

CONTROLLING MITOCHONDRIAL OUTER MEMBRANE PERMEABILIZATION
We are now near the top of the mitochondrial pathway of apoptosis, at the point before the mitochondrial outer membrane permeabilizes to release proteins such as cytochrome c into the cytosol. This is where the major decisions are made that determine whether a cell will die by engaging this pathway. And these decisions depend on interactions among members of the BCL-2 family of proteins.
The BCL-2 proteins, named for the first family member to be described (B cell lymphoma-2), are a collection of related molecules found throughout the animal kingdom. They share only limited sequence similarity, except in short regions called BCL-2 homology (BH) domains, and can be grouped according to which of these domains they carry and, as we will see, by their functions. A list of several BCL-2 proteins in mammals and their BH domains is shown in Figure 1.
Figure 1.

BH domain organization of selected members of the BCL-2 family. TM, transmembrane domain (although in some cases, this is sequestered by the BH groove).
There are three “flavors” of BCL-2 proteins. The pro-apoptotic BCL-2 effectors promote apoptosis by causing mitochondrial outer membrane permeabilization (MOMP). These are the proteins that essentially make the holes in the outer mitochondrial membrane. The anti-apoptotic BCL-2 proteins, which include BCL-2 itself, prevent apoptosis by preventing MOMP. The third group is a subfamily of proteins that promote apoptosis by regulating the other two types of BCL-2 molecules. These are the BH3-only proteins, which share only the BH3 domain, hence their somewhat unfortunate designation.
The BCL-2 proteins are an alphabet soup of names, some sounding nearly identical and others bordering on the unpronounceable (BMF is “bimf,” if that helps). The acronyms have ceased to have any real meaning, and we will treat them here as simple (if confusing) names.1 Nevertheless, these molecules are essential to the mitochondrial pathway of apoptosis.
BAX AND BAK ARE THE EFFECTORS OF MOMP
At first glance, BAX and BAK seem to be quite different proteins, sharing little sequence similarity outside the short BH1, BH2, and BH3 domains. BAX is generally soluble in the cytosol but moves to mitochondria when apoptosis is induced (Fig. 2). In contrast, BAK is tethered to mitochondria in the cell by its carboxy-terminal region.
Figure 2.

BAX moves from the cytosol to the mitochondrial outer membrane during apoptosis. A cell engineered to express both BAX linked to green fluorescent protein (BAX–GFP) and a red fluorescent protein (RFP) that localizes to mitochondria was induced to undergo apoptosis. BAX (green) moves onto the mitochondria (red), revealed as the development of a yellow signal (i.e., merging of red and green emissions) over time. The first image was taken several hours after the initial stress. (Courtesy of Dr. Stephen Tait, University of Glasgow, Scotland, United Kingdom).
When BAX and BAK are activated, they bury themselves in the mitochondrial outer membrane, where they form oligomers of multiple sizes. Studies using artificial membranes have shown that these oligomers can form holes in membranes (technically, these are not pores or channels, both of which are much more organized). These openings are capable of allowing large molecules to pass through the membrane and effect the process of MOMP.
Mice engineered to lack either BAX or BAK develop normally, although BAX-deficient mice have reproductive problems, and cells from these animals undergo MOMP and apoptosis normally. However, animals lacking both BAX and BAK are another story—these have severe developmental defects that are usually lethal to the embryo. Cells from such double-deficient animals do not undergo MOMP or engage the mitochondrial pathway of apoptosis. Therefore, this seems to be a striking case of molecular redundancy—either BAX or BAK can effect MOMP, but at least one must be present.
Another BCL-2 family protein, BOK, is also a pro-apoptotic effector (together with BAX and BAK). Like BAX and BAK, BOK can effect MOMP to cause apoptosis. However, it is regulated in a distinct manner from that of the other effector proteins, and it is not involved in the responses to most of the stimuli that engage BAX and BAK.
The pro-apoptotic BCL-2 effector proteins directly cause MOMP by oligomerizing and inserting into the mitochondrial outer membrane. There are two models for how they do this. One model suggests that the activated proteins form a pore that is lined with the effector molecules arranged in their oligomeric complexes. An alternative model suggests instead that hydrophobic amino acids that are exposed in the activated effectors disrupt the lipids in the membrane, such that they produce a “lipidic pore” (Fig. 3). Although there is evidence for both models, most favors the lipidic pore—although this remains controversial.
Figure 3.

Proteinaceous versus lipidic pores. In a proteinaceous pore, the pore-forming protein “coats” the pore, penetrating the membrane. In a lipidic pore, the protein causes changes in the membrane lipids to produce the opening. Although some models propose a proteinaceous pore comprising the effector proteins (BAX, BAK, or BOK) for MOMP, most evidence supports the idea that these proteins induce the formation of lipidic pores to effect MOMP.
Apoptosis is activated by oncogenic transformation and suppresses cancer unless the apoptotic pathway is disrupted (Green 2022a). From this perspective, it is not surprising that tumors sometimes mutate BAX or BAK and may delete BOK. This is not, however, the only way a cancer can avoid cell death.
ANTI-APOPTOTIC BCL-2 PROTEINS PREVENT MOMP
The anti-apoptotic proteins BCL-2, BCL-xL, MCL-1, and A1 (among others) all act to prevent MOMP and block the mitochondrial pathway of apoptosis. These could work simply by preventing the oligomerization of BAX and BAK, which indeed they do. But how they do this brings us to the edge of waters that were made murky by a controversy that is now resolving into clarity.
At first, everything seems pretty simple. The anti-apoptotic BCL-2 proteins bind to active BAX and BAK. More specifically, they bind to the BH3 regions of BAX and BAK. The structures of BCL-xL bound to a peptide corresponding to the BH3 of BAX or BAK have been solved and are fairly informative (Fig. 4).
Figure 4.
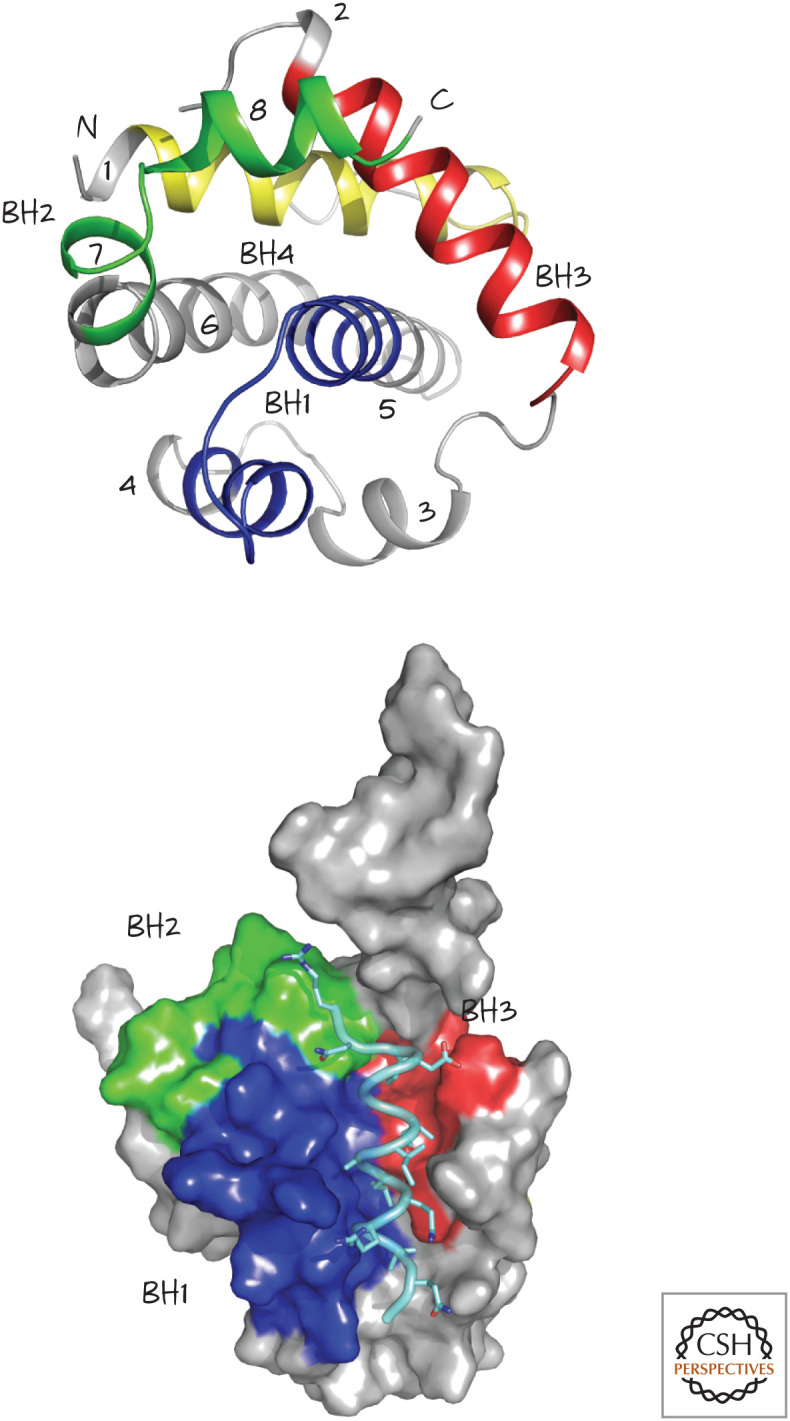
Structure of BCL-xL, a globular protein with eight α-helices (numbered 1–8, top). The four BCL-2 homology (BH) regions, BH1–BH4, are also shown. The structure on the bottom shows the groove formed by BH1, BH2, and BH3, bound to a BH3 peptide from BAK (pale blue). (Top, PDB 1MAZ [Muchmore et al. 1996]; bottom, PDB 1BXL [Sattler et al. 1997].)
The BH3 domains of BAX and BAK bind to a groove in BCL-xL formed from α-helices containing its BH1, BH2, and BH3 domains (the so-called BH groove2). This binding pocket is present in all of the anti-apoptotic BCL-2 proteins and is clearly crucial for their anti-apoptotic function because mutations affecting it can destroy the ability to block MOMP and apoptosis.
Here is the first bit of murk. Given that the BH3 regions of BAX and BAK are not exposed in the native proteins (Fig. 5), how could anti-apoptotic BCL-2 proteins bind to them? Indeed, if anti-apoptotic BCL-2 proteins are mixed with BAX or BAK, they do not bind them. However, if detergents are added, they now bind readily. This is a useful hint to what is going on: Anti-apoptotic BCL-2 proteins and pro-apoptotic effectors can only interact when they are embedded in a hydrophobic environment, such as the outer mitochondrial membrane.
Figure 5.

