

Summary
The deficiency of pancreatic β-cells is the key pathogenesis of diabetes, while glucagon-secreting α-cells are another player in the development of diabetes. Here, we aimed to investigate the effects of glucagon receptor (GCGR) antagonism on β-cell neogenesis in type 2 diabetic (T2D) mice and explore the origins of the neogenic β-cells. We showed that GCGR monoclonal antibody (mAb) elevated plasma insulin level and increased β-cell mass in T2D mice. By using α-cell lineage-tracing (glucagon-cre-β-gal) mice and inducible Ngn3+ pancreatic endocrine progenitor lineage-tracing (Ngn3-CreERT2-tdTomato) mice, we found that GCGR mAb treatment promoted α-cell regression to progenitors, and induced Ngn3+ progenitor reactivation and differentiation toward β-cells. Besides, GCGR mAb upregulated the expression levels of β-cell regeneration-associated genes and promoted insulin secretion in primary mouse islets, indicative of a direct effect on β-cell identity. Our findings suggest that GCGR antagonism not only increases insulin secretion but also promotes pro-α-cell-derived β-cell neogenesis in T2D mice.
Subject areas: Physiology, Cellular physiology, Endocrinology, Cell biology
Graphical abstract
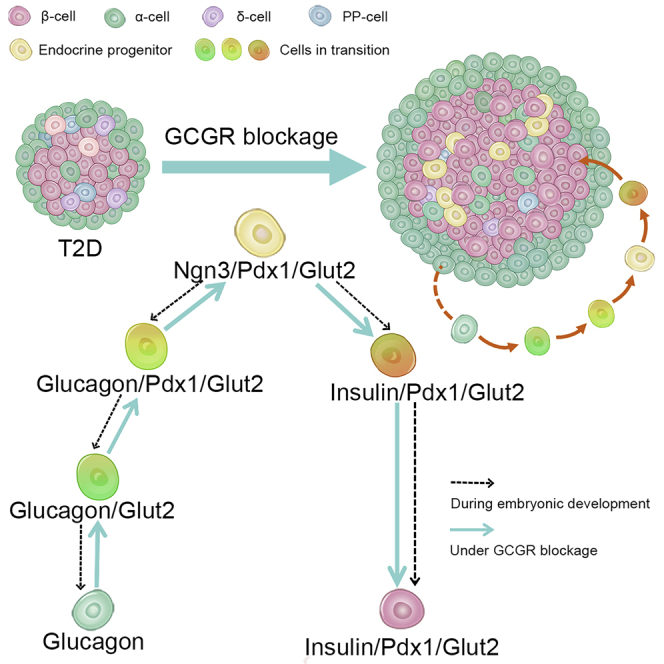
Highlights
-
•
Blockage of α-cell-derived glucagon promotes β-cell regeneration in situ in type 2 diabetic (T2D) mice
-
•
Glucagon receptor (GCGR) mAb induces the trans-differentiation of α-cells to β-cells
-
•
GCGR mAb promotes α-cell regression to pancreatic endocrine progenitors
-
•
GCGR mAb induces Ngn3+ progenitor reactivation and differentiation toward β-cells
Physiology; Cellular physiology; Endocrinology; Cell biology
Introduction
Global prevalence of diabetes has been rising in adults, and 90-95% of diabetes is type 2 diabetes (Li et al., 2020). The deficiency of pancreatic insulin-producing β-cells is an essential factor for the pathophysiology of type 2 diabetes (Kahn, 2003; Weyer et al., 1999). Restoration of functional β-cell mass is a promising therapeutic strategy. So far, however, there is almost no clinically available anti-diabetic agent that can achieve β-cell recovery. Moreover, only a few agents have shown a clinical potential to achieve β-cell regeneration. In patients with type 2 diabetes, treatment with anti-diabetic agents is difficult to maintain the long-term glycemic control on target as they cannot prevent the progressive failure of β-cell function (Turner et al., 1999). Therefore, it is urgent to develop an anti-diabetic agent which can effectively promote β-cell regeneration.
Recently, the role of glucagon-producing α-cells in glucose homeostasis regulation and diabetes development has become increasingly emphasized (Lee et al., 2014). Blockage of glucagon receptor (GCGR) lowers blood glucose level and improves glucose tolerance in type 1 diabetic (T1D) and type 2 diabetic (T2D) animals and humans (Conarello et al., 2007; Lee et al., 2011; Okamoto et al., 2017), which indicates the therapeutic potential in developing GCGR antagonists. Our recent studies have shown that REMD 2.59, an antagonistic GCGR monoclonal antibody (mAb), has a strong hypoglycemic effect in T1D and T2D mice (Lang et al., 2020b; Wei et al., 2019). Interestingly, we and other groups found that GCGR mAb promoted β-cell regeneration by inducing the trans-differentiation of α-cells to β-cells in T1D mice (Wang et al., 2021b; Wei et al., 2019). Whether GCGR mAb also has such beneficial effects on β-cell regeneration in T2D animal models has not been revealed, and the potential mechanisms remain unclear.
In this study, we investigated the effects of GCGR mAb on β-cell mass and function in two mouse models of T2D and traced the source of β-cell regeneration by using several tracing methods. We also determined whether GCGR mAb had a direct effect on islet cell phenotype conversion and β-cell function in cultured primary mouse islets. Our research reveals a novel pharmacological function of GCGR mAb on diabetes control and suggests that blockage of α-cell-derived glucagon can promote pro-α-cell-derived β-cell neogenesis, a new approach to β-cell regeneration, in T2D mice.
Results
Glucagon receptor monoclonal antibody lowers the blood glucose level and improves glucose tolerance in type 2 diabetic mice
During the 4-week treatment, no significant difference was identified between GCGR mAb and IgG control groups in terms of body weight in db/db mice and high-fat diet + streptozotocin (HFD + STZ)-induced T2D mice (Figures 1A and 1G). Compared with baseline or IgG treatment, blood glucose levels at random or fasting state were significantly declined after treatment with GCGR mAb in these two T2D mouse models (Figures 1B and 1H). GCGR mAb significantly decreased the post-load glucose levels during the intraperitoneal glucose tolerance test (IPGTT) in db/db mice (Figures 1C and 1D) and HFD + STZ-induced T2D mice (Figures 1I and 1J). After treatment with GCGR mAb, fasting plasma glucagon levels were significantly increased in these two T2D mouse models, compared with control groups (Figures 1E and 1K). Fasting plasma insulin level in GCGR mAb-treated db/db mice was higher than that in the control group (Figure 1F). Similarly, GCGR mAb had a tendency to increase fasting plasma insulin and C-peptide levels in HFD + STZ-induced T2D mice (p = 0.204 and 0.125, respectively) (Figures 1L and S1A). To further evaluate the β-cell function, we detected the insulin level during IPGTT. Results showed that GCGR mAb increased the glucose-challenged insulin levels, especially 30 min after the glucose loading (Figure S1B).
Figure 1.

Metabolic parameters and hormone levels in two T2D mouse models treated with GCGR mAb or IgG control for 4 weeks
(A–F) Parameters in db/db mice. Age-matched db/m mice treated with IgG were included as a normal control.
