocardiography, and core temperature. Cardiac arrest is induced by potassium chloride injection via a jugular vein catheter. After the no-flow period, resuscitation is accomplished with chest compressions and intravenous epinephrine injection. C: CA/CPR induced AKI-CKD transition. In the early phase of CA/CPR, severe renal injury is observed in histologically and functionally. After the temporal recovery of glomerular filtration, tubulointerstitial fibrosis and renal dysfunction gradually progress (AKI-CKD transition).
Relationship of cardiac flow to development of AKI in CRS-1
Autoregulation of renal filtration pressure and rate were first described nearly a century ago; below the autoregulatory threshold, reduced cardiac output leads to reduced renal filtration. However, the relationship between decreased flow and renal cellular injury is less clear. Preserving/improving renal blood flow seems likely to prevent AKI, and hemodynamic goal-directed therapy is recommended. However, evidence for those strategies is poor and some trials suggest no benefit. For example, in patients with septic shock, maintaining high mean atrial pressure (MAP) (80–85mmHg) did not reduce the incidence of AKI, need for renal replacement therapy, or mortality compared to lower MAP (65–70mmHg) [3]. Similarly, intraoperative hypotension is widely recognized as a harbinger of AKI. However, the timing of postoperative AKI is heterogenous, and other etiologies, such as nephrotoxin exposure, sepsis, and surgical intervention also contribute. These findings indicate that reduced renal blood flow may only partly explain CRS-1 pathophysiology. Therefore, it is necessary to understand the role of non-flow cardiorenal connectors.
CRS-1 Experimental Models
One major challenge in CRS-1 investigation is establishment of models which mimic the pathophysiology of the syndrome. Experimental models of kidney and heart disease are reductive to single systems (for example, coronary artery occlusion used to study heart disease, and renal vascular occlusion to study renal ischemia/reperfusion). While considerable knowledge is afforded by these models, only very limited studies have studied the effects of cardiac injury on renal physiology. Such investigations are most likely to explain clinical findings. A critical step is characterization of cardiac injury models with respect to their effects on kidney function in both the short- and long-term, followed by investigation of possible connecting mechanisms.
To investigate deeply the pathophysiology of heart-kidney interaction in CRS-1, several in vivo models have been used. For example, renal interstitial fibrosis and elevation of urinary and plasma neutrophil gelatinase-associated lipocalin (NGAL) level have been observed in a coronary artery ligation model [4], and podocyte injury was detected in an aortic regurgitation (AR) model [4]. It is also reported that 60% of spontaneous hypertensive rats (SHR) develop heart failure. Since these animals also develop proteinurea and renal dysfunction, acute changes in the heart function of SHR rats may offer another candidate CRS-1 model.[5].
We and others have modeled acute, reversible cardiac dysfunction using cardiac arrest and cardiopulmonary resuscitation in mice (CA/CPR, Fig. 1B–C). CA is induced by potassium chloride injection. Eight minutes later, the animal is resuscitated with chest compressions and epinephrine. After recovery, severe AKI is observed. This model seems to mimic clinical CRS-1 well because CA/CPR-induced AKI is due to systemic ischemia and transient cardiogenic shock. CA/CPR reliably causes renal dysfunction in young and old mice of both sexes, with female protection which corresponds to clinical observations [6]. In the first 24h after resuscitation, reduced urine output and glomerular filtration are observed. Histologically at this time point, tubular epithelial vacuolization, proximal tubular brush border effacement, cell casts, and necrosis, concentrated at the corticomedullary junction, are observed. In addition, 7 weeks after CA/CPR, tubulointerstitial fibrosis, reduced renal function, and proteinuria are seen [7]. Interestingly, glomerular filtration rate temporarily recovers to normal after CA/CPR induced AKI, then gradually decreases. This time course is similar to cases of patients recovered from AKI, known as AKI to subclinical CKD. Another finding of interest is tubular vacuolization, which is rarely seen in renal ischemia/reperfusion, but commonly observed in sepsis models, suggesting that systemic mediators like those which mediate renal injury in sepsis may be involved in CRS-1 due to CA/CPR [8]. Fu et al. have reported similar findings in a rat CA/CPR model. They induced cardiac arrest by alternating current electric shock. Serum creatinine and tubular injury were maximal at 24h, with improvement at later time points [9]. These findings suggest that CA/CPR may well mimic clinical CRS-1.
