

Abstract
Inflammation is a pathological hallmark associated with bacterial and viral infections, autoimmune diseases, genetic disorders, obesity and diabetes, as well as environmental stresses including physical and chemical trauma. Among numerous proteins regulating proinflammatory signaling, very few such as Protein kinase R (PKR), have been shown to play an all-pervading role in inflammation induced by varied stimuli. PKR was initially characterized as an interferon-inducible gene activated by viral double-stranded RNA with a role in protein translation inhibition. However, it has become increasingly clear that PKR is involved in multiple pathways that promote inflammation in response to stress activation, both dependent on and independent of its cellular protein activator PACT. In this review, we discuss the signaling pathways that contribute to the initiation of inflammation, including Toll-like receptor, interferon, and RIG-I-like receptor signaling, as well as inflammasome activation. We go on to discuss the specific roles that PKR and PACT play in such proinflammatory signaling, as well as in metabolic syndrome- and environmental stress-induced inflammation.
Keywords: PACT, PKR, inflammation, inflammasome, RIG-I-like receptors, metaflammation
INTRODUCTION
The cellular response to stimuli that threaten homeostasis involves the induction of programs aimed at the elimination of or adaptation to the continuous presence of those stimuli. These programs often involve the upregulation of genes and increased synthesis of gene products associated with recovery or adaptation [1]. These are in turn accompanied by the inhibition of general protein synthesis to route cellular resources towards these stress response programs [2,3]. Inflammatory signaling is often activated if these programs are insufficient to manage the stimuli’s damaging effects, and cell death mechanisms are engaged if the negative effects cannot be overcome by the adaptive stress response programs, which are unique to individual stress conditions and are reviewed elsewhere [4].
The inhibition of protein synthesis in response to diverse stimuli is mediated by one of four serine/threonine kinases that phosphorylate the α subunit of the eukaryotic initiation factor 2 (eIF2α) [5]. It is well known that stimuli-induced phosphorylation of eIF2α acts a signaling mechanism that initiates a cellular response known as the Integrated Stress Response (ISR) [6]. However, functions of the eIF2α kinases independent of their roles in the ISR have also been reported [7]. Of the eIF2α kinases, protein kinase R (PKR) was initially discovered as an interferon-inducible gene [8] and characterized as the eIF2α kinase activated in response to double-stranded RNA, a common intermediate in several viral infections [9]. Since then, PKR has also been shown to be activated in response to stimuli such as oxidative stress, serum deprivation, heat shock, and ER stress, among others [10–13]. It has also become increasingly apparent that PKR plays an integral role in the activation and initiation of pro-inflammatory signaling in response to pathogens or environmental stressors through Toll-like receptor (TLR)-, nuclear factor-κB (NF-κB)-, and inflammasome-activated signaling pathways [14,15].
While dsRNA can bind to and activate PKR directly [16], activation of PKR in response to several other stress-inducing stimuli depends on its interaction with a cellular protein, the protein activator of PKR (PACT) [17]. The phosphorylation of PACT at serine-287 under stress conditions increases its interaction with PKR, resulting in the activation of PKR, subsequent phosphorylation of eIF2α, and activation of downstream signaling [18]. Recent studies have also elucidated PKR-independent functions of PACT; direct interaction with the retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs) RIG-I, melanoma differentiation-associated gene 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2) is integral to the activation of interferon signaling in response to host cell infection by various viruses of significant consequence to human health [19,20].
In this review, we will highlight emerging evidence that squarely places PKR and PACT in the induction of inflammatory signaling as part of the cellular response to pathogens or chronic physiological stress conditions. We will conclude with reflections on how therapeutic intervention of PKR, PACT or PACT-PKR mediated signaling may be beneficial in various pathological conditions in which their dysregulation has been implicated.
INFLAMMATION: NUTS AND BOLTS
Inflammation is a defense mechanism evolved in higher organisms to respond to extreme insults, such as infection or dramatic changes in pH or temperature that threaten to disrupt homeostasis at the level of the cell, tissue, or the whole organism [21]. The detection of such insults results in the activation of pro-inflammatory gene expression programs. These gene expression programs are typically driven by NF-κB, interferon regulatory factor (IRF), Janus-activated kinase (JAK)-signal transducer and activator of transcription (STAT), or mitogen-activated protein kinase (MAPK)-mediated signaling cascades (Figure 1) [21]. Activation of these pathways results in the synthesis and secretion of inflammatory mediators such as cytokines and chemokines. These mediators together exert influence on the local vasculature to increase blood flow and blood vessel permeability, actively facilitating the passage of plasma and specific mediator-responsive effector cells into affected sites [21–23].
Figure 1.
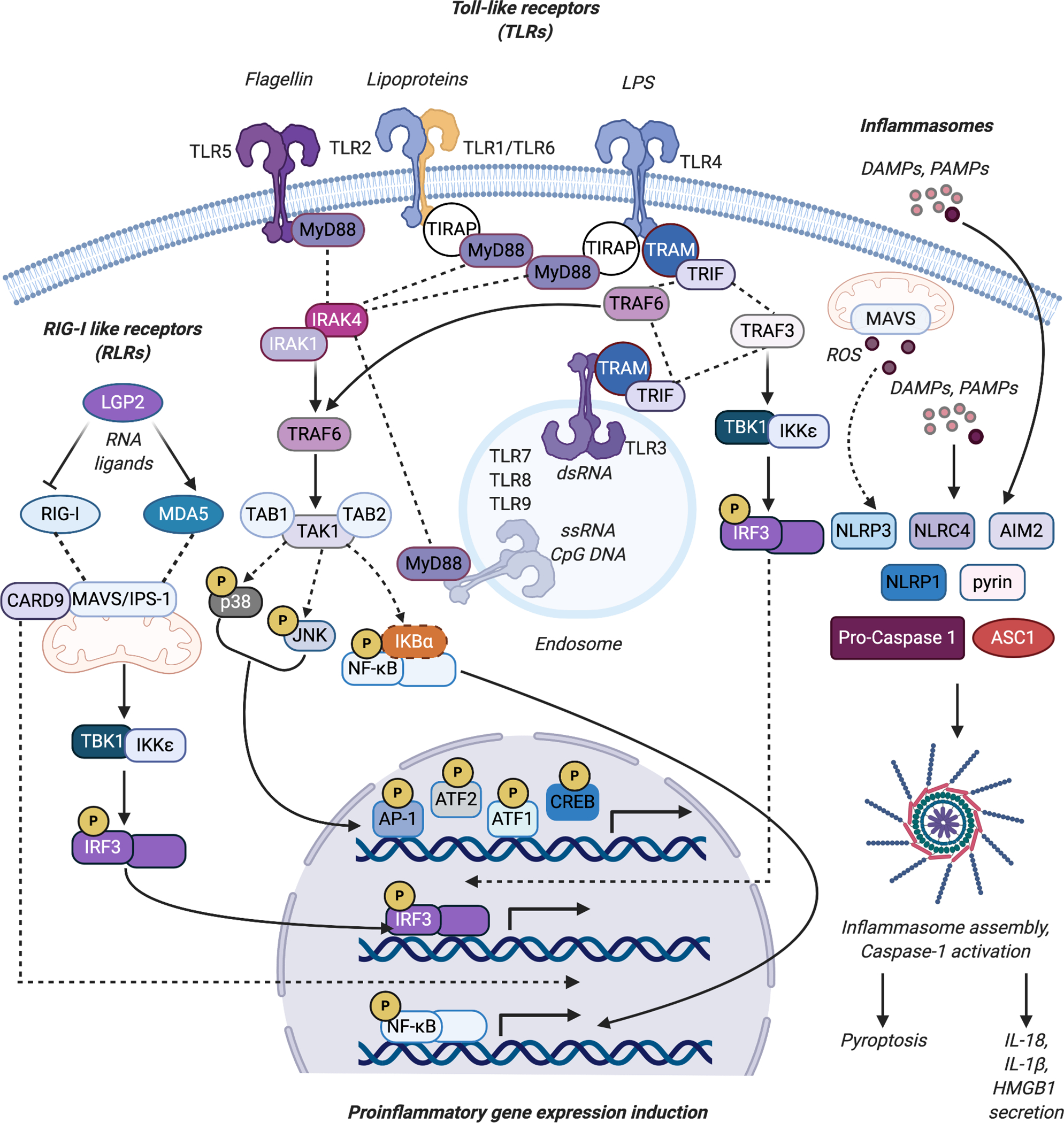
Inflammatory signaling pathways induced by PRRs such as the TLRs and RLRs, as well as the inflammasomes. The binding of cognate ligands to cell surface TLRs (TLR1, TLR2, TLR4, TLR5, TLR6 and TLR10 (not depicted)) and intracellular TLRs (TLR3, TLR7, TLR8, and TLR9) associated with endosomes results in the recruitment of adaptor proteins such as MyD88, TIRAP, TRAM, and TRIF. MyD88 in turn forms a complex with IRAK4 and IRAK1 resulting in IRAK1 activation. This leads to TRAF6-mediated TAK1 and MAPK signaling activation resulting in p38 and JNK -mediated activation of AP-1, ATF1, ATF2, and CREB driven pro-inflammatory signaling. TAK1 activation also leads to nuclear translocation of NF-κB and transcription of KB target genes. TLR engagement can also result in the stimulation of IRF3-driven type I IFN induction through TRAF3-mediated activation of TBK1 and IKKε. The binding of RNA ligands to the RIG-I like receptor (RLR) proteins RIG-I and MDA5 triggers their oligomerization and increased interaction with the adaptor protein MAVS/IPS-1. This leads to TBK1-IKKε-mediated activation of IRF3 and CARD9-mediated activation of NF-κB activity. LGP2 suppresses RIG-I and activates MDA-5-mediated signaling. Recognition of DAMPs and PAMPs by the sensors NLRP1, NLRP3, AIM2, NLRC4 or pyrin results in the assembly of the multiprotein complexed inflammasome, consisting of the oligomerized sensor, the adaptor protein ASC during NLRP3 inflammasome assembly, and pro-caspase 1. MAVS/IPS-1 also facilitates NLRP3 mitochondrial localization and NLRP3 inflammasome activation associated with ROS generation. Caspase 1 activation results in IL-1β and IL-18 maturation from their inactive forms, and stimulates pyroptotic cell death, releasing IL-1β and IL-18 and alarmins like HMGB1 into the extracellular environment. Figure was created using Biorender.com.
The resolution of the inflammatory response upon the successful elimination of the inflammatory insult is achieved at the intracellular level through the expression of feedback regulators of inflammatory signaling [24], and at the intercellular level through the actions of anti-inflammatory soluble mediators such as resolvins, protectins, and maresins [25]. These soluble mediators stop the further recruitment of specialized effector cells and facilitate tissue remodeling via repair and removal of damaged tissue and cellular debris. Conversely, a failure of the acute innate phase inflammatory response to eliminate the inflammatory stimulus engages more specialized effector cells of the adaptive immune system, such as lymphocytes [26]. The persistent inability of these two phases to clear the primary inducers of the inflammatory response, or the continuous presence of such inducers, results in a chronic inflammatory state, which is at the core of several pathological conditions [27].
The presence of pathogenic insults is detected by a variety of plasma membrane or cytosolic receptors collectively referred to as pattern recognition receptors (PRRs), which include RLRs and Toll-like receptors (TLRs), and the cytoplasmic signaling complex, the inflammasome (Figure 1) [28,29]. PRRs recognize a broad range of pathogen-associated molecular patterns (PAMPs) of bacterial, fungal, or viral origin, such as lipopolysaccharide (LPS), CpG DNA, flagellin, or dsRNA [28]. PAMPs are detected by TLRs on the extracellular surface or contained within endosomal compartments, while RLRs, PKR, and inflammasomes function in the cytosol. Material released from dying cells, such as the nuclear protein high-mobility group box protein 1 (HMGB1) categorized as damage-associated molecular patterns (DAMPs), and heat shock proteins (HSPs) can also act as PRR ligands to stimulate proinflammatory signaling [30,31].
Toll-like Receptors (TLRs)
TLRs are type I transmembrane proteins and contain a common extracellular leucine-rich repeat (LRR) motif as well as transmembrane and intracellular Toll and IL-1 (TIR) domains [32]. Ten TLRs have been identified in humans, with TLR1, 2, 4, 5, 6, and 10 expressed on the plasma membrane and recognizing microbial entities, and TLR3, 7, 8, and 9 expressed on endolysosomal membranes and recognizing nucleic acids [33]. The binding of ligands to their cognate TLR receptors triggers TIR-mediated interactions with adaptor proteins myeloid differentiation primary response protein 88 (MyD88), TIR domain-containing adaptor (TIRAP), TIRAP-inducing interferon-β (TRIF), and translocation chain-associated membrane protein (TRAM) [34]. MyD88 and TIRAP, in turn, trigger a signaling cascade involving interleukin 1 receptor-associated kinase 1 (IRAK1), IRAK2, IRAK4, tumor necrosis factor receptor-associated factor 6 (TRAF6), and transforming growth factor β-activated kinase 1 (TAK1) [34]. This culminates in TAK1-mediated activation of NF-κB and MAPK (c-Jun N-terminal kinase [JNK] and p38) activity and the induction of pro-inflammatory cytokines such as interleukin-1β (IL-1β), IL-6, and tumor necrosis factor α (TNFα), chemokines such as monocyte chemoattractant protein 1 (MCP1), and proinflammatory effector enzymes such as inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2), among others [35,36]. Signaling via TRIF and TRAM also results in TRAF6-mediated stimulation of NF-κB transcriptional activity, and additionally results in TRAF3-mediated activation of IRF3 [34]. IRF3 subsequently homodimerizes or heterodimerizes with IRF7 and translocates to the nucleus, where it induces the transcription of genes encoding the type I interferon (IFNα/IFNβ) cytokines [37,38]. The interferons (IFNs) are major components of the inflammatory response to viral infection [39]. IFNs activate interferon-stimulated genes (ISGs) that include several effector proteins, such as PKR or 2’–5’-oligoadenylate synthetase 2 (OAS2), which limit viral proliferation and activity within the host cell [39].
RIG-I-like receptors (RLRs)
The second group of PRRs, the RLRs (RIG-I, MDA5, and LGP2), are a group of cytosolic nucleic acid sensors that trigger the induction of type I interferon expression and NF-κB activation upon interaction with immunostimulatory RNA [40]. Each RLR except for LGP2 contains an N-terminal caspase activation and recruitment domain (CARD), in addition to their common helicase and C-terminal domains [41]. The binding of cognate dsRNA ligands to RIG-I/MDA5 triggers the oligomerization of their CARD domains [42]. This, in turn, facilitates CARD-CARD interaction with the signaling hub mitochondrial antiviral signaling protein (MAVS)/interferon β promoter stimulator 1 (IPS-1)/virus-induced signaling adaptor (VISA), resulting in IκB kinase ε (IKKε)/TRAF family member-associated NF-κB activator-binding kinase 1 (TBK1)-dependent phosphorylation and activation of IRF3 [37,42]. RLR stimulation also results in NF-κB activation through MAVS and CARD9, and subsequent NF-κB-driven inflammatory gene expression [42]. RLR activity is modulated by post-translational modifications such as phosphorylation and small ubiquitin-like modification (SUMOylation), as well as by interactions with the dsRNA binding protein, PACT [43–46]. The interaction of PACT with all three members of the RLR family have been shown to be an essential component of the cellular response to viruses that are of significant clinical consequence to human health, such as the Ebola virus (EBOV) or the herpes simplex virus 1 (HSV1) [47–49]. The mechanisms by which PACT augments the activities of RLRs will be discussed in greater detail in this review.
The inflammasome
The third group of signaling complexes relevant to this review is the multiprotein complex inflammasome, which elicits robust immune responses including the secretion of proinflammatory cytokines and pyroptosis [50]. The prototypical inflammasome consists of a (1) receptor protein which can be a member of the nucleotide-binding oligomerization (NOD) protein family, leucine-rich repeat (LRR)-containing protein (NLR) family members, absent in melanoma 2 (AIM2) like receptors (ALRs), or the pyrin protein; (2) an adaptor protein, ASC (apoptosis-associated speck-like protein containing a CARD); and (3) an effector protein, usually caspase-1, but also caspase-4/5 in the non-canonical inflammasome [50]. In response to PAMP or DAMP recognition, the cognate receptor proteins are activated, undergo oligomerization, and subsequently bind to ASC and pro-caspase-1. The catalytic activation of caspase-1 in this newly assembled inflammasome triggers caspase-1-mediated processing of pro-IL-1β and IL-18 [51]. It has been reported that production of reactive oxygen species (ROS) in the mitochondria can also induce inflammasome activation, and localization of the inflammasome to the mitochondria depends on the presence of the adapter protein MAVS [52]. On the other hand, the non-canonical inflammasome is activated in response to cytosolic LPS, and results in caspase-11 activation, which subsequently activates the NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome to induce caspase-1 activation and concomitant IL-1β and IL-18 processing [53]. The activation of both canonical and noncanonical inflammasomes also induce pyroptosis, a lytic form of cell death in which IL-1β, IL-18, and inflammatory alarmins such as HMGB1 are released into the extracellular environment, where they can exert pro-inflammatory effects on effector cells [54,55]. Similar to the other PRRs, inflammasome assembly and activation is regulated by post-translational modifications and interactions with proteins that further modulate their activity [56]. Evidence exists, though controversial, that indicates that PKR directly interacts with various inflammasome receptor proteins to facilitate inflammasome activation and the induction of pyroptosis and release of HMGB1, IL-1β, and IL-18 from immune cells [15].
