

Abstract
Atherosclerosis is a chronic inflammatory disease driven by hypercholesterolemia. During aging, T cells accumulate cholesterol, potentially affecting inflammation. However, the effect of cholesterol efflux pathways mediated by ATP-binding cassette A1 and G1 (ABCA1/ABCG1) on T cell-dependent age-related inflammation and atherosclerosis remains poorly understood. In this study, we generate mice with T cell-specific Abca1/Abcg1-deficiency on the low-density-lipoprotein-receptor deficient (Ldlr−/−) background. T cell Abca1/Abcg1-deficiency decreases blood, lymph node, and splenic T cells, and increases T cell activation and apoptosis. T cell Abca1/Abcg1-deficiency induces a premature T cell aging phenotype in middle-aged (12–13 months) Ldlr−/− mice, reflected by upregulation of senescence markers. Despite T cell senescence and enhanced T cell activation, T cell Abca1/Abcg1-deficiency decreases atherosclerosis and aortic inflammation in middle-aged Ldlr−/− mice, accompanied by decreased T cells in atherosclerotic plaques. We attribute these effects to T cell apoptosis downstream of T cell activation, compromising T cell functionality. Collectively, we show that T cell cholesterol efflux pathways suppress T cell apoptosis and senescence, and induce atherosclerosis in middle-aged Ldlr−/− mice.
Subject terms: Atherosclerosis, Mechanisms of disease, Lymphocyte differentiation, T cells
Cholesterol efflux is mediated by specific transporters in T cells. Here the authors show that when the ABCA1/ABCG1 cholesterol transporters are absent, peripheral T cell numbers are reduced but activation increased with a premature aging phenotype of T cell senescence and apoptosis in middle aged Ldlr−/− mice.
Introduction
Atherosclerosis is a lipid-driven chronic inflammatory disease of the large- and mid-sized arteries that can lead to myocardial infarction or stroke1. T cells are key contributors to atherosclerosis, by secreting pro- and anti-inflammatory cytokines that affect plaque formation2,3. While initially most T cells in atherosclerotic plaques were reported to be of the T helper 1 (Th1) phenotype that express the transcription factor Tbet and are pro-atherogenic due to secretion of the cytokines interferon γ (IFN-γ) and tumor necrosis factor (TNF), later studies have shown that regulatory T cells (Treg) can be anti-atherogenic by secreting transforming growth factor beta (TGF-β) and interleukin 10 (IL-10)2,4. More recent studies revealed that during atherosclerosis and cardiovascular disease (CVD), Treg cells acquire pro-inflammatory characteristics of Th1 and T follicular helper (TFH) cells5–7. Feeding a cholesterol-rich Western-type diet (WTD) to mice deficient in apolipoprotein E (Apoe−/−) for 15 weeks induces a phenotypic switch from Treg cells to Th1 and TFH cells5. This phenotypic switch was prevented by injections of apolipoprotein A-I (apoA-I), which stimulates cholesterol efflux. Hence, these data suggest that cholesterol accumulation renders Treg cells pro-inflammatory5. Similarly, increased plasma membrane cholesterol accumulation in CD8+ T cells deficient in the enzyme Acetyl-CoA cholesterol Acyltransferase 1 (ACAT1) that esterifies cholesterol, enhances differentiation of naïve T cells into IFN-γ producing T cells8, presumably with pro-atherogenic effects. However, a recent study showed that in advanced atherosclerosis induced by 20 weeks of WTD feeding in Apoe−/− mice, IFN-γ was decreased in CD4+Tbet+(Th1) cells, suggesting that CD4+ T cell functionality is compromised in advanced atherosclerosis9, when these T cells accumulate cholesterol5. While exacerbated cholesterol accumulation in T cells thus has mixed effects on T cell inflammation, its consequences for atherogenesis have not been studied directly.
T cells from aged (~73–76 years old) compared to young (~22–23 years old) humans show increased membrane cholesterol accumulation10,11. During aging, the number of naïve T cells decreases, while activated T cells that show features of cellular senescence and that secrete pro-inflammatory cytokines increase12,13. This process may contribute to inflammaging14, the pro-inflammatory state that develops during aging. Aging is a major risk factor for atherosclerosis, presumably due to inflammaging15. Although thymic atrophy may contribute to the decrease in naïve T cells and increase in activated T cells with features of cellular senescence that promote inflammaging16, data in mice with T cell Acat1 deficiency or Apoe deficiency5,8,9 suggest that cholesterol accumulation in T cells during aging may also directly affect secretion of pro-inflammatory cytokines.
The cholesterol transporters ATP Binding Cassette A1 and G1 (ABCA1 and ABCG1) mediate cholesterol efflux to apoA-I and high-density-lipoprotein (HDL), respectively17–19. Previous studies have shown that activation of the T cell receptor (TCR) by αCD3, which increases T cell proliferation, decreases expression of Abca1 and Abcg1 by >90%20, leading to T cell plasma membrane cholesterol accumulation20,21. It has been suggested that Abcg1 is the most highly expressed cholesterol transporter in T cells20. T cell Abcg1 deficiency increases differentiation of naïve T cells into Treg cells, which suppresses atherosclerosis22. Even though it has been reported that Abca1 expression in T cells is low20, Abcg1−/− T cells show a 6-fold increase in Abca1 expression23, suggesting that similar to macrophages24, the cholesterol transporters ABCA1 and ABCG1 show mutual compensation in T cells.
Here we show that combined T cell Abca1/Abcg1 deficiency, which induces T cell cholesterol and cholesteryl ester (CE) accumulation by ~2.5-fold, decreases peripheral T cell numbers, and induces T cell activation and T cell apoptosis in wild-type and Ldlr−/− mice. T cell Abca1/Abcg1 deficiency decreases T cell functionality in terms of fighting pathogens and mounting an efficient immune response, presumably due to increased T cell apoptosis. Further, T cell Abca1/Abcg1 deficiency induces a premature T cell aging phenotype, reflected by upregulation of senescence markers. While not affecting atherogenesis in young Ldlr−/− mice, T cell Abca1/Abcg1 deficiency decreases atherogenesis in middle-aged (~12–13 months old) Ldlr−/− mice fed a chow diet, accompanied by decreased T cells in atherosclerotic plaques and a decrease in aortic inflammation, which we attribute to increased T cell apoptosis. These data reveal that cholesterol efflux pathways suppress T cell apoptosis, which promotes T cell functionality, but increases atherogenesis during aging.
Results
T cell Abca1/Abcg1 Deficiency Increases Cholesterol Accumulation
We generated a T cell specific Abca1/Abcg1-deficient mouse model by crossbreeding Abca1fl/flAbcg1fl/fl mice with mice expressing the T cell specific CD4Cre promoter. The CD4Cre promoter starts to be expressed at the CD4+CD8+ double positive (DP) stage of thymic T cell maturation and results in deletion of loxP flanked genes in DP and CD4+ or CD8+ single positive (SP) thymocytes25. To assess the deletion of Abca1 and Abcg1 in T cells, we isolated splenic T cells, since the yield of thymic SP thymocytes was too low to properly assess the expression of these transporters. In CD4CreAbca1fl/flAbcg1fl/fl splenic T cells, Abca1 and Abcg1 mRNA expression were decreased by >90% compared to Abca1fl/flAbcg1fl/fl T cells (Supplementary Fig. 1a). To assess the effect of T cell ABCA1 and ABCG1 mediated cholesterol efflux pathways on atherosclerosis, CD4CreAbca1fl/flAbcg1fl/fl mice were crossbred with Ldlr−/− mice to generate CD4CreAbca1fl/flAbcg1fl/flLdlr−/− mice and Abca1fl/flAbcg1fl/flLdlr−/− controls. We refer to these mice as T-AbcdkoLdlr−/− and Ldlr−/− mice, respectively.
To assess the functional consequences of T cell Abca1/Abcg1 deficiency, we performed filipin staining to measure free cholesterol accumulation. T cell Abca1/Abcg1 deficiency increased filipin staining in thymic DP, CD4+, and CD8+ SP cells (Supplementary Fig. 1b–d), and in CD4+ and CD8+ T cells in blood (Supplementary Fig. 1e–g). We next assessed the cellular localization of cholesterol in splenic T cells by confocal microscopy. T cell Abca1/Abcg1 deficiency increased filipin staining at the plasma membrane, reflecting cholesterol accumulation, in Ldlr−/− and Ldlr+/+ T cells (Supplementary Fig. 2a, b). We also observed increased staining of choleratoxin B, suggestive of increased ganglioside GM1, a component of cholesterol-enriched lipid rafts (Supplementary Fig. 2c–e).
To validate the findings on cholesterol accumulation, we performed Gas Chromatography – Mass Spectrometry (GC-MS). T cell Abca1/Abcg1 deficiency increased total cholesterol by 2.5-fold, reflected by increases in free cholesterol and cholesteryl esters (CE) (Supplementary Fig. 2f). In line with CE accumulation, T cell Abca1/Abcg1 deficiency increased Oil Red O staining, reflecting lipid droplets in para-aortic lymph nodes (LNs) (Supplementary Fig. 2g), and BODIPY 493/503 staining in Ldlr−/− and Ldlr+/+ T cells (Supplementary Fig. 3a, b). We also observed BODIPY 493/503 staining reflecting lipid droplets, in Abcg1-deficient T cells, in line with CE accumulation data as shown in23, but not in Abca1-deficient T cells or controls (Supplementary Fig. 3c). To assess whether specific cell organelles were affected in Abca1/Abcg1-deficient T cells, we performed transmission electron microscopy. We confirmed the presence of large lipid droplets in Abca1/Abcg1-deficient CD4+ and CD8+ T cells, and otherwise observed no overt differences (Supplementary Fig. 3d, e). Previous studies have shown that Abcg1 deficiency increases sterol regulatory element binding transcription factor 1 (Srebf1) mRNA expression in CD4+ T cells, while decreasing Ldlr, 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (Hmgcr), HMG-CoA synthase (Hmgcs), and Srebf2 mRNA expression, reflecting a decrease in cholesterol synthesis, consistent with increased cholesterol accumulation in the ER23,26. In line with these data, T cell Abca1/Abcg1 deficiency moderately decreased Srebf2 mRNA expression in CD4+ T cells and Ldlr and Hmgcr mRNA expression in CD8+ T cells (Supplementary Fig. 4a, b).
Collectively, previous studies have shown that Abcg1 deficiency increases membrane cholesterol22 and choleratoxin B staining reflecting increased lipid rafts as well as CE in T cells23. We here show that T cell Abcg1 deficiency increases intracellular lipid droplets, consistent with CE accumulation, while Abca1 deficiency shows no effect, in line with Abcg1 being the predominant T cell cholesterol transporter20. However, Abcg1 deficiency in T cells increases Abca1 expression by 6-fold23, suggesting that Abca1 may contribute to cholesterol accumulation in the setting of Abcg1 deficiency. Consistently, we found that combined deficiency of Abca1 and Abcg1 in T cells increased both free and esterified cholesterol, reflected by plasma membrane cholesterol accumulation and presence of lipid droplets, which was independent of Ldlr expression.
T cell Abca1/Abcg1 deficiency leads to plasma membrane stiffening
High concentrations of plasma membrane cholesterol increase plasma membrane stiffness in model membranes and cells27. To examine whether plasma membrane stiffness was affected by T cell Abca1/Abcg1 deficiency, we stained T cells with the fluorescent dye BODIPY C10 and performed Fluorescence-Lifetime Imaging Microscopy (FLIM). BODIPY C10 is a molecular rotor that has stiffness-dependent fluorescence lifetime28. When BODIPY C10 binds to a fluid membrane, it shows high rotational speed which decreases its fluorescence lifetime, while binding to a stiff membrane leads to increased fluorescence lifetime28. Abca1/Abcg1-deficient CD4+ and CD8+ T cells showed increased BODIPY C10 fluorescence lifetime compared to control CD4+ and CD8+ T cells (Fig. 1a, b and Supplementary Fig. 5a–d), suggesting that T cell Abca1/Abcg1 deficiency increased cell stiffness. In Abca1/Abcg1-deficient CD4+ and CD8+ T cells, structures that resemble CE-rich lipid droplets showed yellow staining, which reflects very high fluorescence lifetime (Fig. 1a, b). When we excluded these from analysis, BODIPY C10 fluorescence lifetime was still increased in Abca1/Abcg1-deficient CD4+ and CD8+ T cells compared to control CD4+ and CD8+ T cells (Fig. 1c–f). These data suggest that cholesterol accumulation in Abca1/Abcg1-deficient T cells leads to plasma membrane stiffening.
Fig. 1. T cell Abca1/Abcg1 deficiency leads to plasma membrane stiffening.

Ldlr−/− and T-AbcdkoLdlr−/− mice were fed a chow diet. Spleens were collected, CD4+ and CD8+ T cells were isolated, stained with BODIPY C10, and analyzed by Fluorescence-Lifetime Imaging Microscopy (FLIM). Representative microscopy pictures for CD4+ (a) and CD8+ (b) T cells. Scale bar represents picosec (ps). Representative fluorescence lifetime decay curves for CD4+ (c) and CD8+ (d) T cells. LDs, lipid droplets. Dashed lines represent fits with mono-exponential decay functions convoluted with the instrument response function (IRF). BODIPY C10 fluorescence lifetime (determined by fitting the fluorescence lifetime decay curves with a mono-exponential decay function) for the plasma membrane of (e) CD4+ (p = 0.00009) and (f) CD8+ (p = 0.013) T cells (without LDs). Per mouse, 43–76 CD4+ and CD8+ T cells were analyzed. n = 4 Ldlr−/− and n = 4 T-AbcdkoLdlr−/− mice. For all panels, error bars represent SEM. Biologically independent samples were included. p value was determined by unpaired two-tailed Student’s t test. *p < 0.05, ***p < 0.001. Source data are provided as a Source Data file.
Effects of T cell Abca1/Abcg1 deficiency on peripheral T cells and T cell activation
LckCreAbca1fl/flAbcg1fl/fl mice with T cell specific Abca1/Abcg1 deficiency have decreased thymic CD4+ and CD8+ SP cells compared to littermate controls and decreased splenic CD4+ and CD8+ T cells29. We thus assessed whether T cell Abca1/Abcg1 deficiency affected the number of T cells in T-AbcdkoLdlr−/− mice. While single T cell Abca1 or Abcg1 deficiency did not affect T cells as a percentage of total blood leukocytes or after correction for total blood leukocyte counts, as measures for T cell concentration in blood (Supplementary Fig. 6a–c), combined T cell Abca1/Abcg1 deficiency decreased blood CD4+ and CD8+ T cells by ~50% in Ldlr−/− (Fig. 2a–c and Supplementary Fig. 6a) and Ldlr+/+ mice (Supplementary Fig. 6d). Because of these decreases, we evaluated thymic cell populations. T cell Abca1/Abcg1 deficiency did not affect thymic weight (Supplementary Fig. 7a). T-AbcdkoLdlr−/− mice showed no changes in thymic CD4−CD8− double negative (DN) cells, thymic CD4+CD8+ DP cells, or thymic CD4+ or CD8+ SP cell populations compared to littermate controls, when shown as percentage of thymocytes, or after correction for total thymic cell numbers (Supplementary Fig. 1b and Supplementary Fig. 7b–d), even though TCRβ+CD24− and TCRβ+CD69− cells, indicative of excessive negative selection30, were decreased compared to Ldlr−/− controls as a percentage of thymocytes (Supplementary Fig. 7e–k). Also, unlike LckCreAbcg1fl/fl mice that showed an increase in thymic Treg cells22, thymic Treg cells were not different between T-AbcdkoLdlr−/− mice and controls (Supplementary Fig. 7l–o). We next examined T cell numbers in spleen and para-aortic LNs. Similar to observations in blood, we found that T cell Abca1/Abcg1 deficiency decreased splenic and para-aortic LN CD4+ and CD8+ T cells by ~50–70% (Fig. 2d–g). Together these data indicate that T cell Abca1/Abcg1 deficiency decreases blood, splenic, and para-aortic LN T cells without affecting thymic DN, DP, CD4+, or CD8+ SP cells.
Fig. 2. T cell Abca1/Abcg1 deficiency decreases blood, splenic, and para-aortic lymph node T cells and increases T cell activation.

Ldlr−/− and T-AbcdkoLdlr−/− mice were fed a chow diet. Blood, spleens, and para-aortic lymph nodes (LNs) were collected. Cells were stained with the indicated antibodies and analyzed by flow cytometry. a Representative flow cytometry plots of T cell receptor β (TCRβ)+ (total T cells), CD4+, and CD8+ T cells gated as in Supplementary Fig. 6a. b Quantification of total (p < 0.000001), CD4+ (p < 0.000001), and CD8+ (p < 0.000001) T cells as percentage of total white blood cells (leukocytes). c Total (p < 0.000001), CD4+ (p < 0.000001), and CD8+ (p < 0.000001) T cell concentration in blood after correction for total blood leukocyte counts. n = 14 Ldlr−/− and n = 15 T-AbcdkoLdlr−/− mice. Total (p = 0.00039), CD4+ (p = 0.0030), and CD8+ (p = 0.00024) T cells as a percentage of total splenic cells (d) and total (p = 0.0087), CD4+ (p = 0.0420), and CD8+ (p = 0.00085) T cells as cells/spleen after correction for total splenic cell number (e). Total (p = 0.000084), CD4+ (p = 0.000037), and CD8+ (p = 0.000016) T cells as a percentage of total para-aortic LN cells (f) and total (p = 0.0044), CD4+ (p = 0.00297), and CD8+ (p = 0.0021) T cells as cells/LN after correction for total para-aortic LN cell number (g). d–g n = 6 Ldlr−/− and n = 7 T-AbcdkoLdlr−/− mice. Representative flow cytometry plots of (h) CD4+CD44−CD62L+ (Tnaive), CD44+CD62L− (Tmemory/effector (Tmem/eff)), CD8+ Tnaive, CD8+CD44+CD62L+ (Tcentral memory (TCM)), and Tmem/eff cells gated as in Supplementary Fig. 7a, (i, j) quantifications of CD4+ Tnaive (p < 0.000001) and Tmem/eff (p < 0.000001) cells (i) or CD8+ Tnaive (p < 0.000001), TCM (p < 0.000001), and Tmem/eff (p = 0.0000027) cells (j) as a percentage of CD4+ (i) or CD8+ (j) T cells, and (k, l) concentrations in blood for CD4+ Tnaive (p < 0.000001) and Tmem/eff cells (k) or CD8+ Tnaive (p < 0.000001), TCM, and Tmem/eff cells (l) after correction for total blood leukocyte counts. n = 14 Ldlr−/− and n = 15 T-AbcdkoLdlr−/− mice. b, i, j The experiments were performed twice with the same results. For all panels, error bars represent SEM. Biologically independent samples were included. p value was determined by unpaired two-tailed Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001. Source data are provided as a Source Data file.
