

Summary
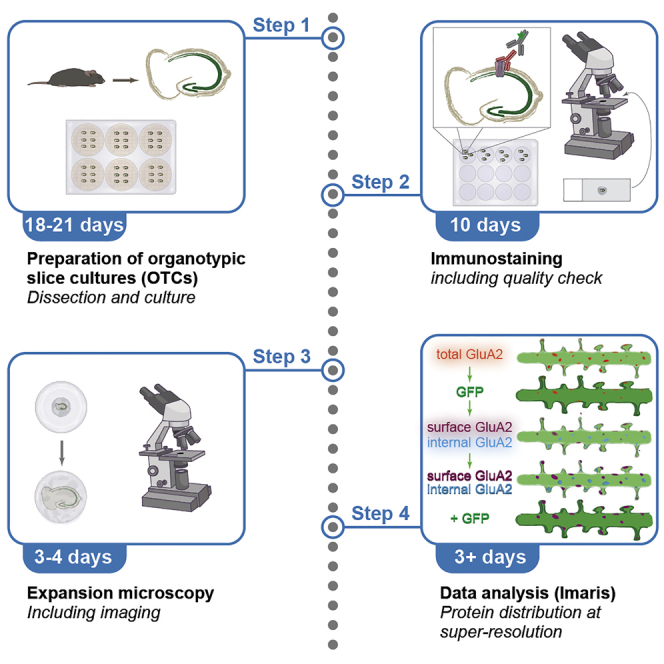
Assessing protein distribution with super-resolution in tissue is often complicated and restrictive. Here, we describe a protocol for immunostaining and expansion microscopy imaging of mouse brain organotypic slice cultures. We detail an Imaris analysis workflow to analyze the surface vs intracellular distribution of AMPA receptors at super-resolution during homeostatic plasticity. We have optimized the protocol for brain organotypic slice culture and tested in acute brain slices. This protocol is suitable to study protein distribution under multiple plasticity paradigms.
For complete details on the use and execution of this protocol, please refer to Bissen et al. (2021).
Subject areas: Cell Biology, Microscopy, Neuroscience
Graphical abstract
Highlights
-
•
Enables immunostaining and visualization of epitopes deep within brain slices
-
•
Utilizes expansion microscopy to increase imaging resolution
-
•
Optimized for brain organotypic slice cultures and tested in acute brain slices
-
•
Analysis workflow for protein distribution (surface vs. intracellular pool) using Imaris
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Assessing protein distribution with super-resolution in tissue is often complicated and restrictive. Here, we describe a protocol for immunostaining and expansion microscopy imaging of mouse brain organotypic slice cultures. We detail an Imaris analysis workflow to analyze the surface vs intracellular distribution of AMPA receptors at super-resolution during homeostatic plasticity. We have optimized the protocol for brain organotypic slice culture and tested in acute brain slices. This protocol is suitable to study protein distribution under multiple plasticity paradigms.
Before you begin
This protocol contains detailed information about the preparation, culture, immunostaining, expansion, and imaging of mouse entorhinal-hippocampal organotypic slice cultures (OTCs), as well as for the analysis of the expanded data to assess protein distribution at super-resolution. Several protocols for OTC preparation and culture have been published (see e.g., Gaehwiler, 1981; Stoppini et al., 1991); we have opted for an air-interface culture in Millipore inserts because of the ease of that protocol for imaging (see “materials and equipment’’ for detailed recipes) (Humpel, 2015; Gaehwiler et al., 1997). We routinely isolate OTCs from pups at postnatal day (P) 3–6 as neurons from young animals recover better from the dissection. Results from younger (even at embryonic stages, see e.g., protocols in Calderon de Anda, 2013; Elias and Kriegstein, 2007) or older mice (e.g., Humpel, 2015; Mewes et al., 2012) have been described. Our protocol could also be adapted to different animal ages or brain areas.
Institutional permissions
All animal use was performed in compliance with the governmental authorities of Hessen and followed the German animal welfare legislation. Researchers should first seek approval from their relevant regulatory institutions for the use of animals in experimental research before following this protocol.
OTC dissection
Timing: ∼ 1 h for preparation and first pup, + ∼ 20 min/additional pup
This protocol is performed in sterile conditions in a culture hood. The microscope for OTC dissection should be in the hood. The vibratome may be placed inside the hood or just next to it.
All animal experiments are carried in compliance with local governmental authorities and animal welfare legislation.
-
1.
Prepare the preparation medium (PM) and the incubation medium (IM) – fresh or in advance. If you prepare the IM in advance, preheat it at 35°C and adjust the pH to 7.29–7.31 (be sure to measure the pH accurately at 35°C).
Note: We recommend preparing 50 mL aliquots of the IM.
-
2.
Prepare a 6-well plate to culture the OTCs in the incubator. Reserve 2 wells per pup and add 1 mL of pre-warmed, pH-adjusted IM per well. Place the plate in the incubator at 35°C and 5% CO2.
-
3.
Calibrate the vibratome. If you use a chiller, turn it on in time to allow time for cooling.
Note: We use a chiller (Julabo) in combination with a vibratome (VT1200S, Leica). Alternatively, you can add ice to the vibratome bath to surround the buffer tray and keep it cool. However, this option is less clean as the ice may fall into the buffer tray.
-
4.
Disinfect the dissection tools with ethanol and flame-sterilize them with a Bunsen burner. Leave them in the hood.
Note: Our dissection tools consist of small scissors (decapitation of the pups and opening of the skull), straight and curved forceps (opening of the skull; OTC dissection), a spatula (brain transport) and two Sichel knives (OTC sectioning at the vibratome and dissection from the brain section).
Note: OTC dissection from the brain slice (step 10) is done under the microscope and requires straight forceps and a Sichel knife (listed above). We avoid using these two tools for other steps to keep them as clean and sharp as possible.
-
5.
Prepare an ice tray and fill two 10 cm Petri dishes with ice-cold PM. Fill the buffer tray with PM and place it in the vibratome.
-
6.Decapitate one pup and dissect its brain under the hood (Figure 1A).
-
a.Hold the head with forceps through the eye sockets, cut the skin on the top of the head longitudinally with microdissection scissors and pull it off with curved forceps.
-
b.Proceed similarly with the skull. Be careful not to cut into the brain tissue.
-
c.Using the spatula, remove the brain from the opened skull into a dish filled with ice-cold PM.
-
d.Cut the cerebellum with a scalpel and turn the brain upside down.Note: This step is specific to the preparation of horizontal sections. For coronal sections, cut the cerebellum and turn the brain so that the midbrain is flat against the dish and the olfactory bulbs point upward. For sagittal sections, cut the cerebellum, hemisect the brain across the midline and turn the brain so that it rests on its most medial surface.
-
e.Place a drop of tissue glue on the specimen tray. Use the spatula to carefully transfer the brain onto the adhesive. For horizontal sections, the brain should be as flat as possible.Note: The acrylic adhesive we use solidifies particularly quickly when in contact with liquids. Since the spatula is covered with PM, it is essential to avoid touching the glue with the spatula when transferring the brain, otherwise the glue will immediately solidify into strands that can damage the brain if they touch it.
-
f.Gently lower the specimen plate into the buffer tray and align the brain as parallel as possible to the blade.
-
a.
-
7.
Section the brain using the appropriate settings on the vibratome. For reference, we use a speed of 0.12 mm/s, an amplitude of 0.8 mm and a section thickness of 300 μm.
CRITICAL: As the tissue is not fixed, slow speeds are recommended to avoid damaging the tissue. The thickness can be varied slightly; however, the viability of the OTCs should be re-evaluated if this parameter is changed.
-
8.
Transfer one slice at a time into a dish filled with ice-cold PM under the microscope. Assess the morphology and keep only sections where the dentate gyrus of the hippocampus shows its typical morphology, usually 5–6 sections (Figure 1B).
