

Abstract
Purpose:
Leiomyosarcoma (LMS) and liposarcoma (LPS) frequently express PD-L1 but are generally resistant to PD-1/PD-L1 inhibition (ICI). Trabectedin is FDA-approved for LMS and LPS. This study aimed to evaluate the safety and efficacy of trabectedin with anti-PD-L1 antibody avelumab in patients with advanced LMS and LPS.
Patients and Methods:
A single-arm, open-label, Phase 1/2 study tested avelumab with trabectedin for advanced LMS and LPS. The phase I portion evaluated safety and feasibility of trabectedin (1, 1.2 and 1.5 mg/m2) with avelumab at standard dosing. Primary endpoint of the phase II portion was objective response rate (ORR) by RECIST 1.1. Correlative studies included T-cell receptor sequencing (TCRseq), multiplex immunohistochemistry, and tumor gene expression.
Results:
33 patients were evaluable; 24 with LMS (6 uterine and 18 non-uterine) and 11 with LPS. In Phase 1, dose limiting toxicities (DLTs) were observed in 2 of 6 patients at both trabectedin 1.2 and 1.5 mg/m2. The recommended Phase 2 dose (RP2D) was 1.0 mg/m2 trabectedin and 800 mg avelumab. Of 23 patients evaluable at RP2D, three (13%) had partial response (PR), ten (43%) had stable disease (SD) as best response. 6-month PFS was 52%; median PFS was 8.3 months. Patients with PR had higher Simpson Clonality score on TCRseq from peripheral blood mononuclear cells (PBMC) versus those with SD (0.182 vs 0.067, p = 0.02) or PD (0.182 vs 0.064, p = 0.01).
Conclusions:
Although the trial did not meet the primary ORR endpoint, PFS compared favorably to prior studies of trabectedin warranting further investigation.
Translational Relevance
This phase 1/2 clinical trial showed that the combination of avelumab and trabectedin was safe and had an overall response rate of 13% at the recommended phase 2 dose, with median progression free survival of 8.3 months and median overall survival of 27.4 months. Higher T-cell receptor clonality on T cell receptor sequencing was significantly associated with partial response by RECIST criteria. Although the trial failed to meet the primary ORR endpoint, additional study is warranted to further test if the combination of avelumab and trabectedin improves progression-free survival, with consideration for stratifying enrollment based on TCR sequencing data or limiting eligibility to patients with a high clonality score.
Introduction
Leiomyosarcoma (LMS) and liposarcoma (LPS) are two of the most common soft tissue sarcoma (STS) subtypes. Treatment for metastatic disease consists of cytotoxic chemotherapy generally with anthracycline and gemcitabine-based regimens.[1] Long term outcomes are poor with median overall survival of 22 months for patients with metastatic disease.[2] Efforts to improve survival for patients with STS with multi-agent chemotherapy with doxorubicin and ifosfamide,[3] or by adding investigational agents such as the hypoxia-activated prodrug evofosfamide[4] and the PDGFR inhibitor olaratumab to standard frontline anthracycline based cytotoxic chemotherapy have been generally unsuccessful.[2] More effective and better tolerated treatments for patients with advanced disease are urgently needed.
Trabectedin is an agent that binds the DNA minor groove and is approved for patients with metastatic or unresectable leiomyosarcoma and liposarcoma.[5] Although trabectedin has been in clinical use for two decades, it was discovered by high through-put drug screen and controversies remain regarding its mechanism of action.[6] In addition to its direct anti-tumor activity, trabectedin also inhibits tumor associated macrophages (TAM) which have been shown to contribute to tumor immune evasion.[7] Thus, trabectedin may actually function both as a direct anti-tumoral cytotoxic therapy and as an immunotherapeutic by overcoming TAM-induced immunosuppression in the tumor microenvironment.
Studies with immune checkpoint inhibitors (ICI) have shown hints of activity in STS, especially in patients with undifferentiated pleomorphic sarcoma[8], alveolar soft parts sarcoma[9], and angiosarcoma[10]. There has been limited activity of ICI in LMS and LPS. Isolated responses in patients with LMS were seen in a study of nivolumab or nivolumab plus ipilimumab for patients with STS, and no responses seen in a small study of 12 patients with uterine LMS treated with nivolumab.[8, 11, 12] Two of ten patients with liposarcoma responded in an initial study of pembrolizumab in STS, no patients with LMS responded.[8] Activity of pembrolizumab in LPS was not confirmed in the expansion cohort, with only 10% of LPS patients responding.[13] Higher expression of TAM markers in both LMS and LPS have been associated with worse prognosis.[14–16] These TAM may prevent ICI activity in spite of strong PD-L1 expression and brisk T cell infiltration in some LMS and LPS tumors.[17]
Recently, we reported that the addition of pembrolizumab to doxorubicin yielded promising PFS (8.1 months) and OS (28 months) versus historical controls, suggesting that combining immune checkpoint inhibition with cytotoxic chemotherapy can be an effective strategy.[18] A small study of trabectedin with durvalumab including soft tissue sarcoma patients showed activity similar to single agent trabectedin, but enrolment was subtype agnostic.[19] Additional small studies of trabectedin and nivolumab and trabectedin and ipilimumab plus nivolumab have been presented in abstract form but not published as a full manuscript.[20, 21] Given its potential to inhibit immunosuppressive macrophages, we hypothesized that trabectedin would enhance the activity of the anti-PD-L1 immune checkpoint inhibitor avelumab in patients with LMS and LPS.
