

Abstract
Although treatment with small molecule epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) is initially efficacious for patients with EGFR mutant lung cancer, drug resistance inevitably develops. Allosteric EGFR inhibitors that bind to a different site on EGFR were developed to treat cancers with EGFR mutations that mediate resistance to existing ATP-competitive EGFR TKIs. In the present study, we identify and study JBJ-09-063, a mutant selective allosteric EGFR inhibitor that is effective in both EGFR TKI sensitive and resistant models including those with EGFR T790M and C797S. We further uncover that homo- or heterodimerization with EGFR, or other ERBB family members, or the EGFRL747S mutation can mediate resistance to JBJ-09-063 but not to ATP-competitive EGFR TKIs. Taken together, our studies highlight the potential clinical utility of JBJ-09-063 as a single agent or in combination with EGFR TKIs in EGFR mutant lung cancer.
One Sentence Summary
The allosteric EGFR inhibitor JBJ-09-063 is effective in both EGFR tyrosine kinase inhibitor sensitive and resistant forms of EGFR L858R non-small cell lung cancer.
Introduction
Lung cancer is the leading cause of cancer-related deaths among both men and women in the United States (1, 2) with non-small cell lung cancer (NSCLC) accounting for 85% of all lung cancers. Epidermal growth factor receptor (EGFR) is one of the most common actionable oncogenic driver mutations in NSCLC. EGFR is a transmembrane protein that belongs to the ERBB family of tyrosine kinase receptors consisting of three other closely related members, ERBB2, ERBB3 and ERBB4. The ERBB family plays a critical role in essential cellular functions and is activated through ligand binding, followed by homo- and/or hetero-dimerization and phosphorylation of receptors. This results in activation of various downstream signaling pathways that regulate cell proliferation, differentiation, survival and migration. Therefore, it is not surprising that the function of these receptors is strictly regulated in normal cells. Mutations that promote constitutive activation, amplification, or overexpression of these receptors lead to uncontrolled cell growth, one of the hallmarks of cancer.
The identification of EGFR as a promising candidate for targeted therapy has led to the development of different strategies to block EGFR activity and its downstream pathways. Activating mutations in exons 18-21 of EGFR, which encode the catalytic domain that regulates EGFR phosphorylation and downstream pathways, are detected in 10-15% of cancers in Caucasian patients and in up to 50% of Asian patients with NSCLC. Therefore, major effort has been put forth in designing effective small molecule inhibitors that compete with the ATP site to inhibit EGFR activity. Phase III clinical trials have shown that patients with advanced EGFR mutant positive NSCLC (EGFR L858R or exon 19 deletion) have an improved progression-free survival (PFS) when treated with first-generation or second-generation EGFR tyrosine kinase inhibitors (TKIs), gefitinib, erlotinib, dacomitinib and afatinib, as a first-line treatment, compared to standard chemotherapy (3–10). Osimertinib, a mutant selective EGFR inhibitor, initially developed to overcome the most common mechanism of resistance to earlier generation EGFR inhibitors, EGFR T790M, leads to an improvement in both progression-free and overall survival compared to gefitinib or erlotinib when used as the first-line therapy in patients with advanced EGFR mutant NSCLC (11–16). It has now become the standard of care first-line EGFR inhibitor for advanced EGFR mutant (L858R and exon 19 deletion) NSCLC worldwide. While dacomitinib has also demonstrated an improved overall survival over gefitinib in EGFR mutant NSCLC, its clinical use remains limited due to its side effect profile (17).
Despite the improved efficacy of osimertinib compared to prior generation EGFR inhibitors, the improvements are disproportionately weighted towards the patients with EGFR exon 19 deletions. The absolute improvement in median PFS is less for patients with EGFR L858R (9.5 vs. 14.4 months) compared to exon 19 deletion (11.0 vs. 21.4 months) and there is no improvement in overall survival in patients with EGFR L858R mutations (18–20). In addition, acquired resistance mechanisms, including mutations in EGFR C797 that mediate resistance to osimertinib, have been identified (21). Depending on the sequence of EGFR inhibitor treatment, these can occur in the presence or absence of EGFR T790M. Although first-generation EGFR inhibitors can retain clinical activity when mutations in EGFR C797 occur after first line osimertinib treatment (i.e. EGFR L858R/C797S), resistance is commonly mediated by the acquisition of the EGFR T790M mutation (i.e. EGFR L858R/T790M/C797S) (22). At present, there are no effective therapies for these triple EGFR mutant cancers.
We have identified and studied allosteric EGFR inhibitors as a strategy to overcome EGFR mutation mediated resistance to ATP-competitive inhibitors (23–25). Allosteric inhibitors bind to a unique site, formed as a result of the EGFR L858R mutation, that is distinct from the ATP site and as such will not be impacted by acquired EGFR mutations that can limit the efficacy of TKIs. The first-generation inhibitor, EAI045, required co-administration of cetuximab in order to disrupt asymmetric EGFR dimerization and as such, allow binding of EAI045 to both halves of the dimer (23). A subsequent allosteric inhibitor, JBJ-04-125-02 was effective as a single agent in engineered models in vitro and in vivo, but not in patient-derived cell lines or xenograft models (24). However, it could co-bind with osimertinib and the co-binding led to an enhanced efficacy in vitro and in vivo compared to each of the single agents (24).
In the current study, we identify a more potent allosteric inhibitor, JBJ-09-063, and study its efficacy in vitro and in vivo. We identify molecular and biological determinants that limit the efficacy of JBJ-09-063 and evaluate approaches to overcome these limitations. Collectively, our findings highlight the clinical potential of treating EGFR L858R mutant cancers including those harboring resistance mutations to ATP-competitive EGFR inhibitors with an allosteric EGFR inhibitor alone or in combination with an ATP competitive inhibitor.
Results
JBJ-09-063 is effective in EGFR TKI resistant cancers in vitro and in vivo.
In order to identify a more potent allosteric inhibitor, we performed a series of alterations focusing on the right-hand side of the previously reported allosteric inhibitor molecule from the isoindolinone series, JBJ-04-125-02 (24) and identified JBJ-09-063 as an exquisitely potent allosteric EGFR inhibitor, with substantial improvement in enzymatic and cellular assays when compared with JBJ-04-125-02 (Fig. 1A).
Figure 1. Characterization of JBJ-09-063 in enzymatic assays, in vitro BaF3 cellular studies and in vivo xenograft models.
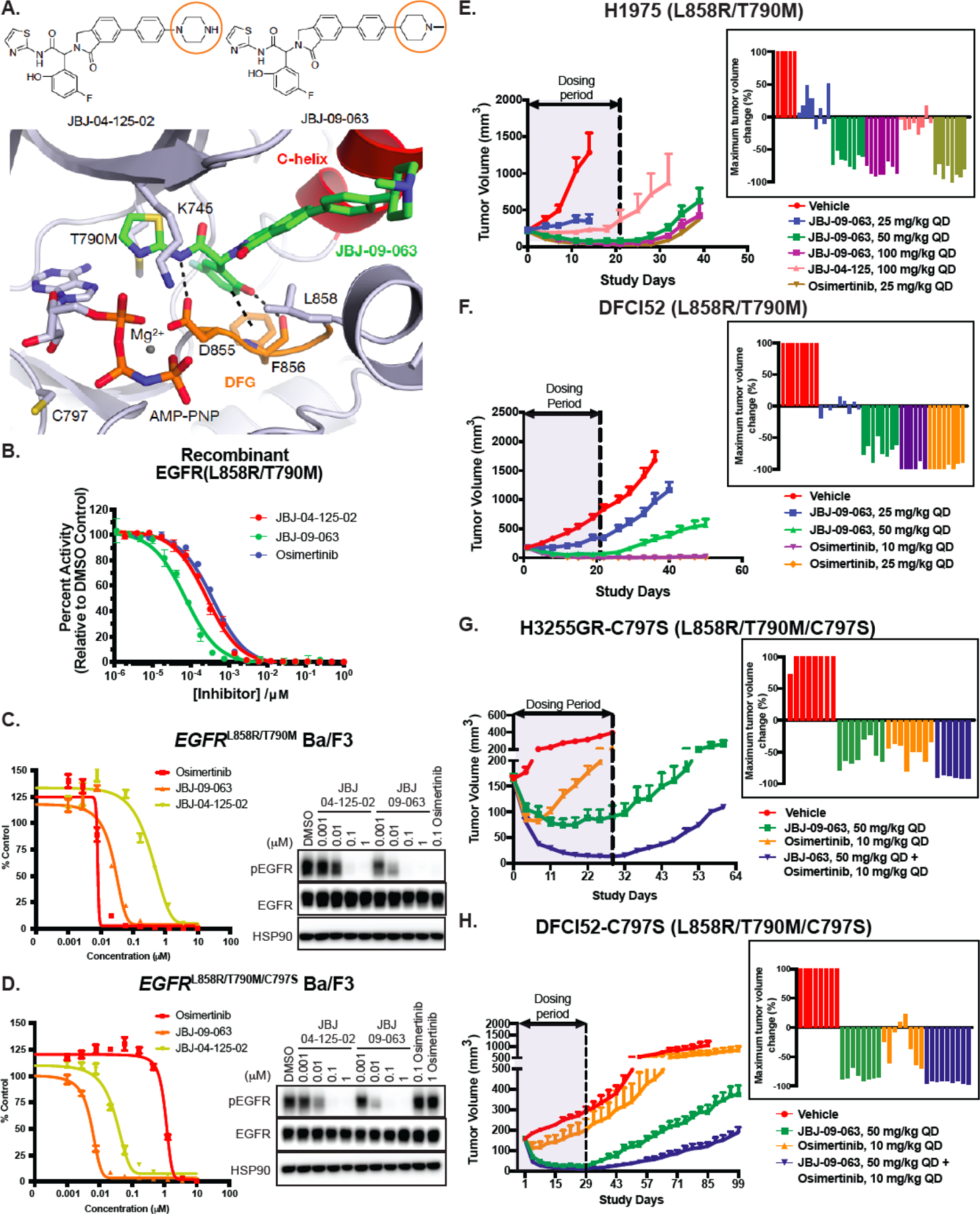
(A) Chemical structures of JBJ-04-125-02 and JBJ-09-063 with orange circles depicting the key difference between the two molecules. The 1.8 Å co-crystal structure of JBJ-09-063 in complex with EGFRT790M/V948R confirming an allosteric binding mode (PDB 7JXQ). JBJ-09-063 forms a critical hydrogen bond with D855 and a pi-stacking interaction with F856 in the DFG motif. (B) In vitro enzymatic inhibition assay of recombinant EGFR L858R/T790M kinase domain treated with increasing concentrations of allosteric inhibitors, JBJ-04-125-02, JBJ-09-063 and osimertinib. Results is graphed as percentage activity relative to DMSO control (mean ± SD). Cell growth and EGFR phosphorylation inhibition activity of JBJ-04-125-02 and JBJ-09-063 in (C) EGFRL858R/T790M and (D) EGFRL858R/T790M/C797S Ba/F3 cells as measured by Cell Titer Glo assay and Western Blot. Cell proliferation was graphed as a percentage relative to DMSO control. Data shown in A-C are representative experiments that were repeated at least three times. Efficacy studies examining the effect of allosteric inhibitors (JBJ-09-063, JBJ-04-125-02) and tyrosine kinase inhibitor (osimertinib) in (E) H1975 and (F) DFCI52 xenograft models harboring the EGFRL858R/T790M mutation. Efficacy studies examining the effect of JBJ-09-063 or osimertinib as a single agent or in combination in (G) H3255GR-C797S and (H) DFCI52-C797S xenograft models harboring the EGFRL858R/T790M/C797S mutation. Data is shown as a group mean of tumor volume in mm3 ± SEM relative to the start of treatment for all available data at the indicated timepoint (Study Days) with corresponding waterfall plots indicating maximum response in each group.