Note: Due to the size of the slices, we use a 5 mL pipette to transfer them from the vibratome to the Petri dish. We cut off about 1 cm of the tips so that the diameter is slightly larger than the usual size of our slices. This has the advantage that the section itself is not touched (compared to e.g., brushes) and therefore no damage is caused.
-
9.
Dissect the hippocampus and entorhinal cortex into a roughly rectangular shape using a scalpel under the microscope (Figures 1B and 1C).
-
10.
When the region of interest has been completely sectioned, assess all available tissue under the microscope to ensure the quality of the sections. Discard any sections with poor quality and/or unclear morphology.
Note: In our case, we routinely have up to 12 hippocampal formations (6 sections × 2 hemispheres), i.e., 12 OTCs. Considering the size of each OTC, we place up to 6 OTCs per well - hence the preparation of 2 wells per pup. If drug treatment is planned, this also offers the advantage of having an untreated control from the same animal. In addition, we usually separate the OTCs so that each well contains a complete arrangement of the hippocampal formation along the anterior-posterior axis.
-
11.Transfer the slices to the 6-well plate (Figure 1D).
-
a.Take out the 6-well plate from the incubator and prepare two wells by adding one Millipore insert per well using forceps. Allow them to soak in IM.
-
b.Transfer the slices to each insert using the 5 mL pipette.
-
c.Arrange the OTCs with a scalpel so that they are more than 3 mm apart from each other, otherwise they may merge as they flatten and enlarge slightly. Troubleshooting.
-
d.Transferring the OTCs will have added IM on top of the Millipore insert. Remove this medium with a 1 mL pipette to prevent additional movement and to facilitate attachment. Be careful not to disturb the OTCs.
-
e.Place the plate back in the incubator.
-
a.
-
12.
Repeat the procedure (steps 6–11) for each pup.
-
13.
Keep the plate in the incubator for the duration of the experiment. Change the IM 3×/week (see steps 14–16).
Figure 1.

OTC dissection and culture
(A) Illustration of the dissection of the brain from the mouse head (step 7). From left to right: cut the skin longitudinally with microdissection scissors and pull it off with curved forceps. Proceed similarly with the skull. Cut the cerebellum with a scalpel.
(B) Horizontal section of a mouse brain. Examine sections under a binocular microscope (step 9) and cut out the region of interest for OTCs according to the red square (step 10). Scale bar: 2 mm.
(C) The entorhinal-hippocampal formation after dissection. Scale bar: 2 mm.
(D) Place the OTCs with appropriate morphology on a Millipore insert in a 6-well plate for subsequent culture (step 12).
(E) Images of different stages during culture of OTCs – DIV 0, 1, 7, 14, 21. OTCs flatten in the first days after their dissection. Up to 6 OTCs can be placed on an insert, safely spaced and with sufficient nutrients from the incubation medium (note that OTCs will have moved slightly after the first image; this may occur during transfer from the hood to the incubator before they are attached to the insert). DIV: day in vitro (where the day of the dissection is DIV0). Scale bar: 2 mm.
OTC culture
This procedure should be repeated every 2–3 days throughout the duration of the experiment. We routinely culture OTCs for up to 6 weeks (3 weeks maturation + 3 weeks experiment) during which they remain healthy (Figure 1E). Troubleshooting.
-
14.
Prewarm the IM to 35°C and adjust the pH to 7.29–7.31 with 1 M NaOH or 1 M HCl.
Note: Calibrate the pH meter regularly to ensure that it is measuring the pH correctly. Be sure to calibrate at 35°C and also pre-warm the calibration buffers.
-
15.
Under the hood, tilt the 6-well plate away from you and remove the old IM from the edge of the Millipore insert. Some IM will remain under the insert.
Note: The IM can be removed with a conventional pipette with a 1 mL tip or with a holder connected to the hood's vacuum pump. As the OTCs are attached to the insert and the IM is removed from the edge of the insert, there is no risk of damaging the OTCs.
-
16.
Add 1 mL of fresh IM to the side of the well. Do not add directly to the insert or to the OTCs - this would affect their air exchange.
Note: We routinely change the IM the day after dissection without any negative impact on the health of the OTCs.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-GluA2 (extracellular epitope, clone 6C4, 1:500) | Millipore | Cat # MAB397; RRID: AB_2113875 |
| Chicken polyclonal anti-GFP (1:1000) | Abcam | Cat # ab13970; RRID: AB_300798 |
| Donkey polyclonal anti-chicken Alexa 488 (1:500) | Dianova | Cat# 703-546-155; RRID: AB_2340376 |
| Donkey polyclonal anti-mouse Alexa 568 (1:500) | Molecular Probes | Cat # A10037; RRID: AB_2534013 |
| Chemicals, peptides, and recombinant proteins | ||
| 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl (4-Hydroxy-TEMPO) | Sigma | Cat# 176141; CAS 2226-96-2 |
| 4-(2-hydroxyethyl)-1- piperazineethanesulfonic acid (HEPES) | Invitrogen | 15630-056 |
| 45% D-(+)-Glucose | Sigma | Cat# G7869; CAS 50-99-7 |
| Acrylamide | AppliChem | Cat# A0951; CAS 79-06-1 |
| Acryloyl-X SE | Thermo Fisher Scientific | Cat# A20770; CAS 63392-86-9 |
| Agarose, low-melt | Roth | Cat# 6351.2: CAS 39346-81-1 |
| Ammonium persulfate (APS) | Bio-Rad | Cat# 161-0700; CAS 7727-54-0 |
| B-27 supplement | Invitrogen | 17504-044 |
| Basal Medium Eagle (BME) | Invitrogen | 41010-026 |
| Dimethylsulfoxide (DMSO), anhydrous | Sigma | 276855 |
| Dulbecco’s phosphate-buffered saline (DPBS) | Invitrogen | 14190-169 |
| Donor Horse serum | Sigma | Cat# 1138 |
| Ethylenediaminetetraacetic acid (EDTA) | Calbiochem | Cat# 1032456; CAS 6381-92-6 |
| GlutaMax | Gibco | 35050-038 |
| Modified Eagle Medium (MEM) | Invitrogen | 21575-022 |
| N-2 supplement | Invitrogen | 17502-048 |
| N,N-methylenebisacrylamide | Sigma | Cat# 146072; CAS 110-26-9 |
| Penicillin/Streptomycin | Gibco | 15140-122 |
| Proteinase K | Roche | Cat# 03115828001; EC 3.4.23.1 |
| Sodium acrylate | Sigma | Cat# 408220; CAS 7446-81-3 |
| Sodium bicarbonate | Roth | HN01.1 |
| Sodium chloride | Roth | HN00.2 |
| Tetramethylethylenediamine (TEMED) | Roth | Cat# 2367.3; CAS 110-18-9 |
| Tris-HCl | Roth | 9090.3 |
| Triton X-100 | Merck | Cat# 1.2298.0101; CAS 9002-93-1 |
| Experimental models: Organisms/strains | ||
| Mouse: Tg(Thy1-EGFP)MJrs (male/female, 3–6 days old) | Feng et al. (2000) | MGI ID: 3766828 |
| Software and algorithms | ||
| Huygens Professional 17.04–19.10 | Scientific Volume Imaging | https://svi.nl/HomePage |
| ImageJ 5.12 | Schneider et al. (2012) | https://imagej.nih.gov/ij/ |
| Imaris 9.0–9.6 | Bitplane | https://imaris.oxinst.com/ |
| Other | ||
| 35 mm dishes, uncoated, coverslip No 1, 14 mm glass diameter, with microwell | MatTek | Cat# P35G-1.0-14-C |
| Acrylate glue | Ellsworth Adhesives | Permabond 102 |
| Coverslips (12/13 mm diameter) | Marienfeld | Cat# 01-115-30 |