We have previously shown that T cell Abca1/Abcg1 deficiency mildly enhanced CD4+ T cell activation in the LckCreAbca1fl/flAbcg1fl/fl model31. We thus assessed the effect of single and combined T cell Abca1/Abcg1 deficiency on T cell activation. Single T cell Abca1 or Abcg1 deficiency did not affect T cell activation in blood (Supplementary Fig. 8a–c). Combined T cell Abca1/Abcg1 deficiency increased Tmemory/effector (Tmem/eff; CD4+CD44+CD62L− and CD8+CD44+CD62L−) and Tcentral memory (TCM; CD8+CD44+CD62L+) cells as a percentage of CD4+ or CD8+ T cells by >3-fold in Ldlr−/− (Fig. 2h–j and Supplementary Fig. 8a) and Ldlr+/+mice (Supplementary Fig. 8d, e). Further, T cell Abca1/Abcg1 deficiency decreased CD4+ and CD8+ Tnaive cells (CD44−CD62L+) cells by ~50% as a percentage of CD4+ or CD8+ T cells in Ldlr−/− (Fig. 2h–j) and Ldlr+/+mice (Supplementary Fig. 8d, e). We observed similar effects in spleen and para-aortic LNs of Ldlr−/− mice (Supplementary Fig. 8f–i). Collectively, as percentages of total CD4+ or CD8+ T cells, T cell Abca1/Abcg1 deficiency increased Tmem/eff and TCM cells in blood, spleen, and para-aortic LNs. However, when corrected for total number of blood leukocytes or total number of cells per LN or spleen, the increases in Tmem/eff and TCM cells were no longer significant between the genotypes (Fig. 2k, l and Supplementary Fig. 8j–l). Moreover, T cell Abca1/Abcg1 deficiency decreased CD4+ and CD8+ Tnaive cells by >70% (Fig. 2k, l, and Supplementary Fig. 8j, k, m). In sum, while increasing Tmem/eff and TCM subsets as percentages of CD4+ and CD8+ T cells, T cell Abca1/Abcg1 deficiency dramatically decreased Tnaive cells in blood, spleen, and para-aortic LNs.
Effects of T cell Abca1/Abcg1 deficiency on T cell exhaustion
Previous studies have shown that T cell cholesterol accumulation induces T cell exhaustion, which was mainly attributed to cholesterol accumulation in the ER32. Exhausted T cells are prone to apoptosis and show reduced proliferative capacity32,33. We therefore investigated whether increased cholesterol accumulation due to loss of Abca1/Abcg1 in T cells would induce T cell exhaustion and thus explain the decrease in T cell numbers.
The percentage of para-aortic LN CD4+ T cells expressing the exhaustion marker programmed cell death 1 (PD-1) was increased in T-AbcdkoLdlr−/− mice compared to Ldlr−/− controls (Supplementary Fig. 9a–c). However, T cell Abca1/Abcg1 deficiency did not affect the total number of CD4+PD-1+ T cells in whole para-aortic LNs (Supplementary Fig. 9d). T cell Abca1/Abcg1 deficiency induced only a modest increase in cytotoxic T-lymphocyte-associated protein 4 (CTLA4) on CD4+ and CD8+ T cells, and T cell immunoglobulin and mucin-domain containing 3 (TIM3) and lymphocyte-activation gene 3 (LAG3) on CD4+ T cells (Supplementary Fig. 9e–l). T cell Abca1/Abcg1 deficiency increased the transcription factor Eomesodermin (Eomes) in CD8+ T cells (Supplementary Fig. 9m–o). We observed similar effects of T cell Abca1/Abcg1 deficiency on splenic CD4+PD-1+ T cell numbers and on expression of CTLA4 or Eomes in splenic T cells (Supplementary Fig. 10a–g). Eomes is an exhaustion marker34, but has also been associated with an increase in TCM cells35. Increased PD-1 on CD4+ T cells may be due to increased T cell activation36. Since TIM3, LAG3, and CTLA4 were minimally increased by T cell Abca1/Abcg1 deficiency, and the increase in the percentage of PD-1+CD4+ T cells and Eomes expression on CD8+ T cells could be attributed to the expansion of Tmem/eff and TCM populations, respectively, these data are suggestive of only minor effects of T cell Abca1/Abcg1 deficiency on T cell exhaustion.
T cell Abca1/Abcg1 deficiency increases T cell apoptosis upon TCR stimulation
We then investigated other mechanisms contributing to the decrease in peripheral T cell numbers. Previous studies have shown that stimulation of the TCR by αCD3 decreases the expression of Abca1 and Abcg1 by >90%, while upregulating the expression of Acat1, Hmgcr, and Ldlr20. These effects were confirmed by a later study8, and suggest that TCR stimulation induces a change in gene expression that favors cholesterol accumulation, likely to generate substrates for cellular growth and proliferation. We confirmed that TCR stimulation decreased Abca1 and Abcg1 mRNA expression in T cells (Supplementary Fig. 11a, b). In genetic models of plasma membrane cholesterol accumulation in T cells, TCR signaling is enhanced8,20,23. Conversely, TCR signaling is suppressed in T cells deficient in SCAP (SREBP cleavage activating protein) that cannot synthesize cholesterol37. We thus assessed whether T cell Abca1/Abcg1 deficiency affected T cell proliferation downstream of TCR signaling. Upon TCR stimulation, Abca1/Abcg1 deficiency increased T cell proliferation in CD4+ and CD8+ Ldlr−/− and Ldlr+/+ T cells (Supplementary Fig. 11c–i). Even though in line with observations of increased T cell proliferation in T cell Abcg1 deficiency20,23, these findings cannot explain the decreased peripheral T cells in T-AbcdkoLdlr−/− mice compared to Ldlr−/− controls. However, these data strongly suggest that T cell Abca1/Abcg1 deficiency enhances TCR signaling. Consistent with increased TCR signaling, we also observed increased Jun N-terminal kinase (JNK)1/2 phosphorylation upon stimulation with αCD3 in CD4+ and CD8+ T cells (Supplementary Fig. 12a, b). We thus investigated whether T cell Abca1/Abcg1 deficiency affects other pathways that regulate peripheral T cell numbers, downstream of TCR signaling.
TCR stimulation enhances the differentiation of Tnaive cells into Tmem/eff and TCM cells38. The increase in Tmem/eff and TCM cells in mice with T cell Abca1/Abcg1 deficiency (Fig. 2h–j and Supplementary Fig. 8f–i) is consistent with increased TCR signaling. Upon TCR stimulation, T cells differentiate into Tmem/eff cells that may undergo apoptosis, in a pathway known as activation-induced cell death (AICD)39. Using αCD3 and interleukin-2 (IL-2) as stimuli, we examined whether T cell Abca1/Abcg1 deficiency enhanced apoptosis downstream of TCR stimulation, by monitoring expression of cleaved caspase 3/7 in T cells over time using the Incucyte system. T cell Abca1/Abcg1 deficiency increased cleaved caspase 3/7 by 3.7-fold in CD4+ T cells and 1.8-fold in CD8+ T cells, reflecting increased T cell apoptosis (Fig. 3a, b and Supplementary Fig. 12c). These experiments were carried out in Ldlr−/− T cells. We found that T cell Abca1/Abcg1 deficiency also increased cleaved caspase 3/7 in Ldlr+/+ T cells, as assessed by flow cytometry (Fig. 3c, d and Supplementary Fig. 12d), while single Abca1 or Abcg1 deficiency showed no effect (Supplementary Fig. 12d–f). We then evaluated whether other modes of cell death may have contributed to the decrease in T cells in mice with T cell Abca1/Abcg1 deficiency. T cell pyroptosis, a highly pro-inflammatory form of lytic programmed cell death is executed by gasdermin D cleavage40, and decreases T cell numbers, at least in the setting of chronic HIV-1 infection41. Upon αCD3/IL-2 stimulation, we detected a very low level of cleaved gasdermin D p30, which was not different between the genotypes (Supplementary Fig. 12g), indicating that T cell pyroptosis was not affected.
Fig. 3. T cell Abca1/Abcg1 deficiency increases T cell apoptosis.

Splenic CD4+ and CD8+ T cells were isolated from Ldlr−/−, T-AbcdkoLdlr−/−, control (Ldlr+/+), and T-Abcdko mice fed chow diet. a–d T cells were stimulated with αCD3 and interleukin 2 (IL-2) for 12 h and stained for cleaved caspase 3/7. a CD4+ (p = 0.0045) and (b) CD8+ (p = 0.0475) T cells acquiring cleaved caspase 3/7 staining over time. n = 4 Ldlr−/− and n = 4 T-AbcdkoLdlr−/− mice. c Representative flow cytometry plots of cleaved caspase 3/7 in CD4+ (p = 0.0101) and CD8+ (p = 0.0315) T cells gated as in Supplementary Fig. 12d, and (d) quantification. n = 5 control and n = 5 T-Abcdko mice. e, f CD4+ T cells were stimulated with αCD3/αCD28 beads, IL-2, and transforming growth factor beta (TGF-β) for 72 h. e Representative flow cytometry plots of CD25+Foxp3+Tregulatory cells (Treg) gated as in Supplementary Fig. 12h, and (f) quantification (p = 0.00096) as a percentage of CD4+ T cells. n = 6 Ldlr−/− and n = 5 T-AbcdkoLdlr−/− mice. g–l T cells were stimulated with αCD3/IL-2 for 12 h, fixed, permeabilized, and stained for B-cell lymphoma 2 (Bcl2). g Representative flow cytometry plots of CD4+Bcl2+ and CD4+Bcl2− T cells gated as in Supplementary Fig. 13c. h Quantification of CD4+Bcl2+ (p = 0.045) and CD4+Bcl2− (p = 0.025) T cells as a percentage of CD4+ T cells. n = 4 Ldlr−/− and n = 4 T-AbcdkoLdlr−/−. Same staining for CD8+ T cells as in (g) in combination with CD44 and CD62L. Representative flow cytometry plots of CD8+Bcl2+, CD8+Bcl2− (i), CD8+Tmem/effBcl2+, and CD8+Tmem/effBcl2− cells (k) gated as in Supplementary Fig. 8a. Quantification of CD8+Bcl2+ (p = 0.00015) and CD8+Bcl2− (p < 0.000001) cells as a percentage of CD8+ T cells (j) and CD8+Tmem/effBcl2+ (p = 0.0017) and CD8+Tmem/effBcl2− (p = 0.0041) cells as a percentage of CD8+Tmem/eff cells (l). n = 4 Ldlr−/− and n = 3 T-AbcdkoLdlr−/−. f, h, j, l Data were corrected for their respective isotype controls. For all panels, error bars represent SEM. Biologically independent samples were included. p value was determined by unpaired two-tailed Student’s t test. For (a, b), p value is based on area under the curve. *p < 0.05, **p < 0.01, ***p < 0.001. Source data are provided as a Source Data file.
Given that Abcg1 deficiency alone enhances differentiation of Tnaive cells in Treg cells22, and Treg cells are athero-protective, we then also studied Treg differentiation in T cells with combined Abca1/Abcg1 deficiency. We used TGF-β, αCD3, and IL-2 to induce CD25+Foxp3+Treg differentiation, similar to previous studies22,42, and found that, in contrast to T cell Abcg1 deficiency22, T cell Abca1/Abcg1 deficiency suppressed Treg differentiation by ~50% (Fig. 3e, f and Supplementary Fig. 12h). Given that, with the exception of TGF-β, stimuli for the apoptosis assay were similar to those used for Treg differentiation, it is likely that the decrease in Treg cells is simply the consequence of increased T cell apoptosis. We thus studied mechanisms that cause T cell apoptosis.
We found that the expression of the death receptor FAS that is highly expressed in lipid rafts and involved in the extrinsic apoptosis pathway43 was not affected by T cell Abca1/Abcg1 deficiency upon stimulation with αCD3 and IL-2 (Supplementary Fig. 12d and and Supplementary Fig. 13a, b). We next measured the expression of the anti-apoptotic protein B-cell lymphoma 2 (Bcl2) that regulates the intrinsic apoptosis pathway44. Upon αCD3/IL-2 stimulation, T cell Abca1/Abcg1 deficiency decreased anti-apoptotic Bcl2+CD4+ and Bcl2+CD8+ T cells by ~20%, while increasing pro-apoptotic Bcl2−CD4+ and Bcl2−CD8+ T cells by ~1.6 and ~3.1-fold, respectively (Fig. 3g–j and Supplementary Fig. 13c). However, less than 20% of Abca1/Abcg1-deficient CD8+ T cells were Bcl2−, while this was ~40% for CD4+ T cells (Fig. 3g–j), which may be due to the expanded CD8+TCM cell population. Indeed, CD8+TCM cells are long-lived45. Since AICD implies apoptosis of the Tmem/eff cell population, we studied the effect of T cell Abca1/Abcg1 deficiency on Bcl2 expression in CD8+TCM and Tmem/eff cells separately. Upon anti-CD3/IL-2 stimulation, ~90% of CD8+TCM cells were Bcl2+ in mice with T cell Abca1/Abcg1 deficiency and controls (Supplementary Fig. 13d, e). T cell Abca1/Abcg1 deficiency decreased anti-apoptotic Bcl2+CD8+Tmem/eff cells by ~30%, while increasing pro-apoptotic Bcl2−CD8+Tmem/eff cells by ~40% (Fig. 3k, l). Once the Bcl2 pathway is activated, mitochondrial ROS (mitoROS) stimulates cytochrome c release from mitochondria, which ultimately leads to caspase activation46. Indeed, upon αCD3/IL-2 stimulation, T cell Abca1/Abcg1 deficiency increased mitoROS levels, mainly in CD8+Tnaive and Tmem/eff cells (Supplementary Fig. 13f–i). There was no effect on CD4+ T cells (Supplementary Fig. 13f–i), perhaps because T cells with high mitoROS had already undergone apoptosis. Collectively, these data indicate that T cell Abca1/Abcg1 deficiency enhances T cell apoptosis, independent of Ldlr expression, and downstream of TCR and CD25, mediated by Bcl2 in the intrinsic apoptosis pathway, especially in Tmem/eff cells. This likely contributed to the ~50% decrease in peripheral T cells in T cell Abca1/Abcg1 deficiency.
We then studied effects of T cell Abca1/Abcg1 deficiency on T cell functionality. Upon stimulation by αCD3/IL-2, T cell Abca1/Abcg1 deficiency increased CD8+granzyme B+ T cells as a percentage of total CD8+ T cells (Fig. 4a, b and Supplementary Fig. 14a), and CD4+IFN-γ+ and CD8+IFN-γ+ T cells as percentages of CD4+ and CD8+ T cells, respectively (Fig. 4c, d and Supplementary Fig. 14a). T cell Abca1/Abcg1 deficiency also increased lysosomal-associated membrane protein 1 (LAMP-1) surface expression on CD8+ T cells (Fig. 4e, f and Supplementary Fig. 14b), indicating T cell degranulation. These findings are suggestive of T cell Abca1/Abcg1 deficiency increasing the TCR response, and increasing T cell functionality. However, in the same assay, T cell Abca1/Abcg1 deficiency decreased IFN-γ secretion into the media (Fig. 4g), presumably due to increased T cell apoptosis. Indeed, Abca1/Abcg1 deficiency decreased T cell counts by ~80% after stimulation by αCD3/IL-2 (Supplementary Fig. 14c), while the number of T cells at the start of the assay was similar between genotypes. Therefore, even though T cell Abca1/Abcg1 deficiency increased the percentage of IFN-γ+ T cells, it decreased IFN-γ secretion presumably due to increased T cell apoptosis. We subsequently investigated T cell mediated effects on macrophage function.
Fig. 4. Effects of T cell Abca1/Abcg1 deficiency on T cell functionality.

Splenic CD4+ a–f, CD8+ (a–f, j), or total T cells (g–i) were isolated from Ldlr−/− and T-AbcdkoLdlr−/− mice. a–f CD4+ and CD8+ T cells were stimulated with αCD3/IL-2 for 12 h, fixed, permeabilized, stained for granzyme B (a, b), or interferon gamma (IFN-γ) (c, d), or alternatively, stained for lysosomal-associated membrane protein-1 (LAMP-1) (e, f). Gating strategies are shown in Supplementary Fig. 14. Representative flow cytometry plots of (a) CD8+granzyme B+ (p = 0.0096), (c) CD4+IFN-γ+ (p = 0.048) and CD8+IFN-γ+ (p = 0.002) T cells and (b, d) quantification as a percentage of CD8+ T cells after correction for isotype control (b) and as a percentage of CD4+ or CD8+ T cells after correction for fluorescence minus one (FMO) control (d). e Representative flow cytometry plots and (f) quantification of LAMP-1 expression on CD4+ and CD8+ (p = 0.027) T cells. b, f n = 7 Ldlr−/− and n = 6 T-AbcdkoLdlr−/− mice. d n = 8 Ldlr−/− and n = 5 T-AbcdkoLdlr−/− mice. g T cells were stimulated with αCD3/IL-2 for 12 h. Medium was collected and IFN-γ levels (p = 0.034) were measured by ELISA. n = 7 Ldlr−/− and n = 6 T-AbcdkoLdlr−/− mice. h Wild-type bone marrow derived macrophages (BMDMs) were incubated with conditioned medium from CD8+ T cells stimulated with αCD3/IL-2 for 12 h, or recombinant IFN-γ (rIFN-γ; positive control) for 3 days. After medium removal, BMDMs were co-incubated with E. coli for 1 h, washed, incubated with gentamicin for 3 h, washed, and lysed. BMDM lysates were plated on agar overnight and E. coli colony forming units (CFU) were counted (Ldlr−/− vs T-AbcdkoLdlr−/−, p = 0.0032; Ldlr−/− or T-AbcdkoLdlr−/− vs rIFN-γ, p < 0.0001). n = 3 Ldlr−/− and n = 3 T-AbcdkoLdlr−/− mice. i BMDMs were co-incubated with T cells in the presence of αCD3/IL-2 (to stimulate T cells) for 24 h. Medium was collected and endogenous lactate dehydrogenase (LDH) was assessed (p = 0.0023). n = 6 Ldlr−/− and n = 5 T-AbcdkoLdlr−/− mice. For all panels, error bars represent SEM. Biologically independent samples were included. p value was determined by unpaired two-tailed Student’s t test (a–i) or one-way ANOVA with Bonferroni post-test (j). *p < 0.05, **p < 0.01, ***p < 0.001. Source data are provided as a Source Data file.