Methods
Patients, Treatment Schedules
This was a Phase I/II trial of trabectedin in combination with avelumab. The protocol and study documents were approved by the Fred Hutchinson Cancer Center Institutional Review Board. All patients provided written informed consent and all study procedures were carried out in accordance with the Helsinki Declaration. The full protocol is provided in the Supplemental material.
Patients must have had a histologically confirmed diagnosis of metastatic or unresectable LMS or LPS, been eligible to receive trabectedin as standard of care in any line of therapy, ECOG performance status ≤ 1 or Karnofsky performance scale ≥ 70, left ventricular ejection fraction (LVEF) >45%, measurable disease by Response Evaluation Criteria in Solid Tumors (RECIST) v1.1, a life expectancy of ≥ 6 months as determined by the treating physician, and adequate end organ function. Patients with active autoimmune disease were excluded. A complete list of eligibility criteria is provided in Supplemental Figure 1. Archival tumor tissue was collected at enrollment. Correlative blood samples were taken prior to starting treatment, on treatment, and at end of treatment.
Trabectedin was given as 24-hour infusion every 3 weeks. Following Cycle 2, patients and providers were allowed to extend the trabectedin interval to 4 weeks, allowing better synchronization with avelumab dosing. Avelumab was initially given at 10mg/kg, and then changed to an 800 mg standardized dose every 2 weeks after updates to the Investigator’s Brochure. Disease status was assessed every 12 weeks by RECIST version 1.1. Patients with an unconfirmed or equivocal progression were allowed to stay on treatment to confirm progression with repeat imaging 6 weeks later as long as they had no clinical decline, no rapid progression as determined by the investigator, and no tumor at critical anatomic sites such as spinal cord. Trial schema and additional protocol information are included in Supplemental Figure 2.
Trial Design, Statistical Basis, and End Points
Three doses of trabectedin were evaluated using a 3+3 design (1.5, 1.2, and 1.0 mg/m2). Dose limiting toxicity was defined as any grade 3 or higher adverse events, at least possibly related to protocol therapy, which occurred during the first 6 weeks of treatment and with pre-specified exceptions as stated in the protocol.
The primary objective of the Phase I component of this study was to evaluate the safety of avelumab given in combination with trabectedin. Primary endpoint for the Phase II component was ORR as assessed by RECIST v1.1. Patients from the Phase I portion treated at the dose ultimately used in the expansion phase were included in the efficacy analysis. Our null hypothesis assumes an ORR of 9.9% for trabectedin monotherapy. An improvement in ORR to 35% would be highly compelling and justify further investigation of the combination. A total of 22 evaluable patients would provide an 85% power to detect this increased ORR assuming a two-sided alpha of 0.05. Six or more patients with objective responses would suggest greater clinical activity of this regimen over trabectedin monotherapy. AEs were graded in severity according to the NCI Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0. Log rank test and Kaplan Meier analysis were done in R version 4.0.2.
TCR Sequencing
Genomic DNA was extracted from pretreatment and on treatment peripheral blood samples and the CDR3 regions of TCRβ chains was sequenced using the immunoSEQ Assay (Adaptive Biotechnologies, Seattle, WA) [22]. T-cell fraction and Simpson Clonality were calculated as previously described.[23] TCR clonality graph was made in Graphpad Prism version 7.
Gene Expression
RNA was extracted from 1 mm thick Formalin fixed paraffin embedded (FFPE) curls. Gene expression was measured on the NanoString nCounter Analysis System (NanoString Technologies, Seattle, WA, USA).[24, 25] A Nanostring nCounter CodeSet (nCounter® PanCancer IO 360™ Panel) with 770 genes was hybridized to extracted RNA and samples were processed in the nCounter Prep Station post-hybridization. nCounter Digital Analyzer was used to quantify mRNA and analyzed using NanoString nSolver Analysis Software v2.0 and Rosalind software. Pathway enrichment analysis was performed using the publicly available Webgestalt[26] analysis tool with comparison against the KEGG[27] and Panther[28] pathway databases using genes identified as differentially expressed in the nSolver analysis.
Multiplex Immunohistochemistry (mIHC)
4-micron FFPE slides were stained on a Leica BOND Rx autostainer (Leica, Buffalo Grove, IL) using a modified Akoya Opal Multiplex IHC protocol (Akoya Biosciences, Menlo Park, CA) as previously described.[29] In short, slides were dewaxed, antigen retrieved, and washed using Leica BOND reagents with an additional high-salt TBST wash after the secondary and tertiary applications (0.05M Tris, 0.3M NaCl, and 0.1% Tween-20, pH 7.2−7.6). 3% H2O2 and TCT buffer protein block (0.05M Tris, 0.15M NaCl, 0.25% Casein, 0.1% Tween 20, pH 7.6 +/− 0.1) were used before each primary antibody step, and the antibodies were incubated for 1 hour at room temperature. Slides were mounted with ProLong Gold and scanned at 20x on an Akoya Polaris after curing for 24 hours. Images were spectrally unmixed using Akoya Phenoptics inForm program and exported as multi-image TIFF’s for analysis using HALO’s High-plex FL software (Indica Labs, Corrales, NM). Comparison of multiplex IHC markers was performed in IBM SPSS v28 using two-sided t-test or ANOVA.
Data Availability
The data generated in this study are available within the article and its supplementary data files. Raw data for the correlative components of this study were generated at Fred Hutchinson Cancer Research Center core facilities (mIHC, NanoString) and at Adaptive Biotechnologies (TCR sequencing). Derived data supporting the findings of this study are available from the corresponding author upon request.
