

OTHER WAYS TO CELL DEATH
Although most apoptosis in mammals occurs by means of the mitochondrial pathway or death receptor ligation, caspase activation can occur by other mechanisms as well. Some of these lead to cell death, and some have other functions.
Apart from the executioner caspases, caspases are activated by the mechanism known as induced proximity. As we have seen, this allows the formation of dimers or higher-order oligomers, which is generally effected by the binding of one or more adapter proteins to the prodomains of the caspase monomers. These adapter proteins help to define the caspase-activation pathway involved. In this review, we discuss the activation and functions of other caspases that can participate in cell death but can also have other functions as well—caspase-1, caspase-2, caspase-4, and caspase-5 (as well as caspase-11 in mice). These caspases are not activated by the pathways described in Green (2022a,b) but, rather, by other activation platforms. And although some of these can induce apoptosis, others cause another form of cell death—pyroptosis (a form of necrosis).
THE “ADAPTERLESS” ACTIVATION OF CASPASES-4, -5, AND -11
So far, we have discussed two ways in which caspases are activated: cleavage in the case of executioner caspases and induced proximity by the binding of initiator caspases to adapter molecules. But the activation of caspases-4 and -5 (in humans), and caspase-11 (in rodents), uses a different approach. Like the initiator caspases, these caspases exist as monomers expressed in myeloid cells (such as macrophages and dendritic cells) and most epithelial cells. And, like initiator caspases, they are activated by induced proximity. However, it appears that this can occur without a requirement for an adapter protein.
Gram-negative bacteria contain lipopolysaccharide (LPS) in their outer coats, which is a molecule composed of a lipid and carbohydrates and is sensed by the immune system as an indication of infection. If such bacteria invade the cytoplasm of a cell, the lipid portion of LPS binds to the caspase-recruitment domain (CARD) of caspases-4 and -5, or caspase-11 in rodents, forcing the caspase to form dimers and thus activating the enzyme. This appears to involve direct binding; it is the LPS that serves as the platform on which the caspase activates. In this sense, these caspases can be thought of as intracellular LPS sensors. At this point, we do not know whether there are other bacterial products that can activate these (or other) caspases through interaction with their prodomains, but this remains a possibility.
The activities of caspases-4, -5, and -11 do not cleave and activate executioner caspases nor promote apoptosis in cells. Instead, they cause cell death by cleaving a specific substrate, gasdermin D. Gasdermin D is auto-inhibited, and this inhibition is disrupted by cleavage. Active gasdermin D then moves to the plasma membrane, where it creates pores, causing the cell to swell and ultimately burst (Fig. 1). It is the action of gasdermin D that promotes this necrotic cell death, and mutation of the cleavage site of gasdermin D prevents cell death by active caspase-11. Mice lacking either caspase-11 or gasdermin D are resistant to LPS shock, a toxic condition caused by intravenous injection of LPS, and to bacterial sepsis. There are several gasdermin proteins, all of which have the pore-forming potential, although only gasdermin D is activated through cleavage by caspase-4, -5, or -11. The functions of the other gasdermin molecules are largely unknown, but it is possible that these proteins participate in other pathways of cell death. In Green (2022c), we will mention a molecule related to gasdermin D, which functions in a form of necrosis.
Figure 1.

Cytosolic lipopolysaccharide (LPS) directly induces caspases and pyroptosis. This appears to be restricted to caspase-4 and caspase-5 in humans and caspase-11 in mice. Other caspases we have discussed are not activated in this direct manner.
Although LPS can activate these caspases without a requirement for an adapter protein, this does not necessarily mean that no such adapter exists. Presently the existence of an adapter protein for caspases-4, -5, and -11 is speculative, there are conditions that appear to activate these caspases without intracellular LPS. One example is the cholera toxin b-chain, which binds to the cell surface and triggers the activation of these caspases.
The activation of caspases-4, -5, or -11 is often referred to as the “noncanonical inflammasome.” Next, we consider the “canonical inflammasomes,” which activate caspase-1.
CASPASE-1 IS ACTIVATED BY INFLAMMASOMES
As outlined in Green (2022d), caspase-1 is involved in the processing of interleukin-1 and interleukin-18, secretion of these and other proteins, and cell death by pyroptosis. The activation of caspase-1 occurs in complexes called inflammasomes, and, unlike the other caspase-activation platforms that we have discussed (the apoptosome and the death-inducing signaling complex (DISC) for caspases-9 and -8, respectively), inflammasomes can comprise different adapter molecules. In most inflammasomes, however, a common feature is a molecule that binds to the CARD in the prodomain of a caspase-1 monomer (Fig. 2).
Figure 2.

