

Graphical abstract
Abbreviations: DBG, De Bruijn Graphs; SVs, Structural Variants; CNV, Copy Number Variation; GO, Gene Ontology; FMT, Fecal Matter Transplantation; CDI, Clostridium Difficile Infection; SRA, Sequence Read Archive; ENA, European Nucleotide Archive; SAM, Sequence Alignment Map; GPL, (GNU) General Public License; TPR, True Positive Rate; FPR, False Positive Rate; ROC, Receiver Operating Curve; MAGs, Metagenome assembled genomes
Keywords: De Bruijn graph, Graph-based analysis, K-core decomposition, Metagenome, Repeats, Unitigs, Functional characterization, Copy number variation (CNV)
Abstract
Characterizing metagenomes via kmer-based, database-dependent taxonomic classification has yielded key insights into underlying microbiome dynamics. However, novel approaches are needed to track community dynamics and genomic flux within metagenomes, particularly in response to perturbations. We describe KOMB, a novel method for tracking genome level dynamics within microbiomes. KOMB utilizes K-core decomposition to identify Structural variations (SVs), specifically, population-level Copy Number Variation (CNV) within microbiomes. K-core decomposition partitions the graph into shells containing nodes of induced degree at least K, yielding reduced computational complexity compared to prior approaches. Through validation on a synthetic community, we show that KOMB recovers and profiles repetitive genomic regions in the sample. KOMB is shown to identify functionally-important regions in Human Microbiome Project datasets, and was used to analyze longitudinal data and identify keystone taxa in Fecal Microbiota Transplantation (FMT) samples. In summary, KOMB represents a novel graph-based, taxonomy-oblivious, and reference-free approach for tracking CNV within microbiomes. KOMB is open source and available for download at https://gitlab.com/treangenlab/komb.
1. Introduction
Metagenomes are known hot spots for genomic diversity [1], [2], [3]. Communities in metagenomes consist of individual organisms whose genomes are dynamic because of Structural Variants (SVs) such as gene duplication, gene loss/gain, horizontal gene transfer, and gene rearrangements [4], [5], [6], [7]. These dynamic events are a results of complex interactions that underpin the microbiome [8], [9]. Therefore, characterizing metagenomic samples from diverse environments and sample types is essential to understanding community structures, interactions, and underlying functional information [10], [11], [12], [13], [14]. The main approaches to analyze metagenomes include functional characterization and taxonomic classification pipelines [15], [16], [17]. These approaches, while informative, do not necessarily capture structural variation events such as sequence-level gene duplication, gene loss/gain or transfer activity found in metagenomic samples over time.
1.1. SVs are important markers for microbial adaptation
Structural Variation, and specifically Copy Number Variation (CNV), has been shown to impact microbial phenotypes. Intra-species CNV in the gut microbiome have been previously linked to specific adaptive functions associated with obesity and inflammatory bowel disease [18]. A recent study found multiple associations between CNV in the gut microbiome and host disease risk factors [19]. Further, several metagenome-wide association studies (M-GWAS) also underscored the effect of host genetics and host environment on the composition and functional potential of the gut microbiota [20], [21], [22]. This complex interplay of the microbial and host environments based on gene copy numbers have indicated the need to look beyond taxonomy-based approaches and additionally focus on microbial functions that are unique, enriched, or depleted [23]. The identification and analysis of CNV in microbiomes is imperative to furthering our understanding of microbial dynamics and functional diversity in complex metagenomes [24].
1.2. Metagenomic assembly is not optimal to track SVs in samples
While metagenomic assembly algorithms that generate metagenome assembled genomes (MAGs) have made great strides and are a natural choice for analyzing microbial communities, there are existing challenges to recovering SVs, and especially CNV, accurately [25]. Much of this stems from the complexity of gene duplications and/or repeats within a same organism (intra-genomic repeats), or shared between distinct organisms (inter-genomic repeats). The shorter relative length of the sequencing reads compared to the length of the repeats, particularly in the intra-genomic case, prohibits fully resolving these repeats [26], [27]. Repeats longer than sequencing reads lead to a computational explosion in the number of possible paths through the assembly graphs. Therefore, most assemblers tend to use heuristics to try and simplify traversal and circumvent the complexity, which ultimately leads to loss of SV information [28]. Although it is possible to tease out variants with a metagenomic short-read scaffolder, such as Bambus2 [29], MaryGold [30] and MetaCarvel [31], and while longer reads can help to resolve repeats within metagenome assembled genomes (MAGs), to the best of our knowledge no method is able to accurately track both CNV and shared genomic content using short-read data alone [4].
1.3. Need for variant-aware methods to track metagenomic SVs
While certain metagenomic communities, including the human microbiome [32], [33], [34], are well studied in different pathological conditions, there exists limited biological insight for a plethora of different microbiomes, including environmental microbial communities [35], [36], [37], [38]. The difficulty in analyzing these metagenomes can often be attributed to the paucity of curated databases and library of reference genomes often required to identify SVs in the sample [39]. The sheer diversity of organisms in these samples that are yet to be identified and annotated further exacerbates this challenge [40], [1].
In order to deal with high-volume metagenomic data from many sample types that may lack an adequate reference, previous efforts have focused on reference-free approaches to quantify structural variants and diversity. Short read assemblers mostly rely on De Bruijn graphs (DBG), assembly graphs, or scaffold graphs to identify structural variation through extraction of specific graph topologies [29], [30], [41], [31]. An overview of the construction of these various graph types including the contributions of this work are illustrated in Fig. 1. These approaches characterize samples by relying on popular graph algorithms like betweenness-centrality to identify repetitive contigs (containing segments with high copy number), or finding 2-vertex cuts to extract end points of bubbles (which represent polymorphic regions), or both. For example, four node bubbles denote the variation between very similar sequences while three-node bubbles potentially represent gene gain/loss events and horizontal gene transfers. In order to reduce the complexity of betweenness-centrality [42], [43], [44], approximation algorithms have sometimes been employed. Another recent approach has focused on allowing end-users to efficiently query neighborhoods of interest in metagenomic–compacted DBGs, specifically by an indexing approach that approximates minimum r-dominating sets [45]. Though the approximation schemes, especially in the case of betweenness centrality, make this calculation more tractable on large metagenomic datasets its sample wide accuracy and sensitivity may still be sub-optimal [41]. Another recent tool, MetaFast [46] compares unitigs in DBGs to find unique regions by comparing a set of “positive” and “negative” samples. To the best of our knowledge, MetaCarvel is the only existing graph-based approach able to track SVs and CNV de novo from metagenomic short read data, which we compare to in this work.
Fig. 1.

Different graph types for metagenomic analyses and their construction. Graphs construction based on a set of five reads are shown. A. Overlap graph [Directed]: built directly from read with an overlap size of 3 base pairs(bp) and transitive edges are removed. B. De Bruijn graph (DBG) with kmer size (k) = 4 bp [Directed]: joins successive kmers obtained from reads having overlap size of length k-1. The kmers in blue represent repeated kmers. C. Unitig scaffold graph [Directed]: joins unitigs according to their relative positions in a DBG D. Hybrid Unitig Graph [Undirected]: An extension of the Unitig scaffold graph but is also repeat-aware and joins unitigs containing repeats of size k-1 where k is the kmer size used to build the DBG. Edge carried forward from the unitig scaffold graph are marked in black and called paired-end edges whereas newly added edges are marked in red and are called repeat edges. 3-mers marked in bold (GTG, GCT and TAA) are the repetitive regions connected by the repeat edge.
1.4. KOMB: a novel approach for identifying CNV in metagenomes
Before we describe our approach, it is important to define what we mean by variation in the context of a broader view of SV detection in metagenomes. Similar to the definition used by Zeevi et al. (2019) [19], we define SVs as genomic segments present in differing copy numbers across a subset of microbial genomes within the community, as well as segments in close proximity to these duplicated regions that may be inserted/deleted (indels) compared to other bacteria in the sample. In our work, we show that through careful graph construction and algorithmic choices, it is possible to retrieve and characterize CNV in metagenomes from a functional perspective.
Previous methods have used DBG types with embeddings or support for efficient repeat retrieval, such as Breakpoint graphs [47], [48], A-bruijn graphs [49], [50], Linked DBGs [51] and SIGAR graphs [52]. But these graph types have been limited in their use for detecting SVs in complex microbial community containing multiple closely-related genomes.
One novelty in the method presented herein is that we add an additional set of edges to the compacted DBG (cDBG). These are called repeat edges, and they allow KOMB to track both intra-genomic and inter-genomic repeats across a given sample. We call the combination the “hybrid unitig graph”, though it retains the canonical adjacency edges that track indels. This leads to formation of densely connected, clique-like regions that can be efficiently recovered in complexity using the K-core decomposition algorithm [53], [54], [94]. We show through validation on simulated datasets that KOMB is able to partition unitigs into shells (bins) based on the copy number of repeats contained as well as the relative proximity of these repeats, which enables capturing a community wide profile of different shared repeats in the sample. Table 1 summarizes the features of previous tools and highlights the contribution of KOMB towards characterization of subtle but previously hidden properties of a metagenome.
Table 1.
Comparison of KOMB to previous tools developed for CNV detection in Metagenomes. KOMB utilizes a fully connected hybrid unitig graph based on repeat linkage to track CNV across samples. Only the central algorithms for repeat detection and/or SV detection are listed as some tools use a combination of algorithms. Abbreviations: BinEM = Bernoulli Mixture Model (BMM) estimated through the Expectation–Maximization (EM), NEM = Neighboring Expectation–Maximization algorithm, Bet. Centrality = Betweenness Centrality
| Tool | Algorithm | Linear | Reference | Detect | Works | Anomaly |
|---|---|---|---|---|---|---|
| Time | free | CNV | on | Detection | ||
| Complexity | Raw Reads | |||||
| PPanGGOLiN [55] | BinEM/NEM | No | No | Yes | No | No |
| Bambus2 [29] | Bet. Centrality | No | Yes | Yes | Yes | No |
| MetaCarvel [31] | Approx. Bet.Centrality | Approx. | Yes | Yes | Yes | No |
| KOMB (this work) | K-core Decomposition | Yes | Yes | Yes | Yes | Yes |
We outline three main results in our work. First, we show that compared to MetaCarvel we can recover a higher number of repeats based on appropriate shell thresholds as well as recover higher number of unitigs that repesent variation (clique-like regions vs bubbles) in a synthetic metagenome. Second, we show that sample-specific difference in Human Microbiome Project (HMP) microbial community structures can be recovered by KOMB repeat profiles. Additionally, KOMB is able to find a statistically significant number of “anomalous” unitigs (high coreness and low degree) that are functionally unique in each HMP body-site. Finally, we also show that KOMB profiles can be used to track changes in longitudinal samples and show that “anomalous” unitigs are representative of keystone taxa required for FMT recovery. Though a more thorough theoretical treatment towards wider applicability and intepretability of these repeat profiles and distinguishing intra- and inter-genomic repeats is an active research question, KOMB shows significant utility in identifying functionally and taxonomically important CNV in metagenomes.
