

Abstract
The V(D)J recombination process rearranges the variable (V), diversity (D), and joining (J) genes in the immunoglobulin (IG) loci to generate antibody repertoires. Annotation of these loci across various species and predicting the V, D, and J genes (IG genes) are critical for studies of the adaptive immune system. However, because the standard gene finding algorithms are not suitable for predicting IG genes, they have been semimanually annotated in very few species. We developed the IGDetective algorithm for predicting IG genes and applied it to species with the assembled IG loci. IGDetective generated the first large collection of IG genes across many species and enabled their evolutionary analysis, including the analysis of the “bat IG diversity” hypothesis. This analysis revealed extremely conserved V genes in evolutionary distant species, indicating that these genes may be subjected to the same selective pressure, for example, pressure driven by common pathogens. IGDetective also revealed extremely diverged V genes and a new family of evolutionary conserved V genes in bats with unusual noncanonical cysteines. Moreover, unlike all other previously reported antibodies, these cysteines are located within complementarity-determining regions. Because cysteines form disulfide bonds, we hypothesize that these cysteine-rich V genes might generate antibodies with noncanonical conformations and could potentially form a unique part of the immune repertoire in bats. We also analyzed the diversity landscape of the recombination signal sequences and revealed their features that trigger the high/low usage of the IG genes.
Introduction
Antibodies (or immunoglobulins [IGs]) are the key components of the immune system of jawed vertebrates that provide adaptive immune response by recognizing and neutralizing antigens. Antibodies are not encoded in the germline genome but rather result from somatic VDJ recombinations (Tonegawa 1983). This process affects IG loci containing the families of the variable (V), diversity (D), and joining (J) genes (referred to as IG genes) by selecting one V gene, one D gene, and one J gene and concatenating them together to generate one of the antibody chains. Antibodies are further diversified by somatic hypermutations (Dudley et al. 2005).
The diversity of the IG loci is driven by the variety of antigens: Different species encounter different antigens and develop their unique ways to fight them through mutations in IG genes. As a result, the IG loci have rapidly evolved independently in different species and resulted in a diverse collection of IG genes that remain largely unknown because both sequencing highly repetitive IG loci and predicting IG genes in these loci are challenging tasks (Das et al. 2012; Pettinello and Dooley 2014).
Mammalian genomes have three IG loci—heavy chain (IGH), kappa light chain (IGK), and lambda light chain (IGL)—as well as four T-cell antigen receptor (TCR) loci (TRA, TRB, TRG, and TRD). In this work, we mainly focus on the IGH locus. The V, D, and J genes in the IGH locus (and the fragments of the IGH locus containing these genes) are also referred to as IGHV, IGHD, and IGHJ, respectively.
Diversity of IG genes
Studies of adaptive immune responses across various vertebrate species open new therapeutic opportunities (Muyldermans and Smider 2016). For example, studies of single-chain camelid antibodies led to the development of nanobodies that are able to diagnose cancer (Rashidian et al. 2015; Keyaerts et al. 2016), whereas studies of ultralong cow antibodies revealed that they recognize various HIV strains (Sok et al. 2017). Understanding the diversity of the adaptive immune systems across various vertebrates can also contribute to analyzing the spread of newly emerged zoonotic pathogens. It is particularly important for bat species that possess a unique immune system capable of neutralizing viruses that are often lethal to other mammals, such as rabies, Ebola, SARS-CoV, MERS-CoV, and SARS-CoV-2 (Teeling et al. 2018).
Schountz et al. (2017) formulated a “bat IG diversity” hypothesis that argues that, because bats have a greater combinatorial diversity of IG genes than other mammals, their large naive antibody repertoires do not need substantial affinity maturation to successfully neutralize antigens. Indeed, although the human IGH locus has only 55 functional V genes, the little brown bat is estimated to have at least 200 V genes (Bratsch et al. 2011), suggesting that bats may be better equipped for clonal selection of B cells responding to viral antigens. However, the IG genes in bats remain poorly characterized; moreover, the only support for the “bat IG diversity” hypothesis comes from a probabilistic model (Bratsch et al. 2011) rather than annotated IG loci in a well-assembled bat genome.
Annotation of IG genes
Annotation of the IG loci (i.e., predicting IG genes) is a prerequisite for most follow-up immunogenomics studies. Because assembly and annotation of the IG loci for novel species are complicated by their highly repetitive structure (Matsuda et al. 1998; Watson et al. 2013; Rodriguez et al. 2020), the sequences of the IG loci are only known for a few species. However, recent advancements in long-read sequencing enabled the first contiguous assemblies of highly repetitive genomic regions, and the Vertebrate Genome Project (VGP) now aims to generate high-quality reference genomes for all vertebrate species (Rhie et al. 2021). So far, the VGP has generated well-assembled genomes of nearly 150 vertebrate species.
Although many IG loci have been assembled in the last two years, their automated annotation remains an open problem. Previous studies of IG genes combined a time-consuming experimental approach with semimanual computational analysis, such as in the studies of the platypus (Gambón-Deza et al. 2009), the cow (Ma et al. 2016), and the ferret (Wong et al. 2020). These studies used the human IG genes for a similarity-based detection of IG genes in the novel species and may have missed diverged IG genes. For example, because most (if not all) V genes in mammalian species were predicted based on their similarities with human V and J genes, it remains unclear whether there exist still-unknown families of V genes that are highly diverged from the human IG genes.
Annotation of recombination signal sequences
De novo prediction of IG genes relies on identifying conserved recombination signal sequences (RSSs) that flank IG genes and enable the VDJ recombination. Because short RSSs have many spurious RSS-like occurrences in the genome (cryptic RSSs), their prediction and downstream IG gene annotation are challenging problems. It is further complicated by the fact that some cryptic RSSs are implicated in unusual genomic rearrangements outside the IG and TCR loci (Messier et al. 2003) and sometimes play an important role in antibody generation. For example, cryptic RSSs flanking the human LAIR1 gene participate in the off-target VDJ recombination and generate new types of antibodies (where LAIR1 represents an additional domain of an unusual VDJ region) that broadly neutralize Plasmodium parasites (Tan et al. 2016). Although Teng et al. (2015) analyzed the impact of off-target VDJ recombinations on lymphocyte genomes, no attempts to analyze the landscape of cryptic RSSs within the IG loci have been made yet.
The problem of identifying RSSs in a genomic sequence was considered by Merelli et al. (2010) (RSSsite tool) and Olivieri et al. (2013) (VgeneExtractor tool). VgeneExtractor further used its RSS predictions for detecting V genes in various species. However, this method does not account for the variations in the RSSs within species and does not report D and J genes. Safonova and Pevzner (2020) recently benchmarked RSSsite and showed that it results in a high false-positive rate. Even though they developed a more accurate SEARCH-D algorithm for detecting RSSs of D genes, the problem of detecting RSSs for all types of IG genes and the follow-up evolutionary analysis of IG genes across multiple species remains open.
Role of cysteines in IGs
Cysteines are structurally important amino acids that form disulfide bonds in IGs (Frangione et al. 1969). Conserved cysteines of human IGs are referred to as canonical cysteines (Tonegawa 1983). Noncanonical cysteines are common in some biomedically important species, such as llamas and cows (de Los Rios et al. 2015; Prabakaran and Chowdhury 2020). The current consensus is that the additional disulfide bonds in IGs increase their stability and enrich the complexity of antigen-binding site topology (Wu et al. 2012). Because cysteine patterns represent a structurally important feature of antibodies, we analyzed noncanonical cysteines in the newly identified IG genes.
IGDetective tool
We describe the IGDetective tool for predicting IG genes, apply it for annotating IG loci in the recently assembled mammalian genomes, generate the largest set of IG genes to date, and reveal the surprising diversity of IG genes across multiple species, such as a new family of unusual cysteine-rich V genes in bats.
Because all previous attempts to annotate V (J) genes in newly sequenced species relied on their similarities with known human IG genes, it remains unclear whether there exist V (J) genes that are not similar to the human ones. In addition to a similarity-based search for IG genes (IterativeIGDetective mode), IGDetective has a BlindIGDetective mode for annotating IG genes in the absence of any prior knowledge about the sequences of IG genes in other species. We benchmark BlindIGDetective on well-annotated human, mouse, and cow genomes; show that it automatically derives nearly all known IG genes in these species in a blind fashion; apply it to newly sequenced genomes to reveal highly diverged IG genes; and reveal new families of highly diverged V genes in bats that evade the similarity-based approach to predicting IG genes.
Results
From predicting RSSs to predicting IG genes
Sequences of human IG genes were inferred as the result of painstaking manual analysis at the dawn of the DNA sequencing era (Li et al. 2002). Consequently, sequences of all nonhuman IG genes were inferred based on similarities with human IG genes (Sitnikova and Su 1998). IterativeIGDetective automates this approach and iteratively extends it by predicting more and more distant IG genes at each iteration. In contrast, BlindIGDetective is designed to predict IG genes even if they share no similarities with currently known IG genes.
Given a newly assembled genome, both IterativeIGDetective and BlindIGDetective start from predicting RSSs in this genome. Each RSS consists of a conserved heptamer followed by a nonconserved spacer (12 nt long in D genes and 23 nt long in V and J genes in the IGH locus) and a conserved nonamer (Fig. 1A). Each V (J) gene has an RSS at the 3′ (5′) end, and each D gene is flanked by RSSs on both ends. We henceforth refer to the 3′ (5′) flanking signal of a V (J) gene as RSSV (RSSJ) and the 5′ (3′) flanking signals of a D gene as RSSDleft (RSSDright).
Figure 1.

