

Abstract
Engineered tissues can model human pathophysiology and be used to test the efficacy and safety of drugs. Yet, to model whole-body physiology and systemic diseases, engineered tissues with preserved phenotypes need to physiologically communicate. Here, we report the development and applicability of a tissue-chip system in which matured human heart, liver, bone and skin tissue niches are linked by recirculating vascular flow, to allow for the recapitulation of interdependent organ functions. Each tissue is cultured in its own optimized environment and is separated from the common vascular flow by a selectively permeable endothelial barrier. The interlinked tissues maintained their molecular, structural and functional phenotypes over 4 weeks of culture, recapitulated the pharmacokinetic and pharmacodynamic profiles of doxorubicin in humans, allowed for the identification of early miRNA biomarkers of cardiotoxicity, and increased the predictive values of clinically observed miRNA responses relative to tissues cultured in isolation and to fluidically interlinked tissues in the absence of endothelial barriers. Vascularly linked and phenotypically stable matured human tissues may facilitate the clinical applicability of tissue chips.
One-sentence editorial summary (to appear right below the title of your Article on the journal’s website): Tissue chips with matured human heart, liver, bone and skin tissue niches linked by recirculating vascular flow recapitulate interdependent functions of these organs.
Drug safety and efficacy are typically evaluated in animal models, which frequently fail to predict clinical responses1–4. Microphysiological systems (MPS), with bioengineered human tissues designed to mimic organ-level functions are being developed to enable modeling human physiology in vitro5–10. Culturing a tissue in isolation can provide meaningful insights into some of the organ functions, but not the systemic interactions that influence organ responses to injury, disease, and therapy5,8,11,12. Organs in the body interact by virtue of (i) boundaries preserving the specificity of their individual environments, (ii) endothelial barriers separating the intratissue from intravascular spaces, and (iii) organ-organ crosstalk via vascular flow. These features are critical for tissue homeostasis in health and disease. Diseases that impact several organs and vascular flow are particularly difficult to emulate in vitro. Systemic diseases remain understudied, including cancer, fibrosis, inflammation and infection that are of high interest.
Although there is a clear need for MPS designs that can model the complexity of human physiology, the functional integration of tissues has been an elusive goal due to the conflicting requirements for maintaining and connecting tissue-specific niches13. Current methods rely on transferring supernatant between the tissues or using of shared (common) media that contain a mix of factors collectively required by all tissues in the system3,5,10. While these models can recapitulate certain aspects of human pathophysiology9, 13, 14, they provide limited phenotypic stability when the same medium is used to cultivate tissues from different germ layers. Common media can also induce committed cells to revert back to more plastic and immature phenotypes. As engineering of physiologically matured tissues continues to advance, methods to preserve the achieved maturity of physiologically linked multi-tissue systems also needs to be developed.
To this end, we designed the InterOrgan tissue chip which uniquely allows physiologically relevant integration of bioengineered tissues by (i) providing each tissue with its own specialized environment, (ii) connecting tissues by vascular flow, and (iii) separating the vascular and tissue compartments by a selectively permeable endothelial barrier. The tissue chip contained four tissues: heart, bone, liver and skin, selected for their distinctly different properties and importance for modeling diseases and testing drugs. The tissues were engineered from human induced pluripotent stem cells (hiPSC) for biological specificity, matured individually for 4–6 weeks under conditions promoting their phenotype, transferred into the tissue chip, and linked by vascular flow.
The maintenance of matured tissues in the InterOrgan tissue chip was compared to the Mixed tissue chip (equivalent to the InterOrgan tissue chip, except for the lack of endothelial barrier, thus representing the common medium culture), and Isolated cultures (tissues cultured individually, with and without endothelial barriers). To demonstrate the tissue chip utility for drug screening, we investigated the multi-organ toxicity of doxorubicin (a drug metabolized by liver) from measured tissue responses at the molecular, cellular, and functional levels, in all tissue chip configurations. The experimental and computational pharmacokinetics (PK)/pharmacodynamics (PD) studies were also conducted for these configurations, enabling further understanding of the contributions of tissues cultured systemically versus individually, with and without endothelial barriers. The tissue outcomes were benchmarked and validated using data from adult and pediatric clinical studies detailing miRNA biomarkers related to doxorubicin induced cardiac toxicity, the most serious side effect of chemotherapy14–19.