2. Methods
2.1. KOMB algorithm
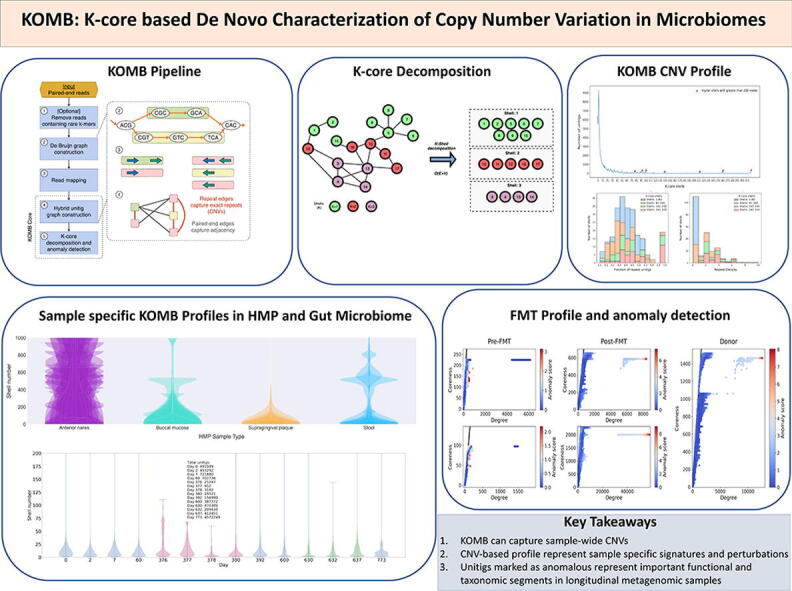
An overview of the KOMB pipeline is given in Fig. 2. The main steps are as follows. First a DBG is constructed from reads in the sample and unitigs are identified from this graph. The reads can be subject to an optional k-mer filter as a preprocessing step. Second, reads are mapped back to unitigs and a graph is constructed on the unitigs by linking them together in two different ways using the read-mapping data (called a “hybrid” graph herein and described in additional detail below). Only unitigs with a GC content between 10% and 90% are considered as nodes for the graph. Finally, the hybrid unitig graph is partitioned using the K-core decomposition into an ordered group of bins (called “shells”), where unitigs in higher shells have a higher copy number. This set of shells along with the unitigs contained in each one is called the KOMB profile, and in what follows we show that it captures a meaningful property of the community.
Fig. 2.

Overview of the KOMB pipeline. 1. As an optional pre-processing step users can use k-mer filtering to discard low-quality erroneous reads. 2. KOMB uses ABySS for memory efficient DBG construction and unitig generation. 3. Paired-end reads are mapped back to the unitigs obtained in 2 in order to connect unitigs. Paired-end reads with just one read mapping are discarded. 4. The hybrid unitig graph is constructed. Edges connecting unitigs mapped by the same read are termed as repeat edges whereas edges between unitigs mapped by paired-end reads are called paired-end edges. The latter are similar to edges in a scaffold graph. 5. The obtained unitig graph is partitioned into K-shells using the K-core decomposition algorithm. Anomalous unitigs are marked using the CORE-A anomaly score algorithm.
KOMB incorporates several widely-used bioinformatics tools as part of its workflow. Raw paired-end reads are input to ABySS [56] for efficient DBG creation and unitig construction, as well as Bowtie2 [57] for fast and accurate read mapping. In addition to this, our tool also relies on the igraph C [58] and OpenMP [59] libraries for the K-core implementation and the fast parallel construction of the hybrid unitig graph, respectively. A k-mer based read filtering tool [60] is also available for use as part of the software for optional pre-processing of reads.
2.1.1. Hybrid unitig graph construction
KOMB constructs a novel hybrid unitig graph to efficiently mine repetitive topologies using K-core graph decomposition. The workflow consists of DBG construction, read mapping, and the KOMB core module as shown in Fig. 2. All reads are initially input to the DBG constuctor ABySS to obtain unitigs. A unitig is a maximal consensus sequence usually obtained from traversing a DBG. By definition, unitigs terminate at branches caused by repeats and variants and, unlike contigs, are non-overlapping. Subsequently, all of the reads are mapped to unitigs using Bowtie 2. We then construct our hybrid unitig graph with two distinct set of edges. First, for each read we create a set of all unitigs that mapped to that read and connect them. We denote these edges as repeat edges, which capture repeats in unitigs. Second, for a given forward and reverse read pair, we check if each individual read in the pair mapped to different unitigs, which would represent potentially adjacent unitigs in the genome. We call these adjacency edges.
2.1.2. K-core decomposition
K-core decomposition is a popular graph-theoretical concept used in network science to identify influential nodes in large networks [61], [62], [63]. The K-core of a graph is defined as the maximal induced subgraph where every node has (induced) degree at least K. A node belongs to the K-shell if it is contained in the K-core but not in the -core. For any given graph, one can iteratively and efficiently decompose it into shells with a complexity proportional to the size of the graph, which is significantly faster than the computation of most exact centrality measures [54]. The shells output as a result of K-core decomposition on the hybrid unitig graph reveal unitigs that are connected either to a similar number of unitigs as a result of their repeat content (via repeat edges) or are adjacent to unitigs with the same properties. At higher shells we observe clique or clique-like behaviours that capture unitigs containing repeats with very high copy number and in some cases appearing very close to each other (e.g., tandem duplications). Both adjacency edges and repeat edges are weighted equally in the graph. A more detailed description of K-core decomposition as well as theoretical analysis of the KOMB K-core profile can be found in Supplementary Figures S1 and S2.
2.1.3. Identifying anomalous unitigs
Identification of biologically important unitigs in a given sample is done through ranking the nodes with a CORE-A anomaly score [64]. The CORE-A anomaly score calculates the deviation from mirror pattern (dmp) as given in Eq. 1 where and denote the rank of degree and coreness (shell that a vertex belongs to). This has been shown to reveal nodes of interest in real-world graphs like social and information networks [64].
| (1) |
2.2. Datasets
We tested KOMB on four different datasets to illustrate various properties of KOMB and underline different use cases while analyzing metagenomes. The datasets and their use cases are briefly described as follows:
-
1.
Simulated data: Reads of length 100 base pairs were simulated using wgsim [65] with no errors, substitutions or indels. The random backone was created with equal probability of observing each base (i.e A, T, G, and C) and validated by running Abyss [56] and obtaining only a single unitig. E.coli and B.cereus reference genomes were downloaded and used as a backbone for validation of inter and intra-genomic repeats.
-
2.
Shakya synthetic metagenome: A well-characterized synthetic metagenome consisting of 64 organisms (48 bacteria and 16 archea)[66]. This dataset is a simple test case to demonstrate how KOMB operates in practice, how to interpret the results, and how the higher shells reflect the structure of repeated regions in the metagenome.
-
3.
Multi-site HMP samples: This dataset contains 50 samples each from four body sites drawn from the Human Microbiome Project (HMP)
[67]. These samples are a useful test case for KOMB because they demonstrate a) that the KOMB profile for samples within a given site are broadly similar to one another, and b) that the overall profile for each body site is characteristic and distinct from other body sites in much the same way that the taxonomic profile is. In other words, it suggests that the KOMB profile is both reproducible and is consistent with what might be expected on highly dissimilar communities. This dataset is also used as an example of how the KOMB profile specifically recovers functionally rich sequences.
-
4.
Longitudinal gut microbiome samples: This data is also from a previous study [68] and contains samples taken from 6 subjects over two years, including one subject that was exposed to antibiotic and bowel cleanse disruption in that time. This is meant to go one step further by showing that the KOMB profile can capture both subject-specific differences at a common body site and variations in an individual community over time as it is subject to perturbations.
-
5.
Fecal microbiota transplantation (FMT): This data has not been previously published and includes samples from two patients undergoing (FMT) from a common donor. Specifically, the samples include both pre- and post-FMT from each patient as well as one sample from the donor. Anomalous unitigs identified in KOMB profiles capture specific taxa that are known to be contributurs to recovery and transition to a disease-free state in Post-FMT samples when compared to both Pre-FMT and Donor samples.
2.3. Running KOMB
The following sections contain detailed descriptions of how KOMB was run on each of the four datasets as well as any steps required for additional analyses discussed in Results below.
2.3.1. Shakya synthetic metagenome
Reads from the Shakya et al. (2014) study were obtained from NCBI Sequence Read Archive (SRA) (SRR606249). Reads were filtered using the kmer-filtering tool packaged as part of Stacks [60] since the dataset contains well characterized contaminants such as Proteiniclasticum in the data [45]. Ground truth for repetitive unitigs was established by using nucmer [93] to map the unitigs to the reference genomes with parameters -c 50 -l 50 as the hybrid unitig graph was built on matching 50 bp exact matches. KOMB was run with the parameters -k (kmer-size) 51 and -l (read length) 101. Fraction of repeat unitigs were calculated by dividing the number of unitigs marked as repetitive by nucmer to the total number of unitigs in the shell. KOMB repeat density calculated for each shell is given by the formula outlined in Eq. 2. We calculate the sum of copy numbers of each repetitive unitig and then divide it by the number of reference genomes these unitigs map to (number between 1–64). This number is then averaged over the number of repetitive unitigs in a shell given by N.
| (2) |
2.3.2. Multi-site HMP samples
HMP 1 data consisting of 50 samples each from four different body sites (anterior nares, stool, supragingival plaque, and buccal mucosa) was downloaded from the HMP website https://www.hmpdacc.org/HMASM/. Prior to running KOMB, we implemented a homogenizing step where only reads having length equal to the longest read length per sample were kept (mostly 100 bp) and the rest were discarded. Functional characterization of unitigs obtained and marked from the anomaly detection stage is done through SeqScreen [69], [70]. Anomalous unitigs are determined by considering all unitigs whose dmp score (see Eq. 1) is above a cutoff score as determined in Eq. 3. In this equation, Q3 represents the third quartile and IQR is the inter-quartile range which is the difference between the third and first quartiles (Q3 - Q1). For the analysis, we combined the anomalous unitigs from each individual sample and, separately, we combined the rest of the unitigs from each of the samples to obtain the set of unique GO terms and set of anomalous GO terms for each body site. Anomalous GO terms refers to the GO terms found in unitigs marked as anomalous by KOMB. Unique GO terms refers to a subset of GO terms found only in the anomalous unitigs but not found in other unitigs in a given body site. Consequently, anomalous GO terms are a superset of unique GO terms. All GO terms are filtered for bacterial specific GO terms using the https://github.com/AstrobioMike/CoV-IRT-Micro python package. Only GO terms belonging to the Biological Process branch were considered for the analysis.
| (3) |
2.3.3. Longitudinal gut microbiome samples
Reads for the dataset were obtained from the European Nucleotide Archive (ENA) website (ID: ERP009422). The reads were filtered using the kmer filter tool packaged as part of Stacks [60].
