

Abstract
A human sialyltransferase ST3GAL II (hST3GAL II) was successfully expressed in Escherichia coli as an active soluble fusion protein with an N-terminal maltose-binding protein (MBP) and a C-terminal hexa-histidine tag. It was used as an efficient catalyst in a one-pot multienzyme (OPME) sialylation system for high-yield production of the glycans of ganglioside GM1b and highly sialylated brain gangliosides GD1a and GT1b. Further sialylation of GM1b and GD1a glycans using a bacterial α2–8-sialyltransferase in another OPME sialylation reaction led to the formation of the glycans of GD1c and brain ganglioside GT1a, respectively. The lower reverse glycosylation activity of the recombinant hST3GAL II compared to its bacterial sialyltransferase counterpart simplifies the handling of enzymatic synthetic reactions and has an advantage for future use in automated chemoenzymatic synthetic processes.
Keywords: biocatalysis, carbohydrate, chemoenzymatic synthesis, ganglioside glycan, human sialyltransferase
Graphical Abstract
A human sialyltransferase ST3GAL II (hST3GAL II) was expressed in E. coli, characterized, and used for highly efficient synthesis of highly sialylated ganglioside glycans in one-pot multienzyme (OPME) sialylation system with high yields.
Gangliosides are sialic acid-containing glycosphingolipids that are ubiquitously found in the plasma membrane of vertebrate cells and are the major sialic acid-containing glycoconjugates in animal nervous systems.[1] N-Acetylneuraminic acid (Neu5Ac) is the most abundant sialic acid form in nature[2] and the major sialic acid form in brain gangliosides.[3]
GM1a (1, also named as GM1) and more highly sialylated gangliosides GD1a (2), GD1b (3), and GT1b (4) (Figure 1) constitute the majority (>87%) of gangliosides in the brains of mammals.[4] Among these four, gangliosides GD1a (2) and GT1b (4) with two and three Neu5Ac residues, respectively, are the most abundant in the mammalian brain and are considered as the “major brain gangliosides”.[5] Alteration in the levels of different brain gangliosides has been linked to Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington disease (HD), and amyotrophic lateral sclerosis (ALS).[4c, 5b, 6] For example, decreased expression of GD1a (2) and GT1b (4) was shown in the substantia nigra of the male patients with PD.[7] Systemic deficiency of GM1a (1) was shown to correlate to sporadic PD. The brains of patients with ALS were found to have decreased levels of GD1b (3), GT1b (4), and GQ1b.[8] On the other hand, ganglioside GM1b (5) has been shown to be an influenza virus receptor[9] and the presence of its antibodies is associated with neurological diseases.[10]
Figure 1.

Structures of major brain gangliosides GM1a (1), GD1a (2), GD1b (3), and GT1b (4) as well as ganglioside GM1b (5) with a representative ceramide (d18:1–18:0) structure (R).
Therapeutic applications of exogenous gangliosides, especially GM1a (1) and analogues, to spinal cord injury, PD, stroke, and AD, have been explored.[4c, 5b, 11] It is worth to note that the glycan component of GM1a (1) was shown to be able to promote tropomyosin receptor kinase A (TrkA)-dependent neurite growth.[12] A GM1a glycan was also shown to reduce the symptoms of sporadic PD in a mouse model.[13] Therefore, ganglioside glycans, in addition to gangliosides, may also have potential therapeutic applications.
Gangliosides in humans are synthesized by a series of glycosyltransferase-catalyzed reactions that sequentially add monosaccharides one at a time to simpler glycosphingolipids.[14] Due to their important biological functions, gangliosides and their corresponding glycans have been attractive synthetic targets for the development of potential therapeutics. Their synthesis is challenging due to their structural complexity and the presence of one or more sialic acids. Chemoenzymatic synthesis of highly sialylated ganglioside glycans reported recently[15] used a bacterial sialyltransferase Pasteurella multocida α2–3-sialyltransferase 1 (PmST1) that has relatively high reverse sialylation activity (previously identified as sialoside product cleavage/sialidase activity).[16] The reactions thus had to be monitored closely and the reactions had to be stopped promptly in order to minimize the product cleavage.[15] The amount of the sialyltransferase used also needed to be controlled precisely. A sialyltransferase with lower reverse sialylation activity is desirable to simplify the synthetic procedures. This is especially important for the development of automated chemoenzymatic synthetic processes.[15b, 17] Herein we show that human ST3GAL II can be expressed in Escherichia coli in a soluble active form. The recombinant human ST3GAL II does not have significant product cleavage activity in the presence of cytidine 5’-monophosphate (CMP) and is highly efficient in a one-pot multienzyme (OPME) α2–3-sialylation system for catalyzing the synthesis of highly sialylated brain ganglioside glycans including the glycans of GD1a, GT1b, and GM1b from GM1a, GD1b, and GA1, respectively.