Results
Patient Demographics
Thirty-five patients enrolled between September 2017 and March 2020. Two patients were not evaluable for response because of death (one patient) or withdrawal of consent (one patient) prior to the first response assessment. 24 had LMS (6 uterine, 18 non-uterine), 11 had LPS (9 dedifferentiated, 1 myxoid/round cell, 1 pleomorphic). The mean age was 59 years. 20 (57%) were female. At the time of enrollment, 5 (14%) patients were treatment naïve, 12 (34%) had received one prior line, 7 (20%) had received two prior lines, and 11 (31%) had received three or more prior lines. Six patients (17%) had received prior chemotherapy in the neoadjuvant/adjuvant setting. Of the 30 patients who were previously treated, 27 received doxorubicin (8 with olaratumab, 10 with ifosfamide, and 9 as a single agent), 17 received gemcitabine (13 with docetaxel, 2 with dacarbazine, 1 with paclitaxel, and 1 single agent), 8 patients received pazopanib, and 6 patients received eribulin. Twenty-three patients were treated at the RP2D. Demographic characteristics are summarized in Table 1.
Table 1.
Demographics of All Enrolled Patients and Best Responses
| Overall (N=35) (%) | |
|---|---|
| Age (years) | |
| Mean (SD) | 59 (13.4) |
| Phase | |
| Phase 1, 1.0 mg/m2 | 7 (20%) |
| Phase 1, 1.2 mg/m2 | 6 (17%) |
| Phase 1, 1.5 mg/m2 | 6 (17%) |
| Phase 2, 1.0 mg/m2 | 16 (46%) |
| Gender | |
| Female | 20 (57%) |
| Male | 15 (43%) |
| Ethnicity | |
| Not Hispanic or Latino | 31 (89%) |
| Unknown or Not Available | 4 (11%) |
| Race | |
| American Indian or Alaska Native | 2 (6%) |
| Asian | 4 (11%) |
| White | 24 (69%) |
| Unknown | 5 (14%) |
| Subtype | |
| Leiomyosarcoma | 24 (69%) |
| Uterine | 6 (51%) |
| Non-uterine | 18 (17%) |
| Liposarcoma | 11 (31%) |
| Dedifferentiated | 9 (26%) |
| Myxoid/Round Cell | 1 (3%) |
| Pleomorphic | 1 (3%) |
| Number of Lines of Prior Therapy | |
| 0 | 5 (14%) |
| 1 | 12 (34%) |
| 2 | 7 (20%) |
| 3 | 5 (8.6%) |
| 4 | 4 (11%) |
| 5 | 2 (5.7%) |
| Best Response | |
| Not Evaluable | 2 (5.7%) |
| Partial Response | 4 (11%) |
| Stable Disease | 16 (46%) |
| Progressive Disease | 13 (37%) |
Safety
Treatment-related adverse events (AEs) occurring with an incidence of at least 5% in the study population are summarized in Supplemental Table 1. Across all patients, 100% experienced an AE of any grade, and 57% experienced at least one grade 3 or higher AE attributed to treatment. The most common toxicities were fatigue (91%), nausea (71%), anorexia (51%), alanine aminotransferase (AST) elevation (43%), and infusion reaction (37%). 89% had immune related events (irAE) of any grade, defined as attributed to avelumab or both drugs, and 17% had irAEs grade 3 or higher. Grade 3–5 treatment-related AEs occurring with any incidence rate in the study population are summarized in Table 2. The most common grade 3 or higher AEs were increased AST (18%), decreased lymphocyte count (12%), and neutropenia (9%). Most (87%) treatment related grade 3–5 events were attributable to trabectedin.
Table 2.
All Treatment-Related Grade 3–5 Adverse Events (N = 35 patients)
| Total | Attributed to Avelumab | Attributed to Trabectedin | Attributed to Both | |||||
|---|---|---|---|---|---|---|---|---|
| AE Term | Count | % | Count | % | Count | % | Count | % |
| Alanine aminotransferase increased | 6 | 18% | 0 | 0% | 6 | 17% | 0 | 0% |
| Lymphocyte Count Decreased | 4 | 12% | 1 | 3% | 4 | 11% | 1 | 3% |
| Neutrophil count decreased | 3 | 9% | 1 | 3% | 1 | 3% | 1 | 3% |
| Anemia | 2 | 6% | 0 | 0% | 2 | 6% | 0 | 0% |
| Ejection fraction decreased | 2 | 6% | 0 | 0% | 2 | 6% | 0 | 0% |
| Fatigue | 1 | 3% | 0 | 0% | 1 | 3% | 0 | 0% |
| Infusion related reaction | 1 | 3% | 1 | 3% | 0 | 0% | 0 | 0% |
| Aspartate aminotransferase increased | 1 | 3% | 1 | 3% | 1 | 3% | 0 | 0% |
| Port Infection/Inflammation/Erythema | 1 | 3% | 0 | 0% | 1 | 3% | 0 | 0% |
| Dyspnea | 1 | 3% | 0 | 0% | 1 | 3% | 0 | 0% |
| Gastrointestinal Disorders – Other | 1 | 3% | 0 | 0% | 1 | 3% | 0 | 0% |
| White blood cell decreased | 1 | 3% | 0 | 0% | 1 | 3% | 0 | 0% |
In the Phase 1 dose finding portion of the trial, there were DLTs in 2 of 6 patients at both higher doses of trabectedin including grade 3 GGT elevation, bilirubin and alanine aminotransferase (ALT) elevation, small bowel obstruction, and reduced ejection fraction. These were attributed to trabectedin and did not require treatment with steroids. The recommended Phase 2 dose was 1.0 mg/m2 trabectedin and 800 mg avelumab. 8 (23%) patients experienced symptoms of port inflammation, infection, or erythema. Of these 8 patients, 5 had their ports removed and 4 had them replaced (1 patient continued treatment via PICC line). One patient required a large resection including a catheter associated thrombus. No patients experienced a reoccurrence of symptoms after port replacement. There were no Grade 4/5 treatment-related AEs at the Phase 2 dose.