Overall, in vivo models vary in terms of difficulty, severity of injury, and mechanism of heart failure (Table 1). For example, CAL and AR cause some permanent heart failure, which means these models potentially include both CRS-1 and 2 [4]. SHR is a genetic and systemic hypertensive model, so the etiology of kidney injury may not be the direct effect of cardiac insult, except in the case of acute worsening of heart failure caused by hypertension [5]. Since the syndrome is heterogenous, disparate models may provide important insight to CRS-1 pathophysiology.
Table 1.
Candidate animal models of CRS-1
| Model | Rodent | Etiology | Advantage | Disadvantage | CRS type |
|---|---|---|---|---|---|
|
| |||||
| Coronary artery ligation (CAL) [4] | Mouse and rat | Acute myocardial infarction | 1) Widely recognized | 1) Variable renal damage 2) Permanent heart failure |
1 and 2 |
| Aortic regurgitation (AR) [4] | Rat | Volume overload | 1) Common etiology | 1) Mild renal damage 2) Permanent heart failure |
Mainly type 2 |
| Spontaneous hypertensive model [5] | Mouse and rat | Essential hypertensive organ failure | 1) Widely recognized | 1) Not direct damage from heart to kidney 2) Mild renal damage |
Mainly type 5 |
| Cardiac arrest and cardiopulmonary resuscitation (CA/CPR) [6] [7] [9] [11] [14] | Mouse and rat | Cardiogenic shock (Cardiac arrest) | 1) Transient acute cardiac injury 2) Severe renal injury |
1) Initially technically challenging | Type 1 |
Candidate Cardiorenal Connectors
Cardiorenal connectors – regulators and mechanisms with interacting actions on heart and kidney function – are central to investigation of acute cardiorenal syndrome (Fig. 1A). Examples of non-flow mediators in CRS-1 are venous pressure, sympathetic outflow, endocrine signals from other organs, and inflammatory signaling. Elevated renal venous pressure likely alters renal function in heart failure and animal models [10]. However, interventions to reduce renal venous pressure may be challenged by low cardiac output in CRS-1. Since some features of CRS-1 mimic those in sepsis (see above), and because other organs besides the kidney are injured when cardiac function is impaired, kidney injury may also be driven by systemic neurological, endocrine, or inflammatory response. Zhang et at. reported that toll-like receptor 4 (TLR4) contributes to CA/CPR induced AKI in mice. They observed that functional and histological severity of AKI after CA/CPR was attenuated in TLR4 knock out mice, in which the expression of TLR4 downstream signals were decreased [11]. A report that the plasma of CRS-1 patients induced monocyte apoptosis and that plasma caspase-8 activity associated with CRS-1 severity further suggests that systemic immune response may be a cardiorenal connector [12]. Similarly, Ramchandra et al. reported that neurohumoral interactions contribute to renal vasoconstriction and reduced renal blood flow in a sheep heart failure model [13]. Our lab’s work in CA/CPR has revealed the importance of cardiac protein in heart-kidney connections, identifying cardiac LIM protein, also known as cysteine-glycine-rich protein-3 (CSRP3), as a putative connector [14]. In the heart, CSRP3 (also known as muscle LIM protein) is a critical myocardial differentiation factor which upregulates transcription of connective tissue genes (such as α-smooth muscle actin) [15]. CSRP3 is dramatically increased in the plasma of human cardiac arrest survivors and of mice after CA/CPR. Assessing renally-filtered CSRP3, we found significant upregulation after CA/CPR. Administration of recombinant CSRP3 to healthy mice led to renal dysfunction, proteinuria, and tubulointerstitial α-smooth muscle actin expression [14]. Based on these observations, renally-filtered, cardiac-specific CSRP-3 seems to be a signaling mechanism in CRS-1. Together, these findings suggest that novel humoral connectors may play important roles in CRS-1, and that interrupting such signaling may provide a pathway to prevention.
Conclusion
It is widely recognized that CRS-1 worsens outcome in acute cardiovascular disease. In addition, recent studies have highlighted the interaction in AKI between the kidney and other organs, which may lead to multiple organ dysfunction. In conclusion, careful and stepwise elucidation of the complex pathophysiology of heart-kidney interaction is required and may be key to discovering novel therapeutic and preventive approaches in CRS-1, potentially improving both short- and long-term outcomes.