IterativeIGDetective (A) and BlindIGDetective (B) pipelines. (A) IterativeIGDetective iteratively extends the set of identified IGHV genes. The “known V genes” box represents known V genes in a reference genome; the “RSSV motif” box represents a profile formed by the reference RSSVs for human V genes. (A1) After identifying a contig containing the IGH locus in the target genome, IterativeIGDetective identifies candidate RSSs for V genes in this contig based on similarities with the human RSS motif. A region preceding a true-positive RSS represents a V gene, whereas a region preceding a false-positive RSS does not. (A2) A region preceding a candidate RSS is classified as a human-like V gene if its similarity with a known human V gene exceeds a similarity threshold. (A3) Target-like V genes in the target genome are identified based on similarities with human-like V genes detected in step (A2). (A3*) Target-like V genes are iteratively identified based on previously detected target-like V genes until no new genes are identified. (B) BlindIGDetective constructs the V-graph, analyzes connected components in this graph, finds clumps of colocalized fragments in each connected component of this graph, and combines the found clumps into clusters that represent candidate families of V genes. (B1) Candidate RSSVs in the entire target genome are identified based on similarities with the human RSSV motif for V genes formed by the reference human RSSVs (represented by the “RSSV motif” box). (B2) The V-graph is constructed on the vertex-set of all RSSVs. Two vertices (RSSVs) in the V-graph are connected by an edge if fragments preceding these RSSVs are similar. True (false)-positive RSSVs form large (small) connected components in the V-graph. (B3) Each connected component in the V-graph is partitioned into clumps of colocated genes. (B4) Nontrivial clumps (containing multiple RSSs from the same connected component and clustered within a short region of the genome) represent putative V genes within putative IG loci. Note that a vertex is not included in a clump if it is not similar to all other vertices in this clump (like the light green vertex in the rightmost clump). At the final step (not shown), BlindIGDetective combines the identified clumps into clusters to reveal IG genes.
Because RSS motifs are highly conserved across all mammals, the human RSS motif (Fig. 1A) can be used for predicting RSS motifs in other mammalian species. IGDetective forms a motif profile from all known RSSs in a reference genome and uses this profile to evaluate the likelihood ratio of an arbitrary string from a target genome to decide whether this string represents a putative RSS (see Methods). For each genomic position, it computes the likelihood ratio that there is an RSS flanking this position and classifies a position as a candidate RSS if this ratio exceeds a likelihood threshold (see Methods).
IterativeIGDetective pipeline
IterativeIGDetective predicts IG genes in the target genome by leveraging the knowledge of IG genes in well-studied reference genomes (human, mouse, and cow). Although the highly repetitive IGH loci in a few other genomes have been semimanually assembled and annotated (e.g., IG loci in pig [Eguchi-Ogawa et al. 2010], goat [Du et al. 2018], and rabbit [Gertz et al. 2013]), it remains unclear how accurate these short-read assemblies are because it is difficult to assemble the IG loci even from long reads (Bankevich and Pevzner 2020), let alone from short reads. In the absence of accurate IG annotation tools, it is also unclear what are the false-positive/false-negative rates of the manually predicted IG genes in these assemblies.
Figure 1A illustrates the IterativeIGDetective pipeline with emphasis on detecting V genes. It starts by identifying a contig (or multiple contigs) containing the IGH locus in the target species and finding candidate RSSs in this locus based on their similarity to the known RSSs in the reference species. Afterward, it analyzes the genomic region flanked by the found RSSs to predict the IG genes themselves. In the case of V and J genes (which are longer and more conserved than D genes), it classifies a region preceding/following the identified RSS as a novel V/J gene if its similarity with a known V/J gene exceeds a similarity threshold determined by percentage of identity and shared k-mers (for parameters specifying similarity thresholds, see Supplemental Methods, “Identification of candidate V and J genes”).
IterativeIGDetective extends the noniterative mode described above (that ends with the similarity-based identification of V/J genes) by the iterative mode for identifying novel V/J genes whose similarity with known V/J genes does not exceed the similarity threshold. The iterative mode extends the set of known V/J genes by the newly identified V/J genes and uses this extended set to iteratively repeat the noniterative mode until no novel V/J genes are found (Fig. 1A).
The challenge of identifying highly divergent IG genes
Although IterativeIGDetective identifies many candidate IG genes in target species, it is not capable of detecting distant IG genes that significantly deviate from all canonical human IG genes, for example, IG genes from a distant family that has not been discovered yet. Reducing the similarity threshold in IterativeIGDetective increases its false-positive rate and does not necessarily lead to identifying distant IG genes.
We hypothesize that, similar to IG genes in known families, highly diverged IG genes from a novel family should (1) display some degree of pairwise similarity to all other IG genes from the same family and (2) be located within a relatively short region of the genome. Based on these two “IG family” criteria, BlindIGDetective identifies novel IG genes that do not resemble known human IG genes by constructing the similarity graph described below.
Similarity graph
Given a position s in a genome, a parameter direction (downstream, “−,” or upstream, “+”), and an integer segment-length (L), we define the s-fragment as the segment of length L either downstream from or upstream of this position depending on the parameter direction. We define the coding length of an s-fragment as the length of the longest out of three open reading frames ending at position s (in the case direction = “−”) or starting at position s (in the case direction = “+”). For simplicity, below we assume that direction = “−.”
Given a set S of positions in a genome, parameter direction, and parameter segment-length, we define an edge-weighted similarity graph (referred to as the S-graph) as follows. Each vertex in this graph corresponds to a position in the set S, and each edge connects similar s-fragments, where similarity is established based on percentage of identity between s-fragments. All isolated vertices are removed from the similarity graph. The edge-weight of an edge (s,s’) is defined as the percent identity between the s-fragment and the s’-fragment. Given a parameter span (default value, 0.5 Mb), vertices in the same connected component of the similarity graph are classified as colocated if they are separated by less than span nucleotides in the genome.
Given a vertex v in a connected component, its clump is defined as the set of all vertices colocated with v after removing all vertices whose percent identity with any other single vertex in the clump does not exceed the similarity threshold (default value 70%). A vertex with a maximum-size clump in a given connected component is called the center of this component (ties are resolved randomly). Two clumps are linked if the distance between their center vertices does not exceed a distance threshold (default value, 1 Mbp). BlindIGDetective constructs clusters using single linkage clustering of linked clumps and analyzes the constructed large clusters (of size larger than the default value smallSize = 3) as putative IG genes within a single IG locus. Clusters are further analyzed as candidates for new IG families as they satisfy the “IG family” criteria specified above.
BlindIGDetective pipeline
Given a position-set S, parameter direction, and parameter segment-length, BlindIGDetective constructs the S-graph (Fig. 1B). Below, we limit attention to the case when S is the set of starting positions of RSSVs predicted by IGDetective, direction = “–”, and segment-length = 350 bp. Because this setting models the search for V genes, we refer to the resulting S-graph (s-fragments) as the V-graph (v-fragments). The default parameters direction and segment-length will need to be modified for D-graphs and J-graphs because they depend on the position of the gene (5′ or 3′ end) with respect to the RSS and the typical length of the gene. Because IG genes in known genomes are located within relatively short regions (e.g., human IGHV genes are located within an 850-kbp-long IGHV locus), a clump of colocated vertices within a connected component of the V-graph may reveal a family of V genes in a newly sequenced genome. To generate such clumps for each connected component, BlindIGDetective forms a clump of the center vertex in this component, removes vertices of the constructed clump from this connected component, and iterates until the component has no vertices left. It further combines clumps into clusters as described above.
We benchmark BlindIGDetective on the entire human genome and show that it reveals the vast majority of human V (J) genes; for example, it finds 52 out of 57 known human IGHV genes without any prior information about their sequences while limiting possible false positives (with respect to known human V genes) to seven. Some of these false positives may represent novel candidate V genes in the human genome (see subsection “BlindIGDetective reveals novel candidate V genes in the human genome”). Although BlindIGDetective missed a small number (five) of 57 human IGHV genes with “weak” RSSs, these genes can be easily identified by slightly reducing the likelihood threshold with a follow-up similarity search against 52 identified IGHVs. In addition to finding IGHV genes, BlindIGDetective finds V genes from other IG and TR loci and even orphan V genes on Chromosomes 15 and 16 (Supplemental Table S1) that resulted from segmental duplications of the human IGH locus (Nagaoka et al. 1994). We thus argue that applying this approach to any mammalian species would reveal most V genes in this species, including highly diverged V genes missed by IterativeIGDetective and unusual genes that are affected by off-target VDJ recombinations.
Data sets
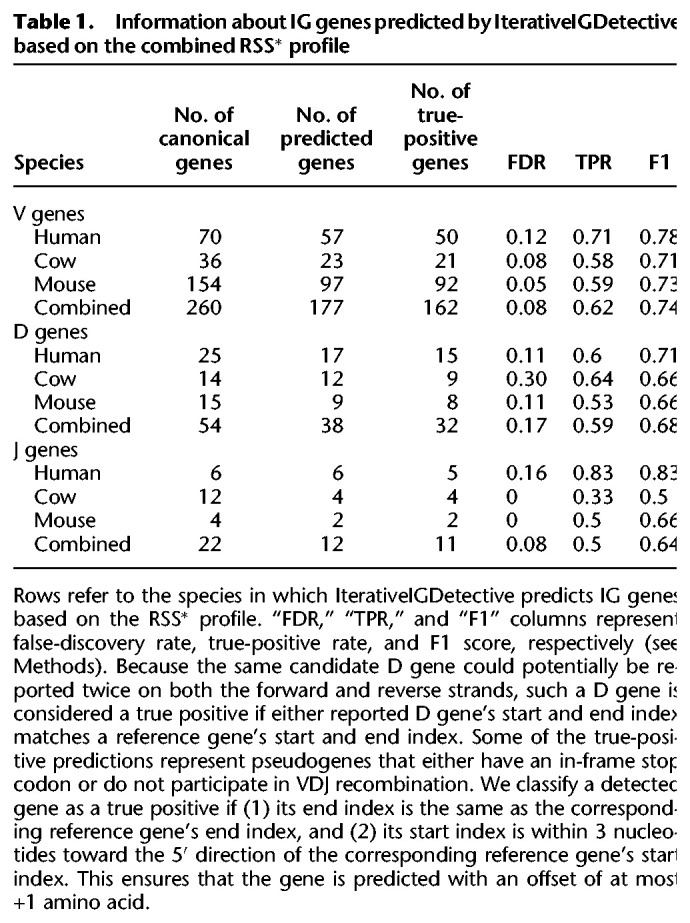
We extracted known IG genes from the ImMunoGeneTics (IMGT) database (Lefranc et al. 2015) for the three reference genomes and mapped them to the IGH locus of the same species as described in the Supplemental Methods, “Annotating IG genes in reference genomes,” and Supplemental Table S2. Table 1 presents information about the mapped IG genes in the reference species that we refer to as canonical IG genes. We also generated a combined set of IG genes by combining human, mouse, and cow IG genes, thus adding one more “reference” to the human, mouse, and cow references. We refer to the profile of all RSSs in this set (for each type of IG gene) as the combined RSS and denote it as RSS*.
Table 1.
