

ABSTRACT
Glioblastoma (GBM) stands out as the most common, aggressive form of primary malignant brain tumor conferring a devastatingly poor prognosis. Despite aggressive standard-of-care in surgical resection and chemoradiation with temozolomide, the median overall survival of patients still remains no longer than 15 months, due to significant tumor heterogeneity, immunosuppression induced by the tumor immune microenvironment and low mutational burden. Advances in immunotherapeutic approaches have revolutionized the treatment of various cancer types and become conceptually attractive for glioblastoma. In this review, we provide an overview of the basic knowledge underlying immune targeting and promising immunotherapeutic strategies including CAR T cells, oncolytic viruses, cancer vaccines, and checkpoint blockade inhibitors that have been recently investigated in glioblastoma. Current clinical trials and previous clinical trial findings are discussed, shedding light on novel strategies to overcome various limitations and challenges.
KEYWORDS: Glioblastoma, immunotherapy, chimeric antigen receptor (CAR) T cell, oncolytic virus
Introduction
Glioblastoma (GBM) remains the most prevalent and malignant glial tumor (WHO grade IV astrocytoma), which represents more than 50% of all primary brain tumors in the United States, with an annual incidence of roughly 3 per 100,000 people.1 Until now, it is highly aggressive and recalcitrant to almost all current standard-of-care treatments, which entail chemotherapy and radiation following complete resection. Despite such aggressive regimens, the median overall survival of patients remains no longer than 15 months from enrollment just prior to radiation therapy and concomitant chemotherapy with temozolomide, with a 5-year survival rate less than 10%.2–4 The success of immunotherapy has been established in various solid tumors, including melanoma, prostate cancer, non-small-cell lung cancer, and renal cell carcinoma, bourgeoning success in the thriving field of immunotherapy.5 According to recent advances in our understanding of GBM, its characteristics of rapid growth rate, proclivity to infiltrate vital brain compartments, molecular heterogeneity, regenerative capability of treatment-resistant cancer cells, and controlling concentrations of chemotherapeutic agents in the central nervous system (CNS) at lower levels have been contributing to its inexorable recurrence, resistance to therapy, and rapid progression.6
This review covers several major immunotherapeutic modalities targeting GBMs, including chimeric antigen receptor (CAR) T cells, oncolytic viruses (OVs), cancer vaccines, checkpoint blockade inhibitors, and combinatorial therapies (Figure 1). We will also briefly discuss the rationale of these approaches, along with its limitations and unique challenges faced when treating GBMs.
Figure 1.
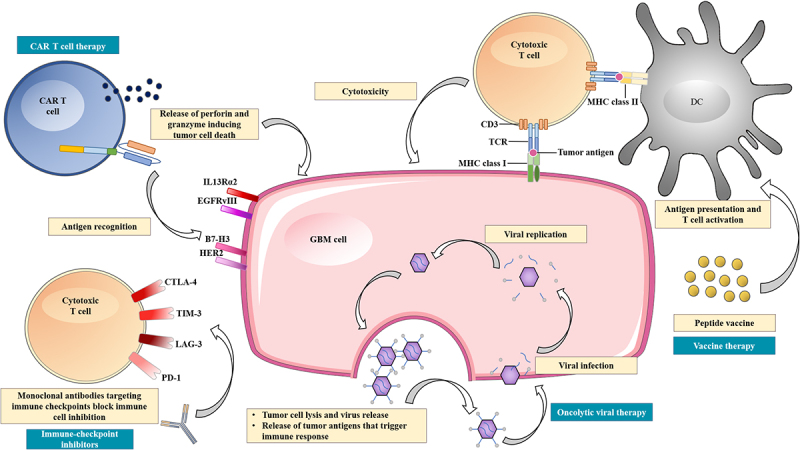
Overview of the current immunotherapeutic modalities for the treatment of glioblastoma.
CAR T cell therapy can target antigens that are highly expressed on GBM cell surfaces, including IL13Rα2, EGFRvIII, B7-H3, and HER2. Oncolytic viral therapy utilizes genetic engineered viruses that can selectively infect and replicate in GBM cells, leading to cell lysis and release of tumor antigens. This can further trigger an adaptive antitumor immune response by stimulating antigen presenting cells. Vaccine therapy depends on dendritic cells, which present antigens or peptides to cytotoxic T cells via MHC class II-TCR interaction resulting in T cell activation. Then, the cytotoxic T cells eradicate GBM cells via MHC class I-TCR interaction. This process, however, can be suppressed by upregulation of immune checkpoint ligands that can bind to various receptors on cytotoxic T cells. Immune checkpoint inhibitors are monoclonal antibodies that target immune checkpoints, such as PD-1, CTLA-4, TIM-3, and LAG-3, thereby blocking immune cell inhibition.
CNS immune privilege
The central nervous system was formerly recognized as an immune privileged site ascribed to the fully developed blood-brain barrier (BBB) and microglial surveillance. The BBB is a network of tissue and blood vessels that consists of non-fenestrated cells, forming endothelial tight junctions.7 These semipermeable connections selectively prevent leakage of hydrophilic solutes whereas allowing the exchange of hydrophobic solutes and active transport of circulating nutrients.8 The BBB also prevents or limits most pathogen invasion, excluding certain bacterial pathogens (Streptococcus pneumoniae, Neisseria meningitidis, group B Streptococcus, Escherichia coli) that adopted specific strategies. The strategies involve not only the release of various substances that bind to cellular receptors, resulting in the necrosis of endothelial cells or disruption of the intercellular junction, but also interactions enabling bacterial transcytosis across the endothelium.9 Given this property, most immune cells such as lymphocytes, monocytes, and dendritic cells are blocked from entry in the quiescent state. Microglia acts as the primary immune cells of the CNS that moves constantly to survey the surrounding parenchyma, presenting antigens to lymphocytes and expressing major histocompatibility complex class II molecules (MHCII).10 Although they can polarize to either a classical proinflammatory phenotype or to an anti-inflammatory phenotype, the immune surveillance of antigens prefers an environment that maintains neuronal homeostasis in the setting of uninflamed CNS.
It is now clear that during an CNS infection, the immune system mounts a full-scale systemic response to antigens.11 Other events including traumatic brain injury, autoimmunity, toxicity of metabolites, or accumulation of misfolded proteins can also induce CNS inflammation, potentiating the ingress of peripheral immune cells across the BBB.12 In 2015, structures similar to conventional lymphatic pathways paralleling the dural venous sinuses were identified in rodents, which changed our understanding of the brain’s immune environment.13 When endogenous danger molecules are released and detected, peripheral immune cells promptly infiltrate the CNS by crossing the BBB, evoking robust inflammatory responses. It is believed that this process provides the essential substrate for immunotherapy directed toward brain tumors.14
Immunosuppressive mechanisms of GBM
GBM is regarded as the most aggressive form of primary brain tumor, attributing to its rapid growth rate, brain tissue infiltrating capacity, molecular heterogeneity, treatment-resistant cancer cells, and the ability to limit the concentration of chemotherapeutic agents in the CNS. Despite immunotherapy being a promising strategy, tapping into the immunological mechanism of the CNS to develop durable antitumor responses remains challenging. In fact, GBM displays similar traits that immunotherapy-responsive tumors display; however, it wields extensive immunosuppressive mechanisms and further benefits from its location in the CNS.15 Overcoming intrinsic resistance, correcting systemic immunosuppression, countering adaptive resistance, and adjusting to acquired resistance are key for reversing the immunosuppressive machinery of GBM.6 Intrinsic resistance reflects the molecular and clinical characteristics, including the location, tissue of origin, and its biological features. A study developed a surgical multisampling scheme that collected spatially distinct tumor fragments from 11 GBM patients, and uncovered extensive intratumor heterogeneity showing different subtypes within the same tumor.16 This brings up a problem as selective eradication of clones susceptible to treatment is followed by progressive accelerating proliferation of resistant ones. Nevertheless, mounting an immune response that targets several diverse antigens is exceedingly risky of antigenic overlap with normal tissue for a cocktail of tumor-associated antigens (TAAs). However, it is not an issue for multiple neoantigens derived from autologous tumor cells. The ‘Three Es Hypothesis’ is an immune editing concept for understanding these interactions.17 It describes immune cells and tumor cells engaging on a continuum of elimination, equilibrium, and escape. The immune surveillance acts as an extrinsic tumor suppressor, eliminating precancerous clones, and sculpting the phenotype of tumors by selecting the least immunogenic ones. This selective pressure leads to equilibrium and escape in the end, causing GBM to evolve and gain adaptive resistance. One study interfered with PD-1 signaling, which broadened T cells’ antitumor responses and provided more target choices via epitope spreading, allowing T cells to adapt to a tumor’s evolving molecular profile.18 Therefore, immunotherapy should target neoantigens that is essential for tumor survival across various subtypes while being absent on healthy tissue. While neoantigens are addressed by intralesional cytolytic virus, autologous tumor antigen-based vaccines, as well as immune checkpoint inhibitors, they are not a strategy for CAR T cells or peptide vaccines.
The GBM microenvironment also plays a fundamental role in tumor immune evasion, leading to immunosuppression (Figure 2). In the uninflamed CNS, tissue-resident microglia, which functions as the main APC, make up the majority of the myeloid composition. Microglia are believed to enhance tumor infiltration in preclinical studies, leading to tumor progression and increased invasiveness. A study of cultured murine brain slices found that microglia increased the activity of metalloprotease-2, which caused increased breakdown of extracellular matrix and thereby promoted tumor invasiveness.19 Another study showed that elevated inflammatory cytokines secreted by microglia contribute to the expansion of GBM.20 As mentioned, the BBB guards the entry of most immune cells in the quiescent state. However, chemo-attractants secreted by the GBM cells, including CCL2, CCL5, CXCL and SDF-1 actively recruit tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs) across the BBB.21 Generally, TAMs have been shown to boost the expression of immune checkpoints, support cancer stem cells, and drive epithelial to mesenchymal transition (EMT), while MDSCs being the main source of TGF-β and PD-L1. Together they account for up to half of the immune compartment, significantly modulating the GBM microenvironment.22–24 Inhibiting CCL2, CCL5 activity has been shown to reduce tumor migration and invasion.25,26 The CCL2 secreted by GBM can induce high influx of regulatory T cells (Tregs), shifting tumor cytokine milieu toward immunosuppression, ultimately preventing the elimination of tumor cells.27,28 GBM cells can also overexpress colony stimulating factor (CSF-1), which acts as chemo-attractants, promoting formation of high-grade gliomas in mice.29 CSF-1 receptor inhibitors were given to patients with recurrent GBM in a phase II study, but showed no efficacy,30 indicating that the resistance is GBM microenvironment driven.31 Most of the newly recruited TAMs are polarized into anti-inflammatory M2 macrophages via the direct influence of GBM cells to produce a pro-tumor microenvironment (TME). The M2 phenotype TAMs can release anti-inflammatory cytokines such as IL-6, IL-10, and TGF-β, contributing to the immunosuppressive microenvironment.32 Furthermore, GBM cells can express complement regulators including Factor H, C1-inactivator, and CD59 to avoid complement attack, enhancing immunosuppression.33,34
Figure 2.
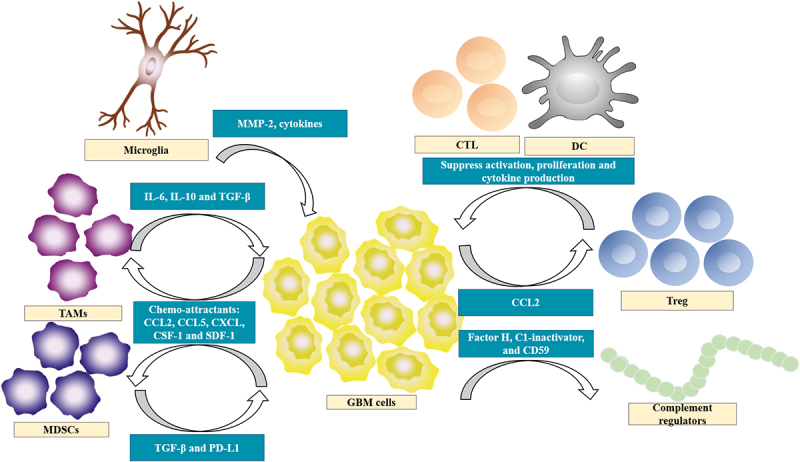
Overview of immunosuppressive pathways in glioblastoma.
Immunotherapy is currently being increasingly pursued due to its potential to address the challenges noted above. Developing a curative immunotherapy for GBM must not only overcome immunotolerance and generate sufficient responses to tumor antigens, but also circumvent the evolving escape mechanisms in the immunosuppressive microenvironment. Despite preclinical data supporting immunotherapy as a viable approach for GBM patients and arduous efforts to treat them, the results have not been translated to patients.35 At present, ongoing studies using combinatory approaches while augmenting current immunotherapeutic strategies are pointing the way for this revolutionary treatment in cancer care.