The basic inflammasome and activation of caspase-1.
Caspase-1 is expressed in myeloid cells, such as macrophages and dendritic cells, as well as epithelial cells, and inflammasomes that activate this caspase assemble in these cells in response to a wide range of signals, most of which are associated with infectious agents. These signals can be proteins, lipids, DNA, or RNA from pathogens, collectively referred to as pathogen-associated molecular patterns (PAMPs). Some inorganic materials, such as some crystals, also induce formation of inflammasomes, as do (probably) some materials released from necrotic cells. Another general term is sometimes used for the latter—damage-associated molecular patterns (DAMPs).1 How PAMPs/DAMPs trigger inflammasome formation and how different inflammasomes are engaged are at the heart of a rapidly emerging area of inflammation research.
SEVERAL INFLAMMASOMES FOR ACTIVATION OF CASPASE-1 INVOLVE ASC
A small CARD-containing protein called ASC (“apoptosis-associated speck-like protein containing a CARD”) can oligomerize and bind caspase-1 through CARD–CARD interactions to activate the protease by induced proximity. In addition to its CARD domain, ASC contains another death fold, a pyrin domain (PyD). If ASC is experimentally overexpressed, it oligomerizes to form large aggregates (appearing as specks in the cytoplasm). These aggregates of ASC are sufficient to bind and activate caspase-1 (Fig. 3). As we will see, however, this artificial situation is not how ASC functions to promote caspase-1 activation. Instead, there are intracellular receptors that interact with ASC to promote the formation of inflammasomes, and the general structure of these has been elucidated in cryoelectron microscopy studies. All of these inflammasomes form disc-like structures when activated, and these, in turn, recruit ASC, which itself forms a cylindrical structure. When the caspase-1 CARD binds to the exposed ASC CARD, the caspase-1 CARD then binds further caspase-1 CARD domains. Filaments of oligomerized caspase-1 then extend from this ASC cylinder (Figs. 4 and 5). Therefore, the structures of inflammasomes are distinct from the structures of other caspase-activation platforms, such as the apoptosome (Green 2022a).
Figure 3.

Overexpression of ASC can activate caspase-1. This is not a physiological mechanism of caspase-1 activation, but the observation is informative.
Figure 4.

General structure of inflammasomes. 1. The intracellular receptor, containing a pyrin domain (PYD), on interaction with its ligand forms a disc-like structure. 2. The center of this structure interacts with ASC through PYD–PYD interactions. 3. The PYD domains of ASC interact with additional ASC PYD domains to form a cylindrical structure. 4. The CARD domains of caspase-1 monomers bind to the CARD of ASC. 5. Bound caspase-1 recruits additional caspase-1 molecules through CARD–CARD interactions, forming a fibril. A “mature” inflammasome is shown in Figure 5.
Figure 5.

Inflammasomes have fibrils of caspase-1.
TLRs INDUCE OTHER INFLAMMASOME COMPONENTS AND CASPASE-1 SUBSTRATES
A set of receptors related to the Drosophila Toll protein is involved in recognizing many PAMPs. These Toll-like receptors (TLRs) recognize components of bacterial cell walls, bacterial RNA, and fungal components. When they interact with PAMPs, TLRs activate nuclear factor-κB (NF-κB) and interferons, which in turn induce the expression of caspases-1, -4, -5 (and caspase-11 in rodents), the interleukin targets of caspase-1, as well as additional inflammasome components that can bind to and oligomerize ASC to facilitate caspase-1 activation. Because of this, cells with potential inflammasome activity often have to be “primed” with agents that activate a TLR. In Figure 6, some of the TLRs in human cells are illustrated, together with some of the PAMPs that they recognize.
Figure 6.

Some TLRs and the PAMPs that activate them.
There is a logic to this arrangement. The production of inflammatory cytokines and the death of cells by pyroptosis can cause tissue damage. By restricting the system to conditions in which an infection is sensed (by TLRs), the activation of the inflammasome is limited.
Unfortunately, however, there are many conditions that can prime myeloid and epithelial cells and also activate caspase-1 in the absence of an overt infection. We return later to these in considering the roles for inflammasomes in promoting disease.
NLRs PARTICIPATE IN INFLAMMASOMES
In addition to the TLRs, there is a large set of intracellular molecules that appear to recognize PAMPs and DAMPs—the Nod-like receptors (NLRs, so named for the first one found, Nod-1). The NLRs are found throughout the animal kingdom and even in plants, where they are involved in host defense against infection. Collectively, TLRs and NLRs are often referred to as “pattern-recognition receptors” (PRRs).
Several NLRs contain death folds, including CARDs and PyDs, and some of these have been shown to participate in the generation of inflammasomes. The most important of these to date are NLRP3 (“NACHT, LRR, and PYD domains-containing protein 3,” also called NALP3 and cryopyrin) and NLRC4 (“NLR family CARD domain-containing protein 4,” also called CARD12 and IPAF). All of the NLRs (including those in plants) also have a NACHT domain (see Green 2022a) and a long tail. In many NLRs, this tail is a leucine-rich region (LRR) believed to be specific for a type of PAMP or DAMP that appears in the cell. Some of the mammalian NLRs and what they appear to recognize are listed in Figure 7.
Figure 7.