Information about IG genes predicted by IterativeIGDetective based on the combined RSS* profile
We selected 20 target mammalian species for prediction of IG genes: three great ape species (Kronenberg et al. 2018) and 17 species assembled by the VGP consortium (Rhie et al. 2021) that represent a wide range of biological orders (Fig. 2A; Supplemental Table S3). For each target species, we identified contigs containing fragments of the IGH loci (IGH-contigs) as described in the Supplemental Methods, “Identifying putative IGH-contigs in a genome assembly,” and Supplemental Table S4.
Figure 2.

Information about the IGH loci in 20 target mammalian species. (A) The phylogenetic tree formed by 20 target and three reference species. The tree was subsampled from the Tree of Life constructed by Hedges et al. (2015). Each species is shown by its common name and a VGP identifier specified in parentheses if available. Each species is encoded by a unique color (left vertical color panel) and a color representing its order (right vertical color panel). The list of orders is shown in the upper left corner. Here and below visualization was performed using the BioRender and Iroki (Moore et al. 2020) tools. (B) Information about the IGH loci of 20 target species. Each line corresponds to a target species and shows fragments of the IGH locus with positions of candidate V (blue), D (orange), J (green), and C (red) genes. For six out of 20 species, the IGH-end covers <80% of the predicted IGH locus length (lynx, blue whale, mastiff bat, horseshoe bat, chimpanzee, and grey squirrel). We showed both the IGH-start and the IGH-end for these six species and only the IGH-end for the remaining species. For a better visualization, all IGH loci are shown as having the same length that does not reflect their real lengths (the bar plots next to the IGH loci show the predicted lengths). The map on the right shows the counts of the productive V, D, J, and C genes identified in the IGH locus. A C gene is classified as productive if its translated regions (defined by the closest human C gene) does not contain stop codons. Nonzero counts are shown in green. The green triangles indicate partially found and (likely) partially missing D and J genes in the sloth IGH locus. Although IterativeIGDetective did not identify any J genes in two species (chimpanzee and spear-nosed bat) within the boundaries of the IGH loci, it found a highly conserved candidate J gene in a short contig in the chimpanzee assembly (denoted as 1* on the map of the right). IterativeIGDetective did not identify any candidate J genes in the spear-nosed bat, presumably because all its RSSJs did not pass the likelihood threshold.
Benchmarking IterativeIGDetective
We benchmark IterativeIGDetective on the IG loci of the human, mouse, and cow genomes by assuming that one of them represents a reference genome and one of the remaining ones represents a target genome. Because the IG genes and RSSs in these species are known, we evaluate IterativeIGDetective based on (1) maximizing the number of the known RSSs and IG genes predicted by IterativeIGDetective and (2) minimizing the number of false RSSs and IG genes predicted by IGDetective. To this end, we attempt to maximize the true-positive rate (TPR) while minimizing the false-discovery rate (FDR).
We tabulated heptamer and nonamer likelihood thresholds (see Methods) (Supplemental Table S5) and visualized the percentage of RSSs and heptamers/nonamers passing the human reference Lmin likelihood thresholds (Supplemental Fig. S1). The vast majority of the heptamers and nonamers in the genome have very low likelihood ratios.
Given the identified likelihood thresholds for each reference (human, mouse, cow, or combined), we first launched IterativeIGDetective with these thresholds on the same species by considering them as target species. We then tabulated the number of detected candidate signals in the human IGH locus (in four cases that represent identifications of putative human RSSs using RSS profiles in human, cow, mouse, and combined) and computed true positives (candidate signals representing canonical signals), false positives (candidate signals that are not canonical signals), and false negatives (canonical signals that are not candidate signals) in Supplemental Table S6. Afterward, we tabulated the RSS detection statistics of IGDetective on all combinations of four references and four targets in Supplemental Methods, “Extended benchmarking of IterativeIGDetective,” and Supplemental Table S7. Finally, we extracted the V, D, and J genes from the candidate RSSs determined by launching IterativeIGDetective in noniterative mode (see subsection “Identification of candidate IG genes” in Methods) with the RSS profile based on the combined reference. Table 1 provides information about the results of IterativeIGDetective on the reference species and using the combined RSS* profile.
Detecting IG genes in target genomes
We applied IterativeIGDetective using the combined RSS* profile to IGH-contigs of all target species (Supplemental Methods, “Analysis of RSSs in reference and target species”; Supplemental Table S8). Because the identified IGH-contigs are usually longer than the IGH loci, the predicted RSSs may include many false positives. For example, the number of predicted RSSV candidates for a single species varies from 69 to 7027 with the median value 995 (Supplemental Table S3). However, further similarity-based filtering (described in Supplemental Methods, “Identification of candidate V and J genes”) of regions flanking these candidate RSSVs greatly reduces the number of false-positive predictions, resulting in three to 64 V genes per species (the median value is 34) (Supplemental Table S3). In total, IterativeIGDetective found 1021 candidate V genes across 20 target species, including 50 target-like V genes (Supplemental Table S9). Thirty-four out of 50 target-like V genes share at least 80% percent identity with other V genes identified at the previous iteration (Supplemental Table S9). After filtering candidates with stop codons in the open reading frame, the number of candidate V genes was reduced to 581.
The number of predicted RSSJs varies from 54 to 3911 and from four to 21 (with the median value, eight) after similarity-based filtering, resulting in a total of 174 candidate J genes. After applying the additional filters based on the conservation of the tryptophan-encoding TGG codon in the candidate J genes (Supplemental Methods, “Comparative analysis of candidate IGHJ genes”; Supplemental Fig. S2), the number of candidate J genes was reduced to 60.
The number of predicted RSSDs varies from one to 17 with a median value of four. IterativeIGDetective identified a total of 137 candidate D genes that were extracted as short regions flanked by the predicted RSSDs (without any filtering). After redefining the boundaries (see Supplemental Methods, “Computing boundaries of the IGH loci using predicted IG genes”) of the IGH loci, we discarded 45 candidate D genes located outside these loci (to minimize the number of false-positive D genes), resulting in a set of 92 candidate D genes.
For each target species, we also found positions of constant (C) IG genes in its assembly by aligning highly conserved human IGHC genes using Bowtie 2 (Langmead and Salzberg 2012). The number of IGHC genes per species varies from two to 19 with the median value seven, resulting in 149 IGHC gene candidates.
The IGH loci widely vary in length across mammalian species
We analyzed the positions of the candidate V, D, J, and C genes within the assembled genome in order to identify the boundaries of the IGH loci and their lengths assuming the standard V→D→J→C ordering (see Methods). Long repeats within the IGHV locus often break its assembly into multiple contigs, with one of the contigs containing the first i V genes (referred to as the IGH-start) and another contig (referred to as an IGH-end) containing all (or some of) the remaining V genes as well as D, J, and C genes (Fig. 2B). See Supplemental Methods, “Unusual IGH locus in the sloth genome,” and Supplemental Figure S3.
The sloth and the spear-nosed bat have the longest IGH loci among analyzed species (6.7 and 4.6 Mbp, respectively), whereas aquatic animals (vaquita, blue whale, platypus, sea lion, and dolphin) have the shortest IGH loci, varying from 311 kbp for the vaquita to 607 kbp for the dolphin.
Our estimate of the length of the platypus IGH locus (457 kbp) is higher than the previous estimate by Gambón-Deza et al. (2009) (271 kbp). The analysis of the IGH-start contig (containing V genes only) and IGH-end contig (containing V, D, J, and C genes) in platypus revealed an unusual feature. It turned out that the IGH-end contig contains the entire IGH locus in platypus, whereas the IGH-start contig contains V genes from the TCRμ locus, a unique TCR locus found only in marsupials and monotremes (Miller 2010). Because previous studies showed that V genes from this locus are more similar to IG V genes than TCR V genes (Miller 2010), this finding illustrates that IGDetective is capable of detecting unusual TCR genes. Nevertheless, to limit analysis to the IGH loci only, we discarded V genes from the platypus TCRμ locus from further analyses.
Comparative analysis reveals highly similar V genes in evolutionarily distant species
We combined the candidate V genes (referred to simply as V genes) across all target species with known V genes in reference species and constructed a phylogenetic tree on their amino acid sequences using Clustal Omega (Sievers et al. 2011). Figure 3A shows the computed tree in which leaves (representing V genes) are colored according to the species they belong to and the order of the species. “Cutting” this tree by a horizontal line at a height threshold results in subtrees formed by clusters of similar V genes. We classified a cluster as large if it contains more than five V genes and as multispecies (multiorder) if it includes V genes from multiple species (orders). Because large multispecies clusters are the main focus of the comparative analysis, we selected the height threshold 1.12, maximizing the number of these clusters. The resulting 219 clusters include 43 large clusters, 25 large multispecies clusters, and seven large multiorder clusters (Supplemental Fig. S4).
Figure 3.

Comparative analysis of mammalian IGHV genes. (A) A phylogenetic tree of IGHV genes in 20 target (581 V genes) and three reference (310 V genes) species. Edges corresponding to clusters C1–C7 described in B are shown in blue. The scale is shown on the left. The upper and lower horizontal bars show colors of species and their orders, respectively. A list of species and their colors is specified below the tree (species from the same order are shown by a colored vertical bar on the left). (B) The plot on the left shows the species distance (x-axis) and the gene distance (y-axis) for 25 large multispecies clusters of V genes. Red and blue dots correspond to single-order and multiorder clusters, respectively. The linear regression line is shown in grey. The Pearson's correlation (r) and P-value (P) are shown on the top of the plot. Each of seven multiorder clusters C1–C7 is represented as a pie chart on the right. An inner (outer) wedge in each pie chart corresponds to a species (an order), and the wedge size is proportional to the number of V genes it contains. (C) Multiple alignment of 21 V genes from the cluster C7. Four human V genes from this cluster are shown on the top. Nonhuman genes are denoted according to the short names of species, and “HS_bat” refers to the horseshoe bat. A position in a nonhuman V gene is shown by a “.” if the amino acid at this position matches the corresponding amino acid in one of four human V genes and by the corresponding amino acid otherwise. Red rectangles show the positions of CDR1 and CDR2 according to the IMGT notation. Green bars show positions of two conserved cysteines (one located close to the start of CDR1 and another located close to the end of the V gene).