2.3.4. Fecal microbiota transplantation (FMT) samples
Sample Collection: Two pediatric patients with a recurrent Clostridium Difficile Infection (CDI) diagnosis received FMT under IRB-approved informed consent (#H-31066) at Baylor College of Medicine. The investigational nature of FMT was highlighted during consenting in accordance with current U.S. Food and Drug Administration (FDA) regulations. CDI diagnosis was based on toxin PCR positivity along with clinical complaints of 3 or more diarrheal stools per day. Patients reported recurrent (return of symptoms within 2 months) or ongoing diarrheal symptoms despite completing at least two courses of CDI-directed antibiotics that included at least one course of metronidazole and vancomycin. Patients received filtered, frozen-thawed fecal preparations from a standardized donor (38–40 year old male during donations) via colonoscopy. The donor screening and fecal preparation procedures were approved by the U.S. FDA (IND#15743). Fecal samples were collected from patients the day prior to FMT and 8–9 weeks following treatment on a follow-up visit. All samples were frozen and kept at −80°C until simultaneously thawed for bacterial DNA extraction using the PowerSoil DNA isolation kit (MO BIO Laboratories, Carlsbad, California, USA). Shotgun metagenomic sequencing was performed with ng of input DNA as previously described in [71] and the sequence was submitted to NCBI BioProject database: PRJNA743023.
Analysis: Reads were mapped to GRCh38p12 using bowtie 2.3.5 to filter out human sequences with preset options bowtie2 –local. Read pairs were extracted from resultant Sequence Alignment Map (SAM) file using samtools 1.9 [72] using flags samtools fastq -f 13 and then run through KOMB. Taxonomic analysis of the anomalous unitigs was done by running the unitigs through Kraken2 [73]. Kraken2 was run with the miniKraken2 database v1 (8 GB). Unitigs that were successfully classified at genus level or below were considered for the analysis. All unitigs classified at species level were assigned to their corresponding genus. For each sample, anomalous unitigs are obtained by selecting those whose dmp score (Eq. 1) is above the cutoff score in Eq. 3. For each genus present in anomalous unitigs, we calculate the Ratio of Ratios score for each genus as given in Eq. 4, where numag and numog are the number of unitigs classified at genus g in the set of anomalous unitigs and other (background) unitigs, respectively. The denominators refer to the sum of all unitigs of all genus present in the set. The total number of unique genus present in both the sets (anomalous and other) are Na and No, respectively. For the analysis, we selected those genera with the ratio of ratios greater than or equal to one () which we term as over-represented genus in the anomalous unitigs.
| (4) |
2.4. Calculating the L1 distance between KOMB Profiles
In order to calculate the distance between two KOMB profiles, we use the L1 norm of the difference between their normalized coreness profiles. More precisely, we first divide the size of each shell by the total number of unitigs in each profile. The shorter of the two profiles is then padded with zeros to equalize the number of shells, i.e., we can represent each profile as a vector of the same size. We then compute the distance between the profiles as the L1 norm of the difference between these two vectors.
3. Results
3.1. Validating KOMB on CNV in simulated data
We tested KOMB ability to recover segments containing CNV through two simulated experiments. First, we embedded two families of repeats 200 × 400 bp and 400 × 200 bp identical repeats in a randomly generated DNA backbone and simulated error-free reads from the genome. We inserted the repeats in the backbone in three different ways to see how the relative placement of repeats effects the identification of CNV. The repeats were inserted (i) randomly over the length of the read (ii) each family of repeats at a different end and (iii) both families at the same end. As the backbone is completely random we expect the unitgs to branch at repeats by definition. We plot the KOMB profile for each of these cases which is the shell number on the x-axis vs the number of unitigs on the y-axis. We see in Fig. 3 that the relative position of the families of repeats in backbone influences the shell number where the repeats are found. In the case where the repeats are inserted at different ends we observe the peaks of unitigs co the repeat families at 201 and 401 which is almost exactly the copy number of the families respectively. The small deviation was caused due to the imperfect unitig output of abyss. When the repeats are interleaved as in (i) or (iii), the peaks were shifted towards the right due to the presence of unitigs with mixed repeats as discussed in Supplementary Figure S2 which increase the shell number of the repeats due to the extra edges.
Fig. 3.

Validation of KOMB on simulated data. KOMB profiles on a random backbone with 200400 bp and 400200 bp identical repeats when randomly inserted (L), inserted at different ends (M), and inserted in the same end (R). The x-axis represents the shell number and the y-axis represents number of nodes (unitigs). In the ideal case (M) where the unitigs have the same repeats at its end we observe peaks at 201 and 401 respectively. Interleaving repeats randomly (L) causes a shift in the peaks towards higher shells to 326 and 449, respectively. Further, inserting both kinds of repeats at the same end (R) results in a similar shift with peaks occurring at 329 and 451.
The second experiment dealt with validation of simulated repeats in a real genomic backbone. We embedded 400 × 400 bp intra-genomic and 500 × 500 bp inter-genomic repeats in an E.coli backbone. Another 200 × 400 bp intra-genomic and 500 × 500 bp inter-genomic repeats were inserted into B.cereus backbone and reads were simulated error-free. The unitigs in the peaks on the KOMB profile were mapped back to genomes to ascertain if they originated from a given family of repeats for validation. Similar to the case with a random backbone, we observed shift in the peaks and additional fragmented peaks around the CNV region. The fragmentation is caused by presence of inherent repeat elements in each of the organisms. We found that the unitigs in the peak around 200 and 400 do indeed correspond to the intra-genomic repeats while the peak around 1000 did indeed correspond to inter-genomic repeats (Fig. 4 and Fig. 5). Under a controlled environment, the KOMB profile allowed for separation and identification of different repeat families in the sample based on their copy number. In addition to these experiments, we also tested KOMB on simulated repeats with low copy numbers (Supplementary Figure S3) that were closer in count to each other (10x400bp and 25x400bp repeats) and we were able to achieve similar results as seen in the previous validation experiments.
Fig. 4.

Validation of KOMB on simulated data with a real genomic backbone. Combined KOMB profile of E. coli (intra: 400400 bp, inter 500500 bp) and B. cereus (intra: 200400 bp, inter 500500 bp) repeats (Left), E. coli single genome (Middle), and B. cereus single genome (Right). For the combined profile of both bacteria (L), there is a clear formation of peaks close to position 1000 (984 and 979), which indicate the inter-genomic repeats, and peaks at shell numbers 277, 396 and 540. We also plot the individual profiles of E.coli and B.cereus. For E.coli (M) we see three peaks at 396, 496 and, 539 given its higher copy number of intra-genomic repeats (400). For B.cereus, we observe peaks at 276 and 492 signalling its intra- and inter-genomic repeats, respectively.
Fig. 5.

Validation of repeat types inE. coli+B. cereussample via mapping unitigs back to the reference using nucmer. Unitigs are labelled based on the repeats they overlap with. Based on ground truth nucmer mapping, the last shell (close to 1000) contains unitigs overlapping exclusively with inter-genomic repeats (FI) whereas the shells around 200 are overlapping B. cereus simulated intra-genomic repeats (F1) and shells around 400 are overlapping E. coli simulated intra-genomic repeats (F2). Finally, the first shells contain background noise (colored black).
3.2. KOMB profile example and interpretation
The Shakya synthetic community was used as a simple example to demonstrate the KOMB profile and provide additional evidence to support the assertion that it captures the pattern of repeated regions in the community.
An input to the DBG construction is k: the exact k-mer size used to join reads. The shells in the KOMB profile are labeled incrementally as they are produced in the K-core decomposition. The number of a given shell is approximately the copy number of a family of exact repeats of size , either within a single genome or across multiple organisms. This is shown in some additional detail below using a simulated example from a community of two microbes. The correspondence between shell number and copy number is not perfect, however, as there are some circumstances where a unitig can wind up in a higher shell for other reasons, also discussed below.
First, Fig. 6(A) shows how the full set of unitigs is distributed according to each shell, with a total of 320 K-core shells obtained after decomposition (for simplicity, we exclude shell 0 which represents isolate unitigs). Early shells (i.e. 1–4) contain the majority of the unitigs and overall the density declines steeply as the shell number grows, similar to what we might expect in a random graph. However, by contrast with a random graph there are a number of small peaks occurring at higher shells after the initial drop-off (marked with red triangles). Most of these peaks are followed by regions of empty cores indicating that these peaks mark dense cliques that all share the 50 bp exact match (as 51 was the k-mer size used). These peaks in higher shells are endemic to the nature of the KOMB profile, and the number and size of the peaks captured in this figure are a simple summary of the KOMB profile for a given community.
Fig. 6.

Characterization of a synthetic metagenome sample using KOMB. A. KOMB profile of the Shakya et al. (2013) dataset representing the shell number on the x-axis and the number of unitigs in the y-axis. Red triangles indicate higher shells with greater than 200 nodes, which represent clique or clique-like regions in the hybrid unitig graph. B. Histogram representing the fraction of unitigs in each of the shells that are repeats as determined by comparing with the nucmer output. C. KOMB repeat density is defined as the average copy number per number of genomes for the repeat unitigs in the shell (see Eq. 2). Larger shells have repeats with high copy number and more specific to a single (or group of related) organisms. For figures B. and C., shell 0 (disconnected nodes) and shells that contained no unitigs are not considered. Shells are split into four different groups (1–80, 81–160, 160–240, 241–320) for visualization.
