

Abstract
Serotonin plays a key role in the development and maintenance of the pathobiology associated with pulmonary arterial hypertension (PAH). Platelet‐driven and locally produced serotonin from lung tissue and arterial endothelial cells induce excessive growth of pulmonary artery smooth muscle cells. The unchecked growth of these cells is a major driver of PAH including the remodeling of pulmonary arteries that dramatically reduces the diameter and flexibility of the arterial lumen. Tryptophan hydroxylase 1 (TPH1) is the rate‐limiting enzyme for biosynthesis of serotonin and is upregulated in PAH arterial endothelial cells, supporting TPH1 inhibition to treat PAH. Targeting the serotonin pathway via inhibition of peripheral serotonin and local production in diseased tissues, rather than individual receptor‐mediated or receptor‐independent mechanisms, may result in the ability to halt or reverse pulmonary vascular remodeling. Rodatristat ethyl, a prodrug for rodatristat, a potent, peripheral inhibitor of TPH1, has demonstrated efficacy in monocrotaline and SUGEN hypoxia nonclinical models of PAH and robust dose‐dependent reductions of 5‐hydroxyindoleacetic acid, the major metabolite of serotonin in plasma and urine of healthy human subjects. ELEVATE 2 (NCT04712669) is a Phase 2b, double‐blind, multicenter trial where patients with PAH are randomized to placebo, 300 or 600 mg twice daily of rodatristat ethyl. The trial incorporates endpoints to generate essential clinical efficacy, safety, pharmacokinetic, and pharmacodynamic data needed to evaluate the ability of rodatristat ethyl to ameliorate PAH by halting or reversing pulmonary vascular remodeling through its unique mechanism of TPH1 inhibition. Herein we describe the experimental design highlighting the trial's unique features.
Keywords: blood vessels, lungs, serotonin, tryptophan hydroxylase 1
INTRODUCTION
ELEVATE 2 is a Phase 2b, double‐blind, international, multicenter trial to evaluate the clinical efficacy, safety, pharmacokinetics, and biomarker pharmacodynamics of rodatristat ethyl administered as an oral twice‐daily (BID) treatment to ameliorate pulmonary arterial hypertension (PAH). Rodatristat ethyl is a first‐in‐class prodrug for rodatristat, a potent inhibitor of tryptophan hydroxylase, the rate limiting enzyme in the biosynthesis of serotonin from dietary tryptophan. Rodatristat ethyl is anticipated to ameliorate PAH by halting or reversing pulmonary vascular remodeling caused in part by increased pulmonary artery endothelial cell serotonin synthesis and subsequent proliferation of pulmonary artery smooth muscle cells (PASMCs).
PAH is one of five pulmonary hypertension subgroups that is classified based on hemodynamic, clinical, and diagnostic data. 1 PAH is a rare, incurable, progressive disease characterized by vasoconstriction, cellular proliferation, and remodeling in the pulmonary arterial bed. These changes lead to elevations in resting mean pulmonary arterial pressure, subsequent right ventricular hypertrophy, and ultimately, right heart failure and death. 2 Serotonin (5‐HT) is a bioactive amine produced via two enzymatic steps, from the dietary amino acid tryptophan. The rate limiting step in the biosynthesis is production of 5‐hydroxytryptophan from tryptophan by isoforms of tryptophan hydroxylase (TPH). Because serotonin does not cross the blood‐brain barrier, two separate pools of serotonin exist and have unique functions. In the central nervous system (CNS), serotonin is a neurotransmitter produced via tryptophan hydroxylase 2 (TPH2), whereas it is an autacoid in the periphery produced via tryptophan hydroxylase 1 (TPH1). The majority of the body's serotonin in healthy individuals (90%–95%) is produced by the enterochromaffin cells of the gastrointestinal tract and to a lesser extent in the cells of blood vessels, bones, immune system, pancreas, lungs, and skin. 3 Imbalance of peripheral serotonin is associated with gastrointestinal disorders, fibrosis, inflammation, anorexigen usage, and PAH. 3 , 4
TPH1 is overexpressed in the lungs and pulmonary arterial endothelial cells of patients with PAH and leads to excess local serotonin production. 5 , 6 Locally produced serotonin from pulmonary arterial endothelial cells, exacerbated by gut‐derived serotonin, induces proliferation of PASMCs. 5 The unchecked growth of PASMCs is a major driver of the pathology of PAH and contributes to the remodeling of pulmonary arteries that dramatically reduces the diameter and flexibility of the arterial lumen (Figure 1). Serotonin has been shown to promote pulmonary smooth muscle cell proliferation and contraction through multiple mechanisms, including receptor mediated (5HT1B) vasoconstriction and receptor independent uptake (serotonin transporter [SERT]) followed by downstream signaling pathways that drive proliferation (Figure 1). 7 , 8 Expression of the 5HT1B receptor is upregulated in PASMCs from female PAH patients potentially exacerbating the impact of increased serotonin levels. 9
Figure 1.
