

Abstract
Healthy development of the gut microbiome provides long-term health benefits. Children raised in countries with high infectious disease burdens are frequently exposed to diarrheal pathogens and antibiotics, which perturb gut microbiome assembly. A recent cluster-randomized trial leveraging >4,000 child observations in Dhaka, Bangladesh, found that automated water chlorination of shared taps effectively reduced child diarrhea and antibiotic use. In this sub-study, we leveraged stool samples collected from 130 children one year after this intervention to examine differences between treatment and control children’s gut microbiota. Water chlorination was associated with increased abundance of several bacterial genera previously linked to improved gut health; however, we observed no effects on the overall richness or diversity of taxa. Several clinically relevant antibiotic resistance genes were relatively more abundant in the gut microbiome of treatment children, possibly due to increases in Enterobacteriaceae. While further studies on the long-term health impacts of drinking chlorinated water would be valuable, we conclude that access to chlorinated water did not substantially impact child gut microbiome development in this setting, supporting the use of chlorination to increase global access to safe drinking water.
Normal assembly of the early-life gut microbiome is critical for human health. The gut microbiome is seeded during birth and stabilizes to an adult-like configuration by the third year of life.1 The progressive, unperturbed colonization of the intestinal tract during this time window is likely essential to the establishment and maturation of multiple developmental pathways related to metabolism, allergy development, weight gain, disease susceptibility, and mental health.2,3
Children raised in low- and middle-income countries (LMICs) are at high risk of early-life environmental insults that might disrupt optimal gut microbiome development. Due to poor sanitation and lack of access to clean drinking water, children living in poverty are frequently exposed to enteric pathogens.4 Pathogen establishment and proliferation in the intestinal tract can perturb the normal gut microbiome by triggering local and systemic inflammation.5,6 In areas with a high infectious disease burden, children also frequently consume antibiotics. In urban Bangladesh, children younger than two years are treated with antibiotics at a rate more than five times higher than that among similarly aged children in the United States.7,8 Frequent antibiotic use early in life diminishes gut microbiota diversity, enriches for antibiotic resistance genes (ARGs), and reduces microbiome richness while increasing variability.9,10 In the United States, these perturbations have been linked to increased risk of multiple childhood-onset health disorders, including asthma, allergy rhinitis, and attention deficit hyperactivity disorder.3
Water chlorination is a promising strategy for reducing exposure to enteric pathogens and associated antibiotic use among young children in LMICs,11 with potential benefits for the gut microbiome and long-term health. Chlorination inactivates many microorganisms present in water and reduces recontamination during transport and storage.12 By reducing children’s exposures to pathogens, chlorination could prevent the early establishment and proliferation of pathogens in the gut and the subsequent use of antibiotics. However, water chlorination could indirectly affect the developing gut microbiome in other ways. Chlorination could systematically impact the types of waterborne organisms that children are exposed to, as it does not inactivate all microorganisms present in drinking water.13 For example, the introduction of chlorine into drinking water systems has been associated with changes in biofilm communities,14 the relative abundance of specific bacterial genera in water,15 and the abundance of ARGs in water systems,16 with the potential for ARGs that function as efflux pumps to be enriched.17 Further, ingested trace chlorine residuals or chlorine disinfection by-products could perturb the gut environment in ways that are not fully understood.18,19
Members of our team recently conducted a double-blinded, placebo-controlled cluster-randomized trial of passive (automated) water chlorination devices installed at shared taps in urban Bangladesh, which disinfected all water used for domestic purposes including drinking, cooking, personal hygiene, and cleaning.11 The passive water chlorination intervention reduced caregiver-reported child diarrhea in the past seven days by 23% and caregiver-reported antibiotic use in the past two months by 7%, relative to controls. The objectives of the present study were to examine the impacts of water chlorination on children’s gut microbiomes, including the ARGs and bacterial pathogens that they harbored, across different phases of gut microbiome development.
Results
Child and stool sample characteristics
We examined fecal metagenomes from 130 children from the control (n=64) and treatment groups (n=66). Samples included in our final analysis were balanced between two study sites (Dhaka Uddan and Tongi) and three pre-specified age strata (6–14 months, 15–30 months, 31 months and older) corresponding to distinct phases of gut microbiome development.1
Characteristics known to impact the early-life gut microbiome were evenly distributed between the treatment and control groups, including child age,1 breastfeeding status,1 recent diarrhea,20 and recent antibiotic use (Table 1).3 Children were exposed to chlorine (treatment) and Vitamin C (active control) doses for an average of 10.5 months (range due to open cohort study design = 1.7 to 14.4 months). Most children (89%) were exposed to chlorine or Vitamin C for at least 6 months. Our average sequencing depth was approximately 6 Gb per sample.
Table 1.
Characteristics of 130 Bangladeshi children participating in a cluster-randomized automated water chlorination trial at the time of stool sample collection.
| Control | Treatment | |
|---|---|---|
|
| ||
| n=64 (%) | n=66 (%) | |
| Female | 40 (63) | 37 (57) |
| Age | ||
| 6–14 months | 13 (20) | 14 (21) |
| 15–30 months | 25 (41) | 26 (39) |
| 31–61 months | 26 (41) | 26 (39) |
| Study site | ||
| Dhaka Uddan | 28 (44) | 28 (42) |
| Tongi | 36 (56) | 38 (58) |
| Currently receiving any human milka,b | 32 (50) | 29 (45) |
| Experienced fever in past seven daysa | 15 (23) | 9 (14) |
| Experienced diarrhea in past seven daysa | 3 (5) | 3 (5) |
| Received antibiotics in past two monthsa | 25 (36) | 23 (35) |
| Months enrolled in trial (mean, SD) | 11.5 (3.0) | 10.8 (3.1) |
Caregiver reported at time of stool collection.
Exclusive breastfeeding was rare at the time of stool sample collection; no children in the control group and only one child in the treatment group (age 14 months) was exclusively breastfed.
Water chlorination impacted the relative abundance of several human enterobacteria
Exposure to chlorinated water significantly impacted the relative abundance of multiple bacterial genera in children’s guts, relative to control children (Figure 1A), when adjusting for age and study site. Consistent with previous studies,1 we observed considerable differences in gut microbiome composition by age group (Figure 2A). We estimated treatment coefficients describing the additive change in the logit-transformed relative abundance of bacterial genera between treatment and control children;21 positive treatment coefficient values indicate elevated levels among children receiving chlorinated water relative to controls. The treatment coefficient generally approximates the log fold change (Extended Data Fig. 1). Overall, several bacterial genera that are frequently reported to colonize humans were significantly more abundant among treatment children (fdr-adjusted p-value<0.05), including Akkermansia (treat. coef: 2.4, 95% confidence interval (CI): 1.9, 3.0), Escherichia (treat. coef: 1.11, 95% CI: 0.7, 1.6), Flavonifractor (treat. coef: 0.89, 95% CI: 0.5, 1.3), and Phascolarctobacterium (treat. coef: 2.1, 95% CI: 1.5, 2.7) (Extended Data Table 1). Each of these genera comprised at least 0.1% of bacterial reads in children’s fecal metagenomes, on average, with the exception of Escherichia, which comprised 4% of bacterial reads.
Figure 1. Differentially abundant gut taxa among children aged 6–61 months who were cluster randomized to an automated chlorinated water intervention in urban Bangladesh and effects on overall richness and diversity.
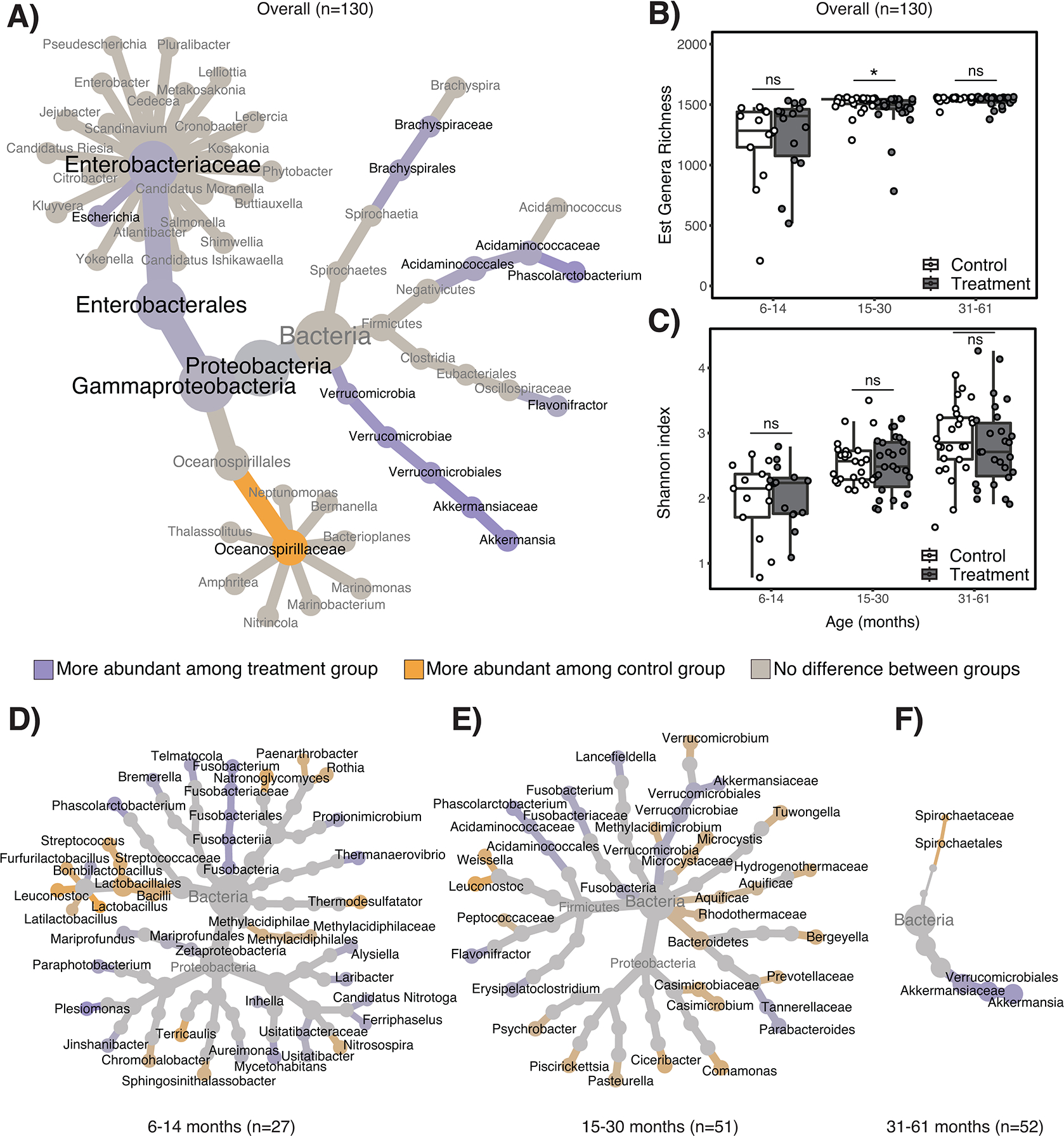
Panel A) is a differential heat tree depicting the taxonomies of bacterial genera and families that significantly differed in their relative abundance between treatment and control children. Genera within differentially abundant families are also depicted. For any given taxonomic level, only taxa that were significantly less abundant (orange) or more abundant (purple) among treatment relative to control children are depicted in color; non-significant taxa are depicted in gray. Panel B) depicts estimated genera richness and Panel C) depicts Shannon diversity indices for treatment and control children, stratified by child age. Estimated genera richness differed between treatment and control children aged 15–30 months by two-sided Wilcoxon signed-rank test (p=0.04); Shannon diversity did not significantly differ for any age stratum using two-sided Wilcoxon signed-rank tests. For all box plots, center line indicates the median; box limits indicate the upper and lower quartiles; and whiskers indicate 1.5x interquartile range. Panels D) – E) are differential heat trees depicting the taxonomies of bacterial genera and families that significantly differed in their relative abundance between treatment and control children aged D) 6–14 months, E) 15–30 months, and F) 31–61 months, controlling for study site.