Inflammation-linked signaling cascades: The NF-κB and MAPK pathways
The NF-κB signaling pathway is highly associated with inflammation and inflammatory signaling in many cell types and organs, and across species [57,58]. Mice deficient in NF-κB signaling are often either embryonic lethal, or deficient in the cellular responses to external inflammatory stimuli, such as viral infection [59]. NF-κB/Rel family dimers are bound in the cytoplasm by inhibitory IκB proteins, which prevent their nuclear translocation. Phosphorylation of the IκB kinase (IKK) protein complex by upstream activators leads to the activation of the complex, which will then phosphorylate IκB proteins at specific serine residues. Phosphorylation of IκB leads to its subsequent ubiquitination and proteasomal degradation, revealing nuclear localization signals of NF-κB and promoting their translocation to the nucleus [60].
There are many ways that NF-κB signaling can be activated, including external stimuli and crosstalk with other signaling pathways [58]. Ligand binding to various TNF receptors, interleukin receptors, TLRs, PRRs, T cell receptors, and B cell receptors can recruit adaptor proteins, such as TRAFs that also act as ubiquitin ligases, and kinases such as receptor-interacting protein (RIP), which bind and activate the IKK complex [60]. Other signal receptors that can activate a non-canonical NF-κB signaling cascade via the NF-κB-inducing kinase (NIK) include lymphotoxin B, B cell activating factor (BAFF), CD27 and CD40 [61,62]. Over 300 NF-κB target genes have been confirmed in various roles related to inflammation, including cytokines/chemokines (IFNG [63], IL-1B [64], IL-6 [65], CCL2 [66], TNFA [67]), immunoreceptors (CD40 [68], TNFR1S1B [69], TLR2 [70], NOD2 [71]), cell adhesion molecules (E-selectin [72], ICAM-1 [73], VCAM-1 [74]), stress response genes (PTGS2 [75], NOS2 [76], SOD2 [77]), growth factors (IGFBP1 [78], VEGFC [79]), and other transcription factors (IRF-1 [80], MYC [81], P53 [82]).
The MAPK signaling pathway is also highly relevant to inflammation and inflammatory signaling. The three classes of MAPKs, extracellular signal-related kinases (ERK), c-Jun N-terminal kinase (JNK), and p38, are activated by upstream MAPK kinases (MAPKKs) MKK1/2, MKK4/7, and MKK3/6, respectively [83]. Upstream activators of the MAPK signaling pathway include a number of inflammatory receptors, such as TNF receptors, B cell and T cell receptors, TLRs, and IL-1 and IL-17 receptors [84–87]. Upon binding of such receptors by appropriate ligands, adaptor proteins are recruited, such as TRAF2, TRAF3, and TRAF6 [83]. These adaptor proteins also interact with other signaling transducing molecules, such as the IKK complex, the cellular inhibitor of apoptosis proteins (cIAP1/2), and TAK1/TAK1 binding protein 2 (TAB2) [83]. Activated MAPK proteins then influence gene expression by activating various other transcription factors, in the case of p38 via binding to mitogen and stress-activated protein kinase 1/2 (MSK1/2), such as activator protein-1 (AP-1), activating transcription factor 2 (ATF2), cyclic-AMP response element binding protein (CREB), ATF1, and NF-κB [88–93]. In the cytoplasm, p38 MAPK can also inhibit KH-type splicing regulatory protein (KSRP) and tristetraprolin (TTP), which in turn inhibit the translation of proinflammatory mRNAs by promoting their degradation [94]. MAPKs also phosphorylate histones, which can contribute to the epigenetic regulation of gene regions controlling inflammation [95–97]. The proinflammatory molecules induced by MAPK signaling include cytokines, such as IL-1, IL-17, IL-6, and others in a cell- and species-specific manner [98–100].
Inflammation in the absence of pathogens: Metabolic inflammation or metaflammation
Within the context of the PRRs described above and the cellular activities they elicit in response to inflammatory stimuli, the traditional view of the inflammatory response as a defense mechanism against infection, sterile foreign bodies, or tissue damage has been largely predominant. However, a modern view of inflammation as a response to dysregulated metabolic processes has emerged in light of an alarming increase in diagnoses in the general population of diseases such as type 2 diabetes, cardiovascular disease, non-alcoholic fatty liver steatosis, and atherosclerosis in recent years [101]. These diseases are defined by low levels of chronic inflammation and metabolic dysfunction, and the risk of developing these diseases is significantly elevated by the presence of a cluster of conditions collectively referred to as metabolic syndrome, consisting of low levels of inflammation throughout the body, hyperglycemia, dyslipidemia, abdominal obesity, and hypertension [102].
Hyperglycemia activates the body’s inflammatory defense mechanism, leading to an inflammatory cytokine cascade, and if unchecked, organ damage [103]. Glucose intake can lead to superoxide radical production, activation of NF-κB and AP-1 transcription factors, and expression of TNF-α and IL-6, while the hormone insulin reduces the expression of TLRs, expression of inflammatory mediators intracellular adhesion molecule-1 (ICAM-1), MCP1, and TNF-α [104–108]. Insulin resistance is highly associated with inflammation, and anti-TNF-α therapy has been shown to improve insulin resistance in rheumatoid arthritis patients [109]. Inflammation is also characterized by changes in lipid metabolism; activation of inflammatory cascades leads to decreased HDL cholesterol and increased triglycerides in the blood, as well as increased levels of C Reactive Protein (CRP), ICAM-1, and E-selectin, which in turn promote inflammatory signaling in a feed-forward loop [110–112]. Both insulin resistance following hyperglycemia and dyslipidemia are associated with obesity, which has been shown to correlate with chronic low-grade inflammation, as well as being underlying causes for diseases such as type 2 diabetes [113]. The increase in adipocytes can lead to overproduction of cytokines, such as TNF-α and IL-6, that induce inflammation [114,115]. It is possible that inflammatory signaling is stimulated by a stress response to the increase of intracellular nutrients, the increase of macrophage infiltration into adipocytes, an increase in hypoxic tissue, or direct adipocyte-mediated activation of inflammatory pathogen sensors [116–120]. Also, build-up of cholesterol and increased triglycerides in the blood contribute to atherosclerosis, the primary cause of cardiovascular disease in obese patients [121]. Regulatory T cells (Tregs), Th17 cells, and dendritic cells in the blood can produce TNF-α, IL-6, IL-17, and MCP1 to induce an inflammatory response in hypertensive patients [121]. Expression of TLRs has been shown to be increased in hypertensive conditions, including diabetes and obesity, while DAMPs produced in these conditions can activate the inflammasome to further increase inflammation [122].
AN INTRODUCTION TO THE dsRNA BINDING PROTEINS PACT AND PKR
Function determined by structure: dsRNA binding motifs (dsRBMs) of PACT and PKR
In line with the established role of PKR as a key effector of the cellular innate immune response to viral infection, the PKR gene (EIF2AK2) is one of several ISGs upregulated as a direct consequence of type I Interferon signaling [123]. Despite increased cellular abundance, PKR remains latent within the cell until it binds dsRNA, an indicator of viral presence within the cell, at which point PKR is activated and subsequently inhibits viral and host cell protein synthesis [124]. Like the other three stress-activated eIF2α kinases (PKR-like ER-resident kinase, PERK; general control non-derepressible 2, GCN2; and heme-regulated inhibitor, HRI), PKR is a serine-threonine kinase containing a conserved C-terminal kinase domain and an N-terminal regulatory domain responsible for fine-tuning its activation in response to its specific stimuli [125].
The N-terminal domain of PKR is made of up two tandem evolutionarily conserved double-stranded RNA binding motifs (dsRBM1 and dsRBM2), which together make up the dsRNA binding domain (dsRBD) [126]. Each of PKR’s dsRBMs consists of ~70 amino acids and is separated from the other by a 20-amino acid flexible linker. Each dsRBM also contains the canonical α-β-β-β-α fold, characteristic of other dsRBMs, with the α-helices packed atop the anti-parallel β-sheets [126]. Information about the interaction between PKR’s dsRBMs and dsRNA has been gleaned from structural studies of other dsRBMs; based on those studies, the two dsRBMs of PKR would predictably bind along both faces of a minor groove in A-form dsRNA [127]. Binding to dsRNA by PKR is not restricted to only dsRNAs of viral origin; endogenous intramolecular dsRNAs such as Alu RNAs and mitochondrial RNAs also bind and activate PKR to regulate various cellular processes [128,129]. Furthermore, PKR can bind ssRNA with secondary dsRNA structural features such as stem loops [130]. In this way, PKR can be activated by non-dsRNA species as is the case with the TAR RNA element in the 5’ ends of human immunodeficiency virus-1 (HIV-1) mRNA transcripts [131]. In addition to mediating interactions between PKR and its cognate RNA ligands, the dsRBMs of PKR also mediate its homomeric interactions with other PKR molecules, as well as its heteromeric interaction with other dsRBPs [132,133]. The catalytic C-terminal domain of PKR consists of a smaller lobe made up of predominantly β-sheets and a larger lobe made of primarily α-helices [134].
Initial observations that PKR could also be activated independently of viral infection and in direct response to a diverse range of stress conditions led investigators to hunt for a stress-stimulated PKR-activating protein. PACT and its murine homolog, PKR-associated protein X (RAX), were identified as two such presumptive PKR activators in two-hybrid screens using human K296R PKR and mouse K271R PKR (kinase activity-deficient) respectively as bait proteins [17]. PACT was subsequently classified as a dsRBP based on the close sequence similarity between the first two dsRBMs of PACT and those of PKR; poly(I:C) binding assays confirmed direct PACT-dsRNA interaction [135]. PACT also has a third dsRBM with significantly less sequence similarity to its first two dsRBMs, much like the TAR RNA-binding protein (TRBP), another dsRBP that binds to both PKR and PACT and inhibits PACT-mediated PKR activity [136–140].
In vitro kinase activity assays and PACT overexpression experiments in cells showed that PACT strongly activated PKR activity and subsequently increased eIF2α phosphorylation and translation inhibition, similar to what was observed during dsRNA-mediated PKR activation [12]. The exposure of cells to stress signals increased the interaction between PACT and PKR in a time course that correlated with phosphorylation of both PKR and eIF2α, indicating that PACT is an endogenous activator of PKR [141]. PKR activity assays using PACT deletion mutants and TRBP-domain swapping experiments showed that PACT’s first two dsRBMs are primarily responsible for mediating the interaction of PACT with PKR, while the third dsRBM is responsible for PACT-mediated PKR activation [135,142].
Also of note was the observation made during in vivo metabolic labeling studies that PACT, like PKR and eIF2α, was also phosphorylated in response to stress signals [18]. Two serine residues in PACT’s third dsRBM were found to be essential to PACT-mediated PKR activation in response to non-viral stress signals. The first, serine 246, is constitutively phosphorylated and is absolutely required for the stress-induced phosphorylation of the second residue serine 287 by an as-yet-unidentified kinase. This stress-induced phosphorylation was eventually shown to serve two purposes; the first to weaken the inhibitory interaction between PACT and TRBP [18,139], and the second to increase PACT homodimer formation and in turn increase PACT-PKR binding and PACT-mediated PKR activation [143,144].
PACT, PKR, AND THE CELLULAR INFLAMMATORY RESPONSE TO PATHOGENS
PKR and the induction of inflammatory signaling during infection
PKR has been shown to play a role in inflammatory signaling following bacterial and viral infections via a number of inflammatory signaling pathways (Figure 2). The role of PKR and its activation have been most studied following viral infection, as dsRNA is historically the primary activator of PKR. Upon binding to viral dsRNA, PKR will become activated and subsequently phosphorylate the α subunit of eukaryotic translation initiation factor 2 (eIF2), leading to the inhibition of synthesis of viral and normal cellular proteins. While inflammatory signaling upon viral infection occurs primarily via the sensing of dsRNA by RIG-I and PAMPs by TLR3 leading to activation of NF-κB and IRF3 transcription factors, PKR also has some direct effects on other inflammatory signaling pathways [145,146]. Following infection with vaccinia virus (VV) or encephalomyocarditis virus (EMCV), PKR has been shown to interact with both RIG-I and MDA5, but was only necessary for IFN induction in an eIF2α phosphorylation-independent manner following MDA5-interaction [147]. However, as cycloheximide can rescue PKR-dependent maximal interferon production downstream of IRF3 activation, it cannot be discounted that PKR also mediates interferon signaling indirectly via inhibition of protein synthesis [148]. Interestingly, both PKR and downstream RLR mediator IPS-1 have been determined to be involved in formation of stress granules following dsRNA sensing [149], although the mechanism is not clear. It is similarly unclear how PKR mediates the phosphorylation of STAT1, leading to interferon gene expression; PKR has been shown to interact with STAT1, preventing its activation prior to viral infection, followed by a separation of this complex upon dsRNA or IFN sensing [150]. It is known that PKR does not directly phosphorylate STAT1, as the interaction is kinase activity-independent and RNA-binding domain-dependent, although in addition to eIF2α, PKR can also phosphorylate the T cell protein tyrosine phosphatase (TC-PTP), which dephosphorylates and deactivates STAT1 [151]. Tissue antigen human leukocyte antigen (HLA)-B27-mediated STAT1 phosphorylation is also dependent on PKR in human monocyte-macrophages [152].
Figure 2.
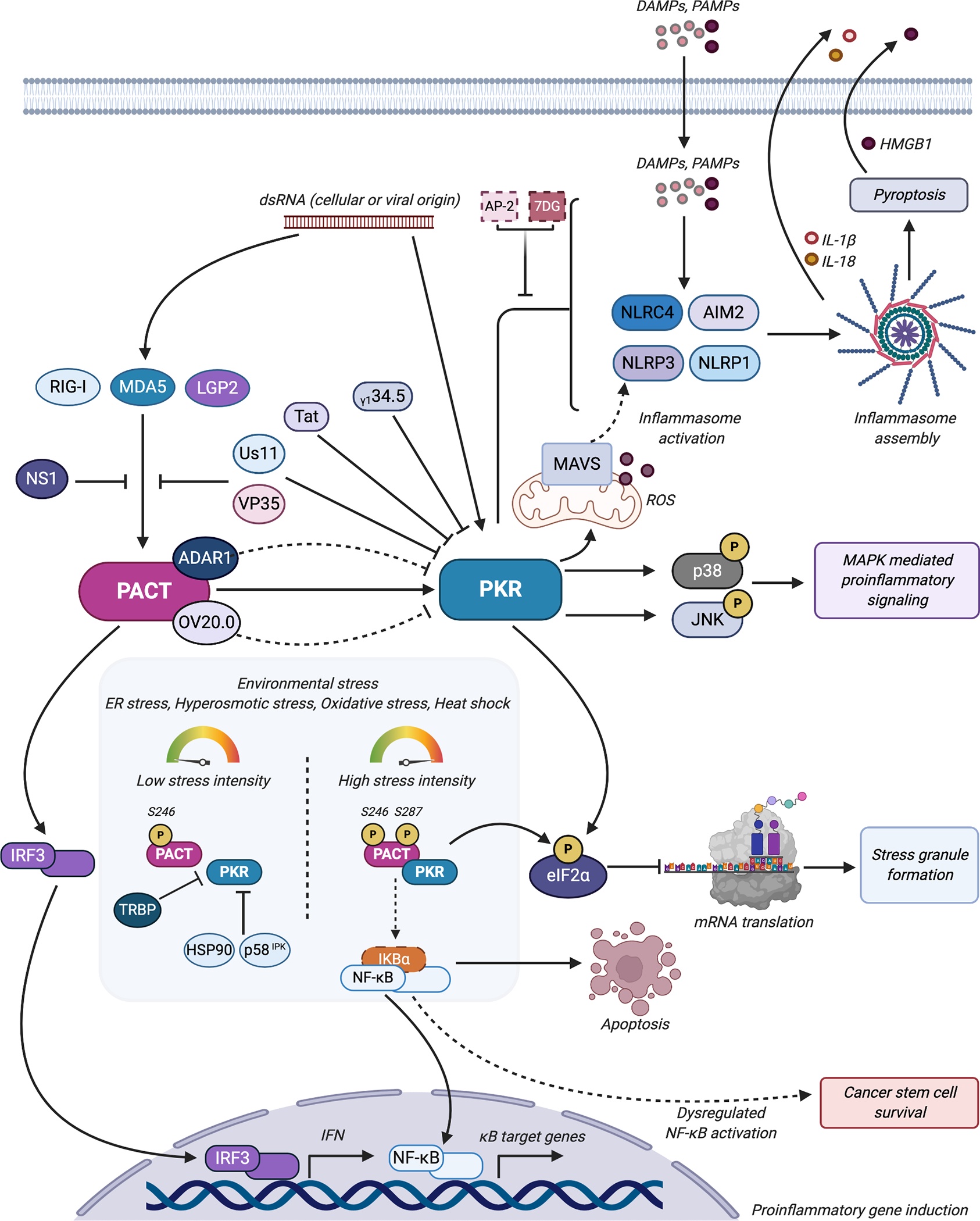
PACT and PKR are involved in several aspects of inflammatory signaling. Long dsRNA of cellular or viral origin activates PKR; PKR subsequently phosphorylates eIF2α and inhibits general protein synthesis and stimulates stress granule formation. PKR also stimulates MAPK mediated proinflammatory signaling through the phosphorylation and activation of the MAPKs p38 and JNK. As such, PKR’s’ antiviral activities are targeted by viral factors such as Us11, Tat, VP35, and γ134.5. Controversial evidence suggests that PKR also stimulates inflammasome activation and assembly through its interactions with DAMP/PAMP sensors NLRP3, NLRP1, NLRC4, and AIM2. PACT-mediated PKR activation generates ROS that drive MAVS-associated NLRP3 inflammasome activation. Inhibitors 2-AP and 7DG appear to inhibit PKR’s inflammasome-stimulatory activity. </P/> PACT modulates RIG-I and MDA5 mediated IFN production through direct interactions and with LGP2; PACT’s stimulatory activity on RLR-mediated pro-inflammatory signaling is targeted by the NS1, Us11, and VP35 viral proteins. Interactions between PACT and the ISG-encoded ADAR1 or the viral-encoded OV20.0 subvert PACT-mediated PKR activation during viral infection. During environmental stress conditions, PACT, which is constitutively phosphorylated on Serine 246, is also phosphorylated on Serine 287 and interacts strongly with PKR, leading to activation of PKR’s kinase activity and subsequent eIF2α phosphorylation and translation inhibition. Increased intensity of environmental stress conditions leads to increased proinflammatory gene induction and apoptosis via NF-κB activation. Dysregulation of NF-κB activation through aberrant PACT-PKR signaling have been implicated in cancer stem cell proliferation and survival. PACT-mediated PKR activation is modulated in these scenarios by other cellular proteins like TRBP, HSP90, and p58IPK. Figure was created using Biorender.com.