T cell IFN-γ secretion enhances the capacity of macrophages to kill bacteria47. To examine whether this was affected by T cell Abca1/Abcg1 deficiency, we co-incubated wild-type bone marrow derived macrophages (BMDMs) with conditioned medium from αCD3/IL-2 stimulated Ldlr−/− or T-AbcdkoLdlr−/− CD8+ T cells prior to infection with E.coli bacteria. Conditioned medium from T-AbcdkoLdlr−/− CD8+ T cells increased the number of E.coli bacteria colony forming units (CFU) compared to medium from Ldlr−/− CD8+ T cells (Fig. 4h), reflecting reduced bacterial killing capacity by macrophages.
We then examined effects of Abca1/Abcg1 deficiency on T cell mediated cytotoxicity by co-incubating αCD3/IL-2 stimulated Ldlr−/− or T-AbcdkoLdlr−/− T cells with wild-type BMDMs. Lactate dehydrogenase (LDH), a cytosolic enzyme released into the medium upon damage of the plasma membrane, reflects cell death40. T-AbcdkoLdlr−/− T cells induced less BMDM LDH release compared to Ldlr−/− T cells (Fig. 4i), indicating less macrophage killing and decreased T cell mediated cytotoxicity.
Therefore, despite increasing IFN-γ+ T cells and CD8+granzyme B+ T cells, T cell Abca1/Abcg1 deficiency decreased the capacity of macrophages to kill bacteria as well as T cell mediated macrophage killing, presumably due to increased T cell apoptosis. These findings indicate that T cell Abca1/Abcg1 deficiency decreases T cell functionality.
Aging increases cholesterol accumulation and apoptosis in T cells
We then asked whether there would be a broader physiological relevance of the phenotype we observed in T cell Abca1/Abcg1 deficiency. Aged humans (~73–76 years old) show increased T cell plasma membrane cholesterol accumulation compared to young humans (~22–23 years old)10,11 and a decrease in peripheral T cells13. The phenotype in T cells from aged individuals resembles the phenotype of mice with T cell Abca1/Abcg1 deficiency and may suggest a broader physiological relevance for T cell plasma membrane cholesterol accumulation in regulating peripheral T cell numbers. Using T cells from aged (~24 months old) and young (~3 months old) mice, we investigated this further.
Similar to findings in aged humans10,11, blood CD4+ and CD8+ T cells from aged mice show an increase in filipin staining compared to T cells from young mice, reflecting an increase in plasma membrane cholesterol accumulation (Fig. 5a, b and Supplementary Fig. 1e). Aging also increased choleratoxin B staining on CD8+ T cells, suggestive of more lipid rafts (Fig. 5c, d and Supplementary Fig. 2c). Further, aging increased total T cell cholesterol content as assessed by GC-MS by ~1.5-fold (Fig. 5e), while there were no signs of lipid droplets based on BODIPY 493/503 staining (Supplementary Fig. 15a), indicating that the increase in cellular cholesterol reflects an increase in free cholesterol. Aging decreases Abca1 and Abcg1 mRNA expression in mouse splenic and peritoneal macrophages48; however, we found no effect of aging on mRNA expression of Abca1 and Abcg1 in T cells, while mRNA expression of other genes affecting cholesterol metabolism such as Hmcgr and Hmgcs was minimally decreased and Ldlr mRNA expression showed a moderate decrease, especially in CD8+ T cells (Supplementary Fig. 15b, c). Ldlr expression does not affect T cell cholesterol accumulation (Supplementary Fig. 2a, b and Supplementary Fig. 3a, b). Therefore, effects of aging on T cell cholesterol accumulation cannot be explained by changes in expression of these genes. Perhaps repeated TCR stimulation increases cholesterol accumulation during aging.
Fig. 5. Aging increases cholesterol accumulation and apoptosis in T cells.

Blood and spleens from young (3 months) and aged (24 months) wild-type mice fed a chow diet were collected. Filipin (a, b) and choleratoxin B (c, d) stainings on blood T cells. a Representative flow cytometry plots of filipin staining on CD4+ (p < 0.000001) and CD8+ (p < 0.000001) T cells gated as in Supplementary Fig. 1e and (b) quantification. c Representative flow cytometry plots of choleratoxin B staining on CD4+ and CD8+ (p = 0.00002) T cells gated as in Supplementary Fig. 2c and (d) quantification. b, d n = 10 young and n = 10 aged wild-type mice. e Splenic T cells were isolated and total cholesterol (p = 0.013) was measured by Gas Chromatography – Mass Spectrometry. n = 5 young and n = 6 aged wild-type mice. f Total (p < 0.000001), CD4+ (p < 0.000001), and CD8+ (p = 0.0026) T cells as a percentage of total blood leukocytes. Tnaive (p < 0.000001) and Tmem/eff (p < 0.000001) cells as a percentage of CD4+ (g) and Tnaive (p < 0.000001), Tmem/eff (p < 0.000001), and TCM (p = 0.00016) cells as a percentage of CD8+ T cells (h) in blood. f–h n = 10 young and n = 10 aged wild-type mice. i–m Splenic total, CD4+, and CD8+ T cells were isolated. i–k T cells were labeled with CFSE and stimulated with αCD3/αCD28 beads. CFSE dilution was measured by flow cytometry at 72 h after stimulation. i Representative CFSE dilutions gated as in Supplementary Fig. 11c and quantification of the number of divisions of (j) CD4+ (division 0, p = 0.030; 3, p = 0.035; 4, p = 0.023; 5, p = 0.026) and (k) CD8+ T cells. n = 4 young and n = 4 aged wild-type mice. l–m CD4+ and CD8+ T cells were stimulated with αCD3/IL-2 for 12 h with concomitant staining for cleaved caspase 3/7. I Representative flow cytometry plots of cleaved caspase 3/7 in CD4+ (p = 0.004) and CD8+ (p = 0.009) T cells gated as in Supplementary Fig. 12d and (m) quantification. n = 6 young and n = 6 aged wild-type mice. For all panels, error bars represent SEM. Biologically independent samples were included. p value was determined by unpaired two-tailed Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001. Source data are provided as a Source Data file.
Aging did not affect plasma cholesterol, or its distribution over lipoproteins, but, in line with previous observations49, decreased plasma triglycerides (TG) by ~42% (P < 0.001), reflected by a decrease in the very low-density lipoprotein (VLDL)-TG fraction (Supplementary Table 1 and Supplementary Fig. 15d, e). Similar to previous observations from aged humans and mice12,50,51, aged mice showed a decrease in peripheral T cells compared to young mice (Fig. 5f), mainly due to decreased Tnaive cells, while Tmem/eff and TCM cells were increased (Fig. 5g, h). CD4+ T cells from aged mice showed a slight decrease in T cell proliferation in response to TCR stimulation, while CD8+ T cell proliferation was not affected by aging (Fig. 5i–k and Supplementary Fig. 11c), in line with previous findings showing that Tnaive cells from aged mice still proliferate efficiently12. We then studied apoptosis in response to αCD3/IL-2 stimulation in young and aged T cells. αCD3/IL-2 stimulation increased cleaved caspase3/7 in aged compared to young T cells (Fig. 5l, m and Supplementary Fig. 12d), reflecting increased apoptosis. The data in aged mice (Fig. 5a–h, l, m) together with our findings in T-AbcdkoLdlr−/− mice suggest that plasma membrane cholesterol accumulation in T cells from aged mice increases T cell activation and apoptosis, which likely contributes to the decrease in T cells during aging.
Further, upon stimulation by αCD3/IL-2, aging increased CD8+granzyme B+cells as percentage of CD8+ T cells (Supplementary Fig. 14a and Supplementary Fig. 16a, b) and CD4+IFN-γ+ and CD8+IFN-γ+ cells as percentages of CD4+ and CD8+ T cells, respectively (Supplementary Fig. 14a and Supplementary Fig. 16c, d). Aging also increased LAMP-1 surface expression on CD4+ and CD8+ T cells (Supplementary Fig. 14b and Supplementary Fig. 16e, f), indicating increased T cell degranulation. These findings suggest that aging increased the TCR response and enhanced T cell functionality. Consistently, aging increased IFN-γ secretion from T cells (Supplementary Fig. 16g), in line with previous studies52,53, but different from mice with T cell Abca1/Abcg1 deficiency. The latter was presumably due to the more pronounced increase of T cell apoptosis upon T cell Abca1/Abcg1 deficiency than upon T cell aging. Still, the immune system loses its ability to mount an effective immune response to pathogens during aging54, and our findings suggest that increased T cell apoptosis may contribute.
T cell Abca1/Abcg1 deficiency induces a premature T cell aging phenotype
We then asked whether aging would affect TCR responses in T-AbcdkoLdlr−/− mice. We aged T-AbcdkoLdlr−/− mice and Ldlr−/− littermate controls until 12–13 months (middle-aged). The phenotype in terms of T cell numbers and activation did not differ between middle-aged and young T-AbcdkoLdlr−/− mice (Supplementary Fig. 17a–c). T cell Abca1/Abcg1 deficiency increased cleaved caspase3/7 upon TCR stimulation in middle-aged mice, reflecting increased apoptosis (Fig. 6a, b and Supplementary Fig. 12d). Strikingly, upon TCR stimulation, T cell Abca1/Abcg1 deficiency almost completely abolished CD4+ and CD8+ T cell proliferation, while T cells from control mice still proliferated, in the absence (Fig. 6c–e and Supplementary Fig. 11c) and presence of Ldlr expression (Fig. 6f–h and Supplementary Fig. 11c). Moreover, treatment with reconstituted HDL (rHDL), which promotes cholesterol efflux in the absence of Abca1/Abcg1 via passive efflux mechanisms, at least in macrophages55, for the whole duration of the proliferation assay (72 h), still further decreased T cell proliferation in control and Abca1/Abcg1-deficient T cells (Supplementary Fig. 17d–h). These data indicate that even when proliferation is almost impaired in middle-aged T cells with Abca1/Abcg1 deficiency, cholesterol depletion still suppresses T cell proliferation. The remaining T cell proliferation is thus cholesterol-dependent. T cells with Abca1/Abcg1 deficiency may not enter the cell cycle due to upregulation of cyclin dependent kinase inhibitors, including Cdkn1a56. Abca1/Abcg1 deficiency increased Cdkn1a mRNA expression by 6-fold in CD4+ T cells and 2.5-fold in CD8+ T cells compared to middle-aged control T cells (Fig. 6i, j), while Cdkn2a mRNA expression was not affected (Supplementary Fig. 17i, j). Of note, unlike in other cell types, in T cells, Cdkn2a rather reflects increased T cell activation than aberrant cell cycling13. When comparing Cdkn1a mRNA expression in T cells from middle-aged T-AbcdkoLdlr−/− mice to T cells from aged wild-type mice, we observed that Abca1/Abcg1 deficiency increased Cdkn1a mRNA expression by ~6-fold in CD4+ and CD8+ T cells (Fig. 6i, j). Together, these data indicate that middle-aged Abca1/Abcg1-deficient T cells upregulate Cdkn1a and do not enter the cell cycle, which is suggestive of T cell senescence and premature T cell aging.
Fig. 6. T cell Abca1/Abcg1 deficiency induces a premature T cell aging phenotype.

Ldlr−/− and T-AbcdkoLdlr−/− mice (a–e, i–j) and control (Ldlr+/+) and T-Abcdko mice (f–h) were fed a chow diet for 12–13 months, and wild-type mice for 3 months (young) or 24 months (aged) (i–j). a–j Spleens were collected and CD4+ and CD8+ T cells were isolated. a, b CD4+ and CD8+ T cells were stimulated with αCD3/IL-2 for 12 h with concomitant staining for cleaved caspase3/7. a Representative flow cytometry plots of cleaved caspase 3/7 in CD4+ (p = 0.015) and CD8+ (p = 0.007) T cells gated as in Supplementary Fig. 12d and (b) quantification. n = 6 Ldlr−/− and n = 5 T-AbcdkoLdlr−/− mice. c–h T cells were labeled with CFSE and stimulated with αCD3/αCD28 beads. CFSE dilution was measured by flow cytometry at 72 h after stimulation. c, f Representative CFSE dilutions gated as in Supplementary Fig. 11c. The number of divisions were quantified for (d) CD4+ (division 0, p = 0.000017; 2, p = 0.00002; 3, p < 0.000001; 4, p < 0.000001) and (e) CD8+ (division 0, p = 0.0029; 1, p = 0.00095; 3, p = 0.0023; 4, p = 0.0000097; 5, p = 0.008) T cells. n = 7 Ldlr−/− and n = 6 T-AbcdkoLdlr−/− mice. The number of divisions were quantified for (g) CD4+ (division 0), p = 0.0002; 1, p = 0.013; 2, p = 0.017; 3, p = 0.00067; 4, p = 0.0031 and (h) CD8+ T cells (division 0, p = 0.0004; 1, p = 0.0056; 3, p = 0.012; 4, p = 0.0003; 5, p = 0.02). n = 4 control and n = 4 T-Abcdko mice. i–j T cells were isolated and RNA was extracted. Cyclin dependent kinase inhibitor 1a (Cdkn1a) mRNA expression was measured in (i) CD4+ (Ldlr−/− or aged wild-type vs T-AbcdkoLdlr−/−, p < 0.0001) and (j) CD8+ (Ldlr−/− vs T-AbcdkoLdlr−/−, p=0.0003; aged wild-type vs T-AbcdkoLdlr−/−, p < 0.0001) T cells by qPCR and shown as fold change compared to young wild-type mice. n = 8 young wild-type, n = 6 aged wild-type, n = 6 Ldlr−/−, and n = 5 T-AbcdkoLdlr−/− mice. For all panels, error bars represent SEM. Biologically independent samples were included. p value was determined by unpaired two-tailed Student’s t test (b, d–e, g, h) or one-way ANOVA with Bonferroni post-test (i, j). *p < 0.05, **p < 0.01, ***p < 0.001. Source data are provided as a Source Data file.
Effects of T cell Abca1/Abcg1 deficiency on T cell subsets and atherosclerosis
Previous studies have shown that T cell Abcg1 deficiency decreases atherosclerosis by enhancing the formation of Treg cells in thymus and LNs of Ldlr−/− mice fed WTD22. In addition, WTD feeding enhances the conversion of Treg cells into TFH and Th1 cells, which was dependent on cholesterol accumulation5. T cell Acat1 deficiency induces plasma membrane cholesterol accumulation in CD8+ T cells, leading to expansion of CD8+IFN-γ+ T cells8 that are pro-atherogenic. We thus investigated whether exacerbated cholesterol accumulation due to loss of Abca1/Abcg1 in T cells would affect T cell subsets.
We assessed these T cell subsets in para-aortic LNs of T-AbcdkoLdlr−/− and Ldlr−/− mice fed a chow diet. T cell Abca1/Abcg1 deficiency did not affect para-aortic LN CD25+Foxp3+Treg cells, but increased CD25−Foxp3+Treg cells as a percentage of CD4+ T cells (Fig. 7a, b and Supplementary Fig. 18a), which may suggest that upon cholesterol accumulation, Treg cells lose their CD25 expression, and start to express markers of TFH and Th1 cells as has been reported in Apoe−/− mice fed WTD5. Both in T-AbcdkoLdlr−/− and Ldlr−/− mice, CD25−Foxp3+T cells were mainly of the activated Tmem/eff (CD44+CD62L−) phenotype, while CD25+Foxp3+cells were mostly of the Tnaive (CD44−CD62L+) phenotype (Supplementary Fig. 18b, c). After correction for total para-aortic LN cell number, T cell Abca1/Abcg1 deficiency decreased CD25+Foxp3+ Treg cells by ~75% in whole para-aortic LNs, while not affecting CD25−Foxp3+ Treg cells (Fig. 7c), consistent with the decrease in CD25+Foxp3+ Treg cells that we observed in vitro (Fig. 3e, f), presumably because there were less total para-aortic LN T cells (Fig. 2g) due to increased T cell apoptosis. T cell Abca1/Abcg1 deficiency increased TFH cells as a percentage of CD4+ Tmem/eff cells in para-aortic LNs (Fig. 7d, e and Supplementary Fig. 18d), but did not affect TFH cells after correction for total para-aortic LN cell number (Fig. 7f), presumably also due to the decrease in total para-aortic LN T cells (Fig. 2g).
Fig. 7. Effects of T cell Abca1/Abcg1 deficiency on para-aortic LN T cell subsets and atherosclerosis in Ldlr−/− mice.

Female Ldlr−/− and T-AbcdkoLdlr−/− mice were fed chow diet for 28 weeks (a–i, k, n–o) or WTD for 10 weeks (j, l–m). a–i Para-aortic LNs were isolated, stained with the indicated antibodies, and analyzed by flow cytometry. Gating strategies are shown in Supplementary Fig. 18. a Representative flow cytometry plots and (b) quantification of CD25−Foxp3+ (p = 0.000001) and CD25+Foxp3+ Treg cells as a percentage of CD4+ T cells after correction for isotype control. c CD25−Foxp3+ and CD25+Foxp3+ Treg cells (p = 0.00026) as cells/LN after correction for total LN cells. n = 10 Ldlr−/− and n = 9 T-AbcdkoLdlr−/− mice. d Representative flow cytometry plots and (e) quantification of CD4+CD44+CD62L− C-X-C chemokine receptor type 5 (CXCR5)+ programmed cell death 1 (PD-1)+ Tfollicular helper (TFH) cells (p = 0.023) as a percentage of CD4+ Tmem/eff cells, and (f) as cells/LN. n = 6 Ldlr−/− and n = 6 T-AbcdkoLdlr−/− mice. g Representative flow cytometry plots and (h) quantification of CD4+Tbet+ (p = 0.00099) and CD8+Tbet+ (p = 0.0036) T cells as percentages of CD4+ and CD8+ T cells after correction for isotype control. i CD4+Tbet+ and CD8+Tbet+ (p = 0.0188) T cells as cells/LN. n = 5 Ldlr−/− and n = 5 T-AbcdkoLdlr−/− mice. Hearts were isolated and paraffin sections of the aortic root were stained for (j, k) haematoxylin-eosin (H&E) or (l-o) CD3. Representative pictures of H&E staining and quantification of atherosclerotic lesion area for WTD-fed (j) or chow diet-fed (k) mice. Scale bar represents 300 μm. Representative pictures and quantification of CD3+ cells per section for WTD-fed (p = 0.027) (l) or chow diet-fed (p = 0.0153) (n) mice. CD3+ cells are depicted by arrows. Scale bar represents 80 μm. CD3+ T cells as percentage of total cells per section for WTD-fed (p = 0.027) (m) or chow diet-fed (p = 0.043) mice (o). j n = 16 Ldlr−/− and n = 16 T-AbcdkoLdlr−/− mice. k, n, o n = 18 Ldlr−/− and n = 16 T-AbcdkoLdlr−/− mice. l–m n = 15 Ldlr−/− and n = 15 T-AbcdkoLdlr−/− mice. For all panels, error bars represent SEM. Biologically independent samples were included. p value was determined by unpaired two-tailed Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001. Source data are provided as a Source Data file.