Acknowledgements / Disclosure Statement / Funding Sources
This material was supported in part by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development (VA Merit Award #1I01BX004288 to MPH).
References
- 1.Ronco C, Haapio M, House AA, Anavekar N, Bellomo R. Cardiorenal syndrome. J Am Coll Cardiol. 2008. Nov 4;52(19):1527–39. [DOI] [PubMed] [Google Scholar]
- 2.Tujjar O, Mineo G, Dell’Anna A, Poyatos-Robles B, Donadello K, Scolletta S, et al. Acute kidney injury after cardiac arrest. Crit Care. 2015. Apr 17;19:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asfar P, Meziani F, Hamel JF, Grelon F, Megarbane B, Anguel N, et al. High versus low blood-pressure target in patients with septic shock. N Engl J Med. 2014. Apr 24;370(17):1583–93. [DOI] [PubMed] [Google Scholar]
- 4.Hewitson TD, Holt SG, Smith ER. Animal Models to Study Links between Cardiovascular Disease and Renal Failure and Their Relevance to Human Pathology. Front Immunol. 2015;6:465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pinto YM, Paul M, Ganten D. Lessons from rat models of hypertension: from Goldblatt to genetic engineering. Cardiovasc Res. 1998. Jul;39(1):77–88. [DOI] [PubMed] [Google Scholar]
- 6.Ikeda M, Wakasaki R, Schenning KJ, Swide T, Lee JH, Miller MB, et al. Determination of renal function and injury using near-infrared fluorimetry in experimental cardiorenal syndrome. Am J Physiol Renal Physiol. 2017. Apr 1;312(4):F629–F39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsushita K, Saritas T, Eiwaz MB, McClellan N, Coe I, Zhu W, et al. The acute kidney injury to chronic kidney disease transition in a mouse model of acute cardiorenal syndrome emphasizes the role of inflammation. Kidney Int. 2020. Jan;97(1):95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doi K, Leelahavanichkul A, Hu X, Sidransky KL, Zhou H, Qin Y, et al. Pre-existing renal disease promotes sepsis-induced acute kidney injury and worsens outcome. Kidney Int. 2008. Oct;74(8):1017–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fu ZY, Wu ZJ, Zheng JH, Qin T, Yang YG, Chen MH. The incidence of acute kidney injury following cardiac arrest and cardiopulmonary resuscitation in a rat model. Ren Fail. 2019. Nov;41(1):278–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li X, Liu M, Bedja D, Thoburn C, Gabrielson K, Racusen L, et al. Acute renal venous obstruction is more detrimental to the kidney than arterial occlusion: implication for murine models of acute kidney injury. Am J Physiol Renal Physiol. 2012. Mar 1;302(5):F519–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Q, Li G, Xu L, Li Q, Wang Q, Zhang Y, et al. Tolllike receptor 4 contributes to acute kidney injury after cardiopulmonary resuscitation in mice. Mol Med Rep. 2016. Oct;14(4):2983–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Virzì GM, Torregrossa R, Cruz DN, Chionh CY, de Cal M, Soni SS, et al. Cardiorenal Syndrome Type 1 May Be Immunologically Mediated: A Pilot Evaluation of Monocyte Apoptosis. Cardiorenal Med. 2012. Feb;2(1):33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramchandra R, Xing DT, Matear M, Lambert G, Allen AM, May CN. Neurohumoral interactions contributing to renal vasoconstriction and decreased renal blood flow in heart failure. Am J Physiol Regul Integr Comp Physiol. 2019. Sep 1;317(3):R386–r96. [DOI] [PubMed] [Google Scholar]
- 14.Wakasaki R, Matsushita K, Golgotiu K, Anderson S, Eiwaz MB, Orton DJ, et al. Glomerular filtrate proteins in acute cardiorenal syndrome. JCI Insight. 2019. Feb 21;4(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vafiadaki E, Arvanitis DA, Sanoudou D. Muscle LIM Protein: Master regulator of cardiac and skeletal muscle functions. Gene. 2015. Jul 15;566(1):1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]