HeLa and amnion U cells deficient in PKR demonstrated a lack of p38, JNK, and ATF2 phosphorylation following infection with the measles virus, which was restored upon reconstitution with wild-type human PKR but not with kinase activity-deficient PKR [153]. It has not been shown that PKR can directly phosphorylate these proteins, but it remains unclear whether PKR phosphorylates activating proteins upstream of these MAPKs or whether they are indirectly activated following PKR phosphorylation of its substrate eIF2α and subsequent viral protein synthesis inhibition. The phosphorylation of human protein tripartite motif-containing protein 28 (TRIM28) at serine-473 has been recently shown to be dependent on PKR-mediated p38 MAPK activation, leading to the inactivation of this repressor of interferon signaling in response to highly pathogenic avian influenza viruses (HPAIV) such as H1N1, a novel alternative pathway of PKR-mediated interferon signaling downstream of p38 [154]. Interestingly, the PKR/p38 signaling axis affects not only inflammation, but may also affect protein synthesis; Newcastle disease virus (NDV) infection induced PKR-dependent activation of p38 MAPK, which subsequently phosphorylated MAPK-interacting protein 1 (MNK1), which promoted eIF4E-mediated cap-dependent translation, boosting viral protein synthesis despite simultaneous PKR-mediated phosphorylation of eIF2α [155].
Many viruses have evolved strategies to bypass or inhibit PKR-mediated inhibition of protein synthesis. The herpes simplex virus type 1 (HSV-1) encodes several proteins that aid in the evasion of PKR, including γ134.5, which recruits a phosphatase to dephosphorylate eIF2α [156], and Us11, which can physically bind to PKR and prevent activation, or bind to activated PKR to prevent binding to eIF2α [157]. HIV-1 encodes the protein Tat, which competes with eIF2α for PKR binding [158]. PKR phosphorylation of Tat also then improves Tat binding to TAR-containing viral mRNA and its subsequent translation elongation [159]. In addition, Tat can activate NF-κB in manners both dependent on and independent of PKR interaction [160,161], leading to the coordinated transcription of viral mRNAs. More recently, it was shown that Tat-mediated PKR activation following HIV infection also promoted the growth of parasitic Leishmania amazonensis in a PKR-dependent manner, by modulating NF-κB activity [162]. Meanwhile, Ebola virus, Lloviu virus, and Marburg virus viral protein 35 (VP35) can also inhibit PKR activity, albeit via a different mechanism. VP35 binds to the dsRNA itself, likely masking recognition by endogenous PKR, RIG-I, or MDA5 proteins [163]. Expression of VP35 not only inhibits PKR-dependent inhibition of protein synthesis, but also STAT1 phosphorylation and induction of interferon gene transcription in a cell type-dependent manner [164–166].
Viruses also directly engage known PKR-associated host proteins such as TRBP to inhibit PKR-mediated inhibition of viral protein synthesis [131]. PACT itself is also a direct target of viral evasive strategies; in addition to binding to dsRNA and PKR, the Orf virus (ORFV)-encoded protein OV20.0 interacts with PACT to block PACT-mediated PKR activation [167]. In contrast, other studies have revealed rather surprising proviral functions of PACT during HIV infection with respect to PKR activity. In this unexpected role of PACT as a PKR inhibitor, PACT, alongside the ISG product adenosine deaminase acting on RNA 1 (ADAR1), forms a multiprotein complex that increases its interaction with PKR over the course of HIV infection [168,169]. This increased interaction serves to inhibit PKR and facilitate virion production and replication in infected cells by enhancing viral protein synthesis. Evidence from other studies involving PACT/RAX knockout mice support the notion that PACT can act as a PKR inhibitor under specific cellular contexts [170]. The mice generated from these studies had severe anterior pituitary hypoplasia, craniofacial defects, and hearing impairments, and a complementary knockout of PKR or functional replacement with the dominant negative PKR mutant (K271R) rescued the observed defects. This strongly suggested that the defects were a direct result of the activation or the absence of inhibition of PKR activity brought on by PACT/RAX ablation. It is currently unknown if PACT/RAX is phosphorylated in either scenario in which PACT inhibits PKR, and it may be possible that an absence of or changes in stress-induced PACT phosphorylation or additional unidentified post-translational modifications are responsible for the switch of PACT from an inhibitor to an activator, and vice versa. It is also possible that the singly phosphorylated PACT isoform (at serine 246), which readily heterodimerizes with TRBP, may act cooperatively to prevent PKR activation [18].
ADAR1 can be expressed along with PKR, and has both pro- and antiviral functions; ADAR1 catalyzes the deamination of adenosine in RNA duplexes, leading to their degradation, and coincidently serving as a negative feedback mechanism of PKR activation [171], while in response to measles virus, NDV, Sendai virus, and influenza A virus (IAV) the p150 segment of ADAR1 displayed protective effects and restricted viral growth [172]. In addition, ADAR1 edits Alu repeat elements in endogenous mRNAs, thereby preventing overactive PKR responses to self RNA and limiting autoimmunity [173]. ADAR1 also antagonizes the activation of PKR independently of its deaminase activity during HIV infection by forming a complex with both PACT and PKR to inhibit PKR-mediated inhibition of viral replication [138]. ADAR1 also attenuates ORFV inhibition of PKR through the OV20.0 protein and stimulates Zika virus replication in infected cells by inhibiting PKR activation and consequently stimulating Zika viral protein synthesis [174,175].
PKR has long been linked to other signaling pathways, including MAPK signaling, following pathogen infection. Following stimulation with bacterial endotoxin, such as LPS, PKR-deficient cells demonstrated a decrease in p38 MAPK activation and production of inflammatory cytokines IL-6 and IL-12 [145]. It is unclear how PKR is activated following bacterial infection, although PKR activation has been observed in human monocyte-derived dendritic cells downstream of TLR4 and MyD88 and dependent on IRE1α following Chlamydia trachomatis infection [176]. In PKR-deficient mouse embryonic fibroblasts, activation of p38 MAPK and subsequent IL-6 and IL-12 production were limited following LPS administration, although not abolished, as p38 can be activated by crosstalk of other signaling pathways independent of PKR [145]. This activity appeared to be independent of dsRNA, as only the bacterial endotoxin was used, although a role for PACT was not investigated.
PKR and the activation of NF-κB pathway
There have been many studies that link PKR to NF-κB activation, although the mechanism remains unclear. Initially, it was determined that PKR could directly phosphorylate the inhibitor of NF-κB, IκBα, in vitro [177]. Later, it was shown that PKR induced IκBα phosphorylation via interaction with the IKK complex, but that kinase-deficient PKR was also able to induce this phosphorylation, indicating an indirect role for PKR in mediating NF-κB activation [178]. Further studies indicated that not only is the N-terminal protein-binding domain of PKR necessary for interaction with IKKβ [179], but this interaction could be stabilized by the leukemia-related protein 16 (LRP16), leading to NF-κB activation following treatment of a colorectal carcinoma cell line with DNA damaging agents [180]. As the understanding of PKR-mediated NF-κB activation evolved, it has become clear that other protein interactions can limit the free PKR available to affect NF-κB activity. It is, however, unclear whether PKR interacts with these proteins as a monomer or as an activated dimer. PKR, its protein activator PACT, and dsRNA can all interact with TRBP in the cytoplasm [140], inhibiting PKR from interacting with these activators and subsequently activating NF-κB signaling [138,181].
Induction of interferon gene expression programs during viral infection: PACT and the RIG-I like receptors (RLRs)
As mentioned previously, PACT was initially revealed as a PKR-interacting protein in a cDNA library screen, and subsequent studies using recombinant proteins in in vitro PKR activity assays and as well as in cultured cells showed that PACT’s interaction with PKR resulted in PKR activation. The first indication that PACT could stimulate an antiviral inflammatory response independent of PKR was provided in a study assessing the induction of pro-inflammatory signaling through IRF3 and NF-κB in response to Newcastle Disease Virus (NDV) [182]. In this study, Iwamura et al. demonstrated that PACT, not PKR, stimulated IFN-β promoter activity in NDV-infected cells. Additional reporter assays showed that this was due to increased IRF3 and IRF7 nuclear translocation and transcriptional activity. The authors also established that PACT-mediated stimulation of IFNα/β gene expression in response to viral infection hinges on coordination between the dsRNA binding domains of PACT and their interactions with the host or viral effector proteins.
Over the next ten years, successive reports revealed that some of these effector proteins were members of the RLR family. The close similarity between the functional DExD/H box RNA helicase domain of Dicer and that of RIG-I, as well as the stimulatory effect of PACT and TRBP on Dicer activity, led researchers to question whether PACT similarly stimulated RIG-I activity. Interaction studies in various systems demonstrated that PACT directly interacted with RIG-I independently of RNA presence in Sendai virus-infected cells [47]. Analysis of IRF3 dimerization in HEK 293 cells overexpressing PACT and RIG-I corroborated results from IFN-β enhancer and IRF3 binding element reporter assays. Taken together, these data strongly suggest that the functional significance of the observed PACT-RIG-I interaction was the enhanced stimulation of RIG-I-mediated IRF3 activation and consequent IFN induction in response to viral infection. Mechanistic studies using a helicase-dead RIG-I mutant and in vitro ATPase activity assays showed that the enhancement of RIG-I activity by PACT was primarily achieved through PACT-mediated stimulation of the ATP-dependent helicase activity of RIG-I [47]. It was also found that defective interfering (DI) RNA from the Hu-191 vaccine strain of the measles virus activated production of IFN-β in a PACT and RIG-I-dependent manner, supporting a model in which PACT and RIG-I work together to sense viral DI RNA and initiate an innate immune response [183].
The important conclusions from these findings were further underscored by reports from subsequent independent investigations into mechanisms employed by the EBOV, HSV-1, IAV, and Mĕnglà virus (MLAV) to evade host innate immune defenses [48,49,184,185]. In two different studies, investigators elucidated mechanisms by which EBOV and MLAV dampened RIG-I-mediated type I IFN induction through the viral-encoded innate immune antagonist, VP35. The authors observed that EBOV VP35 robustly inhibited PACT-mediated activation of RIG-I ATPase activity by directly binding to PACT to preclude its interaction with RIG-I. Similarly, other virally encoded proteins such as the HSV-1 encoded protein Us11 and the IAV-encoded protein non-structural protein 1 (NS1) inhibited RIG-I activation and RIG-I mediated type I IFN induction by preventing PACT-RIG-I interaction in favor of viral protein-PACT interaction [48,184].
Based on the observation that PACT could also stimulate MDA5-mediated IRF3 activation and IFNβ promoter activity, subsequent studies explored the mechanisms through which PACT could augment type I IFN induction in cells infected by RNA viruses recognized by MDA5, such as ECMV [47]. The genetic ablation of PACT expression completely abolished IFNβ expression in EMCV-infected fibroblasts and was accordingly restored upon ectopic expression of PACT. PACT-MDA5-specific involvement in this increased IFNβ expression was determined by IFNβ-promoter reporter assays in cells exposed to an MDA5 ligand mimic [186]. Co-expression of PACT significantly boosted MDA-5 induction of IFNβ promoter activity and IRF3 dimerization, demonstrating that MDA5, like RIG-I, is also directly positively regulated by PACT. MDA5 is known to oligomerize upon stimulation by its ligand, so it was expected that PACT’s direct enhancement of MDA5 activity would be reflected in increased MDA5 oligomerization. Results from non-denaturing native polyacrylamide gel electrophoresis experiments showed that this was the most likely scenario, as PACT stimulated MDA5 oligomerization in the presence of its ligand in a time-dependent manner [187].
PACT was also shown to interact directly with MDA5, and further experimentation with PACT deletion mutants showed that PACT’s stimulation of MDA5 activity was highly dependent on PACT’s dsRNA binding ability [186]. Based on this, it was proposed that PACT activated MDA5 activity by binding to MDA5’s ligand, and in so doing, increased the interaction between the ligand and MDA5 to stimulate MDA5 activity. In line with this mechanism, results from an MDA5 ligand (high molecular weight-poly(I:C)) pull-down assay showed significantly enhanced interaction between MDA5 and its ligand in the presence of PACT, suggesting that PACT may stimulate MDA5 activity by increasing its interaction with its ligand, the result of which was increased oligomerization, IPS-1/VISA interaction, and IRF3 activity. As seen with RIG-I, the interaction of PACT with MDA5 was also shown to be the target of innate immune evasion strategies from the Middle East Respiratory Syndrome Coronavirus (MERS-CoV) [188] and Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) [189]. In these instances, the viruses used the viral-encoded proteins MERS-CoV 4a and SARS-CoV N respectively to inhibit PACT-mediated activation of both RIG-I and MDA5, and in so doing attenuated IFNβ expression.
Finally, a recent study demonstrated that PACT also interacts with the third member of the RLR family, LGP2, to modulate PACT-mediated activation of RIG-I and MDA5 [190]. In similar IFNβ promoter assays as described above, LGP2 overexpression effectively blunted the response of RIG-I to the presence of its activators and viruses recognized by RIG-I. This was in stark contrast to its effect on MDA5, as LGP2 overexpression significantly enhanced MDA5-mediated induction of IFNβ promoter-driven expression in the presence of MDA5-specific ligand and responsive viruses. Unbiased proteomic screening approaches to detect LGP2-interacting proteins that could modulate RIG-I and MDA-5 and account for the markedly different effects of LGP2 on both proteins identified PACT as one such protein. Subsequent interaction validation experiments identified a mutation in the C-terminal domain of LGP2 that disrupted the interaction between PACT and LGP2. The inability of this mutant LGP2 to alter PACT’s aforementioned effects on MDA5 and RIG-I activity established clearly that the interaction between PACT and LGP2 was the determining factor in the LGP2-mediated modulation of RIG-I, MDA5-mediated activation of Type I IFN induction, and ISRE-driven gene expression in response to the distinct ligand of each protein.
While the initial PACT and IRF3 activity study reported that PACT overexpression enhanced NF-κB reporter activity in addition to IRF3/IRF7-driven expression [185], none of the subsequent studies reported a similar effect on NF-κB transcriptional activity. Interaction studies showed that PACT did not promote the increased association of the NF-κB activity stimulating enhancer CARD9 with IPS-1/MAVS/VISA, in line with the observed lack of enhanced NFκB -driven promoter activity [47,191]. It may be possible that there are cell type-specific scenarios in which PACT further stimulates MDA5 or RIG-I mediated NF-κB transcriptional activity. It is also possible that differences in NF-κB subunit species present in the cell types tested could account for the observed inability of PACT to stimulate RIG-I or MDA5-driven NF-κB activity. As yet, the mechanism that limits PACT-mediated enhancement of MDA5 and RIG-I activity leading to IRF3 activation has not been elucidated.
Evidence also exists to suggest that the antiviral activities of PACT are not limited to its stimulation of type I IFN induction through the RLRs; PACT also directly interferes with viral replication and RNA transcription by preventing the formation of a competent RNA-dependent RNA polymerase complex with VP35 as is the case with EBOV [192], or by binding to and inhibiting viral RNA polymerase activity, as is observed in IAV infection [193,194]. It should also be noted that PACT’s inhibition of IAV replication is chiefly independent of PACT-RIG-I mediated IFNβ induction; future studies of other host-virus interactions during infection may unveil other scenarios in which PACT functions as an independent host antiviral factor.
It is currently unclear if and how TRBP acts to directly antagonize or facilitate the PKR-independent antiviral activities of PACT, as has been the case in the cellular response to physiological stressors [137]. Both PACT and TRBP can bind to Dicer via the dsRNA-binding domains, the sequences of which confer some specificity on the size of miRNAs processed by these two non-redundant complexes [195]. Experimental evidence showed that the PACT-Dicer complex inhibited the biogenesis of siRNAs in comparison with the TRBP-Dicer complex [196]. Interestingly, the RIG-I-like sensor LGP2 binds to TRBP via the dsRNA-binding domains, preventing TRBP-mediated siRNA processing [197]. This finding suggests a regulatory convergence of small RNA processing, as interaction between TRBP and LGP2 not only sequesters TRBP to dampen miRNA biogenesis, but LGP2 can also directly associate with Dicer to inhibit RNA cleavage into miRNAs.