Human CMP-N-acetylneuraminate-β-galactosaminide-α-2,3-sialyltransferase 2 (hST3GAL II) (E.C.2.4.99.4)[18] is a type II membrane protein. It shares protein sequence similarity with other mammalian sialyltransferases in the carbohydrate active enzyme (CAZy) database (www.cazy.org)[19] glycosyltransferase GT29 family. Overexpression of hST3GAL II was shown to be related to skin aging.[20] A higher mRNA level of ST3Gal II was found to be related to advanced oral cancer.[21] Mouse[22] ST3Gal II and human ST3GAL II were reported to be the key enzymes involved in the synthesis of GD1a and GT1b.[23] Human ST3GAL II was also shown to be responsible for the synthesis of ganglioside stage-specific embryonic antigen 4 (SSEA4) which is overexpressed in various cancer cells.[24] We have been interested in expressing hST3GAL II in E. coli to explore its application in chemoenzymatic synthesis of oligosaccharides and glycoconjugates.
Among the twenty known human sialyltransferases, only three including hST3GAL I,[25] hST6GAL I,[25–26] hST6GALNAC I[27] have been reported to be expressed in E. coli as soluble active recombinant enzymes. Expressing additional human glycosyltransferases in amounts sufficient for preparative-scale synthesis of carbohydrates can expand the tool box of readily accessible synthetically useful carbohydrate biosynthetic enzymes which may have properties different from currently available enzymes that have been expressed in E. coli.
Based on topological model predictions of protein transmembrane domains, a codon optimized gene for a truncated hST3GAL II with the predicted N-terminal transmembrane domain being removed was cloned in pMAL-c4X vector to express a recombinant fusion protein with an N-terminal maltose-binding protein (MBP) and a C-terminal His6-tag (MBP-Δ27hST3GAL II-His6) (Figure S1). Expression of MBP-Δ27hST3GAL II-His6 in E. coli BL21 (DE3) followed by Ni2+-nitrilotriacetic acid (Ni2+-NTA) affinity column purification provided a soluble active enzyme. About 9 mU of purified enzyme was routinely obtained from E. coli cells cultured in one liter of LB media. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis (Figure S3A) showed that the purified protein has a molecular weight close to the calculated value of 79 kDa although a weaker band with a smaller molecular weight was also observed.
Recombinant hST3GAL II expressed in various mammalian cell cultures has been reported. It was shown to catalyze the transfer of Neu5Ac from CMP-Neu5Ac to the terminal Gal residue in Galβ1–3GalNAc oligosaccharides or glycoconjugates to form an α2–3-sialyl linkage.[28] We carried out acceptor substrate specificity studies for MBP-Δ27hST3GAL II-His6 using a high-performance liquid chromatography (HPLC)-based quantitative assay. Several structurally defined synthetic disaccharides (6–11) and a monosaccharide (12) containing a hydrophobic benzyloxycarbonyl (Cbz)-protected propylamine (ProNHCbz)[29] or a methyl 2-anthranilic acid ester (2AA) tag[30] at the reducing end were used as potential acceptor substrates. As shown in Table 1, MBP-Δ27hST3GAL II-His6 was the most reactive towards type IV glycan Galβ1–3GalNAcβProNHCbz (6) and type III or Core 1 glycan Galβ1–3GalNAcαProNHCbz (7). It was active, but with lower efficiencies, towards type I glycans Galβ1–3GlcNAcβProNHCbz (8a) and Galβ1–3GlcNAcβ2AA (8b) as well as the corresponding disaccharide analogue Galβ1–3GlcNAcαProNHCbz (9) with an α-linkage at the GlcNAc. No activity was observed for type II LacNAcβ2AA (10) and type VI LacβProNHCbz (11) glycans, or monosaccharide glycoside GalNAcαProNHCbz (12). The preference of MBP-Δ27hST3GAL II-His6 towards Galβ1–3GalNAc-type acceptors was in consistent with previous reports regarding acceptor substrate specificity of purified and recombinant human ST3GAL II.[28] Using UV-detectable hydrophobic tag conjugated synthetic glycans, we demonstrated here that Galβ1–3GlcNAc-type glycans are also suitable although weaker acceptor substrates for the recombinant MBP-Δ27hST3GAL II-His6. The preference of β1–3-linked galactoside acceptors by MBP-Δ27hST3GAL II-His6 is complimentary to that of Pasteurella multocida sialyltransferase 3 (PmST3) which prefers β1–4-linked galactoside acceptors.[31] The information learned about acceptor substrate specificity of sialyltransferases will be very useful for choosing proper enzymes for selective sialylation of desired galactosyl branches in complex acceptor substrates containing different types of terminal galactosyl linkages.