Tumor Response
23 patients at the RP2D were evaluable for the primary ORR endpoint. 3 (13%) had partial response (PR) and 10 (43%) had stable disease (SD) as best response (Supplemental Table 2). Clinical benefit rate (PR+SD) at 12 weeks was 56%. Subgroup analysis revealed that in all 24 patients with leiomyosarcoma (Phase 1 and 2), 4 (17%) had PR and 9 (38%) had SD (Table 1). Responses were observed in patients with uterine and non-uterine leiomyosarcoma (Supplemental Figure 3). All 4 patients with a partial response were on therapy for at least 36 weeks and all responses were ongoing at the time of data cutoff. In all patients with LPS (N= 11), there were no RECIST 1.1 responses and 7 (64%) had SD several of which were highly durable including one LPS patient who remained on treatment for over two years (Figure 1).
Figure 1. Swimmers plot (A) and Spider Plot (B) of patients at the RP2D.
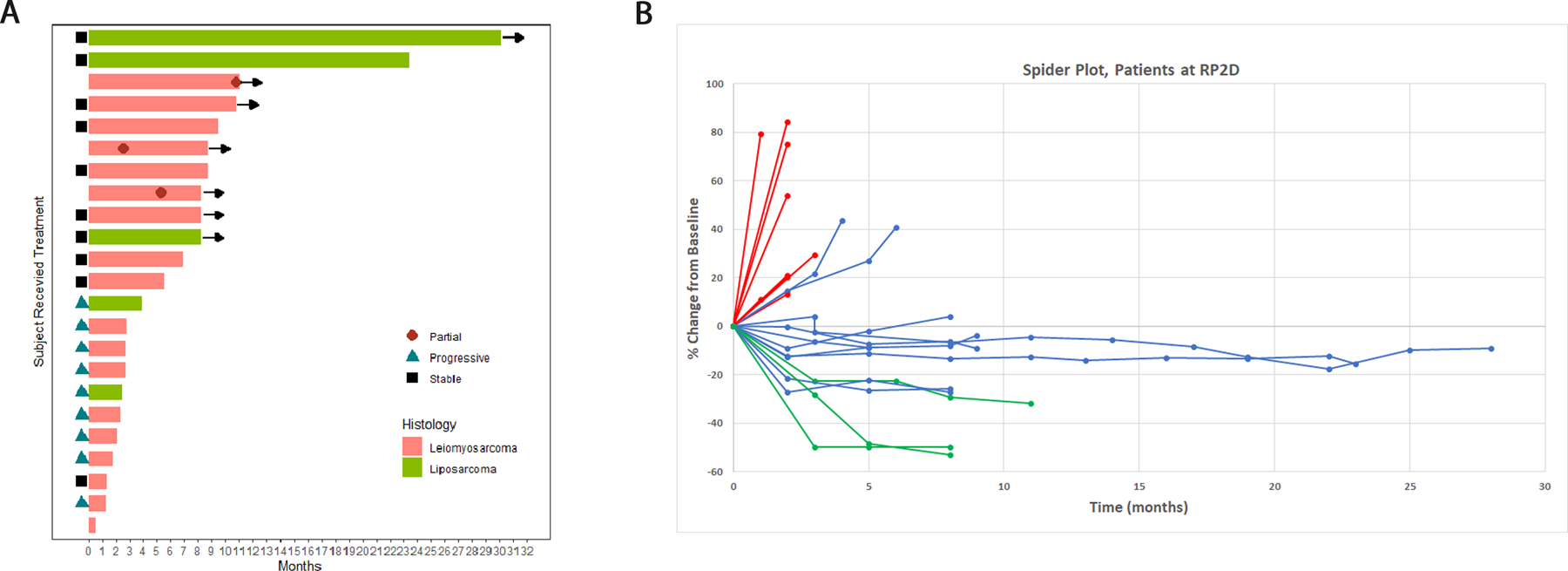
In the swimmer plot, patients with leiomyosarcoma are indicated in pink, and those with liposarcoma are indicated in green. Time to partial response is indicated where marked on the individual patient. Patients still on treatment at the time of data cutoff are indicated by an arrow. In the spider plot (B) patients with progressive disease as best response are marked in red, those with stable disease as best response are blue, and those with partial response are green.
Survival Outcomes
For all patients in Phase I and Phase II, median PFS was 8.1 months (95% CI 4.3-NA/Infintiy (Inf)). 6-month PFS was 50% (95% CI 36−70) (Figure 2A). Median PFS of the patients at the RP2D (N = 23) was 8.3 months (95% CI 2.5-NA/Inf), with a 6-month PFS rate of 52% (95% CI 35−77). There was no difference by histological subtype (p = 0.267 for all patients and p = 0.578 for RP2D patients).
Figure 2.