Immunotherapeutic strategies
Chimeric antigen receptor (CAR) T cells
CAR T cells demonstrate a promising strategy to counter challenges imposed by the BBB and TME as a form of adoptive cell T cell therapy (ACT). ACT is the reinfusion of autologous or allogenic antitumor T cells to attack tumor-specific antigens that are highly expressed on the surface of tumor cells while absent on normal cells. T-cells engineered in vitro with a lentiviral vector allows the construction of an autologous cell product to stably express a high-affinity single-chain fragment variable (scFv) that is specific for a target of interest. The scFv is fused to a transmembrane region, one or more co-stimulatory domains, and an intracellular signaling domain derived from the CD3ζ molecule of the endogenous T cell receptor (TCR). This grants the autologous T cells the ability to be activated upon scFv recognition of antigen, which then induces clustering and immobilization of the CAR molecules. Signals are initiated through the tyrosine kinase ζ-associated protein of 70 kDa (ZAP70) when the ITAM domains on the CD3ζ chain are phosphorylated, thereby evoking T cell effector response that includes releasing of cytokines, proliferation, cytotoxicity, and metabolic transformations.36 The antitumoral function of CAR T cells is thought to be mainly through release of cytokines, the granzyme and perforin axis, as well as the Fas and Fas ligand axis.37 These attributes endow CAR T cells with the ability to overcome the immunosuppression present in the TME. Until now, CAR T cells have demonstrated great clinical efficacy against hematopoietic malignancies, but applicability of CAR T cells for GBM is still being explored (Table 1).38
Table 1.
Summary of CAR T cells and oncolytic viruses used in clinical trials.
| Trial name (ClinicalTrials.gov Identifier) | Treatment | Phase of trial | Number of participants | Primary end point or outcomes | Summary of results |
|---|---|---|---|---|---|
| CAR T-cell therapies | |||||
| NCT00730613 | IL13 Rα2-targeted CAR T cells | I | 3 | Feasibility and safety | Grade 3 adverse events were observed, median OS 11 months |
| NCT02208362 | IL13 Rα2-targeted CAR T cells | I | 92 | AEs and DLT | Ongoing study, estimated completion date 1/2022 |
| NCT04661384 | IL13 Rα2-targeted CAR T cells | I | 30 | AEs and OS | Ongoing study, estimated completion date 12/2023 |
| NCT04003649 | IL13 Rα2-targeted CAR T cells with or without nivolumab and ipilimumab | I | 60 | AEs, DLT, feasibility, and OS | Ongoing study, estimated completion date 12/2022 |
| NCT03726515 | EGFRvIII-directed CAR T cells in combination with pembrolizumab | I | 7 | OS, PFS, and ORR | Study completed, no available info |
| NCT02209376 | EGFRvIII-directed CAR T cells | I | 11 | AEs | No DLT was observed, median OS 8 months |
| NCT01454596 | EGFRvIII-directed CAR T cells with cyclophosphamide, fludarabine and aldesleukin | I/II | 18 | AEs and PFS | DLT was observed at highest dose, median OS 6.9 months |
| NCT04385173 | B7-H3-targeted CAR T cells with temozolomide | I | 12 | AEs, MTD, OS, and PFS | Ongoing study, estimated completion date 6/2024 |
| NCT04077866 | B7-H3-targeted CAR T cells with or without temozolomide | I/II | 40 | OS and PFS | Ongoing study, estimated completion date 5/2023 |
| NCT01109095 | HER2 CMV-specific CAR T cells | I | 16 | DLT | Study completed, no available info |
| Oncolytic viral therapies | |||||
| NCT02798406 | DNX-2401 (Delta-24-RGD adenovirus) and i.v. pembrolizumab (anti-PD-1 antibody) | II | 48 | ORR by interval tumor size change | Ongoing study, expected completion 08/2023 |
| NCT01956734) | DNX-2401 (Delta-24-RGD adenovirus) and TMZ | I | 31 | Number of patients with AEs | No AEs related to DNX-2401 |
| NCT00805376 | DNX-2401 (conditionally replication-competent adenovirus) ± surgery | I | 37 | MTD | Treatment induced tumor infiltration by CD8+ and T-bet+ cells |
| NCT02062827 | M032 (modified strain of HSV-1) by intratumoural infusion | I | 36 | MTD | |
| NCT00390299 | MV-CEA (measles virus expressing carcinoembryonic antigen) before or after surgery | I | 40 | Toxicity and MTD | Study suspended for unknown reason |
| NCT02026271 | Ad-RTS-hIL-12 (INXN-2001, adenovirus modified to deliver hIL-2) and oral veledimex (INXN-1001, activator ligand) | I | 48 | Safety and tolerability | Response correlated with CD8+ (cytotoxic) and FoxP3+ (regulatory) T-cell counts in the peripheral blood |
| NCT02457845 | G207 (modified oncolytic strain of HSV-1) single-dose inoculation | I | 18 | Safety and tolerability | |
| NCT02986178 | PVSRIPO (genetically recombinant nonpathogenic poliovirus:rhinovirus chimera) ± lomustine | II | 62 | OS at 24 months | |
| NCT03152318 | rQNestin34.5v.2 (oncolytic HSV-1) + cyclophosphamide | I | 108 | MTD | Ongoing study, estimated completion 07/2022 |
| NCT03714334 | DNX-2440 conditionally replication-competent adenovirus with O×40 ligand (T-cell stimulator) | I | 24 | Treatment-related adverse events | |
| NCT01470794 | Toca 511 (vocimagene amiretrorepvec) injection to resection cavity of recurrent glioblastoma, and Toca FC (extended-release 5-FU) | I | 58 | DLT | Durable complete responses were observed |
| NCT03330197 | Ad-RTS-hIL-12 + veledimex | I | 25 | Safety and tolerability | |
| NCT03896568 | Ad5-DNX-2401 (oncolytic adenovirus) in bone marrow human mesenchymal stem cells | I | 36 | MTD | Ongoing study, estimated completion 05/2022 |
| NCT03294486 | TG6002 (modified vaccinia virus) and 5-FC | II | 78 | Safety, DLT, and rate of tumor progression at 6 months | |
| NCT01301430 | H-1 parvovirus (H-1PV) | II | 18 | Safety and DLT | No DLTs; 1 patient had a SUSAR that was not considered a DLT |
| NCT1491893 | PVSRIPO (genetically recombinant nonpathogenic poliovirus:rhinovirus chimera) | I | 61 | MTD | 21% long-term survivors at 36 months |
| NCT02197169 | DNX-2401 (Delta-24-RGD adenovirus) ± IFN-γ | I | 37 | ORR by interval tumor size change | No benefit with the addition of IFN/IFN poorly tolerated |
| NCT02414165 | Toca 511 (vocimagene amiretrorepvec) and Toca FC (extended release 5-FC) | II/III | 403 | OS | Stopped prematurely for lack of efficacy |
IL13 Rα2-Specific CAR T cells
The interleukin-13 receptor subunit alpha-2 (IL13 Rα2) is a monomeric receptor for interleukin 13 that is present in up to 60% of GBMs and is associated with the activation of proinflammatory and immune pathways.39,40 Its over-expression in GBM patients activates the phosphatidylinositol-3 kinase/AKT/mammalian target of rapamycin pathway, leading to poor prognosis and increased tumor invasiveness.41,42 In a preclinical study, a second-generation IL13 Rα2-directed CAR T cell comprising a mutated IL13-zetakine extracellular domain (IL13.E13K.R109K) that is linked to a CD28 costimulatory and CD3ζ signaling domain was engineered. By demonstrating reduced but persisting activity against normal tissue, whereas being potently activated in the presence of IL13 Rα2, these CAR T cells proved enhanced selectivity for IL13 Rα2 over IL13Ra1/IL4Ra. Further tests applying single intracranial injections of IL13-zetakine CAR T cells in a human glioma xenograft model resulted in noticeable increases in median overall survival.43 In 2015, a first-in-human pilot safety and feasibility trial evaluating IL13-zetakine CAR T cells was conducted in three patients with recurrent GBM. IL13 Rα2-directed CAR T cells were delivered via an implanted reservoir/catheter system into the resection cavity, inducing controllable temporary brain inflammation. Although the CAR T cells were well tolerated, adverse events were observed, including grade 3 headaches and a transient grade 3 neurologic event. Tumor tissue analysis revealed overall reduction in IL13 Rα2 expression and MRI analysis indicated a persistent increase in tumor necrotic volume, possibly extending duration of overall survival.44 Following this trial, the group developed a second-generation IL13-zetakine CAR T cell with a 4-1BB costimulatory domain and a mutated IgG4-Fc linker to reduce off-target interactions while boosting antitumor potential. A 50-year-old man presented with recurrent GBM and multifocal leptomeningeal disease was enrolled. He was treated with 6 cycles of intracavitary infusions of CAR T cells that were administered into the resected tumor cavity followed by another 10 cycles of infusions into the ventricular system. Despite inhibiting the tumor progression locally during the intracavitary infusions, other intracranial tumors progressed and new spinal lesions were found. Subsequently, the fifth intraventricular infusion resulted in substantial reduction of all intracranial and spinal tumors with a 77–100% decrease in tumor size. However, regression of all tumors lasted for only 7.5 months and recurrence was observed at new locations along with low IL13 Rα2 expression, encouraging combinatory therapies to address the antigen-loss relapse.45
EGFRvIII-Specific CAR T cells
The epidermal growth factor receptor variant III (EGFRvIII), resulting from an in-frame deletion of exons 2–7 of EGFR gene, is the most common mutation of this receptor that occurs heterogeneously in up to 30% of GBM specimens.46,47 EGFRvIII alteration was thought to be associated with shorter survival in GBM until recently suggested otherwise. Data showed that the prognosis for EGFRvIII patients might not differ from that of EGFR-amplified patients.48 One group discovered that EGFRvIII-directed CAR T cells were able to regulate tumor growth in xenogeneic subcutaneous and orthotopic models of human EGFRvIII (+) GBM, leading to a phase I clinical trial involving patients with either residual or recurrent GBM.49 This study was eventually terminated prior to completion as the sponsor decided to pursue combination therapies. A first-in-human study was carried out by treating a single dose of autologous EGFRvIII-directed CAR T cells to 10 patients with recurrent GBM. The results suggested that the infusion was safe and feasible, showing no off-tumor toxicities or cytokine release syndrome and noticeable levels of EGFRvIII-directed CAR T cells in the peripheral blood. Although the study was not designed for efficacy evaluation, tumor regression was not observed in any patients according to magnetic resonance imaging. In total, 7 out of 10 patients underwent surgical intervention following CAR T cell therapy and tumor tissue analysis revealed decreased EGFRvIII expression among five of them. Further investigation of the TME found increased expression of regulatory T cells as well as immunosuppressive molecules, particularly IDO1 and FoxP3, indicating immunoediting.50 Another phase I clinical trial treated 18 patients with recurrent GBM, where the presence of EGFRvIII was confirmed by PCR and biopsy. Third-generation EGFRvIII-directed CAR T cells containing CD28 and 4-1BB costimulatory domains were delivered in a dose-escalating manner. Patients started to develop respiratory symptoms within hours post-infusion at the highest dose levels, implying that dose-limiting toxicity was reached despite showing no signs of clinical benefit. Although median overall survival was 6.9 months, 2 patients survived over a year and a third patient managed to remain alive up to 59 months post-infusion.51 In 2019, an approach was developed to treat EGFRvIII-negative, EGFR-positive glioblastoma by combining a EGFRvIII scFv with a bispecific T-cell engager (BiTE). The BiTEs are bispecific synthetic antibodies that bind to two appropriate target antigens, serving as bridges to enhance the immune interactions and to improve antibody specificity.52 Recently, dual receptor circuits were used in a study to integrate recognition of multiple antigens,53 which enables controlled tumor cell death by targeting antigens that are not totally tumor specific. These SynNotch-CAR T cells improved the specificity, completeness, and persistence over conventional T cell therapy against glioblastoma.
