

Abstract
Cells form hollow, spheroidal structures during the development of many tissues, including the ocular lens, inner ear, and many glands. Therefore, techniques for in vitro formation of hollow spheroids are valued for studying developmental and disease processes. Current in vitro methods require cells to self-organize into hollow morphologies; we explored an alternative strategy based on cell growth in predefined, spherical scaffolds. Our method uses sacrificial, gelatin microbeads to simultaneously template spherical chambers within a hydrogel and deliver cells into the chambers. We use mouse lens epithelial cells to demonstrate that cells can populate the internal surfaces of the chambers within a week to create numerous hollow spheroids. The platform supports manipulation of matrix mechanics, curvature, and biochemical composition to mimic in vivo microenvironments. It also provides a starting point for engineering organoids of tissues that develop from hollow spheroids.
Keywords: 3D culture, Gelatin methacrylate, Spheroid, Lens
1. Introduction
Three-dimensional (3D) cell culture techniques vary the mechanical properties, geometry, and biochemical cues in a cell’s microenvironment in order to influence processes such as differentiation, migration, and morphogenesis [1–4]. Under certain conditions, cells in 3D culture will exhibit in vivo-like physiological behaviors that are not observed using traditional cultures on flat, rigid substrates. As a result, 3D culture techniques are now widely adopted for fundamental cell biology studies [5,6] and for applied research in drug testing [7,8] and tissue engineering [9].
A common aim of 3D culture is to organize cells into certain configurations (e.g., tubes for vessels or layers for stratified tissues) [10,11]. In order to achieve a desired configuration, researchers can impart constraints on cell organization when creating their 3D system [12,13] and/or rely on the inherent ability of cells to self-organize [14]. To form vessels for example, previous studies either grew vascular cells in predefined channels, allowed the cells to self-organize into vascular networks, or combined defined channels with self-organization [15–18].
The configuration we are interested in is the hollow spheroid. Spheroidal vesicles are composed of a layer of cells surrounding a fluid-filled lumen and are frequently observed during development [19]. Examples include Rathke’s pouch and the lens, otic, and renal vesicles, which are hollow precursors to the anterior pituitary, ocular lens, inner ear, and nephron, respectively. Cells organized into hollow spheroids are exposed to cues that they may not experience when organized as 2D monolayers, such as radial and circumferential stresses [20], asymmetric biochemical signals from the luminal and external sides of the sphere [4], and curvature that can alter cytoskeletal assembly and contraction [4,21]. Therefore, methods to create hollow spheroids are important for studying the development and physiology of the cells that compose them.
The prevailing methods for creating hollow spheroids rely on self-organization, especially with stem cells [22–24]. Complementary methods that predefine spherical geometries, analogous to the use of predefined channels for vessels, are limited. Soft lithography techniques can define the shapes of cavities in hydrogels for cell growth, but spherical chambers cannot be attained with these methods [25,26]. Several microfluidic and electro-spraying techniques exist to encapsulate cells in solid or liquid core microbeads [27–30]. However, the beads were designed to generate solid microspheres and often used non-cell adhesive materials (e.g., agarose and alginate). Here we describe a 3D culture technique that combines cell encapsulation in sacrificial microbeads with the formation of spherical chambers in hydrogel matrices. We demonstrate with lens epithelial cells (LECs) that the platform enables the directed formation of hollow, cell-lined spheroids.
2. Methods
2.1. Materials
Culture related reagents were purchased from Gibco including advanced DMEM/F-12, DMEM/F-12 with HEPES, 10x TrypLE, Gluta-MAX, penicillin-streptomycin, fetal bovine serum (FBS), 100x B-27 supplement, 0.05% trypsin-EDTA, Hank’s balanced salt solution with calcium and magnesium (HBSS), phosphate buffered saline (PBS), paraffin oil, LIVE/DEAD viability/cytotoxicity kit. Rat anti-E-cadherin antibody (Invitrogen, cat#13-1900) and fluorescently labelled phalloidins (Thermo-Fisher Scientific, Waltham, MA). Growth factor reduced Matrigel (Corning Life Sciences, Tewksbury, MA). TGF-β1 receptor inhibitor SB-431542 (Stemgent, cat#04-0010-10), and Rho-associated, coiled-coil containing protein kinase inhibitor Y-27632 (Selleck Chemicals, Houston, TX and ApexBio, Houston, TX). Gelatin type A from porcine skin 300 Bloom, dispase, methacrylic anhydride, dichlorodimethylsilane, and 3-(Trimethoxysilyl)propyl methacrylate (Sigma Aldrich, St. Louis, MO). Soybean lecithin and the photoinitiator lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) (TCI America, Portland, OR). Bovine serum albumin (BSA) (Research Products International, Prospect, IL). Normal goat serum and VECTASHIELD antifade mounting medium with 4′,5-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA).
2.2. Mouse strains
Mouse care and breeding were performed according to the Animal Care and Use Committee approved animal protocol (University of California Berkeley, Berkeley, CA, USA). Both wildtype mice and membrane tdTomato/nuclear GFP (mT/nG) transgenic mice in the C57BL/6 background were used. Double transgenic mT/nG mice were generated by mating mice expressing membrane-targeted tdTomato [31] (strain 007676, The Jackson Laboratory, Bar Harbor, ME) with mice expressing histone 2B-green fluorescent protein (H2B-GFP) fusion proteins [32] (strain 006069, The Jackson Laboratory).
2.3. Cell culture
Primary cultured lens epithelial cells (LECs) were harvested from adult mice. Eyes were removed from euthanized mice then the lenses were dissected out into advanced DMEM/F-12, washed in PBS, and transferred to trypsin-EDTA at 37 °C for 5 min with gentle shaking. The lenses were washed 3X with PBS then resuspended in Advanced DMEM/F-12. The lens capsules with attached LECs were then dissected away from the bulk mass of fibers and placed in 2U/mL dispase in HBSS (50 μl/capsule) at 37 °C for 30 min with gentle shaking. An equal volume of 10X TrypLE was then added and the capsules were incubated for an additional 5 min. The mixture was triturated by pipetting, added to advanced DMEM/F-12 (9 times the combined volume of dispase/TrypLE), and centrifuged at ~225 × g for 5 min. All but ~300 μl of supernatant were aspirated away, then the LECs were resuspended in growth media (advanced DMEM/F-12, 0.5X penicillin/streptomycin, 1X Gluta-MAX, 2% FBS, 1X B-27, 5 μM SB-431452, and 10 μM Y-27632) and plated into 6 cm tissue culture dishes that were previously coated with 0.1 mg/ml Matrigel in DMEM/F-12. Typically, LECs from 2 to 4 lenses were used to seed each dish. Cells were grown at 37 °C in 5% CO2, 95% relative humidity conditions and media was changed every 2–3 days.