JBJ-09-063 contains a terminal N-methypiperidine ring in place of a terminal piperazine ring in JBJ-04-125-02, and a co-crystal structure of JBJ-09-063 confirmed its allosteric binding mode as similar to that of JBJ-04-125-02 (Fig. 1A, Table S1; Fig. S1A). HTRF-based enzymatic assays using recombinant EGFRL858R/T790M and EGFRL858R/T790M/C797S kinase domains showed that while osimertinib had similar biochemical efficacy as JBJ-04-125-02 against EGFRL858R/T790M, JBJ-09-063 was approximately 5-fold more potent than the other two compounds and unaffected by C797S (Fig. 1B; Table S2). In EGFRL858R/T790M and EGFRL858R/T790M/C797S Ba/F3 cells, JBJ-09-063 led to more potent (about 10-fold) growth inhibition than JBJ-04-125-02 with comparable efficacy to osimertinib in the EGFRL858R/T790M Ba/F3 cells (Fig. 1C-D; Table S2). In EGFRL858R/C797S Ba/F3 cells that were osimertinib-resistant, the potency of JBJ-09-063 was comparable to that of gefitinib (Fig. S1B-C; Table S2). We evaluated the ability of these compounds to inhibit EGFR phosphorylation. Consistent with the impact on growth inhibition, JBJ-09-063 was able to reduce EGFR phosphorylation at lower concentrations than JBJ 04-125-02 in all three EGFR mutant Ba/F3 cell lines (Fig. 1C-D; Fig. S1B-C).
We next compared the pharmacokinetic properties of JBJ-09-063 with JBJ-04-125-02 ((24); Table S3) and found that JBJ-09-063 had higher IV clearance (5.0 ml/min/kg vs. 15.7 ml/min/kg) but increased bioavailability (14.6% vs. 3%) and the oral AUC values were similar for the two compounds (Table S3). Therefore, we hypothesized that the increased potency of JBJ-09-063 coupled with its improved pharmacological properties would lead to an enhanced in vivo efficacy. In a pharmacodynamic study, using 50 mg/kg daily dosing in the H1975 model, JBJ-09-063 effectively inhibited EGFR, Akt and ERK 1/2 phosphorylation (Fig. S1D). Hence, we first used H1975 (Fig. 1E) and the patient derived DFCI52 xenograft models (Fig. 1F; Fig. S1E) which both harbor the EGFRL858R/T790M mutation to evaluate JBJ-09-063 in vivo. JBJ-09-063 treatment led to a dose-dependent decrease in tumor volume over time and was more potent than the previous efficacious dose of JBJ-04-125-02 at 100 mg/kg in the H1975 in vivo model (Fig. 1E). Notably, the 50 mg/kg and 100 mg/kg doses of JBJ 09-063 were as effective as osimertinib (25 mg/kg) in this model. In the DFCI52 model, the 50 mg/kg dose of JBJ-09-063 was similarly effective as osimertinib although there was a more rapid tumor outgrowth following the end of dosing compared to mice treated with osimertinib (Fig. 1F).
To date, there are very few, if any, reliable human cancer cell lines or patient derived in vivo models that harbor the EGFRL858R/T790M/C797S mutation. In order to generate cells with the C797S mutation, we utilized H3255GR and DFCI52, both of which possess the EGFRL858R/T790M mutation, and introduced the C797S mutation in cis with T790M using CRISPR/CAS9 genome editing followed by selection with 100 nM osimertinib (Fig. S1F). CRISPR sequencing results demonstrated that the C797S mutation was indeed integrated in cis and the relative allelic frequency was 45% and 2.74% in DFCI52 and H3255GR cells, respectively (Fig. S1G). The H3255GR-C797S and DFCI52-C797S cells were resistant to osimertinib compared to their parental H3255GR and DFCI52 counterparts (Fig. S1H). Consistent with these observations, EGFR, Akt and ERK1/2 phosphorylation was no longer inhibited effectively by osimertinib in cells with the C797S mutation (Fig. S1I). We subsequently used the DFCI52-C797S and H3255GR-C797S cells for in vivo studies (Fig. 1G-H). In agreement with the in vitro observations, both models were resistant to osimertinib but sensitive to JBJ-09-063. Interestingly, in the H3255GR-C797S model, but not in the DFCI52-C797S model, osimertinib demonstrated transient efficacy likely due to the lower allelic fraction of the C797S mutation (Fig. 1G-H).
JBJ-09-063 is effective in human EGFRL858R/T790M mutant cancer lines in vitro but only when combined with an ATP competitive EGFR inhibitor
We next evaluated the efficacy of JBJ-09-063 in vitro using human cancer cell lines. To our surprise, viability assay in both DFCI52 (Fig. 2A) and H3255GR (Fig. S2A) cells revealed little difference between JBJ-04-125-02 or JBJ-09-063 treatment, in contrast to the in vivo findings. Furthermore, while JBJ-09-063 was able to inhibit EGFR and Akt phosphorylation slightly better than JBJ-04-125-02, osimertinib at 0.1 µM was still much more effective at abolishing EGFR, Akt and ERK1/2 phosphorylation than JBJ-09-063 in DFCI52 cells and H3255GR cells respectively (Fig. 2B and Fig. S2B).
Figure 2. JBJ-09-063 efficacy in human cancer cells is enhanced when combined with gefitinib.
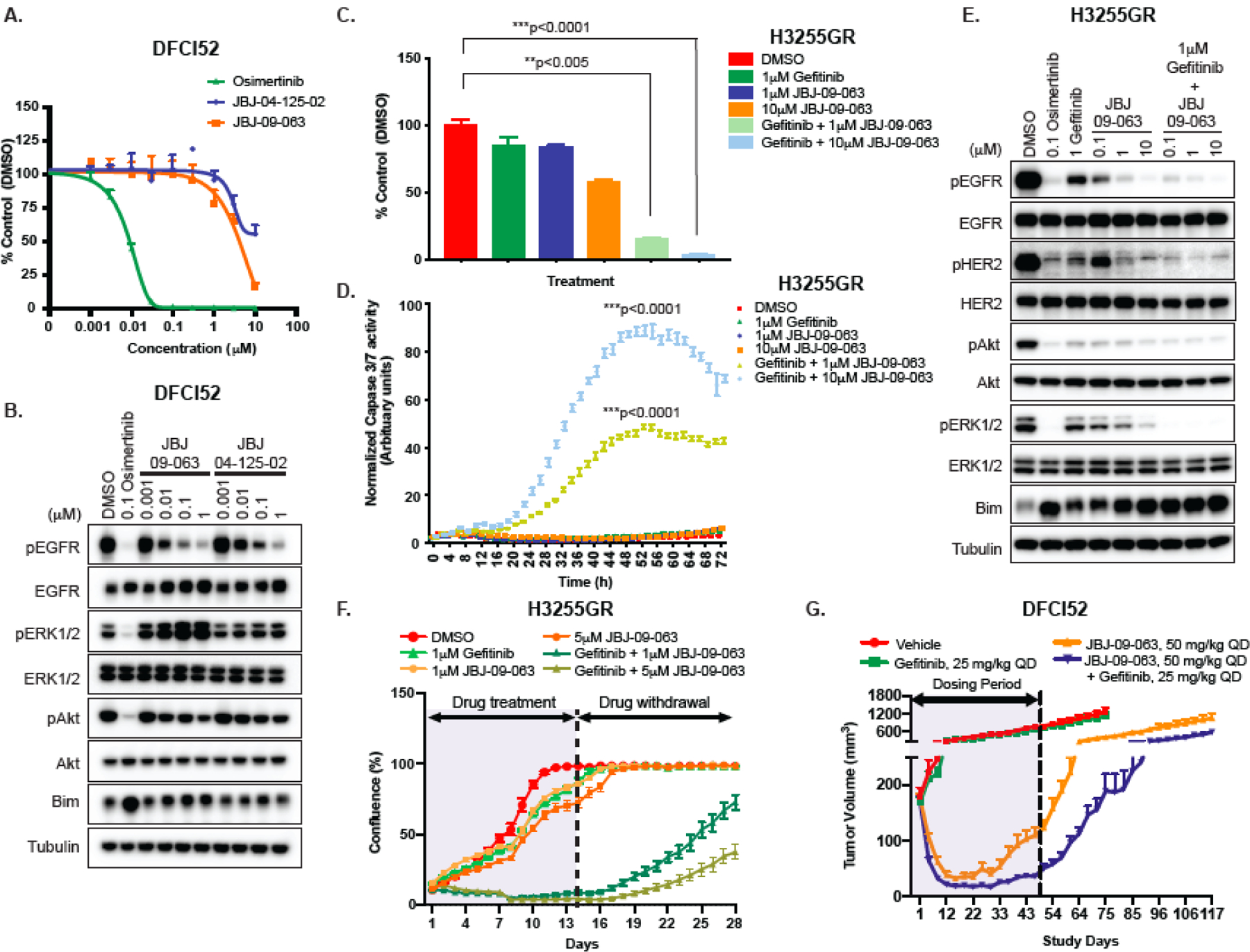
(A) Cell viability and (B) Western Blot analyses of DFCI52 cells treated with indicated concentrations of osimertinib, JBJ-09-063 and JBJ-04-125-02. (C) Cell viability and (D) apoptosis measured as normalized Caspase3/7 activity and (E) Western Blot analyses of H3255GR cells treated with indicated concentrations of gefitinib, JBJ-09-063 and the combination of both agents. (F) Long-term cell growth assay measured as confluency (%) in H3255GR cells treated with indicated concentrations of gefitinib, JBJ-09-063 and the combination of both drugs for two weeks followed by drug withdrawal for an additional two weeks. Data shown in A-F a representative experiment that was repeated at least two times. All cell viability assays were graphed as a percentage of activity relative to DMSO control over indicated concentrations and all apoptosis experiments were graphed as normalized caspase 3/7 activity (in arbitrary units) over time. Statistical significance was determined using a one-way ANOVA on ranks (the Kruskal-Wallis Test) and Dunn’s multiple comparisons test. **P<0.005; ***P<0.0001. (G) Efficacy studies examining the effect of JBJ-09-063 as a single agent or in combination with gefitinib in DFCI52 xenograft model harboring the EGFRL858R/T790M mutation. Data is shown as a group mean of tumor volume (mm3) ± SEM relative to the start of treatment for all available data at the indicated timepoint (Study Days).