| Dumont #7 - Fine Forceps | Fine Science Tools | Cat# 11274-20 |
| Mayo Scissors - ToughCut® | Fine Science Tools | Cat# 14110-17 |
| Millicell cell culture inserts | Merck | Cat# PICMORG50 |
| Sichel knife | Fine Science Tools | Cat# 10073-14 |
| Spatula | Hammacher | Cat# HWN034-15 |
| Spring Scissors - 8 mm Cutting Edge | Fine Science Tools | Cat# 15024-10 |
| Student Dumont #5 Forceps | Fine Science Tools | Cat# 91150-20 |
| Two-channel laser-scanning confocal microscope | Leica | TCS SP8 |
| Vibratome | Leica | VT1200S |
Materials and equipment
Preparation medium (PM) – 500 mL
| Reagent | Final concentration | Amount |
|---|---|---|
| MEM | 94.5% (v/v) | 472.5 mL |
| Penicillin/Streptomycin | 100 U/mL Penicillin 0.1 mg/mL Streptomycin |
5 mL |
| 1 M HEPES | 25 mM | 12.5 mL |
| GlutaMax | 2 mM | 5 mL |
| 45% D-(+)-Glucose | 0.45% (v/v) | 5 mL |
| Total | 500 mL |
Incubation medium (IM) – 100 mL
| Reagent | Final concentration | Amount |
|---|---|---|
| MEM | 42% (v/v) | 42 mL |
| Penicillin/Streptomycin | 100 U/mL Penicillin 0.1 mg/mL Streptomycin |
1 mL |
| 1 M HEPES | 25 mM | 2.5 mL |
| GlutaMax | 2 mM | 1 mL |
| 45% D-(+)-Glucose | 0.65% (v/v) | 1.5 mL |
| Donor Horse Serum | 25% (v/v) | 25 mL |
| Basal Medium Eagle (BME) | 25% (v/v) | 25 mL |
| Sodium bicarbonate, 7.5% | 0.15% (v/v) | 2 mL |
| N-2 Supplement | 1% (v/v) | 1 mL |
| B-27 Supplement | 1% (v/v) | 1 mL |
| Total | 100 mL |
Acryloyl-X SE/anhydrous DMSO
| Reagent | Final concentration | Amount |
|---|---|---|
| Acryloyl-X SE | 35.4 mM | 5 mg |
| Anhydrous DMSO | n/a | 500 μL |
| Total | 500 μL |
Prepare 10 μL (for 5 slices) or 20 μL (for 10 slices) aliquots and store at −20°C in a sealed container with a desiccator. We have used aliquots for at least one year after preparation without problems. Do not refreeze after use.
Digestion buffer (100 mL)
| Reagent | Final concentration | Amount |
|---|---|---|
| Triton X-100 | 0.5% (v/v) | 0.5 mL |
| EDTA, disodium (0.5 M, pH 8) | 1 mM | 0.2 mL |
| Tris-HCl (1 M, aqueous solution, pH8) | 50 mM | 5 mL |
| NaCl | 800 mM | 4.67 g |
| Water | n/a | Add up to 100 mL |
| Proteinase K (800 U/mL) | 8 U/mL | 1:100 dilution |
| Total | 100 mL |
Store the buffer (without Proteinase K) in 10 mL aliquots at −20°C. We have used buffer aliquots for up to three months after preparation without problems. Proteinase K should only be added at the last minute immediately prior to addition to the sample (i.e., 20 μL/2 mL buffer per sample).
Gelling solution (per sample)
| Reagent | Final concentration | Amount (first gelling solution) | Amount (second gelling solution) |
|---|---|---|---|
| Monomer solution∗ | n/a | 188 μL | 141 μL |
| 4-Hydroxy-TEMPO (4-HT) | 0.1 g/l | 4 μL | 3 μL |
| Tetramethylethylenediamine (TEMED) | 2 g/l | 4 μL | 3 μL |
| Ammonium persulfate (APS) | 2 g/l | 4 μL | 3 μL |
| Total | 200 μL | 150 μL |
Prepare on ice and use immediately. Add TEMED or APS last to prevent early gel polymerization.
4-HT: dissolve 0.1 g in 20 mL of water and store in 0.5 mL–1 mL aliquots at –20°C. We have used aliquots for at least one year after preparation without problems.
TEMED: prepare a 10% stock by diluting 1:10 the commercial reagent in water. Store at 4°C. We have used the stock for at least one year after preparation without problems.
APS: prepare a 10% stock and store at 4°C. We have used the stock for at least one year after preparation without problems.
∗Monomer solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Sodium acrylate | 86 g/l | 2.25 mL |
| Acrylamide | 25 g/l | 0.86 mL |
| N,N-methylenebisacrylamide | 1.5 g/l | 0.75 mL |
| NaCl (5 M) | 117 g/l | 4 mL |
| 10× DPBS | 1× | 1 mL |
| Water | n/a | 0.54 mL |
| Total | 9.4 mL |
Divide in 1 mL aliquots and store at −20°C. Do not freeze after thawing an aliquot; a thawed aliquot can be used up to several weeks later.
Sodium acrylate: dissolve 19 g in 50 mL water to obtain a stock solution of 38 g/100 mL, prepare 5 mL aliquots and store at –20°C. We have used aliquots for at least six months after preparation without problems.
Acrylamide: the referenced product is already in solution at the desired stock concentration of 290 g/l. Store at 4°C. We have used this reagent for at least one year without problems.
N,N-methylenebisacrylamide: dissolve 1 g in 50 mL water to obtain a stock solution of 2 g/100 mL, prepare 5 mL aliquots and store at –20°C. We have used aliquots for at least one year after preparation without problems.
Step-by-step method details
Immunostaining
We recommend culturing the OTCs for three weeks to ensure their maturation and stabilization before starting experiments and immunostaining. Immunostaining of OTCs is complicated by their thickness (even if it decreases during cultivation; Guy et al., 2011) and the glial scar caused by dissection, which severely hinders antibody penetration. While it is still possible to stain within a few microns of the surface using classical immunostaining protocols, cells located deep within the OTCs cannot be reliably stained. We aimed at localizing the expression of GluA2 containing AMPA receptors in the different spine subtypes along dendritic stretches determined by the transgenic mouse line (Tg(Thy1-EGFP)MJrs (Feng et al., 2000)). Because GFP-expressing cells are generally localized deeper than a few microns, we optimized a staining protocol combining a short fixation with long antibody incubations that ensured reliable and consistent depth penetration of antibodies by constant permeabilization.
Note: This protocol can also be applied to thick slices we have successfully applied it to 400 μm thick acute slices (see Bissen et al., 2021).
Day 1: Fixation, blocking, and primary antibody incubation
-
1.
Prepare the fixative (4% PFA/1× PBS) and the blocking buffer (3% normal donkey serum + 3% normal goat serum in 1× PBST) fresh and store at 4°C until use. PBST (2% Triton X-100/1× PBS) can be prepared in advance and stored at 22°C.
-
2.
Briefly rinse the OTCs in 1× PBS by completely removing the IM and adding 1 mL 1× PBS between the edge of the well and the insert.
-
3.
Fix the OTCs on ice for 5 min by completely removing the PBS and adding 1 mL of 4% PFA between the well and the insert and a few drops on top of the OTCs.
-
4.
Wash 5 × 10 min in 1× PBS on a rocker at 22°C. After 2–3 washes, gently scrape the OTCs off the insert with two thin spatulas and transfer them to a 24-well plate (Figure 2A).
Pause point: If necessary, OTCs can be stored in 1× PBS at 4°C in the dark and wrapped in parafilm until the protocol can be continued.
Note: We use 24-well plates because our OTCs are quite small; the diameter of the wells should be adjusted to the size of the OTCs. We usually place all OTCs of one insert (up to 6) in the same well.
-
5.
Incubate with blocking buffer for 2 h at 22°C on a rocker (250 μL/well).
-
6.
Incubate with the primary antibody in blocking buffer at the appropriate concentration for 7 days at 4°C on a rocker (250 μL/well). To avoid evaporation, wrap the plate in parafilm.