2.4. Gelatin methacrylate synthesis
Gelatin methacrylate (GelMA) was synthesized similarly to as described previously [33,34]. 10 g of gelatin was dissolved in 100 ml of 100 mM Na2HPO4 at 50 °C. A total of 0.4 ml of methacrylic anhydride per gram of gelatin was added over the course of 3 h with stirring and the temperature was maintained at 50 °C. 1/6 of the total methacrylic anhydride was added every 30 min and the pH was maintained between 7.5 and 8.5 by dropwise addition of 4 M NaOH. The reaction was diluted in 100 ml of H2O then subjected to dialysis with a 12–14 kDa molecular weight cut off membrane. The buffer was changed at least once daily over the course of one week. Dialysis was first against 1X PBS, then against 0.1X PBS, then against H2O for the remainder of the week. The dialyzed GelMA solution was adjusted to pH ~7.5 with 7.5% w/v NaHCO3 then frozen, lyophilized, and stored at −80 °C.
2.5. Cell encapsulation
Confluent monolayers of primary cultured, passage 0 LECs were detached using 1X TrypLE in PBS and resuspended in 12% w/v gelatin in DMEM/F-12 to a concentration of 5–7.5 × 106 cells/ml. 400–500 μL of cells in gelatin were then added to a 25 ml beaker containing 7 g of 0.2% w/w soy lecithin in paraffin oil to begin gelatin microbead synthesis. The beaker was held in a 37 °C water bath while being stirred with a magnetic stir bar at 300 RPM. After 4 min, the beaker was transferred to a room temperature water bath and after another 4 min it was transferred to an ice bath for 4 more minutes. Stirring was maintained at 300 RPM throughout. All subsequent centrifugation steps were performed at ~80 × g and ice cold solutions were used. The microbead emulsion was centrifuged for 3 min, the supernatant was pipetted away, then the microbeads were resuspended in 3 ml of paraffin oil. 6 ml of PBS was added and the mixture was centrifuged for 3 min. The oil layer was then removed, the remaining mixture was thoroughly mixed by pipetting, then centrifuged for 3 more minutes. The supernatant was pipetted away, the beads were resuspended in 10 ml of PBS, and then spun down for 1 min. The previous step was repeated and the beads were suspended in 5 mL of DMEM/F-12. The beads were filtered on a 100 μm nylon mesh to remove small beads and remaining beads were eluted off the mesh with 10 ml of DMEM/F-12 and then spun down for 1 min. Thereafter, 8 ml of the supernatant was removed and the remaining beads were used for encapsulation.
2.6. Microbead encapsulation and cell growth
GelMA was dissolved to 18% w/v in a 1.5 mg/ml solution of LAP in PBS. Microbeads (70 μl per 150 μl total volume), Matrigel (2.75 mg/ml), and PBS were mixed and held on ice. Molds were defined by sandwiching ~0.5 mm thick strips of Parafilm between a bottom glass layer and a top layer of parafilm or dichlorodimethylsilane-treated glass. The bottom glass layer was treated with 3-(Trimethoxysilyl)propyl methacrylate to allow covalent bonding of GelMA hydrogels for time course imaging. GelMA was mixed with the microbead/Matrigel mixture at a 2:1 vol ratio, pipetted into the molds, then immediately photopolymerized by 365 nm UV light for 45 s (4 mW/cm2, UVP UVL-23RW). The top mold layers were then removed and the gels were immersed in DMEM/F-12 at room temperature. After 5 min the gels were moved to the cell culture incubator (37 °C, 95% relative humidity), then the media was replaced with growth media after 1 h. Media was changed every 2–3 days afterwards. Conditioned media was prepared by collecting the media from LECs growing in culture dishes, filtering through a 0.2 μm filter, and mixing 1:1 with growth media. LECs were detached from within chambers using 2U/ml dispase in HBSS for 1 h at 37 °C. The dispase was washed away with DMEM/F-12 and the cells were returned to growth media. Co-culture-like conditions were performed by including LECs in the polymerization mixture at a concentration of 1 × 106 cells/ml and seeding LECs at a density of 5000 cells/cm2 on the gels after polymerization.
2.7. Viability testing, immunostaining and imaging
Images were acquired with an LSM 700 confocal microscope (Zeiss, Jena, Germany). Live mT/nG LECs were maintained at 37 °C with a heated microscope stage during imaging. Cell viability was tested using a LIVE/DEAD kit by incubating the gels in 1 μM calcein AM and 1 μM ethidium homodimer-1 in HBSS for 1 h after embedding and melting of the gelatin beads. Purified microbeads were fixed with cold 1% paraformaldehyde (PFA) in PBS for 10 min while being agitated then washed with PBS 2X. The fixed beads were imaged and the diameters of 100 beads from each of three separate emulsions were measured using ZEN Black software (Zeiss). For staining, cells grown in microchambers were fixed with 4% PFA in PBS for 10 min then washed 3X with PBS. For immunostaining, the gels were covered in permeabilization and blocking (PB) solution (3% wt/vol BSA, 3% vol/vol NGS, 0.3% vol/vol Triton X-100 in PBS) for at least 30 min. Primary antibody diluted 1:100 in PB was applied overnight at 4 °C. After washing 4X in PBS, secondary antibodies and/or phalloidin diluted 1:100 in PB were applied overnight 4 °C. After washing 4X in PBS, the gels were treated with a 50:50 mixture DAPI mounting media and PBS. Fluorescent images were processed with ZEN’s median filter to reduce noise. 3D rendering was performed with ZEN and UCSF Chimera [35].
3. Results
3.1. Overview of 3D culture platform
A schematic of the 3D culture protocol for generating hollow spheroids is shown in Fig. 1. First, cells of interest are encapsulated in gelatin microbeads by a water-in-oil emulsification process. The microbeads are then embedded in a thin hydrogel layer that is cross-linked by photopolymerization. Next, the gelatin beads are melted at 37 °C to leave fluid-filled chambers. Finally, the cells are allowed to proliferate over the inner surfaces of the chambers. As a proof of concept for our platform, we used primary cultures of mouse lens epithelial cells. We initially used LECs from mT/nG mice, which have fluorescently labelled membranes (tdTomato) and nuclei (GFP), so that we could easily monitor the LECs by fluorescent microscopy. Dissociated LECs isolated from one pair of mouse lenses grew over the course of 7–10 days into sufficient cells for encapsulation into more than 1000 microbeads.
Fig. 1. Schematic of process to perform 3D culture in microchambers.