Note: ns=non-significant. *indicates p<0.05 by Wilcoxon signed-rank test.
Figure 2. Effects of automated water chlorination on the taxonomic structure of children’s gut microbiomes in urban Bangladesh.
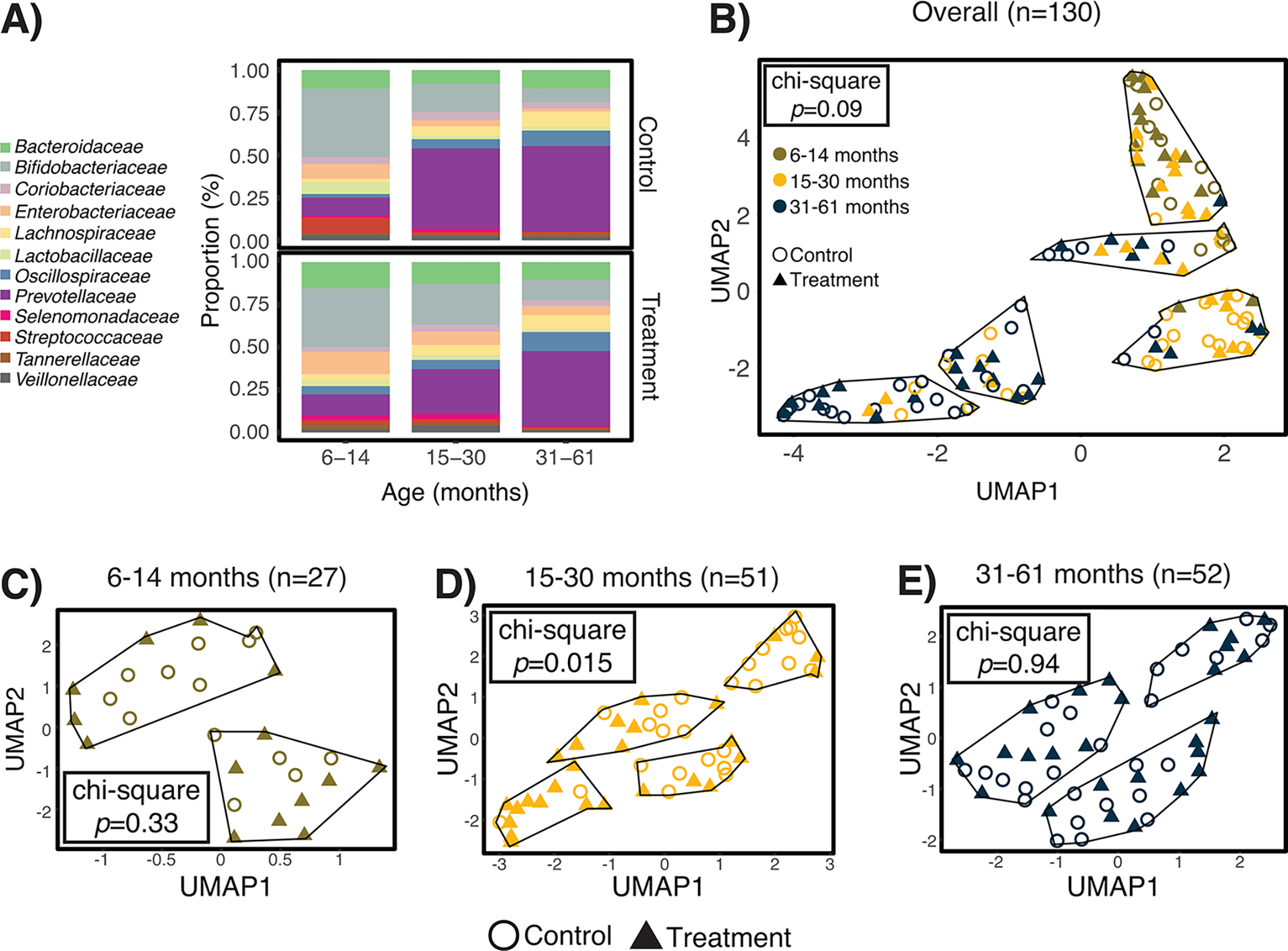
Water chlorination impacted the structure of the gut microbiomes of children aged 15–30 months, but not younger or older children. Panel A) depicts the average relative abundance of bacterial families with ≥1% mean relative abundance across all samples. Panel B) is a two-dimensional representation of the pairwise genomic distances between each sample, as identified by Mash. We observed a marginal association between treatment status and cluster classification by the chi-square test. Panels C-E) depict pairwise distances and resulting clusters when stratified by child age. Treatment status was only associated with cluster classifications among children aged 15–30 months.
The effects of water chlorination on the relative abundance of gut bacteria were modified by age (Figure 1D–E). Among the oldest children (age 31–61 months), there were fewer differentially abundant genera between treatment and controls (i.e., 1 genera compared to 19 differentally abundant genera in children aged 15–30 months and 34 among children aged 6–14 months) (Extended Data Table 1). A substantial portion of metagenomic reads could not be classified to any taxonomy, and the proportion of unclassified reads in each sample increased from an average of 13% at age 6–14 months to 42% at age 31–61 months (Extended Data Fig. 2).
Because of the open cohort study design, study participants were exposed to the intervention for varying durations. To determine whether longer exposure to the intervention, (i.e., ≥6 months) was associated with different impacts on the gut microbiome, we conducted a sub-group analysis among children older than 14 months (since age would be correlated to exposure time for children aged 6–14 months). Among all children older than 14 months (n=103), we identified three genera that were signficantly more abundant among treatment versus controls (Extended Data Table 2). The direction of the effect was maintained for each of these genera among the subset exposed for at least six months (n=91), although only one effect estimate remained significant.
Treatment effects on gut microbial richness were limited to children aged 15–30 months
Estimated genera richness was fairly constant across each age stratum, and we did not observe any association between richness and treatment status among the youngest (6–14 months) or the oldest (31 months and older) children. However, among children aged 15–30 months, water chlorination was associated with lower estimated richness on average (Figure 1B). Shannon diversity was lowest among the youngest children (age 6–14 months) and progressively higher among each successive age stratum, a trend that is well-described.2 We observed no association between Shannon diversity and treatment status within any age stratum (Figure 1C).
We identified clusters of similar gut metagenomes by calculating pairwise distances between each sample. Among all children, metagenome clusters were primarily defined by age group (p<0.001; two-sided chi-square test), rather than exposure to chlorinated water (p=0.09) (Figure 2B). Age group stratified analyses revealed treatment status was associated with clustering only among children aged 15–30 months (p=0.015; Figure 2C–E).
Water chlorination was not associated with presence of specific bacterial pathogens
We observed no impacts of automated water chlorination on the occurrence of several bacterial pathogens that can cause enteric infections in children in Bangladesh,22 after adjusting for child’s age and study site (Table 2). Using a qualitative multiplex pathogen assay, among the children selected for metagenomic sequencing for whom raw stool aliquots were available for analysis (125/130), we found that 75% of children harbored at least one bacterial pathogen, and children harbored 1.3 bacterial pathogens on average (SD: 1.0 pathogens). Campylobacter spp., Salmonella spp., Enterotoxigenic E. coli [ETEC], and Shigella spp. were detected in at least 10% of children’s stool, while Shiga toxin-producing E. coli [STEC] and Clostridioides difficile were rarely detected (<5%). We repeated this analysis using the full set of children’s stool samples from the original trial (n=527) and with a broader set of 14 gastrointestinal pathogens, including viral and protozoan targets. Despite increased power to detect differences, we did not observe significant reductions or increases in any of these pathogens among children receiving chlorinated water (Extended Data Table 3).
Table 2.
Detection of 8 bacterial pathogens in the stool of 125 children participating in a cluster-randomized automated water chlorination trial following 10 months of exposure, on average.a
| Control n=61 (%) |
Treatment n=64 (%) |
RR (95% CI) | Adjusted p-value | |
|---|---|---|---|---|
|
| ||||
| Campylobacter spp. | 10 (16) | 16 (25) | 1.42 (0.65, 3.25) | 0.81 |
| Salmonella spp. | 15 (25) | 25 (39) | 1.56 (0.83, 3.02) | 0.81 |
| Enterotoxigenic E. coli (ETEC) LT/ST | 22 (36) | 20 (31) | 0.87 (0.47, 1.60) | 0.81 |
| Shigella spp. | 19 (31) | 23 (36) | 1.17 (0.64, 2.18) | 0.81 |
| C. difficile | 3 (5) | 3 (5) | 0.90 (0.17, 4.90) | 0.90 |
| Shiga-like toxin-producing E. coli (STEC) stx1/stx2 | 4 (7) | 0 | -- | -- |
| Vibrio cholerae | 0 | 0 | -- | -- |
Note: Relative risk ratios rates (RR) were calculated using Poisson regression models adjusted for child’s age and study site. Resulting two-sided p-values were adjusted for multiple comparisons using the Benjamini–Hochberg method. RRs, associated 95% CIs, and adjusted p-values are only presented for pathogens that were detected among at least 5% of samples. 75% of children harbored at least one bacterial pathogen; 67% of children harbored at least one non-Escherichia-related bacterial pathogen.
Raw stool aliquots from 125 of 130 children were available for pathogen analysis.
ARGs were enriched among children receiving chlorinated water
Study children frequently harbored ARGs in their guts, and resistance to the same antibiotic classes was observed in the treatment and control groups (Extended Data Fig. 3A). Resistance to some antibiotic classes was enriched among treatment children (Extended Data Fig. 4), including sulfonamides and quinolones (all age strata), trimethoprim (children aged 15–30 months), macrolides (15–30 months), and aminoglycosides (31 months and older) (treatment coefficient range for above drug classes: 0.4–1.5). ARGs conferring resistance to beta lactam antibiotics were less abundant among treatment children aged 15–30 months (treat. Coeff: −0.84, 95% CI: −1.37, −0.32), relative to controls.