Note: Shorter incubation times (24–72 h) may be sufficient to yield a consistent and specific staining if the cells of interest are localized at the surface of the OTCs.
Note: We recommend including a sample that is not incubated with the primary antibody with each experiment to evaluate non-specific signals from the sample and/or the secondary antibody.
Figure 2.

Immunostaining
(A) Fixed OTCS are best removed by carefully scraping them from the Millipore insert with a metal spatula (step 4).
(B) The quality of immunostaining prior to expansion microscopy can be assessed by placing an OTC on a slide, drawing a closed circle around it with a PAP pencil, and carefully adding enough PBS to prevent a staining background caused by drying and to allow imaging with a water-dipping objective (step 10).
(C) Representative image of a successful immunostaining for GFP (green) and GluA2 (red). This is a single step within a Z-stack to better visualize GluA2 staining. Scale bar: 5 μm.
Day 7: Secondary antibody incubation
-
7.
Wash 3× 5 min in 1× PBS (1 mL/well) at 22°C on a rocker.
-
8.
Incubate with the secondary antibody in blocking buffer at the appropriate concentration for 3 days at 4°C in the dark (250 μL/well). Wrap the plate in parafilm to avoid evaporation.
Note: Shorter incubation times (3 h at 22°C or overnight (12 h–16 h) at 4°C) may be sufficient to yield a consistent and specific staining if the cells of interest are localized at the surface of the OTCs.
Day 10: Imaging
-
9.
Wash 3× 5 min in 1× PBS (1 mL/well).
Optional: Counterstaining with DAPI (1 μg/mL) can be performed at this step: 30 min at 22°C (250 μL/well) followed by 3× 5 min washes in 1× PBS. DAPI can help visualize the tissue if immediate imaging is performed but it does not survive expansion microscopy.
-
10.Image directly stained OTCs under a confocal microscope.Note: OTCs intended for expansion microscopy should not be mounted and can be imaged using a dry objective for low magnification and/or a water immersion objective for higher magnification pictures.
-
a.Draw a circle with a PAP pen (the diameter should be slightly larger than the size of the OTCs) and fill it with PBS.
-
b.Transfer one OTC in the PBS with a metal spatula (Figure 2B).Note: Optimize the imaging parameters according to the imaging setup (Figure 2C). Troubleshooting.Note: We strongly recommend at this point to assess the quality of the staining before proceeding with expansion microscopy; poor staining will be worse after expansion (where fluorophores are spread over a larger area), and if imaging of the expanded samples is problematic, pre-expansion imaging will help narrow down the cause of the problems to staining or expansion. We have also found that the best way to calculate the expansion factor is to measure the distance between two landmarks in the tissue before and after expansion, and the first measurement should be taken at this stage (see Figure 4A). For instance, we have taken advantage of the sparse labeling of pyramidal neurons by GFP in our transgenic mouse line to use somata of readily identifiable pyramidal neurons as landmarks.Note: PBS evaporates rapidly under the heat of the laser. There should be enough PBS to accommodate the normally larger working distance of water immersion objectives, and fresh PBS can be carefully added directly to the OTC if needed.
-
a.
-
11.
Keep the OTCs in 1× PBS (1 mL/well), wrap the plate in parafilm and aluminum foil (to protect from light), and store at 4°C until use.
Figure 4.

Imaging expanded OTCs
(A) The expansion factor can be calculated by measuring the distance between two orientation points before (left) and after (right) expansion. Scale bar: 100 μm.
(B) Representative pictures of dendritic stretch before (top) and after (bottom) expansion, with GFP in green. Scale bar: 5 μm. Note that the scale bar for expanded pictures corresponds to the physical size of the sample as given directly by the microscope based on the imaging parameters.
Expansion microscopy
To analyze protein distribution at the super-resolution level, we took advantage of expansion microscopy, a recently developed technique that improves spatial resolution by physically increasing sample size (Chen et al., 2015). Briefly, proteins are cross-linked with a polymer-based gel; the tissue is then digested, leaving only the anchored proteins within the gel, which is then expanded by osmosis in multiple water baths. Several variants of the initial protocol have been published, with optimizations for the model (cells, slices) and for the molecules of interest (protein or RNA), and they have been collected in a protocol paper (Asano et al., 2018). The present protocol is adapted from the Basic Protocol 2 from Asano and colleagues, with practical twists optimized for OTCs.
Note: Since samples are stained, all steps should be protected from light.
Day 1: Cross-linking
-
12.
Thaw one Acryloyl-X SE/DMSO aliquot in the fridge. Dilute it in 1× PBS (5 samples: 10 μL in 990 μL PBS; 10 samples: 20 μL in 1980 μL PBS).
-
13.
Transfer the OTC into the microwell of MatTek dishes using spatulas or brushes (1 OTC/microwell; Figure 3A).
Note: Microwells in MatTek dishes provide a favorable environment for gel formation by offering a suitable substitute for the gelation chamber that must otherwise be constructed (e.g., with coverslips; see Asano et al., 2018). Microwells are 700 μm deep and can thus contain any sliced tissue. The diameter of the microwell can also be adapted to the size of the tissue to avoid generating an overly large gel.
-
14.
Add 200 μL Acryloyl-X SE/DMSO/PBS to each microwell; the resulting bubble will cover the entire microwell. Leave the samples overnight (12 h–16 h) at 22°C (Figure 3A).
Figure 3.

Expansion microscopy
(A) Cross-linking: Transfer each OTC into the microwell of a MatTek dish (step 13) and add 200 μL Acryloyl-X into the microwell (step 14).
(B) Gelation: After 2× 15 min PBS washes, add 200 μL fresh gelling solution in the microwell and let stand at 4°C for 30 min (left panel, steps 15–19); replace the old gelling solution with 150 μL fresh gelling solution, carefully place a coverslip on top and let stand at 37°C for 2 h (right panel, steps 21 and 22).
(C) Post-Gelation: Gently pull off the coverslip (Option A); sometimes the coverslip sticks to the gel, in which case both should be removed from the microwell (Option B; step 23).
(D) Digestion: Add 2 mL digestion buffer and allow the gel to stand overnight (12 h–16 h) at 22°C (left panel; steps 24 and 25). The OTCs are already expanding during the digestion: compare the left (before digestion) and right (after digestion) panels (steps 25 and 26). Note that the gel has expanded out of the microwell.
(E) Expansion: Add ∼ 5 mL distilled or MilliQ water to the dish and let the OTC sit for ∼ 20 min; repeat until expansion reaches a plateau (in our hands, after 5–6 baths). The picture shows the same sample from D, now expanded after the fifth bath (step 28). The gel was cut around the sample to facilitate handling.
(F) Embedding: Add viscous agarose around the sample and wait a few minutes for the agarose to gel (1st step), then add a few extra drops to the edges of the gel to prevent movements in Z (2nd step), being careful not to cover the OTC to avoid diffraction artifacts (steps 32 and 33).
Day 2: Gelation and digestion
Gelation.
-
15.
Thaw the digestion buffer and the components for the gelling solution in the fridge.
-
16.
Wash the OTCs 2 × 15 min in 1× PBS.
-
17.
Prepare the gelling solution (200 μL/slice) immediately before use as it solidifies fast.
Note: Add TEMED and APS last to prevent immediate solidification.
-
18.
Remove the PBS and carefully add the gelling solution on top of the sample within the microwell (Figure 3B).
-
19.
Incubate in the dark for 30 min at 4°C.
Note: This step ensures deep penetration of the gelling solution within the tissue. It is recommended for slices thicker than 100 μm, but it is not necessary for thinner slices and cell cultures.
-
20.
Prepare fresh gelling solution (150 μL/slice) immediately before use.
-
21.
Carefully aspirate the old gelling solution from the microwell and replace it with freshly prepared solution. Using forceps, carefully place a 15 μm coverslip on top of the microwell (Figure 3B).