Effector BCL-2 family members BAX and BAK. The BCL-2 homology (BH) regions BH1, BH2, and BH3 are shown, as is the carboxy-terminal tail of BAX (gold). (Left, PDB 1F16 [Suzuki et al. 2000]; right, PDB 2IMS [Moldoveanu et al. 2006].)
So far, so good. Biochemical evidence indicates that, as BAX or BAK are activated, this exposes the BH3 domain of the protein, which opens a groove into which the BH3 domain of another BAX or BAK molecule can bind, forming a dimer. BCL-xL, then, can prevent this step in oligomerization by binding to this exposed BH3 domain and thereby preventing the BAX–BAX or BAK–BAK interactions required for MOMP (Fig. 6).
Figure 6.

BAK activation and inhibition. When BAK is activated, it exposes its BH3 region and appears to create a BH groove. This allows BAK–BAK oligomerization (upper) or binding of anti-apoptotic BCL-2 proteins that block oligomerization (lower).
The binding of anti-apoptotic BCL-2 proteins to BAX and BAK is not universal—there is some specificity to the interactions. The BH3 domain of BAK binds to the BH pockets of BCL-xL and MCL-1 very well, but much less effectively to BCL-2. Conversely, the BH3 domain of BAX binds very well to BCL-xL and BCL-2, but poorly to MCL-1 (Fig. 7).
Figure 7.
BAX/BAK specificity of anti-apoptotic BCL-2 proteins. The binding differences are relative, not absolute.
But here the already murky waters deepen. MCL-1, despite binding poorly to BAX, can block apoptosis very well when apoptosis is mediated by BAX (e.g., when there is no BAK). Similarly, BCL-2 binds poorly to BAK but prevents apoptosis that is mediated by BAK (e.g., when there is no BAX). We can conclude from this that apoptosis is not simply controlled by the balance of anti-apoptotic BCL-2 proteins and the pro-apoptotic effectors BAX and BAK. The latter idea, once called the “rheostat model” (Fig. 8), is not quite right—something is missing.
Figure 8.
The simple rheostat model. An increase in BAX or BAK causes apoptosis, whereas an increase in BCL2, BCL-xL, or MCL-1 results in survival. Problems with this simple model include the specificities of anti-apoptotic proteins for binding to the pro-apoptotic effectors.
BH3-ONLY PROTEINS PROMOTE MOMP AND APOPTOSIS
The “somethings” missing are the BH3-only proteins. Although the BH3 domains of BAX and BAK bind to the anti-apoptotic BCL-2 proteins, the BH3-only proteins also bind to the BH grooves of the anti-apoptotic molecules. The BH3 domain is not well conserved, however, and does not have sufficient sequence characteristics to permit its simple identification by straightforward bioinformatic approaches. Consequently, BH3-only proteins are generally identified by their functions (i.e., binding to anti-apoptotic BCL-2 proteins via a BH3-like region). Therefore, we do not know whether there are more BH3-only proteins waiting to be elucidated or whether other unrelated sequences can have similar functions (we return to this idea later). Some examples of bona fide BH3 sequences in BH3-only proteins are shown in Figure 9.
Figure 9.

(Left) BH3 regions of several human BH3-only proteins. BH3 regions of BAX, BAK, and BOK are shown for comparison. Note the conserved leucine (L) and aspartate (D) residues (*). Hydrophobic residues (h0–h4) are often (but not always) present where indicated. (Right) The binding of the BH3 region of BIM to MCL-1. The hydrophobic residues and the conserved aspartate interact with the BH groove of MCL-1.
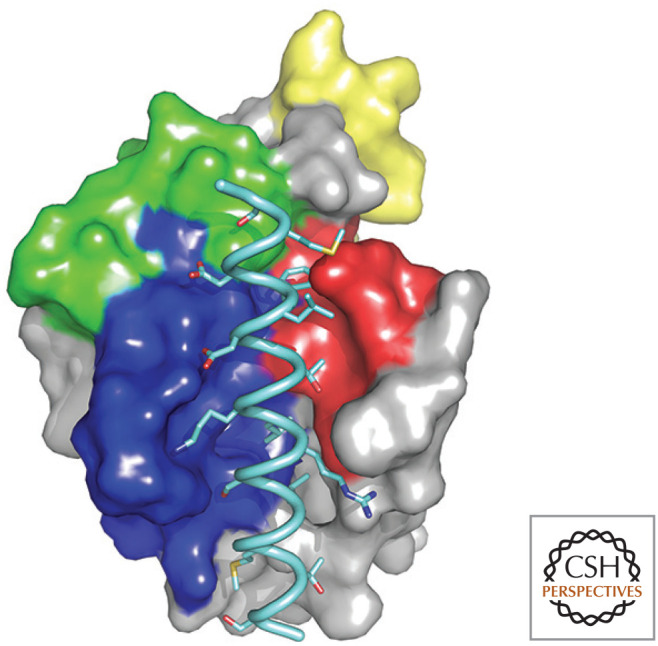
Unlike the pro-apoptotic effectors BAX and BAK, most of the BH3-only proteins are intrinsically unstructured, and their BH3 regions might be readily available for binding once the BH3 proteins are synthesized, rather than requiring activation by another protein. (An exception to this is the BH3-only protein BID, discussed in more detail below.) These BH3-only proteins can therefore interfere with the ability of an anti-apoptotic protein to bind to the BH3 region of other proteins and thus neutralize them. This depends, of course, on how well the BH3-only protein binds to a particular anti-apoptotic BCL-2 protein (and how much of the BH3-only protein is available). Figure 10 shows the relative specificities of several BH3 peptides from BH3-only proteins for different anti-apoptotic BCL-2 proteins.
Figure 10.