Table 1.
Acceptor substrate specificity of MBP-Δ27hST3GAL II-His6.[a]
| Acceptor | 30 min (%) | 16 h (%) |
|---|---|---|
| Galβ1–3GalNAcβProNHCbz (6) | 44.0 ±0.1 | 100 |
| Galβ1–3GalNAcαProNHCbz (7) | 35.1 ±0.7 | 100 |
| Galβ1–3GlcNAcβProNHCbz (8a) | 9.1 ± 0.3 | 83.6 ±0.5 |
| Galβ1–3GlcNAcβPro2AA (8b) | 8.3 ±3.0 | 84.9 ±1.9 |
| Galβ1–3GlcNAcαProNHCbz (9) | 13.6 ± 2.2 | 96.4 ± 0.6 |
| LacβProNHCbz (10) | 0 | 0 |
| LacNAcβPro2AA (11) | 0 | 0 |
| GalNAcαProNHCbz (12) | 0 | 0 |
Quantitative HPLC methods were used to determine the yields.
We also explored the donor substrate specificity of MBP-Δ27hST3GAL II-His6 using in situ-generated CMP-sialic acids and derivatives. A two-step process was established to do this. In the first step, CMP-sialic acid or it derivative was prepared from sialic acid, its derivative, or its precursor using a recombinant Neisseria meningitidis CMP-sialic acid synthetase (NmCSS)[32] with (when a sialic acid precursor was used as the starting material) or without (when sialic acid or its derivative was used as the starting material) Pasteurella multocida sialic acid aldolase (PmAldolase)[33] and sodium pyruvate in the presence of cytidine 5’-triphosphate (CTP). In the second step, MBP-Δ27hST3GAL II-His6 sialylation reactions were carried out for 20 minutes or 16 hours using Galβ1–3GalNAcβProNHCbz (6)[29] as the acceptor substrate and the reaction mixtures from the first step as the sources of CMP-sialic acid donors. As shown in Table 2, MBP-Δ27hST3GAL II-His6 was able to tolerate a variety of CMP-sialic acids and derivatives as donor substrates. CMP-Neu5Ac was generated highly effectively from either Neu5Ac or ManNAc, and was used efficiently by MBP-Δ27hST3GAL II-His6 which was also efficient in using in situ-generated CMP-Kdn (generated from Kdn or mannose) and CMP-Neu5Ac9N3 (generated from ManNAc6N3[16a]). In comparison, CMP-Neu5N3 (generated from Man2N3[32]) and CMP-Neu5Ac7N3 (generated from ManNAc4N3[35]) were suitable but less efficient donor substrates for MBP-Δ27hST3GAL II-His6.
Table 2.
Donor substrate specificity of MBP-Δ27hST3GAL II-His6.
| Donor precursor | CMP-sialic acid yield (%) | Sialoside yield | |
|---|---|---|---|
| 20 min (%) | 16 h (%) | ||
| Neu5Ac | 100 ±1.2 | 62.6 ±1.2 | 100 ±0 |
| Kdn | 91.2 ±1.6 | 41.8 ±2.5 | 87.6 ±2.3 |
| ManNAc | 89.9 ±0.1 | 53.1 ±2.4 | 98.3 ±2.4 |
| Mannose | 86.3 ±2.1 | 40.8 ±3.8 | 86.0 ±0.8 |
| Man2N3 | 78.3 ±4.5 | 28.9 ±1.1 | 46.3 ±5.5 |
| ManNAc | 96.9 ±3.1 | 62.3 ±0.4 | 100 ±0.0 |
| ManNAc4N3 | 85.3 ±2.8 | 27.5 ±2.4 | 64.0 ±1.9 |
| ManNAc6N3 | 90.3 ±1.2 | 48.1 ±1.4 | 95.3 1.2 |
pH profile studies (Figure S4) using Galβ1–3GalNAcβProNHCbz (6)[29] as the acceptor substrate showed that MBP-Δ27hST3GAL II-His6 was active in a broad pH range of 4.5–10.0. It was the most active in a pH range of 4.5–5.5 with an optimal activity at pH 5.0 in MES buffer. About 50% of its maximal activity was observed at pH 6.0 and in the pH range of 8.0–9.5. And 32–43% of its maximal activity was observed in the pH range of 6.5–7.5. The sialyltransferase activity fell to minimal when the pH was at or below 4.0 or when it was at or above 10.5. Reaction temperature profile studies (Figure S5A) showed that MBP-Δ27hST3GAL II-His6 was the most active at 45 °C and 42 °C. More than 50% of the optimal activity was observed in the range of 35–45 °C. Minimal activity was observed when the temperature was at or below 25 °C or at or above 55 °C. Thermostability studies (Figure S5B) showed that the enzyme lost more than 60% activity by incubation at 35 °C for 1 hour and incubation at 37 °C for 1 hour retained only 14% of its activity. Incubation at 40 °C or a higher temperature for 1 hour completely abolished the enzyme activity. Altogether, these data showed that 30 °C is a well suited reaction temperature for MBP-Δ27hST3GAL II-His6 for preparative-scale synthesis of sialosides.