RESULTS
Multi-tissue chips design
The InterOrgan tissue chip was designed to allow physiological communication of engineered human tissues over long culture times (4 weeks) while maintaining their individual phenotypes (Fig. 1A&B, Extended Data Fig. 1A–C and 2). All tissues and endothelium were matured individually prior to integration to establish adult-like functions and connected by vascular flow containing circulating immune cells, cytokines and extracellular vesicles, with endothelial barrier separating the tissues and vascular flow. The tissue chip is manufactured from polysulfone, a biocompatible inert polymer. The tissue chambers are modular, have open access, and can be connected in any order to model different physiological scenarios (Fig. 1B, Extended Data Fig. 1D–F, Supplementary Video 1), by virtue of the same outside geometry and customized interiors, leak-free click connectors and ports. The tissue chip uses a single channel of a peristaltic pump to recirculate culture media at a set flow rate and shear stress (Extended Data Fig. 1G&H, Extended Data Fig. 2). The modular design allows tissues to be placed into the tissue chip after they have passed quality control and enables configurability in terms of the types, numbers and order of the tissues, tailored to the exact experimental design. To validate the tissue chip utility, we evaluated the maintenance of tissue phenotypes over 4 weeks of culture (Fig. 1C) and in response to doxorubicin (Fig. 1D).
Figure 1 |. Integrated multi-organ chip enables maintenance of a tissue-specific niche while allowing for organ cross-talk.
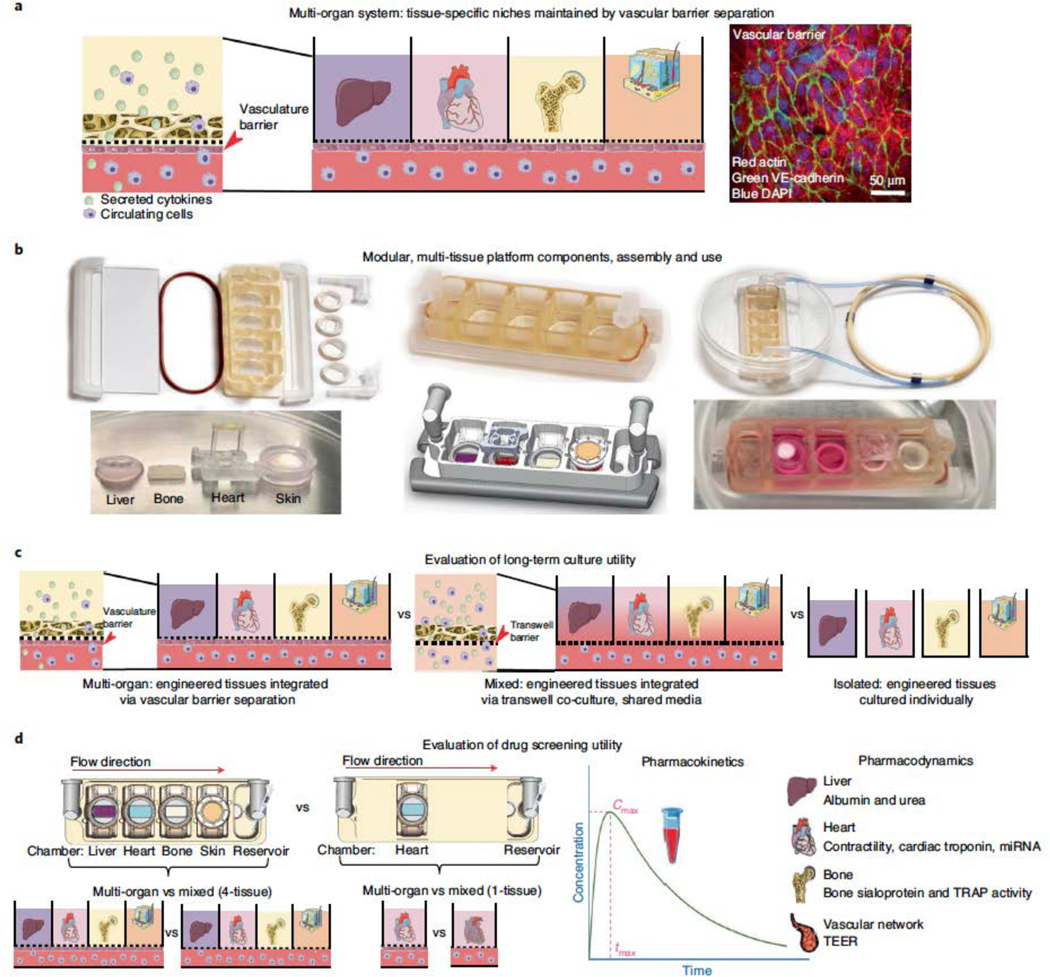
a, Schematic detailing a side view of the multi-tissue chip where integration is enabled by a vascular barrier beneath each tissue which creates a tissue-specific niche in the above chambers for each engineered organ, while enabling cross-talk between organs within the system through the vascular chamber. Immunostain demonstrating expression of actin alpha (red) and VE-cadherin (green) by the endothelial barrier. Samples were counterstained with DAPI (blue). Scale bar, 50 μm. b, Photographs detailing that the engineered chip is easily configurable, allowing for a “plug-and-play” system for individual organ chambers and a vascular flow channel beneath each organ. Engineered tissues are shown before and after being placed into the engineered tissue chip, where the vascular barrier enables maintenance of each specific media, as detailed by the differences in media color within the photograph. c, Schematics of the experimental design for evaluating of tissue chip’s long term culture utility to validate the importance of the vascular barrier within the InterOrgan tissue chip, as compared to the lack of vascular barrier in the Mixed tissue chip, and benchmarked against the tissues cultured for the same length of time in isolation. d, Schematics of the experimental designs for evaluating tissue chip utility for drug screening of doxorubicin, where the drug responses of the engineered tissues cultured in the InterOrgan and Mixed tissue chips were compared directly, for cases where all four tissues or single tissues were cultured in either tissue chip.