Specificities of some BH3-only proteins for binding to anti-apoptotic BCL-2 proteins Bcl-2, BCL-xL, and MCL-1.
In a cell expressing both BCL-2 and MCL-1, neutralization of only BCL-2 (e.g., by BAD) might not be sufficient to promote apoptosis unless MCL-1 is also blocked (e.g., by NOXA). This tells us that there could be cooperation between the BH3-only proteins in controlling apoptosis by the mitochondrial pathway. As we will see, however, inhibiting the anti-apoptotic BCL-2 proteins might not be enough to induce MOMP and apoptosis.
SOME BH3-ONLY PROTEINS ACTIVATE BAX AND BAK
As we saw above, the pro-apoptotic BCL-2 effectors BAX and BAK undergo conformational changes, insert into membranes, and oligomerize when they are activated. This results in MOMP unless anti-apoptotic BCL-2 proteins block them. So, if BH3-only proteins are available to block the anti-apoptotic BCL-2 proteins, what activates the effectors? It turns out that some BH3-only proteins have this function too.
BH3 peptides from the BH3-only proteins BID and BIM can trigger BAX or BAK oligomerization and permeabilization of synthetic membranes or isolated mitochondria. In addition, active forms of BID and BIM proteins can activate BAX and BAK to cause MOMP. For this reason, they are referred to as “direct activators.”
The BH3-only protein PUMA is another direct activator of BAX and BAK. This might be true of other BH3-only proteins as well, but we have yet to show this definitively. However, mice lacking PUMA, BIM, and BID do not have the developmental defects seen in BAX–BAK double-knockout mice. Even more remarkably, cells lacking all known BH3-only proteins can undergo MOMP and engage the mitochondrial pathway of apoptosis. Therefore, it is clear that there are other ways to activate BAX and BAK in addition to the action of the BH3-only proteins. There might also be other proteins or other types of molecular interactions that activate BAX and BAK. One of these non-BCL-2-family direct activators is considered in Green (2022a), but there is clearly more to the activation of BAX and BAK than we know.
ACTIVATION OF BAX AND BAK IS BY A “HIT-AND-RUN” MECHANISM
When BAX or BAK is activated by BID or BIM, the BH3-only direct activators do not remain associated with the pro-apoptotic effector proteins. A physical and transient interaction between BAX and active BID protein has been shown using Förster resonance energy transfer (FRET), a method that can detect interactions between proteins by monitoring the energy transfer between fluorescent tags attached to them. These studies showed that BID and BAX interact only when they are in membranes and that this is rapidly followed by the interaction of BAX with other BAX molecules (i.e., oligomerization) releasing the BH3-only direct activator. This is then followed by permeabilization of the membranes, presumably by BAX oligomers.
Insights into how the interaction of a direct activator with BAX or BAK results in their activation have come from structural studies. A BH3 peptide from the direct activator BID binds to BAX or BAK in solution, causing conformational changes in the effector molecules. The direct activator BID BH3 peptide binds to the BH groove of BAX or BAK, similar in structure to the BH grooves in anti-apoptotic BCL-2 proteins, but, unlike the latter, this moves the “latch” region of the protein (α6–α8) away from the core region defined by α1–α5 in BAX or BAK and exposes the BH3 of the effector (in α2). The bent core then straightens and interacts with the membrane (as do exposed hydrophobic amino acids in α2–α5) (Fig. 11). Meanwhile, the exposed BH3 of the effector binds to the groove of an adjacent effector molecule, forming a dimer (Fig. 12). It is likely that this binding displaces the BH3 of the direct activator BH3-only protein, accounting for the hit-and-run kinetics discussed above. Additional interactions (probably α6–α6, based on biochemical studies) allow these dimers to form higher-order oligomers in the mitochondrial outer membranes.
Figure 11.

(A) Structure of activated BAX. Following activation, the “latch” of the protein (α6–α8) moves away from the core (α2–α5), ultimately straightening α5–α6, and (B) exposed hydrophobic residues in the protein presumably interact with the lipids of the outer mitochondrial membrane. Similar events occur during activation of BAK.
Figure 12.

Activated BAX forms dimers by mutual BH3–groove interactions. The structure of the dimer formed by two BAX α2–α5 proteins (blue and green, respectively) showing the symmetrical binding of the BH3 from one in the groove of the other.
But why does this only happen on the mitochondria? There is evidence that specific lipids on the mitochondrial outer membrane participate in the activation process. Cardiolipin, present in the cell only on the mitochondrial inner membrane, can facilitate this activation of effector molecules, but its presence on the mitochondrial outer membrane is controversial. Conversely, a lipid formed on the mitochondrial outer membrane, sphingosine-1-phosphate, promotes the BH3-induced activation of BAK, whereas another product of the same pathway (the fatty aldehyde 2-trans-hexadecanal) promotes the activation of BAX. Interestingly, this lipid pathway is initiated by enzymes present in the endoplasmic reticulum (ER), suggesting that contact sites between the mitochondria and the ER are important for MOMP, and indeed there is evidence that these contact sites are where MOMP begins.
Unlike BAK, BAX is soluble in the cytosol. In this state, the BH groove of BAX is filled with the carboxy-terminal α9 helix of the protein. Another interaction exposes this α9 helix, which then interacts with the mitochondrial outer membrane, tethering the protein and preparing it for the activation events that follow. Other structural studies suggest a mechanism; the BIM BH3 domain does not bind to BAX in the region corresponding to the BH groove of an anti-apoptotic protein. Instead, it binds to the “back” of BAX (Fig. 13). This causes conformational changes in the BAX protein that might serve to expose the carboxy-terminal α9 helix of BAX, allowing it to tether to the mitochondria, while making the BH groove accessible to the BH3 region of direct activator proteins (Fig. 14). Therefore, it appears that there are two different interactions of the activating BH3-only proteins with BAX—one at the “back” to expose the BH groove and tether BAX to the membrane, then binding to the BH groove to induce the opening of the protein and its dimerization and subsequent oligomerization.
Figure 13.

The BIM BH3 region binds to BAX and induces conformational changes. Two views are shown. A BIM BH3 peptide (yellow) binds to the “back” face of BAX (brown), opposite the BH groove, where it displaces a loop to cause several subtle rearrangements in BAX. Illustrated are regions of the free (green) and BIM-bound (blue) BAX that undergo conformational changes. (PDB 1F16 [Suzuki et al. 2000]; overlay, PDB 2K7W [Gavathiotis et al. 2008].)
Figure 14.

Order of events for BAX activation and MOMP. (1) Active BID binds very rapidly to the mitochondrial outer membrane, and then (2) BID binds to the back of BAX, whereupon (3) BAX inserts its α-helix 9 (α9) into the membrane. Then (4) BID binds to the BH groove of BAX, “unlatching” BAX and exposing its BH3, and finally (5) BAX oligomerizes by binding to additional, active BAX molecules, inducing MOMP.
TWO MODELS OF BH3-ONLY PROTEIN FUNCTION IN APOPTOSIS
Currently, there are at least two different models for how BH3-only proteins cause MOMP and apoptosis. As we will see, these models are fairly similar on reflection, but they might have profoundly different consequences regarding our ability to manipulate apoptosis for therapeutic benefit.
In the first model, apoptosis is triggered when BH3-only proteins disrupt the interactions between anti-apoptotic BCL-2 proteins and the pro-apoptotic effectors BAX and BAK. This model is based on the idea that activation of BAX and/or BAK is neither rate limiting nor a decision point in cell death or survival. When active BAX or BAK are displaced from the anti-apoptotic proteins by the appropriate BH3-only proteins, BAX and/or BAK self-associate and promote MOMP and apoptosis. We refer to this as the “neutralization model” (Fig. 15).
Figure 15.