Some NLRs that function in inflammasomes. The PAMPs and DAMPs that induce them are listed. Toxins from bacteria can also alter potassium levels, thereby indirectly facilitating inflammasome formation.
Whether NLRs respond directly to a DAMP or PAMP has not been formally proven. It remains possible (and in some cases likely) that other molecules participate in this recognition event, as is the case for NLRC4 (discussed below).
THE NLRP3 INFLAMMASOME
NLRP3 contains a PyD that binds to the PyD of ASC. In addition, NLRP3 has a NACHT domain and an LRR. NLRP3 and ASC are required for the formation of inflammasomes in response to a number of PAMPs, including those from Staphylococcus aureus, Listeria monocytogenes, and bacterial RNAs. This does not depend on TLR signaling. However, TLR signals induce the expression of NLRP3 (and other inflammasome components, as we have seen) to increase sensitivity. Once activated, NLRP3 promotes inflammasome formation and caspase-1 activation (Fig. 8).
Figure 8.

Activation of the NLRP3 inflammasome.
Some noninfectious materials also act to engage the NLRP3 inflammasome to activate caspase-1. These include asbestos, crystals of uric acid (the cause of gout), and crystals of calcium pyrophosphate dihydrate (CPPD, the cause of pseudogout), as well as crystals of cholesterol (Fig. 9).
Figure 9.

Inert crystals induce the NLRP3 inflammasome in gout and other diseases. (Bottom left, Reprinted from Gillray 1799.)
It is unlikely that all of these interact directly with NLRP3 to trigger its interaction with ASC and caspase-1. There might be common denominators. One of these appears to be the mitochondria. Upon any of these treatments, mitochondria seem to become stressed, producing reactive oxygen by the action of the mitochondrial electron-transport chain. Indeed, an inhibitor of mitochondrial complex I—rotenone—can induce the activity of the NLRP3 inflammasome through the production of reactive oxygen. Furthermore, cells that lack an electron-transport chain because of loss of mitochondrial DNA do not activate the NLRP3 inflammasome in response to these agents. However, it is not clear whether, or how, reactive oxygen activates the NLRP3 inflammasome. Some studies have suggested that it is not reactive oxygen, but instead oxidized mitochondrial DNA, that performs this function, but again the mechanism underlying this remains obscure.
Another potential common denominator is potassium. The high levels of potassium in cells inhibit the NLRP3 inflammasome. Activation of potassium channels in the plasma membrane allows an efflux of potassium, permitting inflammasome activation.
One such potassium channel is the P2X purinoceptor 7 (P2X7). This is activated by extracellular ATP, which might act as a damage signal that cells (pathogens or host cells) are lysing in the vicinity. Extracellular ATP activates the NLRP3 inflammasome in primed macrophages. Bacteria produce a variety of toxins that bind to the cell surface and also cause potassium efflux. Again, such toxins are potent activators of the NLRP3 inflammasome.
One other way in which potassium levels in a cell can decrease to promote the activation of the NLRP3 inflammasome is when caspase-4, -5, or -11 is activated by intracellular LPS (see above). When gasdermin D is cleaved, and before the cell dies, this effector causes potassium efflux, perhaps as a consequence of disruption of the plasma membrane. Therefore, although these caspases do not process interleukin-1β or interleukin-18 directly, they can promote the activation of the NLRP3 inflammasome to activate caspase-1, which has this cytokine-processing ability.
However, all of this is complicated by the fact that potassium levels also affect mitochondrial function, and therefore it remains possible that it is the mitochondria, and not only potassium levels, that somehow activate the NLRP3 inflammasome. The production of reactive oxygen species from mitochondria has been implicated in NLRP3 activation, although the mechanism for this is obscure. Until we fully understand the roles for potassium and mitochondria in NLRP3 activation, the common denominators that link all of the agents that can induce this inflammasome remain murky.
THE NLRC4 INFLAMMASOME
Another important NLR for inflammasome formation is NLRC4, which is required for the response to bacterial flagellin from Salmonella typhimurium and Legionella pneumophila and to Shigella flexneri infection, as well as the response to pathogenic Escherichia coli.
It turns out that NLRC4 does not recognize these PAMPs directly. Another set of molecules, comprising NAIP1, 2, 5, and 6, is important in the NLRC4 response. These proteins do not have a death fold. Instead, each has a region with three BIR domains. It therefore resembles an IAP protein (see Green 2022d) but without caspase-inhibitory activity (as far as we know). NAIP5 and NAIP6 respond to bacterial flagellin, whereas NAIP1 and NAIP2 respond to components of the bacterial type III secretion system, which functions to inject bacterial components into the cell. One molecule of any of these NAIP proteins, on activation, binds to an NLRC4 molecule, inducing a conformational change in the latter. This activated NLRC4 can then activate another NLRC4 molecule, thus creating a chain reaction that results in the formation of a disc-like structure containing one NAIP protein and several NLRC4 proteins. The NLRC4 complex then recruits ASC, which in turn activates caspase-1 (Fig. 10).
Figure 10.