B7-H3-Specific CAR T cells
The B7-H3 (also known as CD276), a type I transmembrane protein, plays a key role in the activation or inhibition of T-cell function. It was recognized as an important costimulatory molecule belonging to the B7 and CD28 families.54 Until now, challenges still remain such as creation of a unified view on the receptor of the B7-H3 and further elucidation of its regulation of immune responses. Particularly, B7-H3 was found to be highly overexpressed among a wide range of human cancer cells and correlates with negative clinical outcomes as well as poor prognoses in patients, making it a valuable target for immunotherapy.55–57 In our previous study, we constructed a third-generation B7-H3-directed CAR with 4-1BB and CD28 costimulatory domain and transduced it into T cells by lentivirus. In vitro and in vivo assessment of GBM samples from patients and cell lines was performed to analyze the antitumor effects of the engineered CAR T cells. Our results showed 58% B7-H3 positivity among the clinical samples, primary tumor cells and cell lines. B7-H3 expression level was also found to be correlated to the malignancy grade of glioma and the poor survival. In the preclinical models, potent antitumor effects of B7-H3-directed CAR T cells against GBM were demonstrated, leading to a clinical evaluation of the therapeutic potential in treating recurrent GBM at our institution.58 A 56-year-old woman presenting with recurrent GBM in the left frontal and parietal lobe was enrolled. The patient received craniotomy and standard-of-care in the past two years. Pathological analysis revealed a high but heterogeneous B7-H3 expression, which was also confirmed by flow cytometry assay of primary tumor cells. The patient underwent intracavitary infusions of B7-H3 targeted CAR T cells in a in a dose-escalating manner for 7 cycles, after which the patient dropped out of the clinical study. Although no adverse events of grade 3 or higher were observed, the patient suffered from headaches and appeared in altered consciousness as well as drowsiness. Significant reduction of the recurrent GBM was shown by MRI analysis and clinical response sustained for up to 50 days post-infusion. Unfortunately, the tumor became resistant to the therapy and relapsed, suggesting the development of multi-antigen directed CAR T cells or using combinatory therapies.59,60
HER2-Specific CAR T cells
The human epidermal growth factor 2 (HER2), formerly discovered as an overexpressed TAA in certain types of breast cancer, is a transmembrane glycoprotein with an extracellular ligand binding domain and an intracellular tyrosine kinase domain.61 HER2 is expressed across a wide array of biologically diverse CNS tumors including GBM, ependymoma, and medulloblastoma, but it is not expressed on normal CNS tissue, making it an appealing target.62 Monoclonal antibodies, such as trastuzumab, have limited effects in CNS tumors due to the BBB and lower HER2 expression relative to breast cancer.63 A preclinical study of HER2-specific T cells from 10 GBM patients that were generated by transduction with a retroviral vector showed promising results.61 The group then conducted a clinical trial to treat 17 patients with progressive HER2-positive glioblastoma. They utilized a second-generation CAR encoding FRP5 (anti-Her2) scFv and a CD28 signaling endodomain. The treatments were well tolerated with CAR T cells being detected up to 12 months since the infusion while showing no dose-limiting toxic effects. However, lack of expansion of the CAR T cells and no significant survival benefit were observed with a median overall survival of 11.1 months.64 An interim analysis of HER2-specific CAR T cells treatment for 3 young participants suggested that repeated locoregional infusion were viable and that correlative cerebrospinal fluid markers could be helpful for measuring CAR T cell activity in the CNS.65 A recent preclinical study addresses targeting 3 antigens using a single CAR T cell product, including HER2, IL13 Rα2 and ephrin-A2 in 15 primary samples. This highlighted the improved cytokine release and cytotoxicity in comparison to monospecific or bispecific CAR T cells, shedding light on the potential utility of trivalent CAR T cell therapy for GBM.66
Oncolytic viruses
In recent years, OVs are a novel class of therapeutic remedy in various solid tumors treatment including GBM. OVs offer a dual mechanism of antitumor responses of tumor-specific cell killing and the induction of systemic antitumor immunity (innate and adaptive).67 OVs induce immunogenic cell death of tumor cells. In this process, TAAs, damage-associated molecular patterns (DAMPs), and pathogen-associated molecular patterns (PAMPs) are released from the disrupted tumor cells.68 Both DAMPs and PAMPs act as a potent stimulus for the effective innate immunity through activating the pattern recognition receptors, such as toll-like receptors. In addition, DAMPs and PAMPs can further improve the antigen cross-presentation and adaptive immune responses.69,70 Moreover, OVs activate a proinflammatory immune response and increase the production of CXCL9, CXCL10 and CXCL11, which can promote T cells trafficking and tumor infiltration.71
The efficacy of OVs such as adenovirus, herpes simplex virus, measles virus, parvovirus, poliovirus, and zika virus against GBM has been demonstrated in preclinical studies.72–76 In the case of clinical trials, a variety of OVs are currently under investigation in clinical trials with some encouraging data.77 Initial clinical trials of OVs have showed satisfying safety profile and promising efficacy, which also offer some evidence of intratumoral viral replication and lymphocytes infiltration.78–80 Here, we summarize ongoing clinical trials based on virotherapy for the treatment of GBM.
A phase I clinical trial (NCT01470794) showed the safety and efficacy of Toca 511 treating 56 recurrent high-grade gliomas patients. In 23 patients who are eligible for the follow-up phase III study, the median OS was 14.4 months and OS was 65.2% and 34.8% at 1 and 2 years, respectively. More encouragingly, 5 patients had a complete response and survived 33.9–52.2 months after Toca 511 treatment.81 DNX-2401 (Delta-24-RGD; tasadenoturev) is a tumor-selective, replication-competent oncolytic adenovirus. Preclinical studies demonstrated antiglioma efficacy.82 A phase I, dose-escalation clinical trial of DNX-2401 was tested in 37 patients with recurrent malignant glioma. The safety and response were evaluated across 8 dose levels (1 × 10e7-3 × 10e10 vp in 1 mL) in GBM patients who received a single intratumoral injection of DNX-2401. 20% of patients survived over 3 years after treatment and three patients had over 95% reduction of the enhancing tumor with a PFS of over 3 years. In another subgroup, analyses of post-treatment tumor specimens showed that DNX-2401 replicates and spreads within the tumor. In addition, DNX-2401 can induce intratumoral CD8+ and T-bet+ T cells infiltration and decrease the expression of transmembrane immunoglobulin mucin-3.83 In Toca 511 and DNX-2401 trials, both offered encouraging results that approximately 20% of GBM patients show complete response after receiving OVs intratumorally. Meanwhile, virotherapy-associated severe adverse events are rare with no dose-limiting toxicities.81,83
Oncolytic H-1 parvovirus, an attenuated parvovirus, showed a slight improvement in the median OS in GBM patients who received intratumorally administration. Encouragingly, an increasing number of infiltrating lymphocytes and IFN-r levels was observed.84
PVSRIPO, a live attenuated poliovirus type 1 (Sabin) vaccine with its cognate internal ribosome entry site replaced with that of human rhinovirus type 2. PVSRIPO received breakthrough therapy designation from the FDA in May 2016 based on the findings of a phase I study in patients with recurrent GBM.85 PVSRIPO recognizes the poliovirus receptor CD155, which is widely upregulated on malignant cells of solid tumors and in major components of the TME such as antigen-presenting cells.86–88 In a phase I dose-finding and toxicity study in 61 recurrent supratentorial grade IV malignant glioma patients, convection-enhanced and intratumoral delivery of PVSRIPO was evaluated. In total, 19% of the patients had grade 3 or higher PVSRIPO-related adverse events and no neuropathogenicity or virus shedding was observed. Overall survival among the patients who received PVSRIPO reached a plateau of 21% (95% confidence interval, 11 to 33) at 24 months that was sustained at 36 months. Moreover, encouragingly, about 20% of patients remained alive for 57–70 months after the PVSRIPO injection.89 A phase II randomized trial of PVSRIPO alone or in combination with a single cycle of lomustine in patients with recurrent grade IV malignant glioma (NCT02986178) is ongoing.
Herpes simplex virus-1 (HSV-1) is a double-stranded, linear DNA virus. HSV-1 has been widely investigated and applied to the treatment of various solid tumor including GBM.90 HSV-1 has a large genome size of 152kb and most segments are non-essential. Therefore, HSV-1 can incorporate multiple large transgenes into its genome.91 G207 is one genetically engineered HSV-1 variants, which contains deletion of the diploid γ1 34.5 neurovirulence gene and has viral ribonucleotide reductase (UL39) disabled by insertion of Escherichia coli lacZ.92 A phase I clinical trial using G207 alone and with radiation to treat children and adolescents with recurrent or progressive high-grade gliomas showed no dose-limiting toxicity or serious adverse events. G207 converted immunologically “cold” tumors to “hot”.93
G47 delta (DELYTACT) is another oncolytic HSV-1 variant that introduces another deletion mutation based on G207. Therefore, G47 delta has triple mutation within the HSV-1 genome that can selectively kill tumor stem cells derived from GBM.94,95 Recently, a single-arm phase II open-label, non-randomized clinical trial in Japan was completed to evaluate the safety and efficacy of G47 delta (DELYTACT) administered in adult patients with residual or recurrent GBM. Side effects were very limited with only fever observed in 2 out of 16 patients. It is noteworthy that the survival rate of 13 patients after 1 year has reached 92.3% compared with 15% for conventional drugs.96 Based on the results of this phase II clinical trial, G47 delta (Delytact/Teserpaturev) has received conditional and time-approval from Japan Ministry of Health, Labor and Welfare (MHLW) as an OV for the treatment of patients with malignant glioma in Japan.
Some other clinical trials using OV as a treatment agent in patients with recurrent high-grade glioma are ongoing. Although promising, more clinical trials are needed to prove the safety and efficacy of the OV as a therapy for GBM.
Vaccines
Vaccine is another treatment agent against tumor that consists of tumor antigens. Vaccination can induce active immune surveillance against GBM and strengthen the adaptive immune system. Currently, there are four main approaches based on vaccine in GBM, including peptide vaccines, DNA vaccines, cell vaccines, and mRNA vaccines. Peptide or DNA vaccines involves the injection of tumor-specific antigens or DNA to induce the adaptative immune response. Cell vaccines also refer to DC vaccines based on DC cells that are derived from peripheral blood mononuclear cells (PBMCs) and are primed with tumor antigens. mRNA vaccines are some viral vectors loaded with mRNA coding tumor antigens to elicit potent immune responses. This approach has been developed over the years and are still being studied. (Table 2) However, only 3 vaccination agents have reached phase III clinical trial: Rindopepimut, DCvax, and PPV.97–99
Table 2.
Summary of vaccines used in clinical trials.