For each large multispecies cluster formed by V genes g1,…,gn from species s1,…,sm, we computed its gene distance and species distance. The gene distance is computed as the max{GeneDist(gi, gj)} for all pairs of genes g1,…,gn, where GeneDist(x, y) represents the fraction of nonmatching positions in the alignment between genes x and y. The species distance is computed as max{SpeciesDist(si, sj)} for all pairs of species s1,…, sm, where SpeciesDist(x, y) is the distance between species x and y in the tree shown in Figure 2A. Figure 3B illustrates the correlation between species distances and gene distances for 25 large multispecies clusters (Pearson's correlation r = 0.77, P-value = 7.15 × 10–6). Seven multiorder clusters (referred to as C1–C7) are represented in blue in Figure 3B, and for all of them but one (cluster C7 shown as a blue asterisk), the gene distance is either well estimated or underestimated by the regression line. The unusual C7 cluster is formed by V genes from six target species (chimp, gorilla, orangutan, marmoset, red squirrel, and horseshoe bat) that are similar to human genes IGHV3-30, IGHV3-30-3, IGHV3-30-5, and IGHV3-33 (Fig. 3C). This cluster reveals a surprising conservation of V genes across distant species; for example, the amino acid sequence of the gene HS_bat_58 in the horseshoe bat has even fewer differences (eight) with the human V genes in this cluster than some chimpanzee V genes (nine). We thus conjecture that V genes in this cluster are subjected to the same selective pressure, for example, driven by common pathogens that are faced by the species in this cluster.
A new family of cysteine-rich IGHV genes
Two hundred fifty-four out of 309 canonical human V genes (82%) listed in the IMGT database with productive amino acid sequences (including allelic variants) have two canonical cysteines located at conserved positions (Fig. 3C). We classify a V gene as cysteine-rich if it contains four or more cysteines and analyze cysteine-rich V genes among all V genes shown in Figure 3A. There are no human cysteine-rich V genes and only two (one) mouse (cow) cysteine-rich V genes. It remains unclear whether the mouse and cow cysteine-rich V genes represent pseudogenes rather than functional genes; for example, the cysteine-rich V gene in the cow does not contribute to antibody repertoires (Safonova et al. 2022).
Seven hundred fifteen out of 891 V genes (80%) from both reference and target species have two cysteines, and only 61 (7%) are cysteine-rich. Cysteine-rich V genes are grouped together in the phylogenetic tree (shown in dark green in Fig. 4A) and appear only in two out of 25 identified large multispecies clusters, including a multiorder cluster C4 (Fig. 3B) and a single-order cluster that we refer to as C*. Cluster C4 contains 27 V genes from six species: dolphin, blue whale, sloth, horseshoe bat, spear-nosed bat, and mastiff bat (Fig. 4B). Cluster C* consists of 12 V genes from grey and red squirrels (Fig. 4B). Twenty-five out of 27 V genes in cluster C4 are cysteine-rich, and 11 out of 12 V genes in cluster C* are cysteine-rich.
Figure 4.

Two novel cysteine-rich clusters of mammalian IGHV genes. (A) A phylogenetic tree of IGHV genes colored according to the number of cysteines in their amino acid sequences. (B) Only two out of 25 large multispecies clusters of V genes contain cysteine-rich V genes. The description of pie plots is provided in the caption to Figure 3B. The number within parentheses next to the species name indicates the number of V genes from this species. (C) Multiple alignment of genes from two clusters shown in B. Three non-cysteine-rich V genes are marked with a grey circle on the left. The gene on top of each cluster is chosen as the sequence of a V gene with the minimum average distance from other genes in the cluster. Green (purple) bars show positions of canonical (noncanonical) cysteines. Some proteins contain cysteines (that are shown in purple) outside these positions. “HS_bat,” “M_bat,” and “SN_bat” in the top alignment refer to V genes of the horseshoe bat, the mastiff bat, and spear-nosed bat, respectively. “Red_sq” and “grey_sq” in the bottom alignment refer to V genes of the red squirrel and the grey squirrel, respectively.
Figure 4C shows that, in addition to two canonical cysteines at conserved positions, most V genes from clusters C4 and C* have two other cysteines, also at conserved positions (one cysteine in CDR1 and another in CDR2). Although several antibodies with a single cysteine in either CDR1 or CDR2 were reported before (Wu et al. 2012; Prabakaran and Chowdhury 2020), antibodies with cysteines in both CDR1 and CDR2 have not been reported yet. Because cysteines form disulfide bonds, we hypothesize that cysteine-rich V genes might generate unusual antibodies with noncanonical conformations and could potentially form a unique part of bat immunity against the great variety of viruses they host.
The cysteine-rich cluster C4 includes a sloth V gene (Fig. 4C, sloth_38) with the unusual six-aa-long suffix CVLLCE classified as the beginning of CDR3. The vast majority of known V genes have the conserved CAR suffix and thus contribute to, at most, the three first amino acids of CDR3s. Two known exceptions from this rule are the cattle IGHV1-7 gene that contributes to ultralong antibodies in combination with ultralong D gene IGHD8-2 (Wang et al. 2013; Safonova et al. 2022) and the platypus IGHV1-20 gene with unknown functions (Gambón-Deza et al. 2009). We hypothesize that, similar to the cattle IGHV1-7 gene contributing to generation of ultralong antibodies, both sloth mChoDid1_38 and platypus IGHV1-20 V genes with long CDR3 prefixes may generate (alone or in combination with D genes) antibodies with noncanonical structural features. Below we analyze unusual candidate D genes in these species and perform comparative analysis of all detected D genes.
Comparative analysis of mammalian D genes
IGDetective identified 92 candidate D genes in target species (Fig. 5A). For the comparative analysis, we combined these D gene candidates with 81 known D genes of three reference species (27 human, 31 mouse, and 23 cow D genes), resulting in a set of 173 D genes. For the sake of simplicity, we refer to both reference and candidate D genes as simply D genes.
Figure 5.

Comparative analysis of D genes. (A) The distribution of the counts and lengths of D genes for 20 target species. (B) D genes shared among two or more reference and target species. Green cells show species containing the corresponding D gene candidates. Species from left to right: chimp, gorilla, human, orangutan, mastiff bat, horseshoe bat, and otter. (C) The largest connected component of the Hamming graph on amino acid 4-mers of D genes. The component is shown by the subgraph of the Hamming graph (left subpanel) and the amino acid content at each position of the 4-mer (right subpanel). Vertices of the Hamming graph are colored according to the number of species they represent: from two (pale green) to six (dark green). The amino acid sequence of the G/S/Y-rich cow D gene IGHD8-2 is shown on the bottom of the right subpanel. (D–F) Illustration of the analysis of D genes in the platypus genome. (D) Positions of D genes detected by SEARCH-D in the platypus IGH locus. D genes are colored according to their lengths: ≤50 nt (purple), 51–100 nt (green), and 51–150 nt (orange). (E) The dotplot on the left shows the alignment of the ≅60-kbp-long platypus IGHD locus against itself. Positions and sequences of genes from four D gene families with two cysteines are shown on the right. (F) Motif logos of RSSDleft heptamer (L7), RSSDleft nonamer (L9), RSSDright heptamer (R7), and RSSDright nonamer (R9) for families D1–D4. Positions that do not match nucleotides in the consensus RSSs computed using the combined references are highlighted in grey. Consensus RSSs for the combined reference are shown in Supplemental Figure S7.
Figure 5B shows that only five D genes are shared among two or more species. Because, differently from V and J genes, D genes are short and very diverse, it remains unclear whether there exist specific features of D genes that are shared among nearly all mammalian species. To reveal such features, we searched for common substrings in all 173 D genes. We translated each D gene into three reading frames and extracted all its 4-mers in the amino acid alphabet. In total, we collected 738 4-mers, with 128 of them appearing in multiple species. The maximum number of species represented by a single 4-mer is six.
We constructed the Hamming graph on 128 shared 4-mers by connecting two 4-mers by an edge if they differ in a single amino acid (Safonova et al. 2015). It turned out that all connected components in this graph are small (less than five vertices) with exception of two components consisting of 43 and 42 4-mers and covering 68% of all 4-mers appearing in multiple species (Fig. 5C). These two components cover all highly abundant 4-mers (4-mers that are present in three to six species). Below, we focus on the first component because the second component represents the same substrings of D genes as the first component but translated in a different reading frame.
The 4-mers in the first component represents 16 out 23 analyzed species and are mostly formed by amino acids G, S, and Y. We refer to D genes that encode these 4-mers as G/S/Y-rich D genes. A half of all possible single-nucleotide mutations of cysteine-encoding codons (TGT and TGC) result in the codons GGT, AGT, TAT, and TCT encoding amino acids G, S, S, and Y, respectively. These three amino acids are extremely frequent in the longest cattle D gene IGHD8-2 (Fig. 5С), where they play a special cysteine-triggering role in ultralong cattle antibodies: Somatic hypermutations create new cysteines from these amino acids, forming new disulfide bonds and reshaping the resulting antibody (Wang et al. 2013). We conjecture that the G/S/Y-rich D genes may play a similar cysteine-triggering role as the IGHD8-2 gene in cows.
Unusual cysteine-rich D genes in the platypus genome
Platypus and sloth genomes have two V genes with long noncanonical suffixes that represent the beginnings of CDR3s: the platypus gene IGHV1-20 with suffix LAAELLYCR and the sloth gene “sloth_38” with suffix CVLLCE (Supplemental Fig. S5). The only other known V gene with a long noncanonical suffix is the cow gene IGHV1-7, which plays a special role in generating ultralong CDR3s by recombining with the longest known D gene (IGHD8-2) of length 148 nt (Wang et al. 2013; Safonova et al. 2022). This long D gene is highly unusual: All but three amino acids in its translation are either cysteines or the cysteine-triggering amino acids G, S, and Y (Fig. 5C). We thus searched for D genes with similar properties in the platypus and sloth IGH loci. However, IGDetective, which uses rather stringent parameters for RSS search, did not report any unusual (long or C/G/S/Y-rich) D genes. In contrast to IGDetective, SEARCH-D (Safonova and Pevzner 2020) uses relaxed parameters for finding RSSs at the cost of reporting more false-positive D gene candidates.
We thus launched SEARCH-D on the platypus (457 kbp long) and sloth (6.7 Mbp long) IGH loci. SEARCH-D reported 45 and 76 D gene candidates (simply referred to as D genes) for the platypus and the sloth, respectively (including six platypus and one sloth D gene candidates reported by IGDetective). Because the candidate D genes in the sloth are scattered through the entire IGH locus, we were unable to identify the location of the sloth IGHD locus (Supplemental Fig. S6). In contrast, 29 out of 45 candidate D genes in platypus form a dense 60-kbp-long cluster pointing to a previously unknown location of the IGHD locus (Fig. 5D,E).