(G–L) Parameters in HFD + STZ-induced T2D mice.
(A and G) Body weight. (B and H) Random or fasting blood glucose. (C, I) Blood glucose during the intraperitoneal glucose tolerance test (IPGTT). Black triangle indicated 33.3 mmol/L (the upper detection limit of the glucometer). (D, J) The areas under curve (AUC) of blood glucose during the IPGTT. (E, K) Fasting plasma glucagon. (F, L) Fasting plasma insulin.n = 6 in db/m mice and n = 10 per group in db/db mice. n = 6 in control group and n = 9 in GCGR mAb group in HFD + STZ-induced T2D mice. Data represent the mean ± SEM. Statistical analysis was conducted by ANOVA followed by the post hoc Tukey-Kramer test or by Student’s t-test, as appropriate. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 vs. control; §p < 0.05, §§p < 0.01, §§§p < 0.001 vs. db/m; †p < 0.05 vs. pretreatment in the same group. See also Figure S1.
Glucagon receptor monoclonal antibody increases islet number and area, and α-cell and β-cell numbers in type 2 diabetic mice
The islet number and islet area were significantly increased by GCGR mAb in db/db mice and HFD + STZ-induced T2D mice as indicated by histological analysis of the entire pancreata (Figure S2). Notably, GCGR mAb induced a striking expansion in the number of glucagon+ α-cells and insulin+ β-cells in db/db mice compared with IgG control group and db/m mice (Figures 2A and 2B). Similarly, glucagon+ α-cell and insulin+ β-cell numbers were significantly increased in HFD + STZ-induced T2D mice after treatment with GCGR mAb (Figures 2C and 2D).
Figure 2.

Histological analysis of pancreatic α-cell number and β-cell number in the pancreatic tissues of two T2D mouse models treated with GCGR mAb or IgG control for 4 weeks
(A and C) Representative image of an islet immunostained for glucagon and insulin in db/db mice (A) and HFD + STZ-induced T2D mice (C) treated with GCGR mAb or IgG control. The arrows indicate co-labeled cells. The cells in the small box are enlarged at the right of the image. Scale bar = 50 μm.
(B and D) Quantification of the number of α-cells and β-cells per islet slice, and the ratio of β-cell number and α-cell number in db/db mice (B) and HFD + STZ-induced T2D mice (D).n = 3-5 sections/mouse multiplied by 6 mice/group in db/db mice, and n = 3-5 sections/mouse multiplied by 9 mice/group in HFD + STZ-induced T2D mice. Data are expressed as the median (interquartile range). Statistical analysis was conducted by the Mann-Whitney test. ∗∗p < 0.01, ∗∗∗p < 0.001 vs. control; §p < 0.05, §§p < 0.01 vs. db/m. See also Figure S2.
Glucagon receptor monoclonal antibody accelerates progenitor-derived β-cell neogenesis in type 2 diabetic mice
As GCGR mAb treatment increased insulin+ β-cell number and plasma insulin level, we tried to explore the possible origin of the increased β-cells. Interestingly, histological analysis in the pancreata of db/db mice and HFD/STZ-induced T2D mice showed that there were some glucagon+ cells or insulin+ cells located in the ductal region in GCGR mAb treatment groups, but they were rare in IgG control groups (Figures 3A and 3B). These findings implied that GCGR mAb might induce progenitor-derived β-cell neogenesis in T2D mice.
Figure 3.

Immunofluorescent analysis of progenitor-derived β-cell neogenesis in the pancreatic tissues of T2D mice treated with GCGR mAb or IgG control for 4 weeks
(A and B) Representative photograph showing glucagon+ cells and insulin+ cells located in the ductal region of db/db mice (A) and HFD + STZ-induced T2D mice (B). The arrows indicate glucagon+ cells or insulin+ cells in the ductal lining.
(C) Representative image of an islet immunostained with RFP (Ngn3+ cell lineage-tracing marker) and insulin in Ngn3+ cell lineage-tracing T2D mice that were induced by HFD + STZ. The arrows indicate co-labeled cells, and the ductal lumen is outlined with dashed lines. The cells in the small box are enlarged at the right of the image. Scale bar = 50 μm. See also Figures S3 and S4.
Neurogenin 3 (Ngn3), a notch-regulated transcription factor that is required for pancreatic endocrine differentiation during the development, has been shown to be also required for adult endocrine cell neogenesis (Al-Hasani et al., 2013). To confirm that GCGR mAb promoted β-cell neogenesis from pancreatic endocrine progenitors, we used inducible Ngn3+ cell lineage-tracing T2D mice, which labeled reactivated endocrine progenitors by tdTomato (RFP). As expected, there were much more RFP+insulin+ cells in GCGR mAb group than in control group (4.48 ± 0.44% vs. 1.82 ± 0.26%, p < 0.0001) (Figure 3C). These results provided strong evidence that GCGR mAb induced pancreatic endocrine progenitor reactivation and differentiation toward β-cells in T2D mice.
Glucagon receptor monoclonal antibody induced α-cell regression to the progenitor state in type 2 diabetic mice
Subsequently, we explored the source of progenitors which contributed to β-cell neogenesis. Progenitor harbored in pancreatic ducts is one source of β-cell regeneration (Bonner-Weir et al., 2008; Xu et al., 2008). In this study, we found that GCGR mAb induced the appearance of insulin+ cells located in the ductal region (Figures 3A and 3B), but the insulin+ cells were not co-immunostained with cytokeratin 19, a marker of mature duct cells (Figure S3). The progenitor lineage-tracing marker RFP+ cells were rarely located within or near pancreatic ducts in the Ngn3+ cell lineage-tracing T2D mice despite treatment with GCGR mAb or IgG control (Figure S4). These results indicated that duct-derived progenitors might not be the main source of progenitors which were responsible for β-cell neogenesis in T2D mice.
As α-cells were significantly expanded in the pancreatic tissues of these two T2D mouse models after being treated with GCGR mAb (Figure 2), we inferred that α-cells might be the source of progenitors which contributed to β-cell neogenesis. It is generally believed that during embryonic development, islet cells differentiate from pancreatic endocrine progenitors in which pancreatic and duodenal homeobox 1 (Pdx1) expression is an early step to initiate the production of glucose transporter 2 (Glut2) and Ngn3, and is subsequently turned off by Ngn3 before the initiation of islet hormone expression (Serafimidis et al., 2008; Vuguin et al., 2006). In order to confirm our inferences, we analyzed the state of α-cells in the GCGR mAb-treated T2D mice by using pancreatic α-cell lineage-tracing, which labeled pre-existing α-cells by β-galactosidase (β-gal) reporter (Figure S5). As expected, the β-gal+Ngn3+ cells were increased by GCGR mAb treatment, which suggested that Ngn3+ progenitors might derive from the α-cell regression (Figure 4A). In addition, both β-gal+Glut2+ cells and β-gal+Pdx1+ cells were also increased in the GCGR mAb group compared with IgG control group (Figures 4B and 4C). These results strongly indicated that after treatment with GCGR mAb, α-cells regressed to the immature state, and expressed the progenitor’s markers (Ngn3, Glut2, and Pdx1). Therefore, the pro-α-cell-derived progenitors appeared to be the important source of progenitors which were responsible for β-cell neogenesis in T2D mice.