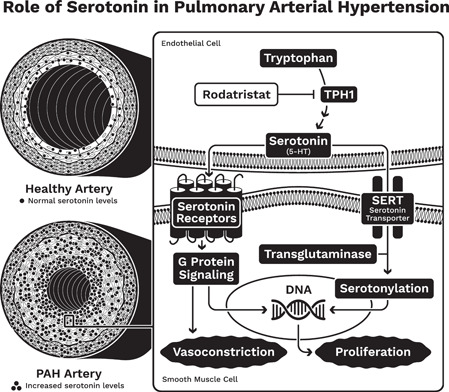
The role of serotonin in the development of pulmonary arterial hypertension (PAH). DNA, deoxyribonucleic acid; TPH1, tryptophan hydroxylase 1
Recent advances highlight the importance of dysregulated signaling with bone morphogenetic protein receptor type 2 (BMPR2), a transforming growth factor‐β superfamily receptor. 10 , 11 Balance of antiproliferative and proproliferative signaling via BMP receptors, including BMPR‐2 and ActR2a, is important for pulmonary vascular homeostasis 11 , 12 and up to 86% of familial PAH and 35% of idiopathic PAH patients show a genetic defect in BMPR2. 11 Dysregulated serotonin signaling is also thought to interact with BMPR2‐associated mechanisms driving the pathogenesis of pulmonary hypertension. Mice lacking a copy of BMPR2 (Bmpr2 +/ −) exhibit increased pulmonary artery systolic pressure and arterial remodeling in response to hypoxia combined with chronic infusion of serotonin. PASMCs from Bmpr2 + / − mice demonstrated an increased proliferative response to serotonin compared with wild‐type cells. Serotonin inhibited BMP signaling in PASMCs and resulted in a decrease of Smad1/5 phosphorylation and BMP2‐stimulated expression of the antiproliferative gene ID3. 13 Thus, inhibition of serotonin signaling by rodatristat may decrease PASMC proliferation in PAH patients with BMPR2 heterozygous mutations.
Current standard‐of‐care therapies for the treatment of PAH mainly aid in the alleviation of symptoms, primarily through vasodilation. These agents do not address the underlying pathology, halt disease progression, nor reverse the disease process. 14 Several serotonin‐targeted approaches have been evaluated including receptor antagonists (e.g., ketanserin and terguride), receptor agonists (e.g., sumatriptan), or transporter reuptake inhibitors (e.g., fluoxetine). 15 , 16 , 17 , 18 Unfortunately, the results have been disappointing and limited by several factors including choice of receptor target, use of a receptor agonist rather than antagonist, or inadequate drug exposure. 7 , 19 , 20 No clinical PAH studies have evaluated reduction in the overall peripheral and lung serotonin pool via TPH1 inhibition. Since TPH1 is the rate‐limiting enzyme in the biosynthesis of serotonin and there is wide acceptance of the role of serotonin in the pathobiology of PAH, inhibiting TPH1 presents as a reasonable approach to treat PAH. 7 , 21 Moreover, a more complete targeting of the serotonin pathway via inhibition of peripheral serotonin and local production in diseased tissues, instead of the individual receptor‐mediated and receptor‐independent mechanisms, may lead to beneficial pulmonary vascular remodeling. 20
Rodatristat, the active component of the oral prodrug rodatristat ethyl has a unique target product profile in that it has a novel mechanism of action distinct from the pathways employed by approved PAH medications; has the potential to halt or reverse remodeling of the pulmonary vasculature, and has low potential for drug‐drug interactions with other approved PAH medications allowing for evaluation in combination with approved therapies. 22
The effects of rodatristat have been evaluated in nonclinical safety pharmacology and tissue distribution studies and both support low potential for pharmacological effects in the CNS. By design, rodatristat has negligible ability to cross the blood‐brain barrier. Quantitative whole‐body autoradiography and multiple dose study data in rats suggest that rodatristat ethyl and rodatristat have sub‐pharmacologically relevant exposure levels in the CNS and do not reduce brain 5‐HT levels. Analysis of lung exposure data supports accumulation of the active drug in the lung. This is important as there is evidence to suggest that serotonin production is upregulated in the lungs of PAH patients. 5 , 6 In repeat‐dose studies characterizing CNS drug exposure at steady state, male Wistro‐Kyoto rats were dosed with rodatristat ethyl once daily (QD) at 30, 100, and 300 mg/kg/day for a period of 1, 5, or 13 days and brain tissues were collected 24 h postdosing. The efficacious 100 mg/kg daily dosing regimen resulted in CNS exposures below the TPH2 IC50, indicating a low probability of a CNS effect. In the lung, rodatristat ethyl and rodatristat concentrations were quantifiable in all animals across all rodatristat ethyl dosing groups on days 1, 6, and 14. Dosing at 100 and 300 mg/kg resulted in unbound rodatristat lung concentrations above the TPH1 IC50, suggesting pharmacologically meaningful exposures are achieved in the lung. 23 Before ELEVATE 2, over 150 healthy subjects had received either single or multiple doses (up to 14 days of 800 mg BID) across three studies. Rodatristat was safe and well tolerated with no serious adverse events, dose‐limiting toxicities or deaths observed. No clinically significant findings for vital signs, electrocardiograms, or the Columbia‐Suicide Severity Rating Scale were detected. The most common treatment‐emergent adverse events (TEAEs) were mild to moderate gastrointestinal‐related and mild elevations in hepatic enzymes. In repeat‐dose studies the TEAEs rarely led to discontinuation of study drug (<5% incidence, n = 4).