Similarly to other mammalian IGHD loci (Safonova and Pevzner 2020), the identified 60-kb-long fragment harboring 29 candidate D genes in platypus is a tandem repeat (Fig. 5D,E), reinforcing the conclusion that this region indeed represents the IGHD locus. We classify a D gene as cysteine-rich if it contains at least two cysteines in one of its reading frames (only three out of 25 human D genes are cysteine-rich). Clustering 29 candidate D genes in platypus revealed four groups of similar D genes with percent identity ≥70% (referred as D1–D4) that include many cysteine-rich D genes: 12 out of 15 D genes in these groups are cysteine-rich, and all genes in these groups, similar to the cow D gene IGHD8-2, have many cysteine-triggering amino acids (Fig. 5E).
Even though these 15 candidate D genes have rather diverged RSSs (Fig. 5F), the high level of their sequence conservation indicates that they are likely functional. We assume that, similar to human IGHD2 genes, these D genes can be responsible for generating antibodies with a disulfide bond inside CDR3s (Prabakaran and Chowdhury 2020). The high number of these D genes suggests that the fraction of such antibodies is likely higher in the platypus repertoires compared with the human repertoires. This finding agrees with a study by Johansson et al. (2002) (which reported an unusually high percentage of cysteine-rich antibodies in platypus antibody repertoires) and extends it by revealing the germline D genes contributing to the cysteine-rich antibodies. Further analysis of platypus Rep-Seq data will help to determine if these cysteine-rich D genes are recombined with IGHV1-20 (with unusual suffix LAAELLYCR) and shed light on their role in antibody repertoires.
Benchmarking BlindIGDetective
BlindIGDetective constructed the human V-graph from 28,394 sites in the human genome that passed the RSSV likelihood threshold. A connected component is classified as either small (of a size of at most smallSize), large (of a size larger than smallSize but smaller than giantSize), or giant (of a size of at least giantSize) for default values smallSize = 3 and giantSize = 500. The vast majority of candidate RSSs in the human genome are false RSSs that often represent isolated vertices or vertices of giant components that likely originated from spurious RSSs within repeated regions. Indeed, the human V-graph contains 6942 isolated vertices and one giant component on 20,768 vertices. The vast majority of vertices in the giant component represent spurious repeats that we have ignored in further analysis. Supplemental Table S10 provides information about the V-graphs for three reference species (human, mouse, and cow) and two selected target species (the spear-nosed bat and the horseshoe bat).
For each vertex in the V-graph, we define the percent identity, coding length, annotation index, and conservation index (Supplemental Methods, see “Analyzing connected components in the similarity graph” and “Speeding-up BlindIGDetective”). The conservation index of a vertex is defined as the percent identity between its v-fragment and the closest predicted gene (predicted genes could either be canonical genes from reference species or be candidate genes detected by IterativeIGDetective). A vertex is annotated if its conservation is at least PIannotation (default value = 90%). For annotating human (cow, mouse) vertices, we define conservation with respect to human (cow, mouse) canonical V genes. However, for annotating the target species, we use either a conservation threshold of PIannotation = 90% with respect to IG genes in this species predicted by IterativeIGDetective or a conservation threshold of PIannotation = 80% with respect to human canonical V genes.
A vertex in the V-graph is classified as accordant if its coding length exceeds the minimum coding length threshold (default value minCL = 200 bp). Because clusters with short coding lengths are likely formed by spurious RSS (for reference species, all V genes, except one, have length exceeding 208 bp), below we focus on accordant clusters (clumps) defined as clusters (clumps) with coding lengths exceeding the minCL threshold. We note that accordant clusters (clumps) may contain both accordant and nonaccordant vertices.
We launched BlindIGDetective with the RSS profile corresponding to a 23-nt spacer. This setting is aimed at finding V genes in IGH, IGL, TRA, TRB, TRD, or TRG loci (that all have RSSs with 23-nt spacer) but not the IGK locus (because RSSs in this locus have a 12-nt rather than a 23-nt-long spacer). However, the IGK locus can be easily identified by simply changing a spacer length from 23 to 12 nt in BlindIGDetective.
An annotated vertex in a cluster is said to be part of a specific V gene locus (IGH, IGL, TRA, TRB, or TRG) defined by its most similar (as measured by percent identity) annotated V gene, where the set of annotated V genes can be sourced either from a canonical set of V genes (if available) or from predictions made by IterativeIGDetective. A cluster is classified as annotated by a specific V gene locus if all its annotated vertices are assigned to this locus.
BlindIGDetective reveals novel candidate V genes in the human genome
BlindIGDetective identified 79 clumps from 179 connected components in the human V-graph on 684 vertices (after removing isolated vertices and the giant component). It further removed clumps with very small spans (<1 kb) as such clumps are typically formed by multiple candidate (likely spurious) RSSs located within a short region. Afterward, it combined the remaining clumps into clusters as described in the subsection “Similarity graph,” resulting in 11 (eight) large (accordant) clusters in the human genome (Table 2). Seven out of eight accordant clusters revealed seven known loci of human IG genes (IGH and IGL loci are represented by the largest clusters of size 59 and 31, respectively).
Table 2.
Information about large clusters derived from the human and spear-nosed bat genomes

Table 2 shows that the cluster representing human IGH (IGL) genes is formed by five (five) clumps, including two (one) unannotated clumps (Supplemental Table S11). There are only two (seven) unannotated fragments contained in IGH (IGL) annotated clumps, including one (three) accordant v-fragments. Moreover, unannotated fragments within annotated clumps still show high median conservation of 83% for both IGH and IGL clusters.
Because two (seven) unannotated fragments in IGH (IGL) annotated clumps have large median conservation, we launched an IgBLAST search (Ye et al. 2013) on them and revealed significant hits with E-values of at most 4 × 10−76 (1 × 10−60) and percent identities of at least 84% (75%) against human V genes IGHV3-48 and IGHV1-46 (for two unannotated fragments in IGHV clumps) and IGLV1-44, IGLV3-22, IGLV3-21, IGLV3-9, IGLV3-31, and IGLV7-46 genes (for seven unannotated fragments in IGLV clumps). Alignment of one (three) out of two (seven) unannotated and accordant IGH (IGL) v-fragments from annotated clusters revealed that they align with their closest human V gene in a reading frame containing a stop codon. We therefore suggest that these accordant unannotated fragments could represent previously undiscovered V genes (or pseudogenes) that may be affected by RAG proteins during VDJ recombination.
The unannotated clumps representing human IGH (IGL) genes accounted for five (two) unannotated v-fragments in the IGH (IGL) cluster and included one accordant v-fragment in the IGH locus (coding length, 597 nt) with a low percent identity with known human genes (under 62%). Unannotated clumps retained a low median conservation under 62% in both IGH and IGL clusters. A similar IgBLAST search of these five (two) unannotated v-fragments in the IGH cluster revealed hits for only three v-fragments against human IGHV genes IGHV3-7, IGHV3-11, and IGHV3-21 with low E-values of 8 × 10–13, 2 × 10–18, and 7 × 10–20 and percent identities of 61%, 62%, and 63%, respectively. Although these three v-fragments share some (albeit low) similarity with known human V genes, they are missing in the IMGT database. Moreover, all these v-fragments are located in the IGH locus within a short distance from the canonical IGHV3 gene (at distance 25 kbp, 11 kbp, and 61 kbp, respectively). We therefore suggest that they represent distant IGHV3 genes missed by earlier methods for annotating V genes.
The remaining two (two) v-fragments in the IGH (IGL) loci had high E-values exceeding 0.42. Supplemental Figure S8 shows two alignments between the two pairs of these v-fragments (which extend through the entire sequence) and suggests these 2 + 2 v-fragments could represent undiscovered genes (or pseudogenes) that are not similar to known human V genes and therefore would not have been discovered through V gene finding methods relying on similarity with previously identified genes.
For details of BlindIGDetective benchmarking on other reference species, see Supplemental Methods, “BlindIGDetective results on cow and mouse genomes,” and Supplemental Table S12.
BlindIGDetective reveals highly diverged candidate V genes in the spear-nosed bat genome
Bratsch et al. (2011) and Schountz et al. (2017) formulated the “bat IG diversity” hypothesis stating that bat's ability to carry many disease-causing pathogens (while themselves being unaffected) could be linked to their large and diverse IG gene set. However, because assembly of IG loci in any species is challenging (Bankevich and Pevzner 2020), accurately assembled (let alone, annotated) IG loci in bats remained unavailable until recently. In fact, the only support for the claim that bats have a very large number of IG genes comes from a probabilistic model rather than an annotated IG loci in an assembled bat genome: Bratsch et al. (2011) used this model to predict approximately 240 V genes in the little brown bat without having access to its genome. IterativeIGDetective results do not support the “bat IG diversity” hypothesis: It reported from only nine, 10, 32, and 63 IGHV genes across four bat species. We thus applied BlindIGDetective to reveal divergent V genes that IterativeIGDetective may have missed. We focused on the spear-nosed bat (only 34 IGHV genes reported by IterativeIGDetective) as the bat species with the longest IGH locus (4.6 Mbp).
BlindIGDetective constructed 72 clusters in the spear-nosed bat genome, including 17 (29) accordant (large) clusters (Table 2). Vertices in these clusters were annotated using the candidate IGHV genes predicted by IterativeIGDetective and canonical human genes to annotate V genes from IGL, TRA, TRB, and TRG loci. A total of 13 large, annotated clusters were observed in the spear-nosed bat genome.
BlindIGDetective identified nine large IGH clusters (three of which are accordant) encompassing 27 clumps and 189 v-fragments. It also identified two large IGL clusters with 142 v-fragments, one TRA cluster with 35 v-fragments, and one TRB cluster with 10 v-fragments (all these clusters are accordant). In addition to the large clusters, it identified a small IGH cluster containing two v-fragments. No small clusters were detected for IGL, TRA, and TRB loci. For each v-fragment, the opening reading frame and the start position of the gene were computed using alignment to the closest human germline V gene. A v-fragment is classified as productive if it has an open reading frame that begins at its start position and terminates at its end position. One hundred forty-seven IGH, 95 IGL, 15 TRA, and one TRB annotated v-fragments were found within the identified clusters; 29 IGH, 58 IGL, six TRA, and zero TRB annotated v-fragments were productive and thus classified as V gene candidates.