Figure 4.

Immunofluorescent analysis of α-cell regression to the progenitor state in the pancreatic tissues of T2D mice treated with GCGR mAb or IgG control for 4 weeks
(A–C) Representative photograph showing β-gal (α-cell lineage-tracing marker)+Ngn3+ cells (A), β-gal+Glut2+ cells (B), and β-gal+Pdx1+ cells (C) in α-cell lineage-tracing T2D mice that were induced by HFD + STZ. The arrows indicate co-labeled cells. The cells in the small box are enlarged at the right of the image. Scale bar = 50 μm. See also Figures S5.
Glucagon receptor monoclonal antibody promotes the trans-differentiation of α-cells to β-cells in type 2 diabetic mice
We noticed that after treatment with GCGR mAb, α-cells were located not only in the islet mantle zone but also in the islet core, where β-cells are generally detected (Figures 2A and 2C). Furthermore, the numbers of glucagon and insulin co-localizing cells were much higher in GCGR mAb groups than in IgG control groups (Figures 2A, 2C, 5A, and 5B). We inferred that these glucagon+insulin+ cells were the intermediate state of α-to-β cell conversion. In support of this inference, we found that some glucagon+ cells were co-immunostained with Nkx6.1 (a transcription factor for β-cell development and function), prohormone convertase 1/3 (PC1/3, an important enzyme for processing proinsulin to insulin in β-cells), or C-peptide in these two T2D mouse models treated with GCGR mAb rather than with IgG (Figures S6 and S7).
Figure 5.

Immunofluorescent analysis of α-to-β cell conversion in the pancreatic tissues of T2D mouse models treated with GCGR mAb or IgG control for 4 weeks
(A and B) Quantification of glucagon+insulin+ cells in db/db mice (A) and HFD + STZ-induced T2D mice (B) as shown in (Figures 2A and 2C).
(C and D) Representative image of an islet immunostained with β-gal (α-cell lineage-tracing marker) and insulin (C), and quantification of β-gal+insulin+ cells (D) in α-cell lineage-tracing T2D mice that were induced by HFD + STZ. The arrows indicate co-labeled cells. The cells in the small box are enlarged at the right of the image. Scale bar = 50 μm. n = 5 sections/mouse multiplied by 6 mice/group in db/db mice, n = 3 sections/mouse multiplied by 9 mice/group in HFD + STZ-induced T2D mice, and n = 3 sections/mouse multiplied by 5 mice/group in the α-cell lineage-tracing T2D mice. Data represent the mean ± SEM. Statistical analysis was conducted by Student’s t-test. ∗∗∗p < 0.001 vs. control. See also Figures S5, S6, and S7.
To confirm that α-cells were the origins of progenitors which contributed to β-cell neogenesis, we tracked the insulin expression of α-cells in pancreatic α-cell lineage-tracing mice. As expected, some β-gal+ cells contained insulin in the islets of the GCGR mAb group. By contrast, the β-gal+insulin+ cells were scarce in the control group (Figures 5C and 5D). The results indicated that pro-α-cell-derived β-cell neogenesis or direct α-to-β cell conversion was an important origin of the regenerated β-cells induced by the GCGR antagonism in T2D mice.
Glucagon receptor monoclonal antibody enhances insulin secretion and regulates islet cell phenotype in cultured primary mouse islets
Subsequently, we tried to clarify whether GCGR mAb treatment could directly affect pancreatic β-cell function and islet cell phenotype. We first detected GCGR expression in β-cells. Results showed that GCGR was expressed in mouse β-cells at both the mRNA and protein levels (Figure S8A, S8B, and S8C), suggesting that GCGR mAb might have direct effects on β-cells. Besides, data from RNA Chip and RNA sequencing analyses indicated that GCGR expression in human and mouse β-cells were comparable between T2D and control individuals (Figures S8D and S8E). We then incubated primary mouse islets with GCGR mAb or IgG for 24 h in high glucose (25 mmol/L) medium. As expected, the intracellular glucagon content and supernatant glucagon level were higher in the islets treated with GCGR mAb than those with IgG control (Figures 6A and 6B). Although the insulin content in the GCGR mAb-treated islets was unchanged (Figure 6C), the insulin level in supernatant was significantly increased by GCGR mAb (Figure 6D). We also detected the glucose-stimulated insulin secretion in primary mouse islets treated with GCGR mAb. Results showed that GCGR mAb enhanced glucose concentration-dependent insulin secretion (Figure 6E). Furthermore, the mRNA levels of Gcg (which encodes proglucagon), Pcsk1 (which encodes PC1/3), Ins1 (which encodes insulin), Pdx1 (a master transcription factor that participates in pancreatic fate determination and β-cell maturation and function) and Ngn3 (a specific marker of pancreatic endocrine progenitors) were upregulated by GCGR mAb. By contrast, the mRNA levels of Pcsk2 (which encodes PC2, an important enzyme for processing proglucagon to glucagon) and Ins2 (which encodes insulin) were unchanged by GCGR mAb (Figure 6F). These results suggested that GCGR mAb had a direct beneficial effect on β-cell function and islet cell phenotype conversion.
Figure 6.

Hormone production and gene expression in primary mouse islets incubated with GCGR mAb (1,000 nmol/L) or IgG (as control) for 24 h in high glucose (25 mmol/L) medium
(A–D) Intracellular glucagon content (A), supernatant glucagon level (B), intracellular insulin content (C), and supernatant insulin level (D) were detected by ELISA. The hormone levels were normalized to total protein content.
(E) Glucose-stimulated insulin secretion assay in the islets after treated with GCGR mAb or IgG. The insulin levels were normalized to total protein content.
(F) Gene expression was determined by quantitative RT-PCR. The experiments were repeated 3 times. Data represent the mean ± SEM. Statistical analysis was conducted by Student’s t-test or two-way ANOVA as appropriate. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 vs. control. See also Figure S8.
Discussion
Loss of functional β-cell mass is the key pathogenesis of diabetes, while glucagon-secreting α-cells also play a role in the development of diabetes. Here, we showed that the inhibition of glucagon function by REMD 2.59, an antagonistic GCGR mAb, not only improved blood glucose control, but also promoted insulin secretion and increased β-cell mass in db/db mice and HFD + STZ-induced T2D mice. Notably, we further revealed that the GCGR mAb treatment promoted α-cell regression to the progenitor state, and induced the progenitor-derived β-cell neogenesis. Besides, the GCGR mAb enhanced insulin secretion and upregulated the expression of genes associated with β-cell regeneration in cultured primary mouse islets, indicative of a direct effect on islet cell phenotype conversion.
