

Abstract
Neonatal encephalopathy (NE) that purportedly arises from hypoxia-ischemia is labeled hypoxic-ischemic encephalopathy (HIE). Perinatal asphyxia is a clinical syndrome involving acidosis, a low Apgar score and the need for resuscitation in the delivery room; asphyxia alerts one to the possibility of NE. In the present systematic review, we focused on the noninflammatory biomarkers in cerebrospinal fluid (CSF) that are involved in the development of possible brain injury in asphyxia or HIE. A literature search in PubMed and EMBASE for case-control studies was conducted and 17 studies were found suitable by a priori criteria. Statistical analysis used the Mantel-Haenszel model for dichotomous data. The pooled mean difference and 95% confidence intervals (CIs) were determined. We identified the best biomarkers, based on the estimation approach in evaluating the biological significance, out of hundreds in three categories: cell adhesion and proliferation, oxidants and antioxidants, and cell damage. The following subtotal-population comparisons were made: perinatal asphyxia versus no asphyxia, asphyxia with HIE versus asphyxia without HIE, asphyxia with HIE versus no asphyxia, and term versus preterm HIE newborn with asphyxia. Biological significance of the biomarkers was determined by using a modification of the estimation approach, by ranking the biomarkers according to the difference in the bounds of the CIs. The most promising CSF biomarkers for prognostication especially for the severest HIE include creatine kinase, xanthine oxidase, vascular endothelial growth factor, neuron-specific enolase, superoxide dismutase, and malondialdehyde. Future studies are recommended using such a combined test to prognosticate the most severely affected patients.
Keywords: asphyxia, biomarker, cerebrospinal fluid, hypoxic-ischemic encephalopathy, perinatal
1 |. INTRODUCTION
Neonatal encephalopathy (NE) putatively due to hypoxic-ischemic brain injury has been labeled as hypoxic-ischemic encephalopathy (HIE). This is mostly in a setting of concurrent perinatal asphyxia, a clinical syndrome, involving a combination of severe acidosis (pH < 7.0, base excess >−12 on blood gases), poor Apgar scores (<5 at 10 min of life), or need for resuscitation at delivery (Locatelli et al., 2020). The incidence of HIE with perinatal hypoxia-ischemia (H-I) is 1–3/1,000 and 1–8/1,000 in live term and live preterm births, respectively (Graham et al., 2008; Manuck et al., 2016). Since childhood diseases carry a high burden due to the lifelong consequences to the patient, family, and society, it is important to identify biomarkers to identify and stratify those infants that might develop brain injury from HIE or asphyxia and consider interventions as early as possible. The potential mechanisms for HIE include energy failure, intracellular calcium accumulation, lipid peroxidation, reactive oxygen and nitrogen species, excitatory amino acid-receptor overactivation, caspase-mediated cell death, and inflammatory lipid mediators (Calvert & Zhang, 2005; Tan & Wu, 2020). Newborns have higher tolerance for hypoxia (Singer, 1999) and greater potential for cell regeneration compared to adults (Wigley & Berry, 1988).
Cerebrospinal fluid (CSF) circulates in the surrounding spaces of central nervous systems and plays a pivotal role in biochemical homeostasis. CSF constituents may include RNAs, proteins, lipids, and hormones, the diffusion and transportation of which can indicate the development and progression of certain diseases (Johanson & Johanson, 2016). The brain–CSF barrier is more permeable to brain proteins and metabolites than the blood-brain barrier (Parrado-Fernández et al., 2018; Zhang et al., 2017), probably making CSF markers a better window into brain injury than blood markers.
There have been a lot of studies in the correlation of CSF biomarkers with neonatal brain injury. There was a lot of variance in the studies, and inflammatory biomarkers were more commonly evaluated. In the present systematic review, we focused on noninflammatory markers to obtain an idea of the pathogenetic pathways of injury in hypoxia-ischemia. A useful biomarker would ideally have very low false positives and false negatives. We prioritized the biomarkers after evaluating the association of noninflammatory CSF biomarkers with clinical outcomes. Herein, to estimate the clinical utility of a biomarker, we looked at not only the statistical significance but estimated the biological significance by estimating the difference between the bounds of the 95% confidence intervals (CIs) of the groups.
2 |. METHODS AND MATERIALS
The review protocol can be obtained from the corresponding author. This review was not registered before the completion of data acquisition. A literature search using the strategy of “(csf OR (cerebrospinal fluid)) AND (brain injury) AND (newborn or neonate or neonatal)” in PubMed and EMBASE was performed on May 28, 2020 without limit on publication period. Inclusion criteria were as follows: case–control studies about the correlation between hypoxic neonatal brain injury during the perinatal period. Exclusion criteria were as follows: reviews, case reports, nonclinical studies, samples collected beyond the neonatal period, studies not in English, or studies with incomplete data.
Two researchers (ZS and KL) performed the initial search, screened the titles and abstracts of candidate studies, and extracted data. Disagreement was solved by the third author (SD). Risk of bias of individual studies was analyzed using the Newcastle–Ottawa Quality Assessment Scale (Lo et al., 2014). Data about study design and methods, inclusion and exclusion criteria, patient characteristics, CSF biomarkers levels, patient outcomes, and follow-up information were extracted. The earliest test result was compared between studies of the same biomarker if multiple time points were reported. Some studies did not exactly follow the generally accepted definition of perinatal asphyxia: metabolic acidosis found in umbilical cord or newborn blood gases (pH < 7.0, base excess >−12), poor Apgar scores (<5 at 10 min of life), or need for resuscitation at delivery (Locatelli et al., 2020). We made a note of the variations of the way asphyxia was defined. For primary outcomes, we estimated the correlation between CSF biomarkers and a loose definition of “asphyxia” or HIE in the newborns. Preterm or term asphyxiated newborns were also compared to show the effect of gestation.
2.1 |. Statistical analysis
We made a distinction based on the estimation approach in evaluating the biological significance separate from just statistical significance of the difference between means () of a particular biomarker. The Mantel–Haenszel model was used for dichotomous data. The 95% CIs were determined in the subtotal populations from the standard deviation and sample size. Targeted populations were compared to a control group. We highlighted the biomarkers that would show a clear difference between the CIs between the two groups (Figure 1).
FIGURE 1.
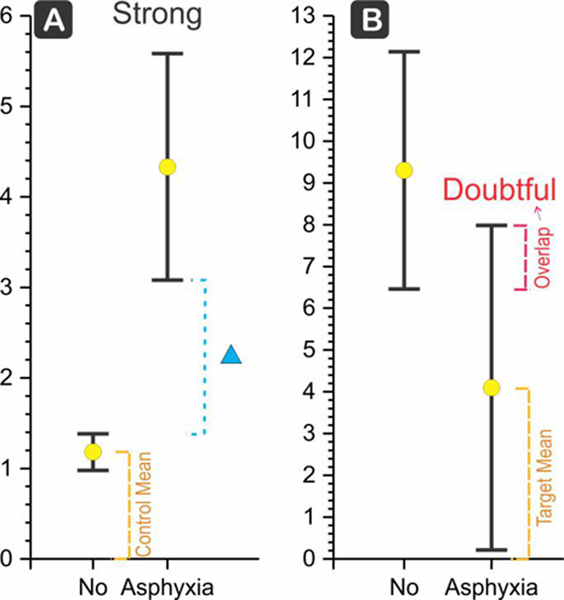
Example of a strong and a doubtful biomarker. (a) The control group (No asphyxia) was compared to a target group (Asphyxia). Data from XO (Batra et al., 1998). Mean () and lower and upper confidence intervals are shown (LCI, UCI). The difference (Δ) between the target LCI and control UCI is calculated and then divided by the control mean and expressed as a percentage(Δ% ). (b) If the CIs overlapped as shown in the dashed line, then the biomarker was considered as doubtful, as meant for a clinician. Data from NGF (Korhonen et al., 1998). Also, note if the biomarker showed a decrease in the target group, the Δ between the control LCI and target UCI was taken. This Δ was expressed as percentage of the target mean (Δ% )
If the target group mean was higher than the control group, then the difference (Δ) between the lower confidence interval (LCI) of the target group and the upper confidence interval (UCI) of the control group was taken as a measure of the biological significance of the biomarker, and this score expressed as percentage of the control mean, Δ% (Figure 1a). Conversely, if the target group mean was lower than the control group, the difference, Δ, between target UCI and control LCI was taken, and this score was again expressed as a percentage of the target mean (the smaller of the two means, Δ% ).
We then prioritized the biomarkers in each category by the magnitude of the Δ expressed as a percentage of the control mean, Δ% , referred henceforth as the “score.” For convenience of the reader, we categorized the biomarkers as strong if score was >100%, moderate if 50%−100%, and weak if 0.5%−50%. If the CIs overlapped, the score was defaulted to zero.
If the CIs overlapped (Figure 1b), the clinical utility of the biomarker became doubtful, as meant for the clinician.
A percentage of the control mean was used for all averages and standard deviations to combine the results from two different studies using the same biomarker and employing different units.
3 |. RESULTS
The a priori search strategy produced 390 publications in PubMed and 603 publications in EMBASE. After screening out duplicated studies and unqualified studies, there were 17 studies (Batra et al., 1998; Blennow et al., 1995; Cao et al., 1993; Dalens, Bezou, et al., 1981; Dalens, Viallard, et al., 1981; Fernandez et al., 1986; Gucuyener et al., 1999; Gulcan et al., 2005; Hussein et al., 2010; Juul et al., 1999; Korhonen et al., 1998; Kumar et al., 2008; Ray et al., 1998; Riikonen et al., 1999; Savman et al., 2013; Talvik et al., 1995; Vasiljevic et al., 2011) included in the final quantitative analysis, most of which reported more than one biomarker (Figure 2). All included studies had 7 out of 8 stars according to the Newcastle–Ottawa Quality Assessment Scale (Table S1). We first examined the situation of perinatal asphyxia since this is the starting point for clinical decision-making in the determination of HIE, all the while noting that the population of asphyxiated cases is not equivalent to the population with definite brain injury.
FIGURE 2.

Flow chart of analysis process
3.1 |. CSF markers in asphyxiated versus non-asphyxiated cases
Thirteen studies (Batra et al., 1998; Blennow et al., 1995; Cao et al., 1993; Dalens, Bezou, et al., 1981; Dalens, Viallard, et al., 1981; Fernandez et al., 1986; Gulcan et al., 2005; Juul et al., 1999; Korhonen et al., 1998; Kumar et al., 2008; Ray et al., 1998; Riikonen et al., 1999; Savman et al., 2013) reported CSF markers in asphyxiated versus non-asphyxiated cases, including 1,048 test results in the asphyxiated group and 801 test results in the non-asphyxiated group (Table 1). All of the non-asphyxiated cases were cases of suspected meningitis or sepsis based on clinical conditions, but were negative for bacterial cultures.
TABLE 1.