Beyond simply showing the KOMB profile for this community, it is worth verifying that higher shells do indeed represent regions with more repeats and of higher repeat-number. Here, we have used nucmer to quantify the repeat number of each unitig, and the stacked bar charts in Fig. 6, Fig. 6(C) show how shells compare to one another according to the fraction of unitigs considered a repeat and average repeat density, respectively. The nucmer repeat quantification is imperfect and the shells are grouped by quartile, but nonetheless the third and fourth quartiles are skewed to the right in each graph, indicating that indeed the higher shells contain unitigs with a heavier density of repeats. This is a fundamental property of the shells in a KOMB profile. Nucmer analysis of repeat unitigs also revealed that the repeats in the higher shells mapped only to a few organism in the sample but had relatively high copy numbers resulting in a higher density. Combining these observations with those from the KOMB profile, we can infer that the majority of shells containing clique/clique-like regions (i.e., repeats and sequences adjacent to similar branching paths in the graph) are likely to lie beyond shell 161. It is important to note here that the topology of the hybrid unitig graph allows KOMB to capture unitigs that are adjacent (as defined by adjacency edges) to repetitive unitigs across copy numbers, in addition to repeats. This is a non-obvious phenonmenon, but the expected result that a small number of the unitigs in higher shells will be there not because of copy number, rather due to their adjacency to a high-copy region.
To evaluate the results obtained by KOMB we compared the repeat unitigs obtained to MetaCarvel [31]. In Supplementary Figure S4 (A), we observe that the contigs marked as repetitive by MetaCarvel have low True Positive Rate (TPR) and False Positive Rate (FPR) of 0.14 and 0.03 respectively. While the low FPR of MetaCarvel is desirable the low TPR can be a bottleneck for analysis. In contrast, KOMB provides higher (TPR) rates at unitig level (with some increase in FPR) going to a maximum of 0.49 (TPR) and 0.14 (FPR) at the cutoff of 20 shells and above. We also observed in Supplementary Figure S4 (B), that KOMB captures far more unitigs as variations in the sample through identification of peaks or K-cores (2229) and high anomaly unitigs (7860) as compared to bubbles (8) and high-centrality (555) contigs provided by MetaCarvel. Here the bubbles from MetaCarvel correspond to the seqments that share the same source and sink node that are representative of variation in the sample while high centrality nodes are contigs marked by the Approximate betweenness centrality algorithm (mean + 3standard deviation). Similarly, the peaks refer to the Clique/Clique like regions found in the KOMB profile above shell 70 (marked in red in Fig. 6 (A). The high anomaly nodes were identified by the anomaly detection algorithm. To ensure that this is not just an artifact of comparing smaller unitigs to contigs we also aligned these peak and anomalous unitigs to the contigs reported as bubbles and high-centrality using nucmer and we observed very little overlap as seen in Supplementary Figure S4 (C). We also compared the runtimes and memory usage of these tools and found that they were comparable with KOMB having a slightly better runtime while MetaCarvel having a slightly better memory efficiency (Supplementary Figure S4 (D)). To further analyze how shell thresholds affect KOMB’s TPR and FPR we plotted a Receiver Operating Curve (ROC) and see that below shell 25 KOMB shell contain low copy number repeats as well as high number of non-repeat unitigs which increased the FPR as observed in Supplementary Figure S5.
3.3. KOMB vis-a-vis beta-diversity and functional annotation
A key test for a novel descriptive profile is whether it is reproducible and whether it shows broad differences where they would intuitively be expected. A key insight about the human microbiome is that the bacterial communities differ substantially by body site, and that communities from the same body site across different individuals are more similar than across body site. We would therefore expect KOMB profiles to follow this same pattern. We considered 50 samples from four different body sites to analyze differences in their KOMB profiles. Supplementary Figure S6 shows the median shell numbers of all samples in a body site and Supplementary Table S1 contains the average and sttandard deviation of the number of reads per body site. Fig. 7(A) shows the same distribution of unitig density by shell number as in the previous dataset, but here it is presented as a violin plot. Specifically, the plots for all 50 samples from the same site are overlaid to visualize their variability. Each site has its own evident shape, and notably the anterior nares site appears to have the largest range of variability for individual samples. We also analyzed the site-specific profiles for intra-site and inter-site distances which are discussed in Supplementary Data SD1.
Fig. 7.

Characterizing community shifts in Human Microbiome Project (HMP) samples. A. KOMB profiles from 4 different body sites containing 50 samples each obtained from HMP datasets. The y-axis of the violin plots represent shell number (cutoff at 1000 for visualization) and the width represents the number of unitigs in each shell. B. Anomaly profiles for each body site (50 samples overlaid), x-axis represents the degree of unitigs and y-axis represents the coreness (or shell number) of the unitigs. The gradient on the color bar represents the CORE-A anomaly score with the white and red representing higher scores within the samples. C. Bar plot showing the percentage of unique GO terms from the set of unitigs marked as anomalous. Black dots represent median of 100,000 random split simulations of GO terms obtained per body site, the whiskers represent (top) and (bottom) percentile indicating significance of the bar plot. D. Jaccard similarity between the set of unique GO term (y-axis) and the entire set of GO terms from the unitig marked as anomalous for each pair of body sites. E. Jaccard similarity between the entire set of anomalous GO terms for each pair of body sites.
This dataset also served as a test case for a hypothesis that the KOMB profile could be used to identify highly “biologically important” segments. The K-core decomposition has been useful for this in other contexts, specifically by identifying anomalous nodes in a social network graph [64]. Here, we hypothesize that “importance” of a unitig could be represented by functional richness.
We utilize the anomaly detection algorithm as proposed by [64]. Fig. 7(B) shows the Coreness vs Degree graph of the unitigs for each body site. The color gradient indicates the CORE-A score with the unitigs having high CORE-A score mainly being high coreness and low degree or low coreness and high degree. Unitigs were separated into those marked as anomalous and those not, then we functionally annotated the unitigs marked as anomalous by assigning GO terms. Then, GO terms occuring only in anomalous unitigs (“unique GO terms”) were expressed as a percentage of all GO terms. For comparison, we conducted simulations in which GO terms were randomly assigned to contigs and ran the same calculation of “unique GO terms”.
Fig. 7(C) shows the results: the bar for each body site is the overall % unique, while the black line (and error bars) represent the values obtained by simulation. The actual values are well above the error bars for all body sites, indicating that anomalous unitigs contain a disproportionate share of gene functions that are found only in these unitigs. Furthermore, previous studies [74], [75], [76] have described the relative evenness and low diversity of the buccal mucosa community especially in comparison to other oral communities like supragingival plaque which is reflected in our functional analysis of anomalous unitigs.
Further, we analyzed how the unique GO terms in a given body site compare with the GO terms found in anomalous unitigs from other body site. We then calculated the jaccard similarities of these sets. We hypothesized that samples from similar regions (eg. oral) would be more similar functionally than others which we recapitulate in a taxonomy-oblivious manner through KOMB. In Fig. 7(D), we see that the jaccard similarities are overall low () indicating that these unique GO terms are generally specific to the microbiome in a given body site. The GO terms in stool were the most dissimilar to those found in anomalous unitigs in other samples (average jaccard similarity = 0.05). The similarity scores of unique GO terms in anterior nares and supragingival plaque had greater similarity with the anomalous GO terms buccal mucosa (0.19). Fig. 7(E) shows the jaccard similarities of anomalous GO terms (but not necessarily unique to anomalous unitigs) between each body site. The oral sites, buccal mucosa and supragingival plaque, had the most similar anomalous GO terms (0.43). Similar to the case with unique GO terms, anterior nares had a higher similarity with buccal mucosa (0.35) than supragingival plaque (0.26). We also observed that anomalous unitigs in stool had the lowest functional similarity to other body sites (average jacaard similarity = 0.186). The GO term ID and names can be found in Supplementary Data SD2.
3.4. KOMB characterizes community shifts in longitudinal samples
3.4.1. Longitudinal gut microbiome samples
To demonstrate KOMB’s ability to derive insights from large scale metagenomic analysis, we considered a temporal gut metagenome study. This study contains gut microbiome samples collected from 7 subjects (5 male and 2 female) at different time points spread over two years. Fig. 8(A) shows the KOMB profiles of each of the 6 analyzed subjects (one subject was excluded because of a missing data point) from the initial three time points (Days 0, 2, 7), each labeled by an alias given in the original study. These violin plots show that the gut samples from the six subjects all have relatively similar KOMB profile distributions, although some idiosyncrasy does appear in subjects Daisy and Bugkiller. To quantify these profiles, the intra-subject and inter-subject sample distances were analyzed and are discussed in Supplementary Data SD3.
Fig. 8.

Characterizing community shifts in longitudinal gut microbiome samples. A. KOMB profiles from 6 different subjects from samples collected Days 0, 2 and 7. The y-axis of the violin plots represent shell number (cutoff at 100 for visualization) and the width represents the number of unitigs in each shell. Alien, Bugkiller, Peacemaker, and Scavenger are male subjects while Daisy and Tigress are female subjects. B. KOMB profile for subject Alien over the course of the 14 different time points in the study. The y-axis (cutoff at 200 for visualization) represent shell number and x-axis represents the day of sample collection. Days 376, 377, 378, and 380 represent profiles during which the subject was exposed to antibiotics, causing compression in the total shell count as well as a significant change in the unitig distribution of the initial shells. Days 630 and 632 indicate time points when the subject underwent a bowel cleanse procedure with a similar but less prominent effect on unitig count and distribution.
To get a more quantitative understanding of the data and the effects of external disruptions on the gut microbiome, we focus our attention on the subject Alien who was the only subject exposed to an antibiotic intervention and bowel cleanse procedure during the course of the study.