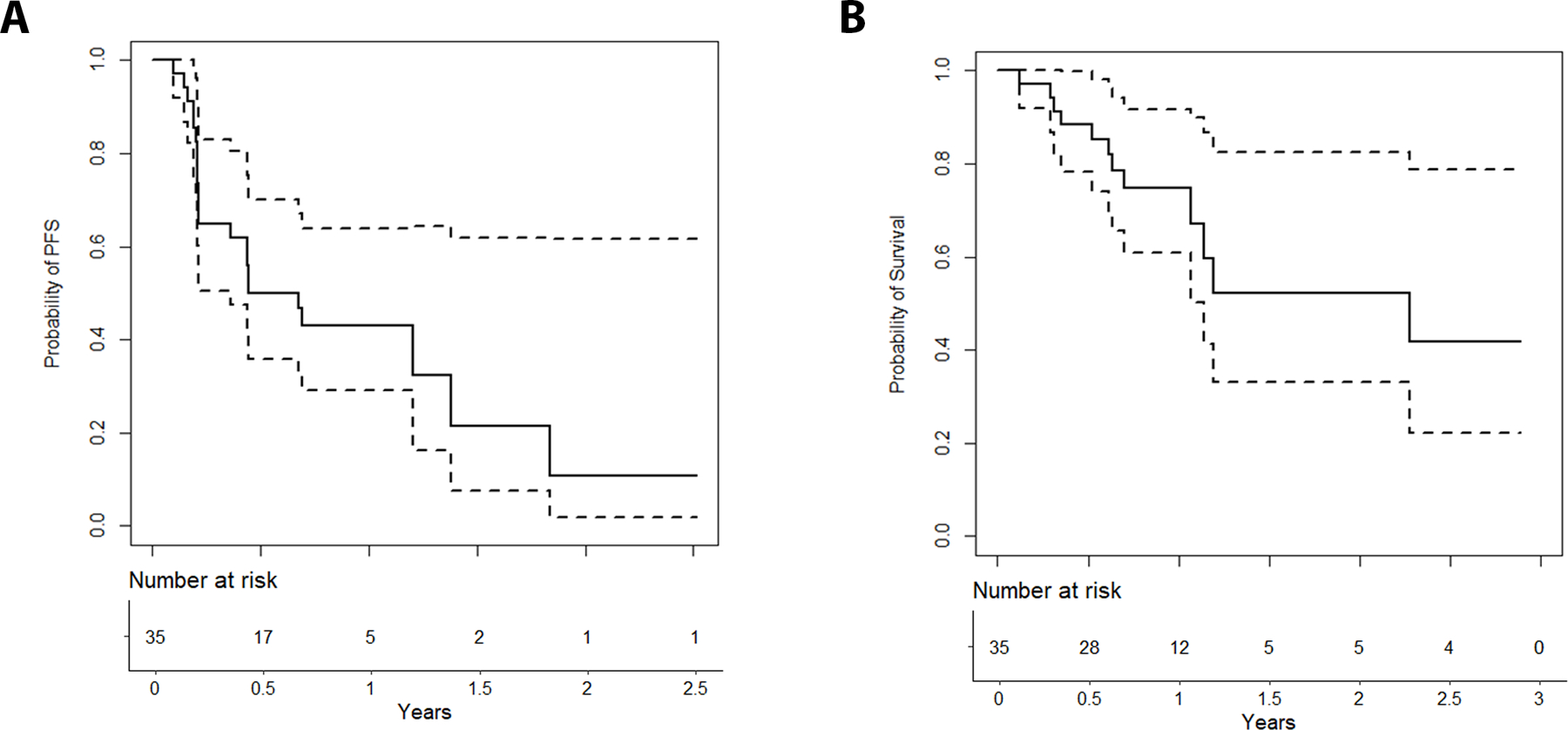
Progression- Free (A) and Overall (B) Survival for all patients (N= 35). Dotted lines represent 95% confidence bands.
Median OS for all patients was 27 months (95% CI 13.7-NA/Inf), with a 6-month OS rate 85% (95% CI 74−98) and 12-month OS rate 75% (95% CI 61–92) (Figure 2B). Median follow-up was 12 months. Median OS for RP2D patients was 27 months (95% CI 13.7-NA/Inf), with a 6-month OS rate of 86% (95% CI 72−100), and 12-month OS rate 81% (95% CI 65−100) with a median follow-up of 11 months.
Correlative Analyses
TCR sequencing
Peripheral blood TCR sequencing data was available for 7 patients with PD as best response, 9 with SD as best response, and 3 with PR. There was no difference in Productive Simpson Clonality between pre-treatment and on-treatment samples. Median Productive Simpson Clonality for all patients was 0.079. Patients with a partial response had significantly higher Productive Simpson Clonality versus those with SD (0.182 vs 0.067, p = 0.02) or progressive disease (PD) (0.182 vs 0.064, p = 0.01) in the on-treatment samples (Supplemental Figure 4).
Multiplex immunohistochemistry
Pre-treatment samples were available for 20 patients with LMS and 9 patients with LPS (29 total). No immunohistochemical markers (PD-1, PDL1, CD3, CD68, CD4, CD8, CD66b, CD14, HLA-DR, GATA-3, or CD163) were statistically associated with response by ANOVA. There was higher PD-L1 expression and lower expression of macrophage markers in the two patients with a partial response for whom adequate tissue was available, compared to patients with SD or PD (Supplemental Figure 5). Patients with LPS had higher relative macrophage infiltration versus those with LMS (Supplemental Figure 6).