Note: The OTC is usually located at the bottom of the microwell and is thus closer to the bottom of the gel. This aspect is important for mounting and imaging (see below).
-
22.
Incubate for 2 h in the dark at 37°C. The gel should be completely polymerized.
Digestion.
-
23.
Carefully peel off the coverslip using straight forceps and check if the gel is completely polymerized (Figure 3C).
Note: Coverslips should be discarded and not reused.
Alternatives: The gel may adhere to the coverslip and detach from the microwell when pulled with forceps (Figure 3C). In this case, do not detach it and place it in the dish facing downward.
-
24.
Prepare a master mix of digestion buffer and proteinase K (2 mL and 20 μL per sample, respectively) and add 2 mL immediately on top of each gel (Figure 3D).
-
25.
Incubate overnight (12 h–16 h) in the dark at 22°C.
Day 3: Expansion and imaging
Expansion.
-
26.
Remove the digestion buffer and (if necessary) the coverslip.
Note: At the end of the digestion, the gel has usually started to expand so that it overflows from the microwell (Figure 3D).
-
27.
Add ∼ 5 mL of distilled water per dish.
Note: Immediate dehydration is not a problem, so all samples can be processed simultaneously rather than sequentially.
-
28.
After ∼ 20 min, renew the water bath with fresh distilled water. The gel is starting to expand, so add enough water to keep it at least completely covered. Repeat this step with fresh water until sample size does not increase anymore, i.e., when maximal expansion is reached. Store at 22°C in the dark (Figure 3E). Troubleshooting.
Note: The speed and extent of the expansion depends on the dish volume, wash time, number, and volume: more washes and/or longer incubation with a higher volume of water in a larger dish will result in greater expansion. The rate of expansion is determined by several factors, such an improper digestion, and most notably the salt concentration in the liquid used for expansion (a high-salt buffer such as 1× PBS will limit expansion and the impact of digestion on expansion compared to diluted PBS or distilled water).The ratio of acrylamide to sodium acrylate also significantly impacts expansion rate (see discussions in Asano et al., 2018; Truckenbrodt et al., 2019). For OTCs, we found that optimal expansion was reached after 5–6× 20 min washes in MatTek dishes. Expansion can be assessed by measuring sample size before digestion and after every bath; in our hands, we routinely reached a 3–3.5× expansion.
Note: Samples become transparent during expansion. Therefore, to facilitate visualization and manipulation, it is best to cut the gel around the sample with a scalpel before the first or second wash and as often as necessary thereafter. This will also facilitate handling during imaging.
Mounting and imaging.
-
29.
Melt 2% agarose in distilled water in the microwave during wash 3 or 4 and set the water bath to 35°C. Keep the agarose in the water bath all the time to prevent solidification.
Alternatives: we describe here embedding with agarose, which can be used for imaging with an upright microscope. Alternatives include glue (for upright imaging) or fixation with poly-D-lysine (for upright or inverted imaging) (see Asano et al., 2018).
-
30.
Remove the agarose from the water bath; it will take a few minutes to cool. In the meantime, remove the water from the sample to be imaged and carefully transfer it to a 3 cm Petri dish.
Note: A 3 cm Petri dish is sufficient to contain expanded OTCs, provided the excess gel was cut off during expansion. However, dish and sample size should be matched.
-
31.
Bring the sample to the confocal microscope. Assess the alignment of the OTC in the gel under the epifluorescence lamp. Troubleshooting.
Note: The epifluorescence lamp also illuminates the gel, which can make it easier to find the sample. However, it is not always sufficient to make sure that the sample is on top of the gel (for upright microscopes), as the illumination field is quite large. Assessing the orientation of the sample within the gel in respect to the Z-axis with confocal lasers is therefore necessary.
-
32.
Once alignment is confirmed and agarose is gelled, add agarose around the sample using a cut 1 mL tip and cover the entire dish to prevent horizontal movement. Make sure the sample is in the center of the dish to facilitate imaging (Figure 3F).
-
33.When the agarose is nearly solidified, place a few drops on the edges of the sample to prevent movement in Z. Cover the sample with distilled water and transfer it to the microscope for imaging with a water immersion objective for high magnification images (Figure 4).Note: Imaging parameters should be adjusted to suit the particular setup. For reference, we used a two-channel laser-scanning confocal (Leica, TCS SP8). This is the protocol we followed:
-
a.We identified our region of interest (the dentate gyrus) using a 10× air objective and switched to a 63× water-dipping objective (NA 0.9) to identify the somata of our cells of interest (granule cells). To avoid diffraction-induced loss of resolution, we selected cells within the upper half of our expanded samples (e.g., less than 200 μm deep).
-
b.We followed the dendritic trees of these cells until the outer molecular layer and imaged secondary dendritic stretches with a 3× zoom.
-
c.We rotated the selected stretch until it was horizontal and determined a rectangular region of interest fitting only this stretch to speed up imaging (1024 × 256 pixels).
-
d.We imaged a Z-stack encompassing the whole stretch with 0.5 μm step size, a frame averaging of 3 and using two lasers simultaneously at 488 nm (for GFP) and 561 nm (for GluA2 labeled with Alexa 568 as secondary antibody) (see Bissen et al., 2021). Troubleshooting.CRITICAL: Do not cover the sample itself with agarose as this will cause diffraction.CRITICAL: Imaging should be performed as soon as possible. The heat from the laser will cause the gel to begin to deposit water and shrink, causing it to separate from the agarose. In our hands, this tended to happen around 30–45 min after mounting. The movements, although small, are sufficient to interfere with imaging. If necessary, remove the agarose and remount the gel with fresh agarose.
-
a.
-
34.
After imaging, discard the water, remove the agarose and carefully return the sample to the original dish.
Note: The agarose can be reused. After a few samples, the agarose stock will become almost completely solid and thus unusable; thaw it briefly in the microwave and allow it cool to 35°C.
-
35.
For longer storage, the sample should be stored in PBS. It will shrink back to its original size and can thus be stored in a 24- or 12-well plate. We have re-used shrunk samples for expansion and imaging for up to two weeks after initial imaging without issues.
Expected outcomes
According to our preparation and culture protocol, OTCs should flatten and clear within a few days, showing clear morphology under the microscope and remaining healthy for several weeks (Figure 1E). After immunostaining, OTCs should show strong and specific staining, even for cells located deep in the tissue (Figure 2C). The expansion factor can be calculated by measuring the distance between two landmarks before and after expansion (Figure 4A). Expansion results in a decrease in signal strength as the fluorophores are stretched over a larger area and possibly lost due to anchoring and digestion; however, the quality of the staining should be sufficient to visualize the proteins of interest with improved spatial resolution (Figure 4B).
Quantification and statistical analysis
Images of stained OTCs before or after expansion were always deconvolved using the dedicated commercial software Huygens (SVI) to improve contrast and resolution. If necessary, motion artifacts in expanded pictures were corrected using the Rigid Body algorithm of the StackReg plugin (EPFL Bioimaging; Thévenaz et al., 1998) in ImageJ (NIH; Schneider et al., 2012). For expanded images, the distribution of the protein of interest was analyzed using the Surface module of the commercial software Imaris (Bitplane). Briefly, we prepared OTCs from mice expressing GFP under the Thy1 promoter (Feng et al., 2000) in a sparse subset of excitatory neurons to visualize neuronal morphology. We reconstructed fluorescent staining of GFP and our protein of interest, GluA2, separately as 3D volumes (to assess the distribution of GluA2-containing AMPA receptors). We used GFP as a mask to distinguish between GluA2 staining outside of the GFP volume (defined as “surface GluA2”) and GluA2 staining inside the GFP volume (defined as “internal GluA2”). We then quantified the two pools separately to assess the relative distribution of GluA2-containing AMPA receptors. Representative images were prepared in ImageJ (NIH) based on snapshots from Imaris, because the software is not well suited for cropping and labeling.