Fig. 8(B) outlines the entire longitudinal trajectory of Alien’s gut microbiome over the course of 14 time points spread across two years. The KOMB profiles as displayed focus on the first 200 shells at each time point. We observe a significant change of shape in the profile on Days 376, 377, 378, and 380 which coincides with samples taken after antibiotic intake and which correspond to a significant perturbation community composition as reported in the study. This is also mirrored by the unitig counts in the samples, which decreases by an order of magnitude. Importantly, the total number of reads in the samples from each time point are similar and, hence, the change in unitig count is most likely caused by a shift in the community composition. Thus, antibiotic intervention causes not only a reduction in the total number of shells but also alters the unitigs present in the initial shells, though this tends to recover slightly towards the end of the antibiotic cycle on Day 380. The distribution of unitigs to shells has returned to form twelve days after the last post-antibiotic sample (Day 392), and the raw number of unitigs has returned to earlier levels by Day 600. We observe similar but less drastic shell compression and quick recovery after a bowel cleanse (Days 630, 632) indicating that antibiotics cause a far greater disruption in microbiome community structure, a finding corroborated by the authors in [68] as well as an earlier study [77]. We also quantified distances between groups of samples at pre, post and during antibiotic treatment time points using L1 distances of KOMB Profiles Supplementary Figure S7. We found that Alien post-antibiotic had greatest pairwise distance to all other samples indicating that antibiotic intervention does in fact cause significant perturbation in KOMB profiles.
3.4.2. FMT samples pre, post, and donor
We analyzed two patient samples at two different time-points namely, Pre-FMT and Post-FMT using KOMB to understand shift in microbiome communities after an FMT procedure. We also compared the KOMB anomaly profiles of Pre-FMT and Post-FMT samples to the Donor sample to track common patterns between them. The Pre-FMT samples were collected from the patients post vancomycin treatment. In Fig. 9(A), we observe that the anomaly profiles of Pre-FMT samples are distinctly shrunk (less coreness) compared to the Post-FMT and Donor samples indicating similar trends previously observed after the antibiotic treatment in the gut microbiome study [68]. We also see that Patient 1 shows some partial recovery towards the Donor profile whereas Patient 2 shows a higher similarity to the Donor in terms of coreness and anomaly score.
Fig. 9.

Characterizing community shifts in fecal microbiota transplantation (FMT) samples. A. (Left) Anomaly profiles of two patients undergoing FMT therapy at two different time points namely Pre-FMT and Post-FMT. (Right) Anomaly profiles of the donor sample, which is common for both patients. The x-axis represents the degree of unitigs and y-axis represents the coreness (or shell number) of the unitigs. The gradient on the colorbar indicates the CORE-A anomaly scores of unitigs in the sample. B. Jaccard similarity between sets of taxa over-represented at the genus level found in unitigs marked as anomalous in each of the 5 samples. The row highlighted in black indicates the jaccard similarities of each patient across time points as compared to Donor. C. Common taxa over-represented in anomalous unitigs for Pre-FMT, Post-FMT and Donor samples. The numbers indicate the ratio of ratios of counts of taxa, indicating the relative level of presence of the corresponding taxa in the anomalous unitig compared to the other unitigs in the sample. The numbers in the figures have been averaged for Pre-FMT and Post-FMT samples from both Patients. The first three genus Akkermansia, Selenomonas and Lactobacillus were common in Pre-FMT while Lactobacillus, Blautia, Veillonella, Paeniclostridium, Ruminoccocus, Oscillibacter, Paenibacillus, Turicibacter, Actinomyces, Dialister, Faecalibacterium were common in Post-FMT samples.
The unitigs obtained after KOMB analysis from one of the Post-FMT samples were too short and fragmented to annotate functionally using SeqScreen. In lieu of this, we examined the taxa represented by anomalous unitigs with the thinking that they may indicate important organisms driving the change in host microbiome post-FMT. For unitigs identified as anomalous (and which could be classified at the genus level, see Methods), over-represented taxa were determined by the score defined in Eq. 4. In Fig. 9(B) we see that, in general, there is a low similarity between over-represented taxa across the samples. We still observe that for both Patients 1 and 2, the Post-FMT samples have a higher taxa similarity to Donor compared to the Pre-FMT samples (highlighted by the black box in Fig. 9(B) as captured in the anomalous unitigs despite a substantial difference in their anomaly profiles.
As seen in Fig. 9(C), Pre-FMT samples had three genera in common; Akkermansia, Selenomonas and Lactobacillus whereas Post-FMT had eleven: Lactobacillus, Blautia, Veillonella, Paeniclostridium, Ruminoccocus, Oscillibacter, Paenibacillus, Turicibacter, Actinomyces, Dialister, Faecalibacterium in common. We compared the relative levels of these taxa in Pre-FMT and Post-FMT and Donor. The values in the heatmap represent the average of the Ratio of Ratios score in both patients. Compared to Pre-FMT levels, we saw a substantial increase in two taxa Akkermansia and Lactobacillus in the Post-FMT anomalous unitigs. Interestingly, previous studies have shown that higher levels of some species belonging to Akkermansia and Lactobacillus were helpful to combat Clostridium difficile infections [78], [79]. In contrast to Pre-FMT and Post-FMT Akkermansia, Selenomonas and Lactobacillus were also present in the anomalous unitigs in the Donor sample but were not over-represented compared to the other (background) unitigs.
Among the taxa common in Post-FMT samples, roughly half (6/11) were similarly over-represented in Donor sample anomalous unitigs, though the levels were much higher in the former. However, Turicibacter and Dialister had a higher level of over-representation. This is noteworthy because Turicibacter is a well-characterized taxa and is one of the most abundant in other reported studies on FMT inoculums and Post-FMT communities [80], [81], [82] whereas the presence of Dialister has been found to be essential in Post-FMT recovery and non-disease states [83], [84]. Kraken 2 outputs and unitig classifications can be found in Supplementary Data SD4.
Supplementary Figure S8 (A,B,C) show the different taxa (at genus level) and their average relative abundances in our Pre-FMT and Post-FMT samples. The taxa shown in bold denote taxa identified as overexpressed in the anomalous unitigs. We observe that KOMB was able to identify sequences belonging to taxa at different abundance levels and thereby not being biased to just the most abundant taxa. To corroborate whether this property of KOMB could help identify keystone species in FMT samples, we compared against RECAST [85] which is a tool specifically targeted to studying sequence-level differences in Pre-FMT and Post-FMT samples. We considered two main outputs from RECAST for comparison. First, we considered those sequences that were marked by RECAST as being absent in the Post-FMT samples. Supplementary Figure S9 (A) shows that KOMB identified 5/7 taxa in this group. Second, we analyzed the output that were indicated by RECAST as being in Post-FMT but derived from Donor. KOMB was able to identify 8/21 genera reported by RECAST including 4/5 of the most abundant in this group (Supplementary Figure S9 (B)). It also important to note here, that some of the taxa found through KOMB but not reported by RECAST are indeed found to be important like Turicibacter as indicated above as well as Actinomyces [86], Oscillibacter [87] and Lactobacillus [88] we found to be keystone in Post-FMT. Relative abundances of FMT samples and RECAST outputs were calculated using MetaPhlAn3 [89].
3.5. Performance
KOMB is written in C++. It uses the igraph C graph library [58] for the unitig construction and K-core decomposition implementations. Table 2 shows the runtime and memory usage of KOMB on the datasets used in our study. The experiments were run on a server with 64 Intel(R) Xeon(R) Gold 5218 CPU @ 2.30 GHz processors having 372 GB of RAM. While analyzing the runtimes of specific stages of the KOMB pipeline we observed that the ABySS unitig generation is the most memory intensive step in the pipeline while read mapping using Bowtie2 is the most computationally intensive step in the pipeline. As KOMB is also extremely memory efficient, one can process multiple metagenomic samples simultaneously on any modern workstation to reduce the runtime on entire datasets even further. Comparison of KOMB’s performance to MetaCarvel can be found in Supplementary Table S2.
Table 2.
Time and memory usage for KOMB. Shakya: Shakya et al. (2013); HMP (Av); average across HMP samples, TGM(Av); average across Temporal Gut Microbiome samples and FMT (Av); average across FMT samples. Read filtering is treated as a pre-processing step, therefore the time and memory usage for it is not reported in this table.For the average, samples having approximately the average number of reads were chosen as representatives for benchmarking. KOMB was run with 20 threads.
| Dataset |
Performance metrics |
|||||
|---|---|---|---|---|---|---|
| Reads | Nodes | Edges | Wall clock | CPU time | RAM | |
| Shakya | 53,997,046 | 96,901 | 1,080,012 | 77m50s | 1296m21s | 25.29 GB |
| HMP (Av) | 16,872,599 | 303,171 | 2,414,541 | 17m44s | 293m8s | 9.59 GB |
| TGM (Av) | 26,520,076 | 776,058 | 7,286,158 | 15m17s | 264m4s | 13.22 GB |
| FMT (Av) | 34,173,634 | 323,431 | 22,994,009 | 57m3s | 971m34s | 13.65 GB |
4. Discussion
KOMB is an attempt at a novel strategy for meaningful characterization of a microbial community. Namely, it amounts to a set of summary statistics that together characterize the broad pattern of gene-sharing within and across the microbes in a microbial community. It goes beyond previous methods both by using a hybrid graph containing both repeat and adjacency signal and by generalizing the graph features to a set of community-level descriptive statistics. This also leads to other open questions about this approach which need further exploration as part of future work. For example, obvious algorithmic questions include additional experimentation with the graph type, graph construction parameters, and other graph decomposition algorithms. Related to the output, further characterizing the repetitive elements in higher shells and anomalous nodes is a clear next step, as well as applying it to environments hypothesized to contain interesting gene-sharing/ horizontal gene transfer (HGT) patterns. Previous studies have shown that CNV and HGT in microbial communities is important to community’s response to stresses as well as dynamics within the ecosystem [90], [91]. In spite of these open questions, KOMB offers advantages over tools requiring reference genomes and coverage analysis in that it avoids any effect of coverage bias. Futhermore, KOMB doesn’t require de novo assembled MAGs, opening the door to CNV detection across a wider range of abundance profiles while avoiding the potential propagation of assembly errors.
Our experimental analysis of KOMB focused on three main priorities. The first was to show that KOMB profiles behave as expected in cases that are well-studied and as a set of community-level descriptive statistics it is capable of telling very different communities apart. Fig. 7(A) shows that the profile distributions for different body sites appear to have a characteristic general shape. Fig. 8 provides two more sanity checks: for a single human subject the KOMB profile is stable across time, with the notable exception of when the subject is taking antibiotic medication where a significant perturbation in the KOMB Profile is observed.