CSF markers in asphyxiated versus non-asphyxiated cases
| Study | Biomarker | Gestation | Insult type | Age sampling | Insult mean | SD | No. | Control mean | SD | No. | Score (Δ% X) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cell adhesion, proliferation | |||||||||||
| Korhonen et al. (1998) | BDNF pg/ml | NA | Asphyxiated | l-14da | 12.4 | 4.5 | 7 | 4 | 0.85 | 8 | 88 |
| Savman et al. (2013) | Galectin-3 ng/ml | ≥37 wk | 5’-Apgar < 6 | NA | 2.64 | 1.67 | 15 | 1.36 | 1.53 | 11 | 0 |
| Korhonen et al. (1998) | NGF pg/ml | NA | asphyxiated | l-14d | 4.1 | 4.2 | 7 | 9.3 | 3.4 | 8 | 0 |
| Riikonen et al. (1999) | NGF pg/ml | 30–42 wk | 5’-Apgar < 4 | 12 hr-14db | 3.76 | 4.13 | 8 | 9.42 | 4.09 | 10 | 0 |
| Combo Korhonen et al. (1998), Riikonen et al. (1999) | NGF pg/ml | 3.9 | 4.5 | 15 | 9.4 | 3.7 | 18 | −28 | |||
| Free radicals | |||||||||||
| Batra et al. (1998) | XO μmol/min/ml | NA | l’-Apgar < 4, dead | 24–48 hr | 8.89 | 2.41 | 5 | 1.18 | 0.49 | 25 | 383 |
| Batra et al. (1998) | XO μmol/min/ml | NA | l’-Apgar < 4, sick | 24–48 hr | 5.11 | 2.36 | 10 | 1.18 | 0.49 | 25 | 173 |
| Batra et al. (1998) | XO μmol/min/ml | NA | l’-Apgar < 4, recovered | 24–48 hr | 2.29 | 1.42 | 15 | 1.18 | 0.49 | 25 | 10 |
| Batra et al. (1998) | XO μmol/min/ml | NA | l’-Apgar < 4, total | 24–48 hr | 4.33 | 3.35 | 30 | 1.18 | 0.49 | 25 | 144 |
| Savman et al. (2013) | QUIN nM | ≥37 wk | Asphyxiated | NA | 335.42 | 249.9 | 18 | 116.56 | 57 | 12 | 50 |
| Batra et al. (1998) | NO μg/ml | NA | l’-Apgar < 4, total | 24–48 hr | 58.31 | 24.86 | 30 | 32.65 | 13.38 | 25 | 33 |
| Kumar et al. (2008) | Malondialdehyde μmol/L | ≥37 wk | l’-Apgar < 3 | 24–48 hr | 1.68 | 0.5 | 50 | 1.03 | 0.29 | 8 | 26 |
| Ray et al. (1998) | LPO nmol/h/dl | NA | Recovered | 12–48 hr | 13.79 | 4.42 | 16 | 10.82 | 3.02 | 25 | 0 |
| Ray et al. (1998) | LPO nmol/h/dl | NA | Sick | 12–48 hr | 17.98 | 4.62 | 8 | 10.82 | 3.02 | 25 | 19 |
| Ray et al. (1998) | LPO nmol/h/dl | NA | Dead | 12–48 hr | 21.43 | 4.36 | 10 | 10.82 | 3.02 | 25 | 58 |
| Ray et al. (1998) | LPO nmol/h/dl | NA | l’-Apgar < 4 | 12–48 hr | 17.02 | 5.46 | 34 | 10.82 | 3.02 | 25 | 28 |
| Batra et al. (1998) | H202 μg/h/ml | NA | l’-Apgar < 4, total | 24–48 hr | 400.73 | 209.09 | 30 | 287.4 | 132.9 | 25 | 0 |
| Ray et al. (1998) | Total calcium mg/dl | NA | Asphyxiated | 12–48 hr | 6.7 | 1.27 | 34 | 5.92 | 1.31 | 25 | 0 |
| Batra et al. (1998) | 02- μmol/min/dl | NA | l’-Apgar < 4, total | 24–48 hr | 1.88 | 1.16 | 30 | 1.42 | 0.55 | 25 | 0 |
| Antioxidants | |||||||||||
| Juul etal. (1999) | Epo mU/ml | mixed | Asphyxiated | NA | 225.9 | 620.8 | 16 | 5.8 | 4.5 | 41 | 0 |
| Ray et al. (1998) | SOD U/ml | NA | Recovered | 12–48 hr | 40.62 | 29.59 | 16 | 23.95 | 5.63 | 25 | 0 |
| Ray et al. (1998) | SOD U/ml | NA | Sick | 12–48 hr | 71.27 | 39.86 | 8 | 23.95 | 5.63 | 25 | 49 |
| Ray et al. (1998) | SOD U/ml | NA | Dead | 12–48 hr | 59.54 | 42.17 | 10 | 23.95 | 5.63 | 25 | 13 |
| Ray et al. (1998) | SOD U/ml | NA | Asphyxiated | 12–48 hr | 53.4 | 37.3 | 34 | 23.95 | 5.63 | 25 | 59 |
| Gulcan et al. (2005) | SOD U/mg protein | ≥37 wk | 5’-Apgar < 6 | <72 hr | 0.45 | 0.25 | 30 | 0.27 | 0.16 | 11 | 0 |
| Combo Ray et al. (1998), Gulcan et al. (2005) | 197 | 181 | 64 | 100 | 64 | 36 | 30 | ||||
| Gulcan et al. (2005) | CAT U/mg protein | ≥37 wk | 5’-Apgar < 6 | <72 hr | 26.33 | 21.19 | 30 | 17.25 | 12.51 | 11 | 0 |
| Gulcan et al. (2005) | GPX U/mg protein | ≥37 wk | 5’-Apgar < 6 | <72 hr | 16.25 | 13.57 | 30 | 10.54 | 7.25 | 11 | 0 |
| Ray et al. (1998) | GPX nmol/min/dl | NA | Recovered | 12–48 hr | 0.82 | 0.51 | 16 | 1.28 | 0.52 | 25 | 0 |
| Ray et al. (1998) | GPX nmol/min/dl | NA | Sick | 12–48 hr | 0.63 | 0.35 | 8 | 1.28 | 0.52 | 25 | −11 |
| Ray et al. (1998) | GPX nmol/min/dl | NA | Dead | 12–48 hr | 0.82 | 0.63 | 10 | 1.28 | 0.52 | 25 | 0 |
| Ray et al. (1998) | GPX nmol/min/dl | NA | Asphyxiated | 12–48 hr | 0.78 | 0.51 | 34 | 1.28 | 0.52 | 25 | −8 |
| Combo Ray et al. (1998); Gulcan et al. (2005) | 105 | 135 | 98 | 100 | 80 | 111 | 0 | ||||
| Neurotransmitter | |||||||||||
| Cao et al. (1993) | ß-EP pg/ml | mixed | Moderate | NA | 103.3 | 236 | 30 | 50.6 | 193 | 40 | 0 |
| Cao et al. (1993) | ß-EP pg/ml | mixed | Severe | NA | 152.1 | 199 | 14 | 50.6 | 193 | 40 | 0 |
| Cao et al. (1993) | p-EP pg/ml | Subtotal | 118.8 | 341 | 44 | 50.6 | 193 | 40 | 0 | ||
| Blennow et al. (1995) | Noradrenaline nM | >35 wk | Subtotal | <24 hr | 4.76 | 7.63 | 21 | 4.27 | 4.3 | 11 | 0 |
| Cao et al. (1993) | DynoAl-13 pg/ml | mixed | Moderate | NA | 106.4 | 198 | 30 | 56.2 | 200 | 40 | 0 |
| Cao et al. (1993) | DynoAl-13 pg/ml | mixed | Severe | NA | 142.4 | 158 | 14 | 56.2 | 200 | 40 | 0 |
| Cao et al. (1993) | DynoAl-13 pg/ml | Subtotal | 117.9 | 274 | 44 | 56.2 | 200 | 40 | 0 | ||
| Cao et al. (1993) | LEK pg/ml | mixed | Moderate | NA | 136.5 | 201 | 30 | 86.9 | 302 | 40 | 0 |
| Cao et al. (1993) | LEK pg/ml | mixed | Severe | NA | 186.1 | 196 | 14 | 86.9 | 302 | 40 | 0 |
| Cao et al. (1993) | LEK pg/ml | Subtotal | 152.3 | 317 | 44 | 86.9 | 302 | 40 | 0 | ||
| Blennow et al. (1995) | DOPACnM | >35 wk | Asphyxiated | <24 hr | 19.93 | 8.04 | 21 | 17.3 | 7.5 | 11 | 0 |
| Blennow et al. (1995) | HIAAnM | >35 wk | Asphyxiated | <24 hr | 436.62 | 137.16 | 21 | 400 | 113 | 11 | 0 |
| Blennow et al. (1995) | HVAnM | >35 wk | Asphyxiated | <24 hr | 545.86 | 209.45 | 21 | 510 | 79 | 11 | 0 |
| Blennow et al. (1995) | MHPACnM | >35 wk | Asphyxiated | <24 hr | 113.1 | 43.55 | 21 | 86 | 17 | 11 | 0 |
| Coagulation | |||||||||||
| Dalens, Viallard, et al., (1981) | FDP μg/ml | ≥37 wk | 5’-Apgar = 3–6 | Id | 5.3 | 9.8 | 18 | 2.53 | 8.9 | 25 | 0 |
| Dalens, Viallard, et al., (1981) | FDP μg/ml | ≥37 wk | 5’-Apgar < 3 | Id | 3.27 | 6.8 | 14 | 2.53 | 8.9 | 25 | 0 |
| Dalens, Viallard, et al., (1981) | FDP μg/ml | ≥37 wk | l’-Apgar < 6 total | 4.4 | 8.6 | 32 | 2.53 | 8.9 | 25 | 0 | |
| Cell damage | |||||||||||
| Fernandez et al. (1986) | LDH3 U/L | ≥37 wk | Asphyxiated | Id | 23.4 | 34.4 | 25 | 12 | 5 | 20 | 0 |
| Fernandez et al. (1986) | LDH2 U/L | ≥37 wk | Asphyxiated | Id | 20.64 | 27.8 | 25 | 12 | 6 | 20 | 0 |
| Dalens, Viallard, et al., (1981) | CKIU/L | ≥37 wk | l’-Apgar < 3 | Id | 1.1 | 1.1 | 14 | 1.3 | 2.5 | 25 | 0 |
| Dalens, Viallard, et al., (1981) | CKIU/L | ≥37 wk | l’-Apgar = 3–6 | Id | 4.1 | 8.1 | 18 | 1.3 | 2.5 | 25 | 0 |
| Dalens, Viallard, et al., (1981) | CKIU/L | ≥37 wk | l’-Apgar < 6 total | Id | 2.8 | 8.1 | 32 | 1.3 | 2.5 | 25 | 0 |
| Ray et al. (1998) | CPKU/I | NA | Recovered | 12–48 hr | 20.16 | 8.6 | 16 | 18.3 | 2.6 | 25 | 0 |
| Ray et al. (1998) | CPKU/I | NA | Sick | 12–48 hr | 21.75 | 9.3 | 8 | 18.3 | 2.6 | 25 | 0 |
| Ray et al. (1998) | CPKU/I | NA | Dead | 12–48 hr | 156 | 56.8 | 10 | 18.3 | 2.6 | 25 | 524 |
| Ray et al. (1998) | CPKU/I | NA | Asphyxiated | 12–48 hr | 60.5 | 69.6 | 34.0 | 18.3 | 2.6 | 25 | 92 |
| Combo Dalens, Viallard, et al. (1981), Ray et al. (1998) | 275 | 732 | 66 | 100 | 193 | 50 | 0 | ||||
| Dalens, Viallard, et al., (1981) | HBD IU/L | ≥37 wk | l’-Apgar < 3 | Id | 28 | 7 | 14 | 38 | 40 | 25 | 0 |
| Dalens, Viallard, et al., (1981) | HBD IU/L | ≥37 wk | l’-Apgar = 3–6 | Id | 48 | 22 | 18 | 38 | 40 | 25 | 0 |
| Dalens, Viallard, et al., (1981) | HBD IU/L | ≥37 wk | l’-Apgar < 6 total | Id | 39.3 | 23 | 32 | 38 | 40 | 25 | 0 |
| Fernandez et al. (1986) | LDH1 U/L | ≥37 wk | Asphyxiated | Id | 11.2 | 21.3 | 25 | 12 | 5 | 20 | 0 |
| Fernandez et al. (1986) | LDH4 U/L | ≥37 wk | Asphyxiated | Id | 6.2 | 21.4 | 25 | 2 | 1 | 20 | 0 |
| Fernandez et al. (1986) | LDH5 U/L | ≥37 wk | Asphyxiated | Id | 2.84 | 13.0 | 25 | 2 | 1 | 20 | 0 |
| Fernandez et al. (1986) | LDH U/L | ≥37 wk | Asphyxiated | Id | 60.2 | 105.0 | 25 | 48 | 18 | 20 | 0 |
| Dalens, Viallard, et al., (1981) | ASATIU/L | ≥37 wk | l’-Apgar < 3 | Id | 21 | 5 | 14 | 22 | 15 | 25 | 0 |
| Dalens, Viallard, et al., (1981) | ASATIU/L | ≥37 wk | l’-Apgar = 3–6 | Id | 26 | 8 | 18 | 22 | 15 | 25 | 0 |
| Dalens, Viallard, et al., (1981) | ASATIU/L | ≥37 wk | l’-Apgar < 6 total | Id | 23.8 | 9 | 32 | 22 | 15 | 25 | 0 |
| Dalens, Viallard, et al., (1981) | LDH IU/L | ≥37 wk | l’-Apgar < 3 | Id | 43 | 13 | 14 | 58 | 56.5 | 25 | 0 |
| Dalens, Viallard, et al., (1981) | LDH IU/L | ≥37 wk | l’-Apgar = 3–6 | Id | 74 | 35 | 18 | 58 | 56.5 | 25 | 0 |
| Dalens, Viallard, et al., (1981) | LDH IU/L | ≥37 wk | l’-Apgar < 6 total | Id | 60.4 | 38 | 32 | 58 | 56.5 | 25 | 0 |
Note: Subtotal = combining previous rows of the same study. Δ% = (LCI of target-UCI of control) × 100/control mean.