BlindIGDetective also identified 44, 47, 20, and nine unannotated v-fragments within the IGH, IGL, TRA, and TRB clusters, respectively (including one IGH, 20 IGL, four TRA, and seven TRB productive v-fragments). The alignments of these productive unannotated v-fragments against the translated human IG and TCR genes revealed high percent identity (in amino acids) varying from 49% to 77% with the average value 68%. To analyze the origin of unannotated candidate V genes, we combined them with productive annotated gene candidates and constructed phylogenetic trees using Clustal Omega (Sievers et al. 2011) for IGH, IGL, and TRA loci. Annotated and unannotated V gene candidates are interspersed in trees for IGH and IGL loci, indicating that unannotated V gene candidates likely represent highly diverged members of the canonical V gene families (Supplemental Fig. S9). In the TRA locus, unannotated V gene candidates form an independent subtree, suggesting that they represent an unknown V gene family.
Prediction of V genes in bats does not support the “bat IG diversity” hypothesis
Our benchmarking of BlindIGDetective revealed nearly all IG genes identified by IterativeIGDetective and more. However, the “bat IG diversity” hypothesis was not supported by our analysis of the spear-nosed bat genome as we identified a much smaller number of IGHV genes than 200+ V genes predicted by Bratsch et al. (2011) using a probabilistic model applied to the little brown bat. There are three possible explanations for a discrepancy between our analysis and the “bat IG diversity” hypothesis: (1) spear-nosed bats have relatively few genes compared with little brown bats; (2) the probabilistic model of Bratsch et al. (2011) does not adequately approximate the number of IGHV genes; and (3) many IGHV genes in spear-nosed bats have “diverged” RSSs that do not pass the default RSS likelihood threshold. The Supplemental Methods, “Applying BlindIGDetective to the horseshoe bat genome,” and Supplemental Table S13 present BlindIGDetective results on the horseshoe bat and also do not support the “bat IG diversity” hypothesis.
Variations in RSSs trigger high/low usage of human D genes
In addition to analyzing variations in IG genes, we also analyzed variations in RSSs and their effect on antibody repertoires.
The usage of a gene in an antibody repertoire is defined as the percentage of antibodies formed by VDJ recombinations in this repertoire that involve this gene. The IG genes have a highly nonuniform usage that may vary by orders of magnitude; for example, whereas the most widely used human D gene (IGHD3-10) is used in ∼15% of all human antibodies, some human D genes hardly ever contribute to formation of human antibodies (Safonova and Pevzner 2019). Because the usage of IG genes is likely affected by the sequence of their RSSs, we analyzed the associations between the gene usage of IG genes and their RSSs. The Supplemental Methods, “Clustering nonamers in RSSVs,” and Supplemental Figures S10 and S11 illustrate that RSSs can be partitioned into subgroups of highly similar signals within the set of all RSSs. By revealing these subgroups of similar RSSs, we can shed light on the correlations between RSSs and the usage of the genes they flank.
Below we focus on analyzing correlations between the usage of D genes and their RSSs by analyzing immunosequencing data sets containing rearranged VDJ sequences from 24 donors from the study by Levin et al. (2017). For each such VDJ sequence, we used the IgScout tool (Safonova and Pevzner 2019) to identify the D gene that contributed to this sequence. The individual usage of a gene for a single data set is defined as the percentage of total VDJ recombinations derived from this gene in this data set. Although the individual usages vary, they have a rather low variance across various individuals in a given species (Safonova and Pevzner 2019). The usage of an IG gene is defined as the average of individual usages across all 24 data sets generated by Levin et al. (2017). The usage of an RSS is defined as the usage of the gene that this signal flanks.
Below we analyze N D-genes in a reference species and consider N pairs (RSSDleft, RSSDright) flanking these genes (similar analysis has been conducted for V and J genes). We refer to heptamers (nonamers) in this pair as l7 and r7 (l9 and r9) and consider 16-mers l7l9 and r7r9 as well as a 32-mer l9l7r7r9, resulting in various signal types. For each signal type, we computed the N × N matrix of Hamming distances between all pairs of signals and performed k-means clustering on N-dimensional vectors formed by rows of this matrix.
We launched the k-means clustering algorithm (20 runs with 200 iterations for each run) for various cluster numbers and selected an optimal number of clusters based on the elbow method (Yuan and Yang 2019) for all signal types. We determined three l7 and three l9 clusters, as well as two r7 and three r9 clusters. Each l9 (r9) cluster can be decomposed into groups of D genes, where each group is a subset of an l7 (l9) cluster and no pair of groups can be merged. For three l9 clusters, the relative sizes of the largest groups with respect to the cluster size are 100%, 100%, and 70% (Supplemental Table S14). For three r9 clusters, the relative sizes of the largest groups are 53%, 75%, and 100%. We hypothesize that clusters l7 and l9 (as well r7 and r9) and the high level of overlap between them can trigger variations in usage patterns of D genes.
Figure 6B illustrates that 12 out of the total 25 human D genes dominate the vast majority (93%) of the usage. We integrate the usage statistics of the D genes into the clustering process. The distribution of usage between the clusters described earlier is recorded for all combinations of the RSSD signals in the Supplemental Methods, “Cluster-based usage of RSSDs,” and Supplemental Table S15. The Kruskal–Wallis test (Kruskal and Wallis 1952) on the usage of the signals belonging to each cluster revealed statistically significant associations for both RSSDleft and RSSDright heptamer clusters (P-values = 0.042 and 0.007, respectively). We also found statistically significant associations for clusters l9l7r7r9, l7r7, and l9r9 (P-values = 0.02, 0.009, and 0.028, respectively). These clusters and their significant usage associations are shown in Figure 6, A and B.
Figure 6.

Clustering and distribution of human RSSDs. (A) Cluster visualization of l7, r7, l9l7r7r9, l7r7, and l9r9 signals. We shall henceforth refer to the red, blue, green, and yellow clusters as clusters 1, 2, 3, and 4, respectively. The consensus of a cluster is noted as the legend label. PC1 and PC2 refer to the first two principal components of the clustering performed on the signals, as described in the Results subsection “Variations in RSSs trigger high/low usage of human D genes.” (B) Usage of D genes with respect to clusters on l7, r7, l9l7r7r9, l7r7, and l9r9 signals. The P-value of correlation is depicted on the top right of each panel. (P*) P-value <0.05; (P**) P-value <0.01. (Bottom, right) Usage of human D genes. Each of 12 highly used human D genes (with usage at least 2%) is represented by a single bar. All remaining low-usage human D genes are represented by a single bar, showing their combined usage equal to 7%.
Analysis of the clusters revealed motifs of RSSs associated with high-usage D genes. Among the heptamers, the highest used RSSDleft cluster (accounting for over 80% of usage) has the general pattern NACTGTG, where “N” stands for an arbitrary nucleotide. The most used RSSDright heptamer cluster (accounting for >94% of usage) has each heptamer equal to CACAGTG. The highest used nonamer cluster for RSSDleft follows a pattern GGTTTNNNN, whereas the RSSDright nonamer cluster follows a pattern NNNAAAACN.
Analysis of the V and J genes did not reveal any associations between RSS clusters and usage patterns (Supplemental Methods, “Finding association between RSSs and usages of V and J genes”; Supplemental Tables S16, S17; Supplemental Figs. S12–S16).
Discussion
IGDetective algorithm
We benchmarked IterativeIGDetective on three well-annotated IGH loci (human, mouse, and cow) and showed that it accurately predicts the known V, D, and J genes in one of these species based on information about RSSs in other species. This observation justifies our comparative immunogenomics approach to annotating the IGH loci in newly sequenced species. Although the IterativeIGDetective analysis in this paper is limited to the IGH loci, we plan to extend it to other IG and TCR loci in a follow-up study. In addition to the three reference species, we applied IterativeIGDetective to 20 mammalian species with the recently assembled IGH loci and predicted 581, 92, and 60 new putative IGHV, IGHD, and IGHJ genes, respectively.
In addition to IterativeIGDetective, we developed a BlindIGDetective algorithm for predicting novel genes that have diverged from currently known IG genes. BlindIGDetective detects most IG genes in the absence of any information about IG genes in other species and thus opens a possibility to identify highly divergent IG genes. The application of BlindIGDetective to detect V genes from three reference species and two bat species (spear-nosed bat and horseshoe bat) revealed that BlindIGDetective was sensitive not only to canonical V genes from the IGH locus but also to other V gene loci driven by RAG proteins, such as V genes in the IGL, TRA, TRB, and TRG loci. Moreover, BlindIGDetective identified multiple highly divergent candidate V genes (or pseudogenes) in various species.
We plan to combine IterativeIGDetective and BlindIGDetective to extend this analysis to immunological model organisms such as rabbits and llamas, as well as nonmammalian vertebrates, with the goal to construct a comprehensive database of predicted IG genes across multiple species.
The diversity of IG genes
We applied IterativeIGDetective to 20 mammalian species with poorly studied IGH loci, performed comparative analysis of the detected IGHV genes, and identified a highly conservative cluster that covers highly divergent species ranging from primates to bats. We hypothesize that V genes in this cluster are subjected to selective pressure driven by common pathogens or the genetic organization of IGH loci. Further investigation of this conservative cluster will require repertoire sequencing (Rep-Seq) data.
In addition to revealing the diversity of IG genes, we also studied the diversity of RSSs, identified clusters of similar RSSDs in humans, revealed associations between these clusters and the usage of the IGHD gene they flank, and found the RSSD motifs triggering high usage of human D genes.
Unusual cysteine-rich V genes
We revealed a new family of unusual cysteine-rich V genes in bats and other species that have cysteines in both CDR1 and CDR2. We hypothesize that cysteine-rich V genes might generate unusual antibodies with noncanonical conformations and could potentially form a unique part of bat immunity against the great variety of viruses they host. Further investigation of antibodies derived from these V genes would require both RepSeq data and protein structures to shed light on the functions and to estimate the therapeutic potential of such antibodies.
G/S/Y-rich motifs in D genes
We showed that, despite being highly diverse, D genes in various mammalian species share the G/S/Y-rich motifs that are formed by cysteine-triggering codons. In cows, the G/S/Y-rich IGHD8-2 gene plays a special cysteine-triggering role in ultralong cattle antibodies, where somatic hypermutations create new cysteines from these three amino acids, forming new disulfide bonds and reshaping the resulting antibody (Wang et al. 2013). We conjecture that found G/S/Y motifs may play a similar cysteine-triggering role in D genes of many mammals.