Generating matured human tissues
Achieving and maintaining tissue maturity, which is necessary for recapitulating adult human physiology, has been difficult to achieve for tissues derived from hiPSC. We formed and matured tissues individually before being linked in the tissue chip, allowing different maturation regimens and durations required for different tissue types as well as quality control of each tissue to maximize consistency. The methods for tissue formation and maturation and the specifications of tissue properties are summarized in Fig. 2, Extended Data Fig. 3, and Supplementary Table 1.
Figure 2 |. Formation, maturation, and characterization of engineered human tissues.

Schematics detailing initial tissue formation (cell types and scaffold or extracellular matrix), tissue-specific maturation protocols, and representative tissue photographs and immunofluorescence or immunohistochemical images for heart (green) (a), liver (b), bone (c), skin (d), and vasculature (e), after the maturation protocol is completed.
Heart muscle was formed from hiPSC-derived cardiomyocytes (CM) and fibroblasts in a fibrin matrix anchored to two auxotonic flexible pillars, and electromechanically matured at an increasing intensity for four weeks (Fig. 2A), as we previously reported11. Electromechanically matured tissues displayed aligned sarcomeres with high expression of alpha-actinin-2 increased maximum capture rate, decreased excitation threshold, and increased contractile force, when compared to non-matured controls (Extended Data Fig. 3 A–C).
Liver tissues were formed from aggregates of hiPSC-derived hepatocytes and supporting human fibroblasts encapsulated in fibrin matrix20 (Fig. 2B, Extended Data Fig. 3D). The liver tissues displayed active metabolism and sustained production of albumin (Extended Data Fig. 3E).
Bone tissues were made by seeding human bone marrow-derived mesenchymal stromal cells (MSC) into decellularized bone scaffolds and inducing the cells towards the osteoblastic phenotype over a period of three weeks21 (Fig. 2C). To recapitulate the osteolytic cycle, primary CD14+ monocytes were seeded into the osteoblastic bone and cultured in osteolytic medium capable of supporting both existing osteoblasts and additional monocytes as they differentiate into osteoclasts21. The resulting bone tissues displayed high presence of bone sialoprotein (BSP) and osteocalcin (OCN), and mature tissue-specific morphology, as detailed by the presence of osteoblast leading edges and tartrate-resistant acid phosphatase (TRAP) positive lacunae (Extended Data Fig. 3F). Micro-computed tomography (μCT) imaging showed increased bone density during osteoblastic maturation and stable bone volume during the induction of the osteolytic phase of bone maturation (Extended Data Fig. 3G).
Skin tissues were produced by seeding human dermal fibroblasts into collagen matrix, adding human keratinocytes, and cultivation at the air liquid interface for three weeks to form and mature stratified epidermal tissue (Fig. 2D, Extended Data Fig. 3H)22. These tissues showed barrier function, as determined by measuring the transepithelial/transedothelial electrical resistance (TEER), consistent with published values for human skin explants (Extended Data Fig. 3I)23.
Generating a mature and selectively permeable vascular barrier
Underneath each tissue chamber is an elastic mesh insert (Fig. 3A&B; Extended Data Fig. 2A) covered with endothelial cells and supporting MSC24,25 forming a vascular barrier (Extended Data Fig.4 A&C). We found that the 20 μm pores best supported the formation of confluent endothelium (Fig. 3C, Extended Data Fig. 4A–C). We further evaluated the effects of shear stress (1.88 to 6.27 dynes cm−2) on vascular endothelium, and determined that the lower shear stress was more conducive to the maintenance of stable vascular barrier (Fig. 3D, Extended Data Fig. 4B–D). We also determined that a slow ramping of the applied shear stress facilitated the establishment of endothelial barrier, and matured the endothelial and mesenchymal cells by gradual exposure to hemodynamic shear7 increasing from 0.5 to 1.88 dynes cm−2 over 60 hours of culture (Fig. 2E). Tight barrier function was maintained at a shear stress of 1.88 dynes cm−2 with the formation of vascular gap junctions and barrier integrity (Fig. 3D, Extended Data Fig. 4C&D). The tissues communicated by cytokines, exosomes, and cells by the circulating flow beneath the vascular barrier (Extended Data Fig. 4E). Additionally, these Is this a place to say that these studies were conducted using hiPSC from a single donor, thereby demonstrating the tissue chip’s utility for “patient on a chip” models. The ability to use matched patient-specific cells in each linked engineered tissue, enables in-vitro human studies of each individual patient and their specific genetic background at both the individual organ and systemic level.