Restoration of functional β-cell mass is a promising therapeutic strategy. Blockage of α-cell-secreted glucagon has an effect on β-cells. In mice with severe insulin resistance induced by the insulin receptor antagonist S961, GCGR mAb not only increased the total amount of α-cells but also increased the total amount of β-cells (Okamoto et al., 2017). We and other groups demonstrated that GCGR antagonism could increase β-cell mass in T1D mice (Wang et al., 2021b; Wei et al., 2019). In this study, we showed that GCGR mAb also increased β-cell mass in T2D mice. There are several ways to increase β-cell mass, including the inhibition of β-cell apoptosis or dedifferentiation, promotion of β-cell proliferation, induction of α-to-β cell conversion, promotion of β-cell differentiation from pancreatic endocrine progenitors or stem cells (Nair et al., 2020; Path et al., 2019; Wang et al., 2021a; Wei and Hong, 2016). Here, we assessed the origins of increased β-cells induced by GCGR mAb in T2D mice and revealed a new approach to β-cell regeneration.
Ngn3+ progenitors are considered to be required for β-cell neogenesis (Wang et al., 2009). Our previous study showed that GCGR mAb induced duct-derived neogenesis in T1D mice (Wei et al., 2019). Here, we conducted a more comprehensive exploration by using Ngn3+ cell lineage-tracing mice to confirm progenitor-derived β-cell neogenesis in T2D mice. Moreover, we demonstrated that the main source of progenitors which contributed to β-cell neogenesis might be α-cell regression to the progenitor state. Notably, human islets could also regenerate from progenitors (Qadir et al., 2020; Yoneda et al., 2013). Therefore, the promoting effects of GCGR mAb on progenitor-derived β-cell neogenesis as shown in this study suggest the potential clinical application of GCGR mAb to regenerate human islets.
Cell trans-differentiation, also known as lineage reprogramming, is a new path for β-cell regeneration (Wei and Hong, 2016). Several strategies were reported to promote the trans-differentiation of α-cells to β-cells (Ben-Othman et al., 2017; Collombat et al., 2009; Courtney et al., 2013; Furuyama et al., 2019; Thorel et al., 2010). However, the extreme β-cell loss conditions and gene modification methods are difficult to be translated into clinical treatment. GABA and artemisinins were reported to induce α-to-β cell conversion (Ben-Othman et al., 2017; Li et al., 2017), albeit there were some controversial results (van der Meulen et al., 2018). Our previous studies indicated that GCGR mAb induced α-to-β cell conversion in two T1D mouse models (Wei et al., 2019). Similarly, this study showed that GCGR mAb also promoted the trans-differentiation of α-cells to β-cells in T2D mice. As α-to-β cell conversion can also occur in human islets (Ben-Othman et al., 2017), we believe that GCGR mAb may have a similar effect in T2D humans.
However, the exact process of α-to-β cell conversion is not yet clear. In this study, we investigated whether α-to-β cell conversion induced by GCGR mAb was direct or indirect trans-differentiation. We found that after treatment with GCGR mAb, α-cells could regress to the immature state, and express the progenitor’s markers (Ngn3, Glut2, and Pdx1) in pancreatic α-cell lineage-tracing T2D mice. During embryonic development, pancreatic islet cells differentiate from endocrine progenitors in which Pdx1 expression is early step for initiating the synthesis of Glut2 and Ngn3 and then is turned off by Ngn3 before the initiation of islet hormone expression (Serafimidis et al., 2008; Vuguin and Charron, 2011). It has been shown that the global deletion of Gcgr inhibits the progression of α-cells to a mature state (Vuguin et al., 2006). In addition, we also revealed that GCGR mAb promoted progenitor-derived β-cell neogenesis in Ngn3+ cell lineage-tracing T2D mice. Taken together, these results suggested that GCGR antagonism not only could directly promote α-to-β cell conversion but also induced α-cell regression to progenitors which were responsible for β-cell neogenesis, thereby contributing to β-cell regeneration in T2D mice.
In conclusion, our study demonstrates that treatment with GCGR mAb in T2D mice ameliorates hyperglycemia and increases functional β-cell mass via inducing pro-α-cell-mediated progenitor-derived β-cell neogenesis. Our study suggests that blockage of α-cell-derived glucagon promotes β-cell regeneration, and provides new insight into the clinical development of GCGR mAb in treating T2D.
Limitation of the study
It has been demonstrated that GCGR mAb displays an improved glycemic control with less need for exogenous insulin in T1D rodents and humans (Pettus et al., 2018; Wang et al., 2021b; Wei et al., 2019). This effect might be accounted for by the enhanced insulin action in both liver and skeletal muscle (Sharma et al., 2018), or by the upregulated circulating glucagon-like peptide-1 (GLP-1) level (Lang et al., 2020a, 2020b). In this study, we found that GCGR mAb increased β-cell mass and elevated plasma insulin levels in T2D mice. Whether this elevation in plasma insulin is owing to altered insulin clearance or other mediators induced by GCGR mAb needs to be clarified.
The levels of endogenous glucagon are highest in the venous drainage of pancreas and hepatic portal vein, and its principal target is liver. GCGR is predominantly expressed in liver, and also lowly expressed in other tissues, including pancreas, kidney, adipose tissue, and brain (Muller et al., 2017). Therefore, the possible mechanism of the GCGR mAb-induced β-cell regeneration may be driven by the loss of GCGR signaling in pancreas per se, or because of the upregulation of the proglucagon-related peptide production that results from loss of GCGR activity in hepatocytes. The exact mechanism deserves further in-depth investigation in the future.