Neutralization model for BH3-only function. BH3-only proteins such as BAD bind to anti-apoptotic BCL-2 proteins (e.g., BCL-xL) to prevent or disrupt the binding of the latter to active BAX or BAK, and MOMP ensues.
The neutralization model is essentially an update of the rheostat model (Fig. 8) discussed earlier, because the relative levels of functional pro-apoptotic and anti-apoptotic BCL-2 proteins determine whether and when apoptosis occurs. In it, the BH3-only proteins drive apoptosis by reducing anti-apoptotic activity. Because BH3-only proteins bind to anti-apoptotic proteins with differing efficiency, the relationships can be complex, but ultimately life and death in this model are determined by the net anti-apoptotic activity in the cell.
In the second model (“direct activator/derepressor model”), activation of BAX and BAK is a crucial component of the process that helps to make the decision to undergo MOMP. Here, the anti-apoptotic BCL-2 proteins sequester direct activators of BAX and BAK if such activators are induced. Other BH3-only proteins drive apoptosis by displacing the direct activators, freeing the direct activators to trigger BAX and BAK. The latter BH3-only proteins, which lack direct activator function, act as sensitizers or derepressors in this model. This scheme is shown in Figure 16.
Figure 16.

Direct activator/derepressor model of BH3-only function. Derepressor BH3-only proteins, such as BAD, bind to anti-apoptotic BCL-2 proteins, preventing or disrupting the binding of the latter to direct activators of BAX and BAK (such as BIM). BAX and BAK are then activated, and MOMP ensues.
According to this second model, inhibition of anti-apoptotic functionality will not necessarily cause MOMP and apoptosis unless molecules with direct activator function are present. The major problem with this model is that, other than BID and BIM (and a few others), we do not know what else activates BAX and BAK.
Although the distinctions between these models are important, the two really are rather similar. BH3-only proteins either displace direct activators of BAX and BAK from anti-apoptotic BCL-2 proteins (model 2) or they displace active BAX or BAK from such proteins (model 1). Unified models of the action of the BH3-only proteins and the inhibition of apoptosis by the anti-apoptotic proteins combine the neutralization and direct activator/derepressor models by suggesting that both happen. Interestingly, the two modes of action of the anti-apoptotic proteins have different consequences for the cell. When anti-apoptotic BCL-2 proteins act predominantly to sequester the direct activator BH3-only proteins (MODE 1), the cells are then poised to rapidly undergo MOMP and apoptosis when this interaction is disrupted (e.g., by other BH3-only proteins). However, when the anti-apoptotic proteins act to sequester the active forms of the effectors, BAX and BAK (MODE 2), the cells appear to be more resistant to derepression (Fig. 17). It is possible that the complexes that form in MODE 2 are more stable than those in MODE 1, perhaps owing to additional interactions between the anti-apoptotic proteins and the effectors.
Figure 17.

Two modes of action of anti-apoptotic BCL-2 proteins. Anti-apoptotic BCL-2 proteins can prevent apoptosis either by binding to BH3-only direct activator proteins (MODE 1) or by binding to active BAX or BAK (MODE 2). Additional BH3-only protein interactions can neutralize the function of the anti-apoptotic BCL-2 protein, allowing apoptosis to proceed. MODE 1 is more readily derepressed than MODE 2 (i.e., MODE 2 is functionally more stable).
BOK IS REGULATED INDEPENDENTLY OF OTHER BCL-2 PROTEINS
Unlike BAX and BAK, BOK appears to be constitutively active, and its activity is not affected by the presence or absence of BH3-only proteins. Also, unlike BAX or BAK, it is not inhibited by BCL-2 or BCL-xL and binds to MCL-1 only very weakly. Instead, BOK seems to be controlled at the level of protein stability. When it is expressed, BOK mostly localizes to the ER, where components of a disposal system, called the endoplasmic reticulum–associated degradation (ERAD) apparatus targets BOK for degradation by the proteasome. If ERAD or the proteasome machinery becomes compromised, BOK accumulates and moves to the mitochondria, where it effects MOMP and apoptosis (Fig. 18).
Figure 18.

Regulation of BOK. BOK is not regulated by other BCL-2 proteins, but is controlled by endoplasmic reticulum–associated degradation (ERAD), which induces degradation of BOK. Disruption of ERAD or the degradation machinery allows BOK to accumulate and oligomerize on the outer mitochondrial membrane to cause mitochondrial outer membrane permeabilization (MOMP).
This helps to explain an apparent paradox. Cells that express high levels of anti-apoptotic proteins, such as some cancers, can nevertheless undergo apoptosis upon treatment with inhibitors of the proteasome. Because BOK functions independently of the anti-apoptotic proteins, it can promote MOMP and apoptosis under conditions in which it is stabilized.
We do not know the physiological conditions under which BOK promotes apoptosis,3 but, as seen in Green (2022b), BOK functions in development to remove unwanted cells.
BH3-ONLY PROTEINS ACT AS “STRESS SENSORS”
We have seen that BH3-only proteins can promote apoptosis in two ways: by directly activating the pro-apoptotic effectors BAX and BAK and by inhibiting the anti-apoptotic BCL-2 proteins. But when do they do this? The different BH3-only proteins have varied tissue distributions, are expressed under different conditions, and are regulated in different ways. They are targets of signal transduction pathways, and therefore we can think of them as “sensors” that connect the environment to the mitochondrial pathway of apoptosis.
The following are a few examples of BH3-only proteins functioning as sensors. In other reviews in this subject collection, we return to specific BH3-only proteins in the context of different physiological or pathological situations.
BID IS A PROTEASE SENSOR
Unlike the other BH3-only proteins that have been examined, BID is structured, and it looks similar to anti-apoptotic BCL-2 proteins and pro-apoptotic effectors (Fig. 19). The BH3 domain of BID in its native state is unavailable for interaction with other BCL-2 family proteins.
Figure 19.

Structure of BID, a structured BH3-only protein with one of the most divergent BCL-2 cores. The BH3 region (red) is next to a protease-susceptible loop, the digestion of which is required for activation. (PDB 2BID [Chou et al. 1999].)
BID has a large flexible loop that can be cleaved by a variety of proteases, including lysosomal proteases (cathepsins), the calcium-activated protease calpain, granzyme B, and caspases (Fig. 20). If BID is cut in this linker, the protein can now insert into mitochondrial membranes, and the BH3 domain presumably becomes exposed. BID can now interact with anti-apoptotic BCL-2 proteins that sequester it, or it can function to activate BAX and BAK, as we have seen. Therefore, if activating proteases appear in the cytoplasm of a cell, BID cleavage can engage the mitochondrial pathway of apoptosis.
Figure 20.

The pathway for cleavage and activation of BID.
BIM AND BAD ARE SENSORS FOR GROWTH FACTOR SIGNALING
When cells are deprived of growth factors, they often undergo apoptosis. In many cases, growth factor receptor signaling activates the serine/threonine-protein kinase AKT that phosphorylates (among other things) the transcription factor FOXO3a. The phosphorylated FOXO3a is sequestered in the cytosol by one of the 14-3-3 proteins that regulate the availability of signaling proteins in the cell, preventing FOXO3a from going to the cell nucleus. When growth factor signaling is disrupted, such as when growth factors become limiting, FOXO3a is released, transits into the nucleus, and triggers the transcription of the gene encoding BIM (Fig. 21). Lymphocytes from mice that lack BIM resist apoptosis caused by growth factor deprivation.
Figure 21.

Growth factor signaling controls BIM expression through the sequestration of FOX3a, a transcription factor required for transcriptional activation of the promoter for the gene encoding BIM.
This is not the only way in which BIM is regulated: The mitogen-activated protein kinase (MAP kinase) ERK is activated by a variety of signaling mechanisms (including growth factor receptor signaling), and this protein phosphorylates BIM, which targets it for degradation, thereby promoting cell survival in some settings. Another MAP kinase, called c-Jun amino-terminal kinase (JNK), has the opposite effect—when JNK phosphorylates BIM, this prevents BIM degradation, and therefore JNK can promote apoptosis by BIM. The interplay of the MAP kinases, therefore, can dictate how extracellular signals cause apoptosis through regulating BIM.
The BH3-only protein BAD is directly phosphorylated by AKT, and the phospho- BAD is then sequestered by 14-3-3 proteins (as we saw for the transcription factor FOXO3a). Following growth factor withdrawal, BAD is released and can now neutralize BCL-2 and BCL-xL, releasing active BIM (Fig. 22).
Figure 22.