Activation of the NLRC4 inflammasome.
NLRC4 has a CARD domain. Following activation, this interacts with ASC through a CARD–CARD interaction. It is likely that this then promotes the PYD–PYD interactions of ASC to form the ASC cylinder, which in turn recruits the CARD of caspase-1. Alternatively, it is possible that the CARD of NLRC4 binds directly to the CARD of caspase-1 in some cases as the activation of caspase-1 by NLRC4 is enhanced by, but does not always require, the presence of ASC. Therefore, if NLRC4 oligomerizes, it can bring together and activate caspase-1 monomers, through direct or indirect interactions.
OTHER INFLAMMASOMES
The lethal bacterial toxin of anthrax activates caspase-1 through another inflammasome, which involves the NLR NLRP1b (also called NALP1b). NLRP1b is different in humans and rodents, and the way in which it activates caspase-1 is somewhat controversial. It has a CARD, and, although some studies indicate that the activation of caspase-1 by NLRP1 requires ASC, others show that this protein can interact directly with caspase-1 through a CARD–CARD interaction.2 These two possibilities are shown in the simplified scheme in Figure 11.
Figure 11.

Two models of the NLRP1 inflammasome. Both may be correct in different settings.
Another inflammasome comprises yet another NLR, NLRP2 (also called NALP2), and ASC. It is not known what NLRP2 recognizes to trigger caspase-1 activation.
There is a type of inflammasome that does not involve an NLR. Double-stranded DNA in the cytosol binds to a protein called AIM2. AIM2 has a PyD that binds to the PyD of ASC. Therefore, when DNA appears in the cytosol, clusters of AIM2 form and, in turn, activate caspase-1 (Fig. 12).
Figure 12.

The AIM2 inflammasome. This is a simplified scheme, and AIM2 facilitates the formation of caspase-1 fibrils, as we saw for other inflammasomes.
AIM2 is a sensor for infection by both RNA and DNA viruses, although it also binds to any other double-stranded DNA, including that from the host (provided it appears in the cytosol). For this reason, the AIM2 inflammasome might have a role in the autoimmune response to double-stranded DNA in systemic lupus erythematosus and related diseases (although other intracellular DNA sensors, not relevant to our discussion here, have also been implicated).
CELL DEATH BY CASPASE-1 AND NONCANONICAL SECRETION
As we have discussed, the activation of caspase-1 can lead to apoptotic cell death, but this is often not the case. Like caspases-4, -5, and -11, caspase-1 can cleave and activate gasdermin D to cause necrotic pyroptosis. If gasdermin D is absent, caspase-1 can cause cell death by cleaving and thereby activating executioner caspases, caspase-7 being more important than caspase-3 in this case. Caspase-1 can also activate BID and thereby engage the mitochondrial pathway of apoptosis (see Green 2022e). Bacteria that infect cells often induce cell death through activation of caspase-1, and, in several cases, the inflammasome responsible for this effect has been identified (as indicated above). For example, Shigella flexneri kills macrophages in a manner that depends on NLRC4, ASC, and caspase-1.
When caspase-1 is activated, it processes interleukin-1β and interleukin-18, activating these cytokines. These are secreted, along with the unprocessed forms, as well as other inflammatory mediators, and this secretion occurs in a manner that is distinct from that of conventional secretion.3
A number of mechanisms have been proposed to explain this noncanonical secretion. However, there is an emerging idea that the release of these and other molecules from cells with active caspase-1 is actually due to the breakdown of the plasma membrane by gasdermin D during pyroptosis. This controversy is difficult to resolve one way or the other as the assays for the bioactive molecules and for cell death are different; if only a few cells die by pyroptosis and release potent factors, it can appear that the release is independent of cell death.
INFLAMMASOMES IN DISEASE
We need our inflammasomes to respond to many types of infections, but, when triggered inappropriately, inflammasomes can cause disease. A wide variety of inflammatory diseases are associated with activation of inflammasomes, especially NLRP3. We have already mentioned its role in gout and pseudogout, where inert crystals activate this inflammasome. The ability of crystals of cholesterol to activate the NLRP3 inflammasome also suggests a role in atherosclerosis, although this is not proven. Diets that are rich in fats (so-called “Western diets”) promote inflammation by inflammasome activation, and this inflammation can have a positive-feedback effect in adipose tissue to cause obesity. Inflammatory conditions associated with aging also have an NLRP3 component, and strikingly animals lacking NLRP3 do not show common pathologies of aging (although they are more susceptible to a wide range of infections, if exposed).
Familial mutations in NLRP3 have been identified that allow it to be activated spontaneously. Patients with such activating mutations often display recurrent fevers (without infection), pain in their joints and extremities, rashes, severe abdominal pain, and conjunctivitis, all of which can be catastrophic.
Many of the consequences of infection are due to inflammation, and, in turn, are due to activation of inflammasomes. The lethal effects of bacterial sepsis by a pathogenic E. coli was shown to depend on NLRC4; for example, interleukin-1, produced by inflammasome activation during infections, can promote damaging inflammation and can act on the vagus nerve to alter feeding behavior, causing anorexia.
But not all inflammasome activation is harmful. Inflammasome activation, in addition to promoting immunity, can also have other beneficial effects for health. For example, interleukin-18 can act to promote healing of the intestine and lungs. The production of this cytokine from macrophages in adipose tissue can prevent muscle wasting, a severe consequence of infection. Therefore, inflammasomes, like many things, present a yin–yang situation: although their activation can cause inflammation and damage, it can also promote immunity and repair.
NLR ACTIVATION AND THE RETURN OF THE JUST-SO STORY
The similarity between APAF1 and the NLRs hopefully has not escaped the reader, and indeed we can include it among the NLRs on the basis of sequence similarity (Fig. 13).
Figure 13.