We also examined chlorination-induced effects on the occurrence and relative abundance of individual ARGs. Several clinically relevant ARGs were detected among both treatment and control children, including blaCTX-M alleles (conferring third-generation cephalosporin resistance), mph(A) (azithromycin resistance), and qnrS1 (low-level fluoroquinolone resistance) (Extended Data Fig. 5). We observed no significant difference (using a fdr-adjusted p-value threshold of 0.05) in the presence/absence of any ARG between treatment and control children when controlling for age and study site. We did, however, observe differences in the relative abundance of several ARGs (Figure 3A). Among all children receiving chlorinated water we observed a higher relative abundance of mdf(A) and tet(A), (multidrug and tetracycline resistance-conferring efflux pumps, respectively), sul2 (sulfonamide resistance) and aadA5 (streptomycin and spectinomycin resistance). Among specific age strata we observed increases in additional ARGs, including tet(X) (tetracycline resistance) among children aged 6–14 months; erm(X) (cross-resistance to macrolides, lincosamides, and streptogramins), mph(A), and nimE (nitroimidazole resistance) among children aged 15–30 months; dfrA17 (trimethoprim resistance) among children aged 31–61 months; and blaTEM (penicillin resistance) among children aged 15 months and older (treatment coefficient range for above ARGs: 0.9–7.3). ARGs that encode efflux pumps were slightly enriched in the treatment group (treat. coef.: 0.3, 95% CI: 0, 0.5), but the direction and significance of this effect was not consistent across age groups. The ant(6’)-Ia gene (aminoglycoside resistance) was less abundant among treatment children aged 15–30 months relative to control children (treat. coeff.: −1.3, 95% CI: −2.2, −0.37).
Figure 3. Effects of automated water chlorination on the richness, diversity, and relative abundance of antibiotic resistance genes (ARGs) harbored by children’s intestinal flora in urban Bangladesh.
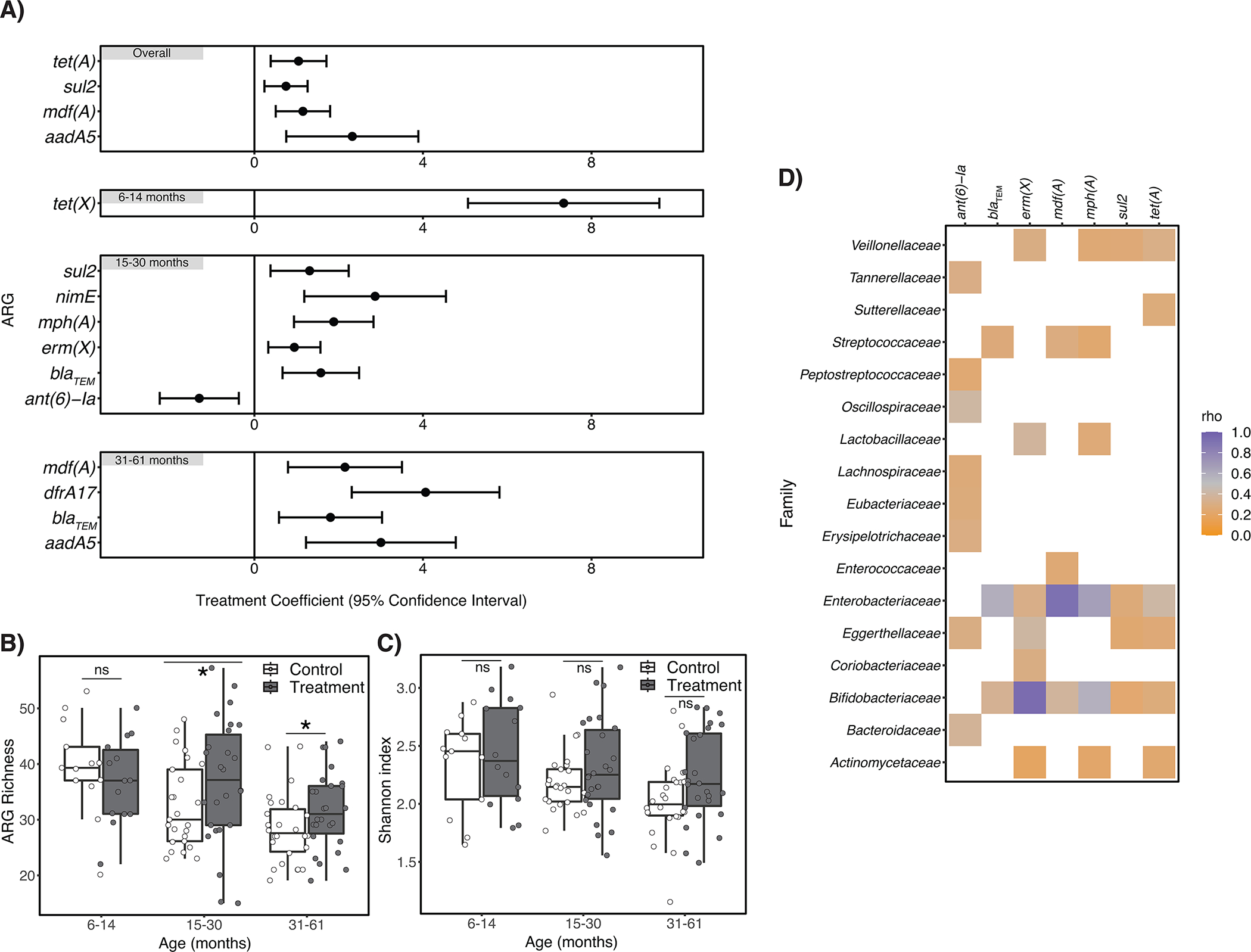
Panel A) describes ARGs that were differentially abundant between treatment and control children (n=130 biologically independent samples), controlling for study site (and child age in the “Overall” panel only). Error bars depict the 95% confidence interval around the treatment coefficient. Positive treatment coefficient values indicate ARGs were more abundant among treatment children relative to controls; negative values indicate ARGs were less abundant. Panel B) depicts the estimated number of unique ARGs (i.e., ARG richness) detected in children’s fecal metagenomes, stratified by child age. Estimated ARG richness significantly differed between treatment and control children aged 15–30 months (p=0.015) and 31–61 months (p=0.016) controlling for study site and using the betta function of the R package breakaway. Panel C) depicts the Shannon diversity indices for treatment and control children, stratified by child age. There was no statistical association between treatment status and ARG diversity for any age stratum by two-sided Wilcoxon signed-rank test. For all box plots, center line indicates the median; box limits indicate the upper and lower quartiles; and whiskers indicate 1.5x interquartile range. For Panels A) – C) the number of biologically independent samples considered for each age strata is as follows: 6–14 months, n=27; 15–30 months, n=51; 31–61 months, n=52. Panel D) is a heatmap depicting the strength of the association between ARGs listed in Panel A) that occurred in at least half of samples and bacterial families that occurred in at least half of samples, as determined by Spearman correlation tests. Resulting rho values are only depicted for statistically significant correlations (p<0.05 after adjustment for multiple testing using the Benjamini–Hochberg method.).
Note: ns=non-significant. *indicates p≤0.05.
Exposure to chlorinated water was also associated with estimated ARG richness in the gut (p=0.019),23,24 when controlling for age and study site. Specifically, we observed higher ARG richness among treatment relative to control children in the 15–30 months and 31–61 months age strata (p=0.015 and p=0.05, respectively) (Figure 3B). We also identified a marginal increase in the Shannon diversity of ARGs among treatment children aged 31–61 months relative to controls (p=0.07), but not among other age strata (Figure 3C).
Antibiotic use in the two months prior to stool collection was most frequently reported among the youngest children (17/27) and least commonly reported among children aged 31–61 months (10/52) (Extended Data Fig. 3B). Caretakers rarely reported the types of antibiotics used. Reported antibiotic use was not associated with ARG richness or ARG Shannon diversity in children’s stool (p=0.30 and p=0.14, respectively, by Wilcoxon signed-rank test).
Differentially abundant ARGs were correlated with specific bacterial taxa
We observed statistically significant Spearman’s correlations (fdr-adjusted p-value<0.05) between the relative abundance of several bacterial families and the ant(6)-1a, blaTEM, erm(X), mdf(A), mph(A), sul2, and tet(A) (Figure 3D) genes, indicating potential origins for these differentially abundant ARGs. In particular, we observed very strong correlations (rho>0.9) between mdf(A) and Enterobacteriaceae25 and erm(X) and Bifidobacteriaceae,26 associations which are well-described. We also observed a strong correlation (rho=0.66) between the relative abundance of mph(A) and Enterobacteriaceae. Of note, Escherichia spp., which belong to the family Enterobacteriaceae, were enriched among children receiving chlorinated water. Other significant correlations were observed between bacterial families and ARGs that were differentially abundant between treatment and control children, but were moderate to weak in strength (i.e., rho<0.6).
Discussion
In this analysis of fecal metagenomes from 130 children who participated in a cluster-randomized water chlorination trial in urban Bangladesh, automated water chlorination at the point of collection impacted children’s gut microbiomes, including the resistance genes they harbored, though shifts in taxa abundance were generally small in magnitude. Children receiving chlorinated water harbored a higher abundance of bacterial genera that are often detected in the human gut, including Akkermansia spp., Flavonifractor spp., Phascolarctobacterium spp., and Escherichia spp. Despite these changes, water chlorination did not impact the overall richness or diversity of children’s gut microbiomes and was not associated with differences between children’s gut microbiome communities, except among children aged 15–30 months. These findings suggest that automated water chlorination, an effective strategy for reducing child diarrhea and associated antibiotic demand,11 does not substantially impact children’s developing gut microbiomes.
Children receiving chlorinated water harbored a higher abundance of several bacterial genera previously linked to healthy guts. Akkermansia was more abundant among treatment children overall, relative to controls, and comprised 0.13% of bacterial reads in children’s fecal metagenomes. A. muciniphila is a mucin-degrading gut commensal that improves intestinal barrier integrity27 and reduces gut inflammation.28 In addition, Flavonifractor was approximately twice as abundant among treatment compared to control children and comprised 0.19% of bacterial reads in children’s fecal metagenomes. F. plautii metabolize flavonoids, which are naturally-occurring compounds common in fruit and vegetables; the by-products of this metabolic activity modulate gut inflammation and weight gain.29,30 However, F. plautii have been associated with some disease states (e.g., colorectal cancer).31 Both A. muciniphila and F. plautii are being explored as probiotic targets.32,33 Phascolarctobacterium spp. are commonly detected in the human gut34 and were four times as abundant among treatment compared to control children. Phascolarctobacterium spp. solely metabolize luminal succinate, which is a key growth substate for enteric pathogens like Clostridioides difficile and Salmonella serovar Typhimurium.35,36 By reducing bioavailable succinate, Phascolarctobacterium spp. may strengthen gut colonization resistance.35 Finally, Escherichia spp. were more abundant in treatment compared to control children. Escherichia spp. include commensal strains that contribute to colonization resistance against bacterial pathogens,37 as well as strains that cause diarrhea or other infections.38 Notably, pathogenic E. coli (e.g., ETEC, STEC) were no more common among treatment relative to control children by the highly sensitive (>90%) and specific (≥99%) qualitative multiplex assay we used,39 suggesting that the higher relative abundance of Escherichia in the treatment group was not driven by increases in pathogenic E. coli. Overall, the higher relative abundance of several human enterobacteria among treatment children compared to controls suggests that chlorination-mediated impacts on the gut microbiome could support engraftment of commensal strains.