The second priority was to show that, as asserted, the shell numbers in the KOMB profile do indeed reflect the copy numbers of the unitigs they contain. This was done first by simulation with a simple case of two community members and fixed levels of gene-sharing, then by looking at a biological dataset and comparing basic measures of repeat-level to the shell numbers (Fig. 6). One outstanding question is under what conditions unitigs can appear in higher shells without reflecting higher copy numbers, and what that might imply.
The final priority was to show that the statistical description of gene-sharing, as hypothesized, correlates with a measure of functional importance within a given environment. The challenge of testing this hypothesis is in defining, let alone measuring, functional importance. Here we have used uniqueness of gene function as a proxy for this, and indeed the measure of this turns out to be over-represented in anomalous unitigs versus simulation of no correlation. This observation is intriguing but also unsatisfying without a deeper exploration of what functions these are and whether the same observations would be reflected by large-scale assembly. But it is central to KOMBs usefulness in the future as it suggests that, possibly, CNV captured by anomalous unitigs might be a way to screen for gene functions that are crucial to species in an environment and that might be specific to it as well. It is important to note here that, in our HMP analysis, anterior nares had the lowest average number of reads per sample and stool had the highest which could influence the number of unique GO terms we observed in the anomalous unitigs. As observed by Lloyd-Price et al. [92] individual anterior nares samples had a very poor clustering compared to other sites when observed using PCoA using Bray–Curtis distances among all microbes at the species level. Also, similar to the observation from KOMB anomalous unitigs, anterior nares had the lowest number of GO terms shared between single and co-assemblies in the samples which could indicate its high degree of variability amongst individual samples compared to other body sites.
Finally, we also investigated KOMB profiles by analyzing community shifts and disruption events in longitudinal samples through results from a FMT study. The small number of samples limit our ability to make generalised conclusions from this study but the graphics suggest how it might be helpful. For one, the comparison in Fig. 9(A) suggests that KOMB profiles may be effective at distinguishing the C. difficile-afflicted and healthy microbiomes. Additionally, the recovery reflects a return to a bi-modal “hourglass” shape, similar to the HMP stool samples in Fig. 7. Comparing the taxonomic representation in anomalous unitigs pre- and post-FMT shows a possible over-representation of taxa known to be associated with healthy and diseased states, although a formal statistical comparison over a large number of samples would be needed. To analyze KOMB’s ability to identify biologically relevant CNV at scale we also ran KOMB on 258 experiments obtained from Greenblum et al. (2015) [18]. Preliminary results from our analysis showed that KOMB was able to identify highly variable Inflammatory Bowel Disease (IBD) associated and Obesity associated Kegg Orthologs that were reported in the study (Supplementary Data SD5). We leave as future work to further investigate how the results obtained from KOMB generalize to other datasets with similar disease phenotypes.
In summary, KOMB can be thought of as a CNV "sensor" for large-scale metagenomic analyses where reference genomes are limited or unavailable. Further characterization of individual CNV and segments output by KOMB and their sample-specific biological significance offers a promising avenue for future research in this space.
5. Conclusions
KOMB enables de novo, reference-free CNV characterization, which captures repetitive DNA contained in microbiomes stemming from gene transfer, gene duplication, and mobile genetic elements. KOMB incorporates a novel hybrid unitig graph and anomaly detection based on K-core decomposition to efficiently identify functionally unique CNV, which may serve as drivers of microbial function and adaptation. As the need to analyze large environmental metagenomes that are scarcely annotated increases, KOMB may open the door to further advances specific to the detection and characterization of CNV in metagenomes.
Declarations
Ethics approval and consent to participate
The fecal specimens of FMT subjects were obtained with parental written consent approved by the Institutional Review Board (#H-31066) at the Baylor College of Medicine.
Competing interests
The authors declare they have no competing interests.
Availability of data and materials
Python jupyter notebooks used for analysis and generating figures can be found here: https://osf.io/r6phg/ KOMB outputs and files used for analysis for the experiments can be found at https://rice.box.com/v/komb-manuscript-data.
Availability and requirements
Project name: KOMB.
Project home page: https://gitlab.com/treangenlab/komb.
Operating system(s): OSX and Linux.
Programming language: C/C++.
Other requirements: Abyss 2.3.1, Bowtie2 2.4.4, Bifrost 1.0.5, igraph 0.8.3, conda version 4.0 or higher. The latest version of KOMB (v1.0), with all requirements installed, is available for download through bioconda at: https://anaconda.org/bioconda/komb. At least 64 GB of RAM recommended.
License: GNU GPL v3.0 or later.
Any restrictions to use by non-academics: No.
Authors contributions
A.B, T.J.T, S.S developed the study. A.B implemented the software, performed the validation, analyzed the data, interpreted the results, generated figures and wrote the manuscript. N.S performed the validation and generated figures. C.S analyzed the data and interpreted the results. R.A.L.E, S.S, T.S and T.J.T contributed to the design of the validation and the interpretation of the results. M.G.N and Y.F helped with the writing of the manuscript. All authors read and approved the final manuscript.
Funding
A.B., N.S., and T.J.T were supported by startup funds from Rice University and the FunGCAT program from the Office of the Director of National Intelligence (ODNI), Intelligence Advanced Research Projects Activity (IARPA), via the Army Research Office (ARO) under Federal Award No. W911NF-17–2-0089. R.A.L.E. was supported by the FunGCAT program from the Office of the Director of National Intelligence (ODNI), Intelligence Advanced Research Projects Activity (IARPA), via the Army Research Office (ARO) under Federal Award No. W911NF-17–2-0089. IARPA: https://www.iarpa.gov/. https://csweb.rice.edu/ C.S and T.S were supported by the P01-AI152999 and U01-AI24290 grants obtained from the National Institutes of Health (NIH). NIH: https://grants.nih.gov/grants/oer.htm. Y.F. is supported in part by funds from Rice University and Ken Kennedy Institute Computer Science Engineering Enhancement Fellowship, funded by the Rice Oil Gas HPC Conference. M.N. was supported by the P01-AI152999 grant obtained from the National Institutes of Health (NIH) as well as a fellowship from the National Library of Medicine Training Program in Biomedical Informatics and Data Science (T15LM007093, PI: Kavraki). NLM: https://www.nlm.nih.gov/ep/GrantTrainInstitute.html. T.J.T and S.S were also supported by National Science Foundation (NSF) grant EF-2126387.
The views and conclusions contained herein are those of the authors and should not be interpreted as necessarily representing the official policies or endorsements, either expressed or implied, of the ODNI, IARPA, ARO, or the US Government.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgement
The authors would like to sincerely thank Dr. Mihai Pop and the Pop Lab at University of Maryland, College Park for their constructive comments and suggestions that helped improve the manuscript and methodology.
Footnotes
Supplementary data associated with this article can be found, in the online version, at: https://doi.org/10.1016/j.csbj.2022.06.019.
Supplementary data
The following are the Supplementary data to this article:
References
- 1.Zhang X., Xu W., Liu Y., Cai M., Luo Z., Li M. Metagenomics reveals microbial diversity and metabolic potentials of seawater and surface sediment from a hadal biosphere at the yap trench. Front Microbiol. 2018;9:2402. doi: 10.3389/fmicb.2018.02402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang S., Yan Z., Wang P., Zheng X., Fan J. Comparative metagenomics reveals the microbial diversity and metabolic potentials in the sediments and surrounding seawaters of qinhuangdao mariculture area. PloS one. 2020;15(6) doi: 10.1371/journal.pone.0234128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vavourakis C.D., Andrei A.-S., Mehrshad M., Ghai R., Sorokin D.Y., Muyzer G. A metagenomics roadmap to the uncultured genome diversity in hypersaline soda lake sediments. Microbiome. 2018;6(1):1–18. doi: 10.1186/s40168-018-0548-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Douglas G.M., Langille M.G. Current and promising approaches to identify horizontal gene transfer events in metagenomes. Genome Biol Evol. 2019;11(10):2750–2766. doi: 10.1093/gbe/evz184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soucy S.M., Huang J., Gogarten J.P. Horizontal gene transfer: building the web of life. Nat Rev Genet. 2015;16(8):472–482. doi: 10.1038/nrg3962. [DOI] [PubMed] [Google Scholar]
- 6.Iranzo J., Wolf Y.I., Koonin E.V., Sela I. Gene gain and loss push prokaryotes beyond the homologous recombination barrier and accelerate genome sequence divergence. Nat Commun. 2019;10(1):1–10. doi: 10.1038/s41467-019-13429-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Treangen T.J., Rocha E.P. Horizontal transfer, not duplication, drives the expansion of protein families in prokaryotes. PLoS Genet. 2011;7(1) doi: 10.1371/journal.pgen.1001284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Faust K., Raes J. Microbial interactions: from networks to models. Nat Rev Microbiol. 2012;10(8):538–550. doi: 10.1038/nrmicro2832. [DOI] [PubMed] [Google Scholar]
- 9.Toft C., Andersson S.G. Evolutionary microbial genomics: insights into bacterial host adaptation. Nat Rev Genet. 2010;11(7):465–475. doi: 10.1038/nrg2798. [DOI] [PubMed] [Google Scholar]