Plasma membrane cholesterol accumulation increases formation of Th1 cells that express the transcription factor Tbet5,8. The percentage of CD4+Tbet+ and CD8+Tbet+ cells was increased in para-aortic LNs of T-AbcdkoLdlr−/− mice compared to Ldlr−/− controls (Fig. 7g, h and Supplementary Fig. 18e), but not affected after correction for total para-aortic LN cell numbers for CD4+Tbet+ cells, while CD8+Tbet+ cells were still ~2-fold increased (Fig. 7i). While CD4+Tbet+ cells were mainly of the Tmem/eff (CD44+CD62L−) phenotype (Supplementary Fig. 18f), CD8+Tbet+ cells were mostly of the long-lived TCM (CD44+CD62L+) phenotype (Supplementary Fig. 18g). In sum, based on total para-aortic LN cell numbers, T cell Abca1/Abcg1 deficiency decreased CD25+Foxp3+Treg cells and increased CD8+Tbet+ cells in para-aortic LNs, while para-aortic LN CD25−Foxp3+Treg cells, TFH, and Th1 cells were not affected (Supplementary Table 2). We also investigated whether these changes in T cells affected myeloid cells in blood. When expressed as a percentage of total leukocytes, T cell Abca1/Abcg1 deficiency increased blood monocytes, Ly6Clo and Ly6Chi monocyte subsets, as well as neutrophils by ~50% (Supplementary Fig. 19a, b). We explain these increases by T cell Abca1/Abcg1 deficiency decreasing T cells by 50% (Fig. 2a, b) and therefore other leukocyte populations, including myeloid cells showing an increase as percentage of total leukocytes. Indeed, the absolute number of blood leukocytes was decreased by T cell Abca1/Abcg1 deficiency (Supplementary Fig. 19c), presumably due to the decrease in blood T cells (Fig. 2a–c), and therefore, when corrected for total blood leukocyte number, T cell Abca1/Abcg1 deficiency no longer affected levels of myeloid cell counts in blood (Supplementary Fig. 19d).
We then investigated the effects of these changes on atherosclerosis. To induce atherogenesis, female T-AbcdkoLdlr−/− mice and Ldlr−/− littermate controls were fed a WTD. Findings on T cell activation and T cell subsets as well as on percentage of blood myeloid cells were similar to mice fed chow diet (Supplementary Fig. 20a–k). However, the increase in total monocytes, even though only ~15%, remained significant after correction for total blood leukocyte numbers in Ldlr−/− mice with T cell Abca1/Abcg1 deficiency compared to controls (Supplementary Fig. 20l). After 10 weeks of WTD, we assessed atherosclerotic lesion size at the level of the aortic root. T cell Abca1/Abcg1 deficiency did not affect atherosclerotic lesion size (Fig. 7j). This was accompanied by a decrease in plasma total cholesterol levels of ~15%, which reflects decreased VLDL and LDL cholesterol, while plasma TG or VLDL-TG was not affected (Supplementary Table 1 and Supplementary Fig. 21a, b). The decrease in plasma VLDL/LDL-cholesterol was likely a consequence of increased total cholesterol accumulation in T cells with Abca1/Abcg1 deficiency. Similarly, previous studies have shown that hematopoietic Abca1/Abcg1 deficiency decreased plasma VLDL/LDL-cholesterol levels in Ldlr−/− mice fed WTD57. However, on a chow diet, hematopoietic Abca1/Abcg1 deficiency, while still inducing cholesterol accumulation in hematopoietic cells, did not affect plasma VLDL/LDL-cholesterol levels55,58, presumably because the clearance of VLDL/LDL particles by hepatic Ldlr is not as much of a limiting factor as in Ldlr−/− mice fed WTD. In an attempt to exclude the confounding factor of decreased VLDL/LDL cholesterol levels to atherogenesis, we fed mice a chow diet for 28 weeks, similar to a study we carried out previously55. Chow diet-fed T-AbcdkoLdlr−/− and Ldlr−/− mice had similar plasma total cholesterol and TG levels, as well as distribution of these lipids over VLDL, LDL, or HDL (Supplementary Table 1 and Supplementary Fig. 21c, d) and developed atherosclerotic lesions similar in size compared to WTD-fed Ldlr−/− mice, but there was no difference in atherosclerotic lesion size between the genotypes (Fig. 7k). In line with observations in blood and secondary lymphoid organs, T-AbcdkoLdlr−/− mice showed a ~50% decrease in plaque CD3+ T cells on both diets compared to Ldlr−/− controls, both when shown as total CD3+ T cells per section or as percentage CD3+ T cells of total cells (nuclei) in lesions (Fig. 7l–o). Further, characterization of atherosclerotic lesions revealed no changes in fibrous cap thickness, collagen content, smooth muscle actin (α-SMA) or galectin-3 (Lgals3 or Mac-2) staining, reflecting predominantly macrophages, after either WTD or chow diet (Supplementary Fig. 22a–g and Supplementary Fig. 23a–g), suggesting no effects on plaque stability. In a separate cohort of mice, we evaluated lipid accumulation in atherosclerotic lesions in mice fed chow diet. After 38 weeks of chow diet (~9 months), T cell Abca1/Abcg1 deficiency did not affect atherosclerotic lesion area, similar to observations at 28 weeks of chow diet (Fig. 7k), but increased Oil Red O area as a percentage of lesion area, reflecting increased lipid accumulation (Supplementary Fig. 22h–j), presumably in both T cells and macrophages.
Hence, despite decreasing T cells in atherosclerotic plaques and decreasing para-aortic LN CD25+Foxp3+ Treg cells, and increasing para-aortic LN CD8+Tbet+ cells (Supplementary Table 2), T cell Abca1/Abcg1 deficiency did not affect atherosclerotic lesion size in Ldlr−/− mice fed chow diet or WTD. Since these subsets have both pro- and anti-inflammatory effects, and the loss of Abca1/Abcg1 in T cells does not affect a single predominant T cell subset, this effect on atherosclerosis could be due to counter-regulatory inflammatory effects in the aorta during sterile inflammation.
T cell Abca1/Abcg1 deficiency decreases atherosclerosis in middle-aged Ldlr−/− mice
We then investigated whether the premature T cell aging phenotype in middle-aged T-AbcdkoLdlr−/− mice affected atherogenesis. T cell Abca1/Abcg1 deficiency in middle-aged Ldlr−/− mice did not affect blood monocytes or neutrophils (Supplementary Fig. 24a, b). Similar to observations in young T-AbcdkoLdlr−/− mice (Fig. 7a–c), T cell Abca1/Abcg1 deficiency increased CD25−Foxp3+ Treg cells but not CD25+Foxp3+ Treg cells as a percentage of CD4+ T cells (Supplementary Fig. 24c, d). T cell Abca1/Abcg1 deficiency did not affect the number of total CD25−Foxp3+ Treg cells in whole para-aortic LNs, but decreased CD25+Foxp3+ Treg cells by ~70% (Supplementary Fig. 24e), similar to findings in young mice (Fig. 7c), and presumably due to the increased T cell apoptosis. Unlike in young mice (Supplementary Fig. 8h), T cell Abca1/Abcg1 deficiency did not affect CD4+ Tmem/eff cells as a percentage of CD4+ T cells (Supplementary Fig. 24f). Moreover, after correction for the total para-aortic LN cell number, T cell Abca1/Abcg1 deficiency decreased this population by ~50% (Supplementary Fig. 24g). CD8+Tbet+ cells were increased both as a percentage and after correction for total para-aortic LN cell number in para-aortic LNs from middle-aged T-AbcdkoLdlr−/− mice compared to controls, while CD4+Tbet+ cells were not affected (Supplementary Fig. 24h–j). Similar to CD25−Foxp3+ Treg cells and the observations in young mice (Fig. 7d–f), T cell Abca1/Abcg1 deficiency increased TFH cells as a percentage of LN CD4+ Tmem/eff cells, but not after correction for total para-aortic LN cell number (Supplementary Fig. 24k–m). Under conditions of similar plasma total cholesterol and TG levels, and similar distribution of these lipids on VLDL, LDL, and HDL (Supplementary Table 1 and Supplementary Fig. 25a, b), T cell Abca1/Abcg1 deficiency decreased atherosclerotic lesion size by ~35% (Fig. 8a), which was accompanied by a ~50% decrease in plaque CD3+ T cells (Fig. 8b, c). While the extent of the decrease in plaque CD3+ T cells was similar to our observations in young mice (Fig. 7l–o), the decrease in atherosclerotic lesion area was not, and also the atherosclerotic lesions of middle-aged mice showed a 2-fold higher T cell content than those of young mice fed chow diet (Fig. 7o), perhaps suggesting a more prominent effect of T cells on atherosclerosis in middle-aged Ldlr−/− mice. Also, the decrease in CD4+ Tmem/eff cells that we observed in middle-aged, but not young mice, may have contributed to the decrease in atherosclerotic lesion size.
Fig. 8. T cell Abca1/Abcg1 deficiency decreases atherosclerosis in middle-aged Ldlr−/− mice.

Ldlr−/− and T-AbcdkoLdlr−/− mice were fed a chow diet for 12–13 months. Hearts were isolated and paraffin sections of the aortic root were stained for (a) H&E or (b, c) CD3. a Representative pictures of H&E staining (left) and quantification (right) of atherosclerotic lesion area (p = 0.003). Scale bar represents 200 μm. n = 11 Ldlr−/− and n = 10 T-AbcdkoLdlr−/− mice. b Representative pictures of CD3 staining on atherosclerotic lesions (left) and quantification (right) of total CD3+ cells per section (p = 0.008). T cells were identified as cells with brown plasma membrane CD3 staining and the hematoxylin staining still visible. CD3+ cells are depicted by arrows. Scale bar represents 80 μm. c CD3+ T cells as percentage of total cells per section (p = 0.0107). b, c n = 11 Ldlr−/− and n = 10 T-AbcdkoLdlr−/− mice. d–i Aortas were collected. d Aortic CD3+ T cells were isolated, RNA was extracted, and Foxp3, Il10, Tgfb1, tumor necrosis factor (Tnf), Bcl2 (p = 0.023), and Cdkn1a (p = 0.025) mRNA expression were measured by qPCR. n = 7 Ldlr−/− and n = 5 T-AbcdkoLdlr−/− mice. e Total aortic cells were labeled with CFSE, stimulated with αCD3/αCD28 beads, and stained for CD4 after 96 h of stimulation. CFSE dilution was measured by flow cytometry and the number of divisions were quantified for aortic CD4+ T cells. n = 8 Ldlr−/− and n = 8 T-AbcdkoLdlr−/− mice. f–i Total aortic cells were stained for CD4, CD44, CD62L, and cleaved caspase 3/7. f Representative flow cytometry plots of cleaved caspase 3/7 in CD4+ T cells gated as in Supplementary Fig. 12d, and (g) quantification (p = 0.015). n = 4 Ldlr−/− and n = 5 T-AbcdkoLdlr−/− mice. h Aortic CD4+ Tmem/eff cells (p = 0.0003) as percentage of aortic CD4+ T cells and (i) cleaved caspase3/7 in aortic CD4+ Tmem/eff cells (p = 0.033). n = 7 Ldlr−/− and n = 7 T-AbcdkoLdlr−/− mice. For all panels, error bars represent SEM. Biologically independent samples were included. p value was determined by unpaired two-tailed Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001. Source data are provided as a Source Data file.
To obtain more insights into the nature of the T cells that were affected by T cell Abca1/Abcg1 deficiency in the aorta, we then further studied total aortic T cells based on mRNA expression. T cell Abca1/Abcg1 deficiency did not affect mRNA expression of the Treg transcription factor Foxp3 (Fig. 8d). Also, mRNA expression of anti-inflammatory Il10 and Tgfb1, and pro-inflammatory Tnf in aortic T cells did not differ between the genotypes (Fig. 8d). In line with findings in splenic T cells (Fig. 6i, j), T cell Abca1/Abcg1 deficiency moderately increased mRNA expression of the senescence marker Cdkn1a in aortic T cells, and decreased Bcl2 mRNA by ~25% (Fig. 8d). However, T cell Abca1/Abcg1 deficiency did not affect αCD3/αCD28 induced proliferation of CD4+ T cells isolated from aortas (Fig. 8e), perhaps due to these cells not showing a high level of proliferation compared to our findings in splenic T cells (Fig. 6c–e). Nevertheless, similar to findings on splenic T cells (Fig. 6a, b), T cell Abca1/Abcg1 deficiency also increased cleaved caspase 3/7 in T cells isolated from the aorta of middle-aged Ldlr−/− mice upon stimulation with αCD3/αCD28, which we only assessed in the CD4+ T cell population (Fig. 8f, g), as we were facing technical challenges to isolate CD8+ T cells from aortas. The increase in aortic CD4+ T cell apoptosis was accompanied by a decrease in the percentage of aortic CD4+ Tmem/eff cells (Fig. 8h) that also showed an increase in cleaved caspase 3/7 (Fig. 8i).
To obtain more insights into the consequences of these processes for atherosclerotic plaques of middle-aged T-AbcdkoLdlr−/− mice, we characterized their atherosclerotic lesions further. Similar to data in young Ldlr−/− mice (Supplementary Fig. 23a, d, e), T cell Abca1/Abcg1 deficiency did not affect α-SMA staining or fibrous cap thickness in middle-aged Ldlr−/− mice (Supplementary Fig. 25c–e), but decreased total collagen area in middle-aged Ldlr−/− mice (Supplementary Fig. 25f). However, after correction for total lesion area, collagen content was not different between the genotypes (Supplementary Fig. 25g). Only few necrotic cores were present in atherosclerotic plaques and the necrotic core area was not different between the genotypes (Supplementary Fig. 25h, i). Although the density of macrophages in atherosclerotic lesions was relatively low (~6–10% of the atherosclerotic lesions), T cell Abca1/Abcg1 deficiency did increase macrophage content by ~60% (Fig. 9a, b). Previous studies have shown that in advanced atherosclerotic plaques, T cells induce macrophage apoptosis, primarily mediated by granzyme B or perforin59. While T cell Abca1/Abcg1 deficiency showed a tendency to decrease the total Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)+Mac-2+ area in middle-aged mice, this decrease became statistically significant when corrected for total Mac-2+ area (Fig. 9c, d), reflecting a decrease in macrophage apoptosis. These data are consistent with T cell Abca1/Abcg1 deficiency inducing less macrophage killing upon TCR stimulation in the T cell macrophage co-incubation experiment (Fig. 4i). We then evaluated mRNA expression of inflammatory cytokines in the aortic T cell negative fraction, consisting of endothelial cells, myeloid cells, and smooth muscle cells (SMCs), in middle-aged Ldlr−/− or T-AbcdkoLdlr−/− mice. T cell Abca1/Abcg1 deficiency decreased mRNA expression of the M1 macrophage markers CC-chemokine ligand 2 (Ccl2), Il1b, and Tnf, and increased mRNA expression of the M2 macrophage marker chitinase-like 3 (Chil3), while not affecting mRNA expression of the myeloid cell marker integrin subunit alpha M (Itgam), or the cytokines Il6, Il10, or Il23a, or intracellular adhesion molecule-1 (Icam1), or the M2 macrophage marker resistin-like alpha (Retnla) (Fig. 9e). These changes in mRNA expression are consistent with a decrease in aortic inflammation, most likely due to increased T cell apoptosis and decreased IFN-γ secretion. Collectively, T cell Abca1/Abcg1 deficiency decreased atherosclerotic lesion size in middle-aged Ldlr−/− mice, presumably due to increased T cell apoptosis and decreased inflammatory gene expression in the aorta. Moreover, T cell Abca1/Abcg1 deficiency increased macrophage lesion content in middle-aged Ldlr−/− mice, likely due to decreased macrophage apoptosis.
Fig. 9. T cell Abca1/Abcg1 deficiency increases macrophage content in atherosclerotic lesions from middle-aged Ldlr−/− mice.

Ldlr−/− and T-AbcdkoLdlr−/− mice were fed a chow diet for 12–13 months. a–d Hearts were isolated and paraffin sections of the aortic root were stained for (a–c) Mac-2 and DAPI, or (d–f) Mac-2, Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), and Hoechst. a Representative pictures of Mac-2 staining (left) and quantification (right) of total Mac-2+ area. b Mac-2+ area as a percentage of total atherosclerotic lesion area (p = 0.046). a, b n = 10 Ldlr−/− and n = 10 T-AbcdkoLdlr−/− mice. c Representative pictures of Mac-2 and TUNEL stainings (left) and quantification (right) of total TUNEL+Mac-2+ area. Apoptotic macrophages were identified as Mac-2+TUNEL+ and are depicted by arrows. d TUNEL+Mac-2+ area as a percentage of total Mac-2+ area (p = 0.030). c, d n = 10 Ldlr−/− and n = 10 T-AbcdkoLdlr−/− mice. a, c Scale bar represents 100 μm. e Aortas from Ldlr−/− and T-AbcdkoLdlr−/− mice were collected, CD3− cells were isolated, RNA was extracted, and integrin subunit alpha M (Itgam), CC-chemokine ligand 2 (Ccl2) (p = 0.028), Il6, Il1b (p = 0.030), Tnf (p = 0.033), Il10, intracellular adhesion molecule-1 (Icam1), Il23a, resistin-like alpha (Retnla), and chitinase-like 3 (Chil3) (p = 0.0056) mRNA expression were measured by qPCR. n = 7 Ldlr−/− and n = 5 T-AbcdkoLdlr−/− mice. For all panels, error bars represent SEM. Biologically independent samples were included. p value was determined by unpaired two-tailed Student’s t test. *p < 0.05. Source data are provided as a Source Data file.
Discussion
These studies provide a link between defective cholesterol efflux and plasma membrane cholesterol accumulation and membrane stiffening in promoting T cell activation and T cell apoptosis with athero-protective effects in middle-aged mice (Fig. 10). Previous studies have shown that T cells accumulate cholesterol during aging10, and that T cell aging induces T cell activation and decreases peripheral T cell numbers. Mechanistically, we now found that T cell Abca1/Abcg1 deficiency induced premature T cell aging, reflected by an almost complete suppression of T cell proliferation and expression of senescence markers, as well as a pronounced increase in T cell apoptosis (Fig. 10).