| Trial name (ClinicalTrials.gov Identifier) | Treatment | Phase of trial | Number of participants | Primary end point or outcomes | Summary of results |
|---|---|---|---|---|---|
| NCT00458601 | EGFRvIII peptide vaccine + TMZ | II | 82 | PFS | Median overall survival of 21.8 months and 36-month survival of 26%. Anti-EGFRvIII antibodies increased ≥4-fold in 85% of patients with duration of treatment |
| NCT01480479 | EGFRvIII peptide vaccine + TMZ | III | 745 | OS | Strong humoral responses; however, no survival advantage and loss of EGFRvIII expression upon recurrence |
| NCT00643097 | EGFRvIII peptide vaccine + DI-TMZ | II | 40 | PFS and hypersensitivity to peptide on the basis of a positive skin test of ≥5mm in diameter | EGFRvIII-expressing cells eradicated and vaccine immunogenic, with DI-TMZ cohort having enhanced humoral response. Median overall survival of 23.6 months |
| NCT00639639 | CMV pp65 DC vaccine + DI-TMZ | I | 16 | Safety and Feasibility | Antigen-specific immune responses and median overall survival of 41.1 months in DI-TMZ cohort. A total of 36% survival 5 years from diagnosis, with four patients remaining progression-free at 59–64 months from diagnosis |
| NCT00045968 | DCVax-L vaccine | III | 348 | PFS | Median overall survival of 23.1 months, with large group (n = 100) reaching 40.5 months |
| NCT02366728 | CMV pp65 DC vaccine +111In-labeled DC vaccine + Td Toxoid + basiliximab | II | 100 | OS | Ongoing, have reported increased DC migration to lymph nodes following Td toxoid pre-conditioning |
| NCT00905060 | HSPPC-96 vaccine + TMZ | II | 46 | Safety profile and survival durations | Median overall survival of 23.8 months. Patients with low PD-L1 expression in myeloid cells had median overall survival of 44.7 months compared to 18 months for those with high expression |
| NCT01280552 | ICT-107 (autologous DC vaccine pulsed with synthetic peptides mimicking GAAs) | II | 124 | OS | Pts in the HLA-A2 subgroup showed increased ICT-107 activity immunologically with a tendency for improved clinical outcome |
| NCT00293423 | HSPPC-96 peptide vaccine | II | 41 | Safety, toxicity, and PFS | Specific immune response in 11 of the 12 patients, responders had median overall survival of 11.8 months |
| NCT02122822 | HSPPC-96 peptide vaccine + TMZ + radiotherapy | I | 20 | Immune response and cardiac effects | Median overall survival of 31.4 months. Patients with high tumor-specific immune responses had median overall survival of >40.5 months compared to 14.6 months for low responders |
| NCT02454634 | IDH1 peptide vaccine | I | 39 | Safety and immunogenicity | A total of 93% vaccine-specific response rate, 84% survival >3 years |
| NCT01498328 | EGFRvIII peptide vaccine + bevacizumab | II | 127 | PFS | 24-month survival of 20% compared to 3% for controls |
| NCT03018288 | pembrolizumab ± HSPPC-96 vaccine | II | 108 | 1-year OS | Ongoing study, estimated completion 01/2025 |
| NCT02149225 | APVAC1 and APVAC2 (personalized peptide vaccine) plus poly-ICLC and GM-CSF | I | 16 | Safety and immunogenicity | Able to generate a strong and lasting immune response |
| NCT02924038 | IMA-950 (peptide vaccine comprising multiple GAAs) and poly-ICLC ± varlilumab (immunostimulatory antiCD27 antibody) | I | 30 | Safety and T cell responses | Ongoing study, estimated completion 12/2022 |
| NCT02287428 | Personalized neoantigen vaccine | I | 16 | Safety and feasibility | Neoantigen selection is feasible and induces immune response |
| NCT02960230 | H3.3K27 M peptide vaccine plus Td and poly-ICLC | I | 29 | Safety and OS | Ongoing study, estimated completion 01/2023 |
| NCT03400917 | AV-GBM-1 (Autologous dendritic cells loaded with tumor associated antigens from a short-term cell culture of autologous tumor cells) | II | 55 | OS | Ongoing study, estimated completion 02/2023 |
| NCT02507583 | IGF-1 R/AS ODN | I | 33 | Safety and tolerability | Well tolerated with an improved PFS compared with standard-of-care |
| NCT02455557 | Montanide ISA 51 VG peptide vaccine + temozolomide | II | 66 | PFS | Improved PFS and OS |
| NCT04116658 | EO2401 peptide vaccine | II | 52 | Safety and tolerability | Ongoing study, estimated completion 8/2023 |
| NCT02465268 | pp65-shLAMP DC dendritic cell vaccine with GM-CSF | II | 175 | OS | Ongoing study, estimated completion 6/2024 |
| NCT00846456 | Tumor stem cell derived mRNA- transfected dendritic cells | I/II | 20 | Adverse events | Progression-free survival (PFS) was 2.9 times longer in vaccinated patients |
| NCT01006044 | Tumor lysate-pulsed autologous dendritic cell vaccine | II | 26 | PFS | Feasible and safe |
| Not Available | Personalized peptide vaccination for human leukocyte antigen (HLA)-A24+ | III | 88 | OS | Met neither the primary nor secondary endpoints |
Rindopepimut (also known as CDX-110 or PEPvIII) is a peptide-based vaccine that targets the EGFR deletion mutation in variant III of the epidermal growth factor receptor (EGFRvIII), which is expressed exclusively in GBM.100 Targeting the EGFRvIII neoantigen that is not expressed on normal cells limits the risk of “on-target, off-tumor” toxicities. However, heterogeneity is a common feature of solid tumor, especially for GBM. EGFRvIII is heterogeneously expressed on glioblastoma cells in vivo. Therefore, there is a risk of tumor progression due to the lack of this antigen.48 EGFRvIII was first tested in a phase II clinical trial ACTIVATE, and subsequently followed up with two additional phase II trials, ACT II and ACT III. These three uncontrolled phase II trials focused in rindopepimut vaccination in selected patients with gross total resection and no evidence of progression after adjuvant chemoradiotherapy. They have shown the evidence of better progression-free survival of 15 months and median survival of 24 months compared with historical controls.101–103 ACT IV is an international, randomized, double-blind phase III clinical trial that examined whether rindopepimut plus standard treatment would prolong overall survival in GBM patients with minimal residual disease (MRD). MRD was defined as the presence of <2 cm2 of enhancing tumor tissue after surgery and chemoradiotherapy. Around 370 patients were randomized into each arm to receive either rindopeptimut or keyhole limpet hemocyanin (KLH; the carrier protein in rindopepimut). At the final analysis, there was no significant difference in overall survival between the rindopeptimut and control group for patients with MRD (median 20.1 months in the rindopepimut group versus 20.0 months in the control group; HR 1.01, 95% CI .79–1.30; P = .93).97
IDH1 peptide is another molecule that has been investigated in clinical trials. Based on promising results of IDH1 peptide vaccines in preclinical experiments,104 two ongoing phase I clinical trials (NOA-16 and RESIST) have been started to investigate its efficacy against GBM patients. This vaccine is engineered to specifically target the IDH1R132 H mutation.105
Survivin is highly expressed in GBM and other cancers. SurVaxM is a peptide vaccine of amino acids 53–67 of the protein, conjugated with keyhole limpet hemocyanin.106 A phase II study (NCT02455557) is currently investigating the treatment of 64 newly diagnosed GBM patients with TMZ and the SurVaxM vaccine. (Survaxm Vaccine Therapy and Temozolomide in Treating Patients With Newly Diagnosed Glioblastoma. Available at: https://ClinicalTrials.gov/show/NCT02455557.) Early results indicate an improved PFS and OS compared to historical controls.107 A phase I clinical trial (NCT02507583) is investigating the use of an antisense oligodeoxynucleotide against insulin-like growth factor (IGF) type I receptor (IMV-001) for treatment of 33 newly diagnosed glioblastoma. The results of this clinical trial indicated that IGV-001 was well tolerated with an improved PFS compared with standard-of-care.108 In an earlier phase I personalized protein vaccine (PPV) immunotherapy trial of 12 recurrent GBM patients who were temozolomide-refractory, the researchers found that PPV monotherapy was safe and active in suppressing cancer progression and may extend patients survival by enhancing immunoreactivity.109 In a randomized, double-blind, phase III trial, 88 HLA-A24-positive GBM patients refractory to temozolomide-based therapy were randomly assigned to receive PPV treatment (58 patients) or best supportive care (BSC; 30 patients). Four peptides chosen from 12 peptide candidates based on pre-vaccination IgG levels specific to each peptide or four corresponding placebos were injected subcutaneously once weekly for 12 times at the first course, followed by biweekly vaccinations until disease progression. However, the results of this trial were unfavorable. The primary endpoint of OS was not met in this clinical trial. Median OS was 8.4 months with PPV and 8 months with BSC.99
Neoantigens are identified by whole exome DNA and RNA sequencing, which are resulted from somatic DNA mutations including nonsynonymous point mutations, insertions or deletions, gene fusions and frameshift mutations.110 Neoantigens are a promising and more personalized cancer treatment approach. Two clinical trials have demonstrated the potential of personalized GBM vaccination.111,112 Three vaccines (APVAC1, APVAC2, and NeoVax) were investigated: 2 (APVAC1 and APVAC2) in the GAPVAC trial and 1 (NeoVax) by Keskin et al. In the GAPVAC study, 16 newly diagnosed GBM patients received two synthesized vaccines, one aimed at unmutated peptides (APVAC1) and the other aimed at neoantigens (APVAC2). In 11 patients, CD4+ and CD8+ T cell response were observed to APVAC1 and APVAC2. However, there are some differences of immunogenicity between these two vaccines. APVAC1 induced 50% immunogenicity while APVAC2 induced 84.7% immunogenicity. APVAC1 elicited mainly CD8+ T cell responses and APVAC2 produced primarily CD4+ T cell responses. The efficacy of these two vaccines was encouraging for which the median PFS and OS was 14.5 and 29 months, respectively.111 In another neoantigen vaccine clinical study, 8 patients with newly diagnosed and MGMT unmethylated GBM received only a neoantigen vaccine (NeoVax). Two patients after receiving NeoVax treatment showed immunogenicity and both types of responses of CD4+ T cells and CD8+ T cells were observed. For survival, A median PFS of 7.6 months and median OS of 16.8 months was reported.112 DCVax-L is a personalized approach to peptide vaccination that uses autologous, or patient-derived, DCs pulsed with resected tumor lysate to target a variety of tumor antigens. In a phase III trial (NCT00045968) analyzing DCVax-L, it showed great potential for this vaccination strategy when combined with standard therapy for a newly diagnosed GBM.98 Some studies explored the feasibility of mRNA-transfected DC vaccines. The mRNA-transfected DCV was suggested to be safe and well-tolerated. Compared to matched controls, progression-free survival (PFS) was 2.9 times longer in vaccinated patients (NCT00846456).113 In a phase II clinical trial, autologous DCs were generated from peripheral blood monocytes and pulsed with autologous whole tumor lysate. This tumor lysate-pulsed DC vaccines in combination with standard chemoradiotherapy was proved feasible and safe in newly diagnosed GBM patients (NCT01006044).114 However, in a multicentric randomized open-label phase II study, vaccination with tumor lysate-charged autologous DCs (Audencel) could not improve the clinical outcomes in newly diagnosed GBM patients.115 AV-GBM-1, which are autologous DCs loaded with TAAs from a short-term cell culture of autologous tumor cells, are given to newly diagnosed GBM patients to evaluate overall survival in an ongoing phase II clinical trial. (NCT03400917).
Dendritic cells (DCs) are antigen presenting cells that can traffic via the tumor draining lymph nodes of the brain to the deep cervical lymph nodes and promote an adaptive antitumor immune response.116 Therefore, DCs are an ideal cell type to develop cellular vaccination against tumors. In preclinical models and early-stage clinical trials, DC vaccinations have shown the effectiveness against gliomas.117,118
A phase I/II clinical trial enrolled 24 patients with recurrent malignant glioma. This trial investigated the use of a DC therapy that was generated with granulocyte macrophage colony-stimulating factor and interleukin 4 with or without OK-432. Dendritic cells were injected intradermally, or both intratumorally and intradermally. This clinical trial demonstrated the tolerance of DC vaccines with no observed adverse events or radiographic evidence of autoimmune reactions. The overall survival of patients with GBM was 480 days which was significantly longer than that in the control group. Moreover, the patients with both intratumoral and intradermal administrations had a better survival time than patients with intradermal administration only. In addition, there was an increased T cell reactivity after DC vaccination.119
ICT-107 is another DC-based vaccine that was specifically designed for newly diagnosed GBM patients. This vaccine consists of DCs incubated ex vivo with TAAs expressed in GBM cells including HER2, IL-13 receptor subunit-α2 (IL-13 Rα2), melanoma-associated antigen 1 (MAGEA1), interferon-inducible protein AIM2, L-dopachrome tautomerase (DCT) and melanocyte protein (PMEL). The phase I clinical trial of ICT-107 enrolled 20 GBM patients and confirmed the safety. The median PFS was 16.9 months and median OS was 38.4 months in the newly diagnosed GBM patients.120 In a double-blind, placebo-controlled phase II clinical trial, the safety and efficacy of ICT-107 were investigated for patients with newly diagnosed GBM. In this trial, 124 GBM patients were randomly assigned to receive ICT-107 or autologous DCs that were not exposed to the TAAs of GBM. Although the overall survival was not significantly improved, ICT-107 has therapeutic activity in HLA-A2-positive patients.121 A randomized, double-blind phase III clinical trial of ICT-107 (NCT02546102) investigated the efficacy of ICT-107 compared to the standard-of-care. However, this study was suspended before achieving the primary endpoint due to a lack of funding.98
Immune checkpoint inhibitors
Immune checkpoint inhibitors (ICIs) are monoclonal antibodies that decrease the activity of negative regulatory pathways that limit T cell activation by targeting surface receptors called immune checkpoints.122,123 Under physiological conditions, immune checkpoint molecules can lower cytotoxic T-cell function, whereas when the immune checkpoint is abnormally activated via ICIs, suppression of tumor immune response occurs and T-cell function is restored, thereby increasing the immunotherapeutic effect. Currently, immune checkpoints are primarily focused on programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) (Table 3).124,125 Considerable improvements in patient outcomes have been achieved for various challenging tumors such as melanoma, lung cancer and renal cancer by utilizing anti-PD-1 and anti-PD-L1 antibodies.126–128 CheckMate-143 (NCT02017717), the first randomized phase I clinical trial in recurrent GBM, evaluated the tolerability and efficacy of nivolumab (PD-1 inhibitor) alone or in combination with ipilimumab (CTLA-4 inhibitor). All enrolled patients received surgical resection, radiotherapy, and temozolomide before being split into three treatment arms and a vast majority of patients received subsequent therapy. Interestingly, nivolumab monotherapy resulted in a greater median overall survival (10.4 months vs 9.2 months or 7.3 months) than NIVO1+IPI3 (nivolumab 1 mg/kg + ipilimumab 3 mg/kg) or NIVO3+IPI1 (nivolumab 3 mg/kg + ipilimumab 1 mg/kg). Grade 3 or higher adverse events were observed only when combined inhibition is administered, suggesting the necessity for further investigation.129 Phase III of this clinical trial enrolled and randomized 369 recurrent GBM patients to receive nivolumab 3 mg/kg or bevacizumab (an antiangiogenic drug targeting VEGF) 10 mg/kg every two weeks until confirmed disease progression. Although median overall survival and toxicity were comparable between nivolumab and bevacizumab treated groups, the latter showed shorter duration of radiologic response.130 Bevacizumab alone and in combination with irinotecan were evaluated for the efficacy in patients with recurrent GBM in a phase II clinical trial. Despite experiencing grade 3 or higher adverse events, both treatment arms were well tolerated with the median overall survival of 9.2 months and 8.7 months, respectively.131 Currently, the CheckMate-548 (NCT02667587) and the CheckMate-498 (NCT02617589) are two ongoing phase III trials investigating nivolumab as a potential treatment for patients with glioblastoma that is MGMT-unmethylated. CheckMate-548 compares temozolomide plus radiation therapy combined with nivolumab or placebo, while CheckMate-498 compares nivolumab vs temozolomide each in combination with radiation therapy.