Figure 3 |. InterOrgan communication via vascularized barrier of tissue-specific niches.
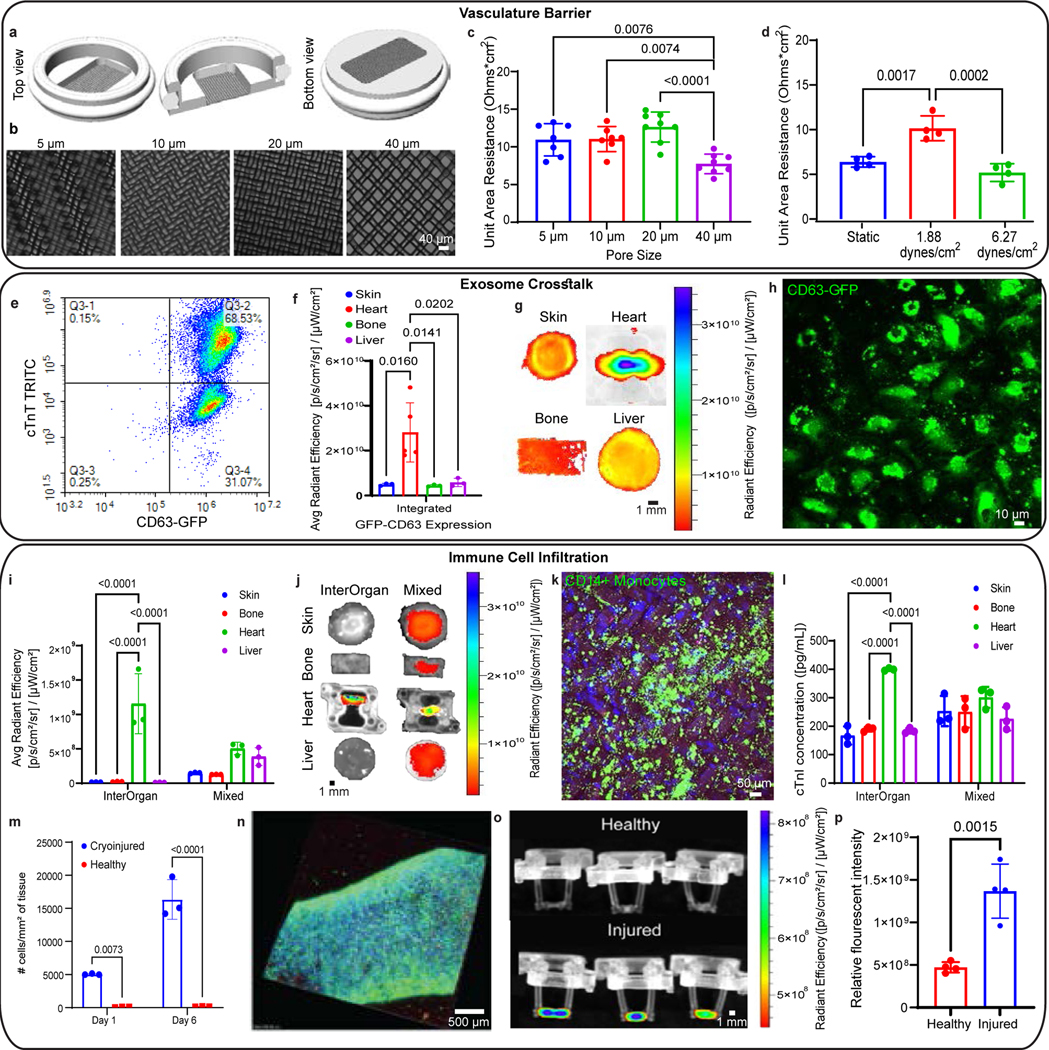
a-c, Characterization of the vascular barrier insert (a) at various pore size (b) as measured by vascular transendothelial electrical resistance (TEER) values (c). (n=7–8 biological replicates) d, TEER measurements as a function of shear stress (n=4 biological replicates). Data is shown as mean ± SD and statistics determined by one-way ANOVA. e, Cardiac tissues contain a majority of GFP+CD63+ cardiomyocytes, as indicated by expression of cardiac troponin (cTnT) and CD63 in quadrant 3 of the flow cytometry scatter plot. f-g, Average expression (f) (n=5 biological replicates) and tissue heat-map expression (g) of GFP+CD63 within all tissues cultured in the InterOrgan tissue chip for 2 weeks. h, Immunofluorescence image of GFP+CD63 expression within the vascular barrier beneath the cardiac tissue after 2 weeks. i-j, Average expression (i) and tissue heat-map expression (j) of labelled monocytes within all tissues cultured in the InterOrgan or Mixed tissue chips for 24 hours (n=3 biological replicates). k, Immunofluorescence image of CD14+ monocytes attached to the vascular barrier beneath the cardiac tissue after 24 hours. Scale bar, 10 μm. l, Cardiac troponin concentration within each tissue chamber 24 hours after cardiac cryoinjury (n=3 biological replicates). m, Monocyte infiltration over time as measured by confocal tracking of labelled monocytes (n=3 biological replicates). n, Immunofluorescence image of CD14+ monocytes (green) attached to a cryoinjured cardiac tissue counterstained with DAPI (blue) after 7 days. Scale bar, 10 μm o-p, Tissue heat-map expression (o) and average expression (p) of labelled monocytes within healthy and cryoinjured cardiac tissues after 7 days (n=4 biological replicates). Data is shown as mean ± SD and statistics determined by unpaired t-test.