In order to decipher the disconnect between the in vivo and in vitro efficacies, we evaluated ATP competitive EGFR inhibitors in combination with JBJ-09-063 in the H3255GR cells. Surprisingly, and unexpectedly, the combination of gefitinib and JBJ-09-063 was remarkably effective at inhibiting cell growth and led to a significant increase in apoptosis, even though H3255GR cells are resistant to gefitinib as a single agent as they contain an EGFRT790M mutation (Fig. 2C-D; (26)). The JBJ-09-063 combination with gefitinib also inhibited EGFR, Akt and ERK1/2 phosphorylation in a dose-dependent fashion and to a similar degree as did osimertinib alone (Fig. 2E). Similar findings were observed in the DFCI52 cells (Fig. S2C-E). Analogously, both H3255GR-C797S and DFCI52-C797S cells were resistant to JBJ-09-063 in vitro despite their sensitivity in vivo. However, cell proliferation and phosphorylation of EGFR and its downstream targets were inhibited when these cells were treated with the combination of JBJ-09-063 and osimertinib (Fig. S2F-H). In addition, administering osimertinib and JBJ-09-063 together in the H3255GR-C797S and DFCI52-C797S xenografts in vivo further potentiated tumor shrinkage, resulting in a delay in tumor regrowth, following treatment cessation at 28 days of dosing (Fig. 1G-H).
We further sought to examine whether the combination of gefitinib and JBJ-09-063 treatment would delay in vitro resistance of chronically treated H3255GR cells (Fig. 2F). Consistent with the inhibition seen on short-term growth and apoptosis studies, the gefitinib and JBJ-09-063 combination was most effective compared to DMSO or either gefitinib or JBJ-09-063 alone and led to a delay in cell regrowth. When we evaluated this approach in vivo using the DFCI52 xenograft model, tumors regressed following treatment with JBJ-09-063 alone and when combined with gefitinib but were resistant to gefitinib alone (Fig. 2G). While the gefitinib and JBJ-09-063 combination led to a greater delay in tumor regrowth after cessation of treatment, it was less impressive than the difference observed in vitro (Fig. 2C-F). Collectively, our findings demonstrate that EGFR TKIs can significantly enhance the in vitro efficacy and downregulation of EGFR signaling imparted by an allosteric EGFR inhibitor even when the cell lines themselves are resistant to the EGFR TKIs. These observations suggest that EGFR TKIs may be inhibiting subsets of EGFR receptors that are not effectively inhibited by the allosteric agent.
Forced homodimerization or heterodimerization of EGFR mutants with ERBB family members can impact the potency of JBJ-09-063
To further elucidate how JBJ-09-063 potency was enhanced in the presence of an EGFR TKI and whether this was driven by EGFR and/or other ERBB family member dimerization, we studied the efficacy of JBJ-09-063 in the presence of different EGFR homo and heterodimers. We utilized the EGFR kinase domain duplication (KDD) mutant as a tool to perform our studies. EGFR-KDD is a rare but recurrent oncogenic mutation that results in the in-frame tandem duplication of the EGFR exons (18–25), which encode the EGFR tyrosine kinase domain. The duplication traps the two EGFR kinase domains in a position that favors spontaneous asymmetric dimerization and persistent intra-molecular EGFR activation (27). We generated a set of such kinase duplications consisting of permutations of mutant EGFR and HER2 tyrosine kinase (TK) domains to study the effect of JBJ-09-063 on EGFR activation (Fig. S3A). We transfected the different constructs into HEK293T/Cl.17 cells, treated the cells with either JBJ-09-063 or osimertinib and evaluated their impact on EGFR phosphorylation. As expected, in cells expressing an EGFR construct with only a single kinase domain harboring EGFRL858R/T790M, both JBJ-09-063 and osimertinib effectively inhibited EGFR phosphorylation (Fig. 3A). However, in cells expressing a tandem of EGFRL858R/T790M TK domains (EGFRKDD-LT/LT) only osimertinib inhibited EGFR phosphorylation. Similar results were obtained when one of the TK domains contained the EGFR mutation and the other was wild type (EGFRKDD-WT/LT or EGFRKDD-LT/WT) regardless of their order (Fig. 3A). We made analogous findings when both TK domains contained an EGFR L858R mutation only (EGFRKDD-L/L) or when one of the TK domains was from HER2 (Fig. S3B-C). We further generated versions of EGFRKDD-WT/LT and ERBBKDD-HER2_WT/EGFR_LT with EGFRI941R and HER2V956R mutations, which render them dimerization-deficient (EGFRKDD-WTdd/LTddERBBKDD-HER2_WTdd/EGFR_LTdd), and noted that in cells expressing these mutants, the inhibitory activity of both osimertinib and JBJ-09-063 was restored. These findings directly implicate dimerization of EGFR with itself or other ERBB family members in the inability of JBJ-09-063 to inhibit EGFR phosphorylation. Consistent with the effect we observed with EGFR phosphorylation, Ba/F3 cells stably expressing EGFRKDD-LT/WT or ERBBKDD-EGFR_LT/HER2 were also significantly more resistant to JBJ-09-063 compared to those expressing just a single kinase domain (EGFRL858R/T790M) while both KDD expressing Ba/F3 cell lines remained sensitive to osimertinib (Fig. 3B-C; Table S2). Finally, EGF, which induces dimerization, also led to resistance to JBJ-09-063 in EGFRL858R or EGFRL858R/T790M expressing Ba/F3 cells (Fig. 3B-C; Fig. S3D-E; Table S2). Collectively, these findings reveal that the dimerization of mutant EGFR with mutant or wild type EGFR or HER2 plays a critical role in mediating resistance to JBJ-09-063 but not to osimertinib.
Figure 3. Forced dimerization of EGFR with other ERBB family members impart resistance to JBJ-09-063.
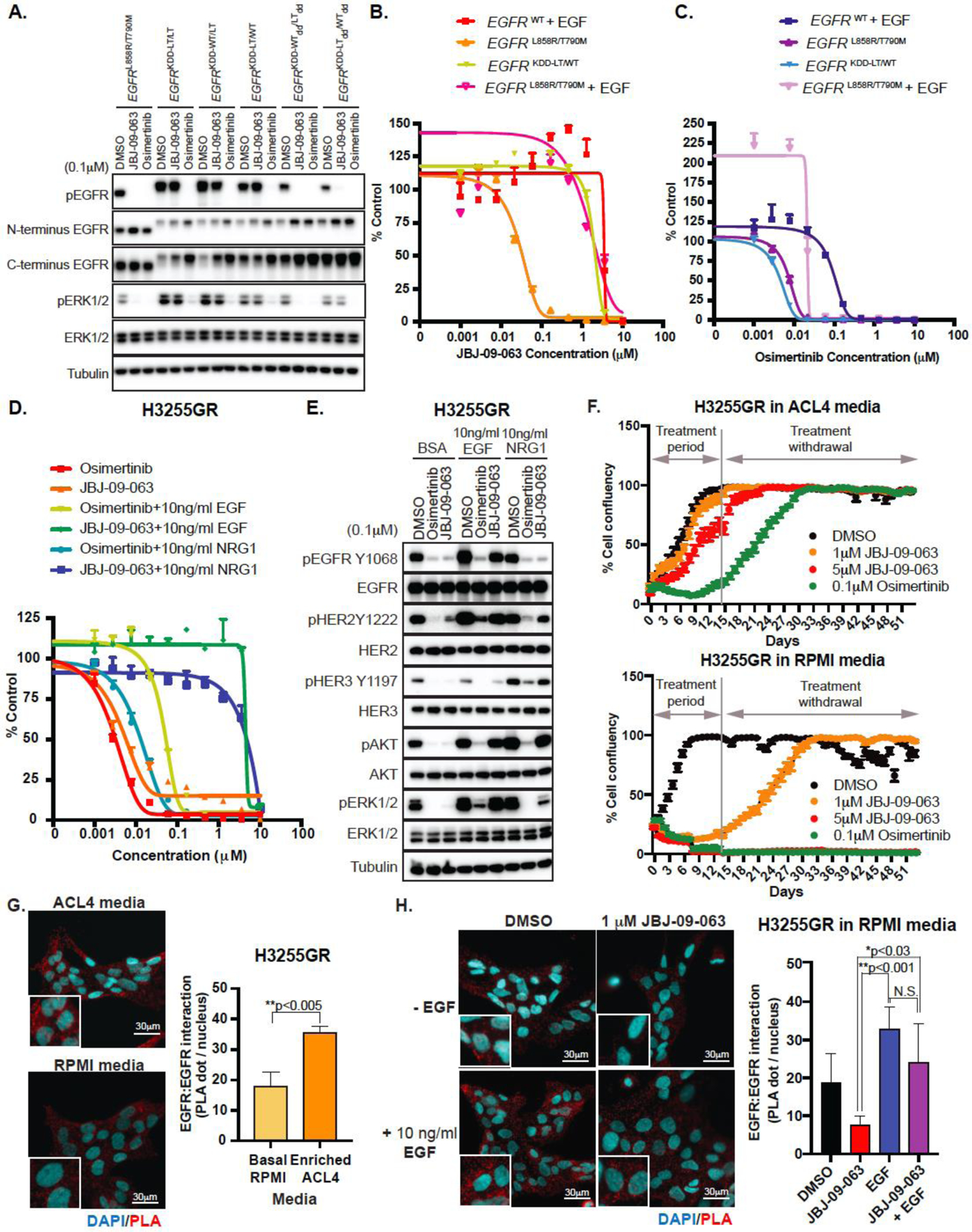
(A) Western Blot analyses of HEK293T/Cl.17 cells transiently transfected with EGFRL858R/T790M or with different EGFRKDD constructs and treated with DMSO, osimertinib or JBJ-09-063. Cell viability in EGFRWT, EGFRKDD-LT/WT, or EGFRL858R/T790M Ba/F3 cells in the presence or absence of EGF treated with increasing concentrations of (B) JBJ-09-063 or (C) osimertinib. (D) Cell viability and (E) Western Blot analyses of H3255GR cells cultured in RPMI media and treated with indicated concentrations of compounds in the presence or absence of EGF or NRG1. Cell viability assays shown in B-D were graphed as a percentage of activity relative to DMSO control over indicated concentrations. (F) Long-term cell growth assay measured as confluency (%) in H3255GR cells cultured in ACL4 media (top panel) versus RPMI media (bottom panel) and treated with DMSO, JBJ-09-063 or osimertinib for two weeks followed by drug withdrawal for an additional two weeks. Cell proliferation was graphed as a percentage relative to DMSO control. (G) Homodimerization of EGFR visualized by proximation ligation assay (PLA) in H3255GR cells cultured in ACL4 media or RPMI media. Statistical significance was determined using the unpaired t-test **P<0.005. (H) EGFR homodimerization in H3255GR cells cultured in RPMI media visualized by PLA after pre-treatment with or without 10 ng/ml of EGF for 15 minutes followed by incubation with DMSO or JBJ-09-063 for 4 hours. Nuclei are stained in cyan and distinct punctate dots in red are PLA signal showing interaction of EGFR homodimers. Statistical significance was determined by ANOVA followed by Tukey’s multiple comparisons test. *P<0.0278; **P<0.0014. Data quantification in 3D-E was performed and graphed as EGFR:EGFR interactions (PLA dot/nucleus) over different culture media. Scale bar = 30 µm. All studies shown here are representative experiments that were repeated at least three times.