Table 3.
Summary of immune checkpoint inhibitors used in clinical trials.
| Trial name (ClinicalTrials.gov Identifier) | Treatment | Phase of trial | Number of participants | Primary end point or outcomes | Summary of results |
|---|---|---|---|---|---|
| NCT02017717 | Nivolumab vs. bevacizumab | III | 530 | OS | Median OS 9.5 months vs 9.8 months |
| NCT02617589 | Nivolumab vs. temozolomide in combination with radiation therapy | III | 560 | OS | Median OS 13.4 months vs 14.88 months |
| NCT02667587 | Temozolomide + radiation therapy in combination with nivolumab or placebo | III | 716 | PFS and OS | No survival advantage over placebo |
| NCT04606316 | Nivolumab in combination with ipilimumab and surgery | I | 60 | Tumor infiltrating T lymphocyte density and safety | Ongoing study, estimated completion date 12/2022 |
| NCT03743662 | Nivolumab with radiation therapy and bevacizumab | II | 94 | OS | Ongoing study, estimated completion date 11/2022 |
| NCT02550249 | Neoadjuvant nivolumab | II | 29 | Efficacy and safety | Median OS 7.3 months |
| NCT04396860 | Ipilimumab and nivolumab plus radiation therapy | II/III | 485 | PFS and OS | Ongoing study, estimated completion date 8/2024 |
| NCT02311920 | Ipilimumab and/or nivolumab in combination with temozolomide | I | 32 | DLT | Ongoing study |
| NCT04145115 | Ipilimumab and nivolumab | II | 37 | ORR | Ongoing study, estimated completion date 5/2023 |
| NCT02337491 | Pembrolizumab with or without bevacizumab | II | 80 | MTD, DLT, and PFS | Median OS 8.8 months together vs 10.3 months for pembrolizumab alone |
| NCT02054806 | Pembrolizumab | I | 477 | Best overall response | Median OS 14.4 months |
| NCT02335918 | Combination of varlilumab and nivolumab | I/II | 175 | DLT, ORR, and OS | Study completed, no available info |
| NCT02336165 | Durvalumab monotherapy, with bevacizumab or with radiaotherapy | II | 159 | OS and PFS | Ongoing study |
| NCT03673787 | Atezolizumab in combination with ipatasertib | I/II | 87 | DLT | Ongoing study, estimated completion date 7/2022 |
| NCT03961971 | Anti-Tim-3 in combination with anti-PD-1 and stereotactic radiosurgery | I | 15 | Serious adverse events | Ongoing study, estimated completion date 9/2022 |
| NCT02658981 | Anti-LAG-3 or urelumab alone and in combination with nivolumab | I | 63 | MTD | Ongoing study |
AEs, adverse events; DLT, dose-limiting toxicity; MTD, maximum tolerated dose; OS, overall survival; PFS, progression-free survival; ORR, objective response rate.
Pembrolizumab, another anti-PD1 checkpoint inhibitor, is currently being investigated as the treatment of gliomas.132 In a phase II trial, neoadjuvant with pembrolizumab administration prior to surgery in addition to post-surgery adjuvant treatment showed increased survival in patients with recurrent GBM.133 CTLA-4 is an immune checkpoint that suppresses T cell function by competitively binding to CD80 and CD86.134 Clinical trials (NCT02311920, NCT02829931) evaluating ipilimumab therapies are still ongoing. LAG-3 is another immune checkpoint receptor that inhibits T-cell activity while promoting the suppressive activity of Tregs.135 A phase I trial (NCT02658981) evaluating BMS -986,016 (LAG-3 inhibitor) alone and in combination with nivolumab in recurrent GBM patients is underway. TIM-3, another receptor expressed on lymphocytes that can induce T-cell exhaustion and suppress immune response, can lead to a poor prognosis.136 A phase I trial (NCT03961971) studying the side effects of stereotactic radiosurgery with MBG453 (TIM-3 inhibitor) and spartalizumab (PD-1 inhibitor) therapy is still in progress.
Limitations
Clinical advances and breakthrough in immunotherapy for other tumors have inspired treatment for GBM from an immunotherapeutic angle. However, GBM presents unique challenges with characteristics including: (1) the BBB that limits the permeability for drug delivery, (2) microglia, TAMs and MDSCs contributing to the immunosuppressive microenvironment, (3) limited cytotoxic T cell activation and function, (4) low mutational burden and limited neoantigen profile. Current explorations in CAR T cells, OVs, vaccines, and immune checkpoint inhibitors have yet to show improvements in survival outcomes. In recurrent presentations, since CAR T cells operate by antigen presentation for T cell activation, CAR T efficacy was limited due to the adaptive resistance of GBM, leading to antigen loss as well as T cell exhaustion. Despite the broad targeting capabilities of vaccines, their applications are mainly affected by the limited somatic mutations of GBM cells, thus making antigen escape a concern. In the application of OVs, achieving sufficient replication for GBM apoptosis without triggering an excessive inflammatory response that leads to early elimination has been greatly challenging. Immune checkpoint inhibitors have been disappointing in GBM, possibly due to inadequate tumor-infiltrating lymphocytes with the presence of excessive Tregs and MDSCs, resulting in unfavorable downstream checkpoint blockade effects.137
Discussion
Immunotherapy has revolutionized cancer care across various tumor types, but failed to generate comparable clinical results for GBM. Here, we introduced basic knowledge underlying immune targeting and promising immunotherapeutic strategies including CAR T cells, OVs, cancer vaccines, and checkpoint blockade inhibitors that have been recently investigated in glioblastoma. As mentioned, GBM cells is presented with a low mutational burden with certain antigen targets being selectively downregulated or not essential for survival, thus providing few therapeutic targets for the immune system.134 GBM’s intratumoral molecular heterogeneity, rapid growth rate, as well as immunosuppressive microenvironment also contribute to the unique challenges when taking an immunotherapeutic approach. Furthermore, significant immunoediting causing an evolution in GBM’s molecular profile can possibly result in an overly suppressive and evasive tumor. This phenomenon creates epitopes that limit the recognition of CAR T cells, thereby resulting in a declining antitumor effect over time. Targeting TAMs and Tregs of the TME has been investigated by inhibition of CSF-1 (NCT02526017) and TGFβ (NCT02423343), hoping for clinical benefits but to no avail.31 Given such findings, it might be possible that tumors present themselves differently in different patients and their susceptibility to immunotherapy may vary from case to case. Identifying which patients are more susceptible can be greatly beneficial for generating a robust response.138 Patients with higher mutation loads suggest higher levels of neoantigens, which may improve the success rate of current immunotherapeutic strategies. Therefore, targeting neoantigens that are absent on healthy tissue while being essential for tumor survival across various GBM subtypes is of great importance. A well-established research design is also crucial to ensure that maximal information can be obtained from every clinical trial, while not exposing patients to futile treatments.
Moreover, supportive treatments including high-dose steroids and chemotherapies contribute to immunosuppression and lymphopenia.139,140 T-cell sequestration in bone marrow also leads to a tumor-adaptive mode of immunocompetent cells dysfunction in the setting of GBM.141 In addition, continual interaction with antigens is likely to induce T-cell exhaustion.142 Together, there is no development of an efficacious or durable antitumor immune response.
Finally, utilizing different immunotherapeutic strategies as combinatory therapies are being actively investigated, yet more than half of the patients with responsive GBM fail to derive clinical benefits. Therefore, immune-oncology research is gradually being directed toward development of strategiesto bypass the tumor resistance mechanisms, including intrinsic resistance (stops the initiation of a response), adaptive resistance (deactivates tumor-infiltrating immune cells) and acquired resistance (protects tumor cells from destruction when attacked by the immune system). This highlights the requirement for an in-depth understanding of the interactions between GBM and the immune system.6
Future prospects
Many current immunotherapeutic strategies are expanding the variety of antigens targeted and modulating the TME to avoid or tackle various mechanisms of resistance. While many advances have been made in the field, there has not been a breakthrough in clinical trials. CAR T therapy should find a way to overcome GBM heterogeneity by engineering multiple antigen specific CAR T cells. ”Off-the-shelf” allogeneic CAR T cells is a promising option such as immediate availability of cryopreserved batches for treatment without requiring the time to manufacture and delay treatment.
However, graft-versus-host disease and quick elimination by the host immune system are issues that need to be addressed in the future development.143 Over the recent years, viral therapy incorporating new genetic constructs that directly involve the protein expression machinery have seen promising results, though uptake efficiencies and larger clinical trials are still issues to be addressed. Vaccine-based immunotherapy proceeds to produce encouraging studies while moving forward to evaluate the durability of antigen combinations. Rapid and enhanced immunogenicity of these treatments as well as individualized vaccines are potentially becoming important. In order to substantially impact clinical outcomes, immune checkpoint inhibitors may require downregulation of counterproductive cytokines and T cell population shifting by certain treatments. Future GBM treatment will likely involve combined approaches with synergistic effects to not only target the tumor but also the reverse the TME suppression as well as preventing antigen escape.
Conclusions
In summary, while the exploration of immunotherapy has dramatically improved the prognosis for numerous types of advanced solid tumors, GBM has demonstrated strong resistance mechanisms. Future combinatory therapies are likely to be delivered at different stages throughout the immunosuppressive cycle and target each tumor resistance mechanisms concurrently to achieve tumor regression. As the search for novel tumor-associated and tumor-specific antigens continues, new discoveries and advancements in therapeutic modalities also boost efficacy and reduce toxicity, conjointly contributing to the clinical efficacy of immunotherapies.
Funding Statement
This work was supported by a grant from the Science and Technology Department of Sichuan Province, P.R. China [2017SZ0015] and National Natural Science Foundation of China (NO. 82073404).