The incretins GLP-1 and glucose-dependent insulinotropic polypeptide (GIP) are gut hormones that potentiate insulin secretion in a glucose-dependent manner. GLP-1 and GIP exert their insulinotropic actions through distinct G-protein-coupled receptors highly expressed on pancreatic β-cells (Campbell and Drucker, 2013). Moreover, it has been reported that GLP-1 and GIP improve β-cell survival in rodents (Drucker, 2013). In our previous studies, we demonstrated that treatment with GCGR mAb could significantly upregulate the GLP-1 production and its circulating level in T2D mice (Lang et al., 2020a, 2020b). Therefore, GLP-1 might participate in the process of β-cell regeneration induced by GCGR mAb. Genetic and pharmacological blockage of the GLP-1 receptor would help to answer the question.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-glucagon | Cell Signaling Technology | Cat#2760; RRID: AB_659831 |
| Mouse monoclonal anti-glucagon | Sigma-Aldrich | Cat#G2654; RRID: AB_259852 |
| Mouse monoclonal anti-insulin | Sigma-Aldrich | Cat#I2018; RRID: AB_260137 |
| Rabbit monoclonal anti-cytokeratin 19 | Abcam | Cat#ab52625; RRID: AB_2281020 |
| Rabbit polyclonal anti-RFP | Abcam | Cat#ab62341; RRID: AB_945213 |
| Rabbit polyclonal anti-β-gal | Abcam | Cat#ab203749; RRID: AB_2920785 |
| Mouse monoclonal anti-Ngn3 | Santa Cruz | Cat# sc-374442; RRID: AB_10988579 |
| Mouse monoclonal anti-Glut2 | Santa Cruz | Cat#sc-518022; RRID: AB_2890905 |
| Rabbit polyclonal anti-Pdx1 | Abcam | Cat#ab47267; RRID: AB_777179 |
| Rabbit monoclonal anti-Nkx6.1 | Abcam | Cat#ab221549; RRID: AB_2754979 |
| Rabbit polyclonal anti-PC1/3 | Millipore | Cat#AB10553; RRID: AB_1977441 |
| Rabbit polyclonal anti-C-peptide | Cell Signaling Technology | Cat#4593; RRID: AB_10691857 |
| Rabbit polyclonal anti-GCGR | Proteintech | Cat# 26784-1-AP; RRID: AB_2880634 |
| Mouse monoclonal anti-GAPDH | Zhongshan Biotechnology | Cat#TA-08; RRID: AB_2107448 |
| Alexa Fluor 488-conjugated AffiniPure goat polyclonal anti-rabbit IgG (H + L) | Jackson ImmunoResearch Laboratories | Cat#115-545-003; RRID: AB_2338046 |
| Alexa Fluor 594-conjugated AffiniPure goat polyclonal anti-mouse IgG (H + L) | Jackson ImmunoResearch Laboratories | Cat#115-585-003; RRID: AB_2338871 |
| Chemicals, peptides, and recombinant proteins | ||
| REMD 2.59 (a human GCGR mAb and competitive antagonist) | REMD Biotherapeutics | N/A |
| High-fat diet (HFD) | Research Diets | Cat#D12492 |
| Streptozocin (STZ) | Sigma-Aldrich | Cat#S0130 |
| Tamoxifen | Sigma-Aldrich | Cat#T5648 |
| Goat serum | Zhongshan Biotechnology | Cat#ZLI-9056 |
| DAPI | Sigma-Aldrich | Cat#D9542 |
| Trizol reagent | Thermo Fisher Scientific | Cat#15596018 |
| SYBR qPCR Mix | TOYOBO | Cat#QPS-201 |
| RIPA lysis buffer | Applygen Technologies Inc. | Cat#C1053 |
| Protease inhibitor | Applygen Technologies Inc. | Cat#P1265 |
| Phosphatase inhibitor | Applygen Technologies Inc. | Cat#P1260 |
| Critical commercial assays | ||
| Insulin ELISA kit | Millipore | Cat#EZRMI-13K |
| Glucagon ELISA kit | R&D Systems | Cat#DGCG0 |
| C-peptide ELISA kit | Millipore | Cat#EZRMCP2-21K |
| Opal 7 color manual kit | Akoya Bioscience | Cat#NEL811001KT |
| RevertAid First Strand cDNA Synthesis kit |
Thermo Fisher Scientific | Cat#K1622 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6N | Vital River Animal Center | Cat#213 |
| Mouse: db/db (BKS.Cg-Dock7m+/+Leprdb/Nju) mice | Nanjing Biomedical Research Institution of Nanjing University | Cat#T001463 |
| Mouse: Tg(Ngn3-cre/ERT2)1Able/J | The Jackson Laboratory | JAX: 028,365 |
| Mouse: B6/JGpt-Rosa26tm1(CAG−LSL-Cas9−tdTomato)/Gpt | GemPharmatech | Cat#T002249 |
| Mouse: B6. Cg-Tg (Gcg-cre)1Herr/Mmnc | Mutant Mouse Resource & Research Centers | MMRRC: 000358-UNC |
| Mouse: B6; 129-Gt (ROSA)26Sortm1Sho/J | The Jackson Laboratory | JAX: 003,504 |
| Software and algorithms | ||
| ImagePro Plus 6.0 | Media Cybernetics | N/A |
| GraphPad Prism v.7.0 | GraphPad Software Inc. | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Tianpei Hong (tpho66@bjmu.edu.cn).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Animals
All animal experimental procedures were conducted at Peking University Health Science Center and approved by the Institutional Animal Care and Use Committee. All the animals were group-housed conventionally and maintained on a 12-h light/dark cycle with free access to food and water. Eight-week-old male db/db (BKS.Cg-Dock7m+/+Leprdb/Nju) mice (Nanjing Biomedical Research Institution of Nanjing University, Nanjing, China) were used as a typical T2D model. The littermate male db/m mice served as normal controls. n = 10 per group in db/db mice, and n = 6 in db/m mice.
To generate high-fat diet + streptozotocin (HFD + STZ)-induced T2D model, 5-week-old male C57BL/6N (Vital River Animal Center, Beijing, China) or genetically modified mice were fed with HFD (fat 60%, carbohydrate 20% and protein 20%; Research Diets, New Brunswick, NJ, USA) for 3–4 months, and then were given 75 mg/kg STZ (Sigma-Aldrich, St. Louis, MO, USA) intraperitoneally. Diabetic condition was confirmed if the fasting blood glucose level was ≥11.1 mmol/L. Mice were sorted into groups having similar distributions based on body weight and blood glucose levels.
Genetically modified mice
Tg(Ngn3-cre/ERT2)1Able/J mice (expressing a tamoxifen-inducible cre recombinase directed to Ngn3-expressing cells; the Jackson Laboratory, Barr Harbor, ME, USA) and B6/JGpt-Rosa26tm1(CAG−LSL-Cas9−tdTomato)/Gpt mice (when crossed to a cre recombinase-expressing strain, tdTomato expression is observed in the cre-expressing tissues; GemPharmatech, Nanjing, China) were crossed to generate pancreatic endocrine progenitor lineage-tracing mice, namely, Ngn3-CreERT2-tdTomato mice. Male mice were selected for subsequent experiments. Tamoxifen (Sigma-Aldrich) was dissolved in corn oil (Sigma-Aldrich), and intraperitoneal injected (20 mg/day) for 5 days since 1 week before being sacrificed. n = three to four per group.
B6.Cg-Tg(Gcg-cre)1Herr/Mmnc mice (cre expression in pancreatic α-cell lineage; Mutant Mouse Resource & Research Centers, Columbia, MO, USA) and B6; 129-Gt(ROSA)26Sortm1Sho/J mice (when crossed to a cre recombinase-expressing strain, lacZ (which encodes β-gal) expression is observed in the cre-expressing tissues; the Jackson Laboratory) were crossed to generate pancreatic α-cell lineage-tracing mice, namely, glucagon-cre-β-gal mice. Male mice were selected for subsequent experiments. n = five to eight per group.
Primary mouse islet isolation and culture
Primary islets were isolated from male C57BL/6N mice aged 8–10 weeks as previously reported (Wang et al., 2014; Wei et al., 2020; Zmuda et al., 2011). Briefly, the pancreas was perfused by collagenase V (Sigma-Aldrich), and individual islets were handpicked to near 100% purity under a dissecting microscope. The islets were cultured for 24 h before treatment in the RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% (v/v) fetal bovine serum (FBS; Hyclone, Logan, UT, USA), 2 mmol/L GlutMax, 1 mmol/L sodium pyruvate, and 1% (v/v) penicillin-streptomycin (Thermo Fisher Scientific, Waltham, MA, USA).
Method details
Animals’ intervention
All mice were treated for 4 weeks via weekly intraperitoneal administration of REMD 2.59 (5 mg/kg; REMD Biotherapeutics, Camarillo, CA, USA), a human antagonistic GCGR mAb, or IgG (5 mg/kg, as control).
Glucose monitoring
Blood samples for glucose detection were collected from the tail vein, and measured by the glucose oxidase method using a hand-held OneTouch Ultra glucometer (LifeScan, Milpitas, CA, USA).