Fig. 10. T cell Abca1/Abcg1 deficiency induces T cell apoptosis and T cell senescence in middle-aged mice.
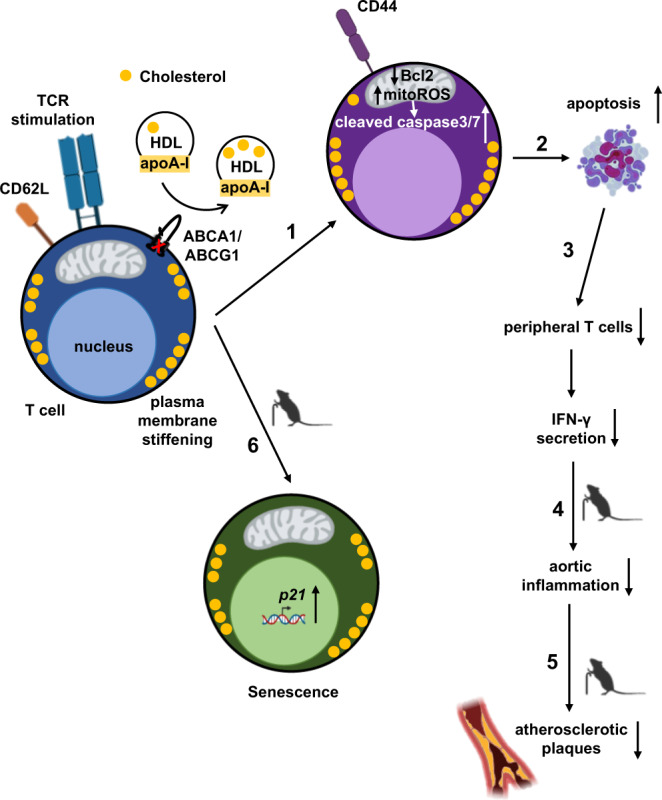
(1) T cell Abca1/Abcg1 deficiency increases T cell activation, leading to (2) T cell apoptosis and (3) decreases in peripheral T cells and interferon gamma (IFN-γ) secretion. Consequently, in middle-aged mice, T cell Abca1/Abcg1 deficiency decreases (4) aortic inflammation leading to (5) a decrease in atherosclerotic lesion area. (6) T cell Abca1/Abcg1 deficiency induces cellular senescence in middle-aged mice. TCR, T cell receptor.
Association studies in the multi-ethnic study of atherosclerosis (MESA) have shown that a low level of CD4+ naïve T cells in blood was associated with increased carotid intima media thickness (cIMT), suggesting that T cell activation enhances atherogenesis in humans60. T cells are numerous in advanced atherosclerotic plaques of human carotids (~50–65% of all plaque cells) and most T cells in atherosclerotic plaques are of the activated CD44+ phenotype2,61,62. Sharing similarities with the MESA study, another study found a positive correlation between blood Tmem/eff cells and cIMT, and elevated blood Tmem/eff cells in patients with chronic stable angina (CSA) or acute myocardial infarction (AMI)63. However, these studies do not take into account the balance between T cell activation, apoptosis, or senescence. Indeed, T cell Abca1/Abcg1 deficiency decreased the expression of pro-inflammatory cytokines in the aorta of middle-aged Ldlr−/− mice despite blood T cells being more activated. However, unlike in blood, T cell Abca1/Abcg1 deficiency decreased the percentage of CD4+ Tmem/eff cells in the aorta, accompanied by increased CD4+ T cell apoptosis. The increase in T cell apoptosis likely accounts for decreased IFN-γ secretion and decreased inflammation in the aorta. A recent study in Apoe−/− mice has also shown that 20 weeks of WTD feeding, which increases T cell cholesterol content5, increases CD4+ T cell apoptosis in T cells from para-aortic LNs and decreases CD4+IFN-γ+ T cells9, consistent with our data. However, this study was done in a global Apoe knockout and therefore effects of T cell cholesterol accumulation on atherosclerotic plaques could not be addressed directly.
T cells in atherosclerotic plaques were increased by 2-fold in middle-aged compared to young Ldlr−/− mice, suggesting a more prominent effect of T cells on atherosclerosis in middle-aged mice. Consistent with this hypothesis, previous studies have found different effects of T cells at different stages of lesion development. Complete CD4+ or CD8+ T cell ablation59,64–66 reduces atherosclerosis, suggesting that T cells in plaques are mainly pro-atherogenic. In early atherogenesis, CD4+ and CD8+ T cells induce macrophage inflammation and monopoiesis in BM mediated by IFN-γ65,66. In advanced lesions, CD8+ T cells induce macrophage apoptosis and necrotic core formation mediated by granzyme B and perforin59. T cell Abca1/Abcg1 deficiency did not affect blood myeloid cells or necrotic core formation, but increased macrophage content in middle-aged Ldlr−/− mice, accompanied by a decrease in macrophage apoptosis. The latter would perhaps not have been expected since T cell Abca1/Abcg1 deficiency increased CD8+granzyme B+ T cells upon αCD3/IL-2 stimulation. However, upon the same stimulus, Abca1/Abcg1 deficiency increased T cell apoptosis, and decreased macrophage death in a T cell mediated cytotoxicity assay. Presumably, the effect of Abca1/Abcg1 deficiency on T cell apoptosis is predominant, and consequently, less granzyme B may have been secreted to mediate macrophage death. The decreased macrophage death in the T cell mediated cytotoxicity assay likely explains the decreased macrophage apoptosis and increased macrophage lesion content in plaques.
T cell Abca1/Abcg1 deficiency also decreased CD25+Foxp3+ Treg cells in para-aortic LNs, which would be suggestive of T cell Abca1/Abcg1 deficiency being pro-atherogenic. However, Foxp3 mRNA expression was not affected in aortic T cells, suggesting no differences in Treg cells in atherosclerotic plaques. Our in vitro studies suggested that the decrease in CD25+Foxp3+Treg cells was entirely due to T cell apoptosis. T cell apoptosis is an anti-inflammatory process67.
One striking observation was that unlike in young mice, middle-aged Abca1/Abcg1-deficient T cells almost completely lost their ability to proliferate upon TCR stimulation. While this could be the consequence of increased T cell apoptosis, the senescence marker Cdkn1a was also increased, suggestive of premature T cell aging. Similar to T cell Abca1/Abcg1 deficiency, aging increases T cell plasma membrane cholesterol accumulation10,11 and increases T cell activation, while decreasing peripheral T cell numbers12,50,51. During aging, thymic involution decreases output of naïve T cells16. However, T cells undergo homeostatic proliferation in the periphery giving rise to new T cells independent of thymic output13,68. We found that T cells from aged wild-type mice showed increased cholesterol accumulation compared to young wild-type mice, and increased apoptosis upon TCR stimulation, similar to our findings in mice with T cell Abca1/Abcg1 deficiency. Together, these data indicate broader physiological relevance of our findings on apoptosis in T cell Abca1/Abcg1 deficiency, and suggest that T cell plasma membrane cholesterol accumulation may account for increased T cell apoptosis during aging, resulting in a decrease in peripheral T cell numbers.
Our finding that T cell Abca1/Abcg1 deficiency only affected atherosclerotic lesion size in middle-aged Ldlr−/− mice is suggestive of a specific effect of Abca1/Abcg1 on T cells during aging that affects atherogenesis. T cell Abca1/Abcg1 deficiency had similar effects on most T cell subsets in young and middle-aged mice, but induced T cell senescence only in middle-aged mice. In humans, T cell aging due to repeated TCR stimulation is characterized by loss of CD2869. Human T cells deficient in CD28 are senescent but still may have an effector function70, and produce high levels of TNF and IFN-γ71. While initial studies reported that CD4+CD28null T cells correlate with unstable angina and acute coronary syndromes in humans71–74, this was recently challenged by a multi-center study with a larger number of patients75. Opposite to the previous findings71–74, in this study75, levels of CD4+CD28null T cells in blood were associated with a lower risk for first time coronary events in a population-based cohort, and did not correlate with cIMT. Based on these findings75, it is unlikely that senescent T cells contributed to lesion size in our study.
In addition to atherosclerotic plaques, peripheral T cells in blood, LNs, and spleen were decreased in T-AbcdkoLdlr−/− mice compared to their littermate controls. Although Ldlr mediated LDL uptake increases TCR stimulation in vitro76, illustrating the importance of membrane cholesterol content for TCR signaling, Ldlr deficiency has not been reported to decrease peripheral T cell numbers in vivo. Consistently, we found that the increased T cell apoptosis and decreased peripheral T cells upon T cell Abca1/Abcg1 deficiency were independent of Ldlr expression. Single deficiency of Abca1 or Abcg1 did not affect peripheral T cells in blood or T cell apoptosis, likely because deficiency of one transporter results in upregulation of the other. Indeed, it has been shown previously that Abcg1 deficiency upregulates Abca1 expression in T cells23.
A previous study has attributed the decrease in peripheral T cells in mice with T cell Abca1/Abcg1 deficiency or T cell deficiency of the transcription factor the liver X receptor (LXR)α and β that induces Abca1 and Abcg1 expression, to increased thymic CD4+and CD8+ T cell apoptosis, while not excluding that extrathymic effects on T cells may also have contributed29. However, in these studies, the Abca1 and Abcg1 floxed genes and also the Lxrα/Lxrβ floxed genes were expressed under control of the LckCre promoter29. In T cells, Lxrβ is more highly expressed than Lxrα20. CD4CreLxrβfl/flmice showed decreased peripheral T cells with only thymic CD4+ T cells and CD4+ Treg cells being decreased, while thymic CD8+ T cells were unchanged21. Perhaps the LckCre promoter resulted in a more complete deficiency of Abca1 and Abcg1 in single positive thymic CD4+ and CD8+ T cells than the CD4Cre promoter that we and others used21, explaining the more pronounced effects on thymic T cells. The decrease in thymic CD4+ and CD8+ T cells in LckCreLxrαfl/flLxrβfl/fl and LckCreAbca1fl/flAbcg1fl/fl mice was attributed to increased surface expression of the cell death receptor FAS29, which is localized in lipid rafts43. Also, upon TCR stimulation, peripheral CD4+ T cells from CD4CreLxrβfl/fl mice showed increased FAS expression and apoptosis; however it was suggested that additional mechanisms may contribute to apoptosis in CD4CreLxrβfl/fl T cells21. Our data in CD4CreAbca1fl/flAbcg1fl/flLdlr−/− mice show that FAS expression was not affected upon TCR stimulation, but Bcl2 expression was decreased. We thus attributed the increased apoptosis in CD4+ and CD8+ T cells (the latter mainly in the CD8+Tmem/eff T cells) from CD4CreAbca1fl/flAbcg1fl/flLdlr−/− mice to a decrease in the anti-apoptotic protein Bcl2. This mechanism may also have contributed to the increased apoptosis in CD4+T cells from CD4CreLxrβfl/fl mice.
Similar to CD4CreAbca1fl/flAbcg1fl/flLdlr−/− mice, CD4CreLxrβfl/flmice showed increased spontaneous T cell activation21. This was attributed to a functional impairment of CD4CreLxrβfl/fl Treg cells, and recapitulated in Foxp3CreLxrβfl/fl mice, indicating that it was a Treg cell-intrinsic effect21. In these specific Foxp3CreLxrβfl/fl T cells, unlike in other CD4+ T cells, Abca1 and Abcg1 expression were not affected by Lxrβ deficiency, suggesting that other LXR target genes contributed to the dysfunctional Treg differentiation21. Although peripheral Treg cells were also decreased in CD4CreAbca1fl/flAbcg1fl/flLdlr−/− mice, there was no specific decrease in the percentage of CD25+Foxp3+ T cells, as in CD4CreLxrβfl/fl mice21, and the decreases in peripheral Treg cells were mainly the consequence of an overall increase in T cell apoptosis. Given that TCR signaling was increased in CD4CreAbca1fl/flAbcg1fl/flLdlr−/− mice, and enhanced TCR signaling promotes T cell activation38, we postulate that this mechanism accounted for the increase in T cell activation in CD4CreAbca1fl/flAbcg1fl/flLdlr−/− mice compared to controls.
Several studies have shown that increased plasma membrane cholesterol accumulation enhances TCR signaling8,20,23,77. Conversely, impaired cholesterol synthesis in the context of SCAP deficiency suppresses TCR signaling37. We found that even when TCR-induced T cell proliferation was almost completely abolished in T cells with Abca1/Abcg1 deficiency, presumably due to T cell senescence, cholesterol depletion by rHDL still further decreased it, substantiating the importance of T cell membrane cholesterol for TCR signaling. Acat1 deficiency increases plasma membrane cholesterol accumulation and TCR nanoclustering, which enhances TCR signaling8. Cholesterol accumulation also increased TCR nanoclustering in a study employing artificial membranes78; however a later study from the same group showed that binding of cholesterol to TCRβ inhibited TCR signaling79, suggesting rather an inhibitory than a stimulating effect. Upon TCR signaling, the plasma membrane cholesterol content increases20,21, presumably due to suppression of Abca1 and Abcg1 expression and increased cholesterol synthesis20. Indeed, our studies have shown that T cell Abca1/Abcg1 deficiency increased plasma membrane cholesterol accumulation leading to plasma membrane stiffening and enhanced TCR signaling. Moreover, a recent study has shown that T cell histone deacetylase 3 (Hdac3) deficiency decreases TCR signaling and CD4+ T cell proliferation, which was attributed to decreased membrane cholesterol content and increased Abca1 and Abcg1 mRNA expression77. This substantiates our data on T cell Abca1/Abcg1 deficiency increasing TCR signaling.
In conclusion, our studies indicate a link between T cell plasma membrane cholesterol accumulation, T cell plasma membrane stiffening, TCR signaling, T cell activation, and T cell apoptosis in T cell Abca1/Abcg1 deficiency, which suppresses aortic inflammation and atherosclerosis in middle-aged, but not young mice. Our studies indicate that plasma membrane cholesterol accumulation not only induces apoptosis, but also a premature T cell aging phenotype characterized by an increase in T cell senescence. This suggests that upregulation of Abca1 and Abcg1 in T cells, for example by an LXR agonist, may suppress T cell apoptosis and senescence, which could be crucial in maintaining T cell numbers and peripheral T cell tolerance, especially during aging. Further, statins, although reported to inhibit TCR induced T cell proliferation8, could also suppress T cell apoptosis and senescence by suppressing cholesterol synthesis in T cells. It would be of interest to investigate this further in a large population cohort.
Methods
Animals
Abca1fl/flAbcg1fl/fl (strain #: 021067), CD4Cre (strain #: 017336), Ldlr−/− (strain #: 002207), and wild-type (strain #: 000664) mice were purchased from Jackson Laboratories (Bar Harbor, ME). All mice were in the C57Bl6/J background. Abca1fl/flAbcg1fl/fl (control; Ldlr+/+) and CD4Cre mice were intercrossed to obtain Abca1fl/flAbcg1fl/fl, Abca1fl/fl, Abcg1fl/fl, CD4CreAbca1fl/fl, CD4CreAbcg1fl/fl, and CD4CreAbca1fl/flAbcg1fl/fl (T-Abcdko) littermates. CD4CreAbca1fl/fl, CD4CreAbcg1fl/fl, and CD4CreAbca1fl/flAbcg1fl/fl mice were intercrossed with Ldlr−/− mice to generate Abca1fl/flLdlr−/−, Abcg1fl/flLdlr−/−, Abca1fl/flAbcg1fl/flLdlr−/− (all Ldlr−/−), CD4CreAbca1fl/fllLdlr−/− (T-Abca1koLdlr−/−), CD4CreAbcg1fl/flLdlr−/− (T-Abcg1koLdlr−/−), and CD4CreAbca1fl/flAbcg1fl/flLdlr−/− (T-AbcdkoLdlr−/−) littermates. Mice were fed a chow diet (10% fat, 23% protein, and 67% carbohydrates; V1554-703, Ssniff Spezialdiäten GmbH) or, starting at 8–12 weeks of age, a WTD (40% fat, 0.15% cholesterol; D12079B, Research Diets).
For all studies, littermates were used, and mice were housed under standard laboratory conditions at 21.5 °C (range: 20.5–22.5 °C) ambient temperature and 55% (range: 30–70%) humidity with a light cycle of 12 h and ad libitum water and food (chow diet or WTD). Water and cages were autoclaved. Cages were changed once weekly, and the health status of the mice was monitored based on body weight, coat, and behavior. The mouse genotype did not cause changes in body weight (mouse body weight between 20–40 g, depending on gender and age) or health. Female littermates (atherosclerosis studies) or littermates from both sexes were randomly assigned to experimental groups, unless stated otherwise. The exact number of the mice used for the experiments are indicated for each experiment in the figure legends. In general, n = 3–18 mice were used per group and experiments were repeated at least once if n = 3 mice were used per experiment to confirm the reproducibility of the results. No inclusion or exclusion criteria were used. All protocols were approved by the Institutional Animal Care and Use Committee from the University of Groningen (Groningen, the Netherlands) under permit number AVD105002015244 and adhered to guidelines set out in the 2010/63/EU directive.
Reagents and resources
Please see Supplementary Table 3.
T cell and thymocyte isolations
Spleens were isolated from control (Ldlr+/+), T-Abcdko, Ldlr−/−, T-AbcdkoLdlr−/−, T-Abca1koLdlr−/−, T-Abcg1koLdlr−/−, and young (3 months) and aged (24 months) wild-type mice, mashed on a 40 μm strainer, and red blood cells (RBCs) were lysed (BD Pharm Lyse, BD Biosciences). Para-aortic LNs were isolated from Ldlr−/− and T-AbcdkoLdlr−/− mice and mashed on a 40 μm strainer in PBS. Splenic and LN homogenates were then centrifuged, washed, and resuspended in MACS buffer (PBS containing 0.5% BSA and 2 mM EDTA). Dead cells were removed using the Dead Cell Removal kit (Miltenyi Biotec), and total T cells were isolated using the Pan T cell kit (Miltenyi Biotec). CD4+ and CD8+ T cells were isolated using CD4 and CD8 coated magnetic beads (Miltenyi Biotec), respectively. For thymocyte isolations, Ldlr−/− and T-AbcdkoLdlr−/− thymi were isolated and mashed on a 40 μm strainer in PBS.