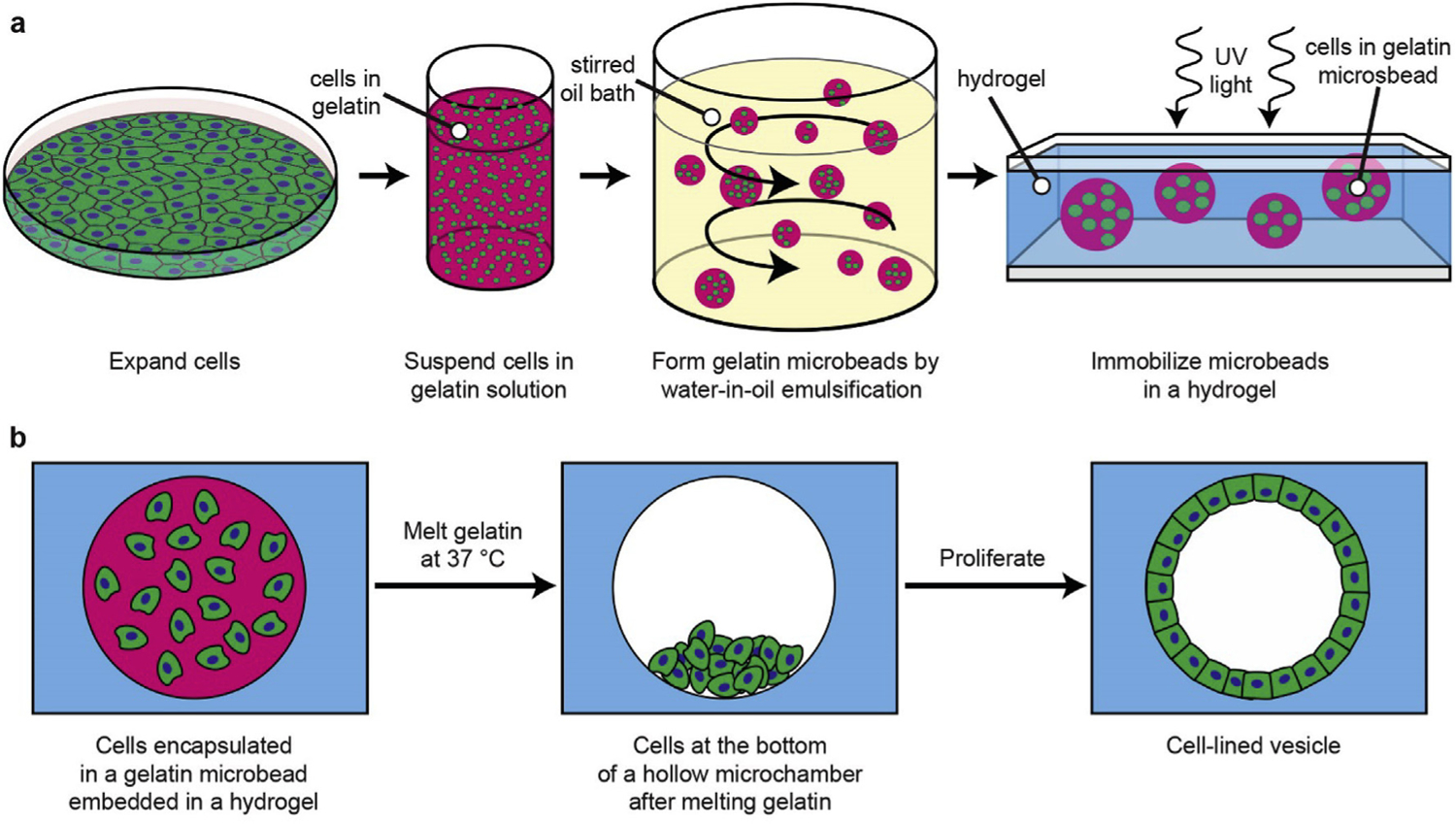
(a) Cells expanded in 2D culture are detached and suspended in melted gelatin. The gelatin mixture is emulsified in an oil bath and solidified on ice. The resulting cell-encapsulating microbead gels are then embedded in a UV-polymerized hydrogel. (b) The encapsulated microbeads are melted at 37 °C, allowing the cells to sink. The cells can then populate the surfaces of the resulting hollow microchamber.
3.2. Cell encapsulation in microbeads
We encapsulated LECs in gelatin microbeads by a water-in-oil emulsification process (Fig. 2a). The water phase was cells suspended at a density of 5–7.5 × 106 per ml in melted 12% w/v gelatin. This density allowed even the smallest beads to encapsulate one or more cells. The continuous phase was paraffin oil with soybean lecithin added as an emulsifier; this combination was previously shown to be cytocompatible with mouse fibroblasts [36]. We tuned the stirring rate and lecithin concentration so that most of the resulting beads were 100–400 μm in diameter (Fig. 2b). That size range was targeted to imitate the lens vesicle, which is approximately 100–200 μm in diameter during early development.
Fig. 2. Characterization of cell encapsulating microbeads.
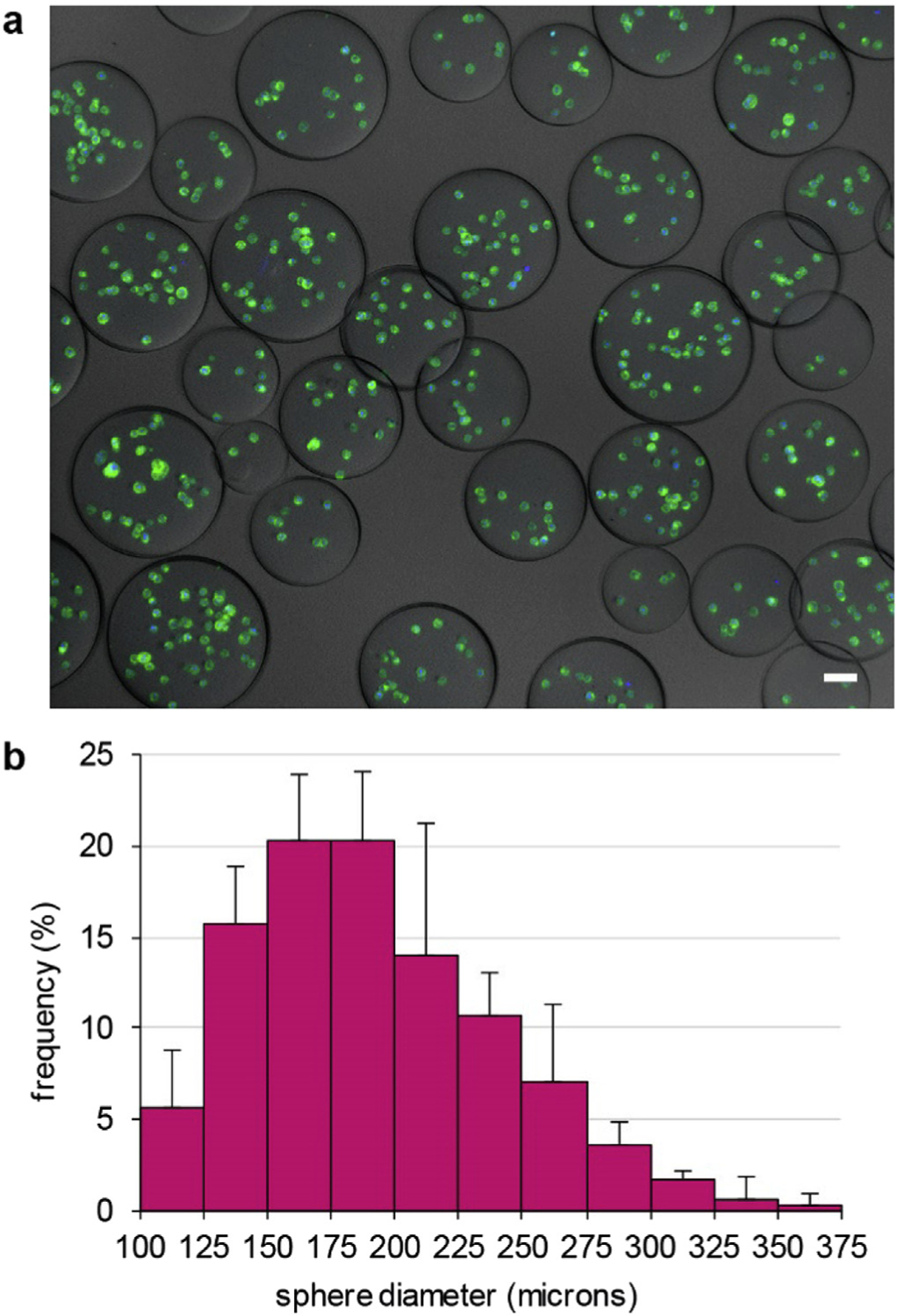
(a) Maximum intensity projection image of mT/nG LECs (membranes: green, nuclei: blue) in gelatin microbeads prepared by emulsification. Scale bar: 50 μm. (b) Histogram of microbead diameters shows that most beads were 100–400 μm in diameter. Error bars indicate standard deviations from measurements on 3 sets of beads.