This analysis can be applied to any protein (provided that the immunostaining is of sufficient quality) and to other types of subcellular distribution (e.g., internal vesicles, such as endosomes, lysosomes, or Golgi apparatus). It can also be implemented in other models; we have successfully used it for protein distributions in acute slices and in neuronal cultures (Bissen et al., 2021).
Deconvolution
-
1.
Deconvolve the images in Huygens Professional (SVI) using the most appropriate parameters. Confocal images should be deconvolved using the classical maximal likelihood estimation (CMLE) algorithm. As a reference, we used a theoretical point spread function (PSF) and the automatic parameters (optimized iteration mode with a maximum of 40 iterations, signal-to-noise ratio of 20, background estimation radius 0.7).
Creation of a GFP mask and masked channels
In our strategy, we create a mask based on GFP staining larger than the neuron to include surface GluA2 staining in the analysis. This mask allows us to focus the analysis on the GluA2 staining associated with the selected dendritic stretch by masking the rest of the staining (step 3). In addition, any GFP staining that is not relevant to the analysis (e.g., additional stretches within the image or a portion of the dendritic stretch that is not currently being analyzed) is excluded (Figure 5A).
-
2.Create a GFP mask.
-
a.Open the deconvolved file in Imaris.
-
b.Select the “Add new Surfaces” and optionally rename the surface as “Mask”.
-
c.In the algorithm settings, select “Segment only a Region of Interest” and uncheck “Classify Surfaces”.Note: We recommend visualizing the data in Maximum Intensity Project (MIP) mode.
-
d.In the next step, define the region of interest (ROI) (Figure 5A).
-
e.Select the source channel from which to define the ROI - in our case “GFP all”. Select smooth and enter 0.120 μm.Note: This value should be adjusted depending on how much detail you want the surface to contain. A small value will allow the surface to match the coloring as closely as possible, but may result in the inclusion of background signals if it is too small.
-
f.Select as thresholding “Absolute Intensity”. To include all signals of the ROI (in our case, GluA2 staining at the dendritic surface located outside from the GFP signal), define the threshold as 0.3 (around 0.4 μm width, i.e., 20% of the dendritic diameter of ∼2 μm). Disable “split touching objects”.
-
g.In Filter Surfaces, set the value so that all signals within your defined ROI are covered by a surface. Click on “Finish: execute all creation steps and terminate wizard” (Figure 5A).
-
a.
-
3.Create masked channels for GFP and GluA2.
-
a.Select the GFP Mask surface from step 1 and go to the “Edit” tab.
-
b.Select “Mask All…”. Keep the default settings and select the GFP channel as the source channel to create a masked GFP channel. “Set voxels outside surface to 0” will exclude all signals outside the GFP Mask surface.
-
c.Repeat this step with the GluA2 channel as the source channel. The masked channels should display now only the signals within the ROI.
-
a.
Figure 5.

Data analysis in Imaris: Creation of a GFP mask (step 2) and GFP/GluA2 reconstruction (steps 3–8)
(A) Schematic illustration of creating a GFP mask in Imaris. First, define the region of interest (ROI; here a dendritic stretch within the picture). Next, create a surface with a low value for the threshold to include all signals within the ROI (here 0.3). Finally, execute all creation steps and terminate wizard. Based on the GFP mask create a masked channel for GFP. Repeat the process for GluA2 (step 3).
(B) Reconstruct the GFP staining so that the signal within the defined ROI is accurately reconstructed (step 4).
(C) Reconstruct the total GluA2 staining. Representative example of a GluA2 reconstruction with GFP staining (step 6).
(D) Separate the surface and internal GluA2 pools by creating two separate channels: set all outer voxels to 1 and inner voxels to 0 for surface GluA2 (magenta) and vice versa for internal GluA2 (blue) by using the masked GluA2 channel as source (steps 7 and 8). Representative example of surface and internal GluA2 reconstruction with GFP staining.
(E) Representative example of surface and internal GluA2 reconstruction with GFP reconstruction. Note that internal GluA2 is within of GFP reconstruction and therefore not visible.
Reconstruction of the GFP staining
-
4.
Create a new surface as described in step 2. Select the masked GFP channel created in step 3 as the source channel. Since a ROI is already defined in this channel, no further ROIs need to be defined in the following steps. Enter a threshold to reconstruct the GFP signal as accurately as possible (Figure 5B).
Creation of subcellular compartments
-
5.
To analyze the subcellular compartments (in our case, the shaft and individual spines) separately, rotate the GFP volume until a spine neck is horizontal. Go to the “Edit” tab and cut at the base of the neck using “Shift + left mouse click” in pointer selection mode. When the cut line is placed correctly and the spine is cut properly, select “Cut Surface” to confirm the cut. Repeat the process for each spine.
Note: We recommend duplicating the entire surface and proceeding with the separation of the subcellular compartments on this new surface. This will allow further analysis of the entire dendritic stretch using the original surface and the different subcellular compartments using the new surface.
Reconstruction of the total GluA2 staining
-
6.
Repeat the procedure from step 4 for GluA2 to obtain a reconstruction of the entire GluA2 pool, regardless of the subcellular pool. For punctate signals, we recommend using background subtraction with 0.3 instead of absolute intensity for thresholding. After reconstruction, all surfaces not in contact with the dendritic stretch should be excluded from reconstruction (Figure 5C).
Separation of surface and internal GluA2 staining and reconstruction of both signals
-
7.To distinguish between surface and internal GluA2 staining, create two additional channels using the masking technique described in step 3.
-
a.Select “masked GFP”, go to the “Edit” tab and select “Mask all…”.
-
b.Select the masked GluA2 channel from step 3 in the channel selection.
-
c.Select “Duplicate channel before applying mask” to create a separate reconstruction for the surface and internal GluA2.
-
d.To create a channel that displays only internal GluA2 staining, select “set voxels outside” to 0 and deselect “set voxels inside”.
-
e.Conversely, to create a channel that displays only surface GluA2 staining, deselect “set voxels outside” and select “set voxels inside” to 1.
-
a.
Note: This step divides the GluA2 staining (not the reconstruction!) into extra- and intracellular pools based on the exact GFP reconstruction as a proxy for the neuron. The masked GluA2 channel is used to focus the GluA2 staining analysis on the dendritic stretch of interest.
Optional: Rename the channels accordingly (e.g., “Surface GluA2” and “Internal GluA2”).
-
8.
Reconstruct these two pools as described in step 6. Create two separate surfaces by using the corresponding channel (“Surface GluA2” or “Internal GluA2”) as the source channel. We recommend using the same parameters as in this step (Figures 5D and 5E).
Note: Surface GluA2 which is not in contact with the GFP surface and therefore not part of the dendritic stretch is not considered in the analysis as described in step 6.
Statistical analysis
-
9.
Extract the volume of GFP or total GluA2 staining from the “Statistics” tab of the corresponding surface and export it to Excel.
Note: Additional information (e.g., position, length, etc.) is provided by Imaris and can be requested from the “Preferences” tab below the “Statistics” tab.
-
10.
For each spine or shaft, calculate the total GluA2/GFP volume by normalizing each GluA2 volume (of the total GluA2 surface, see step 6) to the GFP volume of that spine or shaft.
Note: The expansion factor by which a sample is expanded is likely to vary from sample to sample; in addition, non-uniform expansion (anisotropy) has been reported, especially in fibrous tissue (Pernal et al., 2020; Zhao et al., 2017). This spatial distortion is very limited at synapses (less than 3%; Jiang et al., 2018; Ku et al., 2016), but it is difficult to measure accurately in any sample without other means of super-resolution microscopy. Therefore, to avoid potential bias, we chose to always report our GluA2 data in terms of their GFP volume and compare relative GluA2 distributions.
-
11.
Calculate the average per shaft or spine type using all values from shafts or that spine type, and quantify the total GluA2/GFP distribution as a percentage of total GluA2/GFP by rescaling the averages of mushroom, thin, blunt spines, and shafts to their total sum.
-
12.