Homologs of hST3GAL II have been identified in various higher vertebrates. Differences in acceptor specificity and tissue-specific expression have been observed for hST3GAL II[28a, 28b] versus mouse ST3Gal II.[36] Reversible sialylation activity has been shown previously for both rat and mouse ST3Gal II.[37] Although the reversible glycosylation activities of glycosyltransferases can be used for accessing some targets, they are undesirable for enzymatic synthesis utilizing the main glycosyltransferase functions as if the reaction progresses were not monitored and controlled closely they may lead to low yields due to the cleavage of the target products.[15a, 16c, 38] To investigate whether MBP-Δ27hST3GAL II-His6 has a potential reverse sialylation activity, Neu5Acα2–3Galβ1–3GalNAcβProNHCbz (13) was synthesized from Galβ1–3GalNAcβProNHCbz (6) using a one-pot two-enzyme sialylation system (see ESI and OPME3 in Scheme 1 below) containing NmCSS[32] and MBP-Δ27hST3GAL II-His6. An excellent 96% yield was achieved for a preparative-scale synthetic reaction at 30 °C for 16 h followed by product purification using a simple C18-cartridge-based process. The potential reverse sialylation activity which can be presented as the desialylation of the sialylated product such as Neu5Acα2–3Galβ1–3GalNAcβProNHCbz (13) in the presence of CMP to form asialo-acceptor substrate of the enzyme (e.g. Galβ1–3GalNAcβProNHCbz 6) was then assayed. To our delight, no significant conversion of compound 13 to compound 6 by MBP-Δ27hST3GAL II-His6 was observed when it incubated at different concentrations (190, 19, and 1.9 μU sialyltransferase activities) with sialoside 13 (the product of the sialyltransferase activity) and CMP for 1 h (≤1.4±0.1%) and 20 h (≤3.9±0.1%) (Table 3). This indicated that the sialylated product formed from the reaction catalyzed by MBP-Δ27hST3GAL II-His6 was not hydrolyzed significantly either with an elongated incubation time or with an excess amount of the enzyme, thus minimizing the necessity for close reaction monitoring which is essential for using sialyltransferases with reverse sialylation or product cleavage activity such as PmST1.[15–16]
Scheme 1.

Sequential one-pot multienzyme (OPME) synthesis of highly sialylated ganglioside glycans.
Table 3.
Percentage (%) of sialoside Neu5Acα2–3Galβ1–3GalNAcαProNHCbz (13) (the product of the sialyltransferase activity) that is desialylated by different amounts of MBP-Δ27hST3GAL II-His6 in the presence of CMP for 1 h or 20 h.
The amounts of enzyme used are shown in units for its sialyltransferase activity: 1 unit of the sialyltransferase activity is defined as the amount of the enzyme that catalyzes the formation of 1 μmol of sialylated product Neu5Acα2–3Galβ1–3GalNAcαProNHCbz from Galβ1–3GalNAcαProNHCbz at 37 °C per minute at pH 7.0.
ND, not detected.