3.3. Microbead encapsulation in hydrogels
We next embedded the microbeads into photopolymerized hydrogels made of gelatin methacrylate (GelMA) and Matrigel (Fig. 3a).
Fig. 3. Cell viability after microbead embedding and melting.
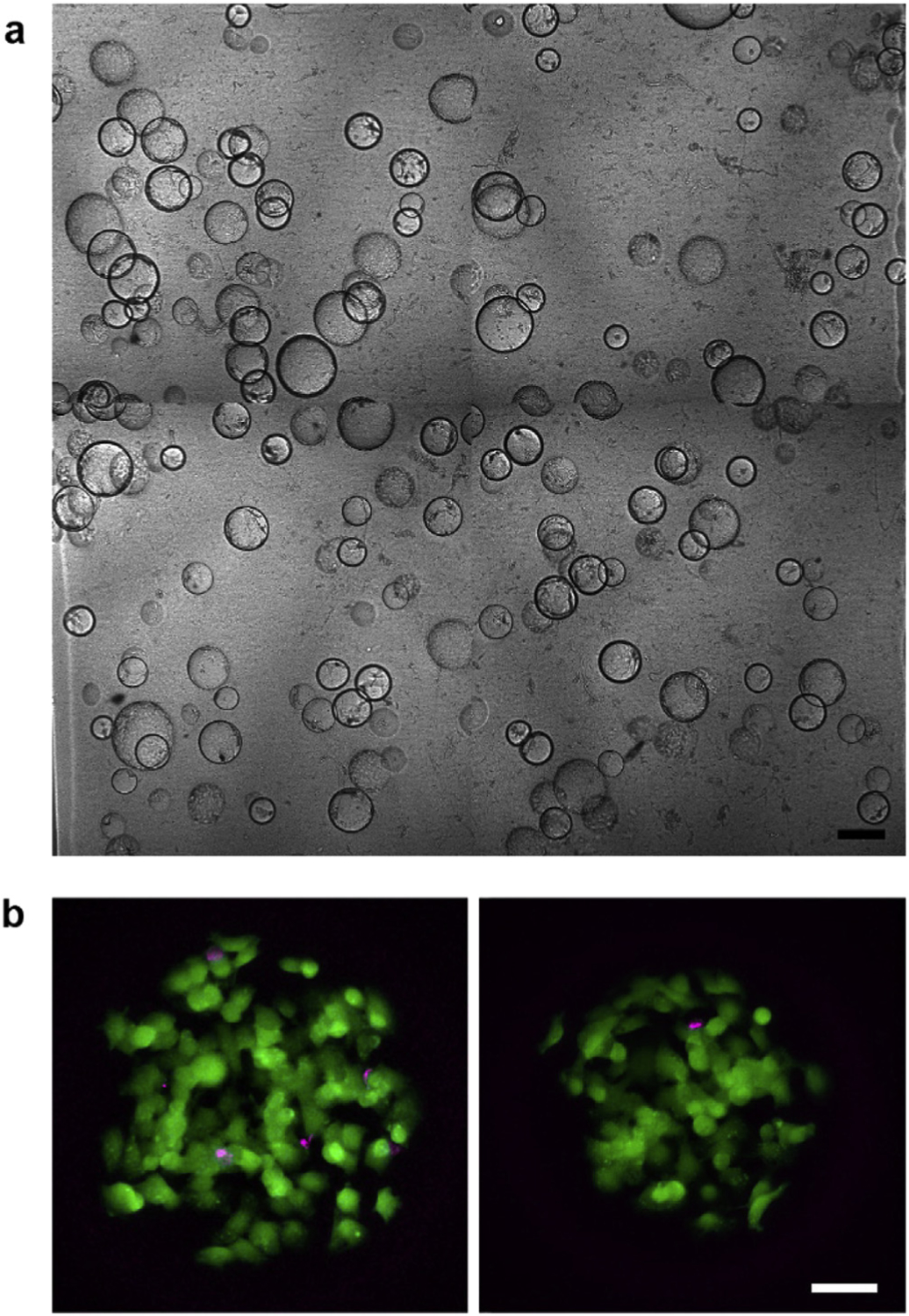
(a) Composite image of microbeads embedded in a photopolymerized GelMA-Matrigel hydrogel. Scale bar: 250 μm. (b) Maximum intensity projection images of two different microchambers after embedding and melting of the gelatin microbeads. Live and dead cells within the chambers were labelled with calcein AM (green) and ethidium homodimer-1 (magenta), respectively. Scale bar: 50 μm.
Matrigel is a basement membrane extract with similar components to the lens capsule [37], the native basement membrane for LECs. Matrigel coatings on rigid substrates significantly increased LEC proliferation [57], but preliminary tests showed that LECs only grew as aggregates when seeded on gels made purely of Matrigel. Therefore, we mixed in gelatin methacrylate to augment the mechanical properties. GelMA is a derivative of gelatin that can be covalently crosslinked by free radical polymerization [33] and was previously used for successful cell encapsulations [38,39]. GelMA was synthesized by treating gelatin with methacrylic anhydride under conditions that lead to complete derivatization of free amines. Having highly substituted GelMA prevents gelation at room temperature [40] so it can be manipulated under temperatures that do not melt the microbeads. After polymerization, the gelatin microbeads were melted by incubation at 37 °C, leaving fluid-filled microchambers behind. The encapsulated LECs are mostly viable after the encapsulation and melting process based on live/dead staining (mean viability = 94%, standard deviation = 3%) (Fig. 3b).