To document cross-talk between tissues, we generated heart tissues from hiPSC-derived CM transfected with a green fluorescent protein (GFP) labelled CD63 exosome reporter (Fig. 3E), thereby enabling the tracking of a non-ubiquitous organ specific marker of known origin. CD63 exosomes secreted by heart tissues were found in all tissues after two weeks of culture in the InterOrgan tissue chip (Fig. 3F&G). Similarly, immunofluorescence imaging of the vascular barrier beneath the heart tissue after two weeks showed exosome uptake by endothelial cells (Fig. 3H).
To include immune cells into the vascular flow, CD14+ monocytes were added into the reservoir. Over 98% of the initial monocytes expressed CD14 and ITGAM (CD11b) markers (Extended Data Fig. 5A, Supplementary Fig. 1), attached to the endothelial barriers (Extended Data Fig. 5B), and remained viable in the tissue chip, with or without endothelial barrier (Extended Data Fig. 5C). Over 4 weeks, monocytes in the InterOrgan tissue chip maintained their classical (CD16−CD14+) phenotype whereas the Mixed and Isolated tissue chip shifted towards an intermediate (CD16+CD14+) phenotype (Extended Data Fig. 5D).
The circulating human CD14+ monocytes extravasated from vascular flow in response to injury signals, and otherwise stayed confined to the vascular compartment. Specifically, in response to a cryo-induced injury to the heart tissue, circulating immune cells extravasated from the vascular flow within 24 hours and targeted the injured tissue, but not the healthy tissues. Selective infiltration of monocytes into the injured tissues was only seen in the InterOrgan tissue chip whereas the Mixed tissue chip showed non-selective infiltration (Fig. 3I&J). Monocytes also accumulated on the endothelial layer underneath the injured tissues (Fig. 3K).
Upon injury, heart tissues secreted troponin - ubiquitous cardiac specific marker, which could be found in other tissue compartments, indicating crosstalk (Fig. 3L). Cardiac troponin concentrations were uniform within the Mixed tissue chip, a non-physiological condition due to the lack of vascular barrier. Culture of healthy and cryoinjured heart tissues in adjacent chambers of the InterOrgan tissue chip showed selective infiltration of monocytes into only the injured heart tissue, a response that was maintained over a week of culture (Fig. 3M–P).
Physiological effects of tissue maturation
The methods we used to achieve maturity utilize tissue-specific mechanisms (chemical, metabolic, electrical, and/or mechanical). Here we highlight the effects of maturation of the heart tissue, achieved by subjecting early hiPSC-derived CM encapsulated in fibrin hydrogel to electromechanical conditioning at a gradually increasing intensity. RNA sequencing of matured heart muscle revealed the role of electromechanical stimulation in promoting the development of contractile function, metabolism, and calcium regulation (Supplementary Fig. 2A–C).
Ingenuity Pathway Analysis (IPA) revealed upregulation of the more mature sarcomere isoforms MYL2 and MYH7 (Supplementary Fig. 2A&C). Gene set enrichment analysis (GSEA) against adult healthy human tissue databases26 showed the enhancement of cardiac myogenesis, metabolic maturation via oxidative phosphorylation and fatty acid metabolism (Supplementary Fig. 2D), and highly upregulated pathways to striated tissue contraction, calcium handling, and relaxation (Supplementary Fig. 2E). Gene set enrichment for gene ontology details that maturation promoted heart muscle contraction, development, and differentiation (Supplementary Fig. 3).