Author contributions
Boyang Yuan and Guoqing Wang contributed equally to original draft preparation, review, and editing. All authors have read and agreed to the published version of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- 1.Ostrom QT, Gittleman H, Stetson L, Virk SM, Barnholtz-Sloan JS.. Epidemiology of gliomas. Cancer Treat Res. 2015;163:1–16. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJB, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase iii study: 5-year analysis of the eortc-ncic trial. Lancet Oncol. 2009;10(5):459–66. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 4.Lapointe S, Perry A, Butowski NA. Primary brain tumours in adults. Lancet. 2018;392(10145):432–46. doi: 10.1016/S0140-6736(18)30990-5. [DOI] [PubMed] [Google Scholar]
- 5.Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from t cell basic science to clinical practice. Nat Rev Immunol. 2020;20(11):651–68. doi: 10.1038/s41577-020-0306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jackson C, Choi J, Lim M. Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat Immunol. 2019;20(9):1100–09. doi: 10.1038/s41590-019-0433-y. [DOI] [PubMed] [Google Scholar]
- 7.Bauer H-C, Krizbai IA, Bauer H, Traweger A. “You shall not pass”-tight junctions of the blood brain barrier. Front Neurosci. 2014;8. doi: 10.3389/fnins.2014.00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campos-Bedolla P, Walter FR, Veszelka S, Deli MA. Role of the blood-brain barrier in the nutrition of the central nervous system. Arch Med Res. 2014;45(8):610–38. doi: 10.1016/j.arcmed.2014.11.018. [DOI] [PubMed] [Google Scholar]
- 9.Le Guennec L, Coureuil M, Nassif X, Bourdoulous S. Strategies used by bacterial pathogens to cross the blood-brain barrier. Cell Microbiol. 2020;22(1):e13132. doi: 10.1111/cmi.13132. [DOI] [PubMed] [Google Scholar]
- 10.Ransohoff RM, Cardona AE. The myeloid cells of the central nervous system parenchyma. Nature. 2010;468(7321):253–62. doi: 10.1038/nature09615. [DOI] [PubMed] [Google Scholar]
- 11.Coureuil M, Lecuyer H, Bourdoulous S, Nassif X. A journey into the brain: insight into how bacterial pathogens cross blood-brain barriers. Nat Rev Microbiol. 2017;15(3):149–59. doi: 10.1038/nrmicro.2016.178. [DOI] [PubMed] [Google Scholar]
- 12.Lecuyer M-A, Kebir H, Prat A. Glial influences on bbb functions and molecular players in immune cell trafficking. Biochim Biophys Acta, Mol Basis Dis. 2016;1862(3):472–82. doi: 10.1016/j.bbadis.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 13.Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, Derecki NC, Castle D, Mandell JW, Lee KS, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523(7560):337±. doi: 10.1038/nature14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marchetti L, Engelhardt B. Immune cell trafficking across the blood-brain barrier in the absence and presence of neuroinflammation. Vasc Biol. 2020;2(1):H1–H18. doi: 10.1530/VB-19-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tomaszewski W, Sanchez-Perez L, Gajewski TF, Sampson JH. Brain tumor microenvironment and host state: implications for immunotherapy. Clin Cancer Res. 2019;25(14):4202–10. doi: 10.1158/1078-0432.CCR-18-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sottoriva A, Spiteri I, Piccirillo SGM, Touloumis A, Collins VP, Marioni JC, Curtis C, Watts C, Tavare S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci U S A. 2013;110(10):4009–14. doi: 10.1073/pnas.1219747110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dunn GP, Old LJ, Schreiber RD. The three es of cancer immunoediting. Annu Rev Immunol. 2004;22(1):329–60. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- 18.Memarnejadian A, Meilleur CE, Shaler CR, Khazaie K, Bennink JR, Schell TD, Haeryfar SMM. Pd-1 blockade promotes epitope spreading in anticancer cd8(+) t cell responses by preventing fratricidal death of subdominant clones to relieve immunodomination. J Immunol. 2017;199(9):3348–59. doi: 10.4049/jimmunol.1700643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Markovic DS, Glass R, Synowitz M, van Rooijen N, Kettenmann H. Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease-2. J Neuropathol Exp Neurol. 2005;64(9):754–62. doi: 10.1097/01.jnen.0000178445.33972.a9. [DOI] [PubMed] [Google Scholar]
- 20.Yeung YT, Bryce NS, Adams S, Braidy N, Konayagi M, McDonald KL, Teo C, Guillemin GJ, Grewal T, Munoz L. P38 mapk inhibitors attenuate pro-inflammatory cytokine production and the invasiveness of human u251 glioblastoma cells. J Neurooncol. 2012;109(1):35–44. doi: 10.1007/s11060-012-0875-7. [DOI] [PubMed] [Google Scholar]
- 21.Rempel SA, Dudas S, Ge SG, Gutierrez JA. Identification and localization of the cytokine sdf1 and its receptor, cxc chemokine receptor 4, to regions of necrosis and angiogenesis in human glioblastoma. Clin Cancer Res. 2000;6:102–11. [PubMed] [Google Scholar]
- 22.Pan Y, Yu Y, Wang X, Zhang T. Tumor-Associated macrophages in tumor immunity. Front Immunol. 2020;11. doi: 10.3389/fimmu.2020.583084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alban TJ, Alvarado AG, Sorensen MD, Bayik D, Volovetz J, Serbinowski E, Mulkearns-Hubert EE, Sinyuk M, Hale JS, Onzi GR, et al. Global immune fingerprinting in glioblastoma patient peripheral blood reveals immune-suppression signatures associated with prognosis. Jci Insight. 2018;3(21). doi: 10.1172/jci.insight.122264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Umemura N, Saio M, Suwa T, Kitoh Y, Bai J, Nonaka K, Ouyang G-F, Okada M, Balazs M, Adany R, et al. Tumor-Infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear m1- and m2-type characteristics. J Leukoc Biol. 2008;83(5):1136–44. doi: 10.1189/jlb.0907611. [DOI] [PubMed] [Google Scholar]
- 25.Zhu X, Fujita M, Snyder LA, Okada H. Systemic delivery of neutralizing antibody targeting ccl2 for glioma therapy. J Neurooncol. 2011;104(1):83–92. doi: 10.1007/s11060-010-0473-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu C-J, Chen C-H, Lin C-Y, Feng L-Y, Lin Y-C, Wei K-C, Huang C-Y, Fang J-Y, Chen P-Y. Ccl5 of glioma-associated microglia/macrophages regulates glioma migration and invasion via calcium-dependent matrix metalloproteinase 2. Neuro-Oncology. 2020;22(2):253–66. doi: 10.1093/neuonc/noz189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jordan JT, Sun W, Hussain SF, DeAngulo G, Prabhu SS, Heimberger AB. Preferential migration of regulatory t cells mediated by glioma-secreted chemokines can be blocked with chemotherapy. Cancer Immunol Immunother. 2008;57(1):123–31. doi: 10.1007/s00262-007-0336-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Humphries W, Wei J, Sampson JH, Heimberger AB. The role of tregs in glioma-mediated immunosuppression: potential target for intervention. Neurosurg Clin N Am. 2010;21(1):125±. doi: 10.1016/j.nec.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De I, Steffen MD, Clark PA, Patros CJ, Sokn E, Bishop SM, Litscher S, Maklakova VI, Kuo JS, Rodriguez FJ, Maklakova VI, et al. Csf1 overexpression promotes high-grade glioma formation without impacting the polarization status of glioma-associated microglia and macrophages. Cancer Res. 2016;76(9):2552–60. doi: 10.1158/0008-5472.CAN-15-2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Butowski N, Colman H, De Groot JF, Omuro AM, Nayak L, Wen PY, Cloughesy TF, Marimuthu A, Haidar S, Perry A, et al. Orally administered colony stimulating factor 1 receptor inhibitor plx3397 in recurrent glioblastoma: an ivy foundation early phase clinical trials consortium phase ii study. Neuro-Oncology. 2016;18(4):557–64. doi: 10.1093/neuonc/nov245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT, Holland EC, Sutton JC, Joyce JA. The tumor microenvironment underlies acquired resistance to csf-1r inhibition in gliomas. Science. 2016;352(6288):352(6288. doi: 10.1126/science.aad3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takenaka MC, Gabriely G, Rothhammer V, Mascanfroni ID, Wheeler MA, Chao C-C, Gutierrez-Vazquez C, Kenison J, Tjon EC, Barroso A, et al. Control of tumor-associated macrophages and t cells in glioblastoma via ahr and cd39. Nat Neurosci. 2019;22(5):729±. doi: 10.1038/s41593-019-0370-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Junnikkala S, Jokiranta TS, Friese MA, Jarva H, Zipfel PF, Meri S. Exceptional resistance of human h2 glioblastoma cells to complement-mediated killing by expression and utilization of factor h and factor h-like protein 1. J Immunol. 2000;164(11):6075–81. doi: 10.4049/jimmunol.164.11.6075. [DOI] [PubMed] [Google Scholar]
- 34.Foernvik K, Maddahi A, Persson O, Osther K, Salford LG, Redebrandt HN. C1-Inactivator is upregulated in glioblastoma. Plos One. 2017;12(9): e0183086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol. 2018;15(7):422–42. doi: 10.1038/s41571-018-0003-5. [DOI] [PubMed] [Google Scholar]
- 36.Larson RC, Maus MV. Recent advances and discoveries in the mechanisms and functions of car t cells. Nat Rev Cancer. 2021;21(3):145–61. doi: 10.1038/s41568-020-00323-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Benmebarek M-R, Karches CH, Cadilha BL, Lesch S, Endres S, Kobold S. Killing mechanisms of chimeric antigen receptor (car) t cells. Int J Mol Sci. 2019;20(6):1283. doi: 10.3390/ijms20061283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, Bleakley M, Brown C, Mgebroff S, Kelly-Spratt KS, et al. Intent-To-Treat leukemia remission by cd19 car t cells of defined formulation and dose in children and young adults. Blood. 2017;129(25):3322–31. doi: 10.1182/blood-2017-02-769208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mintz A, Gibo DM, Slagle-Webb B, Christensen ND, Debinski W. Il-13r alpha 2 is a glioma-restricted receptor for interleukin-13. Neoplasia. 2002;4(5):388–99. doi: 10.1038/sj.neo.7900234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thaci B, Brown CE, Binello E, Werbaneth K, Sampath P, Sengupta S. Significance of interleukin-13 receptor alpha 2-targeted glioblastoma therapy. Neuro-Oncology. 2014;16(10):1304–12. doi: 10.1093/neuonc/nou045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tu M, Wange W, Cai L, Zhu P, Gao Z, Zheng W. Il-13 receptor alpha 2 stimulates human glioma cell growth and metastasis through the src/pi3k/akt/mtor signaling pathway. Tumor Biol. 2016;37(11):14701–09. doi: 10.1007/s13277-016-5346-x. [DOI] [PubMed] [Google Scholar]
- 42.Brown CE, Warden CD, Starr R, Deng X, Badie B, Yuan Y-C, Forman SJ, Barish ME. Glioma IL13Rα2 is associated with mesenchymal signature gene expression and poor patient prognosis. Plos One. 2013;8(10):e77769. doi: 10.1371/journal.pone.0077769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kong S, Sengupta S, Tyler B, Bais AJ, Ma Q, Doucette S, Zhou J, Sahin A, Carter BS, Brem H, et al. Suppression of human glioma xenografts with second-generation il13r-specific chimeric antigen receptor-modified t cells. Clin Cancer Res. 2012;18(21):5949–60. doi: 10.1158/1078-0432.CCR-12-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang W-C, Naranjo A, Starr R, Wagner J, Wright C, et al. Bioactivity and safety of il13r alpha 2-redirected chimeric antigen receptor cd8(+) t cells in patients with recurrent glioblastoma. Clin Cancer Res. 2015;21(18):4062–72. doi: 10.1158/1078-0432.CCR-15-0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, Simpson J, et al. Regression of glioblastoma after chimeric antigen receptor t-cell therapy. N Engl J Med. 2016;375(26):2561–69. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Padfield E, Ellis HP, Kurian KM. Current therapeutic advances targeting egfr and egfrviii in glioblastoma. Front Oncol. 2015;5. doi: 10.3389/fonc.2015.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gan HK, Cvrljevic AN, Johns TG. The epidermal growth factor receptor variant iii (egfrviii): where wild things are altered. Febs J. 2013;280(21):5350–70. doi: 10.1111/febs.12393. [DOI] [PubMed] [Google Scholar]
- 48.Felsberg J, Hentschel B, Kaulich K, Gramatzki D, Zacher A, Malzkorn B, Kamp M, Sabel M, Simon M, Westphal M, et al. Epidermal growth factor receptor variant iii (egfrviii) positivity in egfr-amplified glioblastomas: prognostic role and comparison between primary and recurrent tumors. Clin Cancer Res. 2017;23(22):6846–55. doi: 10.1158/1078-0432.CCR-17-0890. [DOI] [PubMed] [Google Scholar]
- 49.Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, Nace AK, Dentchev T, Thekkat P, Loew A, et al. Rational development and characterization of humanized anti-egfr variant iii chimeric antigen receptor t cells for glioblastoma. Sci Transl Med. 2015;7(275). doi: 10.1126/scitranslmed.aaa4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O’-Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, Martinez-Lage M, Brem S, Maloney E, Shen A, et al. A single dose of peripherally infused egfrviii-directed car t cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399). doi: 10.1126/scitranslmed.aaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, Feldman SA, Lu Y-C, Lu L, Zheng Z, et al. Pilot trial of adoptive transfer of chimeric antigen receptor–transduced T cells targeting EGFRvIII in patients with glioblastoma. J Immunother. 2019;42(4):126–35. doi: 10.1097/CJI.0000000000000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choi BD, Yu X, Castano AP, Bouffard AA, Schmidts A, Larson RC, Bailey SR, Boroughs AC, Frigault MJ, Leick MB, et al. Car-T cells secreting bites circumvent antigen escape without detectable toxicity. Nat Biotechnol. 2019;37(9):1049±. doi: 10.1038/s41587-019-0192-1. [DOI] [PubMed] [Google Scholar]