Gene Expression
RNA was available from 9 LPS and 20 LMS patients. Two patients with PR as best response had RNA samples available. 22 genes were differentially expressed, all of which were more highly expressed in LPS. These were TNFRSF14, CCL8, FCGR3A/B, CD28, CDK2, OASL, CDH5, COL11A2, STC1, SNAI1, SPP1, NCAM1, KAT2B, CD247, TWF1, TAF3, ANLN, CXCR6, ICOSLG, CXCL14, RELA, and IRF8. (Supplemental Figure 7). Over-Representation Analysis (ORA) revealed that T-cell activation genes were differentially expressed with higher enrichment in the LPS samples (Supplemental Figure 7).
Discussion
The RP2D in our study was 1.0 mg/m2 trabectedin, which is below the usual starting dose as a single agent. However, although the response rate in our study did not meet the prespecified threshold of 35%, the ORR of 13% is comparable to prior studies with trabectedin in this population.[5, 30, 31] The median PFS in our study of 8.3 months is higher than the median PFS of 4.2 months, or 5.1 months and 5.6 months reported with single agent trabectedin in patients with LMS and LPS.[30, 31] Another prospective randomized trial demonstrated a median PFS of 4.2 months for trabectedin versus 1.5 months for dacarbazine in pre-treated advanced/ metastatic LMS and LPS.[5] Trabectedin was approved by the FDA at a starting dose of 1.5 mg/m2 for L-sarcomas based on this PFS advantage. A lower dose of 1.2 mg/m2 is standard in Japan based on a study demonstrating efficacy in translocation associated sarcoma.[32] Toulmonde et al report a 6-month PFS rate of 28.6% in sixteen patients with sarcoma treated with trabectedin and durvalumab, perhaps representing the population and treatment regimen most similar to those in our study.[19] Caution must be taken by comparing this single arm phase II to other trials of trabectedin. Factors that may have contributed to our observed favorable PFS compared to other trials include the lack of documented progression as an eligibility requirement for enrollment, and inclusion of patients who were treatment naïve.
Similar to this study, in which the ORR with combined cytotoxic chemotherapy and ICI therapy appears similar to chemotherapy alone, we previously reported that the combination of doxorubicin and pembrolizumab yielded favorable PFS outcomes without meeting the prespecified ORR primary endpoint.[18] This raises the question of whether ORR is the optimal primary endpoint for studies with ICI in sarcomas. PFS may be a more reliable endpoint that more accurately captures the disease control that can be achieved by creating a more favorable immune microenvironment. Moving forward, randomized studies including a comparator arm will be necessary.
There were multiple enrollment holds for DLTs and a higher than expected number of patients with port catheter inflammation requiring intervention. Eight patients on trial experienced port-related infection, inflammation, or erythema and 5 of these patients had their ports removed and replaced (4 replaced, 1 patient continued via peripherally inserted central catheter). AEs related to central venous catheters is a known potential complication with trabectedin[33], and it is possible the addition of avelumab amplified this trabectedin induced inflammation. Transient transaminitis was also seen and led to DLTs, but this is also a relatively common side effect of trabectedin. While autoimmune transaminitis can be an AE of avelumab therapy, the transient nature of it in these cases, resolving without steroid therapy, argues against avelumab as the cause. No patients discontinued drug due to transaminitis. The incidence of grade 3 or higher toxicities was slightly lower in our study compared to what was seen with trabectedin alone, where myelosuppression and transaminitis were the most common grade 3 or higher AEs occurring in about 25% of patients each.[5] The slightly lower rate in our study perhaps is due to the lower RP2D of trabectedin (1.0 mg/m2 versus 1.5 mg/m2) and regular use of granulocyte colony stimulating factor. The combination of trabectedin and avelumab was overall well tolerated at the RP2D.
In exploratory correlative analyses, patients with a partial response had a higher T-cell clonality compared to that of patients with SD or PD, suggesting immune recognition of the tumor prior to study treatment. Higher T-cell clonality has previously been associated with T-cell infiltration and PD-1 and PD-L1 expression levels in sarcoma.[17] The lack of significant change in clonality between pretreatment and post treatment samples in either responders or non-responders suggests that treatment with ICI facilitates a pre-existing cell mediated immunity against the tumor rather than triggering new neo-antigen recognition. It is at least possible that ICI alone would have led to a response in these cases with higher clonality. Future trial design of ICI in sarcoma should consider testing the hypothesis that T-cell clonality correlates with response.
In a previous study of ICI in sarcoma, a higher density of infiltrating immune cells associated with a higher likelihood of objective response.[34] In our analysis, both LMS and LPS had infiltration of immune suppressive macrophages, with a higher proportion in LPS perhaps explaining the lack of objective responses in patients of this subtype. Transcriptomic analysis also suggested a more inflammatory baseline milieu in the LPS patients, consistent with our previous characterization of the LPS immune microenvironment.[16] Although there was an increase in immune markers in LPS, and in particular several markers suggestive of T-cell activation as seen in the overexpression pathway analysis, some of the markers such as CCL8 can also be contributory to aggressive cancer behavior.[35] Additionally, it is important to note that the tissue samples were taken from archival tissue samples and not from fresh biopsies immediately before initiation of study therapy. We recently showed that neoadjuvant treatment modifies the sarcoma microenvironment to enrich for B-cells and myeloid cells, altering the delicate balance of immunostimulatory and immunosuppressive factors.[36] Specifically as it relates to trabectedin, we previously found that a more immunosuppressive microenvironment correlates with a lower likelihood of benefit with trabectedin.[37]
Here, we did not find a correlation with immune markers and response or PFS. One possibility is that additional agents such as a LAG-3 inhibitor as has been recently shown to be efficacious in melanoma[38], are needed to push the immune balance towards an anti-tumor immunostimulatory environment. Ultimately, the true relevance of the disparate immune cells signatures may be elucidated in future studies incorporating serial biopsies so that more direct effects of treatment on the immune microenvironment can be ascertained.