- 10.Moreno-Pino M., Cristi A., Gillooly J.F., Trefault N. Characterizing the microbiomes of antarctic sponges: a functional metagenomic approach. Scientific Rep. 2020;10(1):1–12. doi: 10.1038/s41598-020-57464-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Whittle E., Leonard M.O., Harrison R., Gant T.W., Tonge D.P. Multi-method characterization of the human circulating microbiome. Front Microbiol. 2019;9:3266. doi: 10.3389/fmicb.2018.03266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.E. National Academies of Sciences, Medicine, et al. Microbiomes of the built environment: a research agenda for indoor microbiology, human health, and buildings. National Academies Press; 2017. [PubMed]
- 13.Emmons A.L., Mundorff A.Z., Keenan S.W., Davoren J., Andronowski J., Carter D.O., DeBruyn J.M. Characterizing the postmortem human bone microbiome from surface-decomposed remains. PloS one. 2020;15(7) doi: 10.1371/journal.pone.0218636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu X., Chen X., Wang Z. Characterizing the personalized microbiota dynamics for disease classification by individual-specific edge-network analysis. Front Genet. 2019;10:283. doi: 10.3389/fgene.2019.00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kieser S., Brown J., Zdobnov E.M., Trajkovski M., McCue L.A. Atlas: a snakemake workflow for assembly, annotation, and genomic binning of metagenome sequence data. BMC Bioinf. 2020;21(1):1–8. doi: 10.1186/s12859-020-03585-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li P.-E., Lo C.-C., Anderson J.J., Davenport K.W., Bishop-Lilly K.A., Xu Y., Ahmed S., Feng S., Mokashi V.P., Chain P.S. Enabling the democratization of the genomics revolution with a fully integrated web-based bioinformatics platform. Nucl Acids Res. 2017;45(1):67–80. doi: 10.1093/nar/gkw1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clarke E.L., Taylor L.J., Zhao C., Connell A., Lee J.-J., Fett B., Bushman F.D., Bittinger K. Sunbeam: an extensible pipeline for analyzing metagenomic sequencing experiments. Microbiome. 2019;7(1):1–13. doi: 10.1186/s40168-019-0658-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Greenblum S., Carr R., Borenstein E. Extensive strain-level copy-number variation across human gut microbiome species. Cell. 2015;160(4):583–594. doi: 10.1016/j.cell.2014.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeevi D., Korem T., Godneva A., Bar N., Kurilshikov A., Lotan-Pompan M., Weinberger A., Fu J., Wijmenga C., Zhernakova A., et al. Structural variation in the gut microbiome associates with host health. Nature. 2019;568(7750):43–48. doi: 10.1038/s41586-019-1065-y. [DOI] [PubMed] [Google Scholar]
- 20.Liu X., Tang S., Zhong H., Tong X., Jie Z., Ding Q., Wang D., Guo R., Xiao L., Xu X., et al. A genome-wide association study for gut metagenome in chinese adults illuminates complex diseases. Cell discovery. 2021;7(1):1–15. doi: 10.1038/s41421-020-00239-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bonder M.J., Kurilshikov A., Tigchelaar E.F., Mujagic Z., Imhann F., Vila A.V., Deelen P., Vatanen T., Schirmer M., Smeekens S.P., et al. The effect of host genetics on the gut microbiome. Nat Genet. 2016;48(11):1407–1412. doi: 10.1038/ng.3663. [DOI] [PubMed] [Google Scholar]
- 22.Rothschild D., Weissbrod O., Barkan E., Kurilshikov A., Korem T., Zeevi D., Costea P.I., Godneva A., Kalka I.N., Bar N., et al. Environment dominates over host genetics in shaping human gut microbiota. Nature. 2018;555(7695):210–215. doi: 10.1038/nature25973. [DOI] [PubMed] [Google Scholar]
- 23.Wang J., Jia H. Metagenome-wide association studies: fine-mining the microbiome. Nat Rev Microbiol. 2016;14(8):508–522. doi: 10.1038/nrmicro.2016.83. [DOI] [PubMed] [Google Scholar]
- 24.Durrant M.G., Bhatt A.S. Microbiome genome structure drives function. Nat Microbiol. 2019;4(6):912–913. doi: 10.1038/s41564-019-0473-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lapidus A.L., Korobeynikov A.I. Metagenomic data assembly–the way of decoding unknown microorganisms. Front Microbiol. 2021;12:653. doi: 10.3389/fmicb.2021.613791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kingsford C., Schatz M.C., Pop M. Assembly complexity of prokaryotic genomes using short reads. BMC Bioinf. 2010;11(1):1–11. doi: 10.1186/1471-2105-11-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagarajan N., Pop M. Parametric complexity of sequence assembly: theory and applications to next generation sequencing. J Comput Biol. 2009;16(7):897–908. doi: 10.1089/cmb.2009.0005. [DOI] [PubMed] [Google Scholar]
- 28.Olson N.D., Treangen T.J., Hill C.M., Cepeda-Espinoza V., Ghurye J., Koren S., Pop M. Metagenomic assembly through the lens of validation: recent advances in assessing and improving the quality of genomes assembled from metagenomes. Briefings Bioinf. 2019;20(4):1140–1150. doi: 10.1093/bib/bbx098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koren S., Treangen T.J., Pop M. Bambus 2: scaffolding metagenomes. Bioinformatics. 2011;27(21):2964–2971. doi: 10.1093/bioinformatics/btr520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nijkamp J.F., Pop M., Reinders M.J., de Ridder D. Exploring variation-aware contig graphs for (comparative) metagenomics using marygold. Bioinformatics. 2013;29(22):2826–2834. doi: 10.1093/bioinformatics/btt502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghurye J., Treangen T., Fedarko M., Hervey W.J., Pop M. Metacarvel: linking assembly graph motifs to biological variants. Genome Biol. 2019;20(1):174. doi: 10.1186/s13059-019-1791-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Byrd A.L., Belkaid Y., Segre J.A. The human skin microbiome. Nat Rev Microbiol. 2018;16(3):143. doi: 10.1038/nrmicro.2017.157. [DOI] [PubMed] [Google Scholar]
- 33.Xiao J., Fiscella K.A., Gill S.R. Oral microbiome: possible harbinger for children’s health. Int J Oral Sci. 2020;12(1):1–13. doi: 10.1038/s41368-020-0082-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumpitsch C., Koskinen K., Schöpf V., Moissl-Eichinger C. The microbiome of the upper respiratory tract in health and disease. BMC Biol. 2019;17(1):87. doi: 10.1186/s12915-019-0703-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lombard N., Prestat E., van Elsas J.D., Simonet P. Soil-specific limitations for access and analysis of soil microbial communities by metagenomics. FEMS Microbiol Ecol. 2011;78(1):31–49. doi: 10.1111/j.1574-6941.2011.01140.x. [DOI] [PubMed] [Google Scholar]
- 36.Delmont T.O., Eren A.M., Maccario L., Prestat E., Esen Ö.C., Pelletier E., Le Paslier D., Simonet P., Vogel T.M. Reconstructing rare soil microbial genomes using in situ enrichments and metagenomics. Front Microbiol. 2015;6:358. doi: 10.3389/fmicb.2015.00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Biller S.J., Berube P.M., Dooley K., Williams M., Satinsky B.M., Hackl T., Hogle S.L., Coe A., Bergauer K., Bouman H.A., et al. Marine microbial metagenomes sampled across space and time. Scientific Data. 2018;5 doi: 10.1038/sdata.2018.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kennedy J., Flemer B., Jackson S.A., Lejon D.P., Morrissey J.P., O’gara F., Dobson A.D. Marine metagenomics: new tools for the study and exploitation of marine microbial metabolism. Mar Drugs. 2010;8(3):608–628. doi: 10.3390/md8030608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nayfach S., Rodriguez-Mueller B., Garud N., Pollard K.S. An integrated metagenomics pipeline for strain profiling reveals novel patterns of bacterial transmission and biogeography. Genome Res. 2016;26(11):1612–1625. doi: 10.1101/gr.201863.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Howe A.C., Jansson J.K., Malfatti S.A., Tringe S.G., Tiedje J.M., Brown C.T. Tackling soil diversity with the assembly of large, complex metagenomes. Proc Nat Acad Sci. 2014;111(13):4904–4909. doi: 10.1073/pnas.1402564111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ghurye J., Pop M. WABI; 2016. Better identification of repeats in metagenomic scaffolding. [Google Scholar]
- 42.Freeman L.C. A set of measures of centrality based on betweenness. Sociometry. 1977:35–41. [Google Scholar]
- 43.Brandes U. A faster algorithm for betweenness centrality. J Math Sociol. 2001;25(2):163–177. [Google Scholar]
- 44.Segarra S., Ribeiro A. Stability and continuity of centrality measures in weighted graphs. TSP. 2016;64(3):543–555. [Google Scholar]
- 45.Brown C.T., Moritz D., O’Brien M.P., Reidl F., Reiter T., Sullivan B.D. Exploring neighborhoods in large metagenome assembly graphs using spacegraphcats reveals hidden sequence diversity. Genome Biol. 2020;21(1):1–16. doi: 10.1186/s13059-020-02066-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ulyantsev V.I., Kazakov S.V., Dubinkina V.B., Tyakht A.V., Alexeev D.G. Metafast: fast reference-free graph-based comparison of shotgun metagenomic data. Bioinformatics. 2016;32(18):2760–2767. doi: 10.1093/bioinformatics/btw312. [DOI] [PubMed] [Google Scholar]
- 47.Alekseyev M.A., Pevzner P.A. Breakpoint graphs and ancestral genome reconstructions. Genome Res. 2009;19(5):943–957. doi: 10.1101/gr.082784.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin Y., Nurk S., Pevzner P.A. What is the difference between the breakpoint graph and the de bruijn graph? BMC genomics. 2014;15(6):1–11. doi: 10.1186/1471-2164-15-S6-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pevzner P.A., Tang H., Tesler G. De novo repeat classification and fragment assembly. Genome Res. 2004;14(9):1786–1796. doi: 10.1101/gr.2395204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin Y., Yuan J., Kolmogorov M., Shen M.W., Chaisson M., Pevzner P.A. Assembly of long error-prone reads using de bruijn graphs. Proc Natl Acad Sci. 2016;113(52):E8396–E8405. doi: 10.1073/pnas.1604560113. URL: https://www.pnas.org/content/113/52/E8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Turner I., Garimella K.V., Iqbal Z., McVean G. Integrating long-range connectivity information into de bruijn graphs. Bioinformatics. 2018;34(15):2556–2565. doi: 10.1093/bioinformatics/bty157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Feng Y., Beh L.Y., Chang W.-J., Landweber L.F. Sigar: Inferring features of genome architecture and dna rearrangements by split-read mapping. Genome Biol Evol. 2020;12(10):1711–1718. doi: 10.1093/gbe/evaa147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seidman S.B. Network structure and minimum degree. Social networks. 1983;5(3):269–287. [Google Scholar]
- 54.Batagelj V, Zaversnik M. An o (m) algorithm for cores decomposition of networks, arXiv preprint cs/0310049; 2003.