Water chlorination was associated with microbiota clustering and diminished genera richness among children aged 15–30 months, but this effect was not observed in younger or older children. In a longitudinal study of children born in Europe and the United States, the 15–30 month age range was associated with the “transitional phase” of gut microbiome development, characterized by significant shifts in dominant phyla and Shannon diversity.1 Further studies are needed to examine why children’s gut microbiomes may be affected by water chlorination specifically during this age range and what impacts (if any) this may have on future gut composition. Notably, the impacts of water chlorination on the differential abundance of gut taxa were weakest among children in the oldest age stratum (ages 31 months and older). This could suggest that the microbial gut community is less perturbable by water chlorination later in life. Longitudinal analyses are needed to capture if the subtle impacts of water chlorination on children’s developing gut microbiomes that we describe here have any long-term health effects.
A relationship between the observed microbiome shifts in this study and the finding from the parent trial that water chlorination reduced child diarrhea is possible but not confirmed. The parent trial (in which this study was nested) leveraged >4,000 child observations to detect a 23% relative reduction in diarrhea prevalence over a 1-year time period.11 Among the 130 children included in this study, we observed no differences in the presence of eight diarrheal pathogens between treatment and control children. This could be due to our small sample size, seasonal variability in diarrhea prevalence, or because the presence of enteric pathogens does not necessarily imply a disease state. Instead, growing evidence from high child mortality settings suggests that many enteric pathogens are just as commonly detected among children with no clinical manifestations of diarrhea,40 suggesting that other factors like immunity, exposure dose, inflammation, or intestinal barrier functionality likely play a role in determining disease onset.41 Here, we observed that several gut commensal bacteria were enriched among children receiving chlorinated water; it is possible that the metabolic by-products of these bacteria, their interactions with other gut microbes, or other unmeasured changes to the gut microbiome environment could have improved gut colonization resistance and immunity more broadly.42 Longitudinal stool and environmental sampling in future water chlorination intervention trials could help elucidate whether reductions in child diarrhea from access to chlorinated drinking water are due to disrupted exposures to human, animal, and environmental-origin pathogens, and/or due to improved gut health conferred by shifts in microbiota.42,43
Because children randomized to the water chlorination intervention in the parent trial experienced a 7% reduction in recent antibiotic use, we hypothesized that water chlorination could indirectly reduce the relative abundance of ARGs in children’s guts.9 The water chlorination intervention instead increased the relative abundance of several ARGs in children’s guts, which was likely the result of a higher relative abundance of Enterobacteriaceae spp. among treatment children versus controls. Enterobacteriaceae spp., like E. coli, frequently harbor mobile ARGs, especially in Bangladesh.25,44,45 Our findings suggest that in settings where mobile ARGs are exceptionally common among commensal bacteria,25,44,45 water chlorination alone may be insufficient to reduce the burden of ARGs circulating in the community. Other interventions might be necessary to curb the selection and spread of antibiotic resistance, not just in human communities but also in the food animal production sector, where many of the same antibiotics are used.46 Notably, because we did not use functional genomics to characterize all resistance mechanisms present in children’s fecal microbiomes, we may have missed effects on novel ARGs.
Our findings should be interpreted in the context of this study’s limitations. First, due to the open cohort study design children were exposed to the intervention for varying durations, though most children were exposed for at least six months. Duration of exposure was strongly correlated with age, so we did not control for this covariate in our analyses. Second, stool samples were not collected at the time of enrollment, which could have helped identify preexisting differences in gut community composition between the randomly selected subset of treatment and control children, although major differences are unlikely given the randomized study design. Baseline survey data in the parent trial indicated that the treatment and control groups were well balanced across a range of socioeconomic and child health variables that could plausibly influence gut community composition.11 Third, a substantial proportion of children’s fecal metagenomic reads could not be classified to any known taxonomy, comprising an average of 42% of reads among the oldest children. This may have affected our ability to assess the relative effect of chlorination of children’s gut microbiomes, given that our analyses were based solely on the classifiable fraction. The unclassified portion that we observed is comparable to other studies that have included fecal metagenome data from LMICs (42%–68%),47,48 providing further evidence of biases in existing taxonomy databases towards high-income, Western countries.49 Finally, control children in the parent trial received water dosed with trace amounts of Vitamin C, which could have conferred a nutritional benefit. The impacts of Vitamin C on the gut microbiome are not well-known.50 One pilot trial of healthy adults in Europe51 found that high doses (500 mg/day) administered directly to the colon increased genera richness and the relative abundance of some bacterial taxa, including Akkermansia, but the setting, study population, and administration route are not comparable to the present study. Given that Vitamin C was dosed at very low levels, we anticipate that any subclinical changes to the gut microbiome would not have been distinguishable from natural variation in gut microbiome development among children in this setting.
Conclusion
This study experimentally evaluated the impact of water chlorination, the most common form of drinking water disinfection worldwide, on children’s developing gut microbiomes. Because we leveraged stool samples from a double-blinded, placebo-controlled cluster-randomized automated water chlorination trial, water chlorination can be causally attributed to the outcomes measured here. Our findings suggest that water chlorination did not substantially affect the developing gut microbiomes of children in urban Bangladesh. Specifically, while we observed chlorination-induced shifts in the relative abundance of some bacteria taxa, including beneficial gut commensals, we observed no effects on overall gut genera richness or diversity. However, long-term studies may be needed to confirm that these types of subtle changes do not affect health later in life. Overall, the benefits of automated water chlorination with regards to preventing child diarrhea, reducing antibiotic use, and protecting child health appear to outweigh any potential changes to gut microbiome development in this setting.
Methods
Stool collection
Stool samples were collected from children participating in a double-blinded, cluster-randomized automated water chlorination trial implemented from July 2015-December 2016 in two low-income communities in urban Bangladesh: Tongi, a community outside Dhaka city, and Dhaka Uddan, a community within Dhaka city.11 In brief, shared water taps that served as the primary source of drinking water for children younger than five years old were identified in each community, then randomized to either the treatment (n=50) or control (n=50) arms. Although water taps were selected based on reliance as a primary drinking water source, water from these taps was used for all purposes, including bathing, cleaning, and washing clothes. Over 14 months, treatment water points were automatically dosed with chlorine using a passive water chlorination device, achieving a mean chlorine residual of 0.37 ppm, while control water points were dosed with trace amounts of Vitamin C (active control).11 Vitamin supplementation was included as an active control to improve acceptability to study participants and the local human subjects protection board. Vitamin C was specifically chosen by study investigators because tablets compatible with the dosing devices could be acquired.
Survey data were collected approximately every two months from an open cohort of children younger than five years old, of which 1,036 were enrolled at baseline. The trial used an open cohort study design given high migration rates in both communities. New children living in compounds served by either a treatment or control water tap were continuously enrolled at every follow-up survey round. Field workers confirmed at each study visit that the primary source of drinking water for each enrolled child was either a control or treatment pump. Across all survey rounds in the parent trial, less than 4% of households reported using a secondary water source for drinking.11 Both study participants and researchers processing the samples and performing data analysis were unaware of which households were served by chlorinated taps (double-blinded). This study and the cluster-randomized trial in Bangladesh11 followed the CONSORT checklist for cluster-randomized trials (Extended Data). The study protocol for the original trial was approved by the International Centre for Diarrhoeal Diseases Research, Bangladesh (icddr,b) scientific and ethical review committees (protocol number 14022) and the Stanford University human subjects institutional review board (protocol number 30456). Field staff obtained informed written consent from the owner of each enrolled water point and all study participants, including consent for biospecimens to be used for future unplanned analyses.
Children’s stool samples were collected approximately one year into the trial. Households were provided stool collection kits that included latex gloves and sterile scoops and were instructed on safe collection procedures prior to handling stool. Following stool production, the child’s mother or other caretaker was instructed to immediately transfer a small amount of feces into pre-prepared vials containing 1 mL of RNAlater (Invitrogen), which has been demonstrated to be a suitable preservative for fecal samples,52 and then invert the tube a few times. Field staff conducted up to three follow-up visits per household to retrieve specimens. Field staff transported children’s stool samples to the icddr,b laboratory within 8 hours. Stool aliquots stored in RNAlater were frozen at −80°C upon arrival at icddr,b in Bangladesh and remained frozen during storage and transport on dry ice to Tufts University in the United States for DNA extraction.
Sample selection for metagenomic sequencing
We performed short-read metagenomic sequencing on RNAlater-preserved stool specimens collected from children older than 6 months in the control and treatment groups. Sample selection was done by stratified random sampling of archived child stool samples to balance across treatment and control groups, study community (i.e., Dhaka Uddan versus Tongi), and across three age groups that others have demonstrated to correspond to distinct phases of gut microbiome development (i.e., 6 –14 months, 15–30 months, 31 months and older).1 Total DNA was extracted from approximately 0.25 g of frozen feces at Tufts University using the QIAamp PowerFecal DNA Kit (Qiagen) according to manufacturer’s instructions and quantified using a Qubit 4 fluorometer (Invitrogen). Extraction blanks were included with each batch of extractions; DNA concentrations were below the Qubit level of detection (<0.1 ng/μL) for all. We performed duplicate extractions on two stool samples. DNA extracts were sent to Novogene (UK) Company Limited for short-read, paired-end 150 bp sequencing on an Illumina Novaseq 6000 System using SP4 flow cells to achieve 10 Gb per sample.
Processing metagenomic data
Fastq sequences were trimmed and filtered to remove sequencing adaptors and low quality reads using Trimmomatic v0.36. We tabulated the number of raw reads for each sample and excluded from further analysis any sample with fewer reads than two standard deviations below the mean. Human contaminant sequences were removed from each sample by discarding reads that mapped to a non-redundant version of the Genome Reference Consortium Human Build 38 (GRCh38; www.ncbi.nlm.nih.gov) using Bowtie2 v2.2.3. Human sequence-filtered raw reads were deposited in the Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) under the project number PRJNA726052. We randomly chose one extraction duplicate per sample to include in subsequent metagenomic analyses.
Profiling taxonomy and classifying source
Taxonomic assignment of short reads was performed using Kraken2, using its standard built-in database comprising all complete bacterial, archeal, and viral genomes in NCBI’s RefSeq at the time of build (03 June 2021).53 We used the Bracken species-level sequence abundance estimation algorithm to estimate organism abundance at every taxonomic level.54 For all analyses, we considered organisms classified as bacteria in NCBI’s taxonomy database (https://ftp.ncbi.nlm.nih.gov/pub/taxonomy/), downloaded 23 November 2021.
Identifying ARGs and calculating abundance
We screened for ARGs by mapping short reads to the Resfinder database (v. 3.1.1) using the KMA tool.55 We considered matches with >90% coverage and >95% identity to be true hits. To examine the abundance of ARGs across samples, we calculated the RPKM (Reads Per Kilobase Million) for each ARG in a sample as the number of hits divided by the total number of matched paired-end bacterial reads per million for that sample, then divided by the ARG length in kilobases. To visualize the relative abundance of ARG classes across samples, we summed the RPKM for all ARGs belonging to a class.
Clustering similar metagenomes
All-against-all genomic distances were estimated between each metagenome using Mash.56 We reduced the dimensionality of the resulting matrix using UMAP from the uwot package (https://github.com/jlmelville/uwot) and used the MClust R package v5.4.7 57 to classify samples into clusters. The MClust algorithm chooses the number of clusters and the specific clustering model (of eight models considered) to maximize the BIC; a minimum of one cluster is considered by default and we did not assign a maximum.57,58 We repeated this same process within each age stratum after subsetting the Mash distance matrix. We used chi-square tests to examine associations between treatment status and cluster classification, both within each age group and overall. All p-values are reported in Figure 2.