INVOLVEMENT OF PACT-PKR SIGNALING IN THE ESTABLISHMENT OF STERILE INFLAMMATION
PACT, PKR, and inflammasome activation
Given the established multi-faceted roles of PKR in stimulating proinflammatory signaling in various contexts, reports of an additional stimulatory role for PKR in the activation of inflammasome were not surprising [15,198]. As mentioned previously, the activation of NOD-like receptor proteins (NLRP1, NLRP3, NLRC4) or AIM2 by associated DAMPs or PAMPs initiates the formation of the multi-protein inflammasome. This in turn results in caspase-1/caspase-4/5-mediated proinflammatory cytokine release and the induction of pyroptotic cell death in immunocompetent cells. As more inflammasome-activating stimuli were uncovered, it became increasingly apparent that there was some overlap between these stimuli and certain PKR-activating stimuli [146,199] (Figure 2). This led researchers to speculate about a possible connection and crosstalk between PKR activation and the activation of the inflammasome.
A subsequent study comparing the responses of PKR-deficient macrophages exposed to various inflammasome-activating stimuli to those of wildtype macrophages revealed showed that PKR loss correlated with a marked decrease in inflammasome activation and assembly, as well as with diminished pyroptotic death induction and the secretion of inflammasome-associated cytokines such as HMGB1 [15]. This apparent necessity of PKR for inflammasome activation was further corroborated by similar findings in PKR-depleted bone-marrow derived dendritic cells, in which PKR ablation significantly diminished caspase-1 activation and the concomitant maturation and release of IL-1β and IL-18 [15].
Since several of the inflammasome-activating stimuli used in the aforementioned study specifically stimulate NLRP3 inflammasome activity, it was suggested that PKR directly regulates the activity of the NLRP3 inflammasome. This was supported by results from pull-down assays showing a direct interaction between PKR and NLRP3, which was strengthened in the presence of NLRP3 inflammasome-activating stimuli [15]. However, similar interactions were observed between PKR and the NLRP1, NLRC4 and AIM2 receptor proteins as well, and the ablation of PKR significantly dampened the activation of all three inflammasomes by their cognate stimuli, indicating that PKR may be a central regulatory protein for inflammasome activation [15].
Intriguingly, the kinase activity of PKR was found to be required for caspase-1 activation and the secretion of IL-1β, IL-18 and HMGB1 from macrophages in which each of the aforementioned inflammasomes had been activated. Additional experiments to decipher the necessity of the kinase activity of PKR to NLRP3 inflammasome activation showed that the inhibition of PKR activity (either via 2-aminopurine (2-AP) treatment or expression of K296R PKR) precluded the assembly of the inflammasome in response to activating stimuli [15]. However, none of the inflammasome components were found to be phosphorylated, raising interesting questions about which of the proteins in the complex is a substrate of PKR and if not, how PKR can regulate inflammasome activation as a kinase. It is plausible that the kinase activity of PKR may be important for critical steps preceding NLRP3 inflammasome assembly; NLRP3 activation is a two-step process, the first of which is a priming step in which NLRP3 and IL-1β expression are induced through NF-κB transcriptional activity, and the second in which the NLRP3 inflammasome is activated by cytosolic DAMPs or PAMPs [200]. The loss of PKR, however, had no effect on expression levels of NLRP3 and IL-1β, the step preceding inflammasome activation [15]. The PACT-mediated kinase activity of PKR was also required for an increase in production of cellular ROS, which may also promote MAVS-dependent inflammasome mitochondrial localization [52]. This leaves the purpose of the kinase activity of PKR in inflammasome activation unclear.
This question was reexamined in a subsequent study, in which PKR was identified as a target of a small-molecule inhibitor of anthrax lethal toxin (LT)-induced cell death in macrophages, 7-desacetoxy-6,7-dehydrogedunin (7DG) [201]. 7DG was shown to protect macrophages from LT-induced pyroptosis, as LT is known to trigger the activation of the NLRP1 inflammasome. The authors subsequently performed pull-down assays using biotinylated 7DG to identify protein targets that could account for its protective effects; PKR was identified as one such protein, and PKR knockdown was sufficient to confer protection from LT-induced cell death. Studies into the mechanisms by which 7DG inhibited PKR activity revealed that PKR phosphorylation, which was observed in the previous study, was largely absent in LT-treated macrophages. Furthermore, inhibition of the kinase activity of PKR using 2-AP or the imidazolo-oxindole PKR inhibitor compound 16 (C16) had no effect on LT-induced pyroptotic cell death, contrasting the previous study, that the kinase activity of PKR was not required to stimulate NLRP1 inflammasome-mediated pyroptosis. The conclusions drawn from both of these findings have been called into question by subsequent investigations that do not observe the previously stated activation of inflammasome [202], or even demonstrate that PKR inhibits, rather than stimulates, inflammasome activation [203]. Since significant differences in PKR-mediated cellular signaling have been observed in cells derived from mice with C-terminal or N-terminal PKR deletions, an attempt was made to replicate the findings from the initial study that showed that PKR was required for inflammasome activation [204]. Differences in inflammasome activation in macrophages from either null or wild type mice exposed to various inflammasome-activating stimuli were undetectable, in contrast to those observed in the initial study. These discrepancies in findings stress the need for more uniform genetic models to define the effects of PKR ablation on cellular signaling and especially in inflammasome activation.
PACT-mediated PKR activation and inflammatory signaling during environmental stress
PACT has been shown to activate PKR in response to several stimuli, including IL-3 deprivation, and treatment with thapsigargin, arsenite, and H2O2 in cultured cells [136] (Figure 2). In addition, overexpression of PACT by artificial or genetic means can lead to PKR activation [136]. However, it is not clear what the primary environmental signals are that lead to endogenous in vivo PACT-mediated PKR activation and subsequent induction of proinflammatory signaling.
In addition to infection and other sterile inflammation sources such as metabolic inflammation, PKR can be activated in response to various environmental stresses, including ER stress, oxidative stress, heat shock, and hyperosmotic stress [13,205–207]. PACT has been shown to be phosphorylated in response to thapsigargin- and tunicamycin-induced ER stress, leading to its increased association with PKR and increased apoptosis independent of PERK activation [13,205]. However, it is unclear what signal leads to PACT phosphorylation upstream of this PKR activation. In oxidative stress conditions, it was shown that PKR expression is induced downstream of IFN-γ, and that this expression further contributed to oxidation-mediated apoptosis, although the mechanism by which PKR acted was not investigated [206]. However, overexpression and phosphorylation of TRBP, which forms dimers with both PACT and PKR in homeostatic conditions, by MAPKs ERK1/2 and JNK was able to inhibit PKR activation in oxidative stress conditions [10]. The role of PACT-mediated PKR activation in heat shock conditions is unclear; while PKR can be activated by heat shock [208], PACT has not been determined to be involved, although several heat shock-induced proteins such as P58IPK and HSP90 appear to subsequently inhibit this PKR activation [209,210]. Additionally, PACT has been demonstrated to be phosphorylated in high-intensity hyperosmotic stress conditions, promoting its association with and activation of PKR, subsequently leading to nuclear localization of a subset of NF-κB dimers made up of p65, and p50, while excluding those including c-Rel, promoting proinflammatory gene transcription and apoptosis [208,211]. It is important to note, however, that not all of these signaling mechanisms were corroborated by PACT-deficient mice, suggesting that these signaling pathways may be redundant or act on gradient scales to induce inflammation [11]. PACT-mediated PKR activation appears to have a fine-tuning effect on inflammatory signaling, rather than an all-or-nothing response; inflammation can still arise in the absence of PACT or PKR, but their contribution to inflammatory signaling can be missed in, for example, high-intensity stress conditions [211].
Cellular localization of PKR in response to stress
PKR is generally considered to be active in the cytoplasm, where viral dsRNA is released upon infection. Stress granules (SGs) are a class of membraneless cytoplasmic aggregates containing messenger ribonucleoprotein (mRNP) complexes which are formed in cells in response to environmental stressors such as heat shock and sodium arsenite [212,213]. eIF2α kinase activation and the subsequent inhibition of protein synthesis lead to the accumulation of stalled translation initiation complexes, which are assembled into stress granules facilitated by SG-nucleating proteins such as Ras GTPase-activating protein-binding protein 1 (G3BP1), G3BP2, and T-cell intracellular antigen 1 (TIA1) [214,215]. These SGs keep the mRNAs sequestered from ribosomes, and translation resumes upon the removal of the stressful conditions. SG formation is also induced in response to viral infection, and it has been shown that PKR is involved both in the initial inhibition of viral protein synthesis and is actively recruited into these antiviral SGs (avSGs) through its interaction with G3BP1 or IPS-1 [216,217]. In these avSGs, PKR, alongside the RLR proteins, RIG-I and MDA5, serves as a key member of an antiviral signaling hub inhibiting viral protein synthesis and activating the IFN-induced signaling pathway.
While generally accepted that PERK is the principal eIF2α kinase activated in response to ER stress, it has been shown that PKR is also activated in response to ER stress inducers such as tunicamycin and thapsigargin [13,136,205,218,219]. This raises a number of questions as to how PKR is activated by ER perturbations when it has been shown that it is primarily localized to the cytoplasm. It is possible then that subcellular localization of PKR, as well as PACT, which has been shown to be essential for PKR activation in these instances, may be the key to the activation of PKR’s kinase activity.
Metabolic disease-associated inflammation – Potential involvement of PKR activation
As previously discussed, chronic low-level inflammation is a hallmark of metabolic syndrome, insulin resistance, and related disorders. Several studies have shown that PKR contributes to the proinflammatory signaling in adipocytes and other tissues [220]. Increases in dietary and genetic obesity are generally accompanied by increased PKR activation, while PKR inhibition can reduce metabolic dysregulation, as excess nutrients and energy can be utilized by protein synthesis [198]. Increased leptin receptor also correlated with increased PKR activity in white adipose tissue of obese mice [198]. Reduction in PKR also correlates with a reduction in phosphorylation of insulin receptor substrate-1 (IRS-1) phosphorylation, which led to the discovery that this protein is a substrate of PKR phosphorylation [198]. PKR phosphorylates IRS-1 at the inhibitory site Ser-307, which allows the transcription factor Foxo1 to increase protein expression of IRS-2 [221,222]. In support of PKR-mediated insulin resistance, PKR-deficient mice can display increased insulin sensitivity and glucose tolerance [223]. Downstream effectors of PKR, including JNK, IKK, and protein phosphatase 2A (PP2A) can also contribute to IRS-1 phosphorylation and subsequent insulin resistance [199].
In patients displaying obesity and insulin resistance, although both PKR and TLR4 are active in adipose tissues, PKR activation has been shown to be independent of TLR4 activation [120]. It is unclear how PKR is activated in this condition, as no requirement for dsRNA or PACT has been demonstrated. However, it was shown that PKR could be activated by free fatty acids (FFA); at high concentrations, the long unsaturated FFA palmitate can interact with the kinase domain of PKR to inhibit kinase activity [224]. It was also shown that PKR activation is dependent on interaction with phosphorylated TRBP, and show that inhibition of TRBP can lead to increased glucose sensitivity [225]. Once again, it is important to note that PKR-induced inflammation in obesity and diabetes may be a redundant or gradient pathway, as PKR deletion does not always inhibit inflammation in animal models [226].
CONSEQUENCES OF PACT- AND PKR-MEDIATED INFLAMMATORY SIGNALING IN HEALTH AND DISEASE
In addition to metabolic syndrome, PACT and PKR have been implicated in a number of human diseases, including autoimmune disorders such as systemic lupus erythematosus (SLE), neurodegenerative disorders such as Alzheimer’s disease (AD), genetic disorders such as dystonia, and inflammatory conditions such as inflammatory bowel disease (IBD). Therapies directed against PACT or PKR have also been developed for laboratory research, although none have been approved yet by the FDA for use in humans.
SLE is a disease characterized by disordered immune cell responses that promote autoantibody production [227]. In patients with SLE, PKR protein is preferentially overexpressed in T cells, despite decreased EIF2AK2 mRNA levels, and mRNA translation rates are decreased [228]. No definitive mechanism has been elucidated that leads to an autoimmune response; although it is possible that PKR-dependent inhibition of translation contributes to the deficiency of mitogen-activated T cell proliferation. PKR activity has been shown to be inhibited by the binding of endogenous 16–26 bp circular RNAs (circRNAs) in SLE, which was decreased upon RNAse L-mediated circRNA degradation [229].
IBD, including Crohn’s disease and ulcerative colitis, is an inflammatory condition characterized by lesions in the intestinal epithelial wall and inflammatory signaling. PACT is highly expressed in colonic epithelial cells, particularly at the top of the crypt morphology, where cells are fated to undergo apoptosis [230]. A cell culture model of hyperosmotic stress mimicking IBD showed that this stress condition led to PACT-mediated PKR activation, increased transcription of a subset of proinflammatory genes, and caspase-mediated apoptosis [208]. It is likely that PACT-mediated PKR activation plays a role in IBD in humans, as well as in other pathologies and environmental stress conditions, via similar pathways.
A form of early-onset dystonia (DYT16) has been linked to mutations in PACT. A recessive inherited disease results from the point mutation P222L, while a dominant inherited disease arises from a frameshift-induced missense mutation, both of which leads to robust spontaneous PKR activation and apoptosis [230,231]. These mutant PACT proteins also display altered affinity for TRBP, and release of wild-type PACT from the inhibitory PACT-TRBP complex upon cellular stress signals led to longer-lasting PKR- and caspase-activation in patient-derived lymphoblasts [231,232].
AD is characterized by accumulation of amyloid β plaques and tau fibrillary tangles in degenerating neurons [233]. It was also observed that PKR was highly phosphorylated in these generating neurons, as well as in degenerating neurons in other neurodegenerative diseases such as Parkinson’s disease (PD), Huntington’s disease (HD), and amyloid lateral sclerosis (ALS) [7,234]. It is presumed that PKR-mediated inflammatory signaling and apoptosis may contribute to the phenotype of these diseases. However, in AD, a more direct link was found, as PKR phosphorylation of eIF2α promoted the selective synthesis of beta-secretase 1 precursor protein (BACE1) through upstream open reading frame (uORF) modulation, which cleaves the amyloid precursor protein to amyloid β [235]. In addition, PKR can signal via glycogen synthase kinase 3β (GSK3β) to stimulate tau phosphorylation in AD brains and amyloid β or tunicamycin-treated SH-SY5Y neuronal cell cultures [236].
In neurodegenerative models in particular, the uses of small molecule inhibitors and plant derivatives to inhibit PKR have shown some efficacy. The compound C16, an ATP analog that binds to the ATP-binding pocket of PKR, was able to inhibit neuronal loss in a mouse model of AD [237]. The flavonoid luteolin was able to disrupt the protein-protein interaction between PACT and PKR, leading to reduced PKR activation and inflammatory gene induction in murine microglial cells, although it did lead to increased PKR-mediated inflammasome activation [238]. Various PKR regulators can limit or promote inflammasome activation; for example, amyloid β1–42 can induce activation of the NLRP3 inflammasome dependent on PKR [13], and the inhibitor protein P58IPK can bind to PKR to inhibit NF-κB and JNK-mediated proinflammatory signaling and activation of the NLRP3 inflammasome [239]. In addition, endogenous or pathological conditions that promote PKR-TRBP interaction or inhibit TRBP phosphorylation might also limit PKR-mediated inflammatory activity.
CONCLUSIONS AND FUTURE DIRECTIONS
PKR and PACT are intricately and inextricably linked to inflammation and inflammatory signaling. These two proteins can mediate signaling by TLRs, RLRs, and the inflammasome, in NF-κB and MAPK signaling pathways, in response to infection, environmental stresses, and metabolic syndrome. PKR can have positive or negative effects on homeostasis, depending on stimuli and cellular contexts. PKR activation is necessary to mount a proper antiviral response following infection. Also, proliferation of hematopoietic and mesenchymal stem cells can also be initiated by PKR activation [240,241]. However, prolonged activation can promote inflammatory signaling, apoptosis, and disease pathology. One emerging example of this is in cancer development, where it has recently been shown that dysregulation of PACT-mediated PKR activation induces NF-κB-mediated pro-growth gene expression and can even sensitize resistant HER2+ breast cancers to trastuzumab treatment [242].
It remains to be determined whether drugs that inhibit PACT/PKR interaction or PKR activation, such as C16 or luteolin [238,243], will be effective in treating diseases with an inflammatory component. Understanding the unique molecular targets in homeostatic and stress contexts will be of critical importance in targeted therapeutics. It is possible that inhibiting the activation of PKR in non-stress conditions may additionally represent a means of prevention or delay of the initiation of inflammatory signaling. A case can also be made for stimulating PACT or PKR-mediated pro-inflammatory signaling in certain contexts where such signaling would be beneficial, such as during vaccination and the subsequent build-up of immunity against the target pathogen. Further research may confirm that targeting or disrupting the interaction between these proteins or their interaction with inflammatory signaling molecules could be beneficial in homeostatic conditions (the best of times) and the various pathological conditions in which their dysregulation has been implicated (the worst of times).
ACKNOWLEDGMENTS
This body of work was supported by the following grants: NIH R01DK53307, R01DK060596, and R01DK113196 to M.H., CDDRCC pilot grant NIH DK097948 to M.H, NIH/NIAID grants R01AI116730 and R21AI144264 to P.R.; and CDDRCC pilot grant NIH DK097948 to P.R.