3.4. Cell growth in microchambers
LECs migrated and proliferated along the surfaces of the microchambers. Fig. 4 shows mT/nG cells within the same microbead over the course of 5 days. Cells sank and were found only on the bottom of the chambers immediately after the gelatin microbeads melted. Within 24 h, some cells had migrated across the chamber equator to the top pole. Proliferation led to almost complete coverage by day 3 and complete surface coverage by day 5. Imaging of microchambers revealed a monolayer of confluent cells with squamous (~6–9 μm thick), cobblestone cell morphologies and E-cadherin on cell-cell boundaries according to in situ immunostaining (Fig. 5). Although approximately 60% of beads were less than 200 μm in diameter (Fig. 2b), only 9% of the successful monolayer-lined chambers we found were sub-200 μm (Supplementary Fig. 1a). The poor formation of monolayers in small chambers could be due to the low cell counts in small chambers, but there was no apparent improvement when using conditioned media nor when the initial cell concentration was increased from 5 × 106 to 7.5 × 106 cells/ml during bead synthesis. Instead, the smaller radius of curvature could be influencing the LEC behavior [21,41]. Cells in smaller microchambers often did not spread conformally along the chamber surfaces, they instead attached only at distinct points (Supplementary Fig. 1b). We further tested gels made without Matrigel, but the cells did not adhere, instead they aggregated into solid spheroids (Supplementary Fig. 2). We also tried using beads made of 9% rather than 12% w/v gelatin, but many cells did not sink to the bottoms of the chambers after melting the 9% beads. We attributed this to entrapment by GelMA that had infiltrated and polymerized within the beads.
Fig. 4. Time course of mT/nG LEC growth within a microchamber.
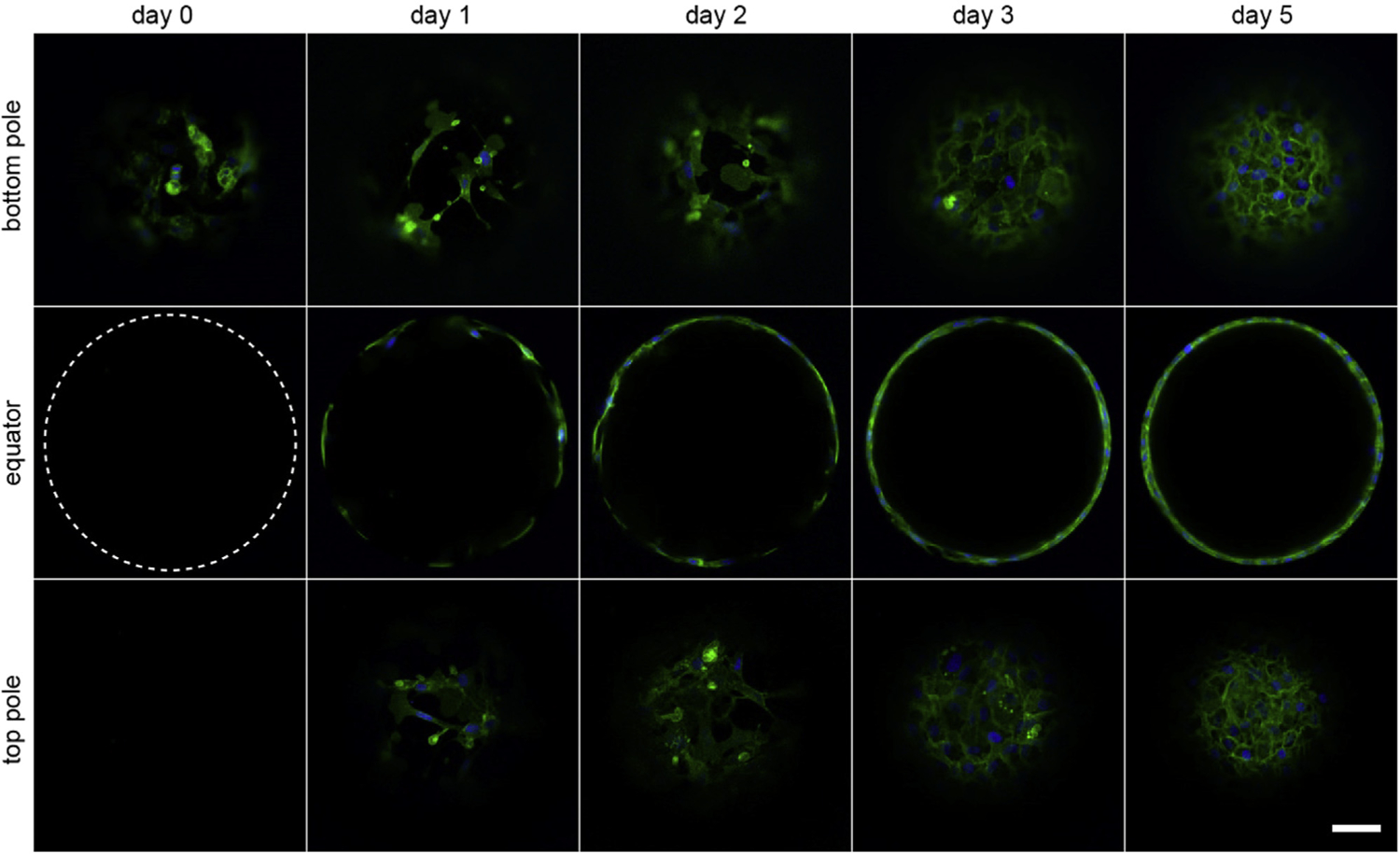
Initially, the cells are confined to the bottom of the microchamber (dashed circle denotes location of bead border). Within one day, cells migrate to the equator and top pole. They proliferate on the internal surfaces, forming a confluent monolayer in 3–5 days (green: membrane, blue: nuclei). Scale bar: 50 μm.
Fig. 5. LEC phenotype in hollow spheroids.

(a) 3D rendering of membrane fluorescent signal from mT/nG LECs in a hollow spheroid with color scale indicating depth. A fourth of the sphere was not rendered so a portion of the internal surface is visible. The LECs establish a confluent cobblestone morphology. (b) Maximum intensity projection image from a portion of a sphere stained with E-cadherin and DAPI. Scale bar: 20 μm.
3.5. Formation of detached hollow spheroids
We next aimed to derive sub-200 μm diameter spheroids from larger spheres since monolayers did not form in small diameter chambers. To do so, we allowed the LECs to form confluent monolayers within the microchambers as before, then gently detached the cells away from the chamber walls using dispase (Fig. 6). The cells in detached spheroids changed from being squamous to being more cuboidal/columnar in morphology. Moreover, they maintained their cell-cell contacts, allowing for conservation of the monolayered, spherical topology.
Fig. 6. Sub-200 μm diameter, detached hollow spheroids.