- 55.Gautreau G., Bazin A., Gachet M., Planel R., Burlot L., Dubois M., Perrin A., Médigue C., Calteau A., Cruveiller S., et al. Ppanggolin: depicting microbial diversity via a partitioned pangenome graph. PLoS Comput Biol. 2020;16(3) doi: 10.1371/journal.pcbi.1007732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jackman S.D., Vandervalk B.P., Mohamadi H., Chu J., Yeo S., Hammond S.A., Jahesh G., Khan H., Coombe L., Warren R.L., et al. Abyss 2.0: resource-efficient assembly of large genomes using a bloom filter. Genome Res. 2017;27(5):768–777. doi: 10.1101/gr.214346.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Langmead B., Salzberg S.L. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Csardi G., Nepusz T., et al. The igraph software package for complex network research. Int J Complex Syst. 2006;1695(5):1–9. [Google Scholar]
- 59.Dagum L., Menon R. Openmp: An industry-standard api for shared-memory programming. Comput Sci Eng. 1998;1:46–55. [Google Scholar]
- 60.Catchen J., Hohenlohe P.A., Bassham S., Amores A., Cresko W.A. Stacks: an analysis tool set for population genomics. Mol Ecol. 2013;22(11):3124–3140. doi: 10.1111/mec.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Alvarez-Hamelin JI, Dall’Asta L, Barrat A, Vespignani A. Large scale networks fingerprinting and visualization using the k-core decomposition. In Advances in neural information processing systems; 2006. pp. 41–50.
- 62.Khaouid W., Barsky M., Srinivasan V., Thomo A. K-core decomposition of large networks on a single pc. Proceedings of the VLDB Endowment. 2015;9(1):13–23. [Google Scholar]
- 63.Zhang H., Zhao H., Cai W., Liu J., Zhou W. Using the k-core decomposition to analyze the static structure of large-scale software systems. J Supercomput. 2010;53(2):352–369. [Google Scholar]
- 64.Shin K., Eliassi-Rad T., Faloutsos C. 2016 IEEE 16th International Conference on Data Mining (ICDM), IEEE. 2016. Corescope: Graph mining using k-core analysis–patterns, anomalies and algorithms; pp. 469–478. [Google Scholar]
- 65.Li H. wgsim-read simulator for next generation sequencing. Github Repository. 2011 https://github.com/lh3/wgsim. [Google Scholar]
- 66.Shakya M., Quince C., Campbell J.H., Yang Z.K., Schadt C.W., Podar M. Comparative metagenomic and rrna microbial diversity characterization using archaeal and bacterial synthetic communities. Environ Microbiol. 2013;15(6):1882–1899. doi: 10.1111/1462-2920.12086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gevers D., Knight R., Petrosino J.F., Huang K., McGuire A.L., Birren B.W., Nelson K.E., White O., Methé B.A., Huttenhower C. The human microbiome project: a community resource for the healthy human microbiome. PLoS Biol. 2012;10(8) doi: 10.1371/journal.pbio.1001377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Voigt A.Y., Costea P.I., Kultima J.R., Li S.S., Zeller G., Sunagawa S., Bork P. Temporal and technical variability of human gut metagenomes. Genome Biol. 2015;16(1):73. doi: 10.1186/s13059-015-0639-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Balaji A, Kille B, Kappell A, Godbold GD, Diep M, Leo Elworth RA, et al. Accurate and sensitive functional screening of pathogenic sequences via ensemble learning. bioRxiv. 2021 doi: 10.1101/2021.05.02.442344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Albin D., Nasko D., Elworth R.L., Lu J., Balaji A., Diaz C., Shah N., Selengut J., Hulme-Lowe C., Muthu P., et al. 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). IEEE. 2019. Seqscreen: a biocuration platform for robust taxonomic and biological process characterization of nucleic acid sequences of interest; pp. 1729–1736. [Google Scholar]
- 71.Hollister E.B., Oezguen N., Chumpitazi B.P., Luna R.A., Weidler E.M., Rubio-Gonzales M., Dahdouli M., Cope J.L., Mistretta T.-A., Raza S., et al. Leveraging human microbiome features to diagnose and stratify children with irritable bowel syndrome. J Mol Diagn. 2019;21(3):449–461. doi: 10.1016/j.jmoldx.2019.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. The sequence alignment/map format and samtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wood D.E., Lu J., Langmead B. Improved metagenomic analysis with kraken 2. Genome Biol. 2019;20(1):1–13. doi: 10.1186/s13059-019-1891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moon J.-H., Lee J.-H. Probing the diversity of healthy oral microbiome with bioinformatics approaches. BMB Rep. 2016;49(12):662. doi: 10.5483/BMBRep.2016.49.12.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Utter D.R., Borisy G.G., Eren A.M., Cavanaugh C.M., Welch J.L.M. Metapangenomics of the oral microbiome provides insights into habitat adaptation and cultivar diversity. Genome Biol. 2020;21(1):1–25. doi: 10.1186/s13059-020-02200-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wei Y., Shi M., Zhen M., Wang C., Hu W., Nie Y., Wu X. Comparison of subgingival and buccal mucosa microbiome in chronic and aggressive periodontitis: a pilot study. Front Cell Infect Microbiol. 2019;9:53. doi: 10.3389/fcimb.2019.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.O’Brien C.L., Allison G.E., Grimpen F., Pavli P. Impact of colonoscopy bowel preparation on intestinal microbiota. PloS one. 2013;8(5) doi: 10.1371/journal.pone.0062815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Goldenberg J.Z., Yap C., Lytvyn L., Lo C.K.-F., Beardsley J., Mertz D., Johnston B.C. Probiotics for the prevention of clostridium difficile-associated diarrhea in adults and children. Cochrane Database of Systematic Reviews (12) 2017 doi: 10.1002/14651858.CD006095.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Deng H., Yang S., Zhang Y., Qian K., Zhang Z., Liu Y., Wang Y., Bai Y., Fan H., Zhao X., et al. Bacteroides fragilis prevents clostridium difficile infection in a mouse model by restoring gut barrier and microbiome regulation. Front Microbiol. 2018;9:2976. doi: 10.3389/fmicb.2018.02976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Siegerstetter S.-C., Petri R.M., Magowan E., Lawlor P.G., Zebeli Q., O’Connell N.E., Metzler-Zebeli B.U. Fecal microbiota transplant from highly feed-efficient donors shows little effect on age-related changes in feed-efficiency-associated fecal microbiota from chickens. Appl Environ Microbiol. 2018;84(2) doi: 10.1128/AEM.02330-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rodriguez D.M., Benninghoff A.D., Aardema N.D., Phatak S., Hintze K.J. Basal diet determined long-term composition of the gut microbiome and mouse phenotype to a greater extent than fecal microbiome transfer from lean or obese human donors. Nutrients. 2019;11(7):1630. doi: 10.3390/nu11071630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lai Z.-L., Tseng C.-H., Ho H.J., Cheung C.K., Lin J.-Y., Chen Y.-J., Cheng F.-C., Hsu Y.-C., Lin J.-T., El-Omar E.M., et al. Fecal microbiota transplantation confers beneficial metabolic effects of diet and exercise on diet-induced obese mice. Scientific Rep. 2018;8(1):1–11. doi: 10.1038/s41598-018-33893-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ohara T. Identification of the microbial diversity after fecal microbiota transplantation therapy for chronic intractable constipation using 16s rrna amplicon sequencing. Plos one. 2019;14(3) doi: 10.1371/journal.pone.0214085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhao H.-J., Luo X., Shi Y.-C., Li J.-F., Pan F., Ren R.-R., Peng L.-H., Shi X.-Y., Yang G., Wang J., et al. The efficacy of fecal microbiota transplantation for children with tourette syndrome: A preliminary study. Front Psychiatry. 2020;11:1520. doi: 10.3389/fpsyt.2020.554441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Olekhnovich E.I., Ivanov A.B., Ulyantsev V.I., Ilina E.N. Separation of donor and recipient microbial diversity allows determination of taxonomic and functional features of gut microbiota restructuring following fecal transplantation. Msystems. 2021;6(4):e00811–21. doi: 10.1128/mSystems.00811-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.De Groot P., Nikolic T., Pellegrini S., Sordi V., Imangaliyev S., Rampanelli E., Hanssen N., Attaye I., Bakker G., Duinkerken G., et al. Faecal microbiota transplantation halts progression of human new-onset type 1 diabetes in a randomised controlled trial. Gut. 2021;70(1):92–105. doi: 10.1136/gutjnl-2020-322630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kazemian N., Ramezankhani M., Sehgal A., Khalid F.M., Kalkhoran A.H.Z., Narayan A., Wong G.K.-S., Kao D., Pakpour S. The trans-kingdom battle between donor and recipient gut microbiome influences fecal microbiota transplantation outcome. Scientific Rep. 2020;10(1):1–10. doi: 10.1038/s41598-020-75162-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Garza-González E., Mendoza-Olazarán S., Morfin-Otero R., Ramírez-Fontes A., Rodríguez-Zulueta P., Flores-Treviño S., Bocanegra-Ibarias P., Maldonado-Garza H., Camacho-Ortiz A. Intestinal microbiome changes in fecal microbiota transplant (fmt) vs. fmt enriched with lactobacillus in the treatment of recurrent clostridioides difficile infection, Canadian. J Gastroenterol Hepatol. 2019 doi: 10.1155/2019/4549298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Beghini F., McIver L.J., Blanco-Míguez A., Dubois L., Asnicar F., Maharjan S., Mailyan A., Manghi P., Scholz M., Thomas A.M., et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with biobakery 3. Elife. 2021;10 doi: 10.7554/eLife.65088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ochman H, Lawrence JG, Groisman EA. Lateral gene transfer and the nature of bacterial innovation. Nature 2000;405(6784):299. [DOI] [PubMed]
- 91.Kondrashov F.A. Gene duplication as a mechanism of genomic adaptation to a changing environment. Proc R Soc B. 2012;279(1749):5048–5057. doi: 10.1098/rspb.2012.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lloyd-Price J., Mahurkar A., Rahnavard G., Crabtree J., Orvis J., Hall A.B., Brady A., Creasy H.H., McCracken C., Giglio M.G., et al. Strains, functions and dynamics in the expanded human microbiome project. Nature. 2017;550(7674):61–66. doi: 10.1038/nature23889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Marçais Guillaume, Delcher Arthur, Phillippy Adam, Coston Rachel, Salzberg Steven, Zimin Aleksey. MUMmer4: A fast and versatile genome alignment system. PLoS computational biology. 2018;14(1) doi: 10.1371/journal.pcbi.1005944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Batagelj Vladimir, Zaveršnik Matjaž. Fast algorithms for determining (generalized) core groups in social networks. Advances in Data Analysis and Classification. 2011;5:129–145. doi: 10.1007/s11634-010-0079-y. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Python jupyter notebooks used for analysis and generating figures can be found here: https://osf.io/r6phg/ KOMB outputs and files used for analysis for the experiments can be found at https://rice.box.com/v/komb-manuscript-data.