- 53.Choe JH, Watchmaker PB, Simic MS, Gilbert RD, Li AW, Krasnow NA, Downey KM, Yu W, Carrera DA, Celli A, et al. Synnotch-Car t cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci Transl Med. 2021;13(591). doi: 10.1126/scitranslmed.abe7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Picarda E, Ohaegbulam KC, Zang X. Molecular pathways: targeting b7-h3 (cd276) for human cancer immunotherapy. Clin Cancer Res. 2016;22(14):3425–31. doi: 10.1158/1078-0432.CCR-15-2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang S, Wei W, Zhao Q. B7-H3, a checkpoint molecule, as a target for cancer immunotherapy. Int J Biol Sci. 2020;16(11):1767–73. doi: 10.7150/ijbs.41105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Majzner RG, Theruvath JL, Nellan A, Heitzeneder S, Cui Y, Mount CW, Rietberg SP, Linde MH, Xu P, Rota C, et al. Car t cells targeting b7-h3, a pan-cancer antigen, demonstrate potent preclinical activity against pediatric solid tumors and brain tumors. Clin Cancer Res. 2019;25(8):2560–74. doi: 10.1158/1078-0432.CCR-18-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou Z, Luther N, Ibrahim GM, Hawkins C, Vibhakar R, Handler MH, Souweidane MM. B7-H3, a potential therapeutic target, is expressed in diffuse intrinsic pontine glioma. J Neurooncol. 2013;111(3):257–64. doi: 10.1007/s11060-012-1021-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tang X, Zhao S, Zhang Y, Wang Y, Zhang Z, Yang M, Zhu Y, Zhang G, Guo G, Tong A, et al. B7-H3 as a novel car-t therapeutic target for glioblastoma. Mol Ther-Oncolytics. 2019;14:279–87. doi: 10.1016/j.omto.2019.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tang X, Wang Y, Huang J, Zhang Z, Liu F, Xu J, Guo G, Wang W, Tong A, Zhou L. Administration of b7-h3 targeted chimeric antigen receptor-t cells induce regression of glioblastoma. Signal Transduction Targeted Ther. 2021;6(1). doi: 10.1038/s41392-021-00505-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang J, Zheng M, Zhang Z, Tang X, Chen Y, Peng A, Peng X, Tong A, Zhou L. Interleukin-7-Loaded oncolytic adenovirus improves car-t cell therapy for glioblastoma. Cancer Immunol Immunother. 2021;70(9):2453–65. doi: 10.1007/s00262-021-02856-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ahmed N, Salsman VS, Kew Y, Shaffer D, Powell S, Zhang YJ, Grossman RG, Heslop HE, Gottschalk S. Her2-Specific t cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res. 2010;16(2):474–85. doi: 10.1158/1078-0432.CCR-09-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang JG, Kruse CA, Driggers L, Hoa N, Wisoff J, Allen JC, Zagzag D, Newcomb EW, Jadus MR. Tumor antigen precursor protein profiles of adult and pediatric brain tumors identify potential targets for immunotherapy. J Neurooncol. 2008;88(1):65–76. doi: 10.1007/s11060-008-9534-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pienkowski T, Zielinski CC. Trastuzumab treatment in patients with breast cancer and metastatic cns disease. Ann Oncol. 2010;21(5):917–24. doi: 10.1093/annonc/mdp353. [DOI] [PubMed] [Google Scholar]
- 64.Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, Robertson C, Gray TL, Diouf O, Wakefield A, et al. Her2-Specific chimeric antigen receptor-modified virus-specific t cells for progressive glioblastoma a phase 1 dose-escalation trial. Jama Oncol. 2017;3(8):1094–101. doi: 10.1001/jamaoncol.2017.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vitanza NA, Johnson AJ, Wilson AL, Brown C, Yokoyama JK, Kunkele A, Chang CA, Rawlings-Rhea S, Huang W, Seidel K, et al. Locoregional infusion of her2-specific car t cells in children and young adults with recurrent or refractory cns tumors: an interim analysis. Nat Med. 2021;27(9):1544±. doi: 10.1038/s41591-021-01404-8. [DOI] [PubMed] [Google Scholar]
- 66.Bielamowicz K, Fousek K, Byrd TT, Samaha H, Mukherjee M, Aware N, Wu M-F, Orange JS, Sumazin P, Man T-K, et al. Trivalent car t cells overcome interpatient antigenic variability in glioblastoma. Neuro-Oncology. 2018;20(4):506–18. doi: 10.1093/neuonc/nox182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer. 2014;14(8):559–67. doi: 10.1038/nrc3770. [DOI] [PubMed] [Google Scholar]
- 68.Russell SJ, Barber GN. Oncolytic viruses as antigen-agnostic cancer vaccines. Cancer Cell. 2018;33(4):599–605. doi: 10.1016/j.ccell.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bartlett DL, Liu Z, Sathaiah M, Ravindranathan R, Guo Z, He Y, Guo ZS. Oncolytic viruses as therapeutic cancer vaccines. Mol Cancer. 2013;12(1):1–16. doi: 10.1186/1476-4598-12-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chiocca EA, Rabkin SD. Chiocca EA and Rabkin SD. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol Res. 2014;2(4):295–300. doi: 10.1158/2326-6066.CIR-14-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brown MC, Holl EK, Boczkowski D, Dobrikova E, Mosaheb M, Chandramohan V, Bigner DD, Gromeier M, Nair SK. Cancer immunotherapy with recombinant poliovirus induces ifn-dominant activation of dendritic cells and tumor antigen-specific ctls. Sci Transl Med. 2017;9(408). doi: 10.1126/scitranslmed.aan4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hardcastle J, Mills L, Malo CS, Jin F, Kurokawa C, Geekiyanage H, Schroeder M, Sarkaria J, Johnson AJ, Galanis E. Immunovirotherapy with measles virus strains in combination with anti-pd-1 antibody blockade enhances antitumor activity in glioblastoma treatment. Neuro-Oncology. 2017;19(4):493–502. doi: 10.1093/neuonc/now179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rajaraman S, Canjuga D, Ghosh M, Codrea MC, Sieger R, Wedekink F, Tatagiba M, Koch M, Lauer UM, Nahnsen S, et al. Measles virus-based treatments trigger a pro-inflammatory cascade and a distinctive immunopeptidome in glioblastoma. Mol Ther-Oncolytics. 2019;12:147–61. doi: 10.1016/j.omto.2018.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kiyokawa J, Wakimoto H. Preclinical and clinical development of oncolytic adenovirus for the treatment of malignant glioma. Oncolytic Virother. 2019;8:27–37. doi: 10.2147/OV.S196403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Alessandrini F, Menotti L, Avitabile E, Appolloni I, Ceresa D, Marubbi D, Campadelli-Fiume G, Malatesta P. Eradication of glioblastoma by immuno-virotherapy with a retargeted oncolytic hsv in a preclinical model. Oncogene. 2019;38(23):4467–79. doi: 10.1038/s41388-019-0737-2. [DOI] [PubMed] [Google Scholar]
- 76.Zhu Z, Mesci P, Bernatchez JA, Gimple RC, Wang X, Schafer ST, Wettersten HI, Beck S, Clark AE, Wu Q, et al. Zika virus targets glioblastoma stem cells through a sox2-integrin alpha(v)beta(5) axis. Cell Stem Cell. 2020;26(2):187±. doi: 10.1016/j.stem.2019.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pol J, Buque A, Aranda F, Bloy N, Cremer I, Eggermont A, Erbs P, Fucikova J, Galon J, Limacher J-M, et al. Trial watch—Oncolytic viruses and cancer therapy. Oncoimmunology. 2016;5(2):e1117740. doi: 10.1080/2162402X.2015.1117740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fulci G, Chiocca EA. Oncolytic viruses for the therapy of brain tumors and other solid malignancies: a review. Front Biosci-Landmark. 2003;8:E346–E360. doi: 10.2741/976. [DOI] [PubMed] [Google Scholar]
- 79.Chiocca EA, Abbed KM, Tatter S, Louis DN, Hochberg FH, Barker F, Kracher J, Grossman SA, Fisher JD, Carson K, et al. A phase i open-label, dose-escalation, multi-institutional trial of injection with an e1b-attenuated adenovirus, onyx-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Mol Ther. 2004;10(5):958–66. doi: 10.1016/j.ymthe.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 80.Kemeny N, Brown K, Covey A, Kim T, Bhargava A, Brody L, Guilfoyle B, Haag NP, Karrasch M, Glasschroeder B, et al. Phase i, open-label, dose-escalating study of a genetically engineered herpes simplex virus, nv1020, in subjects with metastatic colorectal carcinoma to the liver. Hum Gene Ther. 2006;17(12):1214–24. doi: 10.1089/hum.2006.17.1214. [DOI] [PubMed] [Google Scholar]
- 81.Cloughesy TF, Landolfi J, Vogelbaum MA, Ostertag D, Elder JB, Bloomfield S, Carter B, Chen CC, Kalkanis SN, Kesari S, et al. Durable complete responses in some recurrent high grade glioma patients treated with toca 511+toca fc. Neuro-Oncology. 2018;20(10):1383–92. doi: 10.1093/neuonc/noy075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fueyo J, Alemany R, Gomez-Manzano C, Fuller GN, Khan A, Conrad CA, Liu TJ, Jiang H, Lemoine MG, Suzuki K, et al. Preclinical characterization of the antiglioma activity of a tropism-enhanced adenovirus targeted to the retinoblastoma pathway. J Natl Cancer Inst. 2003;95(9):652–60. doi: 10.1093/jnci/95.9.652. [DOI] [PubMed] [Google Scholar]
- 83.Lang FF, Conrad C, Gomez-Manzano C, Yung WKA, Sawaya R, Weinberg JS, Prabhu SS, Rao G, Fuller GN, Aldape KD, et al. Phase i study of dnx-2401 (delta-24-rgd) oncolytic adenovirus: replication and immunotherapeutic effects in recurrent malignant glioma. J Clin Oncol. 2018;36(14):1419±. doi: 10.1200/JCO.2017.75.8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Geletneky K, Hajda J, Angelova AL, Leuchs B, Capper D, Bartsch AJ, Neumann J-O, Schoening T, Huesing J, Beelte B, et al. Oncolytic h-1 parvovirus shows safety and signs of immunogenic activity in a first phase i/iia glioblastoma trial. Mol Ther. 2017;25(12):2620–34. doi: 10.1016/j.ymthe.2017.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gromeier M, Alexander L, Wimmer E. Internal ribosomal entry site substitution eliminates neurovirulence in intergeneric poliovirus recombinants. Proc Natl Acad Sci U S A. 1996;93(6):2370–75. doi: 10.1073/pnas.93.6.2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mendelsohn CL, Wimmer E, Racaniello VR. Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell. 1989;56(5):855–65. doi: 10.1016/0092-8674(89)90690-9. [DOI] [PubMed] [Google Scholar]
- 87.Chandramohan V, Bryant JD, Piao H, Keir ST, Lipp ES, Lefaivre M, Perkinson K, Bigner DD, Gromeier M, McLendon RE. Validation of an immunohistochemistry assay for detection of cd155, the poliovirus receptor, in malignant gliomas. Arch Pathol & Lab Med. 2017;141(12):1697–704. doi: 10.5858/arpa.2016-0580-OA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Takai Y, Miyoshi J, Ikeda W, Ogita H. Nectins and nectin-like molecules: roles in contact inhibition of cell movement and proliferation. Nat Rev Mol Cell Biol. 2008;9(8):603–15. doi: 10.1038/nrm2457. [DOI] [PubMed] [Google Scholar]
- 89.Desjardins A, Gromeier M, Herndon JE II, Beaubier N, Bolognesi DP, Friedman AH, Friedman HS, McSherry F, Muscat AM, Nair S, et al. Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med. 2018;379(2):150–61. doi: 10.1056/NEJMoa1716435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Advani SJ, Markert JM, Sood RF, Samuel S, Gillespie GY, Shao MY, Roizman B, Weichselbaum RR. Increased oncolytic efficacy for high-grade gliomas by optimal integration of ionizing radiation into the replicative cycle of hsv-1. Gene Ther. 2011;18(11):1098–102. doi: 10.1038/gt.2011.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14(9):642±. doi: 10.1038/nrd4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mineta T, Rabkin SD, Yazaki T, Hunter WD, Martuza RL. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat Med. 1995;1(9):938–43. doi: 10.1038/nm0995-938. [DOI] [PubMed] [Google Scholar]
- 93.Friedman GK, Johnston JM, Bag AK, Bernstock JD, Li R, Aban I, Kachurak K, Nan L, Kang KD, Totsch S, et al. Oncolytic hsv-1 g207 immunovirotherapy for pediatric high-grade gliomas. N Engl J Med. 2021;384(17):1613–22. doi: 10.1056/NEJMoa2024947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sugawara K, Iwai M, Yajima S, Tanaka M, Yanagihara K, Seto Y, Todo T. Efficacy of a third-generation oncolytic herpes virus g47 delta in advanced stage models of human gastric cancer. Mol Ther-Oncolytics. 2020;17:205–15. doi: 10.1016/j.omto.2020.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fukuhara H, Ino Y, Todo T. Oncolytic virus therapy: a new era of cancer treatment at dawn. Cancer Sci. 2016;107(10):1373–79. doi: 10.1111/cas.13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Taguchi S, Fukuhara H, Todo T. Oncolytic virus therapy in japan: progress in clinical trials and future perspectives. Jpn J Clin Oncol. 2019;49(3):201–09. doi: 10.1093/jjco/hyy170. [DOI] [PubMed] [Google Scholar]