Using the separate reconstructions of surface and internal GluA2 (see step 8), normalize each individual value to the corresponding GFP volume. For each spine (or shaft), add the total volumes of surface and internal GluA2 to obtain the total GluA2 for that spine, and recalculate the surface and internal GluA2 as a percentage of the total GluA2 for that spine (or shaft). Use all individual values from each spine type (or shaft) to calculate the average for that spine type (or shaft).
-
13.
For statistical analysis, assess the normality of each data set using an appropriate normality test (the Shapiro-Wilke test works well for small sample sizes) and use the appropriate test (Student’s t-test, Mann-Whitney U-test, parametric or nonparametric ANOVA).
Limitations
Staining
Organotypic slice cultures have two major advantages over monolayer neuronal cultures: their thickness allows preservation of the 3D architecture of neuronal networks, and they can be cultured for a much longer period (at least 6 weeks in our case). They are thus closer to the ideal in vivo situation and can be subjected to more physiological paradigms to study neuronal function and plasticity. Immunostaining allows direct visualization of protein distribution, in combination with expansion microscopy at a high-resolution. However, the thickness of the OTC significantly hinders antibody penetration, which can only be compensated by stringent permeabilization. Such a protocol increases the risk of nonspecific staining, and the specificity of primary and secondary antibodies should be tested. If available, a knockout or knockdown line should be used to test the specificity of the antibody in immunostaining following the same protocol. This is the most stringent test, as it eliminates any possible confounding factors coming from proteins of similar size and/or cellular localization that could also be recognized by the antibody (and thus show plausible results by Western blot or immunostaining in wildtype tissue). Depending on the protein of interest, one option is to test the antibodies under non-permeabilized conditions in neuronal cultures - no signal should be visible if the protein is exclusively intracellular, whereas a surface protein should show no difference in distribution between non-permeabilized and permeabilized conditions. As an example, for GluA2-containing AMPA receptors, we determined the amounts of GluA2 under non-permeabilized conditions (surface GluA2 only) and after permeabilization. We compared the amount of surface GluA2 under these two conditions and showed that they were similar, confirming the validity of our analysis (Bissen et al., 2021).
Our immunostaining protocol takes longer than classical paradigms in order to reliably stain epitopes located in cells deep in organotypic cultures. If the cells of interest are located closer to the surface of OTCs, we suggest trying first shorter incubation times (e.g., 24–72 h at 4°C for the primary antibody, 2–4 h at 22°C or overnight (12 h–16 h) at 4°C for the secondary antibody; see e.g., Gogolla et al., 2006b), as they may be sufficient to obtain a reliable staining. Although we have not tried them, alternative methods proposed for cultured cells and fixed slices such as incubating at 37°C could also be considered (see e.g., Ferris et al., 2009). Alternatives for fixation could also be tested; trichloroacetic acid (TCA) and glyoxal did not work well in our hands, but could perform better with other antibodies.
Expansion microscopy
Anisotropic expansion has also been reported, especially for fibrous tissues (Pernal et al., 2020). While this does not apply to OTCs, they do have a glial scar from the dissection which could affect expansion if unequally digested. We did not observe gross anisotropy in our hands, but it is difficult to quantify precisely without other structures with a known size (e.g., microtubules). We therefore recommend avoiding comparison of absolute measurements between samples and referencing data to an intrinsic reference whenever possible (in our case, GFP volume as a proxy for neuronal volume).
In addition, imaging expanded samples can be very difficult due to the size and structure of the gel. This problem is discussed in troubleshooting, and part of the solution is to limit the number of fluorophores imaged per sample, thereby limiting the amount of information that can be obtained per experiment. Furthermore, not every dye survives expansion. For instance, the cyanine family of dyes is degraded and will yield very poor signal; as others, we have successfully used Abberior 635P as far-red dye (Asano et al., 2018). An alternative solution is to perform immunostaining after expansion, as long as the epitopes survive the digestion; we have not tried this variant in OTCs, but see no reason why it should not work (see Asano et al., 2018 for a protocol in fixed slices).
Troubleshooting
Problem 1
OTCs started to merge after culturing for several days (before you begin – step 11c).
Potential solution
If the OTCs are too close together, they will merge as they flatten and slightly enlarge after a few days (see Figures 6A and 6B). Arrange the OTCs as in Figure 6A so that they are sufficiently spaced apart.
Figure 6.

Troubleshooting
(A) Images of different stages during culture of OTCs with an appropriate distance – DIV 0, 1, 7, 14, 21. DIV: day in vitro (where the day of the dissection is DIV0).
(B) OTCs placed too close together will touch when flattening. DIV: day in vitro (where the day of the dissection is DIV0). Scale bar: 2 mm.
(C) Illustration shows correct (left) and incorrect (right) orientation of the gel during embedding for imaging in an upright microscope. Misalignment of the gel and positioning the sample far from the objective will result in significant distortion due to the gel.
Problem 2
Unhealthy OTCs in culture (before you begin - steps 14–16).
Potential solution
OTC health can be directly monitored under the binocular microscope: They may lose their transparency, become thicker, or lose their shape and disintegrate. Another option to check OTC health is to stain them live with propidium iodide, fix them briefly, and assess the propidium iodide staining under a confocal microscope.
Several parameters can be responsible. In particular, we recommend checking:
Whether the OTCs are covered with medium: this may prevent proper oxygen/carbon dioxide exchange with the incubator air and drown the slices.
Whether the temperature and carbon dioxide concentration in the incubator are constant.
If the horse serum is of good quality, and especially if it is to be heat-inactivated.
If the vibratome is properly calibrated. Lack of calibration can significantly affect the initial thickness of the OTCs when cut, and thus their survival.
If bacterial or fungal contamination is present.
Problem 3
Weak or no staining can be observed during imaging (step 10).
Potential solution
Insufficient permeabilization is probably the cause. The following solutions can be tested individually or in combination:
Increase the concentration of detergent or test other detergents (alone or in combination).
Increase the duration of incubation with the primary antibody.
Shorten PFA fixation and increase washing after fixation. An additional glycine wash could also help quench excess PFA, as described for cultured cells in Asano et al. (2018).
Note: Increased permeabilization may also result to increased non-specific staining. Blocking can be enhanced (e.g., by increasing the amount of normal goat or donkey serum or by adding bovine serum albumin) to prevent non-specific binding while allowing sufficient antibody penetration.
Problem 4
Expansion factor is small (steps 27 and 28).
Potential solution
OTCs reliably expand by a factor of ∼ 3–3.5× in our hands. This is lower than what has been reported by the Boyden lab (e.g., Chen et al., 2015) for fixed slices, but similar to other studies (e.g., Hafner et al., 2019). Expansion is limited by water volume and dish size; try expansion in larger dishes (e.g., 6 or 10 cm Petri dishes) and larger water volumes (e.g., 20–70 mL water).
If the expansion factor is consistently less than 3 even in large dishes, extend the digestion time by incubating for an additional 24 h in fresh proteinase K/digestion buffer. Also check that the proteinase K is not too old and has been stored properly. The quality of the water is also important, as salts in the water will affect the osmotic process and the expansion of the gel. Use MilliQ or deionized water, and check if changing the type of water improves the expansion factor.
Problem 5
Signal is distorted in expanded samples (steps 31–33).
Potential solution
The distortion is due to diffraction by the gel. Check the orientation of the gel to ensure that the sample is as close to the top of the gel as possible (Figure 6C), and also make sure that there is no agarose on the sample).
Problem 6
Mounted samples keep drifting during imaging (step 33).