Dimerization-inducing ligands in culture media can also impact the potency of JBJ-09-063
Intrigued by the results of our EGFRKDD and ERBBKDD studies, we hypothesized that the difference between the in vitro and in vivo efficacy of JBJ-09-063 in human cancer cells lines may also be attributed to the extent of homo- or heterodimerization. H3255GR and DFCI52 cells were established and grown in ACL4 media with 5% FBS and RETM media with 10% FBS, respectively. ACL4 media is composed of the basal RPMI media with added growth factors including EGF; RETM media is nutrient-rich media which also contains growth factors such as EGF. The presence of EGF could inadvertently affect dimerization and thus sensitivity of these cell lines to JBJ-09-063. Therefore, we evaluated the efficacy of JBJ-09-063 in RPMI media in the presence or absence of ERBB family ligands EGF and NRG1. In RPMI media, the H3255GR cells were almost equally as sensitive to JBJ-09-063 as to osimertinib (Fig. 3D). The presence of either EGF or NRG1 markedly shifted the IC50 of JBJ-09-063 towards resistance, while the efficacy remained largely unchanged when combined with osimertinib (Table S2). Analogously, the addition of EGF or NRG1 to cells grown in RPMI blocked the ability of JBJ-09-063 to inhibit downstream Akt or ERK1/2 phosphorylation, either because of persistent EGFR phosphorylation (due to EGF) or HER2/HER3 phosphorylation (due to NRG1; Fig. 3E). Identical findings were observed in the DFCI52 cells (Fig. S3F-G). In a long-term in vitro assay, H3255GR cells grown in RPMI media were also more sensitive to JBJ-09-063 compared to those grown in the ACL4 media (Fig. 3F). Interestingly, although they were sensitive to osimertinib in both contexts, the delay in tumor cell regrowth was more evident in RPMI than in ACL4 media. To further study the impact of different media and EGF treatment on dimerization and the efficacy of JBJ-09-063 in NSCLC cell lines, we used a proximity ligation assay (PLA). PLA is an immunofluorescence-based technique that detects the interaction or dimerization of proteins when the probes attached to the antibodies specific for the proteins of interest come in close proximity and become ligated (28). The ligated DNA is then amplified by DNA polymerase to produce a red dot-like signal. When compared to H3255GR cells grown in RPMI, we observed significantly greater number of EGFR dimers in cells grown in ACL4 (Fig. 3G). H3255GR cells grown in RPMI had significantly less EGFR dimers when treated with JBJ-09-063 compared to EGF (Fig. 3H). This persisted even when the cells were co-treated with both JBJ-09-063 and EGF (Fig. 3H). Collectively, these studies highlight how modulation of growth conditions, specifically the presence of ERBB family ligands, can impact the efficacy of an allosteric EGFR inhibitor.
JBJ-09-063 is mutant selective and is also effective in EGFRL858R models.
We next sought to examine whether JBJ-09-063 was also effective in EGFR inhibitor treatment naïve models. Our initial allosteric compounds were discovered in a screen with the EGFRL858R/T790M kinase domain and had little potency against the EGFRL858R mutant. However, biochemical characterization of JBJ-09-063 revealed 0.147 nM potency against L858R in the in vitro kinase assays (Table S2). As one of the future potential therapeutic options for an allosteric EGFR inhibitor would be to co-administer it with osimertinib, we wanted to determine whether JBJ-09-063 also had efficacy on its own against the initiating L858R mutant. In both EGFRL858R Ba/F3 cells and in H3255 cells (which harbor an EGFRL858R mutation), JBJ-09-063 was effective, albeit slightly less than gefitinib or osimertinib (Fig. 4A-B; Table S2). These findings were mirrored by a Western Blot analysis demonstrating that slightly higher concentrations of JBJ-09-063 were necessary to inhibit EGFR, Akt and ERK 1/2 phosphorylation compared to osimertinib alone in H3255 cells (Fig. 4C). In a long-term treatment study in H3255 cells, all three EGFR inhibitors were effective, although regrowth following drug withdrawal occurred earlier with JBJ-09-063 treatment compared to gefitinib or osimertinib (Fig. 4D).
Figure 4. JBJ-09-063 is effective in TKI naïve EGFR L858R mutant models.
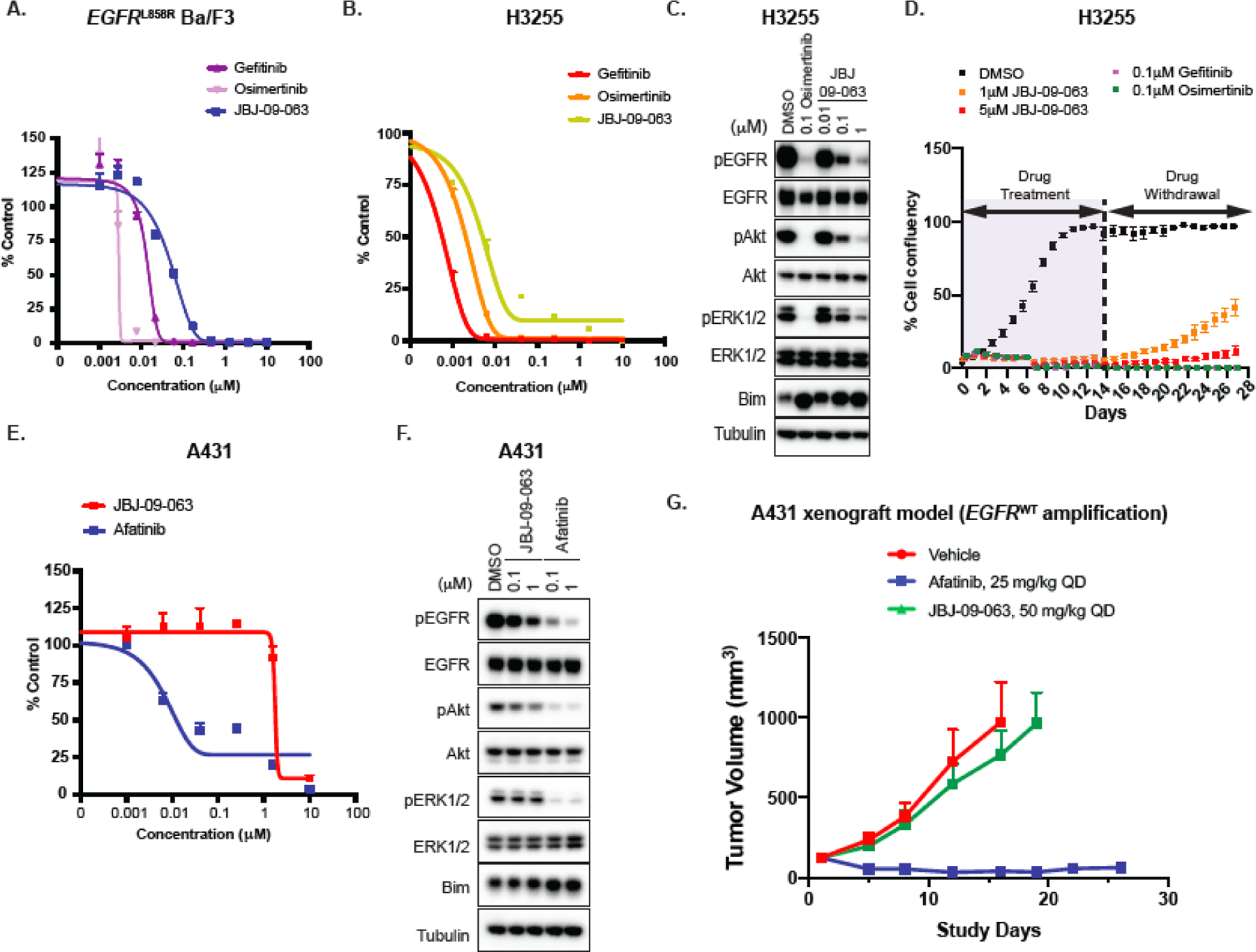
(A) Cell viability of EGFRL858R Ba/F3 cells following treatment with gefitinib, osimertinib or JBJ-09-063. (B) Cell inhibitory activity and (C) Western Blot analyses of H3255 cells cultured in RPMI media and treated with indicated concentrations of specific inhibitors for 72 hours and 24 hours respectively. (D) Long-term cell growth activity measured as confluency (%) in H3255 parental cells cultured in RPMI media and treated with indicated concentrations of JBJ-09-063, gefitinib or osimertinib for two weeks followed by drug withdrawal for another two weeks. (E) Cell viability and (F) Western Blot analyses of A431 cells treated with increasing concentrations of JBJ-09-063 and afatinib. Data shown in A, B, D, E are representative experiments that were repeated at least three times. Cell viability was graphed as a percentage relative to DMSO control. (G) Efficacy studies examining the effect of JBJ-09-063 or afatinib as single agents in the A431 xenograft model. Mice in the in vivo studies were treated after tumor development for 30 days. Data is shown as a group mean of tumor volume ± SEM relative to the start of treatment (Tumor volume (mm3)) for all available data at the indicated timepoint (Study Days).
To examine whether mutant selectivity was maintained in JBJ-09-063, we compared its efficacy in vitro and in vivo using A431 cells (which harbor an EGFRWT amplification). Compared to afatinib, JBJ-09-063 was significantly less effective at inhibiting cell viability or EGFR, Akt and ERK 1/2 phosphorylation (Fig. 4E-F; Table S2). In an in vivo study, using a dose of JBJ-09-063 that was effective in EGFR mutant models as a single agent (50 mg/kg; Fig. 1E-H), we observed no effect on A431 tumor volume during the course of the study (Fig. 4G). In contrast, afatinib (25 mg/kg) led to substantial tumor reduction (Fig. 4G).
JBJ-09-063 is efficacious against non-C797S osimertinib-resistant EGFR mutations
Although C797S mutation is the most commonly acquired osimertinib-resistant mutation, it is not the only resistance mutation identified (29). Other osimertinib-resistant mutations include L718Q in the beta sheet proceeding the turn of the phosphate-binding loop, L792F in the kinase hinge, and G796S that precedes the site of covalent modification by osimertinib (30). Modelling of these mutations revealed steric clashes with osimertinib, with L792F being the least severe (Fig. 5A-B). However, since these mutations are localized to the ATP site, they are not expected to affect the binding of allosteric inhibitors. Therefore, we examined whether JBJ-09-063 was effective in the presence of these osimertinib-resistant mutations. We generated Ba/F3 cells expressing EGFRLT/L718Q, EGFRLT/L792F and EGFRLT/G796S and compared the efficacy of JBJ-09-063 to osimertinib on both cell growth and EGFR signaling. We confirmed that these mutants were indeed resistant to osimertinib. The Ba/F3 cells harboring the EGFRLT/L792Fmutation, were least resistant to osimertinib as predicted by the modeling (Fig. 5C; Table S2). All of the triple EGFR mutant Ba/F3 cells were more sensitive to JBJ-09-063 compared to osimertinib, although the magnitude of sensitivity varied between the mutants. Similarly, JBJ-09-063 was much more effective at inhibiting EGFR, Akt and ERK 1/2 phosphorylation in the triple mutant Ba/F3 cells compared to osimertinib (Fig. 5D). When further introduced the different osimertinib resistance mutant alleles into H1975 cells which harbor the EGFRL858R/T790M mutation. The results (both in growth inhibition and in impact of EGFR and downstream signaling) observed in H1975 cells are consistent with those of the Ba/F3 cells; however, the effect was not as robust (Fig. S4A-B; Table S2). These findings suggest that an allosteric EGFR inhibitor can be effective in a broad range of osimertinib-resistant EGFR mutant cancers.
Figure 5. JBJ-09-063 is effective in a broad range of osimertinib-resistant EGFR mutant contexts.