Abbreviations: ASAT, aminotransferase; BDNF, brain-derived neurotrophic factor; CAT, catalase; CPK, creatine phosphokinase (same as CK, creatine kinase); d, day; DOPAC, 3.4-dihydroxyphenylacetic acid; DynoA1-13, dynorphin A1-13; Epo, erythropoietin; FDP, fibrin-fibrinogen degradation products; GPX, glutathione peroxidase; H2O2, hydrogen peroxide; HBD, hydroxybutyrate dehydrogenase; HIAA, 5-hydroxyindole-3-acetic acid; HVA, homovanillic acid; LDH, lactate dehydrogenase; LEK, leu-enkephalin; LPO, lipid peroxidation; MHPAC, 3-methoxy-4-hydroxy-phenylglycol; NGF, nerve growth factor; NO, nitric oxide; O2−, superoxide anions; QUIN, quinolinic acid; SOD, superoxide dismutase; wk, week; XO, xanthine oxidase; β-EP, β-endorphin.
Postnatal 1–14 days.
Postnatal 12 hr to 14 days.
In biomarkers classified as cell adhesion and proliferation indicators, galectin-3 and brain-derived neurotrophic factor (BDNF) were significantly higher in the asphyxiated group, while nerve growth factor (NGF) was significantly lower in the asphyxiated group (Korhonen et al., 1998; Riikonen et al., 1999; Savman et al., 2013). Despite the statistical significance, the relative biological importance of BDNF and NGF were considered as moderate and weak (scores of 88 and 28), respectively.
Biomarkers involved in the production of free radicals, such as quinolinic acid (QUIN), malondialdehyde, xanthine oxidase (XO), hydrogen peroxide (H2O2), nitric oxide (NO), lipid peroxidation (LPO), and total calcium were significantly higher in the asphyxiated group, while superoxide anions (O2−) were similar between the asphyxiated and non-asphyxiated groups (Batra et al., 1998; Kumar et al., 2008; Ray et al., 1998; Savman et al., 2013). Out of these, XO was a strong biomarker (score 144, Figures 1a and 3), while QUIN was moderate (score 50), and NO and lipid peroxidation products including malondialdehyde were weak biomarkers (scores 33, 26–28, respectively).
FIGURE 3.
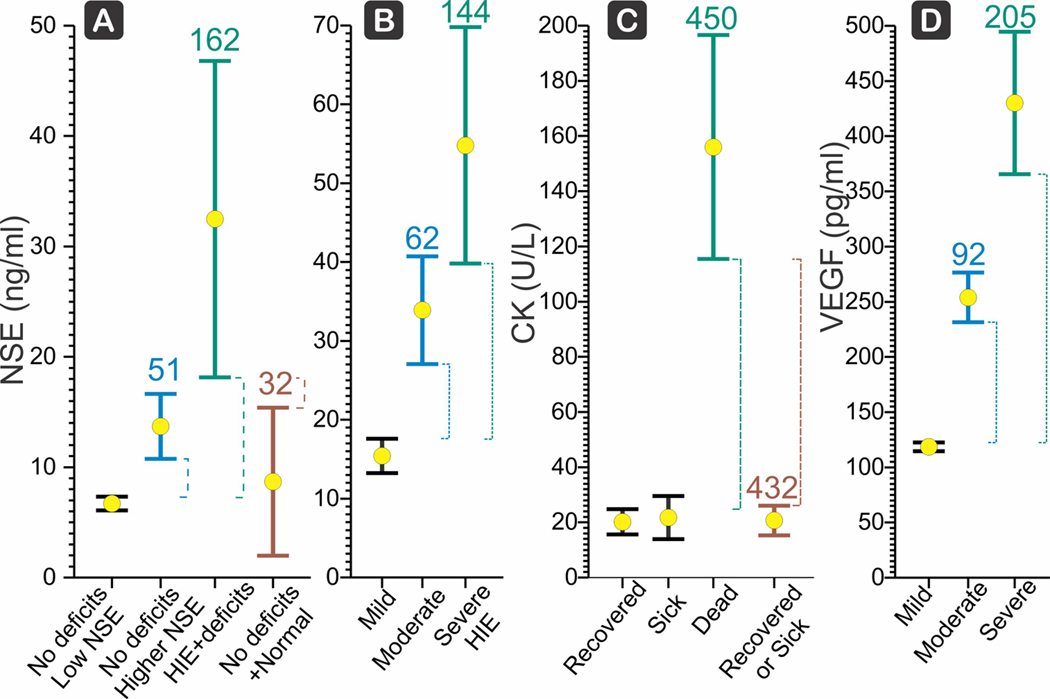
(a) Neuron-specific enolase (NSE) has a moderate score for the subtotal population of hypoxic-ischemic encephalopathy (HIE) with neurobehavioral deficits compared to all others with abnormal Sarnat score after birth, who recovered (brown, score 32) (Hussein et al., 2010). Mean () and LCI, UCI shown. Score is Δ (dashed lines) between the target LCI and control UCI, divided by the control mean, and expressed as a percentage. (b) NSE shows a progression from mild to moderate to severe HIE based on presentation of clinical signs initially (Vasiljevic et al., 2011). Severe HIE had stupor or coma with decerebrate posture, or absent activity, hypotonia, absent reflexes, seizures, nonreactive pupils, abnormal cranial nerve function, and severely abnormal aEEG patterns, NSE score was 144. (c) Creatine kinase (CK) is a strong biomarker (Ray et al., 1998) for death with HIE compared to HIE that recovers with a score of 450 (green) but not for HIE that survives. CK is also a strong biomarker HIE that results in mortality compared to all other perinatal asphyxia, recovered or sick (brown, score 432). (d) Vascular endothelial growth factor (VEGF) score for severe HIE (Vasiljevic et al., 2011) was 205 compared to mild HIE
Biomarkers showing antioxidant effects, such as superoxide dismutase (SOD) and erythropoietin (Epo) were significantly higher in the asphyxiated group, while catalase (CAT) was similar between the asphyxiated and non-asphyxiated groups (Gulcan et al., 2005; Juul et al., 1999; Ray et al., 1998). Glutathione peroxidase (GPX) was similar between the two groups in one study (Gulcan et al., 2005), and was significantly lower in the asphyxiated group in the other study (Ray et al., 1998). In this group, even after combining two studies for SOD and GPX, only SOD was found to be a weak biomarker (score of 30); the rest were considered as doubtful.
Neurotransmitters, such as leu-enkephalin (LEK), β-endorphin (β-EP), dynorphin A1–13 (DynoA1–13), and 3-methoxy-4-hydroxy-phenylglycol (MHPAC), were significantly higher in the asphyxiated group (Blennow et al., 1995; Cao et al., 1993). Fibrin-fibrinogen degradation products (FDP), a coagulation marker, were significantly higher in the asphyxiated group (Dalens, Bezou, et al., 1981). In this group, all neurotransmitters tested were considered doubtful for biomarker utility based on our score criteria.
Biomarkers hinting at cell damage, such as aminotransferase (ASAT) and creatine kinase (CK) were significantly higher in the asphyxiated group, while hydroxybutyrate dehydrogenase (HBD) and lactate dehydrogenase (LDH) remained similar between the asphyxiated and non-asphyxiated groups (Dalens, Viallard, et al., 1981; Fernandez et al., 1986). Interestingly, three isomers of LDH were significantly higher in the asphyxiated group (Fernandez et al., 1986). In this group, all the markers were considered doubtful based on the score. CK (same as CPK) showed promise with a moderate score in one study (Ray et al., 1998), but when we combined the two studies done for CK (Dalens, Viallard, et al., 1981; Ray et al., 1998), this marker was considered doubtful for differentiating between asphyxia and normal.
We next analyzed cases with documented HIE in asphyxia and compared with studies documenting the absence of any overt injury.
3.2 |. CSF markers in asphyxiated HIE versus asphyxiated non-HIE cases
Seven studies (Batra et al., 1998; Cao et al., 1993; Fernandez et al., 1986; Hussein et al., 2010; Kumar et al., 2008; Ray et al., 1998; Vasiljevic et al., 2011) reported CSF markers in asphyxiated HIE (337 test results) versus asphyxiated non-HIE (495 test results) cases (Table 2).
TABLE 2.
CSF markers in asphyxiated HIE versus asphyxiated non-HIE cases
| Study | Biomarker | Gestation | Case type | Age sampling | Case mean | SD | No. | Control type | Control mean | SD | No. | Score (Δ% ) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cell damage | ||||||||||||
| Hussein et al. (2010) | NSE ng/ml | >37 wk | HIE | 5–6d | 32.48 | 11.54 | 5 | l’or 5’-Apgar < 5, normal, low NSE | 6.7 | 1.5 | 25 | 162 |
| Hussein et al. (2010) | NSE ng/ml | >37 wk | l’or 5’-Apgar < 5, normal, high NSE | 5–6d | 13.7 | 4.11 | 10 | l’or 5’-Apgar < 5, normal, low NSE | 6.7 | 1.5 | 25 | 51 |
| Hussein et al. (2010) | NSE ng/ml | >37 wk | HIE | 5–6d | 32.48 | 11.54 | 5 | Total normal | 8.7 | 19.5 | 35 | 32 |
| Vasiljevic et al. (2011) | NSE ng/L | 32–37 wk | HIE severe | 3–5d | 54.8 | 19.5 | 9 | HIE mild | 15.4 | 8.2 | 57 | 144 |
| Vasiljevic et al. (2011) | NSE ng/L | 32–37 wk | HIE moderate | 3–5d | 33.9 | 16.2 | 24 | HIE mild | 15.4 | 8.2 | 57 | 62 |
| Vasiljevic et al. (2011) | NSE ng/L | 32–37 wk | Severe or moderate | 3–5d | 39.6 | 25.4 | 33 | HIE mild | 15.4 | 8.2 | 57 | 85 |
| Combo Hussein et al. (2010), Vasiljevic etal. (2011) | 287 | 238 | 38 | 100 | 58 | 82 | 96 | |||||
| Fernandez et al. (1986) | LDH U/L | >37 wk | Abnormal/died at 1 year | Id | 118 | 96 | 6 | Asphyxiated, normal at 1 year | 42 | 44 | 19 | 0 |
| Fernandez et al. (1986) | LDH1 U/L | >37 wk | Abnormal/died at 1 year | Id | 15 | 12 | 6 | Asphyxiated, normal at 1 year | 10 | 17 | 19 | 0 |
| Fernandez et al. (1986) | LDH2 U/L | >37 wk | Abnormal/died at 1 year | Id | 38.5 | 24 | 6 | Asphyxiated, normal at 1 year | 15 | 13 | 19 | 0 |
| Fernandez et al. (1986) | LDH3 U/L | >37 wk | Abnormal/died at 1 year | Id | 50 | 32 | 6 | Asphyxiated, normal at 1 year | 15 | 13 | 19 | 0 |
| Fernandez et al. (1986) | LDH4 U/L | >37 wk | Abnormal/died at 1 year | Id | 10 | 20 | 6 | Asphyxiated, normal at 1 year | 5 | 9 | 19 | 0 |
| Fernandez et al. (1986) | LDH5 U/L | >37 wk | Abnormal/died at 1 year | Id | 5.5 | 12 | 6 | Asphyxiated, normal at 1 year | 2 | 4 | 19 | 0 |
| Ray et al. (1998) | LPO nmol/h/dl | NA | Mortality | 12–48 hr | 21.43 | 4.36 | 10 | Recovered | 13.79 | 4.42 | 16 | 16 |
| Ray et al. (1998) | LPO nmol/h/dl | NA | Morbidity (HIE-II) | 12–48 hr | 17.98 | 4.62 | 8 | Recovered | 13.79 | 4.42 | 16 | 0 |
| Ray et al. (1998) | LPO nmol/h/dl | NA | Subtotal | 12–48 hr | 19.9 | 4.69 | 18 | Recovered | 13.79 | 4.42 | 16 | 10 |
| Ray et al. (1998) | CPKU/I | NA | Mortality | 12–48 hr | 156 | 56.76 | 10 | Recovered | 20.16 | 8.62 | 16 | 450 |