Potential solution
Drift of expanded samples during imaging is a recurring problem and the most difficult of all to solve. The best way to compensate for sample drift is to perform embedding as quickly as possible by keeping the agarose at a temperature just above the solidification temperature (35°C) and imaging quickly. If possible, we recommend narrowing down the region of interest (e.g., a rectangle 1024 pixels long and 256 pixels high rectangle for dendritic stretches that are first rotated to horizontal) and imaging with a single scan rather than sequential scans of multiple channels. This limits the number of fluorophores imaged and thus the number of proteins that can be assessed simultaneously, but increases the quality of the imaging. The imaging speed can also be increased and the line/frame averaging decreased, but care should be taken not to compromise the quality of the images too much. Depending on the size of the sample, the gel could also be cut at the exact dimensions to fit its container, e.g., a chambered slide (see Gaudreau-Lapierre et al., 2021 as example).
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Amparo Acker-Palmer (acker-palmer@bio.uni-frankfurt.de).
Materials availability
This study did not generate new reagents.
Acknowledgments
This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB 834, SFB1080, SFB1193, FOR2325, EXC 115, and EXC 147), ERC_AdG_Neurovessel (Project Number: 669742), the Max Planck Fellow Program and Gutenberg Research College (GRC) at Johannes Gutenberg University Mainz (A.A-P.). We thank E. Harde, A. Vlachos, and T. Deller for help with the lesion model, P. Donlin-Asp and A-S. Hafner for help with expansion microscopy, U. Bauer, P. Brendel, D. Schmelzer, and T. Belefkih for technical support, M. M. Middeke for taking the experimental pictures, M. Segarra for helpful discussions.
Author contributions
D.B. designed, performed, and supervised experiments; M.K.K. designed and performed the analysis of expansion microscopy experiments; F.F. contributed to the design of experiments; A.A.-P. designed and supervised all stages of the project; and all authors wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Diane Bissen, Email: dianebissen@brandeis.edu.
Amparo Acker-Palmer, Email: acker-palmer@bio.uni-frankfurt.de.
Data and code availability
The datasets supporting this study are available from the corresponding author upon request.
References
- Asano S.M., Gao R., Wassie A.T., Tillberg P.W., Chen F., Boyden E.S. Expansion microscopy: protocols for imaging proteins and RNA in cells and tissues. Curr. Protoc. Cell Biol. 2018;80:e56. doi: 10.1002/cpcb.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissen D., Kracht M.K., Hofmann J., Foss F., Acker-Palmer A. EphrinB2 and GRIP1 stabilize mushroom spines during denervation-induced homeostatic plasticity. Cell Rep. 2021;34:108923. doi: 10.1016/j.celrep.2021.108923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon de Anda F. Organotypic slice culture of embryonic brain sections. Bio Protoc. 2013;3:e327. doi: 10.21769/bioprotoc.327. [DOI] [Google Scholar]
- Chen F.C., Tillberg P.W., Boyden E.S. Expansion microscopy. Science. 2015;347:543–548. doi: 10.1126/science.1260088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Turco D., Deller T. Organotypic entorhino-hippocampal slice cultures — a tool to study the molecular and cellular regulation of axonal regeneration and collateral sprouting in vitro. Methods Mol. Biol. 2007;399:55–66. doi: 10.1007/978-1-59745-504-6_5. [DOI] [PubMed] [Google Scholar]
- Elias L., Kriegstein A. Organotypic slice culture of E18 rat brains. JoVE. 2007;6:235. doi: 10.3791/235. http://www.jove.com/index/Details.stp?ID=235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G., Mellor R.H., Bernstein M., Keller-Peck C., Nguyen Q.T., Wallace M., Nerbonne J.M., Lichtman J.W., Sanes J.R. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Ferris A.M., Giberson R.T., Sanders M.A., Day J.R. Advanced laboratory techniques for sample processing and immunolabeling using microwave radiation. J. Neurosci. Methods. 2009;182:157–164. doi: 10.1016/j.jneumeth.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Gaehwiler B.H. Organotypic monolayer cultures of nervous tissue. J. Neurosci. Methods. 1981;4:329–342. doi: 10.1016/0165-0270(81)90003-0. [DOI] [PubMed] [Google Scholar]
- Gaehwiler B.H., Capogna M., Debanne D., McKinney R.A., Thompson S.M. Organotypic slice cultures: A technique has come of age. Trends Neurosci. 1997;10:471–477. doi: 10.1016/s0166-2236(97)01122-3. [DOI] [PubMed] [Google Scholar]
- Gaudreau-Lapierre A., Mulatz K., Béïque J.C., Trinkle-Mulcahy L. Expansion microscopy-based imaging of nuclear structures in cultured cells. STAR Protoc. 2021;2:100630. doi: 10.1016/j.xpro.2021.100630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogolla N., Galimberti I., DePaola V., Caroni P. Preparation of organotypic hippocampal slice cultures for long-term live imaging. Nat. Protoc. 2006;1:1165–1171. doi: 10.1038/nprot.2006.168. [DOI] [PubMed] [Google Scholar]
- Gogolla N., Galimberti I., DePaola V., Caroni P. Staining protocol for organotypic hippocampal slice cultures. Nat. Protoc. 2006;1:2452–2456. doi: 10.1038/nprot.2006.180. [DOI] [PubMed] [Google Scholar]
- Guy Y., Rupert A.E., Sandberg M., Weber S.G. A simple method for measuring organotypic tissue slice culture thickness. J. Neurosci. Methods. 2011;199:78–81. doi: 10.1016/j.jneumeth.2011.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner A.-S., Donlin-Asp P.G., Leitch B., Herzog E., Schuman E.M. Local protein synthesis is a ubiquitous feature of neuronal pre- and postsynaptic compartments. Science. 2019;364:eaau3644. doi: 10.1126/science.aau3644. [DOI] [PubMed] [Google Scholar]
- Humpel C. Organotypic vibrosections from whole brain adult Alzheimer mice (overexpressing amyloid-precursor-protein with the Swedish-Dutch-Iowa mutations) as a model to study clearance of beta-amyloid plaques. Front. Aging Neurosci. 2015;7:47. doi: 10.3389/fnagi.2015.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang N., Kim H.J., Chozinski T.J., Azpurua J.E., Eaton B.A., Vaughan J.C., Parrish J.Z. Superresolution imaging of Drosophila tissues using expansion microscopy. Mol. Biol. Cell. 2018;29:1413–1421. doi: 10.1091/mbc.e17-10-0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku T., Swaney J., Park J.Y., Albanese A., Murray E., Cho J.H., Park Y.G., Mangena V., Chen J., Chung K. Multiplexed and scalable super-resolution imaging of three-dimensional protein localizationin size-adjustable tissues. Nat. Biotechnol. 2016;34:973–981. doi: 10.1038/nbt.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mewes A., Franke H., Singer D. Organotypic brain slice cultures of adult TransgenicP301S mice—a model for tauopathy studies. PLoS One. 2012;7:e45017. doi: 10.1371/journal.pone.0045017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernal S.P., Liyanaarachchi A., Gatti D.L., Formosa B., Pulvender R., Kuhn E.R., Ramos R., Naik A.R., George K., Arslanturk S., et al. Nanoscale imaging using differential expansion microscopy. Histochem. Cell Biol. 2020;153:469–480. doi: 10.1007/s00418-020-01869-7. [DOI] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L., Buchs P.A., Muller D. A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Thévenaz P., Ruttimann U.E., Unser M. A pyramid approach to subpixel registration based on intensity. IEEE Trans. Image Process. 1998;7:27–41. doi: 10.1109/83.650848. [DOI] [PubMed] [Google Scholar]
- Truckenbrodt S., Sommer C., Rizzoli S.O., Danzl J.G. A practical guide to optimization in X10 expansion microscopy. Nat. Protoc. 2019;14:832–863. doi: 10.1038/s41596-018-0117-3. [DOI] [PubMed] [Google Scholar]
- Zhao Y., Bucur O., Irshad H., Chen F., Weins A., Stancu A.L., Oh E.Y., Distasio M., Torous V., Glass B., et al. Nanoscale imaging of clinical specimens using pathology-optimized expansion microscopy. Nat. Biotechnol. 2017;35:757–764. doi: 10.1038/nbt.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets supporting this study are available from the corresponding author upon request.