Gene ontology (GO) analysis showed upregulation of biological processes related to the development of cardiac and striated muscle, myofibril assembly, and striated muscle contraction (Supplementary Fig. 4A). Gene ontology for cellular components showed upregulation of cardiac specific ultrastructure (sarcomere, myofibril, Z-disc, I-band) and sarcoplasmic reticulum (Supplementary Fig. 4B). Matured tissues recapitulated the clinical EC50 values of several drugs, and the bradycardic effects of calcium channel blockers seen in patients (Supplementary Fig. 4C). Of note, screening of these drugs using hiPSC-derived CM monolayers revealed tachycardic responses27, further supporting the need for maturation of engineered tissues for use in modeling disease.
Tissues maintain their phenotypes over 4 weeks in the InterOrgan tissue chip
The maturity of engineered tissues needs to be maintained after the tissues are connected for sufficient periods of time to conduct physiological studies. The InterOrgan tissue chip was designed to preserve the tissue-specific niche and mature phenotype of each tissue while enabling tissue-tissue communication. To evaluate this critically important capacity, the InterOrgan tissue chip was systematically compared over 4 weeks of culture to the identical tissue chip without endothelial barriers (Mixed condition, corresponding to the co-culture in common medium), and tissues cultured individually (Isolated condition, benchmark for phenotype stability) (Fig. 1C, Fig. 4).
Figure 4 |. InterOrgan tissue chip demonstrates maintenance of structural, functional, and molecular phenotypes for each engineered organ over 4 weeks following linking of tissues by vascular flow.
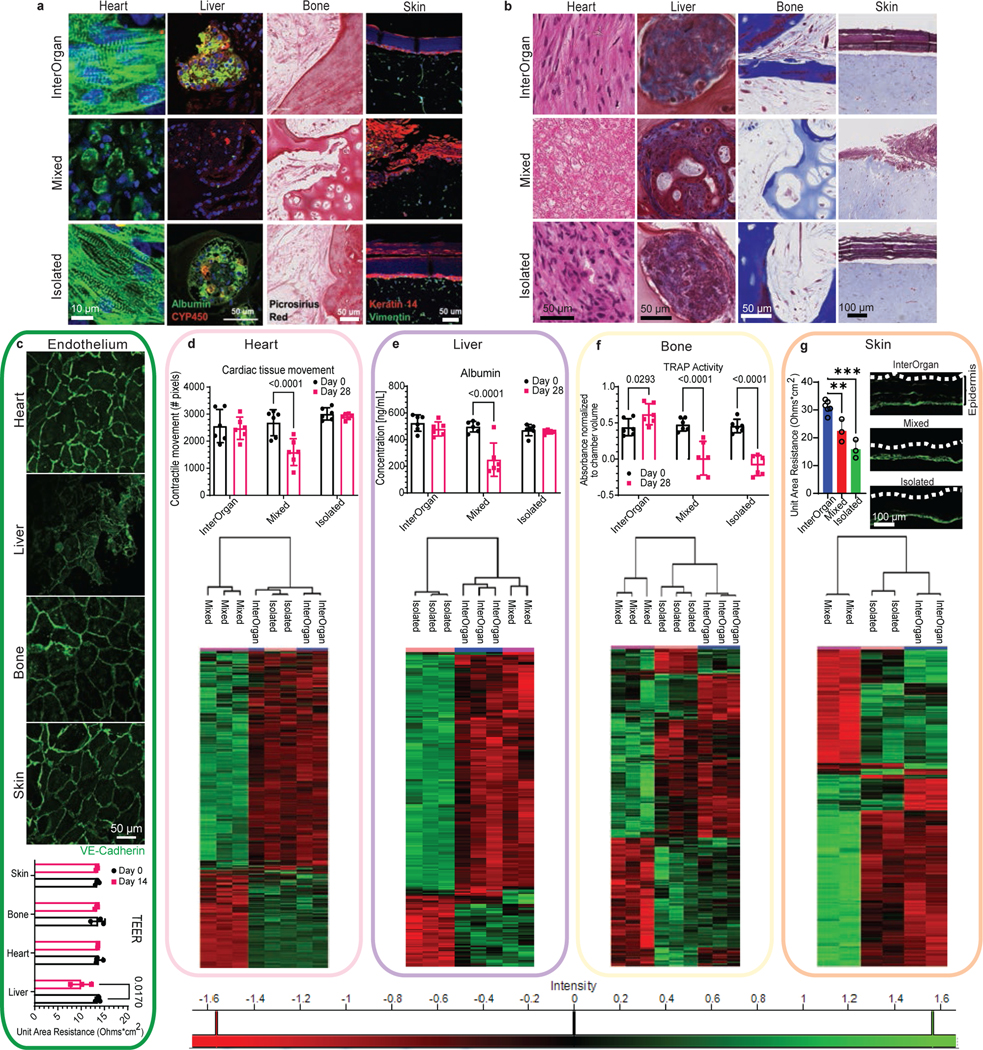
a-b, Representative immunofluorescence staining (a) and trichrome histological staining (b) displays morphological differences between groups (Scale bar, 50 μm for heart, liver, and bone; 100 μm for skin). c, Vascular stability is maintained after 4 weeks in culture as shown by VE-Cadherin expression (green). Scale bar, 50 μm. Transendothelial electrical resistance (TEER) measurements of the endothelial barrier (n=3 biological replicates). d-g, Functional (n=6 biological replicates) and overall proteomic comparison of molecular features (n=2–3 biological replicates) for each engineered organ compared in each condition. Immunostains showing the epidermal layer of the skin. Scale bar, 100 μm, Data is shown as mean ± SD and statistics determined by two-way ANOVA.