| Ray et al. (1998) | CPKU/I | NA | Morbidity | 12–48 hr | 21.75 | 9.31 | 8 | Recovered | 20.16 | 8.62 | 16 | 0 |
| Ray et al. (1998) | CPKU/I | NA | Sick or dead | 12–48 hr | 96.33 | 80.33 | 18 | Recovered | 20.16 | 8.62 | 16 | 157 |
| Ray et al. (1998) | CPKU/I | NA | Dead | 12–48 hr | 156 | 56.76 | 10 | Recovered or sick | 20.69 | 13 | 24 | 432 |
| Ray et al. (1998) | Total calcium mg/dl | NA | Mortality | 12–48 hr | 7.32 | 1.13 | 10 | Recovered | 6.65 | 1.43 | 16 | 0 |
| Ray et al. (1998) | Total calcium mg/dl | NA | Morbidity | 12–48 hr | 6.02 | 0.73 | 8 | Recovered | 6.65 | 1.43 | 16 | 0 |
| Ray et al. (1998) | Total calcium mg/dl | NA | Subtotal | 12–48 hr | 6.74 | 1.16 | 18 | Recovered | 6.65 | 1.43 | 16 | 0 |
| Free radicals | ||||||||||||
| Hussein et al. (2010) | TH carr units | >37 wk | 1’ or 5’-Apgar < 5 | 5–6d | 150.6 | 71.1 | 5 | 1’ or 5’-Apgar < 5, normal, low NSE | 101.8 | 10.5 | 25 | 0 |
| Hussein et al. (2010) | TH carr units | >37 wk | 1’ or 5’-Apgar < 5 | 5–6d | 150.6 | 71.1 | 5 | 1’ or 5’-Apgar < 5, normal, high NSE | 112.4 | 37.9 | 10 | 0 |
| Hussein et al. (2010) | TH carr units | >37 wk | 1’ or 5’-Apgar < 5 | 5–6d | 150.6 | 71.1 | 5 | Total normal | 104.8 | 47.9 | 35 | 0 |
| Kumar et al. (2008) | Malondialdehyde μmol/L | >37 wk | HIE 1 | 24–48 hr | 1.42 | 0.34 | 14 | Perinatal asphyxia, no HIE | 1.25 | 0.28 | 10 | 0 |
| Kumar et al. (2008) | Malondialdehyde μmol/L | >37 wk | HIE II | 24–48 hr | 1.73 | 0.27 | 9 | Perinatal asphyxia, no HIE | 1.25 | 0.28 | 10 | 6 |
| Kumar et al. (2008) | Malondialdehyde μmol/L | >37 wk | HIE III | 24–48 hr | 2.12 | 0.45 | 17 | Perinatal asphyxia, no HIE | 1.25 | 0.28 | 10 | 35 |
| Kumar et al. (2008) | Malondialdehyde μmol/L | >37 wk | Subtotal | 24–48 hr | 1.79 | 0.48 | 40 | Perinatal asphyxia, no HIE | 1.25 | 0.28 | 10 | 15 |
| Batra et al. (1998) | XO μmol/min/ml | NA | HIE dead | 24–48 hr | 8.89 | 2.41 | 5 | Recovered | 2.29 | 1.42 | 15 | 123 |
| Batra et al. (1998) | XO μmol/min/ml | NA | Asphyxia sick | 24–48 hr | 5.11 | 3.36 | 10 | Recovered | 2.29 | 1.42 | 15 | 0 |
| Batra et al. (1998) | 02- μmol/min/dl | NA | HIE dead | 24–48 hr | 1.92 | 0.96 | 5 | Recovered | 1.66 | 1.07 | 15 | 0 |
| Batra et al. (1998) | 02- μmol/min/dl | NA | Asphyxia sick | 24–48 hr | 2.19 | 1.41 | 10 | Recovered | 1.66 | 1.07 | 15 | 0 |
| Batra et al. (1998) | 02- μmol/min/dl | NA | Subtotal | 24–48 hr | 2.1 | 1.25 | 15 | Recovered | 1.66 | 1.07 | 15 | 0 |
| Batra et al. (1998) | H202 μg/h/ml | NA | HIE dead | 24–48 hr | 448.4 | 186.9 | 5 | Recovered | 362.8 | 251.5 | 15 | 0 |
| Batra et al. (1998) | H202 μg/h/ml | NA | Asphyxia sick | 24–48 hr | 433.8 | 148.6 | 10 | Recovered | 362.8 | 251.5 | 15 | 0 |
| Batra et al. (1998) | H202 μg/h/ml | NA | Subtotal | 24–48 hr | 438.7 | 155.6 | 15 | Recovered | 362.8 | 251.5 | 15 | 0 |
| Batra et al. (1998) | NO μg/ml | NA | HIE dead | 24–48 hr | 83.82 | 25.59 | 5 | Recovered | 43.72 | 17.9 | 15 | 0 |
| Batra et al. (1998) | NO μg/ml | NA | Asphyxia sick | 24–48 hr | 67.44 | 19.82 | 10 | Recovered | 43.72 | 17.9 | 15 | 0 |
| Batra et al. (1998) | NO μg/ml | NA | Subtotal | 24–48 hr | 72.9 | 22.4 | 15 | Recovered | 43.72 | 17.9 | 15 | 16 |
| Antioxidants | ||||||||||||
| Hussein et al. (2010) | BAPs μmol/L | >37 wk | 1’ or 5’-Apgar < 5 | 5–6d | 1,330.6 | 479.2 | 5 | 1’ or 5’-Apgar < 5, normal, low NSE | 1,139.4 | 196.5 | 25 | 0 |
| Hussein et al. (2010) | BAPs μmol/L | >37 wk | 1’ or 5’-Apgar < 5 | 5–6d | 1,330.6 | 479.2 | 5 | 1’ or 5’-Apgar < 5, normal, high NSE | 1,458.3 | 717.8 | 10 | 0 |
| Hussein et al. (2010) | BAPs μmol/L | >37 wk | 1’ or 5’-Apgar < 5 | 5–6d | 1,330.6 | 479.2 | 5 | Total normal | 1,230.5 | 1,124.6 | 35 | 0 |
| Ray et al. (1998) | SOD U/ml | NA | Mortality | 12–4 hr | 59.54 | 42.17 | 10 | Recovered | 40.62 | 29.59 | 16 | 0 |
| Ray et al. (1998) | SOD U/ml | NA | Morbidity | 12–48 hr | 71.27 | 39.86 | 8 | Recovered | 40.62 | 29.59 | 16 | 0 |
| Ray et al. (1998) | SOD U/ml | NA | Sub | 12–48 hr | 64.75 | 40.39 | 18 | Recovered | 40.62 | 29.59 | 16 | 0 |
| Ray et al. (1998) | GPX nmol/min/dl | NA | Mortality | 12–48 hr | 0.82 | 0.63 | 10 | Recovered | 0.82 | 0.51 | 16 | 0 |
| Ray et al. (1998) | GPX nmol/min/dl | NA | Morbidity | 12–48 hr | 0.63 | 0.35 | 8 | Recovered | 0.82 | 0.51 | 16 | 0 |
| Ray et al. (1998) | GPX nmol/min/dl | NA | Subtotal | 12–48 hr | 0.74 | 0.52 | 18 | Recovered | 0.82 | 0.51 | 16 | 0 |
| Vasiljevic et al. (2011) | GPX (U/L) | 32–37 wk | HIE severe | 2d | 166.1 | 27.4 | 9 | HIE mild | 106.6 | 29.5 | 57 | 29 |
| Vasiljevic et al. (2011) | GPX (U/L) | 32–37 wk | HIE moderate | 2d | 128.6 | 33.1 | 24 | HIE mild | 106.6 | 29.5 | 57 | 0 |
| Vasiljevic et al. (2011) | GPX (U/L) | 32–37 wk | Severe or moderate | 2d | 138.8 | 43.0 | 33 | HIE mild | 106.6 | 29.5 | 57 | 9 |
| Combo Ray et al. (1998); Vasiljevic et al. (2011) | 116 | 75 | 43 | 100 | 68 | 73 | 0 | |||||
| Neurotransmitter | ||||||||||||
| Cao et al. (1993) | LEK pg/ml | Mixed | Cerebral injury | NA | 204.1 | 342 | 21 | Asphyxiated no cerebral injury | 94.6 | 283 | 23 | 0 |
| Cao et al. (1993) | p-EP pg/ml | Mixed | Cerebral injury | NA | 141.5 | 177 | 21 | Asphyxiated no cerebral injury | 87.8 | 201 | 23 | 0 |
| Cao et al. (1993) | DynoAl-13 pg/ml | Mixed | Cerebral injury | NA | 136.1 | 178 | 21 | Asphyxiated no cerebral injury | 89.3 | 230.7 | 23 | 0 |
| Vasiljevic et al. (2011) | VEGF (pg/ml) | 32–37 wk | HIE severe | 3–5d | 430.2 | 83.9 | 9 | HIE mild | 118.6 | 15.2 | 57 | 205 |
| Vasiljevic et al. (2011) | VEGF (pg/ml) | 32–37 wk | HIE moderate | 3–5d | 253.9 | 53.4 | 24 | HIE mild | 118.6 | 15.2 | 57 | 92 |
| Vasiljevic et al. (2011) | VEGF (pg/ml) | 32–37 wk | Severe or moderate | 3–5d | 302.0 | 99.5 | 33 | HIE mild | 118.6 | 15.2 | 57 | 121 |
Note: Subtotal = combining previous rows of the same study. Δ% = (LCI of target-UCI of control) × 100/control mean.
Abbreviations: BAPs, biological antioxidant potentials; CPK, creatine phosphokinase; d, day; DynoAl-13, dynorphin Al-13; GPX, glutathione peroxidase; H202, hydrogen peroxide; LDH, lactate dehydrogenase; LEK, leu-enkephalin; LPO, lipid peroxidation; NO, nitric oxide; NSE, neuron-specific enolase; O2−, superoxide anions; SOD, superoxide dismutase; TH, total hydroperoxide; wk, week; XO, xanthine oxidase; β-EP, β-endorphin.
Biomarkers showing cell damage, such as neuron-specific enolase (NSE), LDH, LDH1, LDH2, and LDH3, and CK were significantly higher in the asphyxiated HIE group (Fernandez et al., 1986; Hussein et al., 2010; Ray et al., 1998). In this group, NSE at 5–6 days of life was a moderate biomarker for the severe HIE with neurobehavioral deficits with a score of 32 (Figure 3a); in a second study (Vasiljevic et al., 2011) NSE was a strong biomarker for severe HIE defined by clinical signs on presentation and who all had neurological sequelae; with a score of 144 compared to mild HIE (Figure 3b). In this study, mild HIE was defined as altered consciousness, irritability with jitteriness, slight abnormal muscle tone, exaggerated Moro, but absence of autonomic dysfunction and with normal aEEG patterns; thus, we included this in this group of studies as possible asphyxia. Taking the liberty of combining the two studies and using loose definition of the readouts, NSE was still a moderate biomarker with a score of 85. CK was found to be a strong biomarker differentiating HIE with mortality from all other groups with a score of 432 (Figure 3c), but could not differentiate HIE survivors from normal or plain asphyxia newborns. Vascular endothelial growth factor (VEGF165) was a strong biomarker for severe HIE (Figure 3d) compared to mild HIE.
Biomarkers showing free radical activities, such as total hydroperoxide (TH), malondialdehyde, XO, NO, and LPO were significantly higher in the asphyxiated HIE group (Batra et al., 1998; Hussein et al., 2010; Kumar et al., 2008; Ray et al., 1998). In this group, only XO and MDA were found to be useful biomarkers. XO showed a dose-response relationship with clinical severity of patients, progressing from patients who recovered, to those sick and to those who died, and was a strong biomarker for HIE and death, even when compared with asphyxia which recovered or to any live newborn (Figure 4a). MDA had a moderate score for severe HIE compared to asphyxia with no clinical signs (Figure 4b).
FIGURE 4.