Growth factor signaling controls BAD function through the sequestration of BAD.
BIM and BAD are not the only sensors of growth factor deprivation or related forms of cell stress. As we will see, there are other mechanisms whereby anti-apoptotic proteins, MCL-1 in particular, are regulated under growth factor deprivation, and these too contribute to the decision to die.
BIM AND BMF MEDIATE ANOIKIS
Severe disruption of the cytoskeleton induces apoptosis in many cells, and BH3-only proteins are involved in sensing such disturbance. This can happen when adherent cells lose their attachment to a surface or to basement membranes. The resulting apoptosis is called anoikis (“homelessness”) (Fig. 23).
Figure 23.

The pathway of anoikis allowing detached cells to be eliminated.
Dynein motor complexes move along the cytoskeleton in cells. Two BH3-only proteins, BIM and BMF, associate with specific motor complexes, binding to different dynein light chains. BIM appears to associate with cytoskeletal microtubules through this interaction, whereas BMF associates with another component of the cytoskeleton, the actin microfilaments. During anoikis, or in response to pharmacologic agents that disrupt the cytoskeleton, BIM and BMF are released to promote MOMP and apoptosis.
The control of BIM and BMF by the cytoskeleton is not universal, and in some cells, these BH3-only proteins are not associated in this way. Furthermore, it is not established that the cytoskeletal control of BIM and BMF is the only way in which anoikis occurs.4 Nevertheless, the interactions with dynein light chains illustrate one way in which BH3-only proteins act as potential sensors.
The above are only a few ways in which BH3-only proteins sense apoptotic signals and transduce them to the mitochondrial pathway. Additional examples are discussed in Green 2022a,b).
ANTI-APOPTOTIC BCL-2 PROTEINS ARE ALSO CONTROLLED BY SIGNALING
The anti-apoptotic protein MCL-1 is rapidly turned over in cells, and its levels appear to be actively regulated by phosphorylation, ubiquitylation, and degradation. One kinase that phosphorylates MCL-1 to target it for degradation is glycogen synthase kinase 3 (GSK3), which itself is inhibited by AKT. Therefore, when AKT activity is reduced on growth factor withdrawal, MCL-1 levels decline as a consequence of GSK3 function. This is illustrated in Figure 24. There are other kinases that similarly impact the stability of MCL-1 as well.
Figure 24.

Growth factor receptor signaling regulates the stability of MCL-1, either preventing or enhancing mitochondrial outer membrane permeabilization (MOMP), according to the presence (left) or absence (right) of growth factors.
Once MCL-1 is phosphorylated, it interacts with FBW7, an E3-ubiquitin ligase that places the small protein ubiquitin onto proteins, directing them to be degraded by the proteasome. Many cancers lose FBW7, and as a result, these cancers resist apoptosis by increasing MCL-1 levels. Conversely, inhibitors of mTOR, a kinase that acts in complexes to promote cell growth, can sensitize cells for apoptosis by derepressing the activity of GSK3, leading to increased MCL-1 degradation.
Other proteins are involved in control of the stability of MCL-1. MULE (also called HUWE1) is another of the E3-ubiquitin ligases that ubiquitylate MCL-1, targeting it for degradation. Intriguingly, MULE has a BH3 domain that binds to MCL-1, and this might interfere with its enzymatic activity. When it is displaced from MCL-1 by a BH3-only protein such as NOXA, MULE can then cause the degradation of MCL-1 by ubiquitylating it.
BCL-2 and BCL-xL are also phosphorylated, but in this case the role for phosphorylation is less clear. Phosphorylation of BCL-2 on one particular serine (serine 70) has been reported to increase its activity, but phosphorylation at other sites effectively inhibits it. These phosphorylation events occur in the unstructured region of the BCL-2 proteins between BH4 and BH3 (Figs. 1 and 3). This affects the interaction of the disordered region with the core of the protein, influencing the affinity of the BH groove for the BH3 regions of pro-apoptotic BCL-2 proteins.
Another interesting modification of BCL-xL has been described, involving conversion of two asparagine residues into aspartate or isoaspartate in the loop between the BH4 and BH1 regions. This reduces the affinity of the protein for BH3-only proteins, and might represent another mode of regulation. This process, called deamidation, is nonenzymatic, and is affected by intracellular pH, dependent on the sodium-proton antiporter NHE-1. Some cancers have defects in BCL-xL deamidation, making them resistant to DNA-damage-induced apoptosis. Again, this process occurs in the unstructured region of BCL-xL, between BH4 and BH3, affecting its interaction with the core of the protein and the function of the BH groove.
The anti-apoptotic BCL-2 proteins are regulated at the transcriptional level as well, and this might be the major way in which BCL-2 and BCL-xL are controlled. Several transcription factors induce expression of these proteins in different cell types, including nuclear factor-κB (NF-κB), which can produce anti-apoptotic effects in some settings.
BCL-2 PROTEINS IN OTHER ANIMALS
BCL-2 proteins are found throughout the animal kingdom. The situation in nematodes is particularly interesting. In Caenorhabditis elegans, the APAF1 homolog CED4 is held inactive on the mitochondrial outer membrane by CED9. It turns out that CED9 is a homolog of BCL-2 (Fig. 25).
Figure 25.

CED9 is a BCL-2 protein. The BH1–BH4 domains of CED9 and BCL2 are shown for comparison. Structural studies confirm that CED9 and BCL-2 are similar.
Despite this similarity, mammalian BCL-2 proteins do not sequester APAF1 or affect its activity. Therefore, although BCL-2 and CED9 have similar overt functions (inhibition of apoptosis), they perform these functions through different biochemical mechanisms.
CED9 has a BH groove like that of the mammalian anti-apoptotic BCL-2 proteins, but this is not involved in binding to CED4. However, another protein, EGL1, binds to this groove and leads to a conformational change in CED9 that causes the release of CED4, promoting apoptosis. This is shown in Figure 26.
Figure 26.

EGL1 functions to release CED4 from CED9.
EGL1 is a BH3-only protein that is expressed in response to transcription factors that are activated developmentally or in response to DNA damage in germ cells in the adult (considered in more detail in Green 2022b). We can now complete the core signaling pathway for apoptosis in nematodes, as shown in Figure 27.
Figure 27.

The action of participants in apoptotic signaling in nematodes.
As we know, the conformational changes that mammalian BCL-2 proteins undergo during apoptosis require the presence of the outer mitochondrial membrane, and this leads to an intriguing speculation: perhaps the conformational change that is induced by EGL1 in CED9 to release CED4 is facilitated by the mitochondrial membrane itself. This would help to explain the location of CED9 on mitochondria, despite the absence of MOMP upstream of the activation of the APAF1 homolog CED4 in this animal.
In Drosophila, two BCL-2 proteins have been identified on the basis of sequence similarity to mammalian BCL-2 proteins. These are DeBCL (pronounced “debacle”) and Buffy (named for the famous vampire slayer), also called DBorg1 and DBorg2, respectively. However, neither has been shown to have any role in apoptosis in Drosophila, which is intriguing, and their functions in flies are unknown. When we consider other roles for the vertebrate BCL-2 proteins later, it could be interesting to remember these.
Pro-apoptotic BCL-2 family effector proteins resembling BAX and BAK are also found among many invertebrate animal phyla, although not in nematodes or arthropods. This further supports the idea that the mitochondrial pathway of apoptosis in animals might be ancestral, but lost in some lineages.
VIRAL BCL-2 PROTEINS
Because viruses have come up with ways to block apoptosis, it should not be surprising that many viruses that infect mammalian cells express anti-apoptotic proteins related to the BCL-2 proteins. Examples can be found in several types of virus, including adenoviruses, herpes viruses, and pox viruses (Fig. 28). In each case, the anti-apoptotic protein shares weak sequence similarity to BCL-2, but regions can be identified that correspond to the BH domains.
Figure 28.

Viral BCL-2 proteins. All are from human viruses except where indicated. Some of the BH regions are based on structural considerations despite low amino acid homology. (Reprinted from Hardwick et al. 2009, ©2009 with permission from Elsevier.)
The structures of a few viral BCL-2 proteins have been solved, and these very closely resemble those of the cellular anti-apoptotic BCL-2 proteins (Fig. 29). In general, however, they lack the regions where regulatory events tend to occur in the cellular proteins, such as phosphorylation; perhaps this is not surprising.
Figure 29.