Glucose tolerance test and blood sample collection
To perform IPGTT, basal blood glucose levels were first measured after overnight fasting. A 40% (w/v) of glucose solution was administrated by intraperitoneal injection at 2 g/kg, and blood glucose levels were measured at 30, 60 and 120 min after the glucose loading. If the glucose level was higher than 33.3 mmol/L (upper detection limit of the glucometer), the value of 33.3 mmol/L was recorded.
Blood samples for hormone detection were collected from the orbital vein. Aprotinin (1 μg/mL; Sigma-Aldrich) and heparin sodium (1,000 IU/mL; Qianhong Bio-pharma, Changzhou, China) were added to each blood sample.
Immunofluorescent staining and quantification
Pancreata were fixed with 10% (v/v) neutral-buffered formalin and embedded in paraffin, and 5-μm-thick sections were prepared. For immunofluorescence, sections were incubated with primary antibodies at 4 °C overnight and secondary antibodies for 1 h at room temperature, followed by staining with DAPI. Images were captured under Leica TCS SP8 confocal fluorescence microscope (Leica Microsystems, Wetzlar, Germany) or an automatic digital slide scanner (Pannoramic MIDI, 3D HISTECH, Budapest, Hungary).
The primary antibodies were as follows: rabbit polyclonal anti-glucagon (1:800; Cell Signaling Technology, Boston, MA, USA; RRID: AB_659831), mouse monoclonal anti-glucagon (1:400; Sigma-Aldrich; RRID: AB_259852), mouse monoclonal anti-insulin (1:800; Sigma-Aldrich; RRID: AB_260137), rabbit monoclonal anti-cytokeratin 19 (1:200; Abcam, Cambridge, UK; RRID: AB_2281020), rabbit polyclonal anti-RFP (1:200; Abcam; RRID: AB_945213), rabbit polyclonal anti-β-gal (1:100; Abcam; RRID: AB_2920785), mouse monoclonal anti-Ngn3 (1:50; Santa Cruz, CA, USA; RRID: AB_ 10988579), mouse monoclonal anti-Glut2 (1:100; Santa Cruz; RRID: AB_2890905), rabbit polyclonal anti-Pdx1 (1:200; Abcam; RRID: AB_777179), rabbit monoclonal anti-Nkx6.1 (1:400; Abcam; RRID: AB_2754979), rabbit polyclonal anti-PC1/3 (1:400; Millipore, Darmstadt, Germany; RRID: AB_1977441), rabbit polyclonal anti-C-peptide (1:400; Cell Signaling Technology; RRID: AB_10691857), rabbit anti-GCGR (Proteintech, Rosemont, IL, USA; RRID: AB_2880634). The secondary antibodies were as follows: Alexa Fluor 488-conjugated AffiniPure goat polyclonal anti-rabbit IgG (H + L) (RRID: AB_2338046) and Alexa Fluor 594-conjugated AffiniPure goat polyclonal anti-mouse IgG (H + L) (RRID: AB_2338871) (both at 1:800; Jackson ImmunoResearch Laboratories, Philadelphia, PA, USA).
Opal seven color manual kit (Akoya Bioscience, Marlborough, MA, USA) was used for co-immunostaining of Nkx6.1, PC1/3 and C-peptide with glucagon, which was detected by Vectra Polaris (Akoya Bioscience).
For cell quantification in the immunofluorescent staining, three to five equally spaced sections (which covered the entire pancreas) per pancreas were imaged, and the total numbers of positive staining cells from three to nine mice per group were counted manually.
Primary mouse islet culture and intervention
Primary mouse islets were incubated with 1,000 nmol/L REMD 2.59 or human IgG for 24 h in high glucose (25 mmol/L) condition. Islets and supernatants were collected for quantitative RT-PCR analysis and hormone measurement, respectively.
Glucose-stimulated insulin secretion test
After cultured with 1,000 nmol/L REMD 2.59 or IgG for 24 h, five similarly-sized islets were selected from each group. The islets were preincubated in Krebs-Ringer bicarbonate buffer (KRBB) supplemented with 1 g/L BSA and 2.8 mmol/L glucose for 1 h at 37 °C, and then were transferred to another dish with the same buffer. After 1 h of incubation, the buffer was sampled for insulin measurement, and the islets were exposed to a 1-h 16.7 mmol/L glucose challenge followed by another sample for insulin measurement. The insulin secretion in each group was normalized to the total protein content.
Hormone measurement
Blood samples, culture supernatants and islet lysates, and KRBB buffer samples were evaluated with specific ELISA kits for detecting insulin (Millipore), C-peptide (Millipore) and glucagon (R&D System, Minneapolis, MN, USA) according to the manufacturer’s instructions.
Protein extraction and Western blot analysis
The mouse pancreatic β-cell line Min6 cells, kindly gifted by Prof. Yiming Mu from the General Hospital of the People’s Liberation Army (Beijing, China), were cultured in DMEM (25 mmol/L glucose; Invitrogen) supplemented with 15% (v/v) FBS, 2 mmol/L GlutMax and 55 μmol/L β-mercaptoethanol (Thermo Fisher Scientific). Total proteins from Min6 cells were obtained using radioimmunoprecipitation assay (RIPA) lysis buffer, which contained protease inhibitor and phosphatase inhibitor. The denatured proteins (approximately 30 μg) were separated by 12% (w/v) SDS-PAGE electrophoresis and transferred to a nitrocellulose membrane. The membranes were incubated overnight at 4 °C with the primary antibodies (both at 1:1,000 dilution): rabbit anti-GCGR (Proteintech; RRID: AB_2880634) and mouse anti-GAPDH (Zhongshan Biotechnology, Beijing, China; RRID: AB_2107448). After three washes, the blots were incubated for 1 h with RDye 800CW-conjugated goat anti-rabbit IgG or goat anti-mouse IgG (both at 1:10,000 dilutions; LICOR Biosciences, Lincoln, NE, USA). Protein bands were visualized with an Odyssey 290 infrared imaging system (LICOR Biosciences). GAPDH was used as a loading control.
RNA extraction and reverse transcription
Total RNA was extracted with Trizol reagent (Thermo Fisher Scientific) and reversely transcribed to cDNA using a RevertAid First Strand cDNA Synthesis kit (Thermo Fisher Scientific).
Conventional and quantitative RT-PCR
The cDNA was subjected to either the conventional RT-PCR analysis using Taq PCR Mastermix (Tiangen biotechnology, Beijing, China) on an Applied Biosystems Veriti 96 well Thermal Cycler PCR detection system (Thermo Fisher Scientific) or the quantitative RT-PCR analysis using iQ SYBR Green supermix (BioRad Laboratories, Hercules, CA, USA) on a QuantStudio five Real-Time PCR System (Thermo Fisher Scientific). Relative quantification for gene expression was calculated using the 2−ΔΔCT method, which was normalized to the internal reference, β-actin. The primer sequences were summarized in Table S1.