APAF1 is in the Nod-like receptor (NLR) family.
This could mean that we can draw an analogy with APAF1 to explain how NLRs are activated by a ligand. If the LRR regions act like the WD region of APAF1, binding to a ligand might cause conformational changes that expose the nucleotide-binding site in the NACHT domain. Binding of a nucleotide would then, as in APAF1, induce further conformational changes that expose an oligomerization domain, and the protein–protein interaction site (e.g., CARD or PyD) would interact with ASC. At present, however, this is all speculation.
The relationship between APAF1 and the NLRs is tantalizing and brings us back to our just-so story about the evolution of apoptosis. It is easy to imagine that the ancient cell infected by the “mitochondrion to be” had a defense system in place to recognize such infections, using some type of NLR. Perhaps this NLR used bacterial cytochrome c as a kind of PAMP because this protein did not exist in the cell at that time. The recognition of the PAMP could have engaged a cell death mechanism to prevent the spread of the parasite to other cells. In this fantasy, the NLR eventually became APAF1, and the cell death mechanism became the mitochondrial pathway of apoptosis (Fig. 14).
Figure 14.

Return of the just-so story.
THE ACTIVATION OF CASPASE-2
Caspase-2 is a bit of a puzzle. It is the most highly conserved of the caspases throughout the animals and has been implicated in cell death caused by heat shock, cytoskeletal disruption, metabolic perturbation, and, perhaps, DNA damage, among other situations. However, in none of these cases has caspase-2 been shown unambiguously to be required for cell death.
Caspase-2 has a long prodomain containing a CARD and is activated by induced proximity (see Green 2022d). Unlike the other caspases, however, it is a rather poor activator of executioner caspases. Instead, it might require BID to cause cell death by engaging the mitochondrial pathway of apoptosis.
These considerations raise an intriguing question: Is the primary function of caspase-2 to cause apoptosis? Caspase-2 might have another function in cells that has not been identified. But it does seem to be important. Mice lacking caspase-2 are developmentally normal but age prematurely and are more prone to cancer in at least two model systems. Why this might be is considered in more detail below (and in Green 2022f).
The activation of caspase-2 involves an adapter protein called RAIDD, which binds to caspase-2 by a CARD–CARD interaction. RAIDD does not appear itself to oligomerize but instead might require additional molecules. One such molecule is PIDD. PIDD has a death domain (DD), and it binds to RAIDD via a DD–DD interaction. This is illustrated in Figure 15.
Figure 15.

The PIDDosome activates caspase-2. DD, death domain.
The caspase-2 activation platform is called a PIDDosome. The PIDDosome has been resolved at a structural level, which shows it to have multiple PIDD and RAIDD subunits (Fig. 16). This is similar to the structure of the CD95-DD–FADD-DD structure involved in the activation of caspase-8 (see Green 2022b).
Figure 16.