Structures of mammalian BCL-xL and Kaposi sarcoma virus vBCL-2 showing the arrangement of the BH1–BH4 regions. (Left, PDB 1MAZ [Muchmore et al. 1996]; right, PDB 1K3K [Huang et al. 2002].)
Viruses also express proteins that function like anti-apoptotic BCL-2 proteins (e.g., they bind and neutralize active BAX and BAK) but share no sequence or structural similarity with cellular BCL-2. An example is the vMIA protein from human cytomegalovirus. Such proteins can teach us a lot, not only about viral infection, but also how the cellular BCL-2 proteins work.
BACTERIAL TOXINS AND THE RETURN OF THE JUST-SO STORY
In Green (2022d), we discussed a just-so story—a speculation that apoptosis might have arisen with ancient endosymbiosis that resulted in mitochondria. A piece of that story involves BCL-2 proteins.
The structures of BCL-2 proteins BCL-2, BCL-xL, BCL-w, MCL-1, A1, BAX, BAK, BID, and vBCL-2 are remarkably similar to one another. Intriguingly, a similar structure is also found in some bacterial toxins, including diphtheria toxin β-chain and the colicins (Fig. 30). Each of these proteins undergoes a conformational change to bring a hydrophobic core into a membrane. The structural similarity might simply reflect this common function. But it is intriguing to think about this in the context of our just-so story.
Figure 30.

Structures of several pore-forming regions of bacterial toxins, compared with that of BAX. Several α-helical pore-forming toxins have a globular domain reminiscent of the BCL-2 core of BAX. The structures are colored blue to red from the amino to the carboxyl terminus, and the putative pore-forming helices are identified by their side-chain sticks. (Top left, PDB1MAZ [Muchmore et al.1996]; top right, PDB 1F0L [Choe et al. 1992]; lower left, PDB 1CII [Winer et al. 1997]; lower right, PDB 2C9K [Boonserm et al. 2006].)
Bacterial toxins of this type are often encoded in a discrete genetic element that actually makes two components—the toxin that creates holes in membranes and an immunity factor that makes the bacterium that produces it resistant to the toxin. When a bacterium harboring this element is stressed (e.g., by low levels of nutrients), it produces the toxin. This kills any bacteria that do not have the immunity factor, reducing competition for resources.
The original endosymbiosis, as we speculated, might have begun anything but cooperatively, with the bacterium being a parasite in the infected cell. In this scenario, the inner membrane of what would become the mitochondrion is the bacterial membrane (indeed, the inner membranes of modern mitochondria have lipid compositions that more closely resemble bacterial membranes than those of eukaryotes), whereas the outer membrane would have been derived from the infected cell (again, consistent with the composition of modern outer mitochondrial membranes). If stressed, the bacterium might synthesize a toxin that would target the next-nearest membrane—the mitochondrial outer membrane. So, was this the original pro-apoptotic effector?
With time, genetic control of the toxin might have transferred to the nucleus (as did nearly all of the original genes encoding mitochondrial elements, except for those encoding parts of the electron transport chain, ribosomal RNAs, and the tRNAs). Control of the integrity of what became the mitochondrial outer membrane now resided in nuclear genes. And the toxin, perhaps, became the BCL-2 family.
Finally, note that the mitochondrial inner membrane is not permeabilized by active BAX or BAK (we know this, because matrix proteins are not released with the proteins from the intermembrane space). Why not? Is there a distant descendant of the bacterial immunity factor that protects this membrane?
BCL-2-FAMILY PROTEINS HAVE OTHER ROLES IN CELLS
From the perspective of apoptosis, the major role of BCL-2 proteins is to control the integrity of the outer mitochondrial membrane. But these proteins appear to have additional functions in cells, and how these relate to their apoptotic effects and other aspects of cell physiology is emerging as an important area of research. If cell death is the work of the “night crew.” then what are the “day jobs” of the BCL-2 proteins?
Given their limited sequence homologies, BH3-only proteins might have numerous functions beyond apoptosis. In at least one case, a day job for a BH3-only protein has been identified. That protein is BAD. In addition to neutralizing some anti-apoptotic BCL-2 proteins, BAD has a role in regulating glucose metabolism in mammals.
We consider here three other day jobs for BCL-2 proteins: control of calcium homeostasis, regulation of mitochondrial dynamics (fission and fusion), and removal of mitochondria during development of some cells. As we will see, these might all have connections to the control of cell death as well as other effects in cells, and of course, there are probably other non-apoptotic roles yet to be identified.
BCL-2 PROTEINS ACT AT THE ER TO REGULATE CALCIUM
Inositol (1,4,5)-trisphosphate (IP3) is a phospholipid-derived molecule produced in some signaling pathways. IP3 binds to a receptor on the ER (the IP3 receptor), causing an efflux of calcium that regulates many different enzymes in the cytosol.
BCL-2 proteins influence the function of the IP3 receptor. Cells that lack BAX and BAK or overexpress BCL-2 show defective calcium efflux in response to IP3. For example, T lymphocytes lacking BAX and BAK do not elevate calcium in response to T-cell receptor engagement (which induces IP3 production) and are defective for T-cell activation.
How BCL-2 proteins control the IP3 receptor is not entirely clear. BCL-2 binds to the IP3 receptor, but beyond this, the mechanism is obscure. Intriguingly, some BH3- only proteins can induce an increase in intracellular calcium, and it might be that this is through interaction with BCL-2 or another BCL-2 protein. However, we do not know how this contributes to apoptosis or other cellular effects.
MITOCHONDRIAL DYNAMICS ARE INFLUENCED BY BCL-2 PROTEINS
One of the most intriguing day jobs for BCL-2 proteins is in mitochondrial dynamics. Mitochondria do not simply sit around in cells as discrete organelles; instead, they are constantly undergoing active fission and fusion by complex mechanisms that are conserved among the eukaryotes.
The first clue to this function is what happens during MOMP. At about the same time as the mitochondrial outer membrane becomes permeable and proteins from the intermembrane space are released, mitochondria often appear to undergo extensive fragmentation. At first, it was thought that this fission might contribute directly to MOMP, but that does not seem to be the case because MOMP can occur in isolated mitochondria without fission.
A second clue is that, in the absence of BAX and BAK, mitochondria are extensively fragmented, and this turns out not to be due to excess fission but rather to a decrease in mitochondrial fusion. In contrast, increasing the expression of anti-apoptotic BCL-xL increases mitochondrial fusion. Therefore, in two different situations in which MOMP is prevented (increased BCL-xL and decreased BAX and BAK), mitochondria display either more or less fusion. How can these observations and those of mitochondrial fission upon MOMP be reconciled?
Here is one way. If BAX and BAK promote mitochondrial fusion independently of their role in MOMP, when they become engaged during apoptosis (effectively leaving their day jobs to join the night crew), fusion decreases. Because BCL-xL prevents this (e.g., by sequestering activators of BAX and BAK, letting them remain doing their day jobs), fusion is enhanced.
How all this might occur is not known, but BAX and BAK can associate with a protein called mitofusin-2 that participates in mitochondrial fusion. Remarkably, the C. elegans protein CED9 can also associate with mitofusin-2 in mammalian cells and similarly enhances mitochondrial fusion. CED9 cannot block MOMP in mammalian cells but stimulates mitochondrial fusion under these artificial conditions.
When apoptosis occurs in C. elegans, mitochondrial fragmentation is observed, even though there is no MOMP (as we know, MOMP is not upstream in the apoptotic pathway in these animals). EGL1, which disengages CED9 from CED4 (Fig. 22), might therefore also take CED9 away from its day job, that of promoting mitochondrial fusion.
Mitochondrial fragmentation is also observed in Drosophila cells undergoing apoptosis. Here, not only is MOMP not upstream in the apoptotic pathway, but the BCL-2 proteins DeBcl and Buffy do not seem to have a role in cell death. Whether they participate in the mitochondrial fragmentation is not known.
Do these changes in mitochondrial dynamics have anything to do with apoptosis? Some studies have indicated that a protein involved in mitochondrial fission, DRP-1, might help to promote MOMP, and cells in which this protein is blocked can become refractory to MOMP and apoptosis. However, this seems to be the case even when mitochondrial fission and fusion cannot occur, and so the role of this protein in MOMP remains somewhat obscure. However, as we saw above, the carboxyl terminus of BAX must insert into the outer mitochondrial membrane for subsequent activation and MOMP (e.g., Fig. 14). This turns out to depend on the curvature of the mitochondrial membrane, and this, in turn, is affected by mitochondrial dynamics. Smaller, fragmented mitochondria are more curved, and this facilitates the insertion of the BAX carboxy-terminal α9-helix. By promoting mitochondrial fragmentation, active BAX (and BAK) facilitate the insertion and activation of more BAX molecules.
Therefore, the interplay between mitochondrial dynamics and the function of the BCL-2 proteins might go both ways. Other conditions that favor mitochondrial fusion or fission (such as the availability of oxygen or nutrients and the engagement of metabolic pathways) can therefore influence apoptosis.
BCL-2 PROTEINS CAN PARTICIPATE IN THE REMOVAL OF MITOCHONDRIA
Yet another function of the BCL-2 proteins also relates to mitochondria, but in this case it is the way in which excess or damaged mitochondria are removed. This occurs by autophagy, the “self-eating” process that allows cells to survive nutrient deprivation and remove damaged or unwanted organelles. We have much more to say about autophagy in Green (2022e). Autophagy of the mitochondria (mitophagy) involves the creation of a membrane vesicle around the organelle and fusion of the vesicle with a lysosome results in the digestion of the contents of the vesicle.
In at least some cases, the signals that bring the autophagy machinery to the mitochondria are members of the BCL-2 family—in particular, two closely related BH3-only proteins, BNIP3 and NIX. Neither BNIP3 nor NIX seems to have a significant role in apoptosis; instead, they appear to function in mitophagy. In mice lacking NIX, mature red blood cells, which normally lack mitochondria because these have been removed by mitophagy, are found to harbor these organelles (Fig. 31). Another mechanism that promotes mitophagy is discussed in Green (2022e).
Figure 31.