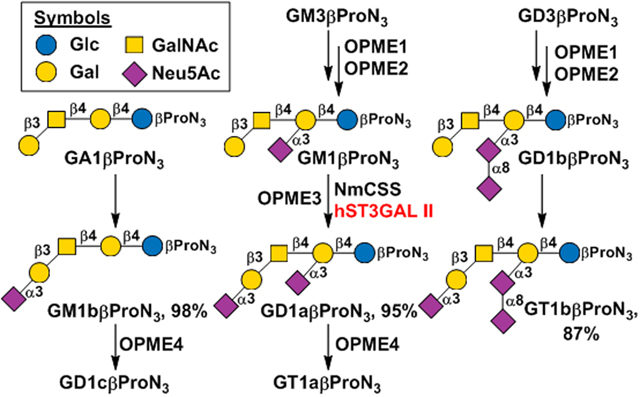
With a general understanding of the properties of MBP-Δ27hST3GAL II-His6, its application in chemoenzymatic synthesis of ganglioside glycans was explored. To synthesize glycans of GM1b, GD1a, and GT1b containing a propylazide aglycone, GM3βProN3 (14) and GD3βProN3 (15)[34] were used as the starting materials. A one-pot four-enzyme N-acetylgalactosamine (GalNAc)-activation and transfer system (OPME1, Scheme 1)[39] containing Bifidobacterium longum strain ATCC55813 N-acetylhexosamine-1-kinase (BLNahK),[40] Pasteurella multocida N-acetylglucosamine uridylyltransferase (PmGlmU),[41] Pasteurella multocida inorganic pyrophosphatase (PmPpA),[42] and Campylobacter jejuni β1–4-N-acetylgalactosaminyltransferase (CjCgtA)[39] was used to add a β1–4-linked GalNAc to GM3βProN3 (14) and GD3βProN3 (15) to form GM2βProN3 (16) and GD2βProN3 (17), respectively, in excellent 98% and 99% yields. A one-pot four-enzyme galactose-activation and transfer system (OPME2, Scheme 1) containing Streptococcus pneumoniae galactokinase (SpGalK),[43] Bifidobacterium longum UDP-sugar pyrophosphorylase (BLUSP),[44] PmPpA, and Campylobacter jejuni β1–3-galactosyltransferase (CjCgtB)[45] was then used to synthesize GM1βProN3 (18) and GD1bβProN3 (19), respectively, with excellent 99% and 94% yields. Similar to the previous reports, we observed that α2–3-sialylated lactosides but not their asialylated forms were suitable acceptor substrates for CjCgtA.[46] Therefore, GA1βProN3 (20) was not able to be obtained directly from LacβProN3 using sequential OPME1 and OPME2 reaction processes described above. It can, however, be readily obtained from GM1βProN3 (18) using a suitable sialidase. We cloned an N-terminal 22 amino acid truncated sialidase from Bacteroides fragilis as a N-terminal His6-tagged fusion protein (His6-Δ22BfGH33C) (Figure S2)[47] (see ESI for cloning, expression, and purification, its SDS-PAGE analysis results are shown in Figure S3B). We found that it was highly efficient in synthesizing GA1βProN3 (20) with an excellent 94% yield from GM1βProN3 (18). Similarly, GA2βProN3 (21) was readily obtained from GM2βProN3 (16) by a His6-Δ22BfGH33C-catalyzed reaction. With GA1βProN3 (20), GM1aβProN3 (18), and GD1bβProN3 (19) in hands, MBP-Δ27hST3GAL II-His6 was used together with NmCSS[32] in a one-pot two-enzyme sialylation system (OPME3, Scheme 1) for the synthesis of GM1bβProN3 (22), GD1aβProN3 (23), and GT1bβProN3 (24), respectively, with 98%, 95%, 87% yields.
Further sialylation of GM1bβProN3 (22) and GD1aβProN3 (23) by adding an α2–8-linked Neu5Ac at the non-reducing end using NmCSS and Campylobacter jejuni α2–3/8-sialyltransferase (CjCstII)[48] in another OPME sialylation reaction (OPME4, Scheme 1) led to the formation of GD1cβProN3 (25), GT1aβProN3 (26) in 78% and 68% yields, respectively. The relatively lower yields in these reactions were due to the formation of over-sialylated byproducts by CjCstII’s capability in adding an additional α2–8-linked Neu5Ac to the desired products 25 and 26, respectively. To purify the desired products, the pH of the reaction mixture was adjusted to 1.0–2.0 before it was loaded to a C18 cartridge to separate the product from nucleotides in the mixture. A second C18 cartridge purification at pH 8.0 was used to separate GD1cβProN3 (25) from the minor byproduct with an additional α2–8-linked Neu5Ac.The same strategy was applied for the purification of GT1aβProN3 (26).