Pharmacodynamic assessments for reduction in serotonin biosynthesis demonstrated robust, dose‐dependent reductions of 5‐hydroxyindoleacetic acid (5‐HIAA), the major biomarker metabolite of serotonin, in plasma and urine. Reductions in serotonin biosynthesis in healthy subjects were observed that exceeded levels associated with efficacy in rat monocrotaline and SUGEN‐hypoxia models of PAH. 24 Rodatristat ethyl doses selected for the ELEVATE 2 Phase 2 trial described herein are anticipated to achieve a clinically meaningful reduction in serotonin in the majority of patients with PAH. 25
METHODS
Trial design
As we enter an era of innovative drug development for PAH, we must recognize increasing patient empowerment and the meaningful involvement of patient communities in the research process and beyond. The physiologic, emotional, and even economic value of symptom alleviation is critical in patient‐centric development. Qualitative patient feedback, coupled with the use of quantitative deidentified clinical data helped to inform the design of ELEVATE 2.
ELEVATE 2 (NCT04712669) is a Phase 2b, randomized, double‐blind, placebo‐controlled, multicenter trial comparing the efficacy, safety, and tolerability of two rodatristat ethyl oral regimens in patients with PAH over a 24‐week treatment period. It is planned to randomize a total of 90 patients in a 1:1:1 ratio to placebo, 300 mg BID or 600 mg BID rodatristat ethyl (Figure 2). The study is being conducted at specialized referral centers experienced in PAH management in approximately 17 countries in North America and Europe. Patients who complete the 24‐week treatment period (Main Study) have the option to enroll into an Open‐Label Extension (OLE). Patients randomized to receive active investigational product (IP) during the Main Study will typically remain on the same active dose in the OLE. Participants receiving placebo will be rerandomized 1:1 to receive 300 or 600 mg BID of rodatristat ethyl. The rodatristat ethyl dose will continue to be blinded during the OLE.
Figure 2.
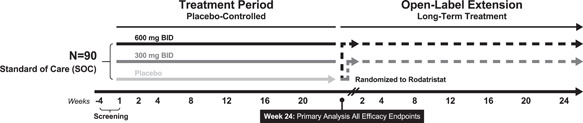
Trial schematic for ELEVATE 2
The Main Study consists of standard clinical visits, as well as the option for domiciliary and telemedicine visits to alleviate travel burden to patients and caregivers and mitigate exposure to severe acute respiratory syndrome coronavirus 2. Being the first study of rodatristat ethyl in PAH patients, it is important to monitor long‐term safety and potential efficacy. The first 24 weeks of the OLE will match that of the Main Study each comprising clinic visits (Day 1 [Week 24 of the Main Study], Weeks 4, 12, and 24), the option of a clinic or home visits (Weeks 8 and 18), and a telemedicine/phone call (Week 2). The total trial duration for patients that either terminate early or do not enroll in the OLE will be ≤32 weeks.
This trial is conducted in accordance with the protocol and the consensus of ethical principles of the Declaration of Helsinki and Council for International Organizations of Medical Sciences International Ethical Guidelines; applicable International Conference on Harmonization Good Clinical Practice Guidelines; and applicable laws and regulations. All relevant documents were approved by the respective institutional review board or independent ethics committee and competent authority before study activation. Written, informed consent will be obtained from all patients before they are enrolled in the study. The trial is registered under the name A Study of Rodatristat Ethyl in Patients with Pulmonary Arterial Hypertension (ELEVATE 2; NCT04712669, EudraCT: 2020‐004971‐42).