Mitochondria in NIX-deficient red blood cells. In developing wild-type red blood cells, mitochondria are removed by mitophagy (left), and these organelles are not present in the mature cells. In NIX-deficient mature red cells, mitochondria persist, many showing signs of damage (right). (Image courtesy of Paul A. Ney, MD, St. Jude Children's Research Hospital, Memphis, Tennessee.)
Other studies have suggested that, during hypoxia (reduced oxygen levels, a situation in which having fewer mitochondria is desirable), a transcription factor called HIF-1 is activated that causes expression of BNIP3. The latter then appears to promote the autophagic removal of mitochondria.
How these BH3-only proteins cause mitophagy is not completely clear, but the process might involve binding to BCL-2 and displacing a protein that is bound to it, Beclin-1. Beclin-1 is part of the machinery that initiates autophagy, and its release from BCL-2 on mitochondria might attract components of the autophagy pathway to these organelles (Fig. 32). It is particularly noteworthy that Beclin-1 contains a BH3 region and binds to the BH groove in BCL-xL (Fig. 33).
Figure 32.

Hypoxia leads to stabilization of hypoxia-induced factor HIF and removal of mitochondria by autophagy.
Figure 33.
Beclin-1 BH3 region bound to BCL-xL. The BH regions of BCL-xL are colored as in Figure 4, and the BH3 peptide is pale blue. (PDB2P1L [Oberstein et al. 2007].)
We return to the role of beclin-1 and BCL-2 in autophagy in Green (2022e). For now, it is sufficient to realize that the day jobs of BCL-2 proteins can involve the type of interactions involved in apoptosis (as in this example) or other activities and interactions.
Footnotes
To make matters a bit worse, many of the BH3-only proteins have distinct gene names (the gene encoding PUMA is BBC3).
This is also called a “BC groove,” with the “B” referring to the binding of BH3 regions of target proteins and the “C” referring to the binding of the protein's own carboxyl terminus in some of the structured BCL-2 proteins. Here, we use “BH groove” because it is formed from the BH1, BH2, and BH3 domains.
It is likely that the regulation of BOK is more complex than indicated here. Some cells in the body (as well as some tumor cells) express stable BOK protein, yet do not spontaneously die. How this can occur is currently unknown.
There is at least one other possibility. When adherent cells lose contact with their substrate, they often stick together (this also applies to daughters of a dividing adherent cell lacking substrate attachment). As a consequence, one cell often engulfs the other, a process called “entosis.” The engulfed cell dies. This could be why many adherent cells in vitro do not grow in semisolid medium; whenever they divide, one cell is killed by entosis. Entosis is considered in more detail in Green (2022c).
From the recent volume Cell Death: Apoptosis and Other Means to an End by Douglas R. Green
Additional Perspectives on Cell Death available at www.cshperspectives.org
ADDITIONAL READING
BCL-2 Proteins and MOMP
Birkinshaw RW, Czabotar PE. 2017. The BCL-2 family of proteins and mitochondrial outer membrane permeabilisation. Semin Cell Dev Biol 72: 152–162.
Kalkavan H, Green DR. 2018. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ 25: 46–55.
Kale J, Osterlund EJ, Andrews DW. 2018. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ 25: 65–80.
Suhaili SH, Karimian H, Stellato M, Lee TH, Aguilar MI. 2017. Mitochondrial outer membrane permeabilization: a focus on the role of mitochondrial membrane structural organization. Biophys Rev 9: 443–457.
Anti-Apoptotic BCL-2 Proteins
Vaux DL, Cory S, Adams JM. 1988. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 335: 440–442.
The first paper to show that BCL-2 preserves cell survival in primary and transformed cells.
Hockenbery D, Nunez G, Milliman C, Schreiber RD, Korsmeyer SJ. 1990. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature 348: 334–336.
The first paper to show that BCL-2 is associated with mitochondria (the original assertion that it is on the inner membrane was due to an artifact) and that BCL-2 inhibits apoptosis.
Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. 1997. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science 275: 1132–1136.
One of two papers (see below) identifying the role of BCL-2 in blocking the release of cytochrome c from mitochondria.
Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. 1997. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 275: 1129–1132.
See above.
Vaux DL, Weissman IL, Kim SK. 1992. Prevention of programmed cell death in Caenorhabditis elegans by human bcl-2. Science 258: 1955–1957.
One of three papers that showed that CED9 and BCL-2 are functionally equivalent in nematodes. Although BCL-2 does not bind to CED4, it can sequester EGL1, which was not shown until later.
Hengartner MO, Horvitz HR. 1994. C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell 76: 665–676.
See above.
Jabbour AM, Puryer MA, Yu JY, Lithgow T, Riffkin CD, Ashley DM, Vaux DL, Ekert PG, Hawkins CJ. 2006. Human Bcl-2 cannot directly inhibit the Caenorhabditis elegans Apaf-1 homologue CED-4, but can interact with EGL-1. J Cell Sci 119: 2572–2582.
See above.
Pro-Apoptotic BCL-2 Effectors
Oltvai ZN, Milliman CL, Korsmeyer SJ. 1993. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 74: 609–619.
The discovery of BAX.
Chittenden T, Harrington EA, O'Connor R, Flemington C, Lutz RJ, Evan GI, Guild BC. 1995. Induction of apoptosis by the Bcl-2 homologue Bak. Nature 374: 733–736.
The discovery of BAK.
Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. 2001. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730.
This paper showed that BAX and BAK are necessary for MOMP and apoptosis via the mitochondrial pathway.
Llambi F, Victor B, Wang Y, Yang M, Schneider DM, Gingras S, Zheng J, Parsons MJ, Brown SA, Pelletier S, et al. 2016. BOK is a non-canonical BCL-2 family effector of apoptosis regulated by ER-associated degradation. Cell 165: 421–433.
This paper shows that Bok can also promote MOMP in a manner that can be independent of other Bcl-2 family proteins.
BH3-Only Proteins
Giam M, Huang DC, Bouillet P. 2008. BH3-only proteins and their roles in programmed cell death. Oncogene (Suppl 1) 27: S128–S136.
Stoka V, Turk B, Schendel SL, Kim TH, Cirman T, Snipas SJ, Ellerby LM, Bredesen D, Freeze H, Abrahamson M, et al. 2001. Lysosomal protease pathways to apoptosis. Cleavage of bid, not pro-caspases, is the most likely route. J Biol Chem 276: 3149–3157.
BID as a protease sensor for engaging the mitochondrial pathway of apoptosis.
Datta SR, Ranger AM, Lin MZ, Sturgill JF, Ma YC, Cowan CW, Dikkes P, Korsmeyer SJ, Greenberg ME. 2002. Survival factor–mediated BAD phosphorylation raises the mitochondrial threshold for apoptosis. Dev Cell 3: 631–643.
The regulation of BAD by survival factor signaling.
Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Kontgen F, Adams JM, Strasser A. 1999. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science 286: 1735–1738.
The BIM knockout mouse illustrates roles for this protein in apoptosis.
Puthalakath H, Huang DC, O'Reilly LA, King SM, Strasser A. 1999. The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol Cell 3: 287–296.
The association of BIM with the cytoskeleton.
Conradt B, Horvitz HR. 1998. The C. elegans protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9. Cell 93: 519–529.
EGL1 is a BH3-only protein in nematodes.