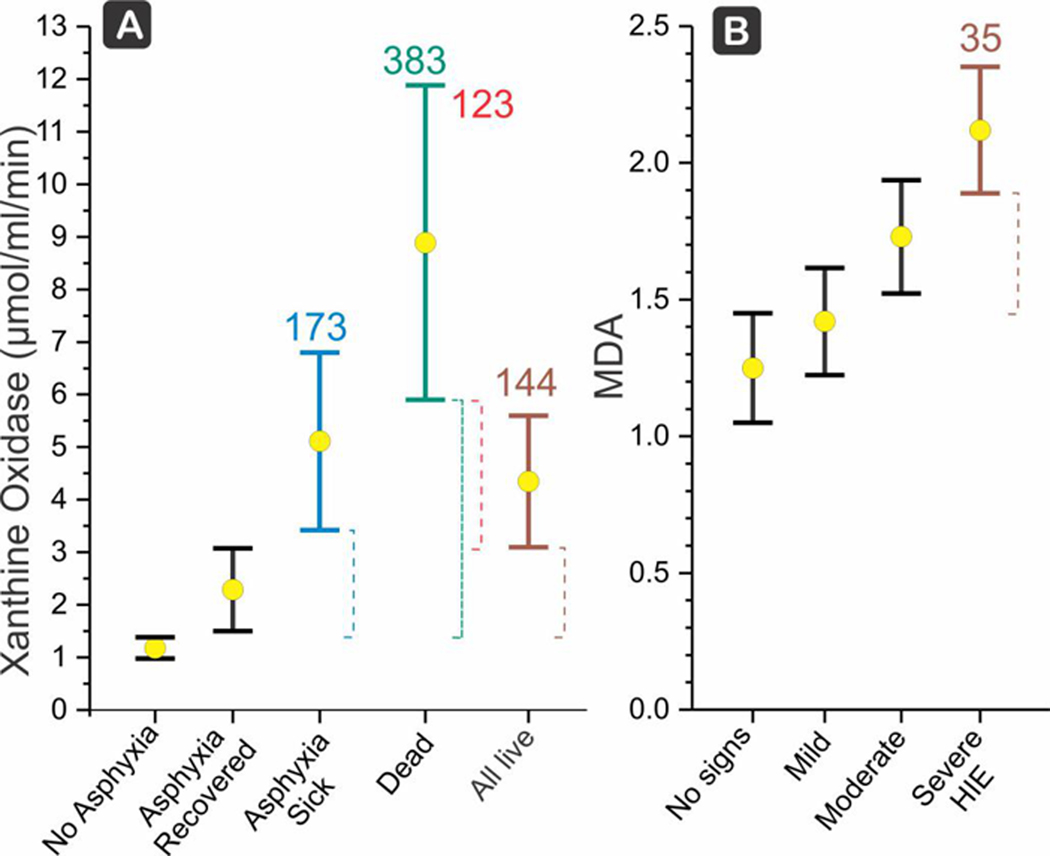
(a) Xanthine oxidase showing a dose response relationship in perinatal asphyxia with increasing values from recovered, to sick (any hypoxic-ischemic encephalopathy [HIE], moderate acidosis, convulsion or bronchopneumonia) to dead (Batra et al., 1998). In brown is any live newborn. XO is a strong biomarker for HIE and death, even when compared with asphyxia which recovered or to any live newborn ‼ (scores 123 and 144, respectively). Mean (X) and LCI, UCI shown. Score is Δ (dashed lines) between the target LCI and control UCI, divided by the control mean, and expressed as a percentage. (b) Malondialdehyde (MDA) in patients with Apgar <3 at 1 min, the subtotal population showing clinical signs of severe HIE by Fenichel classification, namely either stupor, coma, irregular/periodic respirations or ventilated, apnea, convulsions, hypotonia, oculomotor palsies showed a moderate score of 32 compared to a population without any signs of HIE (Kumar et al., 2008)
Biomarkers showing antioxidants activities, such as total hydroperoxide (BAPs), SOD, and GPX were similar between the two groups (Hussein et al., 2010; Ray et al., 1998) and the scores are 0 for all.
Biomarkers showing neurotransmitter activities, such as LEK, β-EP, and DynoA1–13, were significantly higher in the asphyxiated HIE group (Cao et al., 1993). In this group, all the biomarkers tested were considered doubtful. For completion sake, we also compared the patients with brain injury with control normals.
3.3 |. CSF markers in asphyxiated HIE versus non-asphyxiated cases
Five studies (Blennow et al., 1995; Fernandez et al., 1986; Gucuyener et al., 1999; Gulcan et al., 2005; Ray et al., 1998) reported CSF markers in asphyxiated HIE (331 test results) versus non-asphyxiated (369 test results) cases (Table 3).
TABLE 3.
CSF markers in asphyxiated HIE versus non-asphyxiated cases
| Study | Biomarker | Gestation | Case type | Age sampling | Case mean | SD | No. | Control mean | SD | No. | Score (Δ% X) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Free radicals | |||||||||||
| Ray et al. (1998) | LPO nmol/h/dl | NA | Mortality | 12−48 hr | 21.43 | 4.36 | 10 | 10.82 | 3.02 | 25 | 58 |
| Ray et al. (1998) | LPO nmol/h/dl | NA | Morbidity (HIE-II) | 12−48 hr | 17.98 | 4.62 | 8 | 10.82 | 3.02 | 25 | 19 |
| Ray et al. (1998) | LPO nmol/h/dl | NA | Subtotal | 12−48 hr | 19.9 | 4.69 | 18 | 10.82 | 3.02 | 25 | 51 |
| Ray et al. (1998) | CPK U/I | NA | Mortality | 12−48 hr | 156 | 56.76 | 10 | 18.32 | 2.56 | 25 | 524 |
| Ray et al. (1998) | CPK U/I | NA | Morbidity | 12−48 hr | 21.75 | 9.31 | 8 | 18.32 | 2.56 | 25 | 0 |
| Ray et al. (1998) | CPK U/I | NA | Subtotal | 12−48 hr | 96.33 | 80.33 | 18 | 18.32 | 2.56 | 25 | 202 |
| Ray et al. (1998) | Total calcium mg/dl | NA | Mortality | 12−48 hr | 7.32 | 1.13 | 10 | 5.92 | 1.31 | 25 | 1 |
| Ray et al. (1998) | Total calcium mg/dl | NA | Morbidity | 12−48 hr | 6.02 | 0.73 | 8 | 5.92 | 1.31 | 25 | 0 |
| Ray et al. (1998) | Total calcium mg/dl | NA | Subtotal | 12−48 hr | 6.74 | 1.16 | 18 | 5.92 | 1.31 | 25 | 0 |
| Antioxidants | |||||||||||
| Gulcan et al. (2005) | SOD U/mg protein | ≥37 wk | HIE-I | <72 hr | 0.37 | 0.19 | 12 | 0.27 | 0.16 | 11 | 0 |
| Gulcan et al. (2005) | SOD U/mg protein | ≥37 wk | HIE-II | <72 hr | 0.52 | 0.27 | 9 | 0.27 | 0.16 | 11 | 0 |
| Gulcan et al. (2005) | SOD U/mg protein | ≥37 wk | HIE-III | <72 hr | 0.47 | 0.3 | 9 | 0.27 | 0.16 | 11 | 0 |
| Gulcan et al. (2005) | SOD U/mg protein | ≥37 wk | All HIE | <72 hr | 0.45 | 0.25 | 30 | 0.27 | 0.16 | 11 | 0 |
| Ray et al. (1998) | SOD U/ml | 34 wk | Mortality | 12−48 hr | 60 | 42 | 10 | 24 | 6 | 25 | 13 |
| Ray et al. (1998) | SOD U/ml | 34 wk | Morbidity | 12−48 hr | 71 | 40 | 8 | 24 | 6 | 25 | 49 |
| Ray et al. (1998) | SOD U/ml | 34 wk | Subtotal | 12−48 hr | 65 | 40 | 18 | 24 | 6 | 25 | 77 |
| Combo Gulcan et al. (2005), Ray et al. (1998) | 206 | 192 | 48 | 100 | 24 | 36 | 42 | ||||
| Gulcan et al. (2005) | GPX U/mg protein | ≥37 wk | HIE-I | <72 hr | 9.21 | 6.87 | 12 | 10.54 | 7.25 | 11 | 0 |
| Gulcan et al. (2005) | GPX U/mg protein | ≥37 wk | HIE-II | <72 hr | 15.75 | 11.72 | 9 | 10.54 | 7.25 | 11 | 0 |
| Gulcan et al. (2005) | GPX U/mg protein | ≥37 wk | HIE-III | <72 hr | 26.13 | 16.77 | 9 | 10.54 | 7.25 | 11 | 0 |
| Gulcan et al. (2005) | GPX U/mg protein | ≥37 wk | All HIE | <72 hr | 16.25 | 13.57 | 30 | 10.54 | 7.25 | 11 | 0 |
| Ray et al. (1998) | GPX nmol/ min/dl | 34 wk | Mortality | 12−48 hr | 0.82 | 0.63 | 10 | 1.28 | 0.52 | 25 | 0 |
| Ray et al. (1998) | GPX nmol/ min/dl | 34 wk | Morbidity | 12−48 hr | 0.63 | 0.35 | 8 | 1.28 | 0.52 | 25 | −23 |
| Ray et al. (1998) | GPX nmol/ min/dl | 34 wk | Subtotal | 12−48 hr | 0.74 | 0.52 | 18 | 1.28 | 0.52 | 25 | −10 |
| Combo Gulcan et al. (2005), Ray et al. (1998) | 118 | 135 | 48 | 100 | 80 | 36 | 0 | ||||
| Gulcan et al. (2005) | CAT U/mg protein | ≥37 wk | HIE-I | <72 hr | 15.61 | 11.19 | 12 | 17.25 | 12.51 | 11 | 0 |
| Gulcan et al. (2005) | CAT U/mg protein | ≥37 wk | HIE-II | <72 hr | 24.7 | 16.92 | 9 | 17.25 | 12.51 | 11 | 0 |
| Gulcan et al. (2005) | CAT U/mg protein | ≥37 wk | HIE-III | <72 hr | 42.25 | 26.63 | 9 | 17.25 | 12.51 | 11 | 0 |
| Gulcan et al. (2005) | CAT U/mg protein | ≥37 wk | Subtotal | <72 hr | 26.33 | 21.19 | 30 | 17.25 | 12.51 | 11 | 0 |
| Neurotransmitters | |||||||||||
| Blennow et al. (1995) | Noradrenaline nM | >35 wk | Moderate or severe HIE | <24 hr | 2.16 | 2.3 | 11 | 4.27 | 4.3 | 11 | 0 |
| Blennow et al. (1995) | MHPAC nM | >35 wk | Moderate or severe HIE | <24 hr | 105 | 51 | 11 | 86 | 17 | 11 | 0 |
| Blennow et al. (1995) | DOPAC nM | >35 wk | Moderate or severe HIE | <24 hr | 13.5 | 4.7 | 11 | 17.3 | 7.5 | 11 | 0 |
| Blennow et al. (1995) | HVA nM | >35 wk | Moderate or severe HIE | <24 hr | 523 | 109 | 11 | 510 | 79 | 11 | 0 |
| Blennow et al. (1995) | HIAA nM | >35 wk | Moderate or severe HIE | <24 hr | 429 | 54 | 11 | 400 | 113 | 11 | 0 |
| Cell damage | |||||||||||
| Fernandez et al. (1986) | LDH U/L | ≥37 wk | Abnormal or died at 1 year | 1d | 118 | 96 | 6 | 41 | 18 | 20 | 0 |
| Fernandez et al. (1986) | LDH1 U/L | ≥37 wk | Abnormal or died at 1 year | 1d | 15 | 12 | 6 | 12 | 5 | 20 | 0 |
| Fernandez et al. (1986) | LDH2 U/L | ≥37 wk | Abnormal or died at 1 year | 1d | 38.5 | 24 | 6 | 12 | 6 | 20 | 0 |
| Fernandez et al. (1986) | LDH3 U/L | ≥37 wk | Abnormal or died at 1 year | 1d | 50 | 32 | 6 | 12 | 5 | 20 | 19 |
| Fernandez et al. (1986) | LDH4 U/L | ≥37 wk | Abnormal or died at 1 year | 1d | 10 | 20 | 6 | 2 | 1 | 20 | 0 |
| Fernandez et al. (1986) | LDH5 U/L | ≥37 wk | Abnormal or died at 1 year | 1d | 5.5 | 12 | 6 | 2 | 1 | 20 | 0 |
| Gucuyener et al. (1999) | Aspartate μmol/L | 30−39 wk | Abnormal at 3 years | 1−2d | 9.8 | 3.37 | 6 | 3.98 | 1.52 | 12 | 33 |
| Gucuyener et al. (1999) | Aspartate μmol/L | 30−39 wk | Dead | 1−2d | 5.33 | 2.36 | 4 | 3.98 | 1.52 | 12 | 0 |
| Gucuyener et al. (1999) | Aspartate μmol/L | 30−39 wk | Subtotal | 1−2d | 8.01 | 3.67 | 10 | 3.98 | 1.52 | 12 | 11 |
| Gucuyener et al. (1999) | Glutamate μmol/L | 30−39 wk | Abnormal at 3 years | 1−2d | 2.02 | 0.99 | 6 | 1.65 | 1.1 | 12 | 0 |
| Gucuyener et al. (1999) | Glutamate μmol/L | 30−39 wk | Dead | 1−2d | 1.7 | 0.78 | 4 | 1.65 | 1.1 | 12 | 0 |
| Gucuyener et al. (1999) | Glutamate μmol/L | 30−39 wk | Subtotal | 1−2d | 1.89 | 0.88 | 10 | 1.65 | 1.1 | 12 | 0 |
| Gucuyener et al. (1999) | Taurine μmol/L | 30−39 wk | Abnormal at 3 years | 1−2d | 12.58 | 14.24 | 6 | 5.8 | 2.04 | 12 | 0 |
| Gucuyener et al. (1999) | Taurine μmol/L | 30−39 wk | Dead | 1−2d | 14.96 | 11.03 | 4 | 5.8 | 2.04 | 12 | 0 |
| Gucuyener et al. (1999) | Taurine μmol/L | 30−39 wk | Subtotal | 1−2d | 13.53 | 12.44 | 10 | 5.8 | 2.04 | 12 | 0 |
Note: Subtotal = combining previous rows of the same study. Δ% = (LCI of target-UCI of control) × 100/control mean.