Abbreviations:
- 2-AP
2-aminopurine
- 7DG
7-desacetoxy-6,7-dehydrogedunin
- AD
Alzheimer’s Disease
- ADAR1
adenosine deaminase acting on RNA 1
- AIM2
absent in melanoma 2
- ALR
AIM2-like receptor
- ALS
amyotrophic lateral sclerosis
- AP-1
activator protein-1
- ASC
apoptosis associated speck-like protein containing a CARD
- ATF
activating transcription factor
- ATP
adenosine triphosphate
- avSG
antiviral stress granule
- BACE1
beta secretase 1 precursor
- BAFF
B cell activating factor
- C16
PKR inhibitor compound 16
- CARD
caspase recruitment domain
- cDNA
complementary deoxyribonucleic acid
- cIAP
cellular inhibitor of apoptosis
- circRNA
circular ribonucleic acid
- COX-2
cyclooxygenase-2
- CREB
cyclic-AMP response element binding protein
- CRP
C-reactive protein
- DAMP
damage-associated molecular pattern
- DI RNA
defective interfering RNA
- dsRBD
double-stranded RNA binding domain
- dsRBM
double-stranded RNA binding motif
- dsRBP
double-stranded RNA binding protein
- dsRNA
double-stranded ribonucleic acid
- EBOV
ebola virus
- eIF2α
eukaryotic translation initiation factor 2 subunit alpha
- EMCV
encephalomyocarditis virus
- ER
endoplasmic reticulum
- ERK
extracellular signal-related kinase
- FDA
Food and Drug Administration
- FFA
free fatty acid
- G3BP
Ras GTPase-activating protein-binding protein
- GCN2
general control non-derepressible 2
- GSK3β
glycogen synthase kinase 3 beta
- HD
Huntingtin’s Disease
- HDL
high-density lipoprotein
- HIV
human immunodeficiency virus
- HLA-B27
human leukocyte antigen-B27
- HMGB1
high-mobility group box protein 1
- HPAIV
highly pathogenic avian influenza virus
- HRI
heme-regulated inhibitor
- HSP
heat shock protein
- HSV
herpes simplex virus
- IAV
influenza A virus
- IBD
inflammatory bowel disease
- ICAM
intracellular adhesion molecule
- IFN
interferon
- IκB
inhibitor of κB
- IKK
inhibitor of κB kinase
- IL
interleukin
- iNOS
induced nitric oxide synthase
- IPS-1
interferon β promoter stimulator-1
- IRAK
interleukin-1 receptor-associated kinase
- IRF
interferon regulatory factor
- IRS-1
insulin receptor substrate-1
- ISG
interferon-stimulated gene
- ISR
integrated stress response
- JAK
Janus-activated kinase
- JNK
c-Jun N-terminal kinase
- KSRP
KH-type splicing regulatory protein
- LGP2
laboratory of genetics and physiology 2
- LPS
lipopolysaccharide
- LRP
leukemia-related protein
- LRR
leucine-rich repeat
- LT
lethal toxin
- MAPK
mitogen-activated protein kinase
- MAPKK
mitogen-activated protein kinase kinase
- MAVS
mitochondrial antiviral signaling protein
- MCP
monocyte chemoattractant protein
- MDA5
melanoma differentiation-associated gene 5
- MERS-CoV
Middle East respiratory syndrome coronavirus
- MLAV
Mĕnglà virus
- MNK1
MAPK-interacting protein kinase
- mRNP
messenger ribonucleoprotein
- MSK
mitogen and stress-activated protein kinase
- MyD88
myeloid differentiation primary response protein 88
- NDV
Newcastle Disease virus
- NF-κB
nuclear factor kappa B
- NIK
NF-κB-inducing kinase
- NLR
NOD-like receptor
- NLRC4
NLR-family CARD domain-containing protein
- NLRP3
NOD, LRR, and pyrin domain-containing protein
- NOD
nucleotide-binding oligomerization domain
- NS1
non-structural protein 1
- OAS2
2’–5’-oligoadenylate synthase 2
- PACT
protein activator of PKR
- ORFV
Orf virus
- PAMP
pattern-associated molecular pattern
- PD
Parkinson’s Disease
- PERK
PKR-like ER kinase
- PKR
protein kinase R
- PP2a
protein phosphatase 2A
- PRR
pattern recognition receptor
- RAX
PKR-associated protein X
- RIG-I
retinoic acid-inducible gene I
- RIP
receptor interacting-protein
- RLR
RIG-I-like receptor
- RNA
ribonucleic acid
- ROS
reactive oxygen species
- SARS-CoV
sudden acute respiratory syndrome coronavirus
- SG
stress granule
- siRNA
small interfering RNA
- SLE
systemic lupus erythematosus
- ssRNA
single-stranded ribonucleic acid
- STAT
signal transducer and activator of transcription
- SUMO
small ubiquitin-like modifier
- TAB2
TAK1 binding protein 2
- TAK1
TGFβ-activated kinase 1
- TBK-1
TANK-binding kinase-1
- TC-PTP
T-cell protein tyrosine phosphatase
- TIA1
T-cell intracellular antigen 1
- TIR
Toll and IL-1 receptor domain
- TIRAP
Toll and IL-1 receptor domain-containing adaptor
- TLR
Toll-like receptor
- TNF
tumor necrosis factor
- TRAF
TNF receptor associated factor
- TRAM
translocating chain-associated membrane protein
- TRBP
TAR mRNA-binding protein
- Treg
regulatory T cell
- TRIF
TIR domain-containing adaptor-inducing interferon-β
- TRIM
tripartite motif-containing protein
- TTP
tristetraprolin
- uORF
upstream open reading frame
- VCAM
vascular cell adhesion molecule
- VISA
virus-induced signaling adaptor
- VP35
viral protein 35
- VV
vaccinia virus
Footnotes
Conflict of interest: The authors declare no conflicts of interest.
References
- 1.de Nadal E, Ammerer G, Posas F. Controlling gene expression in response to stress. Nat Rev Genet. 2011;12(12):833–845. doi:doi: 10.1038/nrg3055 [DOI] [PubMed] [Google Scholar]
- 2.Liu B, Qian S-B. Translational reprogramming in stress response. Wiley Interdiscip Rev RNA. 2014;5(3):301–305. doi: 10.1002/wrna.1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spriggs KA, Bushell M, Willis AE. Translational regulation of gene expression during conditions of cell stress. Mol Cell. 2010;40(2):228–237. doi: 10.1016/j.molcel.2010.09.028 [DOI] [PubMed] [Google Scholar]
- 4.Fulda S, Gorman AM, Hori O, Samali A. Cellular Stress Responses: Cell Survival and Cell Death. International Journal of Cell Biology. doi: 10.1155/2010/214074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taniuchi S, Miyake M, Tsugawa K, Oyadomari M, Oyadomari S. Integrated stress response of vertebrates is regulated by four eIF2α kinases. Sci Rep. 2016;6(1):32886. doi: 10.1038/srep32886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Costa-Mattioli M, Walter P. The integrated stress response: From mechanism to disease. Science. 2020;368(6489). doi: 10.1126/science.aat5314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gal-Ben-Ari S, Barrera I, Ehrlich M, Rosenblum K. PKR: A Kinase to Remember. Front Mol Neurosci. 2019;11. doi: 10.3389/fnmol.2018.00480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomis DC, Doohan JP, Samuel CE. Mechanism of interferon action: cDNA structure, expression, and regulation of the interferon-induced, RNA-dependent P1/eIF-2 alpha protein kinase from human cells. Virology. 1992;188(1):33–46. doi: 10.1016/0042-6822(92)90732-5 [DOI] [PubMed] [Google Scholar]
- 9.Levin DH, Petryshyn R, London IM. Characterization of double-stranded-RNA-activated kinase that phosphorylates alpha subunit of eukaryotic initiation factor 2 (eIF-2 alpha) in reticulocyte lysates. Proc Natl Acad Sci U S A. 1980;77(2):832–836. doi: 10.1073/pnas.77.2.832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chukwurah E, Patel RC. Stress-induced TRBP phosphorylation enhances its interaction with PKR to regulate cellular survival. Sci Rep. 2018;8(1):1020. doi: 10.1038/s41598-018-19360-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marques JT, White CL, Peters GA, Williams BRG, Sen GC. The Role of PACT in Mediating Gene Induction, PKR Activation, and Apoptosis in Response to Diverse Stimuli. J Interferon Cytokine Res. 2008;28(8):469–475. doi: 10.1089/jir.2007.0006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li S, Peters GA, Ding K, Zhang X, Qin J, Sen GC. Molecular basis for PKR activation by PACT or dsRNA. Proc Natl Acad Sci. 2006;103(26):10005–10010. doi: 10.1073/pnas.0602317103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee E-S, Yoon C-H, Kim Y-S, Bae Y-S. The double-strand RNA-dependent protein kinase PKR plays a significant role in a sustained ER stress-induced apoptosis. FEBS Lett. 2007;581(22):4325–4332. doi: 10.1016/j.febslet.2007.08.001 [DOI] [PubMed] [Google Scholar]
- 14.Cabanski M, Steinmüller M, Marsh LM, Surdziel E, Seeger W, Lohmeyer J. PKR regulates TLR2/TLR4-dependent signaling in murine alveolar macrophages. Am J Respir Cell Mol Biol. 2008;38(1):26–31. doi: 10.1165/rcmb.2007-0010OC [DOI] [PubMed] [Google Scholar]
- 15.Lu B, Nakamura T, Inouye K, et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature. 2012;488(7413):670–674. doi: 10.1038/nature11290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lemaire PA, Anderson E, Lary J, Cole JL. Mechanism of PKR Activation by dsRNA. J Mol Biol. 2008;381(2):351–360. doi: 10.1016/j.jmb.2008.05.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patel RC, Sen GC. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 1998;17(15):4379–4390. doi: 10.1093/emboj/17.15.4379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh M, Castillo D, Patel CV, Patel RC. Stress-induced phosphorylation of PACT reduces its interaction with TRBP and leads to PKR activation. Biochemistry. 2011;50(21):4550–4560. doi: 10.1021/bi200104h [DOI] [PubMed] [Google Scholar]
- 19.Zhang P, Samuel CE. Induction of Protein Kinase PKR-dependent Activation of Interferon Regulatory Factor 3 by Vaccinia Virus Occurs through Adapter IPS-1 Signaling. J Biol Chem. 2008;283(50):34580–34587. doi: 10.1074/jbc.M807029200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dalet A, Gatti E, Pierre P. Integration of PKR-dependent translation inhibition with innate immunity is required for a coordinated anti-viral response. FEBS Lett. 2015;589(14):1539–1545. doi: 10.1016/j.febslet.2015.05.006 [DOI] [PubMed] [Google Scholar]
- 21.Chen L, Deng H, Cui H, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. 2017;9(6):7204–7218. doi: 10.18632/oncotarget.23208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pober JS, Sessa WC. Inflammation and the Blood Microvascular System. Cold Spring Harb Perspect Biol. 2015;7(1). doi: 10.1101/cshperspect.a016345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nathan C Points of control in inflammation. Nature. 2002;420(6917):846–852. doi: 10.1038/nature01320 [DOI] [PubMed] [Google Scholar]
- 24.Winsauer G, de Martin R. Resolution of inflammation: intracellular feedback loops in the endothelium. Thromb Haemost. 2007;97(3):364–369. [PubMed] [Google Scholar]
- 25.Serhan CN, Levy BD. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest. 2018;128(7):2657–2669. doi: 10.1172/JCI97943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cain D, Kondo M, Chen H, Kelsoe G. Effects of Acute and Chronic Inflammation on B-Cell Development and Differentiation. J Invest Dermatol. 2009;129(2):266–277. doi: 10.1038/jid.2008.286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lawrence T, Gilroy DW. Chronic inflammation: a failure of resolution? Int J Exp Pathol. 2007;88(2):85–94. doi: 10.1111/j.1365-2613.2006.00507.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coggins M, Rosenzweig A. The fire within: cardiac inflammatory signaling in health and disease. Circ Res. 2012;110(1):116–125. doi: 10.1161/CIRCRESAHA.111.243196 [DOI] [PubMed] [Google Scholar]
- 29.Kawai T, Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol. 2009;21(4):317–337. doi: 10.1093/intimm/dxp017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klune JR, Dhupar R, Cardinal J, Billiar TR, Tsung A. HMGB1: Endogenous Danger Signaling. Mol Med. 2008;14:476–484. doi: 10.2119/2008-00034.Klune [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murshid A, Theriault J, Gong J, Calderwood SK. Receptors for extracellular heat shock proteins. Methods Mol Biol Clifton NJ. 2011;787:289–302. doi: 10.1007/978-1-61779-295-3_22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Botos I, Segal DM, Davies DR. The structural biology of Toll-like receptors. Struct Lond Engl 1993. 2011;19(4):447–459. doi: 10.1016/j.str.2011.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chaturvedi A, Pierce SK. How location governs Toll like receptor signaling. Traffic Cph Den. 2009;10(6):621–628. doi: 10.1111/j.1600-0854.2009.00899.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawasaki T, Kawai T. Toll-Like Receptor Signaling Pathways. Front Immunol. 2014;5. doi: 10.3389/fimmu.2014.00461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmitz F, Mages J, Heit A, Lang R, Wagner H. Transcriptional activation induced in macrophages by Toll-like receptor (TLR) ligands: from expression profiling to a model of TLR signaling. Eur J Immunol. 2004;34(10):2863–2873. doi: 10.1002/eji.200425228 [DOI] [PubMed] [Google Scholar]
- 36.Buxadé M, Lunazzi G, Minguillón J, et al. Gene expression induced by Toll-like receptors in macrophages requires the transcription factor NFAT5. J Exp Med. 2012;209(2):379–393. doi: 10.1084/jem.20111569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fitzgerald KA, McWhirter SM, Faia KL, et al. IKKε and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4(5):491–496. doi: 10.1038/ni921 [DOI] [PubMed] [Google Scholar]
- 38.Honda K, Takaoka A, Taniguchi T. Type I Inteferon Gene Induction by the Interferon Regulatory Factor Family of Transcription Factors. Immunity. 2006;25(3):349–360. doi: 10.1016/j.immuni.2006.08.009 [DOI] [PubMed] [Google Scholar]
- 39.Schoggins JW. Interferon-stimulated genes: roles in viral pathogenesis. Curr Opin Virol. 2014;6:40–46. doi: 10.1016/j.coviro.2014.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loo Y-M, Gale M Immune Signaling by RIG-I-like Receptors. Immunity. 2011;34(5):680–692. doi: 10.1016/j.immuni.2011.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoneyama M, Kikuchi M, Matsumoto K, et al. Shared and Unique Functions of the DExD/H-Box Helicases RIG-I, MDA5, and LGP2 in Antiviral Innate Immunity. J Immunol. 2005;175(5):2851–2858. doi: 10.4049/jimmunol.175.5.2851 [DOI] [PubMed] [Google Scholar]
- 42.Paz S, Sun Q, Nakhaei P, et al. Induction of IRF-3 and IRF-7 phosphorylation following activation of the RIG-I pathway. Cell Mol Biol Noisy--Gd Fr. 2006;52(1):17–28. [PubMed] [Google Scholar]
- 43.Nistal-Villán E, Gack MU, Martínez-Delgado G, et al. Negative Role of RIG-I Serine 8 Phosphorylation in the Regulation of Interferon-β Production. J Biol Chem. 2010;285(26):20252–20261. doi: 10.1074/jbc.M109.089912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wies E, Wang MK, Maharaj NP, et al. Dephosphorylation of the RNA sensors RIG-I and MDA5 by the phosphatase PP1 is essential for innate immune signaling. Immunity. 2013;38(3):437–449. doi: 10.1016/j.immuni.2012.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takashima K, Oshiumi H, Takaki H, Matsumoto M, Seya T. RIOK3-mediated phosphorylation of MDA5 interferes with its assembly and attenuates the innate immune response. Cell Rep. 2015;11(2):192–200. doi: 10.1016/j.celrep.2015.03.027 [DOI] [PubMed] [Google Scholar]
- 46.Hu M-M, Liao C-Y, Yang Q, Xie X-Q, Shu H-B. Innate immunity to RNA virus is regulated by temporal and reversible sumoylation of RIG-I and MDA5. J Exp Med. 2017;214(4):973–989. doi: 10.1084/jem.20161015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kok K-H, Lui P-Y, Ng M-HJ, Siu K-L, Au SWN, Jin D-Y. The double-stranded RNA-binding protein PACT functions as a cellular activator of RIG-I to facilitate innate antiviral response. Cell Host Microbe. 2011;9(4):299–309. doi: 10.1016/j.chom.2011.03.007 [DOI] [PubMed] [Google Scholar]
- 48.Kew C, Lui P-Y, Chan C-P, et al. Suppression of PACT-Induced Type I Interferon Production by Herpes Simplex Virus 1 Us11 Protein. J Virol. 2013;87(24):13141–13149. doi: 10.1128/JVI.02564-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luthra P, Ramanan P, Mire CE, et al. Mutual antagonism between the Ebola virus VP35 protein and the RIG-I activator PACT determines infection outcome. Cell Host Microbe. 2013;14(1):74–84. doi: 10.1016/j.chom.2013.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16(7):407–420. doi: 10.1038/nri.2016.58 [DOI] [PubMed] [Google Scholar]
- 51.Martinon F, Burns K, Tschopp J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-β. Mol Cell. 2002;10(2):417–426. doi: 10.1016/S1097-2765(02)00599-3 [DOI] [PubMed] [Google Scholar]
- 52.Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adapter MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell. 2013;153(2):348–361. doi: 10.1016/j.cell.2013.02.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kayagaki N, Warming S, Lamkanfi M, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479(7371):117–121. doi: 10.1038/nature10558 [DOI] [PubMed] [Google Scholar]