BCL-2 Protein Interactions
Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. 2002. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2: 183–192.
The proposal of the “direct activator/derepressor” (called “sensitizer”) model of BH3-only protein function.
Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DC. 2005. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 17: 393–403.
The “neutralization” model of BH3-only protein function and specificities of BH3-only proteins for different anti-apoptotic proteins.
Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM, Huang DC. 2005. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev 19: 1294–1305.
The “neutralization” model of BH3-only protein function.
Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. 2002. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111: 331–342.
Biochemical characterization of BAX activation and membrane permeabilization.
Kim H, Tu HC, Ren D, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH. 2009. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell 36: 487–499.
Further analysis of the “direct activator/derepressor” model of BH3-only protein function.
Lovell JF, Billen LP, Bindner S, Shamas-Din A, Fradin C, Leber B, Andrews DW. 2008. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 135: 1074–1084.
Use of FRET technology to characterize derepression and direct activation of BAX.
Leber B, Lin J, Andrews DW. 2007. Embedded together: the life and death consequences of interaction of the Bcl-2 family with membranes. Apoptosis 12: 897–911.
One of two approaches (with below) to unify the neutralization and sensitizer/depression models of BH3-only protein action.
Llambi F, Moldoveanu, T, Tait SW, Bouchier-Hayes L, Temirov J, McCormick LL, Dillon CP, Green, DR. 2011. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol Cell 44: 517–531.
See above.
Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, Tu HC, Kim H, Cheng EH, Tjandra N, et al. 2008. BAX activation is initiated at a novel interaction site. Nature 455: 1076–1081.
Structural analysis of the binding of the BIM BH3 region to BAX and early steps in BAX activation.
Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, Lee EF, Yao S, Robin AY, Smith BJ, et al. 2013. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell 152: 519–531.
Insights into the activation of BAX to form oligomers.
Moldoveanu T, Grace CR, Llambi F, Nourse A, Fitzgerald P, Gehring K, Kriwacki RW, Green DR. 2013. BID-induced structural changes in BAK promote apoptosis. Nat Struct Mol Biol 20: 589–597.
Insights into the activation of BAK.
Other Functions of the BCL-2 Family
Karbowski M, Lee YJ, Gaume B, Jeong SY, Frank S, Nechushtan A, Santel A, Fuller M, Smith CL, Youle RJ. 2002. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol 159: 931–938.
Characterization of interactions between BAX and proteins involved in mitochondrial fission and fusion.
Sheridan C, Delivani P, Cullen SP, Martin SJ. 2008. Bax- or Bak-induced mitochondrial fission can be uncoupled from cytochrome c release. Mol Cell 31: 570–585.
Evidence that mitochondrial fission occurs at the time of MOMP but is not required for permeabilization.
Rong Y, Distelhorst CW. 2008. Bcl-2 protein family members: Versatile regulators of calcium signaling in cell survival and apoptosis. Annu Rev Physiol 70: 73–91.
Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, Berridge MJ, Conway SJ, Holmes AB, Mignery GA, et al. 2004. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol 166: 193–203.
Characterization of the regulation of calcium homeostasis by BCL-2.
Levine B, Sinha S, Kroemer G. 2008. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy 4: 600–606.
Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. 2005. Bcl-2 antiapoptotic proteins inhibit Beclin 1–dependent autophagy. Cell 122: 927–939.
A mechanism for the regulation of autophagy by BCL-2. Autophagy is covered in more detail in Chapter 8.
Cuconati A, White E. 2002. Viral homologs of BCL-2: Role of apoptosis in the regulation of virus infection. Genes Dev 16: 2465–2478.
REFERENCES
*Reference is also in this collection.
- *.Green DR. 2022. a. Cell death and cancer. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a041103 [DOI] [Google Scholar]
- *.Green DR. 2022. b. Cell death in development. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a041095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Green DR. 2022. c. The burial: clearance and consequences. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a041087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Green DR. 2022. d. The mitochondrial pathway of apoptosis, Part 1: MOMP and beyond. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a041038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Green DR. 2021. e. Nonapoptotic cell death pathways. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a041079 [DOI] [PMC free article] [PubMed] [Google Scholar]
FIGURE CREDITS
- Boonserm P, Mo M, Angsuthanasombat C, Lescar J. 2006. Structure of the functional form of the mosquito larvicidal Cry4Aa toxin from Bacillus thuringensis at a 2.8-angstrom resolution. J Bacteriol 188: 3391–3401. 10.1128/JB.111.9.3391-3401.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe S, Bennett M, Fujii G, Curmi PM, Kantardjieff KA, Collier RJ, Eisenberg D. 1992. The crystal structure of diphtheria toxin. Nature 357: 216–222. 10.1038/357216a0 [DOI] [PubMed] [Google Scholar]
- Chou JJ, Li H, Salvesen GS, Yuan J, Wagner G. 1999. Solution structure of BID, an intracellular amplifier of apoptotic signaling. Cell 96: 615–624. 10.1016/S0092-8674(00)80572-3 [DOI] [PubMed] [Google Scholar]
- Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, Tu HC, Kim H, Cheng EH, Tjandra N, et al. 2008. BAX inactivation is initiated at a novel interaction site. Nature 455: 1076–1081. 10.138/nature07396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick JM, Youle RJ. 2009. SnapShot: BCL-2 proteins. Cell 138: 404. 10.1016/j.cell.2009.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q, Petros AM, Virgin HW, Fesik SW, Olejniczak ET. 2002. Solution structure of a Bcl-2 homolog from Kaposi sarcoma virus. Proc Natl Acad Sci 99: 3428–3433. 10.1073/pnas.062525799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldoveanu T, Liu Q, Tocilj A, Watson M, Shore G, Gehring K. 2006. The x-ray structure of a BAK homodimer reveals an inhibitory zinc binding site. Mol Cell 24: 677–688. 10.1016/j.molcell.2006.10.014 [DOI] [PubMed] [Google Scholar]
- Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS, Nettesheim D, Chang BS, Thompson CB, Wong SL, et al. 1996. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature 381: 335–341. 10.1038/381335a0 [DOI] [PubMed] [Google Scholar]
- Oberstein A, Jeffrey PD, Shi Y. 2007. Crystal structure of the Bcl-XL-Beclin 1 peptide complex Beclin 1 is a novel BH3-only protein. J Biol Chem 282: 13123–13132. 10.1074/jbc.M700492200 [DOI] [PubMed] [Google Scholar]
- Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, et al. 1997. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science 275: 983–986. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Youle RJ, Tjandra N. 2000. Structure of Bax: coregulation of dimer formation and intracellular localization. Cell 103: 645–654. 10.1016/S0092-8674(00)00167-7 [DOI] [PubMed] [Google Scholar]
- Wiener M, Freymann D, Ghosh P, Stroud RM. 1997. Crystal formation of colicin Ia. Nature 385: 461–464. [DOI] [PubMed] [Google Scholar]