Abbreviations: CAT, catalase; CPK, creatine phosphokinase; d, day; DOPAC, 3.4-dihydroxyphenylacetic acid; GPX, glutathione peroxidase; HIAA, 5-hydroxyindole-3-acetic acid; HVA, homovanillic acid; LDH, lactate dehydrogenase; LPO, lipid peroxidation; MHPAC, 3-m ethoxy-4-hydroxy-phenylglycol; SOD, superoxide dismutase; wk, week.
LPO was significantly higher in the asphyxiated HIE group (Ray et al., 1998) but was considered doubtful as a biomarker.
Among cell injury markers, CK had a high score demarcating the HIE who died from the control newborns (Ray et al., 1998). The score of the dead and sick was 202 but a closer look shows that CK could not demarcate between asphyxiated sick newborns from control newborns.
Among antioxidants, SOD was significantly higher in the asphyxiated HIE group (Gulcan et al., 2005; Ray et al., 1998), while GPX was similar between the two groups in one study (Gulcan et al., 2005) and was lower in the asphyxiated HIE group in the other study (Ray et al., 1998). In this group, combining two studies, SOD showed it was a weak biomarker with a score of 42.
Biomarkers showing neurotransmitter activities, such as noradrenaline, MHPAC, 3.4-dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), and 5-hydroxyindole-3-acetic acid (HIAA) were all similar between the two groups (Blennow et al., 1995). Biomarkers classified as cell damage indicators, such as LDH, LDH2, LDH3, and LDH4, were significantly higher in the asphyxiated HIE group (Fernandez et al., 1986). In this group, only aspartate was a weak biomarker for either neurobehavioral deficits at 3 years of age or death compared to controls, with a score of 11 (Gucuyener et al., 1999), and the rest were considered doubtful.
3.4 |. CSF markers in preterm versus term asphyxiated or HIE cases
Two studies (Talvik et al., 1995; Vasiljevic et al., 2011) reported CSF markers in asphyxiated HIE versus asphyxiated non-HIE cases, including 214 test results in the preterm asphyxiated group and 225 test results in the term asphyxiated group (Table 4).
TABLE 4.
CSF markers in term versus preterm asphyxiated or HIE cases
| Study | Biomarker | Function | Age sampling | Case type | Mean | SD | No. | Control type | Mean | SD | No. | Score (Δ% ) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Asphyxiated | ||||||||||||
| Talvik et al. (1995) | CK-BB ng/ml | Cell damage | 2–5d | 5’-Apgar < 7, <37 wk | 168 | 22.2 | 79 | ≥37 wk, 5’-Apgar < 7 | 29 | 3.1 | 90 | 460 |
| HIE | ||||||||||||
| Talvik et al. (1995) | CK-BB ng/ml | Cell damage | 2d | HIE 1, <37 wk | 198.7 | 34 | 20 | ≥37 wk, 5’-Apgar < 7 | 29 | 3.1 | 90 | 528 |
| Talvik et al. (1995) | CK-BB ng/ml | Cell damage | 2d | HIE II and III, <37 wk | 317.2 | 33 | 19 | ≥37 wk, 5’-Apgar < 8 | 29 | 3.1 | 90 | 937 |
| Talvik et al. (1995) | CK-BB ng/ml | Cell damage | subtotal | 256.4 | 68.5 | 39 | ≥37 wk, 5’-Apgar < 9 | 29 | 3.1 | 90 | 705 | |
| Talvik et al. (1995) | CK-BB ng/ml | Cell damage | 2d | Premature Moderate Severe | 317.2 | 33 | 19 | Premature mild | 198.7 | 34 | 20 | 44 |
| Talvik et al. (1995) | CK-BB ng/ml | Cell damage | 5d | Premature Moderate Severe | 95 | 48 | 35 | Premature mild | 9 | 3.8 | 6 | 732 |
| Vasiljevic et al. (2011) | NSE ng/L | Neuron destructive | 3–5d | HIE, 32–37 wk | 26.7 | 20.6 | 45 | ≥37 wk | 21.9 | 14.1 | 45 | 0 |
| Vasiljevic et al. (2011) | GPX U/L | Antioxidant | 2d | HIE, 32–37 wk | 137.2 | 33.5 | 45 | ≥37 wk | 99.6 | 25.9 | 45 | 20 |
| Vasiljevic et al. (2011) | VEGFpg/mL | Growth factor | 3–5d | HIE, 32–37 wk | 156.9 | 44.9 | 45 | ≥37 wk | 184 | 53.2 | 45 | 0 |
Note: Subtotal = combining previous rows of the same study.
Abbreviations: CK-BB, creatine kinase brain isoenzyme; d, day; NSE, neuron-specific enolase; GPX, glutathione peroxidase; VEGF, vascular endothelial growth factor; wk, week.
Creatine kinase brain isoenzyme (CK-B B) was significantly higher in both the preterm asphyxiated group and the preterm HIE group, compared with those of the term groups (Talvik et al., 1995). GPX was significantly higher in the preterm HIE group, and VEGF was significantly lower in the preterm HIE group, whereas NSE remained similar between the preterm and term HIE groups (Vasiljevic et al., 2011). The score of CK-BB for premature newborns was 460, indicating it to be a strong biomarker. CK-BB collected at postnatal days 2–5 was significantly increased with severity of impairment diagnosed at 12 months of age (Figure 5). It is notable that the moderate and severe outcome showed higher CK-BB at 2 and 5 days of life compared to mild outcomes with stronger biomarker utility at 5 days (Figure 5).
FIGURE 5.
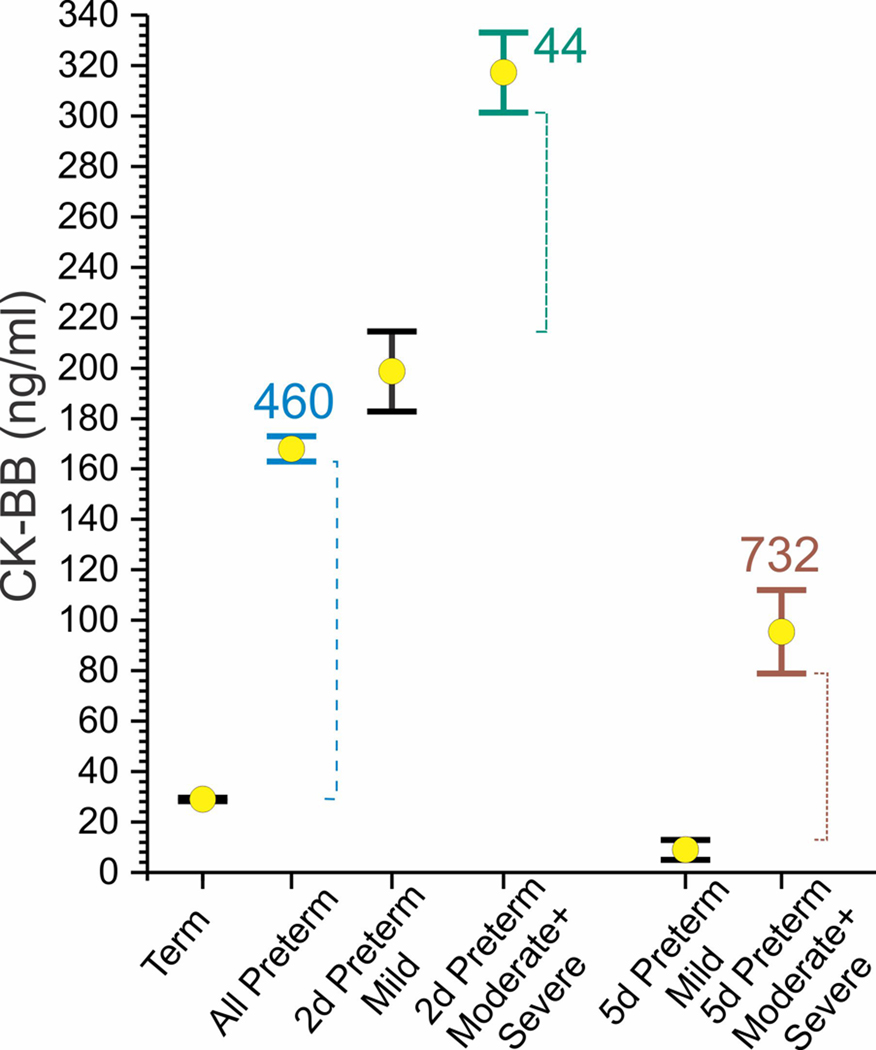
Preterm HIE has much higher creatine kinase brain isoenzyme (CK-BB) than term HIE (Talvik et al., 1995). CK-BB is a weak biomarker for the severity of neurological deficits at 12 months of age if done at 2 days (score 44) but a strong biomarker if done at 5 days of life (score 732)
3.5 |. Effect of checkpoint on CSF biomarkers
The effect of the time of CS collection on the level of biomarkers was tracked in several studies. CK-BB showed a significant lower level when collected at postnatal day 5 (5d) compared with that collected at 2d for all HIE categories (Talvik et al., 1995). The pattern of CK-BB fall suggests that the timing of the insult would be a predominant determinant of the level of CK-BB in the CSF. FDP was highest at P1d, and gradually decreased at 3, 8, and 15d (Dalens, Bezou, et al., 1981).
3.6 |. Effect of level of asphyxia or HIE on CSF biomarkers
LEK, β-EP, and DynoA1–13 were significantly higher in the severely asphyxiated group than in the moderately asphyxiated group (Cao et al., 1993). FDP was significantly lower in the severely asphyxiated group than in the moderately asphyxiated group (Dalens, Bezou, et al., 1981). ASAT, CK, HBD, and LDH were significantly lower in the severely asphyxiated group than in the moderately asphyxiated group (Dalens, Viallard, et al., 1981).
Malondialdehyde was significantly higher in the HIE III group than that in the HIE I (Kumar et al., 2008). XO was significantly higher in the HIE III or died group than that in the HIE I-II group (Batra et al., 1998). CK and total calcium were significantly higher in the mortality group than in the morbidity group (Ray et al., 1998). Aspartate was significantly lower in the 3-year dead group than in the 3-year abnormal group (Gucuyener et al., 1999). NSE, GPX, and VEGF had a linear increase with the increasing metabolic acidosis (Vasiljevic et al., 2011). We did not have enough studies to do a statistical analysis of the dose–response relationship on the severity of the asphyxia components.
4 |. DISCUSSION
Of the noninflammatory CSF biomarkers reported in asphyxiated or HIE neonates, we show that CK, XO, VEGF, NSE, SOD, and malondialdehyde could be useful as clinical biomarkers (ranked in order of strength). The variation in the units of reported biomarkers and methods of measurement as well as the unfocused targets made it difficult to carry out a pooled analysis or meta-analysis, but we presented combined analysis based on each individual study’s control mean if there were two studies reporting on the same biomarker. A strength of this review is pointing out the remarkable fact that even though a lot of biomarkers were statistically significant, the biological significance using estimation approach showed only a few endpoints had clinical utility as a biomarker. An ideal biomarker should have a very low to zero false positive and false negative rate. The utility of a biomarker varies with the endpoint, such as asphyxia versus non-asphyxia, bad neurological outcome versus recovery, etc. This review would be useful to most neonatologists and obstetricians, since the most important endpoint is eventual neurobehavioral outcome and death. In an informal survey of 20 neonatologists, we found that neonatologists would accept a biomarker with a slightly higher false positive rate (14.5% average) than a false negative rate (5.1%) when it comes to long-term neurodevelopmental outcome or death. Using our score based on CIs and means of biomarkers that were higher than controls, it could be argued our strategy minimizes false positive rate more so than false negative rate, but both were very low if the scores are >100. The strength of the biomarkers was evident in the severest HIE cases but much less for the HIE that was moderate or mild when compared to control normals. One could also argue that this is because our score was probably too stringent, given the inherent contradiction of the relationship of false positivity and false negativity. If so, it is still remarkable that we were able to find biomarkers with scores >100, meaning that the Δ was greater than the control mean. It is also notable that CK-BB was a stronger biomarker at postnatal days 5–6 compared to day 2, in differentiating moderate or severe HIE from mild HIE in preterm newborns <37 weeks (Figure 5).
The neonatal brain injury during perinatal H-I evolves gradually. During extended HI, neurons might experience high-energy metabolite depletion, progressive cell depolarization, cytotoxic edema (Gunn et al., 1997), and extracellular accumulation of excitatory amino acids (Tan et al., 1996). While some neurons may die immediately during or soon after HI, some may initially recover at various level, and die later, which is characterized by cerebral energy failure from 6 to 48 hr after insult (Cotten & Shankaran, 2010), and clinically presented as delayed onset of seizures and cytotoxic edema, and resolves over approximately 72 hr after HI. The severity of the secondary failure of oxidative metabolism is closely correlated with neurodevelopmental outcome at 1 and 4 years of age (Roth et al., 1997), and infants with encephalopathy who do not show initial recovery of cerebral oxidative metabolism have extremely poor outcomes (Azzopardi et al., 1989).