PIDDosome structure. The interactions of PIDD (P) and RAIDD (R) DDs are shown on the right. (Left, PDB 2OF5 [Park et al. 2007]; right, reprinted from Park et al. 2007, ©2007 with permission from Elsevier.)
PIDD is an interesting protein that undergoes extensive processing. It has a larger form that does not seem to be involved in activation of caspase-2 but instead activates the transcription factor NF-κB. However, this form of PIDD processes itself to a smaller form that can engage RAIDD. This processing is by an intein mechanism, in which sequences in the protein interact in a manner very similar to that of the function of proteases. This results in autocleavage of the protein to its smaller form. How the relative levels of the different species of PIDD are controlled is not clear.
This dual activity of PIDD is reminiscent of the ability of some of the death receptors to activate caspase-8 (and apoptosis) as well as NF-κB. It is not known whether NF-κB, induced by PIDD, can induce the expression of genes that influence the function of caspase-2, but the idea is intriguing.
Caspase-2 might function to monitor a cell-cycle process. During the cell cycle, the single centrosome in the cell replicates, and these move to opposite poles to orchestrate chromosome separation during mitosis. Only one of these centrosomes is “mature”—that is, it has associated proteins that only appear on the other centrosome after cell division. PIDD is localized to the mature centrosome (Fig. 17), and, under conditions in which extra mature centrosomes appear (such as failed cell division), caspase-2 is activated. This requires both PIDD and RAIDD (Fig. 18). As mentioned above, mice lacking caspase-2 show premature aging and an increased susceptibility to cancer in some model systems. Interestingly, cancers in caspase-2-deficient animals show less chromosomal stability than their caspase-2-sufficient counterparts—that is, they show increased abnormalities in chromosome numbers. These effects would be explained if caspase-2 activation resulted in apoptosis of cells with abnormal centrosomes. However, it seems that caspase-2 does not necessarily cause cell death in such cells, but can instead cause a cell-cycle arrest. This is because caspase-2 can stabilize the tumor-suppressor protein p53, which in turn can cause either apoptosis or cell-cycle arrest. We consider this function of caspase-2 in Green (2022f), in our discussion of the regulation of p53.
Figure 17.
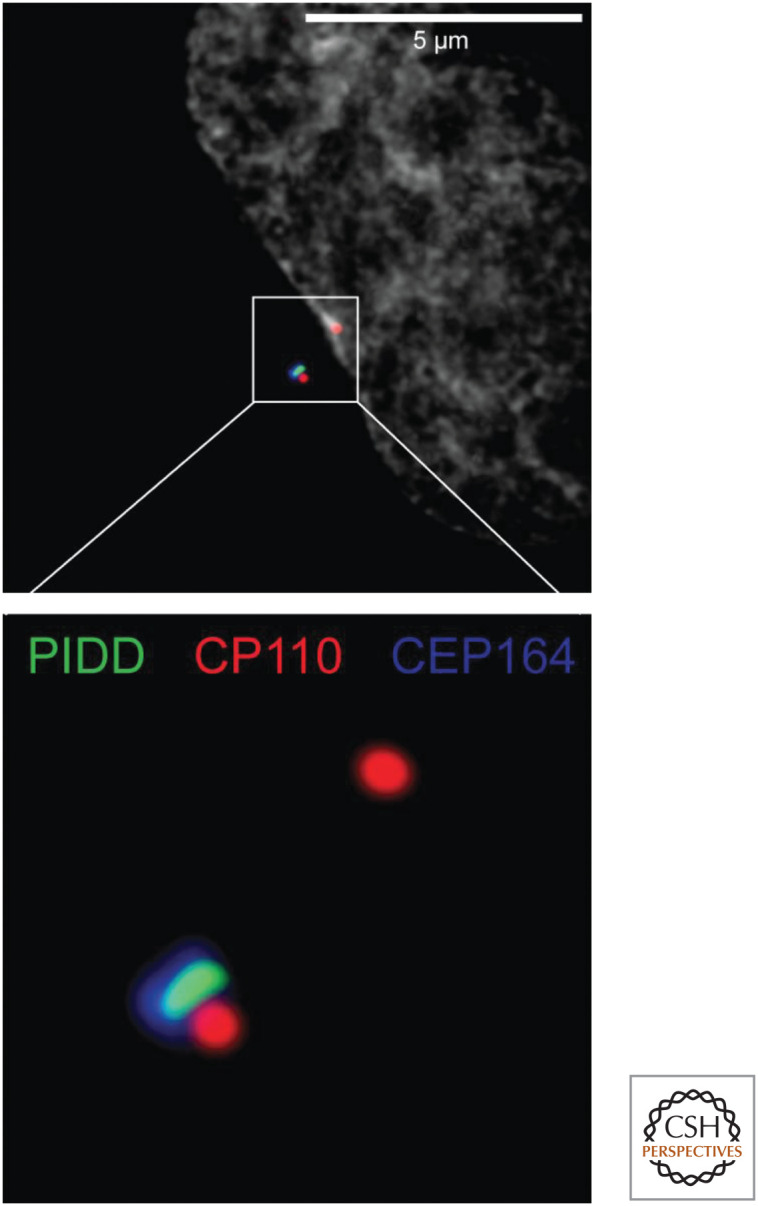
Fluorescence micrograph showing how PIDD localizes to mature centrosomes. PIDD (green) associates with the mature centrosome (which also contains CEP164, blue). The nascent (upper red spot) and mature centrosomes (lower complex of spots) both contain CP110 (red). The lower image is a magnified view of the area enclosed by the white square in the upper image. (Photo courtesy of Dr. Luca Fava and Dr. Andreas Villunger, University of Innsbruck.)
Figure 18.