Aortic cell isolations
Aortas were isolated from middle-aged (12–13 months) female Ldlr−/− and T-AbcdkoLdlr−/− mice. For mRNA expression analyses, aortas were digested for 1 h at 37 °C using an enzyme mixture that contained liberase TH (4 U/mL; Roche), DNAse I (40 U/mL; Sigma-Aldrich), and hyaluronidase (60 U/mL; Sigma-Aldrich). Aortic homogenates were then centrifuged, washed, and resuspended in MACS buffer. Total aortic CD3+ T cells and CD3− cells were isolated using the Pan T cell kit. For in vitro stimulation, aortas were digested for 45 min at 37 °C using an enzyme mixture that contained liberase TM (4 U/mL; Roche), DNAse I (120 U/mL), and hyaluronidase (60 U/mL). Aortic digestion was stopped by adding 10% FBS in RPMI 1640 medium (Gibco). Aortic homogenates were then passed through a 70 μm strainer, centrifuged, washed, and resuspended in RPMI 1640 medium supplemented with 10% FCS, 1% pen-strep, and 50 μM β-mercaptoethanol.
RNA extraction from splenic and aortic cells
Splenic total T cells, CD4+, and CD8+ T cells from 4–5 months old male and female control, T-Abcdko, and Ldlr−/− mice, middle-aged (12–13 months) male and female Ldlr−/− and T-AbcdkoLdlr−/− mice, and young (3 months) and aged (24 months) wild-type male and female mice, and aortic total CD3+ T cells and CD3− cells from middle-aged female Ldlr−/− and T-AbcdkoLdlr−/− mice were isolated as described above and resuspended directly into RLT buffer. RNA was then extracted using the RNeasy Mini Kit (Qiagen) and cDNA was synthesized using the Transcriptor Universal cDNA Master kit (Roche). Abca1, Abcg1 (in splenic total T cells from control and T-Abcdko mice and CD4+ and CD8+ T cells from young wild-type and aged wild-type mice), Ldlr, Hmgcr, Hmgcs, Srebf2, Srebf1, Acat1 (in CD4+ and CD8+ T cells from control, T-Abcdko, young wild-type, and aged wild-type mice), Cdkn2a (in splenic CD4+ and CD8+ T cells from middle-aged Ldlr−/− and T-AbcdkoLdlr−/− mice), Cdkn1a (in splenic CD4+ and CD8+ T cells and aortic total CD3+ T cells from middle-aged Ldlr−/− and T-AbcdkoLdlr−/− mice), Foxp3, Tgfb1, Bcl2, Il10, Tnf (in aortic total CD3+ T cells from middle-aged Ldlr−/− and T-AbcdkoLdlr−/− mice), Itgam, Il6, Il1b, Ccl2, Icam1, Il23a, Retnla, and Chil3 (in aortic CD3− cells from middle-aged Ldlr−/− and T-AbcdkoLdlr−/− mice) mRNA levels were assessed by qPCR using QuantStudio 7 Flex Real-Time PCR System (Applied Biosystems), and initial differences in RNA quantity were corrected for using the housekeeping genes 36b4 and Cyclophilin B. All primer sequences are available in Supplementary Table 3.
Filipin staining—flow cytometry
To analyze T cell membrane cholesterol, blood leukocytes (4–5 months old male and female Ldlr−/− and T-AbcdkoLdlr−/− mice, and young (3 months) and aged (24 months) wild-type male and female mice) and thymocytes (4–5 months old male and female Ldlr−/− and T-AbcdkoLdlr−/ mice) were stained with antibodies against TCRβ-APC, CD8-PE, and CD4-FITC, and thymocytes with antibodies against CD4-APC and CD8-PE. Cells were then centrifuged, washed, and stained with 50 μg/mL Filipin III (Sigma-Aldrich) in 10% FCS/PBS for 45 min at room temperature (RT) and then washed twice with PBS. Samples were analyzed by flow cytometry immediately after staining. Filipin III mfi on blood TCRβ+CD4+ and TCRβ+CD8+ cells, and on thymic CD4+CD8+, CD4+, and CD8+ T cells was assessed on LSRII (BD Biosciences), running FACSDiVa software (BD Biosciences). The data were analyzed using the FlowJo software (FlowJo).
Confocal microscopy, BODIPY, and filipin staining
Splenic CD4+ and CD8+ T cells from 4–5 months old male and female control, T-Abcdko, Ldlr−/−, and T-AbcdkoLdlr−/− mice, and total T cells from 4–5 months old male and female T-Abca1koLdlr−/− and T-Abcg1koLdlr−/− mice, and young (3 months) and aged (24 months) wild-type male and female mice were isolated as described above. Cells were seeded on Poly-L-Lysine (PLL)-coated glass coverslips and incubated at 37 °C for 1 h. For lipid droplet imaging, cells were fixed (4% PFA; 15 min; 4 °C), permeabilized for 30 min at RT using CLSM buffer (PBS containing 3% BSA, 10 mM glycine, 0.1% saponin), and stained for 30 min at RT with BODIPY 493/503 (Invitrogen; 7.5 μM in CLSM buffer). After two PBS washes, cells were mounted using VECTASHIELD Antifade Mounting Medium with DAPI (Vector Laboratories). To assess free cholesterol, cells were fixed (3% PFA; 1 h; RT) and stained with filipin (50 μg/mL in 10% FCS in PBS; 2 h; RT). After three PBS washes, cells were mounted using VECTASHIELD Antifade Mounting Medium (Vector Laboratories).
All samples were imaged on the Zeiss LSM800 microscope with immersion oil lens (63X,1.4 N.A) running Zen software (Zeiss). Background signal was removed using Zen software and fluorescence intensity of filipin or BODIPY was analyzed using ImageJ software (NIH).
Choleratoxin B staining
For analysis of T cell lipid rafts, blood leukocytes from 4–5 months old male and female Ldlr−/− and T-AbcdkoLdlr−/− mice, and young (3 months) and aged (24 months) wild-type male and female mice were stained with a cocktail of antibodies against TCRβ-PB, CD8-PE, and CD4-APC. Cells were then centrifuged, washed, and stained with 200 ng/mL choleratoxin B-FITC (Sigma-Aldrich) for 1 h at RT. Samples were analyzed by flow cytometry immediately after staining. Choleratoxin B mfi on TCRβ+CD4+ and TCRβ+CD8+ cells was assessed by flow cytometry on the LSRII as described above for the filipin staining, and is an indirect indicator of lipid rafts.
Gas chromatography—mass spectrometry
Splenic T cells from female Ldlr−/− and T-AbcdkoLdlr−/− mice fed a chow diet for 28 weeks, and young (3 months) and aged (24 months) wild-type male and female mice were isolated as described above. Cholestanol-D5 was added to the T cells as internal standard and cholesterol was extracted using hexane. For T cells from Ldlr−/− and T-AbcdkoLdlr−/− mice, samples were split for measurement of either total or free cholesterol content using Gas Chromatography—Mass Spectrometry (7890B GS system, 5973 MS system, and 7693 A automatic liquid sampler from Agilent; positive chemical ionization mode with 5% ammonia in methane as reaction gas). A polar DB-WAXetr (30 m × 0.25 mm × 0.25 µm) column was used. CE content was calculated by subtracting free cholesterol from total cholesterol. Cholesterol content was normalized to cellular protein content measured by the Lowry assay. For T cells of young and aged wild-type mice, total cholesterol content was assessed using the same procedure.
Oil red O staining
Female Ldlr−/− and T-AbcdkoLdlr−/−mice were fed a chow diet for 28 or 38 weeks. After the indicated period of time on diet, mice were sacrificed and para-aortic LNs (28 weeks) and hearts (38 weeks) were isolated and embedded into OCT compound on dry ice. Frozen cross-sections (4 μm thickness) were made and stained with Oil Red O (Sigma-Aldrich) to assess neutral lipid accumulation. Sections were counterstained with haematoxylin. Quantification of the number of lipid droplets on sections of para-aortic LN sections (per field) was performed in a blinded fashion, i.e. the observer was unaware of the genotypes. For atherosclerosis analysis, the average from 6 heart sections (40 μm distance between sections) for each animal was used to determine lesion size. Lesion size was quantified by morphometric analysis and Oil Red O+area was measured using ImageJ software (NIH).
Transmission electron microscopy
Splenic CD4+ and CD8+ T cells from 4–5 months old male and female Ldlr−/−and T-AbcdkoLdlr−/− mice were isolated as described above. Cells were seeded on PLL-coated glass coverslips placed in 24-well plates and incubated at 37 °C for 1 h. Cells were then fixed with 2% glutaraldehyde (Sigma-Aldrich) in PB (0.1 M phosphate buffer, pH 7.4) for 60 min at RT. After three PB washes, cells were post-fixed with 1% osmium tetroxide in PB for 60 min at RT. Cells were incubated overnight in 0.5% uranyl acetate, dehydrated with graded steps of ethanol (50%, 70%, 96%, 100%), and embedded in Epon resin. Sections of 70 nm thickness were made and stained with 2% uranyl acetate solution and lead citrate solution. Stained sections were then examined using a CM12 transmission electron microscope (Phillips).
Fluorescence-lifetime imaging microscopy
Splenic CD4+ and CD8+ T cells from 4–5 months old male and female Ldlr−/−and T-AbcdkoLdlr−/− mice were isolated as described above. Cells were stained with BODIPY C10 (kind gift from Dr. Ulf Diederichsen, Georg-August-Universität Göttingen, Germany; 4 μM; 30 min; on ice). Cells were washed twice with phenol red free RPMI 1640 medium and seeded in PLL coated µ-Slide 8 Well Glass Bottom Chambers (Ibidi). Cells were incubated at 37 °C for 30 min, and then BODIPY C10 fluorescence lifetime was assessed by fluorescence-lifetime imaging microscopy (FLIM). All images were collected with a MicroTime 200 microscope (PicoQuant) equipped with an Olympus (100x/1,4) oil immersion objective. Images were acquired using the SymPhoTime 64 software (PicoQuant). Data analysis of the FLIM images was performed using the open-source FLIMfit software80. Further image loading options included 2 × 2 spatial binning and time bins 1563 (32 ps/bin). After loading FLIM images, the software’s default settings were used. BODIPY C10 fluorescence lifetime of a whole cell (with lipid droplets (LDs)) or of the plasma membrane without including LDs was determined by fitting the fluorescence histograms with a mono-exponential decay function, using the tool “Fit Selected Decay”. Per mouse, 43–76 CD4+ and CD8+ T cells were analyzed.
Flow cytometry and total white blood cell counts
Blood samples were collected from 4–5 months old male and female control, T-Abcdko, Ldlr−/−, T-Abca1koLdlr−/− and T-Abcg1koLdlr−/− mice, male and female Ldlr−/− and T-AbcdkoLdlr−/− mice (4–5 or 12–13 months old, or on WTD for 10 weeks), and young (3 months) and aged (24 months) wild-type male and female mice by tail bleeding into Eppendorf tubes containing EDTA. Total white blood cell counts were assessed using the Medonic CD620 hematology analyzer (Boule Medical). For flow cytometry, tubes were kept at 4 °C for the whole procedure unless stated otherwise. RBCs were lysed, and white blood cells were centrifuged, washed, and resuspended in FACS buffer (HBSS containing 0.1% BSA and 0.5 mM EDTA). For all stainings, antibodies listed below were used at a 1/200 dilution, unless stated otherwise. For analysis of blood T cells, cells were stained with a cocktail of antibodies against TCRβ-PB, CD8-FITC, and CD4-APC. CD4+ T cells were identified as TCRβ+CD4+ and CD8+ T cells as TCRβ+CD8+. For analysis of naïve and activated blood T cells, cells were stained with a cocktail of antibodies against TCRβ-PB, CD8-FITC, CD44-PE-Cy7, and CD62L-APC. Among these populations, Tnaive cells were identified as CD44−CD62L+, TCM cells as CD8+CD44+CD62L+, and Tmem/eff cells as CD44+CD62L−. The same stainings were carried out on para-aortic LN and splenic homogenates. For analysis of thymocytes (4–5 months old male and female Ldlr−/− and T-AbcdkoLdlr−/− mice), thymic homogenates were stained with CD4-APC and CD8-FITC or TCRβ-PerCP-Cy5.5, CD24-PE-Cy7, and CD69-APC. For analysis of blood myeloid cells (for male and female Ldlr−/− and T-AbcdkoLdlr−/− mice; 4–5 months old or on WTD for 10 weeks), cells were stained with a cocktail of antibodies against CD45-PE-Cy7, Ly6-C/G-PerCP-Cy5.5, and CD115-PE. Monocytes were identified as CD45+CD115+ and further separated into Ly6-Chi and Ly6-Clo subsets, and neutrophils were identified as CD45+CD115−Ly6-C/Ghi.
For analysis of Treg cells in thymus (4–5 months old male and female Ldlr−/− and T-AbcdkoLdlr−/− mice) and para-aortic LN (female Ldlr−/− and T-AbcdkoLdlr−/− mice fed a chow diet for 28 weeks or a WTD for 10 weeks), tissue homogenates were stained with a cocktail of antibodies against CD4-FITC and CD25-APC (for LN Treg cells; Mouse Regulatory T Cell Staining Kit #1, eBioscience) or CD4-PB, CD8-FITC, and CD25-PECy7 (for thymic Treg cells). Cells were then fixed and permeabilized using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience), and subsequently stained with Foxp3-PE (for LN Treg cells) or Foxp3-APC (for thymic Treg cells) (1/100 dilution) or their isotype control (1/100 dilution). Treg cells were identified as CD8−CD4+CD25+Foxp3+ and CD8−CD4+CD25−Foxp3+. For analysis of naïve and activated Treg cells, LN homogenates were stained with a cocktail of antibodies against CD4-PE, CD25-PECy7, CD44-PB, and CD62L-FITC. Cells were then fixed and permeabilized using the Foxp3/Transcription Factor Staining Buffer Set, and subsequently stained with Foxp3-APC (1/100 dilution) or its isotype control (1/100 dilution). Naïve Treg cells were identified as CD4+CD25+Foxp3+CD44−CD62L+and CD4+CD25−Foxp3+CD44−CD62L+. Activated Treg cells were identified as CD4+CD25+Foxp3+CD44+CD62L− and CD4+CD25−Foxp3+CD44+CD62L− (Tmem/eff). For analysis of TFH cells and total PD1+ T cells, splenic and LN homogenates were stained with a cocktail of antibodies against CD4-APC-Cy7, CD44-PE-Cy7, CD62L-APC, PD1-PB, and CXCR5-FITC (1/400 dilution). TFH cells were defined as CD4+CD44+CD62L−CXCR5+PD1+and total PD1+ T cells as CD4+PD1+.
Splenic and LN homogenates from 4–5 months old male and female Ldlr−/− and T-AbcdkoLdlr−/− mice were analyzed for the exhaustion markers CTLA4, TIM3, LAG3, and Eomes. Tissue homogenates were stained with a cocktail of antibodies against CD4-APC, CD8-FITC, and CTLA4-PB or with a cocktail of antibodies against TCRβ-PerCPCy5.5, CD4-FITC, CD8-PE, and TIM3-APC or LAG3-APC. Alternatively, tissue homogenates were stained with a cocktail of antibodies against TCRβ-PB, CD8-APC, and CD4-FITC, then fixed and permeabilized, and subsequently stained with Eomes-PE (1/100 dilution) or its isotype control (1/100 dilution). CTLA4, TIM3, and LAG3 mean fluorescence intensity (mfi) on CD4+and CD8+T cells, and Eomes mfi in CD8+T cells was assessed by flow cytometry.
For analysis of Tbet+ cells, LN homogenates from female Ldlr−/− and T-AbcdkoLdlr−/− mice fed a chow diet for 28 weeks or a WTD for 10 weeks were stained with a cocktail of antibodies against TCRβ-PB, CD4-APC, and CD8-FITC, then fixed and permeabilized, and subsequently stained with Tbet-PE (1/100 dilution) or its isotype control (1/100 dilution). Tbet+cells were identified as TCRβ+CD4+Tbet+and TCRβ+CD8+Tbet+. For analysis of naïve and activated Tbet+cells, LN homogenates were stained with a cocktail of antibodies against TCRβ-PB, CD8-PE, CD44-PECy7, and CD62L-APC, then fixed and permeabilized, and subsequently stained with Tbet-AF488 (1/100 dilution) or its isotype control (1/100 dilution). Naive Tbet+cells were identified as TCRβ+CD4+Tbet+CD44−CD62L+and TCRβ+CD8+Tbet+CD44−CD62L+. Activated Tbet+cells were identified as TCRβ+CD4+Tbet+CD44+CD62L−, TCRβ+CD8+Tbet+CD44+CD62L− (Tmem/eff) and TCRβ+CD8+Tbet+CD44+CD62L+(TCM) cells.
All samples were analyzed on LSRII (BD Biosciences), running FACSDiVa software (BD Biosciences). The data were analyzed using the FlowJo software (FlowJo).
In vitro T cell proliferation assay
Splenic T cells from male and female control, T-Abcdko, Ldlr−/−, and T-AbcdkoLdlr−/− mice (4–5 or 12–13 months old), young (3 months) and aged (24 months) wild-type male and female mice, and total aortic cells from middle-aged (12–13 months) male and female Ldlr−/− and T-AbcdkoLdlr−/− mice were isolated and labeled with 10 μM CFSE-PB (Invitrogen) and stimulated with Mouse T-Activator CD3/CD28 Dynabeads (ratio beads to cells 1:2 for splenic cells and 10:1 for aortic cells) in RPMI 1640 medium supplemented with 10% FCS, 1% pen-strep, and 50 μM β-mercaptoethanol at 37 °C, 5% CO2. Splenic T cells from Ldlr−/− and T-AbcdkoLdlr−/− mice were also incubated with 50 μg/mL rHDL for the whole duration of the proliferation assay. Cells were incubated at 37 °C for 72 h (splenic) or 96 h (aortic). Subsequently, beads were removed, T cells were stained using CD4 and CD8 antibodies as described above, and CFSE-PB dilution was assessed by flow cytometry. Using the Proliferation Modeling tool in the FlowJo software, histograms of CFSE dilution in CD4+ and CD8+ T cells were obtained where each cell division was depicted as a separate peak. The number of CD4+ and CD8+ T cells in each division was automatically calculated by the software after designing gates for each peak. To calculate the percentage of CD4+ and CD8+ T cells in each division, the number of CD4+ and CD8+ T cells in one division was divided by the total number of CD4+ and CD8+ T cells in all divisions, respectively.