The issue of timing of the brain insult is the single most important factor that affects the suitability of a biomarker but the time after delivery does not mean equivalency to time after insult (Tan, 2014). Unfortunately, timing of the insult, whether antepartum or intrapartum, is mostly unknown in newborn babies. Even fetal demise can be remote from delivery and varies in time after insult (Derrick et al., 2012). Lack of precise timing makes it impossible to categorize observed brain injury as being primary (acute energy failure), secondary (next energy failure and cell death), or tertiary brain injury (sensitization to another round of cell death) (Thornton et al., 2012). Free radicals probably occur the earliest, followed by cell death, then the compensatory response involving antioxidants, growth factors, along with continuing cell damages occur later. Since brain is rich in lipids, it makes sense to use lipid peroxidation products such as malondialdehyde as a free radical biomarker. Unfortunately, caution must be undertaken to rely on the 2-thiobarbituric-acid-reactive substances as a marker of lipid peroxidation (Janero, 1990) as shown in Figure 4, since the then published assay has been supplanted by more reliable methods (Guichardant et al., 2004; Nourooz-Zadeh et al., 1999). Nevertheless, given more sophisticated methods of both reactive oxygen and nitrogen species, it may become possible to improve the free radical biomarkers and narrow the timing of the insult to a more recent time.
Xanthine oxidase (EC 1.17.3.2) is molybdenum-containing enzyme, and is a source of free radicals in the presence of purines. The purines, hypoxanthine, and xanthine are substrates for xanthine oxidase, which increase with hypoxia-ischemia with the breakdown of adenine monophosphate to these purines (Parks & Granger, 1986). XO is a term that actually represents two forms: xanthine oxidoreductase (EC 1.2.3.3) existing in healthy cells as the NAD+-reducing xanthine dehydrogenase, which is converted to oxygen radical-producing xanthine oxidase during ischemia. Interestingly, xanthine oxidase is barely detectable in adult human brain tissues: 1–4 nU/mg protein in different parts of the brain (Michel et al., 2010), or 1 mU/mg protein (Kokoglu et al., 1990). Newborn brain levels are unknown. Circulating levels of xanthine oxidase in newborn are low with total xanthine dehydrogenase and xanthine oxidase to be 8–9 μU/ml and xanthine oxidase alone to be 2.5 μU/ml, performed with a sensitive HPLC assay (Tan et al., 1993); even in adults both are low, 1.88 and 1.66 μU/mg protein (Tan et al., 1995). In Figure 4, the units of XO reported by a spectrophotometric assay were in U/ ml for comparison (Batra et al., 1998), which are orders of magnitude higher. Most likely, the predominant source of xanthine oxidase is probably coming from endothelial cells rather than brain tissue itself. It is unknown what is the status of XO in the choroid plexus. Since xanthine oxidase binds to endothelial cells (Houston et al., 1999), it is possible that high levels of XO could emanate from cerebral vessels.
CK is an enzyme (EC 2.7.3.2) catalyzing the conversion of creatine to create phosphocreatine (PCr), while using adenosine triphosphate (ATP). The PCr serves as an energy reservoir for the rapid buffering and regeneration of ATP. Neurons and astroglia have CK and different isozymes are found. Given that CK reflects energy metabolism, it is surprising that it is not a good biomarker for mild or moderate HIE (Ray et al., 1998). However, it is a strong biomarker for severe HIE (Figure 3c) and it may be possible that a certain threshold of brain injury may be necessary for the release of CK into the CSF. The human brain expresses different combinations of CK isozymes, and a future study using ubiquitous mitochondrial CK isozyme that supports oxidative energy metabolism and cytosolic brain-CK that supports glycolytic processes (Lowe et al., 2013) may tease out the pathogenetic pathways in HIE.
The gamma isoform of NSE is a marker for neurons and peripheral neuroendocrine cells, and involved in glycolytic energy metabolism in the brain. It is released from neurons during injury. NSE seems to be a sensitive marker of neonatal brain injury (Figure 3a) when elevations are seen even in the population that shows no neurobehavioral deficits (Hussein et al., 2010). These investigators have postulated that 10 ng/ml of NSE can be used as a cutoff for the upper bound of normal equal to the mean + 3SEM, but Figure 3a,b suggest that the cutoff should probably be >40 ng/ml.
VEGF is a signal protein produced by cells that stimulates the formation of blood vessels and exhibits neurotrophic and neuroprotective effects. The rise of VEGF with the severity of brain injury is somewhat counterintuitive, especially as it has been found to be a neuroprotectant in animal studies of perinatal H-I (Feng et al., 2008). In a way this is to be expected as the eventual neurodevelopmental outcome is dependent on the final outcome between ongoing brain injury, recovery, and plasticity of the brain. The ability of the brain to mount a regenerative response can be utilized as a biomarker in the right circumstance. In our experience, we have found a similar counterintuitive response even in the structural MRI findings where a postnatal dynamic increase in the fractional anisotropy of the internal capsule was associated with a worse outcome (Drobyshevsky et al., 2007).
The simultaneous evaluation of a panel of biomarkers for acute brain damage might provide a number of advantages over the measure of individual markers. Information about neuronal injury combined with free radical-and cell injury markers would be very useful to a neonatologist understanding the etiology of a patient with NE (Tan & Wu, 2020). In future, it is possible that a panel of neuron-enriched proteins may be more useful as biomarkers as has been shown in patients with traumatic brain injury (Siman et al., 2009), because a change in multiple neuron-enriched biomarkers could not come from extracranial sources unrelated to acute brain injury. This problem of extracranial sources becomes an issue with the testing in serum or blood. Furthermore, serum biomarkers are not as sensitive as CSF to brain injury because many proteins do not cross into the circulation or remain undetected due to dilution in circulating blood. Other candidate biomarkers in translational studies of HIE, such as tetrahydrobiopterin (Vasquez-Vivar et al., 2017, 2020), could be investigated in near future.
Typically, when a patient is suspected of NE, the term babies are under time pressure to get cooling started within 6 hr of birth. The timing of the spinal tap then becomes an issue, as cooling would inhibit getting the procedure done before or during cooling. After 72 hr of cooling, cooling is discontinued, and then a spinal tap could easily be performed. On a mechanistic basis, this would put emphasis on biomarkers that would reflect the late secondary or tertiary phase of injury, which is due to the persistent inflammation and epigenetic changes, and causing a blockade of neurogenesis (Fleiss & Gressens, 2012). Most of the biomarkers noted in this review would still be under consideration other than lipid peroxidation products. In premature babies, cooling is not a consideration so the spinal tap could be done any time after birth. Ideally, the CSF biomarkers could be used to decide on babies that needed to be cooled just after delivery, to diagnose mild from moderate or severe brain injury. Future treatments that could be added on in the tertiary phases of injury are umbilical cord stem cells (Drobyshevsky et al., 2015) or tetrahydrobiopterin (Vasquez-Vivar et al., 2020).
The limitations of this systematic review are mainly due to the limited number of studies for each biomarker. We were not able to analyze publication bias or employ the method of meta-analysis for more quantitative data analysis. Some early studies did not describe if the cases were term or preterm, or reported preterm and term cases together, which might have quite different pathological or developmental prognosis. Confirmation of the biomarkers in preclinical studies was hard to find because of the paucity of animal survival studies on CSF biomarkers correlations with neurobehavioral outcome, and the paucity of animal models manifesting a severe outcome. With more extensive studies in future, we could develop better biomarkers for specific clinical situations.
In conclusion, this review identifies some promising biomarkers that could be put in a panel of biomarkers to be simultaneously tested which would help prognosticate not only the most severe HIE but moderate HIE. Improvements with technological development in different assays would further improve the sensitivity and specificity of these biomarkers.
Supplementary Material
TABLE S1 Analysis of risk of bias of individual studies using Newcastle-Ottawa Quality Assessment Scale
Significance.
In the present systematic review, we focused on the cerebrospinal fluid (CSF) and noninflammatory biomarkers that are involved in the development of possible brain injury in asphyxia or hypoxic-ischemic encephalopathy (HIE). We identified the best biomarkers out of hundreds in three categories: cell adhesion and proliferation, oxidants and antioxidants, and cell damage. Biological significance of the biomarkers was determined by using a modification of the estimation approach, by ranking the biomarkers according to the difference in the bounds of the confidence intervals. The most promising CSF biomarkers for prognostication especially for the severest HIE include creatine kinase, xanthine oxidase, vascular endothelial growth factor, neuron-specific enolase, superoxide dismutase, and malondialdehyde.
Funding information
National Institute of Neurological Disorders and Stroke
Funding information Funded through 1R01NS114972 (ST) from NINDS, NIH. The funder has no role in any step of this systematic review.
Footnotes
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1002/jnr.24801.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
CONFLICT OF INTEREST
No potential conflict of interest was reported by the authors.
REFERENCES
- Azzopardi D, Wyatt JS, Cady EB, Delpy DT, Baudin J, Stewart AL, Hope PL, Hamilton PA, & Reynolds EOR (1989). Prognosis of newborn infants with hypoxic-ischemic brain injury assessed by phosphorus magnetic resonance spectroscopy. Pediatric Research, 25(5), 445–451. 10.1203/00006450-198905000-00004 [DOI] [PubMed] [Google Scholar]
- Batra S, Raj G, & Dutta AK (1998). Free radical production during perinatal birth asphyxia. Medical Science Research, 26, 323–325. [Google Scholar]
- Blennow M, Zeman J, Dahlin I, & Lagercrantz H. (1995). Monoamine neurotransmitters and metabolites in the cerebrospinal fluid following perinatal asphyxia. Biology of the Neonate, 67(6), 407–413. 10.1159/000244193 [DOI] [PubMed] [Google Scholar]
- Calvert JW, & Zhang JH (2005). Pathophysiology of an hypoxic-ischemic insult during the perinatal period. Neurological Research, 27(3), 246–260. [DOI] [PubMed] [Google Scholar]
- Cao L, Qian PD, Jing LJ, Liang QJ, & Zhao ZZ (1993). Endogenous opioid-like substances in perinatal asphyxia and cerebral injury due to anoxia. Chinese Medical Journal, 106(10), 783–787. [PubMed] [Google Scholar]
- Cotten CM, & Shankaran S. (2010). Hypothermia for hypoxic-i schemic encephalopathy. Expert Review of Obstetrics & Gynecology, 5(2), 227–239. 10.1586/eog.10.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalens B, Bezou MJ, Coulet M, & Raynaud EJ (1981). Fibrin-fibrinogen degradation products in cerebrospinal fluid as an indicator of neonatal brain damage. Acta Neurologica Scandinavica, 64(2), 81–87. 10.1111/j.1600-0404.1981.tb04390.x [DOI] [PubMed] [Google Scholar]
- Dalens B, Viallard JL, Raynaud EJ, & Dastugue B. (1981). Enzyme studies and neonatal brain damage. Acta Paediatrica, 70(5), 743–749. 10.1111/j.1651-2227.1981.tb05779.x [DOI] [PubMed] [Google Scholar]