Aberrant extra mature centrosomes activate caspase-2. During the S/G2 phases of the cell cycle, the centrosome duplicates, but PIDD remains associated with only the mature centrosome. If cytokinesis fails, leading to the formation of a tetraploid cell in the following G1 phase, two mature centrosomes appear in the cell, leading to PIDD–PIDD interaction, recruitment of RAIDD, and activation of caspase-2.
During the postnatal development of the liver, hepatocytes can double (or even quadruple) their chromosome numbers because of a failure in cytokinesis (the final step in mitosis). This effect is greatly enhanced in animals lacking PIDD or caspase-2. Again, this appears to be caused by the ability of caspase-2 to activate p53, which prevents further proliferation of hepatocytes with failed cytokinesis (and extra mature centrosomes).
Footnotes
DAMPs are sometimes called danger-associated molecular patterns, based on the “danger hypothesis.” Because danger is rather tautological, defined as anything that elicits a response, we prefer the term “damage,” but this has its own problems.
It is possible that this is a difference between the human and the murine protein.
Canonical secretion involves the transport of the protein into the endoplasmic reticulum, where it is then packaged into secretory vesicles that fuse with the plasma membrane to release the proteins. Interleukin-1β, interleukin-18, and other molecules released following caspase-1 activation are not secreted in this manner.
From the recent volume Cell Death: Apoptosis and Other Means to an End by Douglas R. Green
Additional Perspectives on Cell Death available at www.cshperspectives.org
ADDITIONAL READING The Inflammatory Caspases
Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F. 2014. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514: 187–192.
The activation of caspases-4, -5, and -11 by lipopolysaccharide.
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F. 2015. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526: 660–665.
One of the two original papers (with below) on the role of gasdermin D in pyroptosis.
Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, et al. 2015. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526: 666–671.
See above.
Kersse K, Vanden Berghe T, Lamkanfi M, Vandenabeele P. 2007. A phylogenetic and functional overview of inflammatory caspases and caspase-1-related CARD-only proteins. Biochem Soc Trans 35: 1508–1511.
An overview of the inflammatory caspases and the molecules that interact with them.
Cerretti DP, Kozlosky CJ, Mosley B, Nelson N, Van Ness K, Greenstreet TA, March CJ, Kronheim SR, Druck T, Cannizzaro LA, et al. 1992. Molecular cloning of the interleukin-1 β converting enzyme. Science 256: 97–100.
Two papers describing the identification of caspase-1, the first caspase discovered.
Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J, et al. 1992. A novel heterodimeric cysteine protease is required for interleukin-1 β processing in monocytes. Nature 356: 768–774.
See above.
Dinarello CA. 2018. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev 281: 8–27.
Interleukin-1 is a major target of caspase-1, and an understanding of its biological roles provides a context for appreciating the significance of caspase-1 function.
Keller M, Ruegg A, Werner S, Beer HD. 2008. Active caspase-1 is a regulator of unconventional protein secretion. Cell 132: 818–831.
The role of caspase-1 in secretion of proteins that lack conventional secretory signal sequences.
TLRs and NLRs
Gao D, Li W. 2017. Structures and recognition modes of toll-like receptors. Proteins 85: 3–9.
Meunier E, Broz P. 2017. Evolutionary convergence and divergence in NLR function and structure. Trends Immunol 38: 744–757.
Medzhitov R, Preston-Hurlburt P, Janeway CA Jr. 1997. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388: 394–397.
The original paper describing mammalian Toll-like receptors and their importance in the immune response.
Inflammasomes
Malik A, Kanneganti TD. 2017. Inflammasome activation and assembly at a glance. J Cell Sci 130: 3955–3963.
Lu A, Wu H. 2015. Structural mechanisms of inflammasome assembly. FEBS J 282: 435–444.
Duncan JA, Canna SW. 2018. The NLRC4 inflammasome. Immunol Rev 281: 115–123.
Lugrin J. Martinon F. 2018. The AIM2 inflammasome: sensor of pathogens and cellular perturbations. Immunol Rev 281: 99–114.
Martinon F, Burns K, Tschopp J. 2002. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol Cell 10: 417–426.