- 54.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7(2):99–109. doi: 10.1038/nrmicro2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Man SM, Kanneganti T-D. Regulation of inflammasome activation. Immunol Rev. 2015;265(1):6–21. doi: 10.1111/imr.12296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang J, Liu Z, Xiao TS. Post-translational regulation of inflammasomes. Cell Mol Immunol. 2017;14(1):65–79. doi: 10.1038/cmi.2016.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mussbacher M, Salzmann M, Brostjan C, et al. Cell Type-Specific Roles of NF-κB Linking Inflammation and Thrombosis. Front Immunol. 2019;10. doi: 10.3389/fimmu.2019.00085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer. 2013;12(1):86. doi: 10.1186/1476-4598-12-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gerondakis S, Grossmann M, Nakamura Y, Pohl T, Grumont R. Genetic approaches in mice to understand Rel/NF-kappaB and IkappaB function: transgenics and knockouts. Oncogene. 1999;18(49):6888–6895. doi: 10.1038/sj.onc.1203236 [DOI] [PubMed] [Google Scholar]
- 60.Liu T, Zhang L, Joo D, Sun S-C. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2(1):1–9. doi: 10.1038/sigtrans.2017.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun S-C. The non-canonical NF-κB pathway in immunity and inflammation. Nat Rev Immunol. 2017;17(9):545–558. doi: 10.1038/nri.2017.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ramakrishnan P, Wang W, Wallach D. Receptor-specific signaling for both the alternative and the canonical NF-kappaB activation pathways by NF-kappaB-inducing kinase. Immunity. 2004;21(4):477–89. doi: 10.1016/j.immuni.2004.08.009 [DOI] [PubMed] [Google Scholar]
- 63.Sica A, Tan TH, Rice N, Kretzschmar M, Ghosh P, Young HA. The c-rel protooncogene product c-Rel but not NF-kappa B binds to the intronic region of the human interferon-gamma gene at a site related to an interferon-stimulable response element. Proc Natl Acad Sci U S A. 1992;89(5):1740–1744. doi: 10.1073/pnas.89.5.1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hiscott J, Marois J, Garoufalis J, et al. Characterization of a functional NF-kappa B site in the human interleukin 1 beta promoter: evidence for a positive autoregulatory loop. Mol Cell Biol. 1993;13(10):6231–6240. doi: 10.1128/mcb.13.10.6231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Libermann TA, Baltimore D. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol Cell Biol. 1990;10(5):2327–2334. doi: 10.1128/mcb.10.5.2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Grove M, Plumb M. C/EBP, NF-kappa B, and c-Ets family members and transcriptional regulation of the cell-specific and inducible macrophage inflammatory protein 1 alpha immediate-early gene. Mol Cell Biol. 1993;13(9):5276–5289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shakhov AN, Collart MA, Vassalli P, Nedospasov SA, Jongeneel CV. Kappa B-type enhancers are involved in lipopolysaccharide-mediated transcriptional activation of the tumor necrosis factor alpha gene in primary macrophages. J Exp Med. 1990;171(1):35–47. doi: 10.1084/jem.171.1.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hinz M, Löser P, Mathas S, Krappmann D, Dörken B, Scheidereit C. Constitutive NF-kappaB maintains high expression of a characteristic gene network, including CD40, CD86, and a set of antiapoptotic genes in Hodgkin/Reed-Sternberg cells. Blood. 2001;97(9):2798–2807. doi: 10.1182/blood.v97.9.2798 [DOI] [PubMed] [Google Scholar]
- 69.Santee SM, Owen-Schaub LB. Human Tumor Necrosis Factor Receptor p75/80 (CD120b) Gene Structure and Promoter Characterization. J Biol Chem. 1996;271(35):21151–21159. doi: 10.1074/jbc.271.35.21151 [DOI] [PubMed] [Google Scholar]
- 70.Wang T, Lafuse WP, Zwilling BS. NFκB and Sp1 Elements Are Necessary for Maximal Transcription of Toll-like Receptor 2 Induced by Mycobacterium avium. J Immunol. 2001;167(12):6924–6932. doi: 10.4049/jimmunol.167.12.6924 [DOI] [PubMed] [Google Scholar]
- 71.Gutierrez O, Pipaon C, Inohara N, et al. Induction of Nod2 in myelomonocytic and intestinal epithelial cells via nuclear factor-kappa B activation. J Biol Chem. 2002;277(44):41701–41705. doi: 10.1074/jbc.M206473200 [DOI] [PubMed] [Google Scholar]
- 72.Whelan J, Ghersa P, Hooft van Huijsduijnen R, et al. An NF kappa B-like factor is essential but not sufficient for cytokine induction of endothelial leukocyte adhesion molecule 1 (ELAM-1) gene transcription. Nucleic Acids Res. 1991;19(10):2645–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.van de Stolpe A, Caldenhoven E, Stade BG, et al. 12-O-tetradecanoylphorbol-13-acetate- and tumor necrosis factor alpha-mediated induction of intercellular adhesion molecule-1 is inhibited by dexamethasone. Functional analysis of the human intercellular adhesion molecular-1 promoter. J Biol Chem. 1994;269(8):6185–6192. [PubMed] [Google Scholar]
- 74.Iademarco MF, McQuillan JJ, Rosen GD, Dean DC. Characterization of the promoter for vascular cell adhesion molecule-1 (VCAM-1). J Biol Chem. 1992;267(23):16323–16329. [PubMed] [Google Scholar]
- 75.Yamamoto K, Arakawa T, Ueda N, Yamamoto S. Transcriptional roles of nuclear factor kappa B and nuclear factor-interleukin-6 in the tumor necrosis factor alpha-dependent induction of cyclooxygenase-2 in MC3T3-E1 cells. J Biol Chem. 1995;270(52):31315–31320. doi: 10.1074/jbc.270.52.31315 [DOI] [PubMed] [Google Scholar]
- 76.Geller DA, Nguyen D, Shapiro RA, et al. Cytokine induction of interferon regulatory factor-1 in hepatocytes. Surgery. 1993;114(2):235–242. [PubMed] [Google Scholar]
- 77.Xu Y, Krishnan A, Wan XS, et al. Mutations in the promoter reveal a cause for the reduced expression of the human manganese superoxide dismutase gene in cancer cells. Oncogene. 1999;18(1):93–102. doi: 10.1038/sj.onc.1202265 [DOI] [PubMed] [Google Scholar]
- 78.Lang CH, Nystrom GJ, Frost RA. Regulation of IGF binding protein-1 in Hep G2 cells by cytokines and reactive oxygen species. Am J Physiol-Gastrointest Liver Physiol. 1999;276(3):G719–G727. doi: 10.1152/ajpgi.1999.276.3.G719 [DOI] [PubMed] [Google Scholar]
- 79.Chilov D, Kukk E, Taira S, et al. Genomic organization of human and mouse genes for vascular endothelial growth factor C. J Biol Chem. 1997;272(40):25176–25183. doi: 10.1074/jbc.272.40.25176 [DOI] [PubMed] [Google Scholar]
- 80.Harada H, Kondo T, Ogawa S, et al. Accelerated exon skipping of IRF-1 mRNA in human myelodysplasia/leukemia; a possible mechanism of tumor suppressor inactivation. Oncogene. 1994;9(11):3313–3320. [PubMed] [Google Scholar]
- 81.Duyao MP, Buckler AJ, Sonenshein GE. Interaction of an NF-kappa B-like factor with a site upstream of the c-myc promoter. Proc Natl Acad Sci U S A. 1990;87(12):4727–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wu H, Lozano G. NF-kappa B activation of p53. A potential mechanism for suppressing cell growth in response to stress. J Biol Chem. 1994;269(31):20067–20074. [PubMed] [Google Scholar]
- 83.Johnson GL, Lapadat R. Mitogen-Activated Protein Kinase Pathways Mediated by ERK, JNK, and p38 Protein Kinases. Science. 2002;298(5600):1911–1912. doi: 10.1126/science.1072682 [DOI] [PubMed] [Google Scholar]
- 84.Kawauchi K, Lazarus AH, Sanghera JS, Man GLP, Pelech SL, Delovitch TL. Regulation of BCR- and PKCCa2+-mediated activation of the Raf1/MEK/MAPK pathway by protein-tyrosine kinase and -tyrosine phosphatase activities. Mol Immunol. 1996;33(3):287–296. doi: 10.1016/0161-5890(95)00134-4 [DOI] [PubMed] [Google Scholar]
- 85.Franklin RA, Tordai A, Patel H, Gardner AM, Johnson GL, Gelfand EW. Ligation of the T cell receptor complex results in activation of the Ras/Raf-1/MEK/MAPK cascade in human T lymphocytes. J Clin Invest. 1994;93(5):2134–2140. doi: 10.1172/JCI117209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schröder NW, Pfeil D, Opitz B, et al. Activation of mitogen-activated protein kinases p42/44, p38, and stress-activated protein kinases in myelo-monocytic cells by Treponema lipoteichoic acid. J Biol Chem. 2001;276(13):9713–9719. doi: 10.1074/jbc.M008954200 [DOI] [PubMed] [Google Scholar]
- 87.Dunne A, O’Neill LAJ. The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Sci STKE Signal Transduct Knowl Environ. 2003;2003(171):re3. doi: 10.1126/stke.2003.171.re3 [DOI] [PubMed] [Google Scholar]
- 88.Kefaloyianni E, Gaitanaki C, Beis I. ERK1/2 and p38-MAPK signalling pathways, through MSK1, are involved in NF-kappaB transactivation during oxidative stress in skeletal myoblasts. Cell Signal. 2006;18(12):2238–2251. doi: 10.1016/j.cellsig.2006.05.004 [DOI] [PubMed] [Google Scholar]
- 89.August A, Dupont B. Activation of extracellular signal-regulated protein kinase (ERK/MAP kinase) following CD28 cross-linking: activation in cells lacking p56lck. Tissue Antigens. 1995;46(3 ( Pt 1)):155–162. doi: 10.1111/j.1399-0039.1995.tb03114.x [DOI] [PubMed] [Google Scholar]
- 90.Scheid MP, Foltz IN, Young PR, Schrader JW, Duronio V. Ceramide and cyclic adenosine monophosphate (cAMP) induce cAMP response element binding protein phosphorylation via distinct signaling pathways while having opposite effects on myeloid cell survival. Blood. 1999;93(1):217–225. [PubMed] [Google Scholar]
- 91.Hansen TV, Rehfeld JF, Nielsen FC. Mitogen-activated protein kinase and protein kinase A signaling pathways stimulate cholecystokinin transcription via activation of cyclic adenosine 3’,5’-monophosphate response element-binding protein. Mol Endocrinol Baltim Md. 1999;13(3):466–475. doi: 10.1210/mend.13.3.0257 [DOI] [PubMed] [Google Scholar]
- 92.Kinoshita E, Handa N, Hanada K, Kajiyama G, Sugiyama M. Activation of MAP kinase cascade induced by human pancreatic phospholipase A2 in a human pancreatic cancer cell line. FEBS Lett. 1997;407(3):343–346. doi: 10.1016/s0014-5793(97)00373-6 [DOI] [PubMed] [Google Scholar]
- 93.Vanden Berghe W, Plaisance S, Boone E, et al. p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways are required for nuclear factor-kappaB p65 transactivation mediated by tumor necrosis factor. J Biol Chem. 1998;273(6):3285–3290. doi: 10.1074/jbc.273.6.3285 [DOI] [PubMed] [Google Scholar]
- 94.Briata P, Forcales SV, Ponassi M, et al. p38-dependent phosphorylation of the mRNA decay-promoting factor KSRP controls the stability of select myogenic transcripts. Mol Cell. 2005;20(6):891–903. doi: 10.1016/j.molcel.2005.10.021 [DOI] [PubMed] [Google Scholar]
- 95.Suganuma T, Workman JL. MAP kinases and histone modification. J Mol Cell Biol. 2012;4(5):348–350. doi: 10.1093/jmcb/mjs043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Saccani S, Pantano S, Natoli G. p38-dependent marking of inflammatory genes for increased NF-κB recruitment. Nat Immunol. 2002;3(1):69–75. doi: 10.1038/ni748 [DOI] [PubMed] [Google Scholar]
- 97.De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G. The Histone H3 Lysine-27 Demethylase Jmjd3 Links Inflammation to Inhibition of Polycomb-Mediated Gene Silencing. Cell. 2007;130(6):1083–1094. doi: 10.1016/j.cell.2007.08.019 [DOI] [PubMed] [Google Scholar]
- 98.Klemm C, Bruchhagen C, van Krüchten A, et al. Mitogen-activated protein kinases (MAPKs) regulate IL-6 over-production during concomitant influenza virus and Staphylococcus aureus infection. Sci Rep. 2017;7(1):42473. doi: 10.1038/srep42473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Noubade R, Krementsov DN, del Rio R, et al. Activation of p38 MAPK in CD4 T cells controls IL-17 production and autoimmune encephalomyelitis. Blood. 2011;118(12):3290–3300. doi: 10.1182/blood-2011-02-336552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zheng D, Radziszewska A, Woo P. MicroRNA 497 modulates interleukin 1 signalling via the MAPK/ERK pathway. FEBS Lett. 2012;586(23):4165–4172. doi: 10.1016/j.febslet.2012.10.014 [DOI] [PubMed] [Google Scholar]
- 101.Lonardo A, Nascimbeni F, Mantovani A, Targher G. Hypertension, diabetes, atherosclerosis and NASH: Cause or consequence? J Hepatol. 2018;68(2):335–352. doi: 10.1016/j.jhep.2017.09.021 [DOI] [PubMed] [Google Scholar]
- 102.Nguyen NT, Magno CP, Lane KT, Hinojosa MW, Lane JS. Association of hypertension, diabetes, dyslipidemia, and metabolic syndrome with obesity: findings from the National Health and Nutrition Examination Survey, 1999 to 2004. J Am Coll Surg. 2008;207(6):928–934. doi: 10.1016/j.jamcollsurg.2008.08.022 [DOI] [PubMed] [Google Scholar]
- 103.Sun Q, Li J, Gao F. New insights into insulin: The anti-inflammatory effect and its clinical relevance. World J Diabetes. 2014;5(2):89–96. doi: 10.4239/wjd.v5.i2.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mohanty P, Hamouda W, Garg R, Aljada A, Ghanim H, Dandona P. Glucose challenge stimulates reactive oxygen species (ROS) generation by leucocytes. J Clin Endocrinol Metab. 2000;85(8):2970–2973. doi: 10.1210/jcem.85.8.6854 [DOI] [PubMed] [Google Scholar]
- 105.Dandona P, Chaudhuri A, Ghanim H, Mohanty P. Proinflammatory effects of glucose and anti-inflammatory effect of insulin: relevance to cardiovascular disease. Am J Cardiol. 2007;99(4A):15B–26B. doi: 10.1016/j.amjcard.2006.11.003 [DOI] [PubMed] [Google Scholar]
- 106.Dhindsa S, Tripathy D, Mohanty P, et al. Differential effects of glucose and alcohol on reactive oxygen species generation and intranuclear nuclear factor-kappaB in mononuclear cells. Metabolism. 2004;53(3):330–334. doi: 10.1016/j.metabol.2003.10.013 [DOI] [PubMed] [Google Scholar]
- 107.Aljada A, Friedman J, Ghanim H, et al. Glucose ingestion induces an increase in intranuclear nuclear factor kappaB, a fall in cellular inhibitor kappaB, and an increase in tumor necrosis factor alpha messenger RNA by mononuclear cells in healthy human subjects. Metabolism. 2006;55(9):1177–1185. doi: 10.1016/j.metabol.2006.04.016 [DOI] [PubMed] [Google Scholar]
- 108.Esposito K, Nappo F, Marfella R, et al. Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: role of oxidative stress. Circulation. 2002;106(16):2067–2072. doi: 10.1161/01.cir.0000034509.14906.ae [DOI] [PubMed] [Google Scholar]
- 109.Tam L-S, Tomlinson B, Chu TT, Li TK, Li EK. Impact of TNF inhibition on insulin resistance and lipids levels in patients with rheumatoid arthritis. Clin Rheumatol. 2007;26(9):1495–1498. doi: 10.1007/s10067-007-0539-8 [DOI] [PubMed] [Google Scholar]
- 110.Rost Natalia S, Wolf Philip A, Kase Carlos S, et al. Plasma Concentration of C-Reactive Protein and Risk of Ischemic Stroke and Transient Ischemic Attack. Stroke. 2001;32(11):2575–2579. doi: 10.1161/hs1101.098151 [DOI] [PubMed] [Google Scholar]
- 111.Hackman A, Abe Y, Insull W, et al. Levels of soluble cell adhesion molecules in patients with dyslipidemia. Circulation. 1996;93(7):1334–1338. doi: 10.1161/01.cir.93.7.1334 [DOI] [PubMed] [Google Scholar]
- 112.Esteve E, Ricart W, Fernández-Real JM. Dyslipidemia and inflammation: an evolutionary conserved mechanism. Clin Nutr Edinb Scotl. 2005;24(1):16–31. doi: 10.1016/j.clnu.2004.08.004 [DOI] [PubMed] [Google Scholar]
- 113.Lee H, Lee IS, Choue R. Obesity, Inflammation and Diet. Pediatr Gastroenterol Hepatol Nutr. 2013;16(3):143–152. doi: 10.5223/pghn.2013.16.3.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Koh KK, Han SH, Quon MJ. Inflammatory markers and the metabolic syndrome: insights from therapeutic interventions. J Am Coll Cardiol. 2005;46(11):1978–1985. doi: 10.1016/j.jacc.2005.06.082 [DOI] [PubMed] [Google Scholar]
- 115.Horn F, Henze C, Heidrich K. Interleukin-6 signal transduction and lymphocyte function. Immunobiology. 2000;202(2):151–167. doi: 10.1016/S0171-2985(00)80061-3 [DOI] [PubMed] [Google Scholar]
- 116.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–867. doi: 10.1038/nature05485 [DOI] [PubMed] [Google Scholar]
- 117.Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287(5453):664–666. doi: 10.1126/science.287.5453.664 [DOI] [PubMed] [Google Scholar]
- 118.Surmi BK, Hasty AH. Macrophage infiltration into adipose tissue: initiation, propagation and remodeling. Future Lipidol. 2008;3(5):545–556. doi: 10.2217/17460875.3.5.545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ye J, Gao Z, Yin J, He Q. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am J Physiol Endocrinol Metab. 2007;293(4):E1118–1128. doi: 10.1152/ajpendo.00435.2007 [DOI] [PubMed] [Google Scholar]