Quantification and statistical analysis
Data are presented as mean ± SEM or median (interquartile range). All statistical analyses were performed using GraphPad Prism v.7.0 (GraphPad Software Inc., San Diego, CA, USA). Statistical significance was defined as p < 0.05 and determined by Student’s t tests, one-way or two-way ANOVA followed by the post hoc Tukey-Kramer test, or Mann-Whitney test, when appropriate.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81830022, 81970671, 81900789, and 82170875), China Postdoctoral Science Foundation (2020M670068, and 2019M660369), and Peking University Medicine Fund of Fostering Young Scholars’ Scientific & Technological Innovation.
Author contributions
Conceptualization: X.C., R.W., and T.H.; methodology: X.C., J.F., T.W., L.G., D.W., S.L., and K.Y.; software: T.W.; validation: J.F. and T.W.; formal analysis: X.C., J.F., R.W., and T.H.; investigation: X.C., J.F., D.W., S.L., K.Y., and J.Y.; resources: H.Y.; writing – original draft: X.C. and J.F.; writing – review & editing: R.W. and T.H.; visualization: X.C., J.F. and L.G.; supervision: J.Y., H.Y., R.W., and T.H.; project administration: R.W. and T.H.; funding acquisition: X.C., L.G., R.W., and T.H.
Declaration of interests
Hai Yan is a shareholder of REMD Biotherapeutics. The authors have no additional financial interests.
Published: July 15, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104567.
Contributor Information
Rui Wei, Email: weirui@bjmu.edu.cn.
Tianpei Hong, Email: tpho66@bjmu.edu.cn.
Supplemental information
Data and code availability
Any additional information required to reanalyze the data reported in this paper is available from the lead contact Tianpei Hong (tpho66@bjmu.edu.cn) upon request.
References
- Al-Hasani K., Pfeifer A., Courtney M., Ben-Othman N., Gjernes E., Vieira A., Druelle N., Avolio F., Ravassard P., Leuckx G., et al. Adult duct-lining cells can reprogram into beta-like cells able to counter repeated cycles of toxin-induced diabetes. Dev. Cell. 2013;26:86–100. doi: 10.1016/j.devcel.2013.05.018. [DOI] [PubMed] [Google Scholar]
- Ben-Othman N., Vieira A., Courtney M., Record F., Gjernes E., Avolio F., Hadzic B., Druelle N., Napolitano T., Navarro-Sanz S., et al. Long-term GABA administration induces alpha cell-mediated beta-like cell neogenesis. Cell. 2017;168:73–85.e11. doi: 10.1016/j.cell.2016.11.002. [DOI] [PubMed] [Google Scholar]
- Bonner-Weir S., Inada A., Yatoh S., Li W.C., Aye T., Toschi E., Sharma A. Transdifferentiation of pancreatic ductal cells to endocrine beta-cells. Biochem. Soc. Trans. 2008;36:353–356. doi: 10.1042/BST0360353. [DOI] [PubMed] [Google Scholar]
- Campbell J.E., Drucker D.J. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 2013;17:819–837. doi: 10.1016/j.cmet.2013.04.008. [DOI] [PubMed] [Google Scholar]
- Collombat P., Xu X., Ravassard P., Sosa-Pineda B., Dussaud S., Billestrup N., Madsen O.D., Serup P., Heimberg H., Mansouri A. The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into alpha and subsequently beta cells. Cell. 2009;138:449–462. doi: 10.1016/j.cell.2009.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conarello S.L., Jiang G., Mu J., Li Z., Woods J., Zycband E., Ronan J., Liu F., Roy R.S., Zhu L., et al. Glucagon receptor knockout mice are resistant to diet-induced obesity and streptozotocin-mediated beta cell loss and hyperglycaemia. Diabetologia. 2006;50:142–150. doi: 10.1007/s00125-006-0481-3. [DOI] [PubMed] [Google Scholar]
- Courtney M., Gjernes E., Druelle N., Ravaud C., Vieira A., Ben-Othman N., Pfeifer A., Avolio F., Leuckx G., Lacas-Gervais S., et al. The inactivation of Arx in pancreatic alpha-cells triggers their neogenesis and conversion into functional beta-like cells. PLoS Genet. 2013;9 doi: 10.1371/journal.pgen.1003934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drucker D.J. Incretin action in the pancreas: potential promise, possible perils, and pathological pitfalls. Diabetes. 2013;62:3316–3323. doi: 10.2337/db13-0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuyama K., Chera S., van Gurp L., Oropeza D., Ghila L., Damond N., Vethe H., Paulo J.A., Joosten A.M., Berney T., et al. Diabetes relief in mice by glucose-sensing insulin-secreting human alpha-cells. Nature. 2019;567:43–48. doi: 10.1038/s41586-019-0942-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn S.E. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia. 2003;46:3–19. doi: 10.1007/s00125-002-1009-0. [DOI] [PubMed] [Google Scholar]
- Lang S., Wei R., Wei T., Gu L., Feng J., Yan H., Yang J., Hong T. Glucagon receptor antagonism promotes the production of gut proglucagon-derived peptides in diabetic mice. Peptides. 2020;131 doi: 10.1016/j.peptides.2020.170349. [DOI] [PubMed] [Google Scholar]
- Lang S., Yang J., Yang K., Gu L., Cui X., Wei T., Liu J., Le Y., Wang H., Wei R., Hong T. Glucagon receptor antagonist upregulates circulating GLP-1 level by promoting intestinal L-cell proliferation and GLP-1 production in type 2 diabetes. BMJ Open Diabetes Res. Care. 2020;8 doi: 10.1136/bmjdrc-2019-001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y., Berglund E.D., Yu X., Wang M.Y., Evans M.R., Scherer P.E., Holland W.L., Charron M.J., Roth M.G., Unger R.H. Hyperglycemia in rodent models of type 2 diabetes requires insulin-resistant alpha cells. Proc. Natl. Acad. Sci. USA. 2014;111:13217–13222. doi: 10.1073/pnas.1409638111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y., Wang M.Y., Du X.Q., Charron M.J., Unger R.H. Glucagon receptor knockout prevents insulin-deficient type 1 diabetes in mice. Diabetes. 2011;60:391–397. doi: 10.2337/db10-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Casteels T., Frogne T., Ingvorsen C., Honore C., Courtney M., Huber K.V.M., Schmitner N., Kimmel R.A., Romanov R.A., et al. Artemisinins target GABAA receptor signaling and impair alpha cell identity. Cell. 2017;168:86–100.e15. doi: 10.1016/j.cell.2016.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Teng D., Shi X., Qin G., Qin Y., Quan H., Shi B., Sun H., Ba J., Chen B., et al. Prevalence of diabetes recorded in mainland China using 2018 diagnostic criteria from the American Diabetes Association: national cross sectional study. BMJ. 2020;369:m997. doi: 10.1136/bmj.m997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller T.D., Finan B., Clemmensen C., DiMarchi R.D., Tschöp M.H. The new biology and pharmacology of glucagon. Physiol. Rev. 2017;97:721–766. doi: 10.1152/physrev.00025.2016. [DOI] [PubMed] [Google Scholar]