In conclusion, a human sialyltransferase hST3GAL II was successfully expressed in E. coli as a soluble and active fusion protein with an N-terminal MBP and a C-terminal His6-tag. The recombinant MBP-Δ27hST3GAL II-His6 was highly efficient in chemoenzymatic synthesis of GM1b, GD1a and GT1b ganglioside glycans. A recombinant sialidase from Bacteroides fragilis, His6-Δ22BfGH33c, was shown to be highly effective in the formation of GA1βProN3 (20) and GA2βProN3 (21) from GM1βProN3 (18) and GM2βProN3 (16), respectively. Ganglioside glycans GM1b, GD1a, GT1b, GD1c, and GT1a were chemoenzymatically synthesized with high yields using sequential OPME systems with in situ generation of sugar nucleotides from simple monosaccharides.
Supplementary Material
Acknowledgements
This work was supported by the United States (U.S.) National Institutes of Health (NIH) grant no. U01GM120419. Bruker Avance-800 NMR spectrometer was purchased with a U.S. National Science Foundation grant no. DBI-0722538. The Thermo Scientific Q Exactive HF Orbitrap Mass Spectrometer was purchsed with a U.S. NIH grant no. S10OD025271.
Footnotes
Supporting information for this article is given via a link at the end of the document
References
- [1].a) Furukawa K, Ohmi Y, Ohkawa Y, Tajima O, Furukawa K, Adv. Neurobiol 2014, 9, 307–320; [DOI] [PubMed] [Google Scholar]; b) Schnaar RL, J. Mol. Biol 2016, 428, 3325–3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Angata T, Varki A, Chem. Rev 2002, 102, 439–469; [DOI] [PubMed] [Google Scholar]; b) Chen X, Varki A, ACS Chem. Biol 2010, 5, 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Davies LR, Varki A, Top. Curr. Chem 2015, 366, 31–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Tettamanti G, Bonali F, Marchesini S, Zambotti V, Biochim. Biophy. Acta 1973, 296, 160–170; [DOI] [PubMed] [Google Scholar]; b) Schnaar RL, J. Mol. Biol 2016, 428, 3325–3336; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sipione S, Monyror J, Galleguillos D, Steinberg N, Kadam V, Front. Neurosci 2020, 14, 572965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Sturgill ER, Aoki K, Lopez PH, Colacurcio D, Vajn K, Lorenzini I, Majic S, Yang WH, Heffer M, Tiemeyer M, Marth JD, Schnaar RL, Glycobiology 2012, 22, 1289–1301; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ledeen RW, Wu G, Trends Biochem. Sci 2015, 40, 407–418. [DOI] [PubMed] [Google Scholar]
- [6].Schengrund C-L, Trends Biochem. Sci 2015, 40, 397–406. [DOI] [PubMed] [Google Scholar]
- [7].Seyfried TN, Choi H, Chevalier A, Hogan D, Akgoc Z, Schneider JS, ASN Neuro. 2018, 10, 1759091418781889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Rapport MM, Donnenfeld H, Brunner W, Hungund B, Bartfeld H, Ann. Neurol 1985, 18, 60–67. [DOI] [PubMed] [Google Scholar]
- [9].Suzuki Y, Matsunaga M, Nagao Y, Taki T, Hirabayashi Y, Matsumoto M, Vaccine 1985, 3, 201–203. [DOI] [PubMed] [Google Scholar]
- [10].Tatsumoto M, Koga M, Gilbert M, Odaka M, Hirata K, Kuwabara S, Yuki N, J. Neuroimmunol 2006, 177, 201–208. [DOI] [PubMed] [Google Scholar]
- [11].Magistretti PJ, Geisler FH, Schneider JS, Li PA, Fiumelli H, Sipione S, Front. Neurol 2019, 10, 859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chiricozzi E, Pomè DY, Maggioni M, Di Biase E, Parravicini C, Palazzolo L, Loberto N, Eberini I, Sonnino S, J. Neurochem 2017, 143, 645–659. [DOI] [PubMed] [Google Scholar]
- [13].Chiricozzi E, Mauri L, Lunghi G, Di Biase E, Fazzari M, Maggioni M, Valsecchi M, Prioni S, Loberto N, Pomè DY, Ciampa MG, Fato P, Verlengia G, Cattaneo S, Assini R, Wu G, Alselehdar S, Ledeen RW, Sonnino S, Sci. Rep 2019, 9, 19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Schnaar RL, Kinoshita T, in Essentials of Glycobiology (Eds.: rd, Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH, Schnaar RL, Seeberger PH), Cold Spring Harbor (NY), 2015, pp. 125–135. [PubMed] [Google Scholar]