An external, multidisciplinary Independent Data Monitoring Committee will review the progress of the trial and perform interim reviews of unblinded safety and efficacy data at regular intervals and provide safety recommendations. An external, separate, Independent Adjudication Committee will standardize the review of the clinical worsening endpoint and reduce the bias and variability from investigators involved in the study.
INVESTIGATIONAL PRODUCT
Rodatristat ethyl is provided as a 300‐mg oral tablet manufactured by Patheon Pharmaceuticals. All participants will receive two tablets BID based on their randomization (either 2 × matched placebo (placebo), 1 × 300 mg and 1 × placebo, or 2 × 300 mg). Rodatristat ethyl doses selected for the study are based on prior pharmacodynamic observations in nonclinical models of PAH and pharmacokinetic, pharmacodynamic, safety, and tolerability data in healthy subjects administered rodatristat ethyl for up to 14 days. In addition, a dose‐response model accounting for variabilities in drug exposure and pharmacodynamic response observed in previous clinical studies with healthy subjects was developed, and after conducting 10,000 simulations, doses of 300 and 600 mg BID were identified as having a high probability (61% and 84%, respectively) of achieving the targeted 40% reduction in serotonin biosynthesis that was associated with achieving efficacy endpoints in the nonclinical MCT and SuHx PAH studies. 25 Thus, these simulations support the 300 and 600 mg BID dose regimens having a high probability of lowering serotonin biosynthesis to an extent that has the potential to lower pulmonary vascular resistance (PVR) and improve exercise capacity over a 24‐week treatment period.
Temporary dosage reductions or discontinuations may be made to manage an adverse event including gastrointestinal (e.g., diarrhea, nausea, or vomiting) or liver enzyme increase occurring during the 24 weeks of the study. Judicious use of most antidiarrheal and/or antiemetic medications are permitted. One level of dose reduction is allowed. If an adverse event(s) continues, the patient may stop taking the IP completely for up to seven days before restarting. All determinations of IP dose reduction are to be discussed with the medical monitor before implementing unless the reduction is being implanted for safety and time does not allow for discussion.
OBJECTIVES FOR THE MAIN STUDY
The study endpoints are consistent with clinically meaningful assessments, established in prior Phase 2 PAH trials. Evaluation of the change in PVR as measured by right heart catheterization, is a uniformly accepted parameter to assess disease progression and efficacy of a pharmacological intervention in PAH patients. Accordingly, the percent change from baseline of PVR was selected as the primary endpoint of the study.
Secondary objectives of the study aim to evaluate physiologic function, clinical worsening, biomarkers of disease progression, and importantly, changes in health‐related quality of life. Symptoms commonly associated with the disease have a debilitating impact not only on patients' physical functioning, but also psychosocial well‐being. 26 , 27 The Pulmonary Arterial Hypertension‐Symptoms and Impact (PAH‐SYMPACT®) is a PAH‐specific patient reported outcome instrument that is widely used in clinical practice and clinical trial research, and sensitive to patient improvement. 28 Assessment of the safety, tolerability, pharmacokinetics and pharmacodynamics of rodatristat are included as secondary objectives. The endpoints for the trial objectives are listed in Table 1.
Table 1.
Endpoints for the main study objectives for ELEVATE 2
| Endpoints for the primary objective |
|---|
|
| Endpoints for the Secondary Objectives |
|
| Endpoints for the exploratory objectives |
|
Objective for the OLE
The purpose of the OLE is to provide continuous, uninterrupted access to rodatristat ethyl for patients who participated in the Main Study if the patient appears to benefit from the therapy as determined by the investigator and patient. The OLE also provides patients receiving placebo during the Main Study the opportunity to receive treatment with rodatristat ethyl (300 or 600 mg BID). The objective of the OLE is to further evaluate the long‐term safety, tolerability, and efficacy of rodatristat ethyl in patients with PAH.
Patient recruitment and eligibility
The study population will include patients with World Health Organization Group 1 PAH, and the protocol closely follows the published guidance of the 6th World Symposium on pulmonary hypertension hemodynamic criteria and clinical classifications of pulmonary hypertension. Eligible clinical classifications include idiopathic and heritable PAH, or PAH that is drug‐ or toxin‐induced, or associated with connective tissue disease, congenital systemic pulmonary shunt repaired at least 1 year before screening, or human immunodeficiency virus infection. The inclusion and exclusion criteria are listed in Table 2.