- 120.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid–induced insulin resistance. J Clin Invest. 2006;116(11):3015–3025. doi: 10.1172/JCI28898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.De Miguel C, Rudemiller NP, Abais JM, Mattson DL. Inflammation and hypertension: new understandings and potential therapeutic targets. Curr Hypertens Rep. 2015;17(1):507. doi: 10.1007/s11906-014-0507-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.McCarthy CG, Goulopoulou S, Wenceslau CF, Spitler K, Matsumoto T, Webb RC. Toll-like receptors and damage-associated molecular patterns: novel links between inflammation and hypertension. Am J Physiol - Heart Circ Physiol. 2014;306(2):H184–H196. doi: 10.1152/ajpheart.00328.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Thomis DC, Samuel CE. Mechanism of interferon action: autoregulation of RNA-dependent P1/eIF-2 alpha protein kinase (PKR) expression in transfected mammalian cells. Proc Natl Acad Sci U S A. 1992;89(22):10837–10841. doi: 10.1073/pnas.89.22.10837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Roberts WK, Hovanessian A, Brown RE, Clemens MJ, Kerr IM. Interferon-mediated protein kinase and low-molecular-weight inhibitor of protein synthesis. Nature. 1976;264(5585):477–480. doi: 10.1038/264477a0 [DOI] [PubMed] [Google Scholar]
- 125.Donnelly N, Gorman AM, Gupta S, Samali A. The eIF2α kinases: their structures and functions. Cell Mol Life Sci. 2013;70(19):3493–3511. doi: 10.1007/s00018-012-1252-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nanduri S, Carpick BW, Yang Y, Williams BR, Qin J. Structure of the double-stranded RNA-binding domain of the protein kinase PKR reveals the molecular basis of its dsRNA-mediated activation. EMBO J. 1998;17(18):5458–5465. doi: 10.1093/emboj/17.18.5458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Masliah G, Barraud P, Allain FH-T. RNA recognition by double-stranded RNA binding domains: a matter of shape and sequence. Cell Mol Life Sci. 2013;70(11):1875–1895. doi: 10.1007/s00018-012-1119-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kim Y, Park J, Kim S, et al. PKR Senses Nuclear and Mitochondrial Signals by Interacting with Endogenous Double-Stranded RNAs. Mol Cell. 2018;71(6):1051–1063.e6. doi: 10.1016/j.molcel.2018.07.029 [DOI] [PubMed] [Google Scholar]
- 129.Kim Y, Lee JH, Park J-E, Cho J, Yi H, Kim VN. PKR is activated by cellular dsRNAs during mitosis and acts as a mitotic regulator. Genes Dev. 2014;28(12):1310–1322. doi: 10.1101/gad.242644.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mayo CB, Wong CJ, Lopez PE, Lary JW, Cole JL. Activation of PKR by short stem–loop RNAs containing single-stranded arms. RNA. 2016;22(7):1065–1075. doi: 10.1261/rna.053348.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sanghvi VR, Steel LF. The Cellular TAR RNA Binding Protein, TRBP, Promotes HIV-1 Replication Primarily by Inhibiting the Activation of Double-Stranded RNA-Dependent Kinase PKR▿. J Virol. 2011;85(23):12614–12621. doi: 10.1128/JVI.05240-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Patel RC, Stanton P, McMillan NM, Williams BR, Sen GC. The interferon-inducible double-stranded RNA-activated protein kinase self-associates in vitro and in vivo. Proc Natl Acad Sci U S A. 1995;92(18):8283–8287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dey M, Cao C, Dar AC, et al. Mechanistic link between PKR dimerization, autophosphorylation, and eIF2alpha substrate recognition. Cell. 2005;122(6):901–913. doi: 10.1016/j.cell.2005.06.041 [DOI] [PubMed] [Google Scholar]
- 134.Dar AC, Dever TE, Sicheri F. Higher-Order Substrate Recognition of eIF2α by the RNA-Dependent Protein Kinase PKR. Cell. 2005;122(6):887–900. doi: 10.1016/j.cell.2005.06.044 [DOI] [PubMed] [Google Scholar]
- 135.Peters GA, Hartmann R, Qin J, Sen GC. Modular Structure of PACT: Distinct Domains for Binding and Activating PKR. Mol Cell Biol. 2001;21(6):1908–1920. doi: 10.1128/MCB.21.6.1908-1920.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ito T, Yang M, May WS. RAX, a Cellular Activator for Double-stranded RNA-dependent Protein Kinase during Stress Signaling. J Biol Chem. 1999;274(22):15427–15432. doi: 10.1074/jbc.274.22.15427 [DOI] [PubMed] [Google Scholar]
- 137.Gatignol A, Buckler-White A, Berkhout B, Jeang KT. Characterization of a human TAR RNA-binding protein that activates the HIV-1 LTR. Science. 1991;251(5001):1597–1600. doi: 10.1126/science.2011739 [DOI] [PubMed] [Google Scholar]
- 138.Daher A, Longuet M, Dorin D, et al. Two Dimerization Domains in the Trans-activation Response RNA-binding Protein (TRBP) Individually Reverse the Protein Kinase R Inhibition of HIV-1 Long Terminal Repeat Expression. J Biol Chem. 2001;276(36):33899–33905. doi: 10.1074/jbc.M103584200 [DOI] [PubMed] [Google Scholar]
- 139.Daher A, Laraki G, Singh M, et al. TRBP Control of PACT-Induced Phosphorylation of Protein Kinase R Is Reversed by Stress. Mol Cell Biol. 2009;29(1):254–265. doi: 10.1128/MCB.01030-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Benkirane M, Neuveut C, Chun RF, et al. Oncogenic potential of TAR RNA binding protein TRBP and its regulatory interaction with RNA-dependent protein kinase PKR. EMBO J. 1997;16(3):611–624. doi: 10.1093/emboj/16.3.611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Patel CV, Handy I, Goldsmith T, Patel RC. PACT, a stress-modulated cellular activator of interferon-induced double-stranded RNA-activated protein kinase, PKR. J Biol Chem. 2000;275(48):37993–37998. doi: 10.1074/jbc.M004762200 [DOI] [PubMed] [Google Scholar]
- 142.Gupta V, Huang X, Patel RC. The carboxy-terminal, M3 motifs of PACT and TRBP have opposite effects on PKR activity. Virology. 2003;315(2):283–291. doi: 10.1016/s0042-6822(03)00589-0 [DOI] [PubMed] [Google Scholar]
- 143.Singh M, Patel RC. Increased interaction between PACT molecules in response to stress signals is required for PKR activation. J Cell Biochem. 2012;113(8):2754–2764. doi: 10.1002/jcb.24152 [DOI] [PubMed] [Google Scholar]
- 144.Chukwurah E, Willingham V, Singh M, Castillo-Azofeifa D, Patel RC. Contribution of the two dsRBM motifs to the double-stranded RNA binding and protein interactions of PACT. J Cell Biochem. 2018;119(4):3598–3607. doi: 10.1002/jcb.26561 [DOI] [PubMed] [Google Scholar]
- 145.Goh KC, deVeer MJ, Williams BRG. The protein kinase PKR is required for p38 MAPK activation and the innate immune response to bacterial endotoxin. EMBO J. 2000;19(16):4292–4297. doi: 10.1093/emboj/19.16.4292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hsu L-C, Park JM, Zhang K, et al. The protein kinase PKR is required for macrophage apoptosis after activation of Toll-like receptor 4. Nature. 2004;428(6980):341–345. doi: 10.1038/nature02405 [DOI] [PubMed] [Google Scholar]
- 147.Pham AM, Santa Maria FG, Lahiri T, Friedman E, Marié IJ, Levy DE. PKR Transduces MDA5-Dependent Signals for Type I IFN Induction. PLoS Pathog. 2016;12(3):e1005489. doi: 10.1371/journal.ppat.1005489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.McAllister CS, Taghavi N, Samuel CE. Protein Kinase PKR Amplification of Interferon β Induction Occurs through Initiation Factor eIF-2α-mediated Translational Control. J Biol Chem. 2012;287(43):36384–36392. doi: 10.1074/jbc.M112.390039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhang P, Li Y, Xia J, et al. IPS-1 plays an essential role in dsRNA-induced stress granule formation by interacting with PKR and promoting its activation. J Cell Sci. 2014;127(Pt 11):2471–2482. doi: 10.1242/jcs.139626 [DOI] [PubMed] [Google Scholar]
- 150.Wong AH, Tam NW, Yang YL, et al. Physical association between STAT1 and the interferon-inducible protein kinase PKR and implications for interferon and double-stranded RNA signaling pathways. EMBO J. 1997;16(6):1291–1304. doi: 10.1093/emboj/16.6.1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Raven JF, Wang S, Kazemi S, et al. The eIF2α kinase PKR is a negative regulator of Stat1 and Stat3. FASEB J. 2006;20(4):A496–A496. doi: 10.1096/fasebj.20.4.A496-c [DOI] [Google Scholar]
- 152.Ruuska M, Sahlberg AS, Colbert RA, Granfors K, Penttinen MA. Enhanced phosphorylation of STAT-1 is dependent on double-stranded RNA–dependent protein kinase signaling in HLA–B27–expressing U937 monocytic cells. Arthritis Rheum. 2012;64(3):772–777. doi: 10.1002/art.33391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Taghavi N, Samuel CE. Protein kinase PKR catalytic activity is required for the PKR-dependent activation of mitogen-activated protein kinases and amplification of interferon beta induction following virus infection. Virology. 2012;427(2):208–216. doi: 10.1016/j.virol.2012.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Krischuns T, Günl F, Henschel L, et al. Phosphorylation of TRIM28 Enhances the Expression of IFN-β and Proinflammatory Cytokines During HPAIV Infection of Human Lung Epithelial Cells. Front Immunol. 2018;9. doi: 10.3389/fimmu.2018.02229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zhan Y, Yu S, Yang S, et al. Newcastle Disease virus infection activates PI3K/Akt/mTOR and p38 MAPK/Mnk1 pathways to benefit viral mRNA translation via interaction of the viral NP protein and host eIF4E. PLoS Pathog. 2020;16(6). doi: 10.1371/journal.ppat.1008610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.He B, Gross M, Roizman B. The γ134.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1α to dephosphorylate the α subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci. 1997;94(3):843–848. doi: 10.1073/pnas.94.3.843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cassady KA, Gross M. The Herpes Simplex Virus Type 1 US11 Protein Interacts with Protein Kinase R in Infected Cells and Requires a 30-Amino-Acid Sequence Adjacent to a Kinase Substrate Domain. J Virol. 2002;76(5):2029–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Cai R, Carpick B, Chun RF, Jeang KT, Williams BR. HIV-I TAT inhibits PKR activity by both RNA-dependent and RNA-independent mechanisms. Arch Biochem Biophys. 2000;373(2):361–367. doi: 10.1006/abbi.1999.1583 [DOI] [PubMed] [Google Scholar]
- 159.Endo-Munoz L, Warby T, Harrich D, McMillan NA. Phosphorylation of HIV Tat by PKR increases interaction with TAR RNA and enhances transcription. Virol J. 2005;2:17. doi: 10.1186/1743-422X-2-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Demarchi F, d’Adda di Fagagna F, Falaschi A, Giacca M. Activation of transcription factor NF-kappaB by the Tat protein of human immunodeficiency virus type 1. J Virol. 1996;70(7):4427–4437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Demarchi F, Gutierrez MI, Giacca M. Human Immunodeficiency Virus Type 1 Tat Protein Activates Transcription Factor NF-κB through the Cellular Interferon-Inducible, Double-Stranded RNA-Dependent Protein Kinase, PKR. J Virol. 1999;73(8):7080–7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Vivarini Á de C, Santos Pereira R de M, Barreto-de-Souza V, et al. HIV-1 Tat protein enhances the intracellular growth of Leishmania amazonensis via the ds-RNA induced protein PKR. Sci Rep. 2015;5. doi: 10.1038/srep16777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Feng Z, Cerveny M, Yan Z, He B. The VP35 Protein of Ebola Virus Inhibits the Antiviral Effect Mediated by Double-Stranded RNA-Dependent Protein Kinase PKR. J Virol. 2007;81(1):182–192. doi: 10.1128/JVI.01006-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kimberlin CR, Bornholdt ZA, Li S, Woods VL, MacRae IJ, Saphire EO. Ebolavirus VP35 uses a bimodal strategy to bind dsRNA for innate immune suppression. Proc Natl Acad Sci U S A. 2010;107(1):314–319. doi: 10.1073/pnas.0910547107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Feagins AR, Basler CF. Lloviu virus VP24 and VP35 proteins function as innate immune antagonists in human and bat cells. Virology. 2015;485:145–152. doi: 10.1016/j.virol.2015.07.010 [DOI] [PubMed] [Google Scholar]
- 166.Hume A, Mühlberger E. Marburg Virus Viral Protein 35 Inhibits Protein Kinase R Activation in a Cell Type–Specific Manner. J Infect Dis. 2018;218(Suppl 5):S403–S408. doi: 10.1093/infdis/jiy473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tseng YY, Liao GR, Sen GC, Lin FY, Hsu WL. Regulation of PACT-Mediated Protein Kinase Activation by the OV20.0 Protein of Orf Virus. J Virol. 2015;89:11619–11629. doi: 10.1128/JVI.01739-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Clerzius G, Shaw E, Daher A et al. The PKR activator, PACT, becomes a PKR inhibitor during HIV-1 replication. Retrovirology. 2013;10(96). doi: 10.1186/1742-4690-10-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Chukwurah E, Handy I, Patel RC. ADAR1 and PACT contribute to efficient translation of transcripts containing HIV-1 trans-activating response (TAR) element. Biochem J. 2017;474(7):1241–1257. doi: 10.1042/BCJ20160964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Dickerman BK, White CL, Kessler PM, Sadler AJ, Williams BRG, Sen GC. The protein activator of protein kinase R, PACT/RAX, negatively regulates protein kinase R during mouse anterior pituitary development. The FEBS Journal. 282(24):4766–4781. doi: 10.1111/febs.13533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Pfaller CK, Li Z, George CX, Samuel CE. Protein kinase PKR and RNA adenosine deaminase ADAR1: new roles for old players as modulators of the interferon response. Curr Opin Immunol. 2011;23(5):573–582. doi: 10.1016/j.coi.2011.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ward SV, George CX, Welch MJ, et al. RNA editing enzyme adenosine deaminase is a restriction factor for controlling measles virus replication that also is required for embryogenesis. Proc Natl Acad Sci. 2011;108(1):331–336. doi: 10.1073/pnas.1017241108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Chung H, Calis JJA, Wu X, et al. Human ADAR1 prevents endogenous RNA from triggering translational shutdown. Cell. 2018;172(4):811–824.e14. doi: 10.1016/j.cell.2017.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Liao GR, Tseng YY, Tseng CY, Lin FY, Yamada Y, Liu HP, Kuan CY, Hsu WL. Adenosine deaminase acting on RNA 1 associates with Orf virus OV20.0 and enhances viral replication. J Virol. 2019;93(7):e0912–18. doi: 10.1128/JVI.01912-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhou S, Yang C, Zhao F, Huang Y, Lin Y, Huang C, Ma X, Du J, Wang Y, Long G, He J, Liu C, Zhang P. Double-stranded RNA deaminase ADAR1 promotes the Zika virus replication by inhibiting the activation of protein kinase PKR. J Biol Chem. 2019; 294(88):18168–18180. doi: 10.1074/jbc.RA119.009113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Webster SJ, Ellis L, O’Brien LM, et al. IRE1α mediates PKR activation in response to Chlamydia trachomatis infection. Microbes Infect. 2016;18(7):472–483. doi: 10.1016/j.micinf.2016.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kumar A, Haque J, Lacoste J, Hiscott J, Williams BR. Double-stranded RNA-dependent protein kinase activates transcription factor NF-kappa B by phosphorylating I kappa B. Proc Natl Acad Sci. 1994;91(14):6288–6292. doi: 10.1073/pnas.91.14.6288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ishii T, Kwon H, Hiscott J, Mosialos G, Koromilas AE. Activation of the I kappa B alpha kinase (IKK) complex by double-stranded RNA-binding defective and catalytic inactive mutants of the interferon-inducible protein kinase PKR. Oncogene. 2001;20(15):1900–1912. doi: 10.1038/sj.onc.1204267 [DOI] [PubMed] [Google Scholar]
- 179.Bonnet MC, Daurat C, Ottone C, Meurs EF. The N-terminus of PKR is responsible for the activation of the NF-kappaB signaling pathway by interacting with the IKK complex. Cell Signal. 2006;18(11):1865–1875. doi: 10.1016/j.cellsig.2006.02.010 [DOI] [PubMed] [Google Scholar]
- 180.Li X, Wu Z, An X, et al. Blockade of the LRP16-PKR-NF-κB signaling axis sensitizes colorectal carcinoma cells to DNA-damaging cytotoxic therapy. eLife. 2017;6. doi: 10.7554/eLife.27301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Park H, Davies MV, Langland JO, et al. TAR RNA-binding protein is an inhibitor of the interferon-induced protein kinase PKR. Proc Natl Acad Sci. 1994;91(11):4713–4717. doi: 10.1073/pnas.91.11.4713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Iwamura T, Yoneyama M, Koizumi N, et al. PACT, a Double-Stranded RNA Binding Protein Acts as a Positive Regulator for Type I Interferon Gene Induced by Newcastle Disease Virus. Biochem Biophys Res Commun. 2001;282(2):515–523. doi: 10.1006/bbrc.2001.4606 [DOI] [PubMed] [Google Scholar]