Limitations and future developments
In the past three decades, gene prediction algorithms have evolved from relatively simple statistical tests to sophisticated machine learning approaches (Mathé et al. 2002). However, because even the state-of-the-art gene prediction algorithms generate some false-positive genes, they require validation using complementary experimental approaches, such as transcriptome sequencing (Allen et al. 2004) and mass spectrometry (Tanner et al. 2007). Likewise, because IGDetective is merely the first step toward uncovering the diversity of IG genes across vertebrate species, it has to be complemented by complementary experimental approaches, such as antibody repertoire sequencing. Our next goal is to modify IGDetective for working with both genome assemblies and Rep-Seq data.
IterativeIGDetective is currently based on construction of the profile matrices of known RSSs, detection of novel RSSs using these profile matrices, and follow-up analysis of genomic sequences flanked by these RSSs. In the future, we plan to develop a hidden Markov model for joint analysis of RSSs and the flanking genes. We also plan to complement the existing analysis (based on only three reference genomes without retraining) by bootstrapping when the original model is trained on the references only but the set of references is later extended (based on the reliable predictions of IG genes in new species) for further retraining.
IterativeIGDetective failed to identify any J genes in the spear-nosed bat genome, presumably because all its RSSJ motifs did not pass the default likelihood threshold. Because our current goal is to focus on highly conserved RSSs and minimize the false-positive rate, we did not recompute all IG gene predictions in the spear-nosed bat with a lower likelihood threshold. In the future, we plan to develop a version of IterativeIGDetective that iteratively relaxes the RSS search parameters.
BlindIGDetective currently starts from identifying highly conserved RSS in the entire genome and thus misses all IG genes flanked by less conservative RSSs. Lowering the RSS likelihood threshold leads to explosion of false RSSs, thus making BlindIGDetective prohibitively slow. We plan to modify BlindIGDetective (with the goal of identifying the missed IG genes with less conservative RSSs) by first identifying the IG-contigs, enriching the set of putative RSSs in these contigs by adding less conservative RSSs, and combining BlindIGDetective and IterativeIGDetective in a single pipeline.
Methods
Gene nomenclature
Following the guidelines of the IMGT nomenclature (https://www.imgt.org/IMGTScientificChart/Nomenclature/IMGTnomenclature.html), we have not italicized genes of IG and TCR genes.
Profiles and likelihood ratio of strings
Given a set of k-mers (k-nucleotide-long strings), their profile matrix is a 4 × k matrix Profile defined below. The jth column in the profile matrix represents the jth position in the k-mer, and the four rows represent the four nucleotide bases (A, C, G, and T). Profile(i,j) represents the frequency of occurrence of the ith base at the jth position in the input k-mers adjusted for pseudocounts as described by Compeau and Pevzner (2018). The consensus string (referred to as consensus = consensus(Profile)) is a k-mer generated by taking a nucleotide with the highest frequency at each position of the profile (ties are broken arbitrarily). As opposed to numerous V and D genes, there are few J genes in the reference species, leading to profile matrices with higher entropies (when compared with V or D gene profiles) owing to pseudocounts.
Given a k-mer S = s1…sk and a 4 × k profile Profile, the probability that Profile generates a string S is defined as prob(S|Profile) ∏j = 1,k Profile(sj,j). Because prob(S|Profile) is maximized when the string S is a consensus of Profile, we define the likelihood ratio L(S) of S as
Given a profile Profile and a likelihood ratio threshold Lmin, a k-mer S is classified as a candidate for Profile if its likelihood ratio L(S) exceeds Lmin.
RSS detection algorithm
Given a set of RSSs for each of a reference species (human, mouse, cow, or combined), we compute their profile matrix (with pseudocounts equal to one) for heptamers and nonamers across all signals (RSSV, RSSDleft, RSSDright, and RSSJ). Given a profile-pair (Profile,Profile’) of 4 × 7 and 4 × 9 stochastic matrices and the associated thresholds Lmin and L′min for a heptamer and a nonamer, a string-pair (heptamer, nonamer) is classified as a candidate RSS for a given signal type if both heptamer and nonamer are candidate strings.
We say that a candidate RSSDleft and a candidate RSSDright together form a paired candidate RSSD if the RSSDright is located within at most MaxLengthD positions to the right from the RSSDleft. The condition is important for D genes because they are flanked by RSSDleft and RSSDright and because the vast majority of known D genes are short (maximum lengths are 37 nt and 29 nt in human and mouse genomes, respectively). Because the longest currently known D gene is the 148-nt-long D gene in cows, we set the default value MaxLengthD = 150.
Selection of the likelihood ratio threshold
We select the Lmin thresholds for heptamers or nonamers based on the reference genome chosen. We launch IterativeIGDetective on the reference species, selecting the heptamer Lmin and nonamer L′min thresholds for all signal types (RSSV, RSSDleft, RSSDright, and RSSJ) by performing a grid search within a range [0,0.8] in steps of 0.005. We compute the TPR (the fraction of known RSSs in the target species detected by IGDetective), the FDR (the fraction of erroneous RSSs among all RSSs reported by IGDetective), and the F1 score defined as
For any given reference, the heptamer Lmin and nonamer L′min thresholds (determined through grid search) that maximize the F1 score are selected as that references’ heptamer Lmin and nonamer L’min. More details on parameter selection are described in the Supplemental Methods, “Parameter selection for identifying candidate RSSs.”
Identification of candidate IG genes
Details of procedures for identification of candidate V, D, and J genes, including selection of parameters of alignments, are described in the Supplemental Methods, “Identification of candidate V and J genes,” “Identification of candidate D genes,” “Iterative extension of the set of candidate IG genes,” “Parameter selection for identifying candidate IG genes,” “Aligning candidate IG genes,” and “Alternate similarity thresholds for detection of V genes” (Supplemental Table S18; Supplemental Figs. S17–S19).
Software availability
IGDetective is available as Supplemental Code and at GitHub (https://github.com/Immunotools/IgDetective). Sequences of identified IGHV, IGHD, and IGHJ genes are available as Supplemental Tables S19–S21 and at GitHub (https://github.com/Immunotools/IgDetective_results).
Supplementary Material
Acknowledgments
This work was supported by the National Science Foundation EAGER award (no. 2032783).
Author contributions: All authors contributed to the development of the IGDetective algorithms. V.S. and Y.S. implemented the IGDetective algorithm. V.S. and Y.S. analyzed the data and performed computational experiments. All authors conceived the study and wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276676.122.
Competing interest statement
The authors declare no competing interests.
References
- Allen JE, Pertea M, Salzberg SL. 2004. Computational gene prediction using multiple sources of evidence. Genome Res 14: 142–148. 10.1101/gr.1562804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A, Pevzner P. 2020. mosaicFlye: resolving long mosaic repeats using long error-prone reads. bioRxiv 10.1101/2020.01.15.908285 [DOI]
- Bratsch S, Wertz N, Chaloner K, Kunz TH, Butler JE. 2011. The little brown bat, M. lucifugus, displays a highly diverse VH, DH and JH repertoire but little evidence of somatic hypermutation. Dev Comp Immunol 35: 421–430. 10.1016/j.dci.2010.06.004 [DOI] [PubMed] [Google Scholar]
- Compeau PCA, Pevzner PA. 2018. Bioinformatics algorithms: an active learning approach, 3rd ed. Active Learning Publishers, Victoria, British Columbia, Canada. [Google Scholar]
- Das S, Hirano M, Tako R, McCallister C, Nikolaidis N. 2012. Evolutionary genomics of immunoglobulin-encoding loci in vertebrates. Curr Genomics 13: 95–102. 10.2174/138920212799860652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Los Rios M, Criscitiello MF, Smider VV. 2015. Structural and genetic diversity in antibody repertoires from diverse species. Curr Opin Struct Biol 33: 27–41. 10.1016/j.sbi.2015.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L, Wang S, Zhu Y, Zhao H, Basit A, Yu X, Li Q, Sun X. 2018. Immunoglobulin heavy chain variable region analysis in dairy goats. Immunobiology 223: 599–607. 10.1016/j.imbio.2018.07.005 [DOI] [PubMed] [Google Scholar]
- Dudley DD, Chaudhuri J, Bassing CH, Alt FW. 2005. Mechanism and control of V(D)J recombination versus class switch recombination: similarities and differences. Adv Immunol 86: 43–112. 10.1016/S0065-2776(04)86002-4 [DOI] [PubMed] [Google Scholar]
- Eguchi-Ogawa T, Wertz N, Sun Z, Piumi F, Uenishi H, Wells K, Chardon P, Tobin GJ, Butler JE. 2010. Antibody repertoire development in fetal and neonatal piglets. XI. The relationship of variable heavy chain gene usage and the genomic organization of the variable heavy chain locus. J Immunol 184: 3734–3742. 10.4049/jimmunol.0903616 [DOI] [PubMed] [Google Scholar]
- Frangione B, Milstein C, Pink JR. 1969. Immunoglobulins: structural studies of immunoglobulin G. Nature 221: 145–148. 10.1038/221145a0 [DOI] [PubMed] [Google Scholar]
- Gambón-Deza F, Sánchez-Espinel C, Magadán-Mompó S. 2009. The immunoglobulin heavy chain locus in the platypus (Ornithorhynchus anatinus). Mol Immunol 46: 2515–2523. 10.1016/j.molimm.2009.05.025 [DOI] [PubMed] [Google Scholar]
- Gertz EM, Schäffer AA, Agarwala R, Bonnet-Garnier A, Rogel-Gaillard C, Hayes H, Mage RG. 2013. Accuracy and coverage assessment of Oryctolagus cuniculus (rabbit) genes encoding immunoglobulins in the whole genome sequence assembly (OryCun2.0) and localization of the IGH locus to chromosome 20. Immunogenetics 65: 749–762. 10.1007/s00251-013-0722-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges SB, Marin J, Suleski M, Paymer M, Kumar S. 2015. Tree of life reveals clock-like speciation and diversification. Mol Biol Evol 32: 835–845. 10.1093/molbev/msv037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson J, Aveskogh M, Munday B, Hellman L. 2002. Heavy chain V region diversity in the duck-billed platypus (Ornithorhynchus anatinus): long and highly variable complementarity-determining region 3 compensates for limited germline diversity. J Immunol 168: 5155–5162. 10.4049/jimmunol.168.10.5155 [DOI] [PubMed] [Google Scholar]