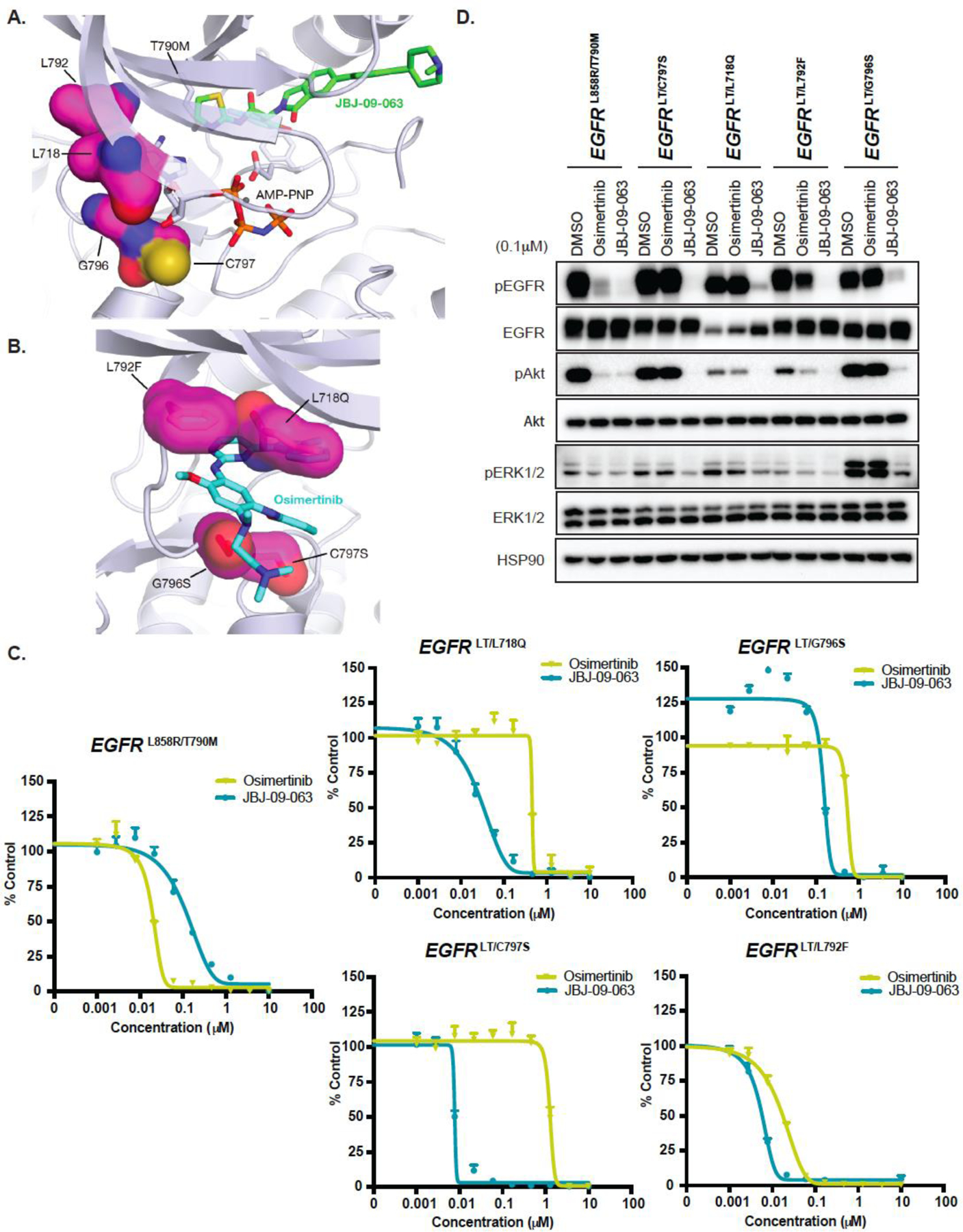
Crystal structure of EGFR in complex with (A) JBJ-09-063 (PDB 7JXQ) or (B) osimertinib (bottom, PDB 4ZAU). Sites of mutation are localized to the ATP binding site and should not affect allosteric inhibitor binding. Modeling of resistance mutations reveals steric clashes with osimertinib with the exception of C797S, which prevents osimertinib from forming a covalent adduct with the protein. (C) Cell proliferation and (D) Western Blot analyses of EGFRL858R/T790M, EGFRLT/C797S, EGFRLT/L718Q, EGFRLT/L792F, and EGFRLT/G796S Ba/F3 cells treated with DMSO, osimertinib or JBJ-09-063. All data shown is a representative experiment that was repeated at least three times. Cell proliferation was graphed as a percentage relative to DMSO control. LT = L858R/T790M.
EGFR L747S mutation is an on-target resistance mechanism for JBJ-09-063 but not to osimertinib
Although JBJ-09-063 is still effective even in the presence of EGFR mutations that lead to osimertinib resistance, it is likely that a different set of EGFR mutations mediate resistance to JBJ-09-063. To identify JBJ-09-063 resistant mutations, we treated EGFRL858R/T790M Ba/F3 cells with N-ethyl-N-nitrosourea (ENU) followed by chronic treatment of 1 µM of JBJ-09-063, 1µM of osimertinib or with the combination of both compounds. We plated a total of 900 clones from each treatment condition and 76/900 and 13/900 JBJ-09-063- and osimertinib-treated cells grew, respectively. However, we were not able to identify any resistant colonies when cells were treated with both drugs together (Fig. 6A). We sequenced the EGFR tyrosine kinase domain of each of these resistant colonies and found that out of 76 JBJ-09-063 resistant colonies, 3 (3.94%) harbored a L747S mutation, while all 13 colonies (100%) in the osimertinib-resistant population carried the C797S mutation. We did not observe any EGFR TK domain mutations in the other 73 JBJ-09-063 resistant colonies. We next performed modelling analyses to examine how L747S mutation may impact the binding and function of JBJ-09-063. The side chain of L747 forms favorable hydrophobic contacts with the phenyl ring of JBJ-09-063 and modeling of the L747S mutation decreased these contacts and widened the putative entrance tunnel for the allosteric pocket. This change may lead to a decrease in drug potency but should not abolish inhibitor binding like substitution to a bulkier amino acid would (Fig. 6B-C). We conducted in vitro enzyme inhibition experiments using purified EGFRLT/L747S kinase domain. While osimertinib effectively inhibited the activity of both EGFRL858R/T790M and EGFRLT/L747S, the efficacy of JBJ-09-063 was substantially reduced (Fig. 6D; Table S2). Similarly, the efficacy of JBJ-09-063 was reduced in EGFRLT/L747S expressing Ba/F3 cells (Fig. 6E; Table S2). Furthermore, and consistent with the enzymatic and growth assays, JBJ-09-063 was less effective at inhibiting EGFR phosphorylation in EGFRLT/L747S expressing Ba/F3 cells while osimertinib was equally effective in EGFRLT and EGFRLT/L747S expressing Ba/F3 cells (Fig. 6F). These results were recapitulated in the H1975 cells engineered to express the EGFRLT/L747S mutation (Fig. S4C-D; Table S2).
Figure 6. L747S mutation is an on-target resistance mechanism to JBJ-09-063 but not to osimertinib.
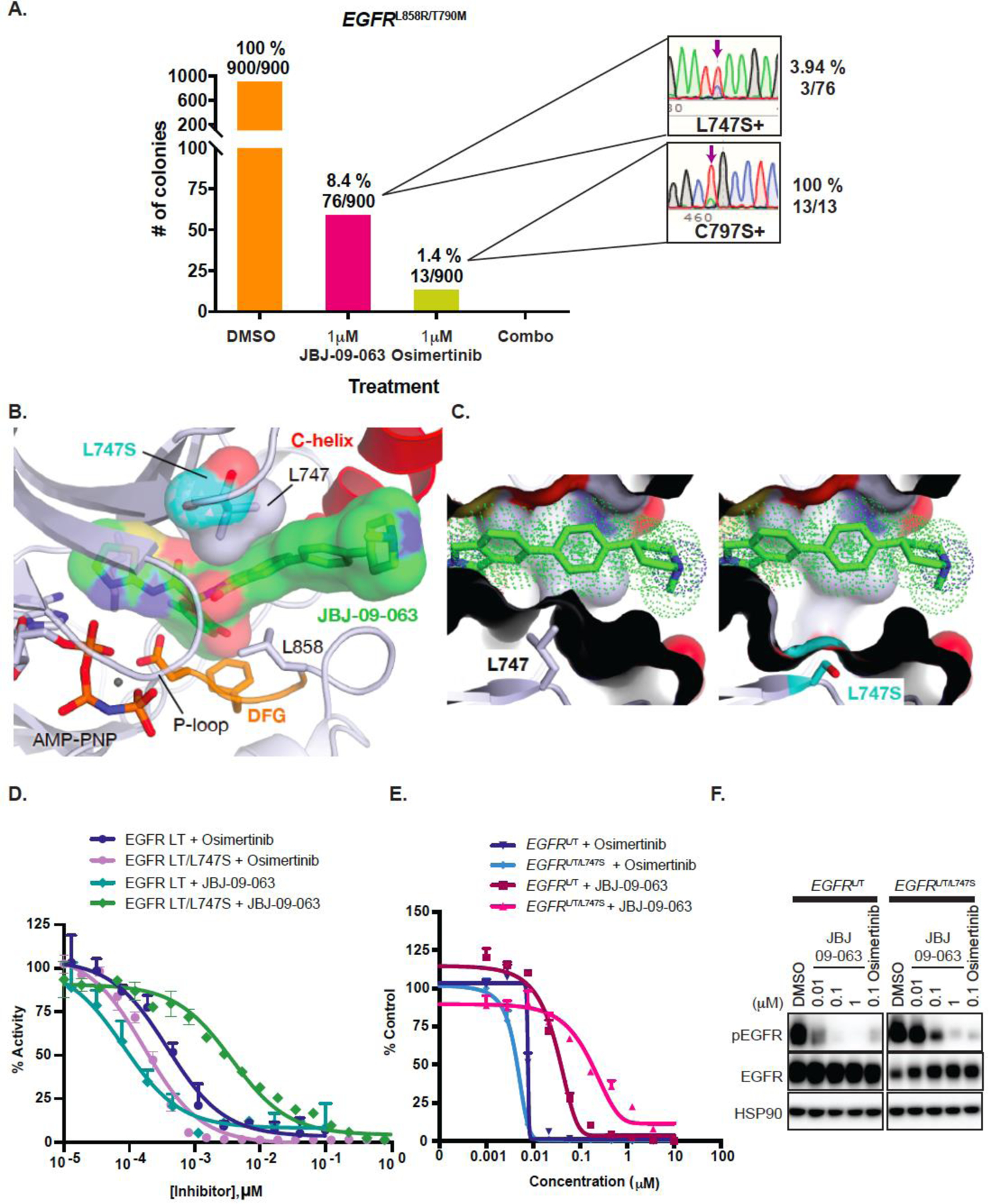
(A) Mutagenesis studies showing the number of colonies that arose when EGFRL858R/T790M Ba/F3 cells that were exposed to N-ethyl-N-nitrosourea (ENU) to induce sporadic mutation were treated with either DMSO, JBJ-09-063, osimertinib or combination treatment for 4 weeks. Sequence tracing and the percentage of L747S and C797S mutations frequencies present in JBJ-09-063- and osimertinib-resistant colonies were shown with purple arrows respectively. (B) Modeling of the L747S resistance mutation (PDB 7JXQ) using the most likely serine rotamer. (C) The side chain of L747 forms favorable hydrophobic contacts with the phenyl ring of the allosteric inhibitor that are lost in the L747S variant. (D) In vitro inhibition of EGFR L858R/T790M and EGFR L858R/T790M/L747S kinases with increasing concentrations of JBJ-09-063 and osimertinib were measured using an HTRF-based assay. Percentage of activity is relative to a 1% DMSO control. (E) Cell growth inhibition and (F) EGFR phosphorylation activity of EGFRL858R/T790M or EGFRLT/L747S Ba/F3 cells treated with JBJ-09-063 or osimertinib was measured by Cell Titer Glo assay and analyzed by Western Blot. Data shown is a representative experiment that was repeated at least three times. Cell viability was graphed as a percentage relative to DMSO control. LT=L858R/T790M.