- 183.Ho TH, Kew C, Lui PY, Chan CP, Satoh T, Aira S, Jin DY, Kok KH. PACT- and RIG-I-dependent activation of type I interferon production by a defective interfering RNA derived from measles virus vaccine. J Virol. 2015;90(3):1557–68. doi: 10.1128/JVI.02161-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Tawaratsumida K, Phan V, Hrincius ER, High AA, Webby R, Redecke V, Hacker H. Quantitative proteomic analysis of the influenza A virus nonstructrual proteins NS1 and NS2 during natural cell infection identifies PACT as an NS1 target protein and antiviral host factor. J Virol. 2014;88:9038–9048. doi: 10.1128/JVI.00830-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Williams CG, Gibbons JS, Keiffer TR, Luthra P, Edwards MR, Basler CF. Impact of Mĕnglà virus proteins on human and bat innate immune pathways. J Virol. 2020;94:e00191–20. doi: 10.1128/JVI.00191-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Lui P-Y, Wong L-YR, Ho T-H, et al. PACT Facilitates RNA-Induced Activation of MDA5 by Promoting MDA5 Oligomerization. J Immunol Baltim Md 1950. 2017;199(5):1846–1855. doi: 10.4049/jimmunol.1601493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Peisley A, Lin C, Wu B, et al. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc Natl Acad Sci. 2011;108(52):21010–21015. doi: 10.1073/pnas.1113651108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Siu K-L, Yeung ML, Kok K-H, et al. Middle east respiratory syndrome coronavirus 4a protein is a double-stranded RNA-binding protein that suppresses PACT-induced activation of RIG-I and MDA5 in the innate antiviral response. J Virol. 2014;88(9):4866–4876. doi: 10.1128/JVI.03649-13 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 189.Ding Z, Fang L, Yuan S, et al. The nucleocapsid proteins of mouse hepatitis virus and severe acute respiratory syndrome coronavirus share the same IFN-β antagonizing mechanism: attenuation of PACT-mediated RIG-I/ MDA5 activation. Oncotarget. 2017;8(30):49655–49670. doi: 10.18632/oncotarget.17912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Sanchez David RY, Combredet C, Najburg V, et al. LGP2 binds to PACT to regulate RIG-I- and MDA5-mediated antiviral responses. Sci Signal. 2019;12(601). doi: 10.1126/scisignal.aar3993 [DOI] [PubMed] [Google Scholar]
- 191.Smale ST. Hierarchies of NF-κB target-gene regulation. Nat Immunol. 2011;12(8):689–694. doi: 10.1038/ni.2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Luthra P, Ramanan P, Mire CE, Ebihara H, Amarasinghe GK, Basler CF. Mutual antagonism between the Ebola virus VP35 protein and the RIG-I activator PACT determines infection outcome. Cell Host & Microbe. 2013;14(1):74–84. doi: 10.1016/j.chom.2013.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Chan CP, Yuen CK, Cheung PHH, Fung SY, Lui PY, Chen H, Kok KH, Jin DY. Antiviral activity of double-stranded RNA-binding protein PACT agains influenza A virus mediated via suppression of viral RNA polymerase. The FASEB Journal. 2018;32(8):4380–4393. doi. 10.1096/fj.210701361R [DOI] [PubMed] [Google Scholar]
- 194.Tafforeau L, Chantier T, Pradezynski F, Pellet J, Mangeot PE, Vidalain PO, Andre P, Rabourdin-Combe-C, Lotteau V. Generation and comprehensive analysis of an influenza virus polymerase cellular interaction network. J Virol. 2011;85(24):13010–13018. doi: 10.1128/JVI.02651-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Takahashi T, Nakano Y, Onooto K, Murakami F, Komori C, Suzuki Y, Yoneyama M, Ui-Tei K. LPG2 virus sensor regulates gene expression network mediated by TRBP-bound microRNAs. Nucleic Acids Res. 2018;46(17):9134–9147. doi: 10.1093/nar/gky575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.van der Veen AG, Maillard PV, Schmidt JM, Lee SA, Deddouche-Grass S, Kjaer S, Snijders AP, e Sousa CR. The RIG-I-like receptor LGP2 inhibits Dicer-dependent processing of long double-stranded RNA and blocks RNA interference in mammalian cells. EMBO J. 2018;37(4):e97479. doi: 10.15252/embj.201797479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Lee HY, Zhou K, Smith AM, Noland CL, Doudna JA. Differential roles of human Dicer-binding proteins TRBP and PACT in small RNA processing. Nucleic Acids Res. 2013; 41(13):6568–6576. doi: 10.1093/nar/gkt361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Zhang J, Zhang L, Zhang S, et al. HMGB1, an innate alarmin, plays a critical role in chronic inflammation of adipose tissue in obesity. Mol Cell Endocrinol. 2017;454:103–111. doi: 10.1016/j.mce.2017.06.012 [DOI] [PubMed] [Google Scholar]
- 199.Nakamura T, Furuhashi M, Li P, et al. Double-stranded RNA-dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell. 2010;140(3):338–348. doi: 10.1016/j.cell.2010.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Swanson KV, Deng M, Ting JP-Y. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19(8):477–489. doi: 10.1038/s41577-019-0165-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Hett EC, Slater LH, Mark KG, et al. Chemical Genetics Reveals a Kinase-Independent Role For Protein Kinase R In Pyroptosis. Nat Chem Biol. 2013;9(6):398–405. doi: 10.1038/nchembio.1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.He Y, Franchi L, Núñez G. The protein kinase PKR is critical for LPS-induced iNOS production but dispensable for inflammasome activation in macrophages. Eur J Immunol. 2013;43(5):1147–1152. doi: 10.1002/eji.201243187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Yim HC, Wang D, Yu L, et al. The kinase activity of PKR represses inflammasome activity. Cell Res. 2016;26(3):367–379. doi: 10.1038/cr.2016.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Baltzis D, Li S, Koromilas AE. Functional characterization of pkr gene products expressed in cells from mice with a targeted deletion of the N terminus or C terminus domain of PKR. J Biol Chem. 2002;277(41):38364–38372. doi: 10.1074/jbc.M203564200 [DOI] [PubMed] [Google Scholar]
- 205.Singh M, Fowlkes V, Handy I, Patel CV, Patel RC. Essential Role of PACT-Mediated PKR Activation in Tunicamycin-induced Apoptosis. J Mol Biol. 2009;385(2):457–468. doi: 10.1016/j.jmb.2008.10.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Pyo C-W, Lee S-H, Choi S-Y. Oxidative stress induces PKR-dependent apoptosis via IFN-gamma activation signaling in Jurkat T cells. Biochem Biophys Res Commun. 2008;377(3):1001–1006. doi: 10.1016/j.bbrc.2008.10.103 [DOI] [PubMed] [Google Scholar]
- 207.Murtha-Riel P, Davies MV, Scherer BJ, Choi SY, Hershey JW, Kaufman RJ. Expression of a phosphorylation-resistant eukaryotic initiation factor 2 alpha-subunit mitigates heat shock inhibition of protein synthesis. J Biol Chem. 1993;268(17):12946–12951. [PubMed] [Google Scholar]
- 208.Farabaugh KT, Majumder M, Guan B-J, et al. Protein Kinase R Mediates the Inflammatory Response Induced by Hyperosmotic Stress. Mol Cell Biol. 2017;37(4). doi: 10.1128/MCB.00521-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Donzé O, Abbas-Terki T, Picard D. The Hsp90 chaperone complex is both a facilitator and a repressor of the dsRNA-dependent kinase PKR. EMBO J. 2001;20(14):3771–3780. doi: 10.1093/emboj/20.14.3771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Melville MW, Tan SL, Wambach M, Song J, Morimoto RI, Katze MG. The cellular inhibitor of the PKR protein kinase, P58(IPK), is an influenza virus-activated co-chaperone that modulates heat shock protein 70 activity. J Biol Chem. 1999;274(6):3797–3803. doi: 10.1074/jbc.274.6.3797 [DOI] [PubMed] [Google Scholar]
- 211.Farabaugh KT, Krokowski D, Guan B-J, et al. PACT-mediated PKR activation acts as a hyperosmotic stress intensity sensor weakening osmoadaptation and enhancing inflammation. O’Riordan MX, Taniguchi T, eds. eLife. 2020;9:e52241. doi: 10.7554/eLife.52241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Collier NC, Heuser J, Levy MA, Schlesinger MJ. Ultrastructural and biochemical analysis of the stress granule in chicken embryo fibroblasts. J Cell Biol. 1988;106(4):1131–1139. doi: 10.1083/jcb.106.4.1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Collier NC, Schlesinger MJ. The dynamic state of heat shock proteins in chicken embryo fibroblasts. J Cell Biol. 1986;103(4):1495–1507. doi: 10.1083/jcb.103.4.1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Kedersha N, Chen S, Gilks N, et al. Evidence that ternary complex (eIF2-GTP-tRNA(i)(Met))-deficient preinitiation complexes are core constituents of mammalian stress granules. Mol Biol Cell. 2002;13(1):195–210. doi: 10.1091/mbc.01-05-0221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Matsuki H, Takahashi M, Higuchi M, Makokha GN, Oie M, Fujii M. Both G3BP1 and G3BP2 contribute to stress granule formation. Genes Cells Devoted Mol Cell Mech. 2013;18(2):135–146. doi: 10.1111/gtc.12023 [DOI] [PubMed] [Google Scholar]
- 216.Reineke LC, Lloyd RE. The stress granule protein G3BP1 recruits protein kinase R to promote multiple innate immune antiviral responses. J Virol. 2015;89(5):2575–2589. doi: 10.1128/JVI.02791-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Onomoto K, Jogi M, Yoo J-S, et al. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PloS One. 2012;7(8):e43031. doi: 10.1371/journal.pone.0043031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Li G, Scull C, Ozcan L, Tabas I. NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J Cell Biol. 2010;191(6):1113–1125. doi: 10.1083/jcb.201006121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Vaughn LS, Snee B, Patel RC. Inhibition of PKR protects against tunicamycin-induced apoptosis in neuroblastoma cells. Gene. 2014;536(1):90–96. doi: 10.1016/j.gene.2013.11.074 [DOI] [PubMed] [Google Scholar]
- 220.Monteiro R, Azevedo I. Chronic Inflammation in Obesity and the Metabolic Syndrome. Mediators Inflamm. 2010;2010. doi: 10.1155/2010/289645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Copps KD, White MF. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia. 2012;55(10):2565–2582. doi: 10.1007/s00125-012-2644-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Copps KD, Hancer NJ, Opare-Ado L, Qiu W, Walsh C, White MF. Irs1 Serine 307 Promotes Insulin Sensitivity in Mice. Cell Metab. 2010;11(1):84–92. doi: 10.1016/j.cmet.2009.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Carvalho-Filho MA, Carvalho BM, Oliveira AG, et al. Double-Stranded RNA-Activated Protein Kinase Is a Key Modulator of Insulin Sensitivity in Physiological Conditions and in Obesity in Mice. Endocrinology. 2012;153(11):5261–5274. doi: 10.1210/en.2012-1400 [DOI] [PubMed] [Google Scholar]
- 224.Fang L, Cho HJ, Chan C, Feig M. Binding site multiplicity with fatty acid ligands: implications for the regulation of PKR kinase autophosphorylation with palmitate. Proteins. 2014;82(10):2429–2442. doi: 10.1002/prot.24607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Nakamura T, Kunz RC, Zhang C, et al. A critical role for PKR complexes with TRBP in Immunometabolic regulation and eIF2α phosphorylation in obesity. Cell Rep. 2015;11(2):295–307. doi: 10.1016/j.celrep.2015.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Lancaster GI, Kammoun HL, Kraakman MJ, Kowalski GM, Bruce CR, Febbraio MA. PKR is not obligatory for high-fat diet-induced obesity and its associated metabolic and inflammatory complications. Nat Commun. 2016;7. doi: 10.1038/ncomms10626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Herrada AA, Escobedo N, Iruretagoyena M, et al. Innate Immune Cells’ Contribution to Systemic Lupus Erythematosus. Front Immunol. 2019;10. doi: 10.3389/fimmu.2019.00772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Grolleau A, Kaplan MJ, Hanash SM, Beretta L, Richardson B. Impaired translational response and increased protein kinase PKR expression in T cells from lupus patients. J Clin Invest. 2000;106(12):1561–1568. doi: 10.1172/JCI9352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Liu C-X, Li X, Nan F, et al. Structure and Degradation of Circular RNAs Regulate PKR Activation in Innate Immunity. Cell. 2019;177(4):865–880.e21. doi: 10.1016/j.cell.2019.03.046 [DOI] [PubMed] [Google Scholar]
- 230.Gupta V, Patel RC. Proapoptotic protein PACT is expressed at high levels in colonic epithelial cells in mice. Am J Physiol Gastrointest Liver Physiol. 2002;283(3):G801–808. doi: 10.1152/ajpgi.00498.2001 [DOI] [PubMed] [Google Scholar]
- 231.Burnett SB, Vaughn LS, Strom JM, Francois A, Patel RC. A truncated PACT protein resulting from a frameshift mutation reported in movement disorder DYT16 triggers caspase activation and apoptosis. J Cell Biochem. 2019;120(11):19004–19018. doi: 10.1002/jcb.29223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Vaughn LS, Bragg DC, Sharma N, Camargos S, Cardoso F, Patel RC. Altered Activation of Protein Kinase PKR and Enhanced Apoptosis in Dystonia Cells Carrying a Mutation in PKR Activator Protein PACT. J Biol Chem. 2015;290(37):22543–22557. doi: 10.1074/jbc.M115.669408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener. 2019;14(1):32. doi: 10.1186/s13024-019-0333-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Marchal JA, Lopez GJ, Peran M, et al. The impact of PKR activation: from neurodegeneration to cancer. FASEB J. 2014;28(5):1965–1974. doi: 10.1096/fj.13-248294 [DOI] [PubMed] [Google Scholar]
- 235.Mouton-Liger F, Paquet C, Dumurgier J, et al. Oxidative stress increases BACE1 protein levels through activation of the PKR-eIF2α pathway. Biochim Biophys Acta BBA - Mol Basis Dis. 2012;1822(6):885–896. doi: 10.1016/j.bbadis.2012.01.009 [DOI] [PubMed] [Google Scholar]
- 236.Bose A, Mouton-Liger F, Paquet C, et al. Modulation of tau phosphorylation by the kinase PKR: implications in Alzheimer’s disease. Brain Pathol Zurich Switz. 2011;21(2):189–200. doi: 10.1111/j.1750-3639.2010.00437.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Ingrand S, Barrier L, Lafay-Chebassier C, Fauconneau B, Page G, Hugon J. The oxindole/imidazole derivative C16 reduces in vivo brain PKR activation. FEBS Lett. 2007;581(23):4473–4478. doi: 10.1016/j.febslet.2007.08.022 [DOI] [PubMed] [Google Scholar]
- 238.Dabo S, Maillard P, Collados Rodriguez M, et al. Inhibition of the inflammatory response to stress by targeting interaction between PKR and its cellular activator PACT. Sci Rep. 2017;7(1):16129. doi: 10.1038/s41598-017-16089-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Boriushkin E, Wang JJ, Li J, Bhatta M, Zhang SX. p58 IPK suppresses NLRP3 inflammasome activation and IL-1β production via inhibition of PKR in macrophages. Sci Rep. 2016;6(1):25013. doi: 10.1038/srep25013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Crop MJ, Baan CC, Korevaar SS, IJzermans JNM, Pescatori M, Stubbs AP, van IJcken WFJ, Dahlke MH, Eggenhofer E, Weimar W, Hoogduijn MJ. Inflammatory conditions affect gene expression and function of human adipose tissue-derived mesenchymal stem cells. Clin Exp Immunol. 2010;162(3):474–486. doi: 10.1111/j.1365-2249.2010.04256.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Liu X, Bennett RL, Cheng X, Byrne M, Reinhard MK, May WS Jr. PKR regulates proliferation, differentiation, and survival of murine hematopoietic stem/progenitor cells. Blood. 2013;121(17):3364–3374. doi: 10.1182/blood-2012-09-456400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Darini C, Ghaddar N, Chabot C, Assaker G, Sabri S, Wang S, Krishnamoorthy J, Buchanan M, Aguilar-Mahecha A, Abdulkarim B, Deschenes J, Torres J, Ursini-Siegel J, Basik M, Koromilas AE. An integrated stress response via PKR suppresses HER2+ cancers and improves trastuzumab therapy. Nat Commun. 2019;10(1):2139. doi: 10.1038/s41467-019010138-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Xiao J, Tan Y, Li Y, Luo Y. The specific protein kinase R (PKR) inhibitor C16 protects neonatal hypoxia-ischemia brain damages by inhibiting neuroinflammation in a neonatal rat model. Med Sci Monit. 2016;22:5074–5081. doi: 10.12659/MSM.898139 [DOI] [PMC free article] [PubMed] [Google Scholar]