- Derrick M, Englof I, Drobyshevsky A, Luo K, Yu L, & Tan S. (2012). Intrauterine fetal demise can be remote from the inciting insult in an animal model of hypoxia-ischemia. Pediatric Research, 72(2), 154–160. 10.1038/pr.2012.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drobyshevsky A, Bregman J, Storey P, Meyer J, Prasad PV, Derrick M, MacKendrick W, & Tan S. (2007). Serial diffusion tensor imaging detects white matter changes that correlate with motor outcome in premature infants. Developmental Neuroscience, 29(4–5), 289–301. 10.1159/000105470 [DOI] [PubMed] [Google Scholar]
- Drobyshevsky A, Cotten CM, Shi Z, Luo K, Jiang R, Derrick M, Tracy ET, Gentry T, Goldberg RN, Kurtzberg J, & Tan S. (2015). Human umbilical cord blood cells ameliorate motor deficits in rabbits in a cerebral palsy model. Developmental Neuroscience, 37(4–5), 349–362. 10.1159/000374107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Rhodes PG, & Bhatt AJ (2008). Neuroprotective effects of vascular endothelial growth factor following hypoxic ischemic brain injury in neonatal rats. Pediatric Research, 64(4), 370–374. 10.1203/PDR.0b013e318180ebe6 [DOI] [PubMed] [Google Scholar]
- Fernandez F, Quero J, Verdu A, Ferreiros MC, Daimiel E, & Roche MC (1986). LDH isoenzymes in CSF in the diagnosis of neonatal brain damage. Acta Neurologica Scandinavica, 74(1), 30–33. 10.1111/j.1600-0404.1986.tb04621.x [DOI] [PubMed] [Google Scholar]
- Fleiss B, & Gressens P. (2012). Tertiary mechanisms of brain damage: A new hope for treatment of cerebral palsy? Lancet Neurology, 11(6), 556–566. 10.1016/S1474-4422(12)70058-3 [DOI] [PubMed] [Google Scholar]
- Graham EM, Ruis KA, Hartman AL, Northington FJ, & Fox HE (2008). A systematic review of the role of intrapartum hypoxia-ischemia in the causation of neonatal encephalopathy. American Journal of Obstetrics and Gynecology, 199(6), 587–595. 10.1016/j.ajog.2008.06.094 [DOI] [PubMed] [Google Scholar]
- Gucuyener K, Atalay Y, Aral YZ, Hasanoglu A, Turkyilmaz C, & Biberoglu G. (1999). Excitatory amino acids and taurine levels in cerebrospinal fluid of hypoxic ischemic encephalopathy in newborn. Clinical Neurology and Neurosurgery, 101(3), 171–174. 10.1016/s0303-8467(99)00035-9 [DOI] [PubMed] [Google Scholar]
- Guichardant M, Chantegrel B, Deshayes C, Doutheau A, Moliere P, & Lagarde M. (2004). Specific markers of lipid peroxidation issued from n-3 and n-6 fatty acids. Biochemical Society Transactions, 32(Pt 1), 139–140. 10.1042/bst0320139 [DOI] [PubMed] [Google Scholar]
- Gulcan H, Ozturk IC, & Arslan S. (2005). Alterations in antioxidant enzyme activities in cerebrospinal fluid related with severity of hypoxic ischemic encephalopathy in newborns. Biology of the Neonate, 88(2), 87–91. 10.1159/000084905 [DOI] [PubMed] [Google Scholar]
- Gunn AJ, Gunn TR, de Haan HH, Williams CE, & Gluckman PD (1997). Dramatic neuronal rescue with prolonged selective head cooling after ischemia in fetal lambs. Journal of Clinical Investigation, 99(2), 248–256. 10.1172/JCI119153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houston M, Estevez A, Chumley P, Aslan M, Marklund S, Parks DA, & Freeman BA (1999). Binding of xanthine oxidase to vascular endothelium. Kinetic characterization and oxidative impairment of nitric oxide-dependent signaling. Journal of Biological Chemistry, 274(8), 4985–4994. 10.1074/jbc.274.8.4985 [DOI] [PubMed] [Google Scholar]
- Hussein MH, Daoud GA, Kakita H, Kato S, Goto T, Kamei M, Goto K, Nobata M, Ozaki Y, Ito T, Fukuda S, Kato I, Suzuki S, Sobajima H, Hara F, Hashimoto T, & Togari H. (2010). High cerebrospinal fluid antioxidants and interleukin 8 are protective of hypoxic brain damage in newborns. Free Radical Research, 44(4), 422–429. 10.3109/10715760903548245 [DOI] [PubMed] [Google Scholar]
- Janero DR (1990). Malondialdehyde and thiobarbituric acid-reactivity as diagnostic indices of lipid peroxidation and peroxidative tissue injury. Free Radical Biology and Medicine, 9(6), 515–540. 10.1016/0891-5849(90)90131-2 [DOI] [PubMed] [Google Scholar]
- Johanson C, & Johanson N. (2016). Merging transport data for choroid plexus with blood-brain barrier to model CNS homeostasis and disease more effectively. CNS & Neurological Disorders: Drug Targets, 15(9), 1151–1180. 10.2174/1871527315666160915120758 [DOI] [PubMed] [Google Scholar]
- Juul SE, Stallings SA, & Christensen RD (1999). Erythropoietin in the cerebrospinal fluid of neonates who sustained CNS injury. Pediatric Research, 46(5), 543–547. 10.1203/00006450-199911000-00009 [DOI] [PubMed] [Google Scholar]
- Kokoglu E, Belce A, Ozyurt E, & Tepeler Z. (1990). Xanthine oxidase levels in human brain tumors. Cancer Letters, 50(3), 179–181. 10.1016/0304-3835(90)90262-v [DOI] [PubMed] [Google Scholar]
- Korhonen L, Riikonen R, Nawa H, & Lindholm D. (1998). Brain derived neurotrophic factor is increased in cerebrospinal fluid of children suffering from asphyxia. Neuroscience Letters, 240(3), 151–154. 10.1016/s0304-3940(97)00937-3 [DOI] [PubMed] [Google Scholar]
- Kumar A, Ramakrishna SV, Basu S, & Rao GR (2008). Oxidative stress in perinatal asphyxia. Pediatric Neurology, 38(3), 181–185. 10.1016/j.pediatrneurol.2007.10.008 [DOI] [PubMed] [Google Scholar]
- Lo CK, Mertz D, & Loeb M. (2014). Newcastle-Ottawa Scale: Comparing reviewers’ to authors’ assessments. BMC Medical Research Methodology, 14, 45. 10.1186/1471-2288-14-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locatelli A, Lambicchi L, Incerti M, Bonati F, Ferdico M, Malguzzi S, Torcasio F, Calzi P, Varisco T, & Paterlini G. (2020). Is perinatal asphyxia predictable? BMC Pregnancy Childbirth, 20(1), 186. 10.1186/s12884-020-02876-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe MT, Kim EH, Faull RL, Christie DL, & Waldvogel HJ (2013). Dissociated expression of mitochondrial and cytosolic creatine kinases in the human brain: A new perspective on the role of creatine in brain energy metabolism. Journal of Cerebral Blood Flow and Metabolism, 33(8), 1295–1306. 10.1038/jcbfm.2013.84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuck TA, Rice MM, Bailit JL, Grobman WA, Reddy UM, Wapner RJ, Thorp JM, Caritis SN, Prasad M, Tita ATN, Saade GR, Sorokin Y, Rouse DJ, Blackwell SC, Tolosa JE, Varner M, Hill K, Sowles A, Postma J, … VanDorsten JP (2016). Preterm neonatal morbidity and mortality by gestational age: A contemporary cohort. American Journal of Obstetrics and Gynecology, 215(1), 103.e101–103.e114. 10.1016/j.ajog.2016.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel TM, Camara S, Tatschner T, Frangou S, Sheldrick AJ, Riederer P, & Grunblatt E. (2010). Increased xanthine oxidase in the thalamus and putamen in depression. World Journal of Biological Psychiatry, 11(2 Pt 2), 314–320. 10.3109/15622970802123695 [DOI] [PubMed] [Google Scholar]
- Nourooz-Zadeh J, Liu EH, Yhlen B, Anggard EE, & Halliwell B. (1999). F4-isoprostanes as specific marker of docosahexaenoic acid peroxidation in Alzheimer’s disease. Journal of Neurochemistry, 72(2), 734–740. 10.1046/j.1471-4159.1999.0720734.x [DOI] [PubMed] [Google Scholar]
- Parks DA, & Granger DN (1986). Xanthine oxidase: Biochemistry, distribution and physiology. Acta Physiologica Scandinavica Supplementum, 548, 87–99. [PubMed] [Google Scholar]
- Parrado-Fernández C, Blennow K, Hansson M, Leoni V, Cedazo-Minguez A, & Björkhem I. (2018). Evidence for sex difference in the CSF/plasma albumin ratio in ~20 000 patients and 335 healthy volunteers. Journal of Cellular and Molecular Medicine, 22(10), 5151–5154. 10.1111/jcmm.13767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray G, Husain SA, Dutta AK, & Batra S. (1998). Lipid peroxidation and antioxidant status in asphyxiated neonates. Medical Science Research, 26, 201–202. [Google Scholar]
- Riikonen RS, Korhonen LT, & Lindholm DB (1999). Cerebrospinal nerve growth factor— A marker of asphyxia? Pediatric Neurology, 20(2), 137–141. 10.1016/s0887-8994(98)00122-2 [DOI] [PubMed] [Google Scholar]
- Roth SC, Baudin J, Cady E, Johal K, Townsend JP, Wyatt JS, Reynolds EOR, & Stewart AL (1997). Relation of deranged neonatal cerebral oxidative metabolism with neurodevelopmental outcome and head circumference at 4 years. Developmental Medicine and Child Neurology, 39(11), 718–725. 10.1111/j.1469-8749.1997.tb07372.x [DOI] [PubMed] [Google Scholar]
- Savman K, Heyes MP, Svedin P, & Karlsson A. (2013). Microglia/macrophage-derived inflammatory mediators galectin-3 and quinolinic acid are elevated in cerebrospinal fluid from newborn infants after birth asphyxia. Translational Stroke Research, 4(2), 228–235. 10.1007/s12975-012-0216-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siman R, Toraskar N, Dang A, McNeil E, McGarvey M, Plaum J, Maloney E, & Grady MS (2009). A panel of neuron-enriched proteins as markers for traumatic brain injury in humans. Journal of Neurotrauma, 26(11), 1867–1877. 10.1089/neu.2009.0882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer D. (1999). Neonatal tolerance to hypoxia: A comparative-physiological approach. Comparative Biochemistry and Physiology Part A Molecular Integrative Physiology, 123(3), 221–234. 10.1016/s1095-6433(99)00057-4 [DOI] [PubMed] [Google Scholar]
- Talvik T, Haldre S, Soot A, Hamarik M, Piirsoo A, & Mikelsaar AV (1995). Creatine kinase isoenzyme BB concentrations in cerebrospinal fluid in asphyxiated preterm neonates. Acta Paediatrica, 84(10), 1183–1187. 10.1111/j.1651-2227.1995.tb13521.x [DOI] [PubMed] [Google Scholar]
- Tan S. (2014). Fault and blame, insults to the perinatal brain may be remote from time of birth. Clinics in Perinatology, 41(1), 105–117. 10.1016/j.clp.2013.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S, Gelman S, Wheat JK, & Parks DA (1995). Circulating xanthine oxidase in human ischemia reperfusion. Southern Medical Journal, 88(4), 479–482. 10.1097/00007611-199504000-00021 [DOI] [PubMed] [Google Scholar]
- Tan S, Radi R, Gaudier F, Evans RA, Rivera A, Kirk KA, & Parks DA (1993). Physiologic levels of uric acid inhibit xanthine oxidase in human plasma. Pediatric Research, 34(3), 303–307. 10.1203/00006450-199309000-00013 [DOI] [PubMed] [Google Scholar]
- Tan S, & Wu Y. (2020). Etiology and pathogenesis of neonatal encephalopathy (Post TW, Ed.). UpToDate. [Google Scholar]
- Tan WK, Williams CE, During MJ, Mallard CE, Gunning MI, Gunn AJ, & Gluckman PD (1996). Accumulation of cytotoxins during the development of seizures and edema after hypoxic-ischemic injury in late gestation fetal sheep. Pediatric Research, 39(5), 791–797. 10.1203/00006450-199605000-00008 [DOI] [PubMed] [Google Scholar]
- Thornton C, Rousset CI, Kichev A, Miyakuni Y, Vontell R, Baburamani AA, Fleiss B, Gressens P, & Hagberg H. (2012). Molecular mechanisms of neonatal brain injury. Neurology Research International, 2012, 506320. 10.1155/2012/506320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiljevic B, Maglajlic-D jukic S, Gojnic M, Stankovic S, Ignjatovic S, & Lutovac D. (2011). New insights into the pathogenesis of perinatal hypoxic-ischemic brain injury. Pediatrics International, 53(4), 454–462. 10.1111/j.1442-200X.2010.03290.x [DOI] [PubMed] [Google Scholar]
- Vasquez-Vivar J, Shi Z, Jeong JW, Luo K, Sharma A, Thirugnanam K, & Tan S. (2020). Neuronal vulnerability to fetal hypoxia-reoxygenation injury and motor deficit development relies on regional brain tetrahydrobiopterin levels. Redox Biology, 29, 101407. 10.1016/j.redox.2019.101407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez-Vivar J, Shi Z, Luo K, Thirugnanam K, & Tan S. (2017). Tetrahydrobiopterin in antenatal brain hypoxia-ischemia-induced motor impairments and cerebral palsy. Redox Biology, 13, 594–599. 10.1016/j.redox.2017.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigley CB, & Berry M. (1988). Regeneration of adult rat retinal ganglion cell processes in monolayer culture: Comparisons between cultures of adult and neonatal neurons. Brain Research, 470(1), 85–98. 10.1016/0165-3806(88)90204-0 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Fan F, Zeng G, Zhou L, Zhang Y, Zhang J, Jiao HE, Zhang T, Su D, Yang C, Wang X, Xiao K, Li H, & Zhong Z. (2017). Temporal analysis of blood-brain barrier disruption and cerebrospinal fluid matrix metalloproteinases in rhesus monkeys subjected to transient ischemic stroke. Journal of Cerebral Blood Flow and Metabolism, 37(8), 2963–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 Analysis of risk of bias of individual studies using Newcastle-Ottawa Quality Assessment Scale