Discussion
The identification of EGFR as an actionable oncogenic driver mutation target and the successful development of effective ATP-competitive EGFR TKIs has had a tremendous impact on the treatment of patients with EGFR mutant NSCLC. However, the development of acquired drug resistance limits the long-term efficacy of EGFR TKIs. There is currently a need to develop novel treatment strategies especially for cancers that develop EGFR mutations as a mechanism of osimertinib resistance.
In the current study, we identify and study an allosteric EGFR inhibitor that is effective against a broad range of osimertinib resistance mutations in vitro, and in vivo as a single agent in models of osimertinib resistance mediated by EGFR C797S. Coupled with its mutant selectivity over wild type EGFR in vitro and in vivo, allosteric EGFR inhibition may thus be a viable therapeutic approach to targeting drug resistant EGFR L858R mutant cancer. However, additional development of clinically deployable allosteric EGFR inhibitors is still needed. Allosteric EGFR inhibitors may also be therapeutically valuable in EGFR TKI naïve EGFR L858R mutant cancers, given their inferior PFS and OS outcomes following osimertinib treatment relative to EGFR exon 19 deletion cancers (18–20). Our prior studies demonstrated that osimertinib and allosteric EGFR inhibitors can co-bind EGFR L858R at the same time (24). Whether this dual EGFR inhibition can lead to enhanced clinical efficacy compared to osimertinib alone in patients with newly diagnosed advanced EGFR L858R cancers needs to be tested in future clinical trials. We also do not at present have data on the ability of JBJ-09-063 to penetrate the central nervous system which has been a clinically desirable feature of osimertinib. Alternative therapeutic approaches, such as the development of ATP competitive EGFR inhibitors able to inhibit C797S, BLU-945 (NCT04862780) and BBT-176 (NCT04820023) are also being clinically developed. The optimal therapeutic strategy against EGFR C797S will await the findings from the ATP competitive and allosteric EGFR inhibitor clinical studies.
A unique characteristic of allosteric, but not of ATP competitive EGFR inhibitors, is the therapeutic limitation conferred by ERBB family dimerization. This can be induced by ERBB family ligands (including EGF and NRG1 (Fig. 3D-E)) or through forced dimerization (Fig. 3A-C). In the presence of an asymmetric EGFR homo- or heterodimer, an allosteric inhibitor can only access one of the allosteric binding sites. In contrast, both ATP binding sites remain accessible. Intriguingly, EGFR amplification alone is not sufficient to blunt the efficacy of JBJ-09-063. This is exemplified by H3255 (Fig. 4B-D) and H3255GR (Fig. 3D-F) cells both of which contain EGFR amplifications but retain sensitivity to JBJ-09-063 when grown in RPMI media. How these observations would impact the potential clinical efficacy of an allosteric EGFR inhibitor is currently unknown. Very little is known about the presence of ERBB family ligands in EGFR mutant cancers and/or their microenvironment and these are not routinely assayed clinically. Recent studies do however highlight the role of cancer associated fibroblasts or macrophages in secreting ligands that may mediate drug resistance (31, 32). One strategy to mitigate the potential impact of ligand mediated dimerization is to develop an allosteric EGFR inhibitor together with an ATP competitive inhibitor (Fig. 2C-G). EGFR mutations that mediate resistance to JBJ-09-063 retain sensitivity to osimertinib (Fig. 6D-F), providing a further rationale for a co-administration strategy. Finally, the synergy we observed with osimertinib and our allosteric agents suggests the possibility that co-administration of a suitable allosteric inhibitor with osimertinib at a reduced dose might yield equivalent or improved efficacy, but with improved tolerability, as the side-effect profile of the allosteric agent is likely to differ from that of ATP-site directed TKIs. An alternative strategy would be to evaluate a series of anti-ERBB family member antibodies to block dimerization and/or ligand binding. Although preclinical studies suggest this is feasible and therapeutically efficacious, the ability to translate these observations into the clinic maybe limited by toxicity as these approaches are not mutant selective (33, 34).
Allosteric kinase inhibitors exist only for a minority of human kinases. Ascinimib (ABL001), binds the myristol site of BCR-ABL and induces ABL into an inactive conformation (35). In pre-clinical models, it is active in models harboring secondary ABL mutations, including T315I, that mediate resistance to ATP competitive ABL inhibitors. In the phase I clinical trial, ascinimib led to clinical responses in the majority of patients (36). Additionally, pre-clinical studies have revealed that the combination of ascinimib and ponatinib is effective against ABL mutants which are resistant to single agent ascinimib (37). One difference between ascinimib and JBJ-09-063 is their binding location. While JBJ-09-063 binds an allosteric site adjacent to the ATP binding site, ascinimib binds a myristoyl site distant from the ATP binding site. The clinical success of ascinimib coupled with the pre-clinical studies in the present study suggest that the approach of targeting allosteric sites in kinases could be broadly active in drug resistant forms both as single agents and in combination with ATP competitive inhibitors.
In summary, we have developed an allosteric EGFR inhibitor effective in vivo as a single agent in EGFR mutant drug resistant cancers. The next step is to determine whether JBJ-09-063 can be improved upon to become a candidate clinical treatment, whereby addressing the unmet need of an effective therapy for cancers with EGFR mutation mediated resistance to osimertinib.
Materials and Methods
Synthesis of JBJ-09-063
JBJ-09-063 was synthesized using similar procedures to those previously described for the synthesis of JBJ-04-125-02 (24). 1H NMR (500 MHz, DMSO-d6) δ 7.91 (s, 1H), 7.89 (d, J = 7.9 Hz, 1H), 7.69 – 7.62 (m, 3H), 7.48 (d, J = 3.6 Hz, 1H), 7.36 (d, J = 8.2 Hz, 2H), 7.26 (d, J = 3.6 Hz, 1H), 7.11 (td, J = 8.6, 3.1 Hz, 1H), 6.91 (dd, J = 9.0, 4.8 Hz, 1H), 6.87 (dd, J = 9.2, 3.0 Hz, 1H), 6.33 (s, 1H), 4.65 (d, J = 17.6 Hz, 1H), 4.03 (d, J = 17.6 Hz, 1H), 2.92 (d, J = 11.0 Hz, 2H), 2.57 – 2.50 (m, 1H), 2.24 (s, 3H), 2.05 (t, J = 10.8 Hz, 2H), 1.82 – 1.65 (m, 4H); 13C NMR (125 MHz, DMSO-d6) δ 168.9, 167.9, 158.3, 156.5, 154.6, 152.7, 146.2, 141.7, 140.6, 138.2, 137.7, 132.9, 130.7, 127.9, 127.4, 124.7, 122.7, 122.6, 120.8, 117.1, 117.0, 116.9, 116.7, 116.0, 115.8, 114.4, 56.1, 54.6, 48.9, 46.3, 41.1, 33.1. MS m/z: 557.27 [M+1]+.
Kinase inhibition assays
Inhibition assays were performed using the HTRF KinEASE tyrosine kinase assay kit (Cisbio) according to the manufacturer’s protocol. Inhibitors from 10 mM DMSO stocks were dispensed into black 384-well plates using an D300e dispenser (Hewlett-Packard) and normalized to 1% final DMSO concentration. Assay buffer containing purified EGFR kinase domain at a final concentration of 0.02 nM were dispensed using a Multidrop Combi dispenser (ThermoFisher) and incubated with the inhibitors at room temperature for 30 minutes. Reactions were initiated with 100 µM ATP and allowed to proceed for 30 minutes at room temperature before being quenched using the detection reagent from the KinEASE assay kit. The FRET signal ratio was measured at 665 and 620 nm using a PHERAstar microplate reader (BMG LABTECH). Data were processed using GraphPad Prism and fit to a three-parameter dose-response model.
Protein expression, purification, and crystallization
The kinase domain of EGFR (residues 696-1022) was expressed and purified as previously described (24, 38). Briefly, insect cells infected with recombinant baculovirus were lysed, clarified via ultracentrifugation, and purified using Ni-NTA agarose beads. Following cleavage of the 6xHis-GST tag, the protein was further purified through size exclusion chromatography and concentrated to 3-4 mg/mL.
Purified EGFRT790M/V948R kinase domain was crystallized at approximately 3 mg/mL with 10 mM MgCl2, 1 mM adenylyl-imidodiphosphate (AMP-PNP), and 0.25 mM JBJ-09-063 via hanging drop vapor diffusion at room temperature over wells consisting of 0.1 M Bis-Tris pH 5.0-6.0 and 20-30% (w/v) PEG 3,350. Crystals were briefly cryoprotected in well solution containing 20% ethylene glycol. Diffraction data were collected at the Advanced Photon Source at the Argonne National Laboratory on NE-CAT beamline 24-ID-E at 100 K. Data were indexed, integrated, and scaled using Dials via xia2 (39–41). Structures were phased via molecular replacement with PDB 6DUK (24). Refinement was performed using Phenix with iterative rounds of manual model building in Coot (42, 43) The resulting structure has been deposited in the Protein Data Bank (PDB) with the accession code 7JXQ.
Patients
All patient-related specimens and data were collected in accordance with the Declaration of Helsinki and approved by the Dana-Farber Cancer Institutional Review Board. Written informed consent was attained from the patients. All cell lines and in vivo models that were derived from the patient’s samples were de-identified to protect patient’s privacy.
Cell lines and reagents
Ba/F3 cells were a generous gift from Dr. David Weinstock (in 2014). Ba/F3 cells harboring the EGFRL858R, EGFRL858R/T790M, EGFRL858R/T790M/C797S, EGFRL858R/T790M/L718Q, EGFRWT, HER2WT were previously generated and characterized (23, 24, 44–46). All Ba/F3 cells were maintained in RPMI1640 media with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin (P/S). Wildtype EGFR Ba/F3 cells were additionally supplemented with 10 ng/ml of EGF purchased from Life Technologies. DFCI52 cells were established from xenograft tumors derived from the malignant pleural effusion of a patient using previously described methods (47) and maintained in Renaissance Essential Tumor Medium (RETM) supplemented with B-27, 10% FBS and 1% P/S in low-attachment plates. H3255 and H32555GR were previously characterized extensively (26, 48) and are cultured in ACL4 media with 5% FBS and 1% P/S. HEK293T/17 and A431 cells were purchased from ATCC and were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) in 10% FBS and 1% P/S. All cell lines were tested negative for Mycoplasma using Mycoplasma Plus PCR Primer Set (Agilent) and were passaged and/or used for no longer than 4 weeks for all experiments. Details of reagents and chemicals used in the study are listed in Table S4. JBJ-04-125-02 were synthesized in-house as described previously (24).