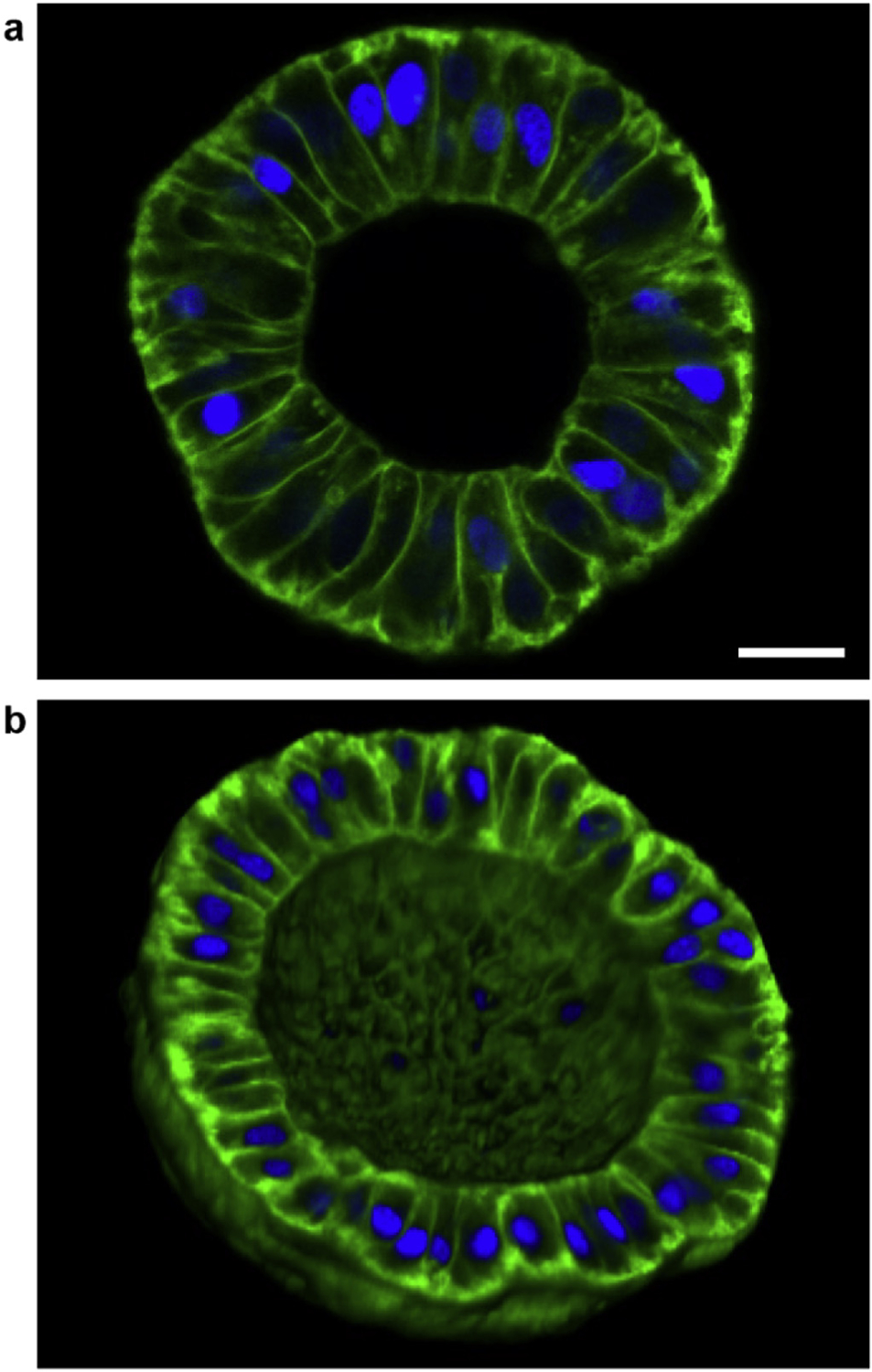
(a) Image of a plane going through the equator of an ~120 μm diameter spheroid showing that the LECs become more cuboidal/columnar after detachment. Scale bar: 20 μm. (b) 3D rendering of one hemisphere of an ~160 μm diameter spheroid showing that the spherical topology is maintained after dispase treatment. (green: membrane; blue: nuclei).
3.6. Co-culture-like conditions
Cells in hollow spheroids are affected by other cells in their microenvironment. For instance, asymmetric lens differentiation is driven by inductive factors originating from the retina [42] and acinar mammary epithelial cells are influenced by surrounding stromal fibroblasts [43]. 3D culture systems that are compatible with co-culture allow such interactions to be examined in vitro. We tested the feasibility of using the hollow spheroid system for co-culture experiments by incorporating LECs not only in the microchambers but also within the gel and on the gel surface. We observed that LECs on the surface and within the microchambers continually proliferated, but LECs embedded in the gel did not grow (Supplementary Fig. 3).
4. Discussion
We demonstrated the directed 3D growth of hollow, cell-lined spheroids using primary mouse LECs as a proof-of-concept. We chose LECs because the ocular lens derives from a hollow sphere, the lens vesicle [42], and, to our knowledge, a lens vesicle-like structure has not been achieved by self-organization [44]. Also, multiple groups have reported that LECs remaining after surgical removal of other lens cells could proliferate, differentiate, and partially regenerate lens-like structures [45,46]. Thus, in vitro systems such as ours for studying lens development and lens regeneration processes are desirable. The current gold standard for in vitro lens culture uses LECs explants grown on their native capsule. These systems have proven extremely valuable, but are inherently low throughput and do not provide a spheroidal environment that mimics the native, primary lens vesicle [47,48]. Our platform provides a complementary technique for studies in 3D environments in a size range that mimics the early developing lens. Still, future efforts are needed to better recapitulate the properties of the lens capsule and to create fully defined medias for LEC growth and differentiation.
Previous studies used gelatin to template channels in hydrogels [10] and to create interconnected pores in tissue engineering scaffolds [49]. Similarly, gelatin served as a convenient and inexpensive sacrificial material for our system. Microbeads were synthesized with common laboratory equipment (heated stir plate and centrifuge) and removal did not require the addition of enzymes or small molecules that would need to be washed away, only 37 °C incubation. Bead synthesis by emulsion generated broadly distributed bead sizes, which was useful for this study, but techniques for synthesis of low size dispersity gelatin spheres exist if required for future studies [36]. Furthermore, bead materials other than gelatin could be compatible with our technique if they can prevent the surrounding material from polymerizing within them during the embedding process and if they can be dissolved/degraded away in a timely and cytocompatible manner without negatively affecting the surrounding hydrogel.
LEC adhesion and growth required Matrigel and GelMA. Matrigel contains components such as laminin and collagen IV that provided sites for LEC adhesion. Matrigel may also have promoted cell growth in the beads because it contains residual growth factors and can absorb additional growth factors present in the media. However, Matrigel alone was unable to support monolayer formation, likely due to its low elastic modulus (on the order of hundreds of pascals [50] versus tens of kilopascals for the primate lens capsule [51]) and thus required mixing with GelMA. GelMA likely served as a relatively stiffer substrate, rather than as a source of additional cell adhesion sites based on the observation of solid spheroid formation in scaffolds lacking Matrigel. GelMA is a suitable hydrogel material for our system because it can be polymerized in less than 1 min, is biodegradable, and has tunable stiffness [33]. Regardless, our technique could be compatible with other biomaterials as long as they can form without destroying the microbeads and without significantly diffusing into and forming a network inside the beads. Most types of agarose are unsuitable, for example, because they require handling at temperatures that would melt the gelatin beads.