There were several limitations to this study. Our analysis was limited by a small sample size. Moreover, only two patients with a partial response for whom adequate tissue were available for evaluation. This was a non-randomized single arm study performed at a single center. There was no control group, making it difficult to directly compare the potential benefit of the combination over standard of care trabectedin alone. Not all patients had tissue available and many of the samples that were available were core needle biopsies, limiting the scope and generalizability of the correlative analyses.
In conclusion, we demonstrate that administration of avelumab and trabectedin in combination is feasible with acceptable toxicity. The RP2D was 1.0 mg/m2 trabectedin and 800 mg avelumab. The trial did not meet the primary endpoint of ORR. However, the PFS is favorable compared to prior studies of trabectedin in this population and warrants further investigation, especially in leiomyosarcoma.
Supplementary Material
Acknowledgements
This research was financially supported by the healthcare business of Merck KGaA, Darmstadt, Germany (CrossRef Funder ID: 10.13039/100004755), as part of an alliance between the healthcare business of Merck KGaA, Darmstadt, Germany and Pfizer. It was also supported by the Raisbeck Endowed Chair for Collaborative Research. The healthcare business of Merck KGaA, Darmstadt, Germany and Pfizer reviewed the manuscript for medical accuracy only before journal submission. The authors are fully responsible for the content of this manuscript, and the views and opinions described in the publication reflect solely those of the authors.
Footnotes
Trial Registration: ClinicalTrials.gov NCT03074318
References
- 1.Wagner MJ, Amodu LI, Duh MS et al. A retrospective chart review of drug treatment patterns and clinical outcomes among patients with metastatic or recurrent soft tissue sarcoma refractory to one or more prior chemotherapy treatments. BMC Cancer 2015; 15: 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tap WD, Wagner AJ, Schoffski P et al. Effect of Doxorubicin Plus Olaratumab vs Doxorubicin Plus Placebo on Survival in Patients With Advanced Soft Tissue Sarcomas: The ANNOUNCE Randomized Clinical Trial. JAMA 2020; 323: 1266–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Judson I, Verweij J, Gelderblom H et al. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: a randomised controlled phase 3 trial. Lancet Oncol 2014; 15: 415–423. [DOI] [PubMed] [Google Scholar]
- 4.Tap WD, Papai Z, Van Tine BA et al. Doxorubicin plus evofosfamide versus doxorubicin alone in locally advanced, unresectable or metastatic soft-tissue sarcoma (TH CR-406/SARC021): an international, multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2017; 18: 1089–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Demetri GD, von Mehren M, Jones RL et al. Efficacy and Safety of Trabectedin or Dacarbazine for Metastatic Liposarcoma or Leiomyosarcoma After Failure of Conventional Chemotherapy: Results of a Phase III Randomized Multicenter Clinical Trial. J Clin Oncol 2016; 34: 786–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D’Incalci M, Badri N, Galmarini CM, Allavena P. Trabectedin, a drug acting on both cancer cells and the tumour microenvironment. Br J Cancer 2014; 111: 646–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Germano G, Frapolli R, Belgiovine C et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 2013; 23: 249–262. [DOI] [PubMed] [Google Scholar]
- 8.Tawbi HA, Burgess M, Bolejack V et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol 2017; 18: 1493–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilky BA, Trucco MM, Subhawong TK et al. Axitinib plus pembrolizumab in patients with advanced sarcomas including alveolar soft-part sarcoma: a single-centre, single-arm, phase 2 trial. Lancet Oncol 2019; 20: 837–848. [DOI] [PubMed] [Google Scholar]
- 10.Wagner MJ, Othus M, Patel SP et al. Multicenter phase II trial (SWOG S1609, cohort 51) of ipilimumab and nivolumab in metastatic or unresectable angiosarcoma: a substudy of dual anti-CTLA-4 and anti-PD-1 blockade in rare tumors (DART). J Immunother Cancer 2021; 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D’Angelo SP, Mahoney MR, Van Tine BA et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol 2018; 19: 416–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ben-Ami E, Barysauskas CM, Solomon S et al. Immunotherapy with single agent nivolumab for advanced leiomyosarcoma of the uterus: Results of a phase 2 study. Cancer 2017; 123: 3285–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burgess MA, Bolejack V, Schuetze S et al. Clinical activity of pembrolizumab (P) in undifferentiated pleomorphic sarcoma (UPS) and dedifferentiated/pleomorphic liposarcoma (LPS): Final results of SARC028 expansion cohorts. Journal of Clinical Oncology 2019; 37: 11015–11015. [Google Scholar]