Plasmids construction
JP1540-EGFRL858R/T790M/G796S and JP1540-EGFRL858R/T790M/L792F retroviral plasmids were made by introducing EGFR G796S and L792F mutations into our pDNR-dual- EGFRL858R/T790M amplification vector that was previously generated in the laboratory using the QuikChange II XL Site Mutagenesis Kit (Agilent). To generate retroviral and lentiviral plasmids expressing EGFRL858R/T790M, EGFRL858R/T790M/C797S, EGFRL858R/T790M/L718Q, EGFRL858R/T790M/L792F, EGFRL858R/T790M/G796S, and EGFRL858R/T790M/L747S, we shuttled the pDNR-dual vectors with these mutants into either the JP1540 or JP1722 lentiviral destination vectors, respectively, using the In-Fusion HD Cloning Plus Kit (Takara) according to the manufacturer’s protocol. Plasmids were all sequenced at the Massachusetts General Hospital DNA Core to confirm that mutations of interest were successfully transferred.
To construct the pMSCV-EGFRKDD and ERBBKDD plasmids, the original EGFR double kinase expression vector pMSCV-EGFR-TDM, a generous gift from Dr. Christine Lovly, was modified and used as a template for all double-kinase (KDD) derivatives used in our study. First, pMSCV-EGFR-TDM was mutagenized using the QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Agilent) following the manufacturer’s protocols and using mutagenic primers sets outlined in Table S3. EGFR mutations were introduced sequentially to generate: EGFRL858R; EGFRL858R/T790M; EGFRL858R/T790M/I941R. HER2-pRetro-IRES-DsRedExpress previously generated in our laboratory was used as a template to generate the wild type and mutant HER2 (variant 1) constituents of our KDD constructs. The HER2V956R mutation was introduced using the mutagenic primers listed in Table S5. Successful mutagenesis was confirmed by complete plasmid NGS provided by the MGH CCIB DNA Core. The mutagenized intermediates were then used as templates for PCR amplification of individual constituents of final KDD constructs, which were assembled via a series of In-fusion reactions using the Takara Bio In-Fusion HD Cloning Kit and inserted into the original BlpI/NotI digested pMSCV-EGFR-TDM. The primers to amplify each In-fusion component are listed in Table S3. N-terminal EGFR kinase fragments span EGFR Met1 - Leu1038; C-terminal EGFR kinase fragments span EGFR Leu688 - Ala1210*; N-terminal HER2 kinase fragments cover HER2 Met1 - Arg1046; C-terminal HER2 kinase fragments comprise HER2 Leu696 - Val1255*.
To generate the pBABE-EGFRL858R/T790M plasmid, the T790M mutation was introduced to the pBABE-EGFRL858R (PMID: 30952700) provided as a generous gift from the Elenius Laboratory.
Generation of stable Ba/F3 and H1975 EGFR mutant cell lines and CrispR cell lines with C797S mutation
Ba/F3 cells were infected as previously described (44) to generate stable cell lines that express EGFRL858R/T790M/G796S, EGFRL858R/T790M/L792F, EGFRKDD-LT/WT and ERBBKDD-EGFR_LT/HER2. For H1975 stable cell lines, lentiviral particles were generated by transfecting HEK293T cells with JP1722-EGFR mutant vectors along with psPAX2 and pMD2.G helper plasmids using FuGENE HD as the transfection reagent. Lentiviral supernatant was harvested 48 hours following transfection and was cleared by passing through a 0.45 mm filter. H1975 cells were transduced with lentivirus in the presence of 10 µg/mL polybrene. 48 hours post-transduction, cells were treated with 2 µg/mL puromycin to select for stable integrants over the span of 1 week. To create the EGFRC797S mutation in DFCI52 and H3255GR cell lines, sgRNA and donor template were designed using Deskgen (deskgen.com). crRNAs (Integrated DNA Technologies, IDT) were hybridized with tracrRNAs and then ribonucleoprotein complex was formed with Cas9 Nuclease (IDT). The reaction mixtures were nucleofected using Lonza 4D-Nucleofector. After treatment with 100 nM Osimertinib for 1 week, DNA was extracted using the Qiagen DNeasy Mini kit and induced mutations were confirmed by CRISPR sequencing through the MGH DNA core. sgRNA, donor template, and primers are listed in Table S5. Broad/GATK best practices was followed for pre-processing of fastq files. The fastq files were aligned to the human genome version 38 (hg38) using bwa-mem (v0.7.17) and converted to sorted BAM files with samtools (v1.10). Duplicate reads in the BAM files were marked and additional read quality tags were calculated using Picard functionalities (v2.23.4). The base qualities of each read were recalibrated against know SNP databases using GATK (v4.1.6.0). The analysis ready BAM files were loaded into the IGV browser to visualize any variants present in the samples.
Cell viability assays
Ba/F3, H1975, H3255, H3255GR, H3255GR-C797S, DFCI52, DFCI52-C797S cells were plated and treated with increasing concentrations of inhibitors for 72 hours and growth or the inhibition of growth were assessed by Cell Titer Glo reagents from Promega, per manufacturer’s protocol. For experiments that investigate the effect of compounds in wildtype EGFR Ba/F3 cells, 10 ng/ml EGF was added during cell plating. For Ba/F3 mutant cells that were cultured in RPMI media cells, 10 ng/ml EGF and/or 10 ng/ml NRG1 were added at the time of drug treatment. For combination studies, drugs were administered at the same time unless otherwise stated in the figure legend.
Transfection and Western Blotting
Ba/F3, H1975, H3255, H3255GR, H3255GR-C797S, DFCI52, DFCI52-C797S cells were plated and treated for the time and with inhibitors indicated in the figure legends. Cells were harvested and lysed in RIPA or NP-40 lysis buffer followed by BCA protein assay to quantitate and normalize protein levels. Lysates were then processed for Western Blot analyses. For combination treatment with ligands such as EGF and NRG1, ligands were added for 15 minutes before drug treatment. For combination treatments with two drugs, both compounds were added at the same time. For transient overexpression studies, HEK293T/Cl.17 cells were transfected with 2 µg of indicated plasmids using FuGENE HD Transfection Reagent from Promega in OPTI-MEM media, as per manufacturer’s protocol. Media was changed after 24 hours and cells were treated 48 hours post-transfection with inhibitors for 4 hours before they were harvested. Cells were lysed in RIPA buffer followed by Western Blotting. Details of antibodies are listed in Table S4.
IncuCyte assays
For short-term studies assessing cell viability and apoptosis activity, H3255GR and DFCI52 cells were plated and treated with inhibitors in media containing the CellEvent Casapse 3/7 GreenReadyProbes reagent (Thermo Fisher Scientific). Cells were then placed in the IncuCyte chamber where their confluency and level of green fluorescence (measured as object count) were monitored and recorded by automated microscopy every two hours for 72 hours using the IncuCyte Live-Cell Imaging Analysis System. Cell viability was then assessed at the end of the 72 hours using the Cell Titer Glo reagents. For long-term assays examining tumor regrowth over time after drug treatment, H3255 and H3255GR cells were plated, treated with inhibitors and incubated in the Incucyte chamber where confluency is measured every 24 hours for 4 weeks. Cells were first treated for two weeks with drug and media change every week, followed by drug withdrawal where media containing no drug was changed weekly to examine tumor regrowth.
Pharmacokinetic studies
Pharmacokinetic studies were carried out as previously described (24). All procedures described are covered under existing protocols and have been approved by the Scripps Florida IACUC to be conducted in the Scripps vivarium, which is fully AAALAC accredited. Pharmacokinetics was determined in n=3 male C57Bl/6 mice. Compounds were dosed as indicated in the text via intravenous tail vein injection or by oral gavage. Blood was collected using minimal sampling techniques, where ~25 µL blood is collected from a small nick in the tail using Liheparin-coated hematocrit tubes at 5min, 15min, 30min, 1h, 2h, 4h, 6h, and 8h. Plasma was generated via centrifugation using a hematocrit rotor. Plasma concentration was determined via LC-MS/MS by comparison of the analyte/IS peak area using a nine-point standard curve between 0.4ng/mL and 2000ng/mL prepared in mouse plasma. Pharmacokinetic analysis was done with WinNonlin, Centara inc. using a noncompartmental model.
In vivo studies
All mouse husbandry and in vivo experiments were carried out with the approval of Dana-Farber Cancer Institute Animal Care and Use Committee. Female NCr nude mice were purchased from Taconic Bioscience, NY and female NSG mice from Jackson Laboratory, Inc, ME. Mice were implanted with expanded cells or tumor fragments of H1975 and A431 in NCr nude mice and H3255GR, DFCI52, H3255GR-C797S, DFCI52-C797S in NSG mice subcutaneously and randomly grouped into treatment cohorts with at least 8-10 mice in each cohort. Tumor volume was monitored, and treatment was initiated when tumor volume reached 100 – 200mm3. For pharmacodynamic (PD) studies, mice received a single dose of 50 mg/kg JBJ-09-063 and tumors were harvested at 2, 8, 16, and 24 hours after the dose. For single agent and combination agent efficacy studies, mice were dosed for 21 or 28 days and monitored daily. Drugs were then withdrawn, and tumors were monitored daily for up to 100 days unless indicated otherwise in figure legend. Tumor sizes of all mice were monitored, and volumes were calculated using the following formula: (mm3) = length × width × width × 0.5. All mice were euthanized when tumor volume reached approximately 2000mm3 or if the tumors became necrotic or ulcerated. The vehicles used for each compound were as follows: JBJ-09-063 and osimertinib, 0.5% HPMC (hydroxy propyl methylcellulose) in water; JBJ-04-125-02, 5% NMP (5% N-methyl-2-pyrrolidone: 95% PEG-300); Gefitinib, 0.5% HPMC and 0.1% Tween 80 in water and Afatinib, 0.5% methylcellulose and 0.4% Tween-80 in water.
E-ethyl-N-nitrosourea (ENU) Mutagenesis
ENU was purchased from Sigma Aldrich and mutagenesis studies were carried out as described previously (49). Briefly, cells were treated with 50 µg/ml of ENU overnight before cells were washed with RPMI and allowed to expand. Cells were then plated in 96 wells and 5 plates were plated per condition. These cells were treated with 1 μM JBJ-09-063, osimertinib or the combination of both compounds continuously with media and drug change once a week. Cell colonies were monitored, and resistant colonies were counted, expanded and sequenced for novel EGFR mutations in the tyrosine kinase domain.
Proximity Ligation Assays
H3255GR cells were plated on coverslips and treated with inhibitors in culture media detailed in the figure legend. After treatment incubation, cells were fixed with 4 % PFA and permeabilized with 0.1 % Triton X-100. Proximity ligation assay was performed using the manufacturer’s protocol (Sigma Aldrich) with 1:50 000 EGFR antibodies (listed in Table S4). Nuclei were stained with 1 µg/ml DAPI (Cell Signaling Technology) for 10 minutes and the slides were mounted with Immu-Mount (Fisher Scientific). PLA quantification was performed with ImageJ software with the count particles function.
Statistical Analyses
All experimental data shown represent the outcomes of at least three biological replicate studies, with technical replicates varying by study, as described in figure legends. Sample sizes and statistical parameters applied to quantify variation and significance of the data are described in corresponding figure legends.
Supplementary Material
Figure S1. Structure, potency, pharmacodynamic studies of JBJ-09-063 and the development and validation of H3255GR-C797S and DFIC52-C797S models in vitro.