Identifying gastrointestinal pathogens
Given that rare taxa like gastrointestinal pathogens are difficult to capture using metagenomic sequencing approaches, and that pathogenic E. coli (e.g., ETEC, STEC) may be impossible to distinguish from commensal strains using short-read sequencing data, we used a qualitative multiplex assay with >90% sensitivity and ≥99% specificity, the Luminex xTAG® Gastrointestinal Pathogen Panel, to determine pathogen occurrence.39 For children whose stool samples were analyzed using short-read metagenomic sequencing, we examined additional raw aliquots of their stool specimens for the presence of 8 bacterial pathogens, including Campylobacter spp.; Clostridioides difficile; Enterotoxigenic E. coli (ETEC) LT/ST; Shiga-like toxin-producing E. coli (STEC) stx1/stx2; Salmonella spp.; Shigella spp.; Yersinia enterocolitica, and Vibrio cholerae. We also examined all stool specimens collected during the parent trial for a broader set of 14 pathogens, including the aforementioned bacterial pathogens as well as adenovirus 40/41; norovirus GI/GII; rotavirus A; Giardia spp.; Cryptosporidium spp.; and Entamoeba histolytica. Methods have been described in detail elsewhere.59 Briefly, we followed manufacturer instructions’ for the pretreatment, extraction, and analysis of stool samples by the Luminex xTAG® Gastrointestinal Pathogen Panel. DNA and RNA were extracted using the QIAcube HT platform and the QIAamp 96 Virus QIAcube HT Kit (Qiagen, Hilden, Germany).
Comparing extraction duplicates
We compared the taxonomic profiles of both sets of extraction duplicates (Extended Data Fig. 9) and observed no clear differences in the relative abundance of any bacterial family or genera that comprised at least 1% of bacterial reads, on average, across all of the samples we sequenced (n=132). We observed some discordance in the genera that were identified within each extraction pair (3 discordant genera versus 1552 concordant genera among extraction duplicates for Sample A; 85 discordant genera versus 1063 concordant genera among extraction duplicates for Sample B); however, all discordant genera were of exceptionally low abundance (<0.007%). We randomly chose one extraction duplicate per sample to include in subsequent metagenomic analyses.
Data Analysis
We examined differences in bacterial taxa (primary outcome), ARGs (secondary outcome), and the occurrence of gastrointestinal pathogens (secondary outcome) between treatment and control children using several approaches. Analysis of differences in bacteria taxa and ARGs were not planned as part of the original trial, though differences in the occurrence of gastrointestinal pathogens was pre-specified as a secondary outcome in the original trial’s registration and statistical analysis plan (available at: https://osf.io/t98bv/). The original trial was registered with ClinicalTrials.gov, number NCT02606981. Because nearly all children (126/130) lived in different households, each child’s fecal metagenome was considered as an independent data point for these analyses.
For bacterial taxa, we used beta-binomial regression models that account for variable sequencing depth to identify taxa that were differentially abundant between treatment and control children with the R package corncob v0.1.0,21 while controlling for the child’s age and study site. This was the primary outcome we examined. The Benjamini-Hochberg method was used to correct for multiple comparisons (default method for corncob).60 Only taxa that were present in at least 20% of samples were included in these analyses. Given frequent discordance in species-level taxonomy assignments among commonly used taxonomic labeling tools for short-read sequencing data,61 we only examined differences at the genera level and above. We filtered differentially abundant taxa that were exclusively driven by influential points from further analyses. Differentially abundant genera and families (as identified by corncob) were visualized using the metacoder R package v0.3.4.62 We also calculated two estimates of bacterial alpha diversity for each sample: richness, which describes the number of bacterial genera, and the Shannon index. Genera richness was estimated using the breakaway R package v4.7.3.24 Shannon diversity was calculated using the phyloseq R package v1.34.0.63 Differences between treatment and control children for each of these metrics were examined within each age stratum using Wilcoxon signed-rank tests.
We used logistic regression models to examine differences in the presence/absence of ARGs between treatment and control children, while controlling for age and study site. Because allelic variants of beta lactamase genes can differ by as few as one single nucleotide polymorphism, we considered these genes as groups (e.g., blaCTX, blaOXA, blaTEM) rather than as individual variants. Resulting p-values were adjusted for multiple comparisons using the Benjamini–Hochberg method. For ARGs that were common among children in this setting (present in at least 20% of samples), we used the R package corncob to identify differentially abundant ARGs between treatment and control children, while controlling for age and study site (secondary outcome). We separately used corncob to evaluate whether ARGs that function as efflux pumps were differentially abundant between treatment and control children, by creating a composite indicator variable for all efflux pump ARGs in our dataset. We determined the functional annotation of detected ARGs by cross-referencing with the Comprehensive Antibiotic Resistance Database (CARD).64 For any ARGs not listed in CARD (i.e., nimE, nimJ, mefA, tet(O/32/O), tet(O/W), tet(W/32/O), we determined their function by searching the literature. We used breakaway’s betta function23 to examine the effect of chlorination on estimated ARG richness while controlling for study site, both overall (analysis also controlled for age) and within each age stratum (i.e., 6 −14 months, 15–30 months, 31 months and older). We examined differences in ARG diversity between treatment and control children using Wilcoxon signed-rank tests.
To examine differences in gastrointestinal pathogen burden between treatment and control children, we used Poisson regression models to examine associations between treatment status and the presence of any pathogen that was harbored by at least 5% of children, while controlling for child’s age and study site (secondary outcome). The resulting p-values were adjusted for multiple comparisons using the Benjamini–Hochberg method.
We conducted a Spearman’s correlation analysis to investigate how changes in gut taxonomic structure might be driving changes in ARG relative abundance between treatment and control children. Only correlations between ARGs and bacterial families that occurred in at least half of samples were examined. The rho and p-values were calculated using the rcorr function of the Hmisc R package v4.4–2 (https://github.com/harrelfe/Hmisc/) and resulting p-values were adjusted for multiple comparisons using the Benjamini–Hochberg method.
All analyses were conducted in R v. 3.5.0 and R Studio v. 1.1.463. Unless otherwise specified, all figures were created using tidyverse v1.3.0 and ggplot2 v3.3.3.65 All p-values were two-sided and considered to be statistically significant at the 0.05 level.
Extended Data
Extended Data. Fig. 1. Treatment coefficients generated by the R package corncob, representing the additive change in the logit-transformed relative abundance of bacterial genera between treatment and control children, compared to the log of the ratio of the mean relative abundance among treatment children to the mean relative abundance among control children, i.e., the log fold change.
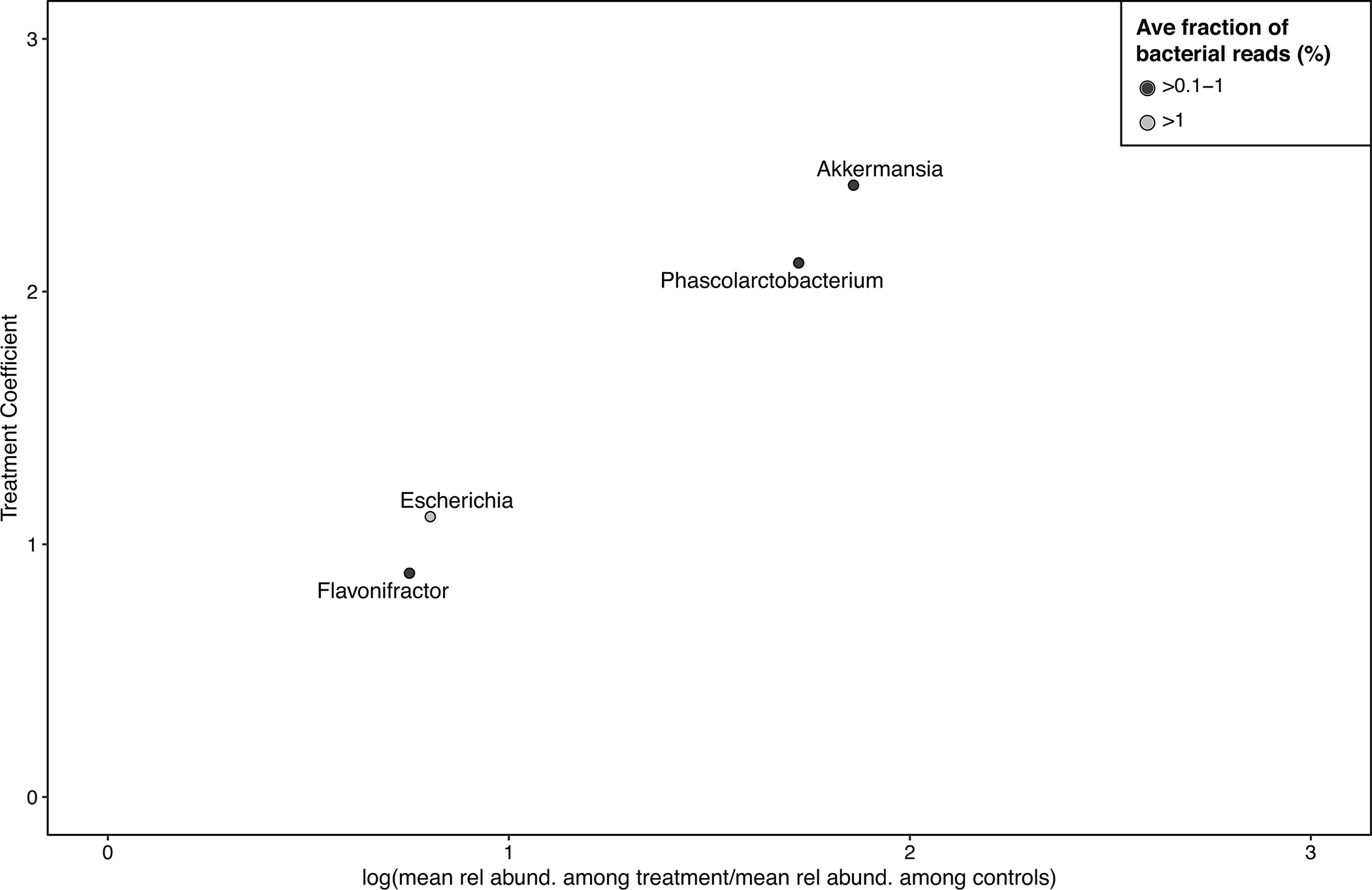
Treatment coefficients generated by corncob generally approximate the log fold change.
Extended Data Fig. 2. Average fraction of reads from 130 Bangladeshi children’s gut metagenomes that were not classified to any taxonomy by Kraken2, stratified by age.