- 14.Espinosa I, Beck AH, Lee CH et al. Coordinate expression of colony-stimulating factor-1 and colony-stimulating factor-1-related proteins is associated with poor prognosis in gynecological and nongynecological leiomyosarcoma. Am J Pathol 2009; 174: 2347–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ganjoo KN, Witten D, Patel M et al. The prognostic value of tumor-associated macrophages in leiomyosarcoma: a single institution study. Am J Clin Oncol 2011; 34: 82–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schroeder BA, LaFranzo NA, LaFleur BJ et al. CD4+ T cell and M2 macrophage infiltration predict dedifferentiated liposarcoma patient outcomes. J Immunother Cancer 2021; 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pollack SM, He Q, Yearley JH et al. T-cell infiltration and clonality correlate with programmed cell death protein 1 and programmed death-ligand 1 expression in patients with soft tissue sarcomas. Cancer 2017; 123: 3291–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pollack SM, Redman MW, Baker KK et al. Assessment of Doxorubicin and Pembrolizumab in Patients With Advanced Anthracycline-Naive Sarcoma: A Phase 1/2 Nonrandomized Clinical Trial. JAMA Oncol 2020; 6: 1778–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toulmonde M, Brahmi M, Giraud A et al. Trabectedin plus durvalumab in patients with advanced pretreated soft tissue sarcoma and ovarian carcinoma (TRAMUNE): an open-label, multicenter phase Ib study. Clin Cancer Res 2021. [DOI] [PubMed]
- 20.Gordon EM, Chua-Alcala VS, Kim K et al. SAINT: Results of an expanded phase II study using safe amounts of ipilimumab (I), nivolumab (N), and trabectedin (T) as first-line treatment of advanced soft tissue sarcoma [NCT03138161]. Journal of Clinical Oncology 2020; 38: 11520–11520. [Google Scholar]
- 21.Müller DW, Reichardt P, Grünwald V et al. 1669TiP A non-randomized, open-label phase II trial evaluating efficacy and feasibility of combined treatment with trabectedin and nivolumab in patients with metastatic or inoperable soft tissue sarcomas (STS) after failure of an anthracycline-containing regimen. Annals of Oncology 2020; 31: S991. [Google Scholar]
- 22.Carlson CS, Emerson RO, Sherwood AM et al. Using synthetic templates to design an unbiased multiplex PCR assay. Nature Communications 2013; 4: 2680. [DOI] [PubMed] [Google Scholar]
- 23.Robins Harlan S, Srivastava Santosh K, Campregher Paulo V et al. Overlap and Effective Size of the Human CD8+ T Cell Receptor Repertoire. Science Translational Medicine 2010; 2: 47ra64–47ra64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geiss GK, Bumgarner RE, Birditt B et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol 2008; 26: 317–325. [DOI] [PubMed] [Google Scholar]
- 25.Payton JE, Grieselhuber NR, Chang LW et al. High throughput digital quantification of mRNA abundance in primary human acute myeloid leukemia samples. J Clin Invest 2009; 119: 1714–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liao Y, Wang J, Jaehnig EJ et al. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res 2019; 47: W199–W205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanehisa M, Furumichi M, Tanabe M et al. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 2017; 45: D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomas PD, Campbell MJ, Kejariwal A et al. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res 2003; 13: 2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seo YD, Jiang X, Sullivan KM et al. Mobilization of CD8(+) T Cells via CXCR4 Blockade Facilitates PD-1 Checkpoint Therapy in Human Pancreatic Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2019; 25: 3934–3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hensley ML, Patel SR, von Mehren M et al. Efficacy and safety of trabectedin or dacarbazine in patients with advanced uterine leiomyosarcoma after failure of anthracycline-based chemotherapy: Subgroup analysis of a phase 3, randomized clinical trial. Gynecol Oncol 2017; 146: 531–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Le Cesne A, Blay JY, Cupissol D et al. A randomized phase III trial comparing trabectedin to best supportive care in patients with pre-treated soft tissue sarcoma: T-SAR, a French Sarcoma Group trial. Ann Oncol 2021. [DOI] [PubMed]
- 32.Kawai A, Araki N, Sugiura H et al. Trabectedin monotherapy after standard chemotherapy versus best supportive care in patients with advanced, translocation-related sarcoma: a randomised, open-label, phase 2 study. Lancet Oncol 2015; 16: 406–416. [DOI] [PubMed] [Google Scholar]
- 33.Verboom MC, Ouwerkerk J, Steeghs N et al. Central venous access related adverse events after trabectedin infusions in soft tissue sarcoma patients; experience and management in a nationwide multi-center study. Clin Sarcoma Res 2017; 7: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keung EZ, Burgess M, Salazar R et al. Correlative Analyses of the SARC028 Trial Reveal an Association Between Sarcoma-Associated Immune Infiltrate and Response to Pembrolizumab. Clin Cancer Res 2020; 26: 1258–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farmaki E, Chatzistamou I, Kaza V, Kiaris H. A CCL8 gradient drives breast cancer cell dissemination. Oncogene 2016; 35: 6309–6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goff PH, Riolobos L, LaFleur BJ et al. Neoadjuvant therapy induces a potent immune response to sarcoma, dominated by myeloid and B cells. Clin Cancer Res 2022. [DOI] [PMC free article] [PubMed]
- 37.Schroeder BA, Zhang Y, Smythe KS et al. Immunologic Gene Signature Analysis Correlates Myeloid Cells and M2 Macrophages with Time to Trabectedin Failure in Sarcoma Patients. Cancers 2022; 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tawbi HA, Schadendorf D, Lipson EJ et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. New England Journal of Medicine 2022; 386: 24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available within the article and its supplementary data files. Raw data for the correlative components of this study were generated at Fred Hutchinson Cancer Research Center core facilities (mIHC, NanoString) and at Adaptive Biotechnologies (TCR sequencing). Derived data supporting the findings of this study are available from the corresponding author upon request.