All tissues in the InterOrgan tissue chip maintained the structural, functional, and molecular stability of the “gold standard” (Isolated group), and markedly exceeded the corresponding tissue properties in the Mixed group over 4 weeks of culture following integration by vascular flow (Fig. 4). Specifically, heart muscle showed increased cell elongation and striations in the InterOrgan and Isolated groups as compared to the Mixed group. Bone tissues in the InterOrgan and Isolated groups displayed mature osteolytic phenotype, while the Mixed group showed decreased collagen deposition. Liver tissue morphology was comparable for the InterOrgan and Isolated groups and inferior in the Mixed group. The epidermal layer of the skin remained intact in the InterOrgan and Isolated groups, whereas the Mixed tissue chip showed disruptions (Fig. 4A,B,G).
The vascular endothelium in the InterOrgan tissue chip has maintained barrier function, with the exception of the liver endothelium that assumed decreased TEER value already by two weeks of cultivation (Fig. 4C). This result is consistent with the characteristically leaky liver endothelium that has lower VE-cadherin expression than other tissues. Further studies are needed for a more definitive characterization of tissue-specific endothelial commitment.
Tissue functions were comparable after 4 weeks of culture for the InterOrgan and Isolated groups. Heart tissues maintained the beating rate (Fig. 4D), liver tissues maintained albumin secretion (Fig. 4E), and skin tissues maintained epidermal thicknesses (Fig. 4G). Bone tissues maintained the mature osteolytic TRAP activity only in the InterOrgan group, consistent with the notion that bone function requires interaction with the other tissues that are not provided in the Isolated cultures (Fig. 4F). Connecting tissues without establishing physiologic separation between the vascular and tissue compartments (Mixed condition) did not support mature osteolytic function 4 weeks after connecting tissues in the tissue chip, suggesting that the maintenance of tissue specific niches is critical for dynamic functions that rely on tissue communication. The Mixed tissue chip was unable to maintain cardiac contractility (Fig. 4D) or liver albumin production (Fig. 4E). Immune cell cytokine secretions remained stable over 4 weeks in the InterOrgan group, and decreased in the Mixed and Isolated groups (Extended Data Fig. 6A–B), suggesting that the vascular barriers may maintain immune cell function and homeostasis.
By whole proteome analysis, we detected thousands of proteins in each engineered tissue (heart: ~6,000; liver: ~4,000; bone: ~5,000; skin: ~2,000) that were differentially expressed between the InterOrgan and Mixed media conditions (Fig. 4D–G, Extended Data Fig. 7&8). Through IPA and GO analysis, we identified common genes expressed in all tissues among the three conditions (InterOrgan, Mixed, and Isolated, Fig. 1C, Fig. 5A). Notably, the top genes shared among all tissues in the InterOrgan and Isolated cultures were primarily related to normal physiological functions (e.g., metabolism), while the shared genes under the Mixed condition were primarily related to the off-target organ functions (Fig. 5A).
Figure 5 |. Proteomic analysis confirms biological fidelity of InterOrgan tissue chip in comparison to gold standards and adult organs.

a, When comparing all engineered organs within each experimental group, GO analysis identified gene pathways shared amongst the different organs. b, Comparison between published adult data and engineered tissues demonstrates high similarities in the shared expression of genes to native tissue (left); additional comparisons in how the expression level of these shared genes within each experimental group correlates to adult tissue is presented as well (right). c, Within each organ system, top proteins of interest were identified via the Human Protein Atlas and compared to adult tissue. d, As cardiac tissues were of highest biological fidelity, we identified a number of candidate proteins that are often found in off-target tissues, including in development of epithelial, osteochondral, and neural tissues, for example. e, GO analysis identified cardiac-specific, adult-like structural components present in the InterOrgan (dark blue) and Isolated (light blue) groups, but not at all in the Mixed condition (gray).