Western blot of JNK and gasdermin D
Splenic CD4+ and CD8+ T cells from 4–5 months old male and female Ldlr−/− and T-AbcdkoLdlr−/− mice were isolated as described above. Cell culture conditions were the same as described above. To assess JNK1/2 phosphorylation, ~1.5 million CD4+ and CD8+ T cells were stimulated with 5 μg/mL αCD3 (Biolegend) for 0, 5, and 10 min. To assess gasdermin D cleavage, ~2 million CD4+ T cells were stimulated with 5 μg/mL plate-bound αCD3 and 100 U/mL IL-2 (Peprotech) for 12 h. After stimulation, cells were harvested and lysed in radio immuno precipitation assay (RIPA) buffer (1% IGEPAL, 0.1% SDS, and 0.5% sodium deoxycholate in PBS) supplemented with complete protease inhibitor cocktail (Sigma-Aldrich), and phosphatase inhibitor cocktail 2 and cocktail 3 (Sigma-Aldrich). Cell lysates were separated by SDS-PAGE gel electrophoresis and immunoblotted with primary antibodies against total JNK1/2 (1:1000; Cell Signaling Technology), phosphorylated JNK1/2 (1:1000; Cell Signaling Technology), or gasdermin D (1:1000; a kind gift from Genentech). Heat shock protein 90 (HSP90) (1:1000; Cell Signaling Technology) was used as loading control. Goat anti-mouse (1:2000; Invitrogen; for phosphorylated JNK1/2), goat anti-rabbit (1:2000; Invitrogen; for total JNK1/2 and HSP90), and goat anti-rat (1:1000; Cell Signaling Technology; for gasdermin D) HRP-conjugated secondary antibodies were used. Blots were developed using the Amersham Imager 600 (GE Healthcare) and bands were quantified using ImageJ software (NIH). Uncropped and unprocessed scans of blots are shown in the Source Data file.
In vitro T cell apoptosis assays
Splenic CD4+ and CD8+ T cells from 4–5 months old male and female Ldlr−/− and T-AbcdkoLdlr−/− mice were isolated and labeled using Caspase3/7 Red (APC) Reagent (Sartorius). Cells were cultured as described above, and stimulated with 5 μg/mL plate-bound αCD3 and 100 U/mL IL-2 for 12 h. For Incucyte experiments, images were taken every 2 h. For image analysis, background signal was removed using the Incucyte ZOOM software (Sartorius) and the number of total cells and cleaved caspase3/7+ cells per image were quantified using ImageJ software (NIH) in a blinded fashion, i.e. the observer was unaware of the genotypes. Alternatively, splenic T cells from 4–5 month old male and female control (Ldlr+/+), T-Abcdko, T-Abca1koLdlr−/− and T-Abcg1koLdlr−/− mice, middle-aged (12–13 months) male and female Ldlr−/− and T-AbcdkoLdlr−/− mice, and young (3 months) and aged (24 months) wild-type male and female mice were isolated and labeled using caspase3/7 reagent (1/1000 dilution), stimulated as described for the Incucyte assay, harvested, stained with CD4-PB (1/200 dilution) or CD8-PB (1/200 dilution), and analyzed by flow cytometry for cleaved caspase 3/7 mfi in splenic CD4+ or CD8+ T cells. Alternatively, total aortic cells from middle-aged (12–13 months) female Ldlr−/− and T-AbcdkoLdlr−/− mice were isolated as described above and stimulated with Mouse T-Activator CD3/CD28 Dynabeads (ratio beads to cells 10:1) in RPMI 1640 medium supplemented with 10% FCS, 1% pen-strep, and 50 μM β-mercaptoethanol for 96 h. Beads were then removed and T cells were stained with caspase3/7 reagent for 1 h at 37 °C. Cells were then centrifuged, washed, resuspended in FACS buffer, and stained with a cocktail of antibodies against CD4-FITC (1/200 dilution), CD44-PE-Cy7 (1/200 dilution), and CD62L-PB (1/200 dilution), and analyzed by flow cytometry. Cleaved caspase3/7 mfi in aortic CD4+ or CD4+Tmem/eff T cells, and the percentages of aortic CD4+Tmem/eff cells were assessed by flow cytometry.
In vitro Treg differentiation assay
Splenic T cells from 4–5 months old male and female Ldlr−/− and T-AbcdkoLdlr−/− mice were isolated and stimulated with Mouse T-Activator CD3/CD28 Dynabeads (ratio beads to cells 1:2) in the presence of 100 U/mL IL-2 and 5 ng/mL TGF-β1 (Peprotech) for 72 h. Then, beads were removed, T cells were stained using CD4, CD25, and Foxp3 antibodies as described above, and samples were analyzed by flow cytometry.
FAS and Bcl2 assays
For all stainings, antibodies listed below were used at a 1/200 dilution, unless stated otherwise. Splenic T cells from 4–5 months old male and female Ldlr−/− and T-AbcdkoLdlr−/− mice were isolated as described above and stimulated as described for the Incucyte caspase 3/7 assay. Cells were harvested and stained with a cocktail of antibodies against CD4-PB, CD8-PE, and FAS-AF488 (for FAS analysis), or CD4-PB (for Bcl2 analysis in CD4+ T cells), or TCRβ-PB, CD8-PE, CD44-PE-Cy7, and CD62L-APC (for Bcl2 analysis in CD8+ T cells). For Bcl2 analysis, cells were then fixed and permeabilized, and subsequently stained with Bcl2-FITC (1/40 dilution) or its isotype control (1/40 dilution). Samples were analyzed by flow cytometry. FAS mfi on CD4+ and CD8+ T cells was assessed. Percentages of CD4+Bcl2+, CD4+Bcl2−, CD8+Bcl2+, CD8+Bcl2−, CD8+TCM Bcl2+, and CD8+TCM Bcl2−, CD8+Tmem/effBcl2+, and CD8+Tmem/effBcl2− T cells were quantified.
Mitochondrial ROS assay
Splenic T cells from 4–5 months old male and female Ldlr−/− and T-AbcdkoLdlr−/− mice were isolated as described above and stimulated as described for the Incucyte caspase 3/7 assay. Cells were harvested and stained with 2 μM MitoSOX Red Mitochondrial Superoxide Indicator or 50 nM MitoTracker Green FM (Invitrogen; carbocyanine-based dye) for 30 min at 37 °C. For all stainings, antibodies were used at a 1/200 dilution. For MitoSOX (eBioscience) analysis, cells were stained with a cocktail of antibodies against CD44-PE-Cy7, CD62L-APC, CD4-PB or CD8-PB. For MitoTracker analysis, cells were stained with a cocktail of antibodies against CD44-PB, CD62L-APC, CD4-PE or CD8-PE. MitoSOX and Mitotracker mfi on CD4+and CD8+Tnaive, CD4+and CD8+Tmem/eff, and CD8+TCM cells was assessed by flow cytometry. To correct the levels of mitochondrial ROS for mitochondrial mass, the MitoSOX/Mitotracker ratio was calculated.
Analysis of T cell granzyme B, IFN-γ, and LAMP-1
Splenic CD4+ and CD8+ T cells from 4–5 months old male and female Ldlr−/− and T-AbcdkoLdlr−/− mice, and young (3 months) and aged (24 months) wild-type male and female mice were isolated as described above and stimulated as described for the Incucyte caspase 3/7 assay. Cells were harvested and stained with a cocktail of antibodies against CD4-PB (1/200 dilution) and CD8-APC (1/200 dilution), and in some assays LAMP-1-FITC (1/200 dilution), or subsequently fixed and permeabilized, and stained with granzyme B-FITC (1/100 dilution) or its isotype control (1/100 dilution), or IFN-γ-FITC (1/100 dilution). Samples were analyzed by flow cytometry. LAMP-1 surface expression on CD4+ and CD8+ T cells was assessed as mfi. Percentages of CD8+granzyme B+, CD4+IFN-γ+, and CD8+IFN-γ+T cells were quantified. For IFN-γ analysis, IFN-γ+T cells were selected based on the fluorescence minus one (FMO) control.
IFN-γ secretion
Splenic T cells from 4–5 months old male and female Ldlr−/− and T-AbcdkoLdlr−/− mice, and young (3 months) and aged (24 months) wild-type male and female mice were isolated as described above and stimulated as described for the Incucyte caspase 3/7 assay Medium was collected after stimulation and the concentration of IFN-γ was determined by ELISA (R&D Systems).
T cell mediated cytotoxicity
Bone marrow (BM) cells were isolated from the femurs and the tibias of young (3 months) wild-type mice and were cultured at 37 °C, 5% CO2 in DMEM (Gibco) supplemented with 10% FCS, 1% pen strep, and 20% L-cell conditioned medium to induce differentiation into BM derived macrophages (BMDMs).
Splenic total T cells from 4–5 months old male and female Ldlr−/− and T-AbcdkoLdlr−/− mice were isolated as described above. To study the effect of Abca1/Abcg1 deficiency on T cell mediated cytotoxicity on macrophages, T cells, while being stimulated with 5 μg/mL αCD3 and 100 U/mL IL-2, were incubated with BMDMs (T cell to BMDM ratio 6:1). After 24 h, medium was collected and macrophage death as a measure for T cell mediated cytotoxicity, was assessed by quantifying endogenous lactate dehydrogenase (LDH) release using the CyQUANT LDH Cytotoxicity Assay kit (Invitrogen) according to the manufacturer’s instructions. To correct for LDH release from T cells, concomitantly T cells from the same mice were stimulated with αCD3 and IL-2 for 24 h, LDH release in medium was measured, and subtracted from levels of LDH in medium from co-cultures.
In vitro macrophage bacteria killing assay
BMDMs from young (3 months) wild-type mice were generated as described above. Splenic CD8+ T cells from 4–5 months old male and female Ldlr−/− and T-AbcdkoLdlr−/− mice were isolated as described above and stimulated as described for the Incucyte caspase 3/7 assay. Then, CD8+ T cell medium was collected. Subsequently, BMDMs were cultured at 37 °C, 5% CO2 in 100 µL DMEM supplemented with 10% FCS, 1% pen strep, and 500 µL CD8+ T cell conditioned medium. As a positive control, BMDMs were incubated with 600 µL DMEM supplemented with 10% FCS and 1% pen strep containing recombinant IFN-γ (420 U/mL; Peprotech). After 3 days, medium was aspirated, BMDMs were washed and co-incubated with E. coli K-12 MG1655 bacteria (BMDM to E.coli ratio 50:1; kind gift from Dr. Marjon G.J. de Vos, University of Groningen, the Netherlands) in DMEM supplemented with 10% FCS, without antibiotics for 1 h. After another wash, BMDMs were incubated in DMEM supplemented with 10% FCS and 200 μg/mL gentamicin (Gibco) for 1 h, washed for removal of dead bacteria, and incubated in DMEM supplemented with 10% FCS and 20 μg/mL gentamicin for 2 h. BMDMs were then washed extensively (three times) and lysed in 0.1% Triton X-100 in PBS. To assess bacterial viability, BMDM cell lysates were plated on agar and incubated o/n at 37 °C. Bacterial colony forming units (CFU) were counted manually in a blinded fashion, i.e. the observer was unaware of the genotypes, and were quantified for macrophages incubated with conditioned medium from Ldlr−/− or T-AbcdkoLdlr−/− CD8+ T cells. The number of bacterial CFU directly reflects the macrophage killing capacity (i.e. the lower the number of CFU, the higher the macrophage killing capacity).
Plasma lipid and lipoprotein analyses
Blood samples were collected by tail bleeding. Plasma was separated by centrifugation and assayed for cholesterol and TG using enzymatic kits (Roche and Diasys Diagnostic Systems, respectively). The Cholesterol standard FS (DiaSys Diagnostic Systems) and Precimat Glycerol (Roche) were used for the cholesterol and TG calibration curves, respectively. To assess lipoprotein cholesterol distribution in plasma from female Ldlr−/− and T-AbcdkoLdlr−/− mice fed a WTD (10 weeks) by fast performance liquid chromatography (FPLC), a system containing a PU-980 pump with a linear degasser and a UV-975 UV/VIS detector (Jasco) was used. Pooled plasma (n = 15 per pool) was injected onto a Superose 6 HR 10/300 GL column (GE Healthcare) and eluted at a constant flow rate of 500 μL/min in PBS (pH 7.4). Plasma fractions were assayed for cholesterol using an enzymatic kit as described above. To assess lipoprotein cholesterol and TG distribution in female Ldlr−/− and T-AbcdkoLdlr−/− mice fed a chow diet (28 weeks), young (3 months) and aged (24 months) male and female wild-type mice, and lipoprotein TG distribution in female Ldlr−/− and T-AbcdkoLdlr−/− mice fed a WTD (10 weeks) by FPLC, a system containing a PU-4180 pump with a linear degasser and a UV-4075 UV/VIS detector (Jasco) was used. Pooled plasma (n = 10–18 per pool) was injected onto a Superose 6 Increase 10/300 GL column (GE Healthcare) and eluted at a constant flow rate of 0.31 mL/min in PBS (pH 7.4). Cholesterol and TG were measured in line by the addition of cholesterol and TG reagents, respectively, at a constant flow rate of 0.1 mL/min using an additional PU-4080i infusion pump (Jasco). Data acquisition and analysis were performed using ChromNav software (Jasco).
Atherosclerotic lesion analysis
Female Ldlr−/− and T-AbcdkoLdlr−/−mice were fed a WTD for 10 weeks (starting at 8–12 weeks of age; n = 16 Ldlr−/− and n = 16 T-AbcdkoLdlr−/−mice) or a chow diet for 28 weeks (n = 18 Ldlr−/− and n = 16 T-AbcdkoLdlr−/−mice) or 12–13 months (n = 11 Ldlr−/− and n = 10 T-AbcdkoLdlr−/−mice). After the indicated period of time on diet, mice were sacrificed and the hearts were isolated and fixed in phosphate-buffered formalin. Hearts were dehydrated and embedded in paraffin, and cross-sectioned throughout the aortic root area (4 µm sections). Haematoxylin-eosin (H&E) staining was performed on the sections and the average from 6 sections (40 μm distance between sections) for each animal was used to determine lesion size. Lesion size was quantified by morphometric analysis. Necrotic core area was determined as acellular area, lacking nuclei and cytoplasm, under the fibrous cap of lesions from H&E stained sections. Necrotic core area was differentiated from regions of dense fibrous scars by the presence of cell debris, as reported previously81. To assess the number of T cells per aortic root section, for antigen retrieval, paraffin sections were incubated in 10 mM citrate buffer at pH 6.0 (15 min; microwave), and blocked in 10% goat serum for 30 min at RT. Subsequently, sections were incubated o/n with rabbit anti-human CD3 antibody at 4 °C (1/250 dilution; Dako), which also cross-reacts with mouse, and subsequently with biotinylated goat anti-rabbit secondary antibody for 30 min at RT (1/250 dilution; Vector Laboratories), and Vectastain Elite ABC Kit-peroxidase kit (Vector Laboratories) according to the manufacturer’s instructions. Sections were then stained with 3,3′-diaminobenzidine (DAB) for 10 min at RT and counterstained with haematoxylin. T cells were identified as cells with brown plasma membrane CD3 staining and the hematoxylin staining still visible. To assess fibrous cap thickness and collagen content in atherosclerotic lesions, aortic root sections (one per mouse) taken from similar regions of the lesions (mouse to mouse) were stained with Weigert’s haematoxylin for 8 min to stain the nuclei and subsequently with Sirius Red (0.1% (w/v)) in 1.3% aqueous picric acid solution for 1 h at RT. Slides were then washed twice 10 sec in acidified water (1% glacial acetic acid solution). Fibrous cap thickness was measured in the largest section at even intervals 2 µm apart. Then the average thickness of lesion was reported in length units, as described82. Total collagen area (whole area staining positive for Sirius Red) was also measured. To assess SMC area, sections were blocked in 10% goat serum for 30 min at RT, incubated o/n at 4 °C with rabbit anti-mouse α-SMA primary antibody (1/200 dilution; Lab Vision) and subsequently with biotinylated goat anti-rabbit secondary antibody for 30 min at RT (1/250 dilution), and Vectastain ABC-peroxidase kit (Vector Laboratories) according to the manufacturer’s instructions. Sections were then stained with DAB for 10 min at RT and counterstained with haematoxylin. SMC positive area was assessed. To assess Mac-2 (Galectin-3) area, mainly reflecting macrophages, for antigen retrieval, paraffin sections were incubated with proteinase K for 9 min at RT (20 μg/mL in PBS; Merck) (immunohistochemistry (IHC)) or Tris-Base EDTA at pH 9.0 (15–20 min; microwave) (immunofluorescence (IF)), and blocked in 10% goat serum for 30 min at RT. Sections were then incubated o/n at 4 °C with rat anti-mouse/human Mac-2 (Galectin-3) primary antibody (1/10000 dilution; Cedarlane). For IHC, sections were subsequently stained with biotinylated goat anti-rat secondary antibody for 30 min at RT (1/125 dilution) and Vectastain ABC-peroxidase kit according to the manufacturer’s instructions, followed by staining with DAB for 10 min at RT and counterstaining with haematoxylin. For IF, sections were stained with anti-rat IgG-AF488 secondary antibody for 30 min at RT (1/200 dilution; Invitrogen) and mounted using ProLong Gold Antifade Mountant with DAPI (Invitrogen). To assess apoptotic macrophages, sections were stained with the In Situ Cell Death Detection Kit TMR red (Roche). After perm wash (BD Biosciences), slides were incubated with proteinase K for 9 min at RT (20 μg/mL in PBS), and further staining was performed according to the manufacturer’s instructions, in combination with Mac-2 staining as described above. Nuclei were stained with Hoechst 33342 (Invitrogen) for 10 min at RT and sections were mounted using VECTASHIELD Antifade Mounting Medium. For IF, sections were imaged using an AxioObserver Z1 compound microscope (10x objective; AxioCam MRm3 CCD camera; Carl Zeiss) running Zen software. The total area of lesions (based on H&E staining), fibrous cap thickness, total collagen and SMC positive areas, Mac-2+area and TUNEL+Mac-2+area were assessed using ImageJ software (NIH). Quantification of all parameters was performed in a blinded fashion, i.e. the observer was unaware of the genotypes.
Quantification and statistical analysis
All data are presented as means ± SEM. In each experiment, n defines the number of mice included. The statistical parameters (n, mean, SEM) can be found within the figure legends. Two-tailed Student’s t test was used to define differences between two datasets. To define differences between three or four datasets, One-way Analysis of Variance (ANOVA) was used with a Bonferroni multiple comparison post-test. The criterion for significance was set at p < 0.05. Statistical analyses were performed using GraphPad Prism 9 (San Diego, California).