- [15].a) Li T, Wolfert MA, Wei N, Huizinga R, Jacobs BC, Boons GJ, J. Am. Chem. Soc 2020, 142, 19611–19621; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li T, Liu L, Wei N, Yang JY, Chapla DG, Moremen KW, Boons GJ, Nat. Chem 2019, 11, 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Yu H, Chokhawala H, Karpel R, Yu H, Wu B, Zhang J, Zhang Y, Jia Q, Chen X, J. Am. Chem. Soc 2005, 127, 17618–17619; [DOI] [PubMed] [Google Scholar]; b) McArthur JB, Yu H, Zeng J, Chen X, Org. Biomol. Chem 2017, 15, 1700–1709; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mehr K, Withers SG, Glycobiology 2016, 26, 353–359. [DOI] [PubMed] [Google Scholar]
- [17].a) Zhang J, Chen C, Gadi MR, Gibbons C, Guo Y, Cao X, Edmunds G, Wang S, Liu D, Yu J, Wen L, Wang PG, Angew. Chem. Int. Ed. Engl 2018, 57, 16638–16642; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang J, Liu D, Saikam V, Gadi MR, Gibbons C, Fu X, Song H, Yu J, Kondengaden SM, Wang PG, Wen L, Angew. Chem. Int. Ed. Engl 2020, 59, 19825–19829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Strausberg RL, Feingold EA, Grouse LH, Derge JG, Klausner RD, Collins FS, Wagner L, Shenmen CM, Schuler GD, Altschul SF, Zeeberg B, Buetow KH, Schaefer CF, Bhat NK, Hopkins RF, Jordan H, Moore T, Max SI, Wang J, Hsieh F, Diatchenko L, Marusina K, Farmer AA, Rubin GM, Hong L, Stapleton M, Soares MB, Bonaldo MF, Casavant TL, Scheetz TE, Brownstein MJ, Usdin TB, Toshiyuki S, Carninci P, Prange C, Raha SS, Loquellano NA, Peters GJ, Abramson RD, Mullahy SJ, Bosak SA, McEwan PJ, McKernan KJ, Malek JA, Gunaratne PH, Richards S, Worley KC, Hale S, Garcia AM, Gay LJ, Hulyk SW, Villalon DK, Muzny DM, Sodergren EJ, Lu X, Gibbs RA, Fahey J, Helton E, Ketteman M, Madan A, Rodrigues S, Sanchez A, Whiting M, Madan A, Young AC, Shevchenko Y, Bouffard GG, Blakesley RW, Touchman JW, Green ED, Dickson MC, Rodriguez AC, Grimwood J, Schmutz J, Myers RM, Butterfield YS, Krzywinski MI, Skalska U, Smailus DE, Schnerch A, Schein JE, Jones SJ, Marra T. MA, Proc. Natl. Acad. Sci. U. S. A 2002, 99, 16899–16903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].a) Campbell JA, Davies GJ, Bulone V, Henrissat B, Biochem. J 1997, 326 ( Pt 3), 929–939; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Coutinho PM, Deleury E, Davies GJ, Henrissat B, J. Mol. Biol 2003, 328, 307–317. [DOI] [PubMed] [Google Scholar]
- [20].Oinam L, Changarathil G, Raja E, Ngo YX, Tateno H, Sada A, Yanagisawa H, Aging Cell 2020, 19, e13190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mehta KA, Patel KA, Pandya SJ, Patel PS, J. Oral Pathol. Med 2020, 49, 253–259. [DOI] [PubMed] [Google Scholar]
- [22].Lopez PH, Aja S, Aoki K, Seldin MM, Lei X, Ronnett GV, Wong GW, Schnaar RL, Glycobiology 2017, 27, 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Schneider JS, PLoS One 2018, 13, e0199189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Aloia A, Petrova E, Tomiuk S, Bissels U, Deas O, Saini M, Zickgraf FM, Wagner S, Spaich S, Sutterlin M, Schneeweiss A, Reitberger M, Ruberg S, Gerstmayer B, Agorku D, Knobel S, Terranegra A, Falleni M, Soldati L, Sprick MR, Trumpp A, Judde JG, Bosio A, Cairo S, Hardt O, Breast Cancer Res. 2015, 17, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ortiz-Soto ME, Seibel J, PLoS One 2016, 11, e0155410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hidari KI, Horie N, Murata T, Miyamoto D, Suzuki T, Usui T, Suzuki Y, Glycoconj. J 2005, 22, 1–11. [DOI] [PubMed] [Google Scholar]