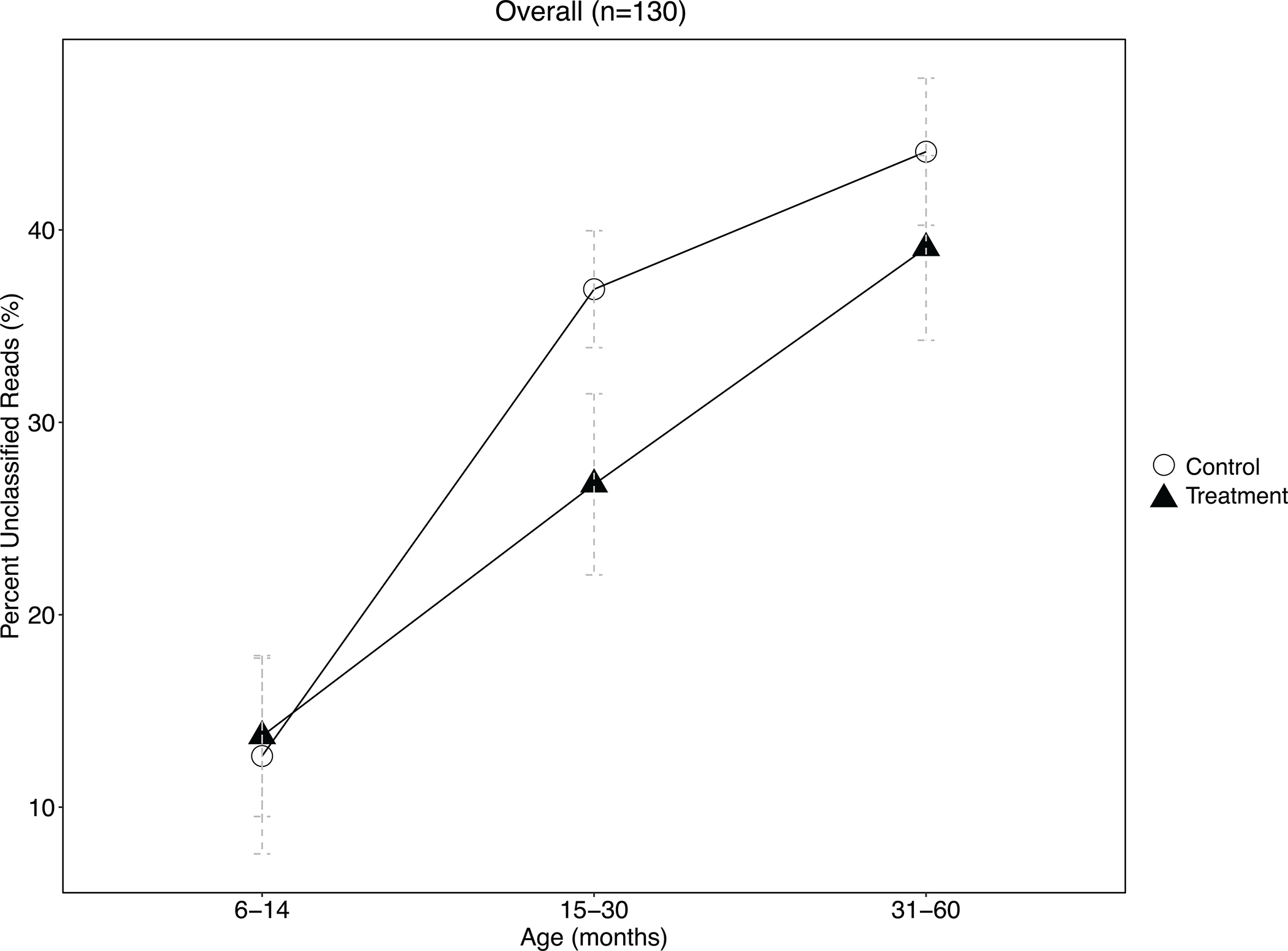
Error bars represent the 95% confidence interval around the mean. The proportion of unclassified reads significantly differed at the p=0.05 level between treatment and control children aged 15–30 months by a two-sided, two-sample t-test, but not among other age strata. The number of biologically independent samples considered for each age strata is as follows: 6–14 months, n=27; 15–30 months, n=51; 31–61 months, n=52.
Extended Data Fig. 3. Gut resistomes and antibiotic consumption patterns among 130 children participating in an automated water chlorination trial in urban Bangladesh.
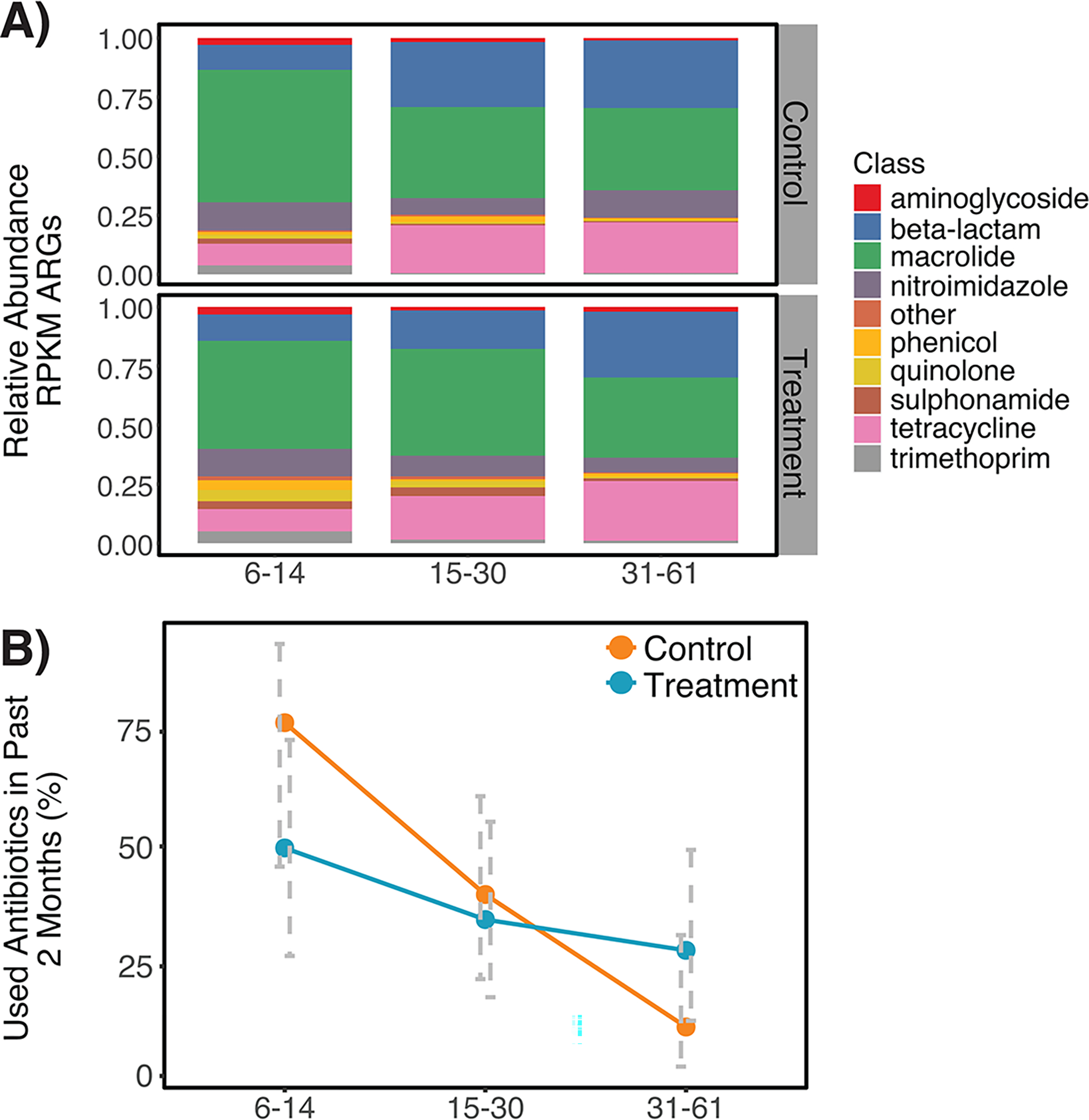
Panel A) depicts relative abundance of antibiotic resistance genes (ARGs) belonging to each ARG class harbored by the fecal metagenomes of control and treatment children, expressed as reads per kilobase of read per million (RPKM). Panel B) depicts the average proportion of control and treatment children whose caretakers reported they had consumed antibiotics in the two months prior to stool collection, stratified by age. Error bars represent the 95% confidence interval around the mean. Antibiotic use was significantly associated with age strata (p<0.001 by chi-square) but was not associated with treatment status in this subset of children from the parent trial. For both panels, the number of biologically independent samples considered for each age strata is as follows: 6–14 months, n=27; 15–30 months, n=51; 31–61 months, n=52.
Extended Data Fig. 4. Differences in resistance to specific antibiotic classes as detected in the fecal metagenomes of 130 children participating in an automated water chlorination trial in urban Bangladesh.
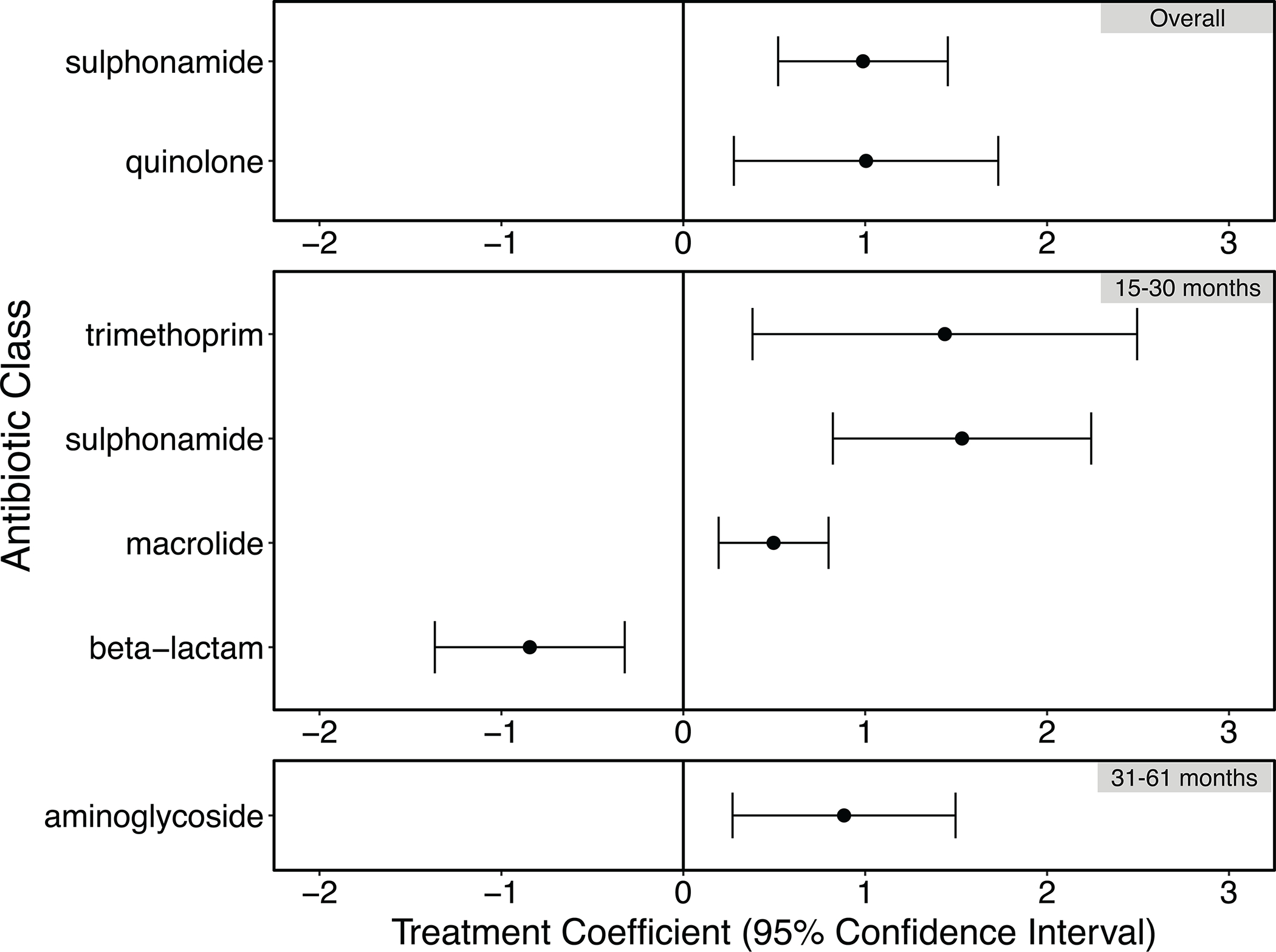
Treatment coefficients were generated by the R package corncob. Positive treatment coefficients indicate that ARGs belonging to the given antibiotic class were relatively more abundant among treatment children relative to controls; negative treatment coefficients indicate ARGs belonging to the given antibiotic class were relatively less abundant. Error bars depict the 95% confidence interval around the treatment coefficient. The number of biologically independent samples examined for each age strata is as follows: 15–30 months, n=51; 31–61 months, n=52. The “Overall” category included 130 biologically independent samples.