Table 2.
Participant inclusion and exclusion criteria for ELEVATE 2
| Main Study |
|---|
| Inclusion criteria |
|
| Exclusion criteria |
|
Placebo‐controlled randomized studies provide the most robust results and are thus considered the most appropriate design. However, approved therapies for PAH are available, making a placebo‐controlled rodatristat ethyl monotherapy trial unethical. Therefore, all participating patients are required to be on a stable background therapy for PAH with their regimens stabilized for ≥12 weeks and their dose(s) stabilized for ≥8 weeks before their screening right heart catheterization. Oral, inhaled, or parenteral prostanoids are permitted. Patients' standard‐of‐care regimens at entry will be maintained for the duration of the trial. Eligible patients are stratified based on the number of background PAH therapies they are receiving (1 vs. 2 vs. 3). The number of patients who are on a prostanoid infusion or selexipag will be capped at 50% and 20%, respectively, of the total patients enrolled.
Sample size justification
Thirty patients per cohort will provide at least 80% power at a significance level of 0.05 (2‐sided hypothesis) to detect a treatment difference of 0.75 times of standard deviation in the percentage change for PVR from baseline between an active cohort and the placebo cohort.
Efficacy‐related assessments
This study will compare the efficacy, safety, and tolerability of two dosing regimens of rodatristat ethyl to placebo in patients with PAH. The primary endpoint is the percent change from baseline in PVR for each of the two active arms versus the placebo arm, calculated from measurements obtained during right heart catheterization over 24 weeks of treatment. Main secondary endpoints are the change from baseline in World Health Organization Functional Class, 6‐min walk distance, and N‐terminal‐pro‐brain natriuretic peptide. Other endpoints are shown in Table 1. In prior studies of patients treated with PAH therapies, up to 24 weeks of treatment with study medication were necessary to demonstrate efficacy as well as allow for assessment of safety and tolerability. 10 Change in safety parameters including adverse events, laboratory values, electrocardiograms, and other clinical assessments will be evaluated from baseline to Week 24.
Pharmacokinetics of rodatristat ethyl and rodatristat
A population PK approach will be utilized to characterize the PK of rodatristat ethyl and rodatristat in subjects with PAH. In vitro experiments and in silico modeling to evaluate the drug‐drug interaction potential with rodatristat ethyl and rodatristat have not identified high or even moderate risks for interactions. There is the potential for weak interactions with substrates of the drug metabolizing enzymes cytochrome P450, CYP2C8, and CYP3A, and drug transporters P‐glycoprotein or organic anion‐transporting polypeptide. Of relevance to the current study, selexipag and its active metabolite, ACT‐333679, are prostacyclin receptor agonists approved for the treatment of PAH and are sensitive substrates of cytochrome P450 CYP2C8. A selexipag‐rodatristat ethyl clinical interaction study in healthy subjects was performed and steady‐state of rodatristat 600 mg BID co‐administered with single‐dose selexipag 400 µg, increased selexipag area under the concentration‐time curve 1.18‐fold and reduced ACT‐333679 area under the concentration‐time curve by 40.5%. Safety data from the study show that diarrhea, headache, nausea, and stomachache were reported (in ≥2 subjects) at approximately the same frequency during exposure to rodatristat ethyl alone and coadministration of selexipag and rodatristat ethyl. There were no changes in laboratory parameters, vital signs, or electrocardiograms of clinical significance. The current understanding of the magnitude of this interaction in patients with PAH and the clinical relevance of these changes in exposure are unknown. Thus, pharmacokinetic samples for assessment of selexipag and ACT‐333679 trough concentrations are collected in ELEVATE 2 to further investigate this finding.
Blood samples for pharmacokinetic analysis of plasma rodatristat ethyl, rodatristat, selexipag, and ACT‐333679 will be assayed using validated liquid chromatography with tandem mass spectrometry methods. During the Main Study, blood samples for evaluation of plasma rodatristat ethyl and rodatristat are collected predose on Day 1 and Week 4, postdose on Week 12 (blood sample will be collected between 1 and 8 h after the morning dose), and a trough sample following the last dose Week 24 (if the patient enrolls in the OLE, this blood sample will be collected before the morning rodatristat ethyl dose). Blood samples for evaluation of selexipag and ACT‐333679 are collected during the Main Study (predose on Day 1 and Week 4) and during the first 24 weeks of the OLE (OLE baseline visit and Week 4). Patients will take their selexipag in the clinic at these visits allowing the pharmacokinetic samples to be collected approximately 12 h after the prior dose. Blood samples for the pharmacokinetic analysis of selexipag and ACT‐333679 are only collected in patients whose standard‐of‐care therapy includes selexipag.