- [27].Skretas G, Carroll S, DeFrees S, Schwartz MF, Johnson KF, Georgiou G, Microb. Cell Fact 2009, 8, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].a) Kim YJ, Kim KS, Kim SH, Kim CH, Ko JH, Choe IS, Tsuji S, Lee YC, Biochem. Biophys. Res. Commun 1996, 228, 324–327; [DOI] [PubMed] [Google Scholar]; b) Giordanengo V, Bannwarth S, Laffont C, Van Miegem V, Harduin-Lepers A, Delannoy P, Lefebvre JC, Eur J Biochem 1997, 247, 558–566; [DOI] [PubMed] [Google Scholar]; c) Gupta R, Matta KL, Neelamegham S, Biochem. Biophys. Res. Commun 2016, 469, 606–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kooner AS, Diaz S, Yu H, Santra A, Varki A, Chen X, J. Org. Chem 2021, 86, 14381–14397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zhao C, Wu Y, Yu H, Shah IM, Li Y, Zeng J, Liu B, Mills DA, Chen X, Chem. Commun 2016, 52, 3899–3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Thon V, Li Y, Yu H, Lau K, Chen X, Appl. Microbiol. Biotechnol 2012, 94, 977–985. [DOI] [PubMed] [Google Scholar]
- [32].Yu H, Yu H, Karpel R, Chen X, Bioorg. Med. Chem 2004, 12, 6427–6435. [DOI] [PubMed] [Google Scholar]
- [33].Li Y, Yu H, Cao H, Lau K, Muthana S, Tiwari VK, Son B, Chen X, Appl. Microbiol. Biotechnol 2008, 79, 963–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Khedri Z, Li Y, Muthana S, Muthana MM, Hsiao CW, Yu H, Chen X, Carbohydr. Res 2014, 389, 100–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lee YC, Kojima N, Wada E, Kurosawa N, Nakaoka T, Hamamoto T, Tsuji S, J. Biol. Chem 1994, 269, 10028–10033. [PubMed] [Google Scholar]
- [36].a) Chandrasekaran EV, Xue J, Xia J, Locke RD, Matta KL, Neelamegham S, Biochemistry 2008, 47, 320–330; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chandrasekaran EV, Xue J, Xia J, Locke RD, Patil SA, Neelamegham S, Matta KL, Biochemistry 2011, 50, 9475–9487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].a) Sugiarto G, Lau K, Li Y, Khedri Z, Yu H, Le DT, Chen X, Mol. Biosyst 2011, 7, 3021–3027 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Na L, Yu H, McArthur JB, Ghosh T, Asbell T, Chen X, ACS Catal. 2020, 10, 6113–6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yu H, Cheng J, Ding L, Khedri Z, Chen Y, Chin S, Lau K, Tiwari VK, Chen X, J. Am. Chem. Soc 2009, 131, 18467–18477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yu H, Li Y, Zeng J, Thon V, Nguyen DM, Ly T, Kuang HY, Ngo A, Chen X, J. Org. Chem 2016, 81, 10809–10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Li Y, Yu H, Chen Y, Lau K, Cai L, Cao H, Tiwari VK, Qu J, Thon V, Wang PG, Chen X, Molecules 2011, 16, 6396–6407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chen Y, Thon V, Li Y, Yu H, Ding L, Lau K, Qu J, Hie L, Chen X, Chem. Commun 2011, 47, 10815–10817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lau K, Thon V, Yu H, Ding L, Chen Y, Muthana MM, Wong D, Huang R, Chen X, Chem. Commun 2010, 46, 6066–6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chen M, Chen LL, Zou Y, Xue M, Liang M, Jin L, Guan WY, Shen J, Wang W, Wang L, Liu J, Wang PG, Carbohydr. Res 2011, 346, 2421–2425. [DOI] [PubMed] [Google Scholar]
- [44].Muthana MM, Qu J, Li Y, Zhang L, Yu H, Ding L, Malekan H, Chen X, Chem. Commun 2012, 48, 2728–2730. [DOI] [PubMed] [Google Scholar]
- [45].Malekan H, Fung G, Thon V, Khedri Z, Yu H, Qu J, Li Y, Ding L, Lam KS, Chen X, Bioorg. Med. Chem 2013, 21, 4778–4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wen L, Zheng Y, Jiang K, Zhang M, Kondengaden SM, Li S, Huang K, Li J, Song J, Wang PG, J. Am. Chem. Soc 2016, 138, 11473–11476. [DOI] [PubMed] [Google Scholar]
- [47].Guo L, Chen X, Xu L, Xiao M, Lu L, Appl. Environ. Microbiol 2018, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Cheng J, Yu H, Lau K, Huang S, Chokhawala HA, Li Y, Tiwari VK, Chen X, Glycobiology 2008, 18, 686–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.