- 97.Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, Ashby L, Mechtler L, Goldlust SA, Iwamoto F, et al. Rindopepimut with temozolomide for patients with newly diagnosed, egfrviii-expressing glioblastoma (act iv): a randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017;18(10):1373–85. doi: 10.1016/S1470-2045(17)30517-X. [DOI] [PubMed] [Google Scholar]
- 98.Liau LM, Ashkan K, Tran DD, Campian JL, Trusheim JE, Cobbs CS, Heth JA, Salacz M, Taylor S, D’-Andre SD, et al. First results on survival from a large phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Transl Med. 2018;16 (1) 142 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Narita Y, Arakawa Y, Yamasaki F, Nishikawa R, Aoki T, Kanamori M, Nagane M, Kumabe T, Hirose Y, Ichikawa T, et al. A randomized, double-blind, phase iii trial of personalized peptide vaccination for recurrent glioblastoma. Neuro Oncol. 2019;21(3):348–59. doi: 10.1093/neuonc/noy200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Weller M, Kaulich K, Hentschel B, Felsberg J, Gramatzki D, Pietsch T, Simon M, Westphal M, Schackert G, Tonn JC, et al. Assessment and prognostic significance of the epidermal growth factor receptor viii mutation in glioblastoma patients treated with concurrent and adjuvant temozolomide radiochemotherapy. Int J Cancer. 2014;134(10):2437–47. doi: 10.1002/ijc.28576. [DOI] [PubMed] [Google Scholar]
- 101.Schuster J, Lai RK, Recht LD, Reardon DA, Paleologos NA, Groves MD, Mrugala MM, Jensen R, Baehring JM, Sloan A, et al. A phase ii, multicenter trial of rindopepimut (cdx-110) in newly diagnosed glioblastoma: the act iii study. Neuro-Oncology. 2015;17(6):854–61. doi: 10.1093/neuonc/nou348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, Gilbert MR, Herndon JE II, McLendon RE, Mitchell DA, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant iii peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28(31):4722–29. doi: 10.1200/JCO.2010.28.6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sampson JH, Aldape KD, Archer GE, Coan A, Desjardins A, Friedman AH, Friedman HS, Gilbert MR, Herndon JE, McLendon RE, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate egfrviii-expressing tumor cells in patients with glioblastoma. Neuro-Oncology. 2011;13(3):324–33. doi: 10.1093/neuonc/noq157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J, Menn O, Osswald M, Oezen I, Ott M, et al. A vaccine targeting mutant idh1 induces antitumour immunity. Nature. 2014;512(7514):324±. doi: 10.1038/nature13387. [DOI] [PubMed] [Google Scholar]
- 105.Platten M, Schilling D, Bunse L, Wick A, Bunse T, Riehl D, Green E, Sanghvi K, Karapanagiotou-Schenkel I, Harting I, et al. Os6.4 noa-16: a first-in-man multicenter phase i clinical trial of the German neurooncology working group evaluating a mutation-specific peptide vaccine targeting idh1r132h in patients with newly diagnosed malignant astrocytomas. Neuro-Oncology. 2018;20:iii226–iii227. doi: 10.1093/neuonc/noy139.041. [DOI] [Google Scholar]
- 106.Fenstermaker RA, Ciesielski MJ, Qiu J, Yang N, Frank CL, Lee KP, Mechtler LR, Belal A, Ahluwalia MS, Hutson AD. Clinical study of a survivin long peptide vaccine (survaxm) in patients with recurrent malignant glioma. Cancer Immunol Immunother. 2016;65(11):1339–52. doi: 10.1007/s00262-016-1890-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ahluwalia M, Reardon D, Abad A, Curry W, Wong E, Peereboom D, Belal A, Qiu J, Mogensen K, Schilero C, et al. Atim-41. Phase ii trial of a survivin vaccine (survaxm) for newly diagnosed glioblastoma. Neuro-Oncology. 2018;20(Suppl 6):vi10–vi11. doi: 10.1093/neuonc/noy148.036. [DOI] [Google Scholar]
- 108.Andrews DW, Judy KD, Scott CB, Garcia S, Hooper DC. Phase ib clinical trial of igv-001 for patients with newly diagnosed glioblastoma. Clin Cancer Res. 2021;27(7):clincanres.3805.2020. doi: 10.1158/1078-0432.CCR-20-3805. [DOI] [PubMed] [Google Scholar]
- 109.Terasaki M, Soichiro S, Narita Y, Fujimaki T, Aoki T. Phase I trial of a personalized peptide vaccine for patients positive for human leukocyte antigen--A24 with recurrent or progressive glioblastoma multiforme. J Clin Oncol. 2011;29(3):337–44. doi: 10.1200/JCO.2010.29.7499. [DOI] [PubMed] [Google Scholar]
- 110.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 111.Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanovic S, Gouttefangeas C, Platten M, Tabatabai G, Dutoit V, van der Burg SH, et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature. 2019;565(7738):240±. doi: 10.1038/s41586-018-0810-y. [DOI] [PubMed] [Google Scholar]
- 112.Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, Oliveira G, Giobbie-Hurder A, Felt K, Gjini E, et al. Neoantigen vaccine generates intratumoral t cell responses in phase ib glioblastoma trial. Nature. 2019;565(7738):234±. doi: 10.1038/s41586-018-0792-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Vik-Mo EO, Nyakas M, Mikkelsen BV, Moe MC, Due-Tonnesen P, Suso EMI, Saeboe-Larssen S, Sandberg C, Brinchmann JE, Helseth E, et al. Therapeutic vaccination against autologous cancer stem cells with mrna-transfected dendritic cells in patients with glioblastoma. Cancer Immunol Immunother. 2013;62(9):1499–509. doi: 10.1007/s00262-013-1453-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Inoges S, Tejada S, de Cerio A-D, Perez-Larraya JG, Espinos J, Idoate MA, Dominguez PD, de Eulate RG, Aristu J, Bendandi M, et al. A phase ii trial of autologous dendritic cell vaccination and radiochemotherapy following fluorescence-guided surgery in newly diagnosed glioblastoma patients. J Transl Med. 2017;15. doi: 10.1186/s12967-017-1202-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Buchroithner J, Erhart F, Pichler J, Widhalm G, Preusser M, Stockhammer G, Nowosielski M, Iglseder S, Freyschlag CE, Oberndorfer S, et al. Audencel immunotherapy based on dendritic cells has no effect on overall and progression-free survival in newly diagnosed glioblastoma: a phase ii randomized trial. Cancers. 2018;10(10):372. doi: 10.3390/cancers10100372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Patente TA, Pinho MP, Oliveira AA, Evangelista GCM, Bergami-Santos PC, Barbuto JAM. Human dendritic cells: their heterogeneity and clinical application potential in cancer immunotherapy. Front Immunol. 2019;9. doi: 10.3389/fimmu.2018.03176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rutkowski S, De Vleeschouwer S, Kaempgen E, Wolff JEA, Kuhl J, Demaerel P, Warmuth-Metz M, Flamen P, Van Calenbergh F, Plets C, et al. Surgery and adjuvant dendritic cell-based tumour vaccination for patients with relapsed malignant glioma, a feasibility study. Br J Cancer. 2004;91(9):1656–62. doi: 10.1038/sj.bjc.6602195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Liau LM, Prins RM, Kiertscher SM, Odesa SK, Kremen TJ, Giovannone AJ, Lin JW, Chute DJ, Mischel PS, Cloughesy TF, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial t-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res. 2005;11(15):5515–25. doi: 10.1158/1078-0432.CCR-05-0464. [DOI] [PubMed] [Google Scholar]
- 119.Yamanaka R, Homma J, Yajima N, Tsuchiya N, Sano M, Kobayashi T, Yoshida S, Abe T, Narita M, Takahashi M, et al. Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: results of a clinical phase i/ii trial. Clin Cancer Res. 2005;11(11):4160–67. doi: 10.1158/1078-0432.CCR-05-0120. [DOI] [PubMed] [Google Scholar]
- 120.Phuphanich S, Wheeler CJ, Rudnick JD, Mazer M, Wang H, Nuno MA, Richardson JE, Fan X, Ji J, Chu RM, et al. Phase i trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol Immunother. 2013;62(1):125–35. doi: 10.1007/s00262-012-1319-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wen PY, Reardon DA, Armstrong TS, Phuphanich S, Aiken RD, Landolfi JC, Curry WT, Zhu J-J, Glantz M, Peereboom DM, et al. A randomized double-blind placebo-controlled phase ii trial of dendritic cell vaccine ict-107 in newly diagnosed patients with glioblastoma. Clin Cancer Res. 2019;25(19):5799–807. doi: 10.1158/1078-0432.CCR-19-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shen S, Chen L, Liu J, Yang L, Zhang M, Wang L, Zhang R, Uemura Y, Wu Q, Yu X, et al. Current state and future of co-inhibitory immune checkpoints for the treatment of glioblastoma. Cancer Biol & Med. 2020;17(3):555–68. doi: 10.20892/j.issn.2095-3941.2020.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhang N, Wei L, Ye M, Kang C, You H. Treatment progress of immune checkpoint blockade therapy for glioblastoma. Front Immunol. 2020;11:592612. doi: 10.3389/fimmu.2020.592612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8(9):1069–86. doi: 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- 125.Zappasodi R, Merghoub T, Wolchok JD. Emerging concepts for immune checkpoint blockade-based combination therapies. Cancer Cell. 2018;33(4):581–98. doi: 10.1016/j.ccell.2018.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373(19):1803–13. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373(17):1627–39. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Omuro A, Vlahovic G, Lim M, Sahebjam S, Baehring J, Cloughesy T, Voloschin A, Ramkissoon SH, Ligon KL, Latek R, et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase i cohorts of checkmate 143. Neuro-Oncology. 2018;20(5):674–86. doi: 10.1093/neuonc/nox208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, Baehring J, Ahluwalia MS, Roth P, Baehr O, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma the checkmate 143 phase 3 randomized clinical trial. Jama Oncol. 2020;6(7):1003–10. doi: 10.1001/jamaoncol.2020.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, Yung WKA, Paleologos N, Nicholas MK, Jensen R, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27(28):4733–40. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 132.Sahebjam S, Forsyth PA, Tran ND, Arrington JA, Macaulay R, Etame AB, Walko CM, Boyle T, Peguero EN, Jaglal M, et al. Hypofractionated stereotactic re-irradiation with pembrolizumab and bevacizumab in patients with recurrent high-grade gliomas: results from a phase i study. Neuro-Oncology. 2021;23(4):677–86. doi: 10.1093/neuonc/noaa260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, Wang AC, Ellingson BM, Rytlewski JA, Sanders CM, et al. Neoadjuvant anti-pd-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25(3):477±. doi: 10.1038/s41591-018-0337-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Fife BT, Bluestone JA. Control of peripheral t-cell tolerance and autoimmunity via the ctla-4 and pd-1 pathways. Immunol Rev. 2008;224:166–82. doi: 10.1111/j.1600-065X.2008.00662.x. [DOI] [PubMed] [Google Scholar]
- 135.Harris-Bookman S, Mathios D, Martin AM, Xia Y, Kim E, Xu H, Belcaid Z, Polanczyk M, Barberi T, Theodros D, et al. Expression of lag-3 and efficacy of combination treatment with anti-lag-3 and anti-pd-1 monoclonal antibodies in glioblastoma. Int J Cancer. 2018;143(12):3201–08. doi: 10.1002/ijc.31661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kim JE, Patel MA, Mangraviti A, Kim ES, Theodros D, Velarde E, Liu A, Sankey EW, Tam A, Xu H, et al. Combination therapy with anti-pd-1, anti-tim-3, and focal radiation results in regression of murine gliomas. Clin Cancer Res. 2017;23(1):124–36. doi: 10.1158/1078-0432.CCR-15-1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhang M, Choi J, Lim M. Advances in immunotherapies for gliomas. Curr Neurol Neurosci Rep. 2022. doi: 10.1007/s11910-022-01176-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, Bordbar D, Shan D, Samanamud J, Mahajan A, et al. Immune and genomic correlates of response to anti-pd-1 immunotherapy in glioblastoma. Nat Med. 2019;25(3):462±. doi: 10.1038/s41591-019-0349-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Grossman SA, Ye X, Lesser G, Sloan A, Carraway H, Desideri S, Piantadosi S, Consortium NC. Immunosuppression in patients with high-grade gliomas treated with radiation and temozolomide. Clin Cancer Res. 2011;17(16):5473–80. doi: 10.1158/1078-0432.CCR-11-0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pitter KL, Tamagno I, Alikhanyan K, Hosni-Ahmed A, Pattwell SS, Donnola S, Dai C, Ozawa T, Chang M, Chan TA, et al. Corticosteroids compromise survival in glioblastoma. Brain. 2016;139:1458–71. doi: 10.1093/brain/aww046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chongsathidkiet P, Jackson C, Koyama S, Loebel F, Cui X, Farber SH, Woroniecka K, Elsamadicy AA, Dechant CA, Kemeny HR, et al. Sequestration of t cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med. 2018;24(9):1459±. doi: 10.1038/s41591-018-0135-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yi JS, Cox MA, Zajac AJ. T-Cell exhaustion: characteristics, causes and conversion. Immunology. 2010;129(4):474–81. doi: 10.1111/j.1365-2567.2010.03255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. ‘Off-The-Shelf’ allogeneic car t cells: development and challenges. Nat Rev Drug Discov. 2020;19(3):185–99. doi: 10.1038/s41573-019-0051-2. [DOI] [PubMed] [Google Scholar]