Extended Data Fig. 5. Relative abundance of genes conferring resistance to medically important antibiotics in the fecal metagenomes of 130 children participating in an automated chlorinated water intervention trial in urban Bangladesh.
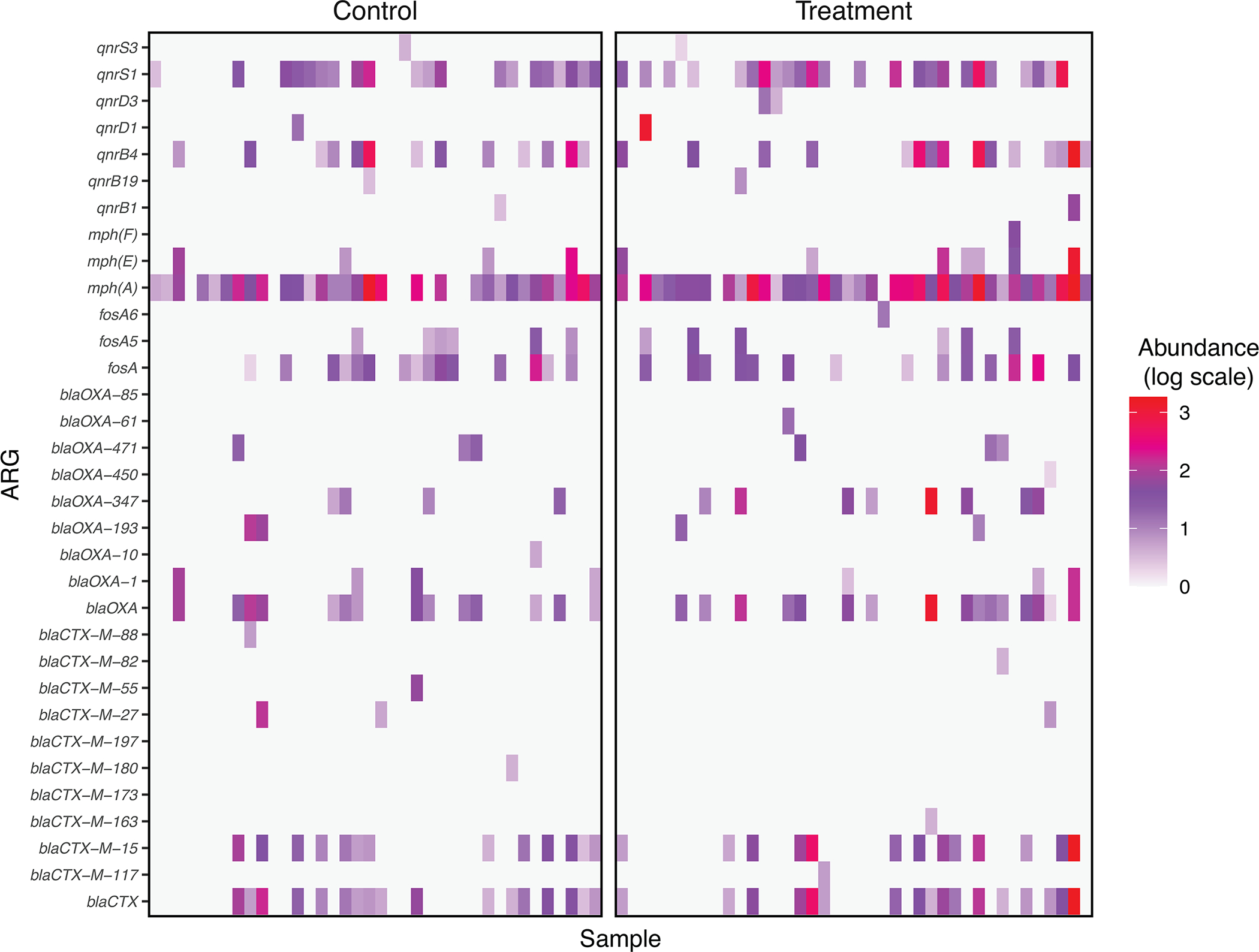
Genes conferring resistance to fluoroquinolones (qnr), azithromycin (mph), fosfomycin (fos), beta lactams (blaOXA), and third-generation cephalosporins (blaCTX) were detected. Genes conferring resistance to colistin and carbapenems, which are considered “last-resort” antibiotics, were not detected.
Extended Data Fig. 6. Comparison of two sets of extraction controls, extracted from the stool of a child aged 6–14 months (Sample A) and 31–61 months (Sample B).
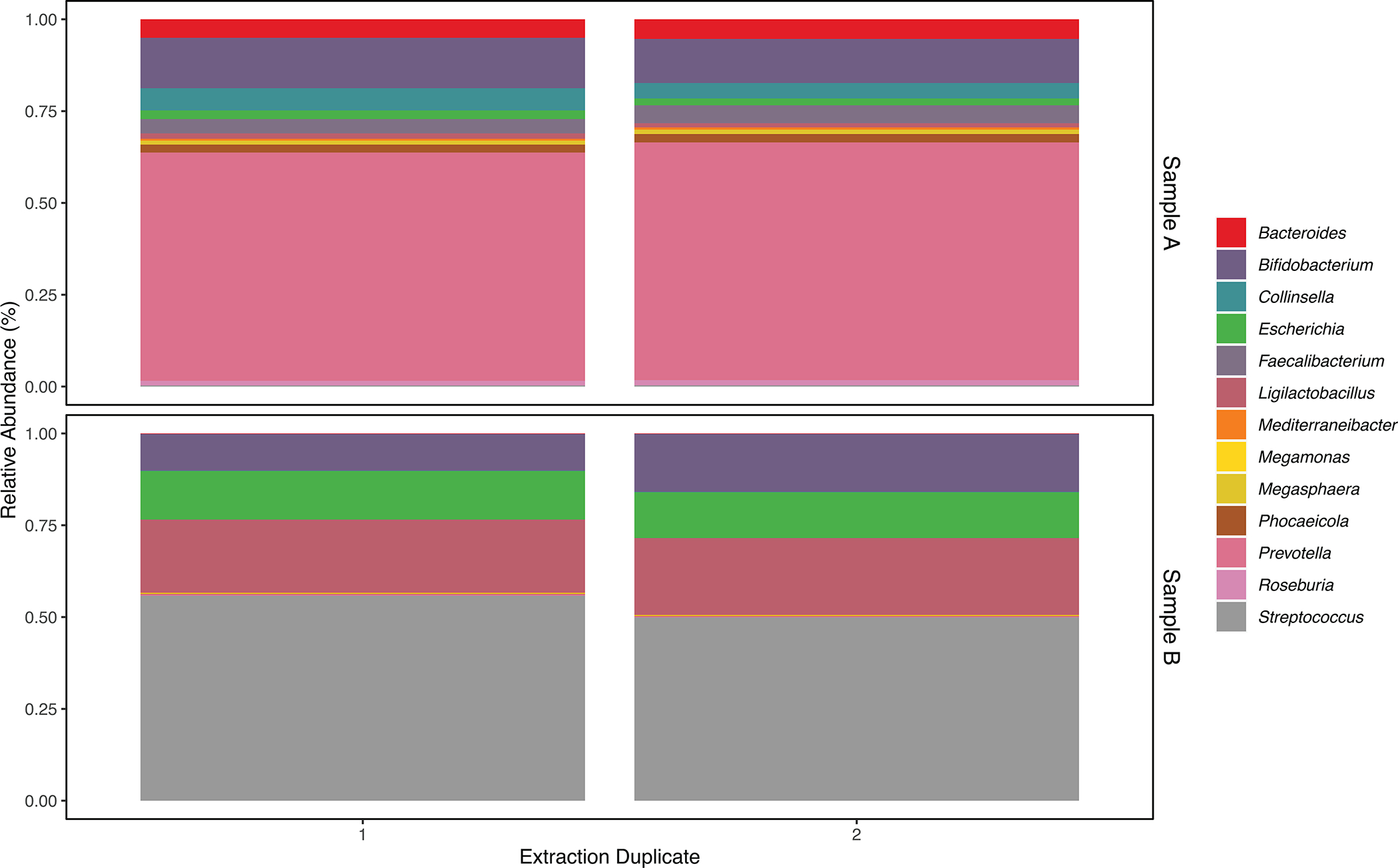
Within each set of duplicates, we observed a similar relative abundance of bacterial families and genera that comprised at least 1% of bacterial reads among all fecal metagenomes sequenced for this study. We observed some discordance in the genera that were identified within each extraction pair (3 discordant genera versus 1552 concordant genera among extraction duplicates for Sample A; 85 discordant genera versus 1063 concordant genera among extraction duplicates for Sample B); however, all discordant genera were of exceptionally low abundance (<0.007%).
Supplementary Material
Acknowledgements
We thank Nazrin Akter for excellent field management. This work was funded by the Thrasher Research Fund (#14205) and The World Bank Strategic Impact Evaluation Fund. M.L.N. was supported by NIH award KL2TR002545 and the Stuart B. Levy Center for Integrated Management of Antimicrobial Resistance at Tufts (Levy CIMAR), a collaboration of Tufts Medical Center and the Tufts University Office of the Vice Provost for Research (OVPR) Research and Scholarship Strategic Plan (RSSP). C.J.W. and A.M.E were supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (NIH) under award no. U19AI110818 to the Broad Institute. E.R.F. was supported by the NSF Postdoctoral Research Fellowships in Biology Program under Grant No. 1906957. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of any of the aforementioned funding organizations.
Role of the funding source
The funder had no role in data collection, data analysis, data interpretation, or writing of this report.
Footnotes
Competing interests statement
The authors report no relevant financial or non-financial competing interests.