Pharmacodynamics
5‐HIAA is a stable metabolite of serotonin and a target‐specific biomarker of its biosynthesis. Blood samples and urine samples are being collected for determination of plasma and urine creatinine‐corrected 5‐HIAA concentrations at pre‐dose on Day 1 and at Weeks 4, 12, and 24. Plasma and urine concentrations of 5‐HIAA will be determined using validated liquid chromatography with tandem mass spectrometry methods. Urine creatinine will be measured by means of a Roche Cobas (Roche Diagnostics) using the Roche Jaffé creatinine method as part of the clinical chemistry panel.
Statistical analyses
Primary and secondary efficacy analyses will be performed using the modified intent‐to‐treat and per protocol populations which ensure patients completed at least one postbaseline efficacy assessment and completed the Week 24 visit without major protocol deviations, respectively. Percent change from baseline in PVR will be analyzed using an analysis of covariance with baseline PVR as the covariate. Additional baseline characteristics may also be evaluated as covariates in the model. The estimated between‐treatment differences, 95% confidence intervals, and p values will be determined. For patients who discontinue from the trial early, the last‐observation‐carried‐forward method will be used to impute the PVR at Week 24.
For the secondary efficacy endpoints, the change from baseline to Week 24 will be analyzed using an analysis of covariance model with treatment as a fixed effect, randomization stratum and baseline assessment as covariates. The estimated between‐treatments differences, 95% confidence intervals, and p values will be determined.
The change in 6‐min walk distance from baseline to Week 24 will be compared between cohorts using nonparametric analysis of covariance within the framework of the extended Cochran‐Mantel‐Haenszel test. For endpoints measured over time, a mixed effect of repeated measure model may be used as sensitivity analyses. The time from the first dose of IP until the first clinical worsening event will be summarized using Kaplan–Meier estimates and compared between treatment cohorts using the log‐rank test.
Safety analyses will be performed using the safety population. Safety and tolerability will be evaluated by assessment of adverse event incidence and changes in clinical laboratory tests, physical examinations, vital signs measurements, electrocardiogram readings, and suicidal ideation and behavior ratings at various time points during the trial.
CONCLUSION
TPH inhibitors, including rodatristat, reduce the cellular release of serotonin that promotes proliferation of pulmonary smooth muscle cells, 22 ELEVATE 2 is a clinical trial to further establish and target serotonin as a critical effector of PAH disease pathogenesis and is the first PAH study to address aberrant production of peripheral serotonin by treating with rodatristat ethyl to inhibit TPH1. All patients in ELEVATE 2 will be receiving background SOC treatment. Nonclinical studies in the rat SuHx model of PAH indicated at least an additive effect of combination rodatristat ethyl and ambrisentan treatment in reducing vessel wall thickness and mean pulmonary arterial pressure and was superior to the combination of ambrisentan and tadalafil. Thus, it is possible that the addition of one agent to another could confer additive or synergistic benefits.
AUTHOR CONTRIBUTIONS
Howard M. Lazarus, Jill Denning, Stephen Wring, William Symonds, and Jeremy Feldman: contributed to the design of the study and preparation of the study protocol. Michelle Palacios and Katelyn Crizer: provided nonclinical input and bioanalytical expertize during the design of the study. Howard M. Lazarus, Jill Denning, Sidra Hoffman, and Watiri Kamau‐Kelley: supported conduct of the study. All authors and Dr. Deborah Piscitelli (medical writer): contributed to the preparation and review of this manuscript.
CONFLICTS OF INTEREST
The authors and Dr. Deborah Piscitelli (medical writer) are responsible for the content and writing of this manuscript. All Altavant Sciences Inc. affiliated authors and the medical writer are paid employees or contractors of Altavant Sciences Inc. Jeremy Feldman is a consultant for Altavant Sciences Inc. in addition to being a consultant and speaker for Janssen, Bayer Pharmaceuticals, and United Therapeutics Corporation.
ETHICS STATEMENT
This trial is conducted in full compliance with the ethical principles derived from international guidelines including the Declaration of Helsinki and Council for International Organizations of Medical Sciences International Ethical Guidelines; applicable International Conference on Harmonisation Good Clinical Practice Guidelines; and applicable laws and regulations. Before patient enrollment, the protocol and amendment(s) are approved by the central institutional review board in the United States, Advarra (Columbia, MD, USA; Pro00048419); outside the United States, the protocol and amendment(s) are approved by the central and local independent ethics committee specific to the study site. Patient participation is voluntary, and the patient or their legally authorized representative is required to sign a statement of informed consent that meets the requirements of 21 Code of Federation 50, local regulations, International Conference on Harmonisation guidelines, Health Insurance Portability and Accountability Act requirements, where applicable, and the central and if applicable, site‐specific institutional review board or independent ethics committee.