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Acknowledgements
This work is supported by VIDI (917.15.350) and Aspasia grants from the Netherlands Organization of Scientific Research (NWO) (M.W.), and a Rosalind Franklin Fellowship from the University of Groningen with EU Co-Fund attached (M.W.).
Source data
Author contributions
V.B.: conceived, designed, and performed experiments, carried out data analyses, and wrote the original draft of the manuscript. A.M.L.R., B.H., A.G.G., A.M.-G., A.T.P.: performed experiments and carried out data analyses. S.M., F.B., R.d.B., D.D.: performed experiments, carried out data analyses, and provided reagents. E.G., A.F.-S., M.H.K., N.J.K., R.H., M.L.-M.: performed experiments. A.d.B., B.v.d.S., A.K., L.Y.-C., G.v.d.B.: provided intellectual input and reviewed the original and revised manuscript. M.W.: conceived, designed, and performed experiments, carried out data analyses, revised the original draft of the manuscript, supervised the study, and obtained funding for the study.
Peer review
Peer review information
Nature Communications thanks Mary Sorci-Thomas and the other anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its supplementary information files. All the raw data generated in this study are provided in the Source Data file. Source data are provided with this paper.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-022-31135-4.
References
- 1.Libby P, et al. Atherosclerosis. Nat. Rev. Dis. Prim. 2019;5:56. doi: 10.1038/s41572-019-0106-z. [DOI] [PubMed] [Google Scholar]
- 2.Saigusa R, Winkels H, Ley K. T cell subsets and functions in atherosclerosis. Nat. Rev. Cardiol. 2020;17:387–401. doi: 10.1038/s41569-020-0352-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Winkels H, Wolf D. Heterogeneity of T cells in atherosclerosis defined by single-cell RNA-sequencing and cytometry by time of flight. Arteriosclerosis, Thrombosis, Vasc. Biol. 2021;41:549–563. doi: 10.1161/ATVBAHA.120.312137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ait-Oufella H, et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat. Med. 2006;12:178–180. doi: 10.1038/nm1343. [DOI] [PubMed] [Google Scholar]
- 5.Gaddis DE, et al. Apolipoprotein AI prevents regulatory to follicular helper T cell switching during atherosclerosis. Nat. Commun. 2018;9:1095. doi: 10.1038/s41467-018-03493-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kimura T, et al. Regulatory CD4+ T cells recognize major histocompatibility complex class II molecule–restricted peptide epitopes of apolipoprotein B. Circulation. 2018;138:1130–1143. doi: 10.1161/CIRCULATIONAHA.117.031420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Butcher MJ, et al. Atherosclerosis-driven treg plasticity results in formation of a dysfunctional subset of plastic IFNγ+ Th1/Tregs. Circulation Res. 2016;119:1190–1203. doi: 10.1161/CIRCRESAHA.116.309764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang W, et al. Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nature. 2016;531:651–655. doi: 10.1038/nature17412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaddis DE, et al. Atherosclerosis impairs naive CD4 T-cell responses via disruption of glycolysis. Arteriosclerosis, Thrombosis, Vasc. Biol. 2021;41:2387–2398. doi: 10.1161/ATVBAHA.120.314189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larbi A, et al. Differential role of lipid rafts in the functions of CD4+ and CD8+ human T lymphocytes with aging. Cell. Signal. 2006;18:1017–1030. doi: 10.1016/j.cellsig.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 11.Larbi A, et al. Immunomodulatory role of high-density lipoproteins: impact on immunosenescence. Age (Omaha) 2014;36:9712. doi: 10.1007/s11357-014-9712-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quinn KM, et al. Age-related decline in primary CD8+ T cell responses is associated with the development of senescence in virtual memory CD8+ T cells. Cell Rep. 2018;23:3512–3524. doi: 10.1016/j.celrep.2018.05.057. [DOI] [PubMed] [Google Scholar]
- 13.Goronzy JJ, Weyand CM. Mechanisms underlying T cell ageing. Nat. Rev. Immunol. 2019;19:573–583. doi: 10.1038/s41577-019-0180-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Franceschi C, Campisi J. Chronic inflammation (Inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. - Ser. A Biol. Sci. Med. Sci. 2014;69:S4–S9. doi: 10.1093/gerona/glu057. [DOI] [PubMed] [Google Scholar]
- 15.Wang JC, Bennett M. Aging and atherosclerosis: Mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circulation Res. 2012;111:245–259. doi: 10.1161/CIRCRESAHA.111.261388. [DOI] [PubMed] [Google Scholar]
- 16.Palmer DB. The effect of age on thymic function. Front. Immunol. 2013;4:316. doi: 10.3389/fimmu.2013.00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kennedy MA, et al. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab. 2005;1:121–131. doi: 10.1016/j.cmet.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 18.Wang N, Silver DL, Costet P, Tall AR. Specific binding of ApoA-I, enhanced cholesterol efflux, and altered plasma membrane morphology in cells expressing ABC1. J. Biol. Chem. 2000;275:33053–33058. doi: 10.1074/jbc.M005438200. [DOI] [PubMed] [Google Scholar]
- 19.Wang N, Lan D, Chen W, Matsuura F, Tall AR. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc. Natl Acad. Sci. 2004;101:9774–9779. doi: 10.1073/pnas.0403506101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bensinger SJ, et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell. 2008;134:97–111. doi: 10.1016/j.cell.2008.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Michaels AJ, Campbell C, Bou-Puerto R, Rudensky AY. Nuclear receptor LXRβ controls fitness and functionality of activated T cells. J. Exp. Med. 2021;218:e20201311. doi: 10.1084/jem.20201311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng HY, et al. Loss of ABCG1 influences regulatory T cell differentiation and atherosclerosis. J. Clin. Investig. 2016;126:3236–3246. doi: 10.1172/JCI83136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Armstrong AJ, Gebre AK, Parks JS, Hedrick CC. ATP-binding cassette transporter G1 negatively regulates thymocyte and peripheral lymphocyte proliferation. J. Immunol. 2010;184:173–183. doi: 10.4049/jimmunol.0902372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yvan-Charvet L, et al. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J. Clin. Investig. 2007;117:3900–3908. doi: 10.1172/JCI33372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee PP, et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 2001;15:763–774. doi: 10.1016/S1074-7613(01)00227-8. [DOI] [PubMed] [Google Scholar]
- 26.Radhakrishnan A, Goldstein JL, McDonald JG, Brown MS. Switch-like Control of SREBP-2 Transport Triggered by Small Changes in ER Cholesterol: A Delicate Balance. Cell Metab. 2008;8:512–521. doi: 10.1016/j.cmet.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fernández-Pérez EJ, et al. Effect of cholesterol on membrane fluidity and association of Aβ oligomers and subsequent neuronal damage: A Double-Edged Sword. Front. Aging Neurosci. 2018;10:226. doi: 10.3389/fnagi.2018.00226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vyšniauskas A, Qurashi M, Kuimova MK. A molecular rotor that measures dynamic changes of lipid bilayer viscosity caused by oxidative stress. Chem. - A Eur. J. 2016;22:13210–13217. doi: 10.1002/chem.201601925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan CT, et al. Liver X receptors are required for thymic resilience and T cell output. J. Exp. Med. 2020;217:e20200318. doi: 10.1084/jem.20200318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu X, et al. Maturation and emigration of single-positive thymocytes. Clin. Developmental Immunol. 2013;2013:282870. doi: 10.1155/2013/282870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Westerterp M, et al. Cholesterol accumulation in dendritic cells links the inflammasome to acquired immunity. Cell Metab. 2017;65:3176–3185. doi: 10.1016/j.cmet.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma X, et al. Cholesterol induces CD8+ T cell exhaustion in the tumor microenvironment. Cell Metab. 2019;30:143–156.e5. doi: 10.1016/j.cmet.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015;15:486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McKinney EF, Smith KGC. Metabolic exhaustion in infection, cancer and autoimmunity review-article. Nat. Immunol. 2018;19:213–221. doi: 10.1038/s41590-018-0045-y. [DOI] [PubMed] [Google Scholar]
- 35.Banerjee A, et al. Cutting edge: the transcription factor eomesodermin enables CD8+ T cells to compete for the memory cell niche. J. Immunol. 2010;185:4988–4992. doi: 10.4049/jimmunol.1002042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simon S, Labarriere N. PD-1 expression on tumor-specific T cells: Friend or foe for immunotherapy? OncoImmunology. 2018;7:e1364828. doi: 10.1080/2162402X.2017.1364828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kidani Y, et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat. Immunol. 2013;14:489–499. doi: 10.1038/ni.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hwang JR, Byeon Y, Kim D, Park SG. Recent insights of T cell receptor-mediated signaling pathways for T cell activation and development. Exp. Mol. Med. 2020;52:750–761. doi: 10.1038/s12276-020-0435-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bouillet P, O’Reilly LA. CD95, BIM and T cell homeostasis. Nat. Rev. Immunol. 2009;9:514–519. doi: 10.1038/nri2570. [DOI] [PubMed] [Google Scholar]
- 40.Shi J, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 41.Zhang C, et al. NLRP3 inflammasome induces CD4+ T cell loss in chronically HIV-1-infected patients. J. Clin. Investig. 2021;131:e138861. doi: 10.1172/JCI138861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Q, Kim YC, Laurence A, Punkosdy GA, Shevach EM. IL-2 controls the stability of Foxp3 expression in TGF-β–induced Foxp3 + T cells in vivo. J. Immunol. 2011;186:6329–6337. doi: 10.4049/jimmunol.1100061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gajate C, Mollinedo F. Lipid raft-mediated Fas/CD95 apoptotic signaling in leukemic cells and normal leukocytes and therapeutic implications. J. Leukoc. Biol. 2015;98:739–759. doi: 10.1189/jlb.2MR0215-055R. [DOI] [PubMed] [Google Scholar]
- 44.Tang D, et al. The molecular machinery of regulated cell death. Cell Res. 2019;29:347–364. doi: 10.1038/s41422-019-0164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kurtulus S, et al. Bcl-2 allows effector and memory CD8+ T cells to tolerate higher expression of bim. J. Immunol. 2011;186:5729–5737. doi: 10.4049/jimmunol.1100102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chong SJF, Low ICC, Pervaiz S. Mitochondrial ROS and involvement of Bcl-2 as a mitochondrial ROS regulator. Mitochondrion. 2014;19:39–48. doi: 10.1016/j.mito.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 47.Greenlee-Wacker MC, Nauseef WM. IFN-γ targets macrophage-mediated immune responses toward Staphylococcus aureus. J. Leukoc. Biol. 2017;101:751–758. doi: 10.1189/jlb.4A1215-565RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sene A, et al. Impaired cholesterol efflux in senescent macrophages promotes age-related macular degeneration. Cell Metab. 2013;17:549–561. doi: 10.1016/j.cmet.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Houtkooper RH, et al. The metabolic footprint of aging in mice. Sci. Rep. 2011;1:134. doi: 10.1038/srep00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harpaz I, Bhattacharya U, Elyahu Y, Strominger I, Monsonego A. Old mice accumulate activated effector CD4 T cells refractory to regulatory T cell-induced immunosuppression. Front. Immunol. 2017;8:283. doi: 10.3389/fimmu.2017.00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moro-García MA, Alonso-Arias R, López-Larrea C. When aging reaches CD4+ T-cells: Phenotypic and functional changes. Front. Immunol. 2013;4:107. doi: 10.3389/fimmu.2013.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Engwerda CR, Fox BS, Handwerger BS. Cytokine production by T lymphocytes from young and aged mice. J. Immunol. 1996;156:3621–3630. [PubMed] [Google Scholar]
- 53.Lages CS, Lewkowich I, Sproles A, Wills-Karp M, Chougnet C. Partial restoration of T-cell function in aged mice by in vitro blockade of the PD-1/PD-L1 pathway. Aging Cell. 2010;9:785–798. doi: 10.1111/j.1474-9726.2010.00611.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nikolich-Žugich J. Ageing and life-long maintenance of T-cell subsets in the face of latent persistent infections. Nat. Rev. Immunol. 2008;8:512–522. doi: 10.1038/nri2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Westerterp M, et al. Deficiency of ABCA1 and ABCG1 in macrophages increases inflammation and accelerates atherosclerosis in mice. Circ. Res. 2013;112:1456–1465. doi: 10.1161/CIRCRESAHA.113.301086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of cellular senescence. Trends Cell Biol. 2018;28:436–453. doi: 10.1016/j.tcb.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 57.Out R, et al. Combined deletion of macrophage ABCA1 and ABCG1 leads to massive lipid accumulation in tissue macrophages and distinct atherosclerosis at relatively low plasma cholesterol levels. Arteriosclerosis, Thrombosis, Vasc. Biol. 2008;28:258–264. doi: 10.1161/ATVBAHA.107.156935. [DOI] [PubMed] [Google Scholar]
- 58.Yvan-Charvet L, et al. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science. 2010;328:1689–1693. doi: 10.1126/science.1189731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kyaw T, et al. Cytotoxic and proinflammatory CD8+ T lymphocytes promote development of vulnerable atherosclerotic plaques in ApoE-deficient mice. Circulation. 2013;127:1028–1039. doi: 10.1161/CIRCULATIONAHA.112.001347. [DOI] [PubMed] [Google Scholar]
- 60.Olson NC, et al. Decreased naive and increased memory CD4+ T cells are associated with subclinical atherosclerosis: the multi-ethnic study of atherosclerosis. PLoS ONE. 2013;8:e71498. doi: 10.1371/journal.pone.0071498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Depuydt MAC, et al. Microanatomy of the human atherosclerotic plaque by single-cell transcriptomics. Circulation Res. 2020;127:1437–1455. doi: 10.1161/CIRCRESAHA.120.316770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fernandez DM, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat. Med. 2019;25:1576–1588. doi: 10.1038/s41591-019-0590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ammirati E, et al. Effector memory T cells are associated with atherosclerosis in humans and animal models. J. Am. Heart Assoc. 2012;1:27–41. doi: 10.1161/xJAHA.111.000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Emeson EE, Shen ML, Bell CG, Qureshi A. Inhibition of atherosclerosis in CD4 T-cell-ablated and nude (nu/nu) C57BL/6 hyperlipidemic mice. Am. J. Pathol. 1996;149:675–685. [PMC free article] [PubMed] [Google Scholar]
- 65.Zhou X, Robertson AKL, Rudling M, Parini P, Hansson GK. Lesion development and response to immunization reveal a complex role for CD4 in atherosclerosis. Circulation Res. 2005;96:427–434. doi: 10.1161/01.RES.0000156889.22364.f1. [DOI] [PubMed] [Google Scholar]
- 66.Cochain C, et al. CD8+ T cells regulate monopoiesis and circulating Ly6Chigh monocyte levels in atherosclerosis in mice. Circulation Res. 2015;117:244–253. doi: 10.1161/CIRCRESAHA.117.304611. [DOI] [PubMed] [Google Scholar]
- 67.Szondy Z, Sarang Z, Kiss B, Garabuczi É, Köröskényi K. Anti-inflammatory mechanisms triggered by apoptotic cells during their clearance. Front. Immunol. 2017;8:909. doi: 10.3389/fimmu.2017.00909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Goronzy JJ, Fang F, Cavanagh MM, Qi Q, Weyand CM. Naive T cell maintenance and function in human aging. J. Immunol. 2015;194:4073–4080. doi: 10.4049/jimmunol.1500046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vallejo AN, Brandes JC, Weyand CM, Goronzy JJ. Modulation of CD28 expression: distinct regulatory pathways during activation and replicative senescence. J. Immunol. 1999;162:6572–6579. [PubMed] [Google Scholar]
- 70.Maly K, Schirmer M. The story of CD4+CD28– T cells revisited: Solved or still ongoing? J. Immunol. Res. 2015;2015:348746. doi: 10.1155/2015/348746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liuzzo G, et al. Perturbation of the T-cell repertoire in patients with unstable angina. Circulation. 1999;100:2135–2139. doi: 10.1161/01.CIR.100.21.2135. [DOI] [PubMed] [Google Scholar]
- 72.Liuzzo G, et al. Unusual CD4+CD28null T lymphocytes and recurrence of acute coronary events. J. Am. Coll. Cardiol. 2007;50:1450–1458. doi: 10.1016/j.jacc.2007.06.040. [DOI] [PubMed] [Google Scholar]
- 73.Nakajima T, et al. T-cell-mediated lysis of endothelial cells in acute coronary syndromes. Circulation. 2002;105:570–575. doi: 10.1161/hc0502.103348. [DOI] [PubMed] [Google Scholar]
- 74.Dumitriu IE, et al. High levels of costimulatory receptors OX40 and 4-1BB characterize CD4+CD28 null T cells in patients with acute coronary syndrome. Circulation Res. 2012;110:857–869. doi: 10.1161/CIRCRESAHA.111.261933. [DOI] [PubMed] [Google Scholar]
- 75.Tomas L, et al. Low levels of CD4+CD28null T cells at baseline are associated with first-time coronary events in a prospective population-based case-control cohort. Arteriosclerosis, Thrombosis, Vasc. Biol. 2020;40:426–436. doi: 10.1161/ATVBAHA.119.313032. [DOI] [PubMed] [Google Scholar]
- 76.Yuan J, et al. Potentiating CD8+ T cell antitumor activity by inhibiting PCSK9 to promote LDLR-mediated TCR recycling and signaling. Protein Cell. 2021;12:240–260. doi: 10.1007/s13238-021-00821-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wilfahrt D, et al. Histone deacetylase 3 represses cholesterol efflux during CD4+ T-cell activation. Elife. 2021;10:e70978. doi: 10.7554/eLife.70978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Molnár E, et al. Cholesterol and sphingomyelin drive ligand-independent T-cell antigen receptor nanoclustering. J. Biol. Chem. 2012;287:42664–42674. doi: 10.1074/jbc.M112.386045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Swamy M, et al. A cholesterol-based allostery model of T cell receptor phosphorylation. Immunity. 2016;44:1091–1101. doi: 10.1016/j.immuni.2016.04.011. [DOI] [PubMed] [Google Scholar]
- 80.Warren SC, et al. Rapid global fitting of large fluorescence lifetime imaging microscopy datasets. PLoS ONE. 2013;8:e70687. doi: 10.1371/journal.pone.0070687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Han S, et al. Macrophage insulin receptor deficiency increases ER stress-induced apoptosis and necrotic core formation in advanced atherosclerotic lesions. Cell Metab. 2006;3:257–266. doi: 10.1016/j.cmet.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 82.Fidler TP, et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature. 2021;592:296–301. doi: 10.1038/s41586-021-03341-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the paper and its supplementary information files. All the raw data generated in this study are provided in the Source Data file. Source data are provided with this paper.