Figure S2. JBJ-09-063 efficacy is reduced in vitro unless combined with gefitinib in H3255GR and DFCI52 cells or with osimertinib in H3255GR-C797S and DFCI52-C797S cells.
Figure S3. Forced or ligand induced dimerization can impart resistance to JBJ-09-063.
Figure S4. JBJ-09-063 is effective in H1975 cells exogenously expressing the osimertinib resistant mutations while osimertinib is effective in H1975 cells exogenously expressing the JBJ-09-063 mutation, L747S.
Table S1. Crystallographic data collection and refinement statistics
Table S2. IC50 of JBJ-04-125-02 and JBJ-09-063 in enzymatic assays, Ba/F3 cells, H1975 cells and human cancer cells
Table S3. Pharmacokinetic profile of JBJ-09-063
Table S4. Reagents and Antibodies
Table S5. Sequences and primers for plasmids and cell lines generation
Acknowledgments
The authors would like to thank Prof. Klaus Elenius (University of Turku, Finland) for generously providing the pBABE constructs, the MGH CCI DNA Core for their next generation sequencing services; Elizabeth Cohen from the Molecular Biology Core Facilities at the Dana-Farber Cancer Institute for processing the DFCI52-C797S and H3255GR-C797S cell line validation FASTA data in IVG; and all staff at the Dana-Farber Cancer Institute Animal Resources Facility for their mouse husbandry services. This work used NE-CAT beamlines (P30 GM124165) and an Eiger detector (S10OD021527) at the APS (DE-AC02-06CH11357).
Funding
This study was supported by the National Cancer Institute grants R35CA220497 (P.A.J.), RO1 CA201049 (M.J.E., N.S.G and P.A.J), PO1CA154303 (M.J.E., N.S.G and P.A.J), The American Cancer Society (CRP-17-111-01-CDD (P.A.J.), The Balassiano Family Fund for Lung Cancer Research (P.A.J.), The Gohl Family Lung Cancer Research Fund (P.A.J.) and by Takeda. T.S.B is supported by a Ruth L. Kirschstein National Research Service Award (1F32CA247198-01). Y.K. is supported in part by JSPS Overseas Research Fellowships and Research Fellowship from Uehara Memorial Foundation.
Footnotes
Competing interests
P.A.J. has received consulting fees from AstraZeneca, Boehringer-Ingelheim, Pfizer, Roche/Genentech, Takeda Oncology, ACEA Biosciences, Eli Lilly and Company, Araxes Pharma, Ignyta, Mirati Therapeutics, Novartis, LOXO Oncology, Daiichi Sankyo, Sanofi Oncology, Voronoi, SFJ Pharmaceuticals, Biocartis, Novartis Oncology, Nuvalent, Esai, Bayer, Transcenta, Silicon Therapeutics, Allorion Therapeutics, Accutar Biotech and AbbVie; receives post-marketing royalties from DFCI owned intellectual property on EGFR mutations licensed to Lab Corp; receives or has received sponsored research funding from AstraZeneca, Astellas, Daichi-Sankyo, PUMA, Boehringer Ingelheim, Eli Lilly and Company, Revolution Medicines, and Takeda; and has stock ownership in Gatekeeper Pharmaceuticals. N.S.G. is a founder, science advisory board member (SAB) and equity holder in Gatekeeper, Syros, Petra, C4, Allorion, Jengu, B2S, Inception, EoCys, Larkspur (board member) and Soltego (board member). The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Jansen, Kinogen, Arbella, Deerfield and Sanofi. M.J.E. has served as a paid consultant to Novartis Institutes for Biomedical Research and H3 Biomedicine. M.J.E. receives sponsored research support from Novartis, Sanofi, and Takeda. D.E.H. is a consultant for Logos Capital and the Jefferies Group. The series of compounds to which JBJ-04-125-02 and JBJ-09-063 belong is described in US patent 10,836,722 B2. All other authors declare no competing interests.
Data and materials availability:
All data in this study are presented in the manuscript and supplement. All materials are available upon request and through a material transfer agreement.
References
- 1.Bray F et al. , Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68, 394–424 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A, Cancer statistics, 2020. CA Cancer J Clin 70, 7–30 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Maemondo M et al. , Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med 362, 2380–2388 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Mitsudomi T et al. , Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 11, 121–128 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Zhou C et al. , Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 12, 735–742 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Rosell R et al. , Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 13, 239–246 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Park K et al. , Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): a phase 2B, open-label, randomised controlled trial. Lancet Oncol 17, 577–589 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Sequist LV et al. , Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 31, 3327–3334 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Wu YL et al. , Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): a randomised, open-label, phase 3 trial. Lancet Oncol 18, 1454–1466 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Wu YL et al. , Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol 15, 213–222 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Soria JC et al. , Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N Engl J Med 378, 113–125 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Janne PA et al. , AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med 372, 1689–1699 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Mok TS et al. , Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N Engl J Med 376, 629–640 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cho BC et al. , Osimertinib versus Standard of Care EGFR TKI as First-Line Treatment in Patients with EGFRm Advanced NSCLC: FLAURA Asian Subset. J Thorac Oncol 14, 99–106 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Ramalingam SS et al. , Osimertinib As First-Line Treatment of EGFR Mutation-Positive Advanced Non-Small-Cell Lung Cancer. J Clin Oncol 36, 841–849 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Soria JC, Ramalingam SS, Osimertinib in EGFR Mutation-Positive Advanced NSCLC. N Engl J Med 378, 1262–1263 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Mok TS et al. , Improvement in Overall Survival in a Randomized Study That Compared Dacomitinib With Gefitinib in Patients With Advanced Non-Small-Cell Lung Cancer and EGFR-Activating Mutations. J Clin Oncol 36, 2244–2250 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Ahn MJ et al. , Osimertinib in patients with T790M mutation-positive, advanced non-small cell lung cancer: Long-term follow-up from a pooled analysis of 2 phase 2 studies. Cancer 125, 892–901 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Auliac JB et al. , Real-life efficacy of osimertinib in pretreated patients with advanced non-small cell lung cancer harboring EGFR T790M mutation. Lung Cancer 127, 96–102 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Igawa S et al. , Impact of EGFR genotype on the efficacy of osimertinib in EGFR tyrosine kinase inhibitor-resistant patients with non-small cell lung cancer: a prospective observational study. Cancer Manag Res 11, 4883–4892 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thress KS et al. , Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med 21, 560–562 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rangachari D et al. , EGFR-Mutated Lung Cancers Resistant to Osimertinib through EGFR C797S Respond to First-Generation Reversible EGFR Inhibitors but Eventually Acquire EGFR T790M/C797S in Preclinical Models and Clinical Samples. J Thorac Oncol 14, 1995–2002 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jia Y et al. , Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 534, 129–132 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.To C et al. , Single and Dual Targeting of Mutant EGFR with an Allosteric Inhibitor. Cancer Discov 9, 926–943 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Clercq DJH et al. , Discovery and Optimization of Dibenzodiazepinones as Allosteric Mutant-Selective EGFR Inhibitors. ACS Med Chem Lett 10, 1549–1553 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Engelman JA et al. , Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest 116, 2695–2706 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gallant JN et al. , EGFR Kinase Domain Duplication (EGFR-KDD) Is a Novel Oncogenic Driver in Lung Cancer That Is Clinically Responsive to Afatinib. Cancer Discov 5, 1155–1163 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alam MS, Proximity Ligation Assay (PLA). Curr Protoc Immunol 123, e58 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leonetti A et al. , Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br J Cancer 121, 725–737 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin L et al. , Acquired rare recurrent EGFR mutations as mechanisms of resistance to Osimertinib in lung cancer and in silico structural modelling. Am J Cancer Res 10, 4005–4015 (2020). [PMC free article] [PubMed] [Google Scholar]
- 31.Hu H et al. , Three subtypes of lung cancer fibroblasts define distinct therapeutic paradigms. Cancer Cell, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma S et al. , Epiregulin confers EGFR-TKI resistance via EGFR/ErbB2 heterodimer in non-small cell lung cancer. Oncogene 40, 2596–2609 (2021). [DOI] [PubMed] [Google Scholar]
- 33.Mancini M et al. , An oligoclonal antibody durably overcomes resistance of lung cancer to third-generation EGFR inhibitors. EMBO Mol Med 10, 294–308 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cho J et al. , Cetuximab response of lung cancer-derived EGF receptor mutants is associated with asymmetric dimerization. Cancer Res 73, 6770–6779 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wylie AA et al. , The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature 543, 733–737 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Hughes TP et al. , Asciminib in Chronic Myeloid Leukemia after ABL Kinase Inhibitor Failure. N Engl J Med 381, 2315–2326 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eide CA et al. , Combining the Allosteric Inhibitor Asciminib with Ponatinib Suppresses Emergence of and Restores Efficacy against Highly Resistant BCR-ABL1 Mutants. Cancer Cell 36, 431–443 e435 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yun CH et al. , The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A 105, 2070–2075 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Winter G et al. , DIALS: implementation and evaluation of a new integration package. Acta Crystallogr D Struct Biol 74, 85–97 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morin A et al. , Collaboration gets the most out of software. Elife 2, e01456 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Winter G, xia2: an expert system for macromolecular crystallography data reduction. Journal of Applied Crystallography 43, 186–190 (2010). [Google Scholar]
- 42.Adams PD et al. , PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Emsley P, Lohkamp B, Scott WG, Cowtan K, Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou W et al. , Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 462, 1070–1074 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kosaka T et al. , Response Heterogeneity of EGFR and HER2 Exon 20 Insertions to Covalent EGFR and HER2 Inhibitors. Cancer Res 77, 2712–2721 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jang J et al. , Mutant-Selective Allosteric EGFR Degraders are Effective Against a Broad Range of Drug-Resistant Mutations. Angew Chem Int Ed Engl 59, 14481–14489 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bahcall M et al. , Amplification of Wild-type KRAS Imparts Resistance to Crizotinib in MET Exon 14 Mutant Non-Small Cell Lung Cancer. Clin Cancer Res 24, 5963–5976 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tracy S et al. , Gefitinib induces apoptosis in the EGFRL858R non-small-cell lung cancer cell line H3255. Cancer Res 64, 7241–7244 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Ercan D et al. , EGFR Mutations and Resistance to Irreversible Pyrimidine-Based EGFR Inhibitors. Clin Cancer Res 21, 3913–3923 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Structure, potency, pharmacodynamic studies of JBJ-09-063 and the development and validation of H3255GR-C797S and DFIC52-C797S models in vitro.
Figure S2. JBJ-09-063 efficacy is reduced in vitro unless combined with gefitinib in H3255GR and DFCI52 cells or with osimertinib in H3255GR-C797S and DFCI52-C797S cells.
Figure S3. Forced or ligand induced dimerization can impart resistance to JBJ-09-063.
Figure S4. JBJ-09-063 is effective in H1975 cells exogenously expressing the osimertinib resistant mutations while osimertinib is effective in H1975 cells exogenously expressing the JBJ-09-063 mutation, L747S.
Table S1. Crystallographic data collection and refinement statistics
Table S2. IC50 of JBJ-04-125-02 and JBJ-09-063 in enzymatic assays, Ba/F3 cells, H1975 cells and human cancer cells
Table S3. Pharmacokinetic profile of JBJ-09-063
Table S4. Reagents and Antibodies
Table S5. Sequences and primers for plasmids and cell lines generation