ACKNOWLEDGMENTS
The authors thank and acknowledge the help of Dr. Deborah Piscitelli in the preparation and editing of this manuscript. They also thank the patients, clinical research personnel, and clinical investigators for helpful contributions to the design of this protocol. The work and ELEVATE 2 are funded by Altavant Sciences Inc.
Lazarus HM, Denning J, Wring S, Palacios M, Hoffman S, Crizer K, Kamau‐Kelley W, Symonds W, Feldman J. A Trial Design to Maximize Knowledge of the Effects of Rodatristat Ethyl in the Treatment of Pulmonary Arterial Hypertension (ELEVATE 2). Pulmonary Circulation. 2022;12:e12088. 10.1002/pul2.12088
REFERENCES
- 1. Maron BA, Abman SH, Elliott CG, Frantz RP, Hopper RK, Horn, EM, Nicolls MR, Shlobin OA, Shah SJ, Kovacs G, Olschewski H, Rosenzweig EBet.al. Pulmonary arterial hypertension: diagnosis, treatment, and novel advances. Am J Respir Crit Care Med. 2021;Epub ahead of print 15, 10.1164/rccm.202012-4317SO [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Agarwal R, Gomberg‐Maitland M. Current therapeutics and practical management strategies for pulmonary arterial hypertension. Am Heart J. 2011;162:201–13. [DOI] [PubMed] [Google Scholar]
- 3. Berger M, Gray JA, Roth BL. The expanded biology of serotonin. Annu Rev Med. 2009;60:355–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2012;122:4306–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eddahibi S, Guignabert C, Barlier‐Mur AM, Dewachter L, Fadel E, Dartevelle P, Humbert M, Simonneau G, Hanoun N, Saurini F, Hamon M, Adnot S. Cross talk between endothelial and smooth muscle cells in pulmonary hypertension: critical role for serotonin‐induced smooth muscle hyperplasia. Circulation. 2006;113:1857–64. [DOI] [PubMed] [Google Scholar]
- 6. Humbert M, Labrune P, Sitbon O, Le Gall C, Callebert J, Hervé P, Samuel D, Machado R, Trembath R, Drouet L, Launay JM, Simonneau G. Pulmonary arterial hypertension and type‐I glycogen‐storage disease: the serotonin hypothesis. Eur Respir J. 2002;20:59–65. [DOI] [PubMed] [Google Scholar]
- 7. MacLean MR. The serotonin hypothesis in pulmonary hypertension revisited: targets for novel therapies (2017 Grover Conference Series). Pulm Circ. 2018;8:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Penumatsa K, Abualkhair S, Wei L, Warburton R, Preston I, Hill NS, Watts SW, Fanburg BL, Toksoz D. Tissue transglutaminase promotes serotonin‐induced AKT signaling and mitogenesis in pulmonary vascular smooth muscle cells. Cell Signal. 2014;26:2818–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wallace E, Morrell NW, Yang XD, Long L, Stevens H, Nilsen M, Loughlin L, Mair KM, Baker AH, MacLean MR. A sex‐specific microRNA‐96/5‐hydroxytryptamine 1B axis influences development of pulmonary hypertension. Am J Respir Crit Care Med. 2015;191:1432–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sitbon O, Gomberg‐Maitland M, Granton J, Lewis MI, Mathai SC, Rainisio M, Stockbridge NL, Wilkins MR, Zamanian RT, Rubin LJ. Clinical trial design and new therapies for pulmonary arterial hypertension. Eur Respir J. 2019;53:1801908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guignabert C, Humbert M. Targeting transforming growth factor‐β receptors in pulmonary hypertension. Eur Respir J. 2021;57:2002341. [DOI] [PubMed] [Google Scholar]
- 12. Humbert M, McLaughlin V, Gibbs JSR, Gomberg‐Maitland M, Hoeper MM, Preston IR, Souza R, Waxman A, Escribano Subias P, Feldman J, Meyer G, Montani D, Olsson KM, Manimaran S, Barnes J, Linde PG, de Oliveira Pena J, Badesch DB, PULSAR Trial I. Sotatercept for the treatment of pulmonary arterial hypertension. N Engl J Med. 2021;384:1204–15. [DOI] [PubMed] [Google Scholar]
- 13. Long L, MacLean MR, Jeffery TK, Morecroft I, Yang X, Rudarakanchana N, Southwood M, James V, Trembath RC, Morrell NW. Serotonin increases susceptibility to pulmonary hypertension n BMPR2‐deficient mice. Circ Res. 2006;98:818–27. [DOI] [PubMed] [Google Scholar]