The first paper describing an inflammasome, which in this case involved NLRP1 (called “Nalp1”). Although this inflammasome was suggested to include caspase-5, all others described do not, and the details in this paper may not be entirely correct.
Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. 2004. NALP3 forms an IL-1β-processing inflammasome with increased activity in Muckle–Wells autoinflammatory disorder. Immunity 20: 319–325.
A description of the NLRP3 (called “NALP3”) inflammasome.
Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, Dixit VM. 2004. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430: 213–218.
A description of the NLRC4 (called IPAF) inflammasome.
Youm YH, Grant RW, McCabe LR, Albarado DC, Nguyen KY, Ravussin A, Pistell P, Newman S, Carter R, Laque A, et al. 2013. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab 18: 519–532.
A role for NLRP3 in aging.
Cell Death and Caspase-1 Activation
Gaidt, MM, Hornung V. 2017. The NLRP3 inflammasome renders cell death pro-inflammatory. J Mol Biol 430: 133–141.
A useful overview of the interplay of cell death pathways.
Kovacs SB, Miao EA. 2017. Gasdermins: Effectors of pyroptosis. Trends Cell Biol 27: 673–684.
Lamkanfi M, Moreira LO, Makena P, Spierings DC, Boyd K, Murray PJ, Green DR, Kanneganti TD. 2009. Caspase-7 deficiency protects from endotoxin-induced lymphocyte apoptosis and improves survival. Blood 113: 2742–2745.
One of two papers (see below) suggesting that the activation of caspase-7 by caspase-1 results in cell death. Note that this work preceded the discovery of gasdermin D.
Lamkanfi M, Kanneganti TD, Van Damme P, Vanden Berghe T, Vanoverberghe I, Vandekerckhove J, Vandenabeele P, Gevaert K, Nunez G. 2008. Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol Cell Proteomics 7: 2350–2363.
See above.
Caspase-2
Sladky V, Schuler F, Fava LL, Villunger, A. 2017. The resurrection of the PIDDosome—emerging roles in the DNA-damage response and centrosome surveillance. J Cell Sci 130: 3779–3787.
Roles for caspase-2 in apoptosis and other phenomena.
Forsberg J, Zhivotovsky B, Olsson M. 2017. Caspase-2: An orphan enzyme out of the shadows. Oncogene 36: 5441–5444.
Tinel A, Tschopp J. 2004. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science 304: 843–846.
The first characterization of a complex responsible for activating caspase-2.
Park HH, Logette E, Raunser S, Cuenin S, Walz T, Tschopp J, Wu H. 2007. Death domain assembly mechanism revealed by crystal structure of the oligomeric PIDDosome core complex. Cell 128: 533–546.
Bouchier-Hayes L, Oberst A, McStay GP, Connell S, Tait SW, Dillon CP, Flanagan JM, Beere HM, Green DR. 2009. Characterization of cytoplasmic caspase-2 activation by induced proximity. Mol Cell 35: 830–840.
The activation of caspase-2 by several stressors, analyzed using a live-cell-imaging technique.
Fava LL, Schuler F, Sladky V, Haschka MD, Soratroi C, Eiterer L, Demetz E, Weiss G, Geley S, Nigg EA, et al. 2017. The PIDDosome activates p53 in response to supernumerary centrosomes. Genes Dev 31: 34–45.
Ando K, Parsons MJ, Shah RB, Charendoff CI, Paris SL, Liu PH, Fassio SR, Rohrman BA, Thompson R, Oberst A, et al. 2017. NPM1 directs PIDDosome-dependent caspase-2 activation in the nucleolus. J Cell Biol 216: 1795–1810.
FIGURE CREDITS
- Gillray J. 1799. The Gout. [Etching] [Google Scholar]
- Park HH, Logette E, Raunser S, Cuenin S, Walz T, Tschopp J, Wu H. 2007. Death domain assembly mechanism revealed by crystal structure of the oligomeric PIDDosome core complex. Cell 128: 533–546. 10.1016/j.cell.2007.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
REFERENCES
*Reference is also in this collection.
- *.Green DR. 2022. a. The mitochondrial pathway of apoptosis, Part 1: MOMP and beyond. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a041038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Green DR. 2022. b. The death receptor pathway of apoptosis. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a041053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Green DR. 2022. c. Nonapoptotic cell death pathways. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a041079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Green DR. 2022. d. Caspase activation and inhibition. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a041020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Green DR. 2022. e. The mitochondrial pathway of apoptosis, Part II: the BCL-2 protein family. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a041046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Green DR. 2022. f. Cell death and cancer. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a041103 [DOI] [Google Scholar]