The morphology, dimensions, and E-cadherin expression of LECs within microchamber monolayers are similar to those observed in the central lens epithelium of adult mice [52,53]. In contrast to adult LECs, cells in the embryonic lens vesicle are cuboidal and columnar in morphology. The hollow spheroids formed by dispase treatment more closely resemble the lens vesicle in terms of cell shape and vesicle dimensions. Therefore, the attached spheres may provide a good model system for lens regeneration and secondary cataract formation after surgical lens fiber removal, while detached spheres may be a better model for embryonic lens development. Future studies using our platform for the lens can focus on the effects of LEC age, the use of LECs derived from stem cells [54], and the differentiation of LECs with growth factor gradients. Although we focused on the lens, other tissue vesicles have similar dimensions, making them suitable targets for our technique.
Our culture system using immobilized chambers has advantages and disadvantages compared to 3D culture within free-floating spheres. Free floating spheres are more conveniently distributed into multi-well plates for high-throughput testing [14]. It is also simpler to isolate cells from single spheres that are not grouped with others. Synthesizing individual immobilized chambers may be achievable by combination with techniques such as stop-flow lithography [55]. Meanwhile, immobilized chambers are more convenient for processes that require stable positioning of cells over extended times, such as time course imaging or exposure to sustained gradients [56]. This is because the orientation and position of each chamber is fixed and will not change with handling, media changes, or fluid flow.
In conclusion, we implemented a 3D cell culture platform for the parallel growth of hollow spheroidal monolayers within hydrogels by predefining spherical, cell-laden microchambers. The efficacy of the technique was demonstrated with LECs that formed structures similar to those observed in the adult or embryonic lens. This method allows for modifications of mechanical, biochemical, and geometric cues and can be applied to the numerous cell types that form hollow spheroids for the study of developmental and disease processes.
Supplementary Material
Acknowledgments
Chun-hong Xia for providing mice. Ellen Robey lab for providing H2B-GFP mice. Lucy Li and Nikki Tjahjono for helping with photopolymerization. Funding: Postdoctoral fellowships to EW from the California Institute for Regenerative Medicine and the UC Berkeley Vision Science Program (NEI T32EY007043), NEI R01EY013849 (XG). Tsinghua Berkeley Shenzhen Institute.
Abbreviations:
- 3D
three-dimensional
- LEC
lens epithelial cell
- mT/nG
membrane Tomato/nuclear GFP
- GelMA
gelatin methacrylate
Footnotes
Conflicts of interest
None.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.biomaterials.2017.07.031.
References
- [1].Pampaloni F, Reynaud EG, Stelzer EHK, The third dimension bridges the gap between cell culture and live tissue, Nat. Rev. Mol. Cell Biol 8 (2007) 839–845. [DOI] [PubMed] [Google Scholar]
- [2].Shamir ER, Ewald AJ, Three-dimensional organotypic culture: experimental models of mammalian biology and disease, Nat. Rev. Mol. Cell Biol 15 (2014) 647–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Griffith LG, Swartz MA, Capturing complex 3D tissue physiology in vitro, Nat. Rev. Mol. Cell Biol 7 (2006) 211–224. [DOI] [PubMed] [Google Scholar]
- [4].Baker BM, Chen CS, Deconstructing the third dimension – how 3D culture microenvironments alter cellular cues, J. Cell Sci 125 (2012) 3015–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Allazetta S, Kolb L, Zerbib S, Bardy JA, Lutolf MP, Cell-instructive microgels with tailor-made physicochemical properties, Small 11 (2015) 5647–5656. [DOI] [PubMed] [Google Scholar]
- [6].Schmeichel KL, Bissell MJ, Modeling tissue-specific signaling and organ function in three dimensions, J. Cell Sci 116 (2003) 2377–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Huh D, Hamilton GA, Ingber DE, From three-dimensional cell culture to organs-on-chips, Trends Cell Biol. 21 (2011) 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Breslin S, O’Driscoll L, Three-dimensional cell culture: the missing link in drug discovery, Drug Discov. Today 18 (2013) 240–249. [DOI] [PubMed] [Google Scholar]
- [9].Murphy SV, Atala A, 3D bioprinting of tissues and organs, Nat. Biotech 32 (2014) 773–785. [DOI] [PubMed] [Google Scholar]
- [10].Golden AP, Tien J, Fabrication of microfluidic hydrogels using molded gelatin as a sacrificial element, Lab a Chip 7 (2007) 720–725. [DOI] [PubMed] [Google Scholar]
- [11].Carlson MW, Alt-Holland A, Egles C, Garlick JA, In Current Protocols in Cell Biology, John Wiley & Sons, Inc, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yuan X, Zhou M, Gough J, Glidle A, Yin H, A novel culture system for modulating single cell geometry in 3D, Acta Biomater. 24 (2015) 228–240. [DOI] [PubMed] [Google Scholar]
- [13].Zhu W, et al. , 3D printing of functional biomaterials for tissue engineering, Curr. Opin. Biotechnol 40 (2016) 103–112. [DOI] [PubMed] [Google Scholar]
- [14].Dolega ME, Abeille F, Picollet-D’hahan N, Gidrol X, Controlled 3D culture in Matrigel microbeads to analyze clonal acinar development, Biomaterials 52 (2015) 347–357. [DOI] [PubMed] [Google Scholar]
- [15].Kusuma S, et al. , Self-organized vascular networks from human pluripotent stem cells in a synthetic matrix, Proc. Natl. Acad. Sci 110 (2013) 12601–12606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kolesky DB, Homan KA, Skylar-Scott MA, Lewis JA, Three-dimensional bioprinting of thick vascularized tissues, Proc. Natl. Acad. Sci 113 (2016) 3179–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wang X, et al. , Engineering anastomosis between living capillary networks and endothelial cell-lined microfluidic channels, Lab a Chip 16 (2016) 282–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Nishiguchi A, Yoshida H, Matsusaki M, Akashi M, Rapid construction of three-dimensional multilayered tissues with endothelial tube networks by the cell-accumulation technique, Adv. Mater 23 (2011) 3506–3510. [DOI] [PubMed] [Google Scholar]
- [19].Durruthy-Durruthy R, Gottlieb A, Heller S, 3D computational reconstruction of tissues with hollow spherical morphologies using single-cell gene expression data, Nat. Protoc 10 (2015) 459–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kurup A, et al. , Novel insights from 3D models: the pivotal role of physical symmetry in epithelial organization, Sci. Rep 5 (2015) 15153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sanz-Herrera JA, Moreo P, García-Aznar JM, Doblaré M, On the effect of substrate curvature on cell mechanics, Biomaterials 30 (2009) 6674–6686. [DOI] [PubMed] [Google Scholar]
- [22].Koehler KR, Mikosz AM, Molosh AI, Patel D, Hashino E, Generation of inner ear sensory epithelia from pluripotent stem cells in 3D culture, Nature 500 (2013) 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Morizane R, et al. , Nephron organoids derived from human pluripotent stem cells model kidney development and injury, Nat. Biotech 33 (2015) 1193–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Debnath J, Muthuswamy SK, Brugge JS, Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures, Methods 30 (2003) 256–268. [DOI] [PubMed] [Google Scholar]