- 14. Mayeux JD, Pan IZ, Dechand J, Jacobs JA, Jones TL, McKellar SH, Beck E, Hatton ND, Ryan JJ. Management of pulmonary arterial hypertension. Curr Cardiovasc Risk Rep. 2021;15:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McGoon MD, Vlietstra RE. Acute hemodynamic response to the S2‐serotonergic receptor antagonist, ketanserin, in patients with primary pulmonary hypertension. Int J Cardiol. 1987;14:303–309. [DOI] [PubMed] [Google Scholar]
- 16. Antoniu SA. Terguride for pulmonary arterial hypertension. Expert Opin Ther Targets. 2011;15:1333–35. [DOI] [PubMed] [Google Scholar]
- 17. MacIntyre PD, Bhargava B, Hogg KJ, Gemmill JD, Hillis WS. Effect of subcutaneous sumatriptan, a selective 5HT1 agonist, on the systemic, pulmonary, and coronary circulation. Circulation. 1993;87:401–405. [DOI] [PubMed] [Google Scholar]
- 18. Sodimu A, Bartolome S, Igenoza OP, Chin KM. Hemodynamic effects of fluoxetine in pulmonary arterial hypertension: an open label pilot study. Pulm Circ. 2020;10:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lythgoe MP, Rhodes CJ, Ghataorhe P, Attard M, Wharton J, Wilkins MR. Why drugs fail in clinical trials in pulmonary arterial hypertension, and strategies to succeed in the future. Pharmacol Ther. 2016;164:195–203. [DOI] [PubMed] [Google Scholar]
- 20. MacLean MR, Dempsie Y. The serotonin hypothesis of pulmonary hypertension revisited. Adv Exp Med Biol. 2010;661:309–22. [DOI] [PubMed] [Google Scholar]
- 21. Matthes S, Bader M. Peripheral serotonin synthesis as a new drug target. Trends Pharmacol Sci. 2018;39:560–72. [DOI] [PubMed] [Google Scholar]
- 22. Aiello RJ, Bourassa PA, Zhang Q, Dubins J, Goldberg DR, De Lombaert S, Humbert M, Guignabert C, Cavasin MA, McKinsey TA, Paralkar V. Tryptophan hydroxylase 1 inhibition impacts pulmonary vascular remodeling in two rat models of pulmonary hypertension. J Pharmacol Exp Ther. 2017;360:360–279. [DOI] [PubMed] [Google Scholar]
- 23. Wring S, Gaukel E, Crizer K. Tissue distribution and biomarker data for rodatristat, a novel serotonin (5‐HT) synthesis inhibitor for PAH, demonstrate negligible blood‐brain barrier penetration and pharmacologically meaningful exposure in lung. Am J Respir Crit Care Med. 2020;201:A2095. [Google Scholar]
- 24. Carpenter D, Keller L, Palacios M, Rurka J, Crizer K, Pack T, Snyder M, Wring S. Once daily oral dosing of rodatristat ethyl (RVT‐1201) achieves reductions in serotonin biosynthesis comparable to those associated with reversal of vascular remodeling in PAH animal models. Chest. 2019;156:A1175–76. [Google Scholar]
- 25. Johnson B, Palacios M, Zhou J, Schmith VD, Wring S. A pharmacokinetic/pharmacodynamic‐based rationale for dose selection of the TPH inhibitor rodatristat ethyl in ELEVATE2—a phase 2b study in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2021;203:A3604. [Google Scholar]
- 26. Chen H, Taichman DB, Doyle RL. Health‐related quality of life and patient‐reported outcomes in pulmonary arterial hypertension. Pro Am Thorac Soc. 2008;5:623–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guillevin L, Armstrong I, Aldrighetti R, Howard LS, Ryftenius H, Fischer A, Lombardi S, Studer S, Ferrari P. Understanding impact of pulmonary arterial hypertension on patients' and carers' lives. Eur Respir Rev. 2013;22:535–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chin KM, Gomberg‐Maitland M, Channick RN, Cuttica MJ, Fischer A, Frantz RP, Hunsche E, Kleinman L, McConnell JW, McLaughlin VV, Miller CE, Zamanian RT, Zastrow MS, Badesch DB. Psychometric validation of the pulmonary arterial hypertension‐symptoms and impact (PAH‐SYMPACT) questionnaire: results of the SYMPHONY trial. Chest. 2018;154:848–61. [DOI] [PubMed] [Google Scholar]