- Keyaerts M, Xavier C, Heemskerk J, Devoogdt N, Everaert H, Ackaert C, Vanhoeij M, Duhoux FP, Gevaert T, Simon P, et al. 2016. Phase I study of 68Ga-HER2-nanobody for PET/CT assessment of HER2 expression in breast carcinoma. J Nucl Med 57: 27–33. 10.2967/jnumed.115.162024 [DOI] [PubMed] [Google Scholar]
- Kronenberg ZN, Fiddes IT, Gordon D, Murali S, Cantsilieris S, Meyerson OS, Underwood JG, Nelson BJ, Chaisson MJP, Dougherty ML, et al. 2018. High-resolution comparative analysis of great ape genomes. Science 360: eaar6343. 10.1126/science.aar6343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruskal WH, Wallis WW. 1952. Use of ranks in one-criterion variance analysis. J Am Stat Assoc 47: 583–621. 10.1080/01621459.1952.10483441 [DOI] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefranc M-P, Giudicelli V, Duroux P, Jabado-Michaloud J, Folch G, Aouinti S, Carillon E, Duvergey H, Houles A, Paysan-Lafosse T, et al. 2015. IMGT®, the international ImMunoGeneTics information system® 25 years on. Nucleic Acids Res 43: D413–D422. 10.1093/nar/gku1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin M, Levander F, Palmason R, Greiff L, Ohlin M. 2017. Antibody-encoding repertoires of bone marrow and peripheral blood—a focus on IgE. J Allergy Clin Immunol 139: 1026–1030. 10.1016/j.jaci.2016.06.040 [DOI] [PubMed] [Google Scholar]
- Li H, Cui X, Pramanik S, Chimge NO. 2002. Genetic diversity of the human immunoglobulin heavy chain VH region. Immunol Rev 190: 53–68. 10.1034/j.1600-065X.2002.19005.x [DOI] [PubMed] [Google Scholar]
- Ma L, Qin T, Chu D, Cheng X, Wang J, Wang X, Wang P, Han H, Ren L, Aitken R, et al. 2016. Internal duplications of DH, JH, and C region genes create an unusual IgH gene locus in cattle. J Immunol 196: 4358–4366. 10.4049/jimmunol.1600158 [DOI] [PubMed] [Google Scholar]
- Mathé M, Sagot MF, Schiex T, Rouzé P. 2002. Current methods of gene prediction, their strengths and weaknesses. Nucleic Acids Res 30: 4103–4117. 10.1093/nar/gkf543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda F, Ishii K, Bourvagnet P, Kuma KI, Hayashida H, Miyata T, Honjo T. 1998. The complete nucleotide sequence of the human immunoglobulin heavy chain variable region locus. J Exp Med 188: 2151–2162. 10.1084/jem.188.11.2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merelli I, Guffanti A, Fabbri M, Cocito A, Furia L, Grazini U, Bonnal RJ, Milanesi L, McBlane F. 2010. RSSsite: a reference database and prediction tool for the identification of cryptic recombination signal sequences in human and murine genomes. Nucleic Acids Res 38: W262–W267. 10.1093/nar/gkq391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messier TL, O'Neill JP, Hou S-M, Nicklas JA, Finette BA. 2003. In vivo transposition mediated by V(D)J recombinase in human T lymphocytes. EMBO J 22: 1381–1388. 10.1093/emboj/cdg137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RD. 2010. Those other mammals: the immunoglobulins and T cell receptors of marsupials and monotremes. Semin Immunol 22: 3–9. 10.1016/j.smim.2009.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RM, Harrison AO, McAllister SM, Polson SW, Wommack KE. 2020. Iroki: automatic customization and visualization of phylogenetic trees. PeerJ 8: e8584. 10.7717/peerj.8584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muyldermans S, Smider VV. 2016. Distinct antibody species: structural differences creating therapeutic opportunities. Curr Opin Immunol 40: 7–13. 10.1016/j.coi.2016.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaoka H, Ozawa K, Matsuda F, Hayashida H, Matsumura R, Haino M, Shin EK, Fukita Y, Imai T, Anand R, et al. 1994. Recent translocation of variable and diversity segments of the human immunoglobulin heavy chain from chromosome 14 to chromosomes 15 and 16. Genomics 22: 189–197. 10.1006/geno.1994.1360 [DOI] [PubMed] [Google Scholar]
- Olivieri D, Faro J, von Haeften B, Sánchez-Espinel C, Gambón-Deza F. 2013. An automated algorithm for extracting functional immunologic V-genes from genomes in jawed vertebrates. Immunogenetics 65: 691–702. 10.1007/s00251-013-0715-8 [DOI] [PubMed] [Google Scholar]
- Pettinello R, Dooley H. 2014. The immunoglobulins of cold-blooded vertebrates. Biomolecules 4: 1045–1069. 10.3390/biom4041045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabakaran P, Chowdhury PS. 2020. Landscape of non-canonical cysteines in human VH repertoire revealed by immunogenetic analysis. Cell Rep 31: 107831. 10.1016/j.celrep.2020.107831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashidian M, Keliher EJ, Bilate AM, Duarte JN, Wojtkiewicz GR, Jacobsen JT, Cragnolini J, Swee LK, Victora GD, Weissleder R, et al. 2015. Noninvasive imaging of immune responses. Proc Natl Acad Sci 112: 6146–6151. 10.1073/pnas.1502609112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhie A, McCarthy SA, Fedrigo O, Damas J, Formenti G, Koren S, Uliano-Silva M, Chow W, Fungtammasan A, Kim J, et al. 2021. Towards complete and error-free genome assemblies of all vertebrate species. Nature 592: 737–746. 10.1038/s41586-021-03451-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez OL, Gibson WS, Parks T, Emery M, Powell J, Strahl M, Deikus G, Auckland K, Eichler EE, Marasco WA, et al. 2020. A novel framework for characterizing genomic haplotype diversity in the human immunoglobulin heavy chain locus. Front Immunol 11: 2136. 10.3389/fimmu.2020.02136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safonova Y, Pevzner PA. 2019. De novo inference of diversity genes and analysis of non-canonical V(DD)J recombination in immunoglobulins. Front Immunol 10: 987. 10.3389/fimmu.2019.00987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safonova Y, Pevzner PA. 2020. V(DD)J recombination is an important and evolutionarily conserved mechanism for generating antibodies with unusually long CDR3s. Genome Res 30: 1547–1558. 10.1101/gr.259598.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safonova Y, Bonissone S, Kurpilyansky E, Starostina E, Lapidus A, Stinson J, DePalatis L, Sandoval W, Lill J, Pevzner PA. 2015. IgRepertoireConstructor: a novel algorithm for antibody repertoire construction and immunoproteogenomics analysis. Bioinformatics 31: i53–i61. 10.1093/bioinformatics/btv238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safonova Y, Shin SB, Kramer L, Reecy J, Watson CT, Smith TP, Pevzner PA. 2022. Variations in antibody repertoires correlate with vaccine responses. Genome Res 32: 791–804. 10.1101/gr.276027.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schountz T, Baker ML, Butler J, Munster V. 2017. Immunological control of viral infections in bats and the emergence of viruses highly pathogenic to humans. Front Immunol 8: 1098. 10.3389/fimmu.2017.01098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, et al. 2011. Fast, scalable generation of high quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7: 539. 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitnikova T, Su C. 1998. Coevolution of immunoglobulin heavy- and light-chain variable-region gene families. Mol Biol Evol 15: 617–625. 10.1093/oxfordjournals.molbev.a025965 [DOI] [PubMed] [Google Scholar]
- Sok D, Le KM, Vadnais M, Saye-Francisco KL, Jardine JG, Torres JL, Berndsen ZT, Kong L, Stanfield R, Ruiz J, et al. 2017. Rapid elicitation of broadly neutralizing antibodies to HIV by immunization in cows. Nature 548: 108–111. 10.1038/nature23301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Pieper K, Piccoli L, Abdi A, Foglierini M, Geiger R, Tully CM, Jarrossay D, Ndungu FM, Wambua J, et al. 2016. A LAIR1 insertion generates broadly reactive antibodies against malaria variant antigens. Nature 529: 105–109. 10.1038/nature16450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner S, Shen Z, Ng J, Florea L, Guigó R, Briggs SP, Bafna V. 2007. Improving gene annotation using peptide mass spectrometry. Genome Res 17: 231–239. 10.1101/gr.5646507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teeling EC, Vernes SC, Dávalos LM, Ray DA, Gilbert MT, Myers E, Bat1K Consortium. 2018. Bat biology, genomes, and the Bat1K project: to generate chromosome-level genomes for all living bat species. Annu Rev Anim Biosci 6: 23–46. 10.1146/annurev-animal-022516-022811 [DOI] [PubMed] [Google Scholar]
- Teng G, Maman Y, Resch W, Kim M, Yamane A, Qian J, Kieffer-Kwon KR, Mandal M, Ji Y, Meffre E, et al. 2015. RAG represents a widespread threat to the lymphocyte genome. Cell 162: 751–765. 10.1016/j.cell.2015.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonegawa S. 1983. Somatic generation of antibody diversity. Nature 302: 575–581. 10.1038/302575a0 [DOI] [PubMed] [Google Scholar]
- Wang F, Ekiert DC, Ahmad I, Yu W, Zhang Y, Bazirgan O, Torkamani A, Raudsepp T, Mwangi W, Criscitiello MF, et al. 2013. Reshaping antibody diversity. Cell 153: 1379–1393. 10.1016/j.cell.2013.04.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson CT, Steinberg KM, Huddleston J, Warren RL, Malig M, Schein J, Willsey AJ, Joy JB, Scott JK, Graves TA, et al. 2013. Complete haplotype sequence of the human immunoglobulin heavy-chain variable, diversity, and joining genes and characterization of allelic and copy-number variation. Am J Hum Genet 92: 530–546. 10.1016/j.ajhg.2013.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong J, Tai CM, Hurt AC, Tan HX, Kent SJ, Wheatley AK. 2020. Sequencing B cell receptors from ferrets (Mustela putorius furo). PLoS One 15: e0233794. 10.1371/journal.pone.0233794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Oficjalska K, Lambert M, Fennell BJ, Darmanin-Sheehan A, Shúilleabháin DN, Autin B, Cummins E, Tchistiakova L, Bloom L, et al. 2012. Fundamental characteristics of the immunoglobulin VH repertoire of chickens in comparison with those of humans, mice, and camelids. J Immunol 188: 322–333. 10.4049/jimmunol.1102466 [DOI] [PubMed] [Google Scholar]
- Ye J, Ma N, Madden TL, Ostell JM. 2013. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res 41: W34–W40. 10.1093/nar/gkt382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan C, Yang H. 2019. Research on K-value selection method of K-means clustering algorithm. J Multidiscip Sci J 2: 226–235. 10.3390/j2020016 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.