- Nair G.G., Tzanakakis E.S., Hebrok M. Emerging routes to the generation of functional beta-cells for diabetes mellitus cell therapy. Nat. Rev. Endocrinol. 2020;16:506–518. doi: 10.1038/s41574-020-0375-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto H., Cavino K., Na E., Krumm E., Kim S.Y., Cheng X., Murphy A.J., Yancopoulos G.D., Gromada J. Glucagon receptor inhibition normalizes blood glucose in severe insulin-resistant mice. Proc. Natl. Acad. Sci. USA. 2017;114:2753–2758. doi: 10.1073/pnas.1621069114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Päth G., Perakakis N., Mantzoros C.S., Seufert J. Stem cells in the treatment of diabetes mellitus - focus on mesenchymal stem cells. Metabolism. 2019;90:1–15. doi: 10.1016/j.metabol.2018.10.005. [DOI] [PubMed] [Google Scholar]
- Pettus J., Reeds D., Cavaiola T.S., Boeder S., Levin M., Tobin G., Cava E., Thai D., Shi J., Yan H., et al. Effect of a glucagon receptor antibody (REMD-477) in type 1 diabetes: a randomized controlled trial. Diabetes Obes. Metab. 2018;20:1302–1305. doi: 10.1111/dom.13202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qadir M.M.F., Alvarez-Cubela S., Klein D., van Dijk J., Muñiz-Anquela R., Muniz-Anquela R., Moreno-Hernández Y.B., Moreno-Hernandez Y.B., Lanzoni G., Sadiq S., et al. Single-cell resolution analysis of the human pancreatic ductal progenitor cell niche. Proc. Natl. Acad. Sci. USA. 2020;117:10876–10887. doi: 10.1073/pnas.1918314117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafimidis I., Rakatzi I., Episkopou V., Gouti M., Gavalas A. Novel effectors of directed and Ngn3-mediated differentiation of mouse embryonic stem cells into endocrine pancreas progenitors. Stem Cells. 2008;26:3–16. doi: 10.1634/stemcells.2007-0194. [DOI] [PubMed] [Google Scholar]
- Sharma A.X., Quittner-Strom E.B., Lee Y., Johnson J.A., Martin S.A., Yu X., Li J., Lu J., Cai Z., Chen S., et al. Glucagon receptor antagonism improves glucose metabolism and cardiac function by promoting AMP-mediated protein kinase in diabetic mice. Cell Rep. 2018;22:1760–1773. doi: 10.1016/j.celrep.2018.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorel F., Népote V., Avril I., Kohno K., Desgraz R., Chera S., Herrera P.L. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature. 2010;464:1149–1154. doi: 10.1038/nature08894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner R.C., Cull C.A., Frighi V., Holman R.R. Glycemic control with diet, sulfonylurea, metformin, or insulin in patients with type 2 diabetes mellitus: progressive requirement for multiple therapies (UKPDS 49). UK Prospective Diabetes Study (UKPDS) Group. JAMA. 1999;281:2005–2012. doi: 10.1001/jama.281.21.2005. [DOI] [PubMed] [Google Scholar]
- van der Meulen T., Lee S., Noordeloos E., Donaldson C.J., Adams M.W., Noguchi G.M., Mawla A.M., Huising M.O. Artemether does not turn alpha cells into beta cells. Cell Metab. 2018;27:218–225.e4. doi: 10.1016/j.cmet.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuguin P.M., Charron M.J. Novel insight into glucagon receptor action: lessons from knockout and transgenic mouse models. Diabetes Obes. Metab. 2011;13(Suppl 1(0 1)):144–150. doi: 10.1111/j.1463-1326.2011.01447.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuguin P.M., Kedees M.H., Cui L., Guz Y., Gelling R.W., Nejathaim M., Charron M.J., Teitelman G. Ablation of the glucagon receptor gene increases fetal lethality and produces alterations in islet development and maturation. Endocrinology. 2006;147:3995–4006. doi: 10.1210/en.2005-1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K.L., Tao M., Wei T.J., Wei R. Pancreatic beta cell regeneration induced by clinical and preclinical agents. World J. Stem Cells. 2021;13:64–77. doi: 10.4252/wjsc.v13.i1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Liu Y., Yang J., Zhao H., Ke J., Tian Q., Zhang L., Wen J., Wei R., Hong T. GLP-1 analog liraglutide enhances proinsulin processing in pancreatic beta-cells via a PKA-dependent pathway. Endocrinology. 2014;155:3817–3828. doi: 10.1210/en.2014-1218. [DOI] [PubMed] [Google Scholar]
- Wang M.Y., Dean E.D., Quittner-Strom E., Zhu Y., Chowdhury K.H., Zhang Z., Zhao S., Li N., Ye R., Lee Y., et al. Glucagon blockade restores functional beta-cell mass in type 1 diabetic mice and enhances function of human islets. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2022142118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Jensen J.N., Seymour P.A., Hsu W., Dor Y., Sander M., Magnuson M.A., Serup P., Gu G. Sustained Neurog3 expression in hormone-expressing islet cells is required for endocrine maturation and function. Proc. Natl. Acad. Sci. USA. 2009;106:9715–9720. doi: 10.1073/pnas.0904247106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei R., Cui X., Feng J., Gu L., Lang S., Wei T., Yang J., Liu J., Le Y., Wang H., et al. Dapagliflozin promotes beta cell regeneration by inducing pancreatic endocrine cell phenotype conversion in type 2 diabetic mice. Metabolism. 2020;111 doi: 10.1016/j.metabol.2020.154324. [DOI] [PubMed] [Google Scholar]
- Wei R., Gu L., Yang J., Yang K., Liu J., Le Y., Lang S., Wang H., Thai D., Yan H., Hong T. Antagonistic glucagon receptor antibody promotes alpha-cell proliferation and increases beta-cell mass in diabetic mice. iScience. 2019;16:326–339. doi: 10.1016/j.isci.2019.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei R., Hong T. Lineage reprogramming: a promising road for pancreatic beta cell regeneration. Trends. Endocrinol. Metab. 2016;27:163–176. doi: 10.1016/j.tem.2016.01.002. [DOI] [PubMed] [Google Scholar]
- Weyer C., Bogardus C., Mott D.M., Pratley R.E. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J. Clin. Invest. 1999;104:787–794. doi: 10.1172/JCI7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., D'Hoker J., Stangé G., Bonné S., De Leu N., Xiao X., Van de Casteele M., Mellitzer G., Ling Z., Pipeleers D., et al. β cells can Be generated from endogenous progenitors in injured adult mouse pancreas. Cell. 2008;132:197–207. doi: 10.1016/j.cell.2007.12.015. [DOI] [PubMed] [Google Scholar]
- Yoneda S., Uno S., Iwahashi H., Fujita Y., Yoshikawa A., Kozawa J., Okita K., Takiuchi D., Eguchi H., Nagano H., et al. Predominance of beta-cell neogenesis rather than replication in humans with an impaired glucose tolerance and newly diagnosed diabetes. J. Clin. Endocrinol. Metab. 2013;98:2053–2061. doi: 10.1210/jc.2012-3832. [DOI] [PubMed] [Google Scholar]
- Zmuda E.J., Powell C.A., Hai T. A method for murine islet isolation and subcapsular kidney transplantation. J. Vis. Exp. 2011;50 doi: 10.3791/2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Any additional information required to reanalyze the data reported in this paper is available from the lead contact Tianpei Hong (tpho66@bjmu.edu.cn) upon request.