- [25].Nelson CM, Inman JL, Bissell MJ, Three-dimensional lithographically defined organotypic tissue arrays for quantitative analysis of morphogenesis and neoplastic progression, Nat. Protoc 3 (2008) 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tang MD, Golden AP, Tien J, Fabrication of collagen gels that contain patterned, micrometer-scale cavities, Adv. Mater 16 (2004) 1345–1348. [Google Scholar]
- [27].Jiang W, Li M, Chen Z, Leong KW, Cell-laden microfluidic microgels for tissue regeneration, Lab a Chip 16 (2016) 4482–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Agarwal P, et al. , A biomimetic core–shell platform for miniaturized 3D cell and tissue engineering, Part. Part. Syst. Charact 32 (2015) 809–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Alessandri K, et al. , Cellular capsules as a tool for multicellular spheroid production and for investigating the mechanics of tumor progression in vitro, Proc. Natl. Acad. Sci 110 (2013) 14843–14848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sakai S, Hashimoto I, Kawakami K, Production of cell-enclosing hollow-core agarose microcapsules via jetting in water-immiscible liquid paraffin and formation of embryoid body-like spherical tissues from mouse ES cells enclosed within these microcapsules, Biotechnol. Bioeng 99 (2008) 235–243. [DOI] [PubMed] [Google Scholar]
- [31].Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L, A global double-fluorescent Cre reporter mouse, genesis 45 (2007) 593–605. [DOI] [PubMed] [Google Scholar]
- [32].Hadjantonakis A-K, Papaioannou VE, Dynamic in vivo imaging and cell tracking using a histone fluorescent protein fusion in mice, BMC Biotechnol. 4 (2004) 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Loessner D, et al. , Functionalization, preparation and use of cell-laden gelatin methacryloyl-based hydrogels as modular tissue culture platforms, Nat. Protoc 11 (2016) 727–746. [DOI] [PubMed] [Google Scholar]
- [34].Lee BH, Shirahama H, Cho N-J, Tan LP, Efficient and controllable synthesis of highly substituted gelatin methacrylamide for mechanically stiff hydrogels, RSC Adv. 5 (2015) 106094–106097. [Google Scholar]
- [35].Pettersen EF, et al. , UCSF Chimera–a visualization system for exploratory research and analysis, J. Comput. Chem 25 (2004) 1605–1612. [DOI] [PubMed] [Google Scholar]
- [36].Sakai S, et al. , Cell-enclosing gelatin-based microcapsule production for tissue engineering using a microfluidic flow-focusing system, Biomicrofluidics 5 (2011) 013402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Danysh BP, Duncan MK, The lens capsule, Exp. Eye Res 88 (2009) 151–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lin H, Cheng AW-M, Alexander PG, Beck AM, Tuan RS, Cartilage tissue engineering application of injectable gelatin hydrogel with in situ visible-light-activated gelation capability in both air and aqueous solution, Tissue Eng. Part A 20 (2014) 2402–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Nichol JW, et al. , Cell-laden microengineered gelatin methacrylate hydrogels, Biomaterials 31 (2010) 5536–5544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hoch E, Schuh C, Hirth T, Tovar GEM, Borchers K, Stiff gelatin hydrogels can be photo-chemically synthesized from low viscous gelatin solutions using molecularly functionalized gelatin with a high degree of methacrylation, J. Mater. Sci. Mater. Med 23 (2012) 2607–2617. [DOI] [PubMed] [Google Scholar]
- [41].Kim M-C, et al. , Integrating focal adhesion dynamics, cytoskeleton remodeling, and actin motor activity for predicting cell migration on 3D curved surfaces of the extracellular matrix, Integr. Biol 4 (2012) 1386–1397. [DOI] [PubMed] [Google Scholar]
- [42].McAvoy JW, Chamberlain CG, de Longh RU, Hales AM, Lovicu FJ, Lens development, Eye 13 (1999) 425–437. [DOI] [PubMed] [Google Scholar]
- [43].Lühr I, et al. , Mammary fibroblasts regulate morphogenesis of normal and tumorigenic breast epithelial cells by mechanical and paracrine signals, Cancer Lett. 325 (2012) 175–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hoffmann A, Nakamura K, Tsonis PA, Intrinsic lens forming potential of mouse lens epithelial versus newt Iris pigment epithelial cells in three-dimensional culture, Tissue Eng. Part C. Methods 20 (2014) 91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Gwon A, Lens regeneration in mammals: a review, Surv. Ophthalmol 51 (2006) 51–62. [DOI] [PubMed] [Google Scholar]
- [46].Zhou K-J, Li Y-N, Huang F-R, Wang Q-M, Yu AY, In vivo observation of lens regeneration in rat using ultra-long scan depth optical coherence tomography, Investig. Ophthalmol. Vis. Sci 57 (2016) 6615–6623. [DOI] [PubMed] [Google Scholar]
- [47].O’Connor MD, McAvoy JW, In vitro generation of functional lens-like structures with relevance to age-related nuclear cataract, Investig. Ophthalmol. Vis. Sci 48 (2007) 1245–1252. [DOI] [PubMed] [Google Scholar]
- [48].West-Mays JA, Pino G, Lovicu FJ, Development and use of the lens epithelial explant system to study lens differentiation and cataractogenesis, Prog. Retin. Eye Res 29 (2010) 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Han L-H, Lai JH, Yu S, Yang F, Dynamic tissue engineering scaffolds with stimuli-responsive macroporosity formation, Biomaterials 34 (2013) 4251–4258. [DOI] [PubMed] [Google Scholar]
- [50].Soofi SS, Last JA, Liliensiek SJ, Nealey PF, Murphy CJ, The elastic modulus of Matrigel™ as determined by atomic force microscopy, J. Struct. Biol 167 (2009) 216–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ziebarth NM, et al. , Primate lens capsule elasticity assessed using Atomic Force Microscopy, Exp. Eye Res 92 (2011) 490–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Shi Y, De Maria A, Lubura S, Šikić H, Bassnett S, The penny pusher: a cellular model of lens GrowthThe penny pusher: a cellular model of lens growth, Investig. Ophthalmol. Vis. Sci 56 (2015) 799–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Cheng C, Gong X, Diverse roles of eph/ephrin signaling in the mouse lens, PLoS One 6 (2011) e28147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Fu Q, et al. , Generation of functional lentoid bodies from human induced pluripotent stem cells derived from urinary CellsGeneration of functional LB from human-induced PSCs, Investig. Ophthalmol. Vis. Sci 58 (2017) 517–527. [DOI] [PubMed] [Google Scholar]
- [55].Dendukuri D, Pregibon DC, Collins J, Hatton TA, Doyle PS, Continuous-flow lithography for high-throughput microparticle synthesis, Nat. Mater 5 (2006) 365–369. [DOI] [PubMed] [Google Scholar]
- [56].Shin Y, et al. , Microfluidic assay for simultaneous culture of multiple cell types on surfaces or within hydrogels, Nat. Protoc 7 (2012) 1247–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wang D, et al. , Sci. Rep in press. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.