Data Availability
All raw reads (human sequences removed) were deposited in the Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) under the project number PRJNA726052. Metadata are publicly available at the following link: https://osf.io/wb3pv/. We accessed the following publicly available database to conduct our analyses: NCBI’s taxonomy database (https://ftp.ncbi.nlm.nih.gov/pub/taxonomy/), downloaded 23 November 2021.
References
- 1.Stewart CJ et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 562, 583–588 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robertson RC, Manges AR, Finlay BB & Prendergast AJ The Human Microbiome and Child Growth – First 1000 Days and Beyond. Trends in Microbiology 27, 131–147 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Aversa Z et al. Association of Infant Antibiotic Exposure With Childhood Health Outcomes. Mayo Clinic Proceedings 96, 66–77 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Platts-Mills JA et al. Use of quantitative molecular diagnostic methods to assess the aetiology, burden, and clinical characteristics of diarrhoea in children in low-resource settings: a reanalysis of the MAL-ED cohort study. Lancet Glob Health 6, e1309–e1318 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carding S, Verbeke K, Vipond DT, Corfe BM & Owen LJ Dysbiosis of the gut microbiota in disease. Microb Ecol Health Dis 26, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borton MA et al. Chemical and pathogen-induced inflammation disrupt the murine intestinal microbiome. Microbiome 5, 47 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vaz LE et al. Recent trends in outpatient antibiotic use in children. Pediatrics 133, 375–385 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rogawski ET et al. Use of antibiotics in children younger than two years in eight countries: a prospective cohort study. Bull. World Health Organ 95, 49–61 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwartz DJ, Langdon AE & Dantas G Understanding the impact of antibiotic perturbation on the human microbiome. Genome Medicine 12, 82 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McDonnell L et al. Association between antibiotics and gut microbiome dysbiosis in children: systematic review and meta-analysis. Gut Microbes 13, 1–18 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pickering AJ et al. Effect of in-line drinking water chlorination at the point of collection on child diarrhoea in urban Bangladesh: a double-blind, cluster-randomised controlled trial. The Lancet Global Health 7, e1247–e1256 (2019). [DOI] [PubMed] [Google Scholar]
- 12.World Health Organization. Guidelines for drinking-water quality: fourth edition incorporating the first addendum. https://www.who.int/publications-detail-redirect/9789241549950. [PubMed]
- 13.Dai Z et al. Disinfection exhibits systematic impacts on the drinking water microbiome. Microbiome 8, 42 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Waak MB, Hozalski RM, Hallé C & LaPara TM Comparison of the microbiomes of two drinking water distribution systems-with and without residual chloramine disinfection. Microbiome 7, 87 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chiao T-H, Clancy TM, Pinto A, Xi C & Raskin L Differential resistance of drinking water bacterial populations to monochloramine disinfection. Environ Sci Technol 48, 4038–4047 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Shi P et al. Metagenomic insights into chlorination effects on microbial antibiotic resistance in drinking water. Water Res 47, 111–120 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Hou A-M et al. Chlorine injury enhances antibiotic resistance in Pseudomonas aeruginosa through over expression of drug efflux pumps. Water Res 156, 366–371 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Li D & Gu AZ Antimicrobial resistance: A new threat from disinfection byproducts and disinfection of drinking water? Current Opinion in Environmental Science & Health 7, 83–91 (2019). [Google Scholar]
- 19.Martino D The Effects of Chlorinated Drinking Water on the Assembly of the Intestinal Microbiome. Challenges 10, (2019). [Google Scholar]
- 20.Pop M et al. Diarrhea in young children from low-income countries leads to large-scale alterations in intestinal microbiota composition. Genome Biol 15, R76 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin BD, Witten D & Willis AD Modeling microbial abundances and dysbiosis with beta-binomial regression. Ann Appl Stat 14, 94–115 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharif N et al. Molecular and Epidemiologic Analysis of Diarrheal Pathogens in Children With Acute Gastroenteritis in Bangladesh During 2014–2019. Pediatr Infect Dis J 39, 580–585 (2020). [DOI] [PubMed] [Google Scholar]
- 23.Willis A, Bunge J & Whitman T Improved detection of changes in species richness in high diversity microbial communities. Journal of the Royal Statistical Society: Series C (Applied Statistics) 66, 963–977 (2017). [Google Scholar]
- 24.Willis A & Bunge J Estimating diversity via frequency ratios. Biometrics 71, 1042–1049 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Montealegre MC et al. High Genomic Diversity and Heterogenous Origins of Pathogenic and Antibiotic-Resistant Escherichia coli in Household Settings Represent a Challenge to Reducing Transmission in Low-Income Settings. mSphere 5, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao L et al. Literature-Based Phenotype Survey and In Silico Genotype Investigation of Antibiotic Resistance in the Genus Bifidobacterium. Curr Microbiol 77, 4104–4113 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Chelakkot C et al. Akkermansia muciniphila-derived extracellular vesicles influence gut permeability through the regulation of tight junctions. Experimental & Molecular Medicine 50, e450–e450 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao S et al. Akkermansia muciniphila improves metabolic profiles by reducing inflammation in chow diet-fed mice. J Mol Endocrinol 58, 1–14 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Borgo F et al. Body Mass Index and Sex Affect Diverse Microbial Niches within the Gut. Front. Microbiol 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mikami A et al. Oral Administration of Flavonifractor plautii, a Bacteria Increased With Green Tea Consumption, Promotes Recovery From Acute Colitis in Mice via Suppression of IL-17. Front. Nutr 7, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gupta A et al. Association of Flavonifractor plautii, a Flavonoid-Degrading Bacterium, with the Gut Microbiome of Colorectal Cancer Patients in India. mSystems 4, e00438–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Depommier C et al. Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: a proof-of-concept exploratory study. Nature Medicine 25, 1096–1103 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagao T, Hase T & Tokimitsu I A Green Tea Extract High in Catechins Reduces Body Fat and Cardiovascular Risks in Humans. Obesity 15, 1473–1483 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Wu F et al. Phascolarctobacterium faecium abundant colonization in human gastrointestinal tract. Exp Ther Med 14, 3122–3126 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nagao-Kitamoto H et al. Interleukin-22-mediated host glycosylation prevents Clostridioides difficile infection by modulating the metabolic activity of the gut microbiota. Nat Med 26, 608–617 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spiga L et al. An Oxidative Central Metabolism Enables Salmonella to Utilize Microbiota-Derived Succinate. Cell Host & Microbe 22, 291–301.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eberl C et al. E. coli enhance colonization resistance against Salmonella Typhimurium by competing for galactitol, a context-dependent limiting carbon source. Cell Host & Microbe 29, 1680–1692.e7 (2021). [DOI] [PubMed] [Google Scholar]
- 38.Kaper JB, Nataro JP & Mobley HLT Pathogenic Escherichia coli. Nature Reviews Microbiology 2, 123–140 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Navidad JF, Griswold DJ, Gradus MS & Bhattacharyya S Evaluation of Luminex xTAG gastrointestinal pathogen analyte-specific reagents for high-throughput, simultaneous detection of bacteria, viruses, and parasites of clinical and public health importance. J Clin Microbiol 51, 3018–3024 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rogawski ET et al. Use of quantitative molecular diagnostic methods to investigate the effect of enteropathogen infections on linear growth in children in low-resource settings: longitudinal analysis of results from the MAL-ED cohort study. The Lancet Global Health 6, e1319–e1328 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baker JM et al. Association of enteropathogen detection with diarrhoea by age and high versus low child mortality settings: a systematic review and meta-analysis. The Lancet Global Health 9, e1402–e1410 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sassone-Corsi M & Raffatellu M No Vacancy: How beneficial microbes cooperate with immunity to provide colonization resistance to pathogens. J Immunol 194, 4081–4087 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buffie CG & Pamer EG Microbiota-mediated colonization resistance against intestinal pathogens. Nat. Rev. Immunol 13, 790–801 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ahmed S et al. High prevalence of mcr-1 -encoded colistin resistance in commensal Escherichia coli from broiler chicken in Bangladesh. Scientific Reports 10, 18637 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rousham Emily K et al. Human Colonization with Extended-Spectrum Beta-Lactamase-Producing E. coli in Relation to Animal and Environmental Exposures in Bangladesh: An Observational One Health Study. Environmental Health Perspectives 129, 037001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Masud AA et al. Drivers of Antibiotic Use in Poultry Production in Bangladesh: Dependencies and Dynamics of a Patron-Client Relationship. Front. Vet. Sci 7, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nordahl Petersen T et al. Meta-genomic analysis of toilet waste from long distance flights; a step towards global surveillance of infectious diseases and antimicrobial resistance. Sci Rep 5, 11444 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hendriksen RS et al. Global monitoring of antimicrobial resistance based on metagenomics analyses of urban sewage. Nature Communications 10, 1124 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Porras AM & Brito IL The internationalization of human microbiome research. Curr. Opin. Microbiol 50, 50–55 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang Q et al. Role of Dietary Nutrients in the Modulation of Gut Microbiota: A Narrative Review. Nutrients 12, 381 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pham VT et al. Effects of colon-targeted vitamins on the composition and metabolic activity of the human gut microbiome– a pilot study. Gut Microbes 13, 1875774 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tang Q et al. Current Sampling Methods for Gut Microbiota: A Call for More Precise Devices. Front. Cell. Infect. Microbiol 10, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wood DE, Lu J & Langmead B Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lu J, Breitwieser FP, Thielen P & Salzberg SL Bracken: estimating species abundance in metagenomics data. PeerJ Computer Science 3, e104 (2017). [Google Scholar]
- 55.Clausen PTLC, Aarestrup FM & Lund O Rapid and precise alignment of raw reads against redundant databases with KMA. BMC Bioinformatics 19, 307 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ondov BD et al. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol 17, 132 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fraley C & Raftery AE MCLUST: Software for rvlodel-Based Clustering, Density Estimation and Discriminant Analysis. 50 (2002). [Google Scholar]
- 58.Fraley C & Raftery AE Model-Based Clustering, Discriminant Analysis, and Density Estimation. Journal of the American Statistical Association 97, 611–631 (2002). [Google Scholar]
- 59.Knee J et al. Effects of an urban sanitation intervention on childhood enteric infection and diarrhea in Maputo, Mozambique: A controlled before-and-after trial. eLife 10, e62278 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Benjamini Y & Hochberg Y Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. Series B (Methodological) 57, 289–300 (1995). [Google Scholar]
- 61.Konstantinidis KT & Tiedje JM Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci U S A 102, 2567–2572 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Foster ZSL, Sharpton TJ & Grünwald NJ Metacoder: An R package for visualization and manipulation of community taxonomic diversity data. PLOS Computational Biology 13, e1005404 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McMurdie PJ & Holmes S phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLOS ONE 8, e61217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alcock BP et al. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res 48, D517–D525 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wickham H ggplot2. WIREs Computational Statistics 3, 180–185 (2011). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw reads (human sequences removed) were deposited in the Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) under the project number PRJNA726052. Metadata are publicly available at the following link: https://osf.io/wb3pv/. We accessed the following publicly available database to conduct our analyses: NCBI’s taxonomy database (https://ftp.ncbi.nlm.nih.gov/pub/taxonomy/), downloaded 23 November 2021.