Tissue-specific proteins expressed in adult and engineered tissues were identified and tertiled into high, medium, and low expression levels based on the Human Protein Atlas28. Engineered tissues showed high overlap of gene expression with adult human tissues (Fig. 5B). Further comparisons of the expression levels of these overlapping genes between matched adult and engineered tissues revealed that the heart, bone, and skin tissues in the InterOrgan and Isolated tissue chips more closely matched the published data for adult donors (Fig. 5B), outperforming the Mixed tissue chip. Additional analysis showed that engineered tissues in the InterOrgan tissue chip better matched the repertoire of highly abundant genes in adult heart, liver, bone, and skin (Fig. 5C), confirming the enhanced ability of the InterOrgan tissue chip to preserve the biological fidelity of connected tissues over 4 weeks of culture. Proteins associated with epithelial, neurogenic, and osteochondral development were markedly higher in heart tissues cultured under the Mixed conditions, presumably due to the presence of growth factors from adjacent tissues in the absence of an endothelial barrier (Fig. 5D).
Mixed culture conditions showed upregulated biological process pathways related to skin epidermis development within the bone tissues (Extended Data Fig. 7D), and upregulated collagen deposition, calcification, and aberrant extracellular matrix reorganization (Extended Data Fig. 7G). Within the cardiac tissues, the Mixed culture conditions clustered separately (Extended Data Fig. 8A) from the other conditions, and showed downregulation of genes related to cardiac structure, energetics, and calcium handling (Extended Data Fig. 8C). Mixed culture conditions suppressed the pathways related to cardiac conduction, muscle contraction, striated muscle tissue development, action potential, muscle adaptation and cardiac cell development within the cardiac tissues (Extended Data Fig. 8D). GO analysis of cellular components revealed that only InterOrgan and Isolated conditions maintained the mature cardiac phenotype, as evidenced by enriched contractile proteins and T-tubules (Fig. 5E), further supporting the need to preserve tissue-specific niches.
Mechanistic multi-compartment model of the tissue chip for drug studies
PK models are typically formulated as a multi-compartment model (flow-rate limited) or a blood-tissue model (permeability-rate limited)29–32. To enable translation to in vivo, we developed a mechanistic multicompartment model of the InterOrgan tissue chip (Extended Data Fig. 9A) using ordinary differential equations (ODE) for distributed multi-compartment reduced-order (MCRO) model that simulated the tissue chambers fluidically connected by a closed vascular flow loop. Each chamber was modeled with a fluidic inflow segment (IFC), fluidic perfusion segment (FC), endothelialized membrane (MM), and a tissue tank (T).
The topology of each tissue was designed to replicate its morphology and location. The heart tissue was suspended by vertical pillars, the skin tissue was cultured in the top section to maintain air-liquid interface, and the liver and bone tissues were cultured in the lower sections (Extended Data Fig. 9B–F). The endothelial barrier was modeled to replicate the porous membrane and a layer of cells at each side (Supplementary Fig. 6A). For simulation of studies without endothelial barrier (Mixed conditions), the membrane separating the tank from the fluidic channel consists of a single layer (Supplementary Fig. 6D–E). A set of flux equations was used to calculate drug transport across the tissue chip. As tubing permits partitioning of lipophilic compounds, we solved for species mass transport between the fluid and the tubing wall.
In a previous study, we showed relatively slow diffusive distribution of a drug within the tissue chamber33. For better resolution of concentration gradients, each tissue chamber was vertically divided into three sub-compartments. The reservoir had a time-dependent volume due to sampling. The entire fluidic loop was represented as a virtual linear model with a number of segments in the external flow loop. The mathematical models were solved for the time-dependent fluid mass and species transport assuming constant flow rates. We validated the model for mass and volume conservation over the entire duration of simulation (Supplementary Fig. 6B–C).
Fluorescein isothiocyanate (FITC)-labeled dextran was circulated for 72 hours without measurable absorption by the transwells (Supplementary Fig 7A). In previous studies, we had already tested FITC absorption by the tissue chip with similar results33. We also assessed diffusion through the transwell and observed FITC-dextran reached a uniform concentration after 72 hours, without flow rate (Supplementary Fig. 7B). FITC-dextran introduced into the circulation at a 3.3 mL min−1 flow rate reached uniform concentration between all chambers within 24 hours (Supplementary Fig. 7C–H).
In vitro PK model of doxorubicin
Having established tissue-specific functionality along with tissue cross-talk over 4 weeks of culture, the InterOrgan tissue chip provided an in vitro mimic of human physiology for drug testing. To show capability for elucidating the PK and PD of therapeutic agents, we modeled cardiotoxicity of doxorubicin, a drug used in treating several types of cancer. Doxorubicin is metabolized in liver into doxorubicinol, which we also observed in doxorubicin exposed liver tissues (Supplementary Fig. 8). A doxorubicin dose of 30 μM (17,400 ng/mL) was determined according to the surface area of the tissues to correspond to the clinically administered cumulative dose shown to induce cardiotoxicity17,18 and was delivered to the tissues through the vascular channel.
We compared the InterOrgan condition against the Mixed condition and the Isolated single-tissue tissue chips with and without endothelium (Fig. 1D), to elucidate the effects of endothelial barrier and tissue communication. The predictive power of the computational model was determined against experimental data. The time-concentration profiles of doxorubicin and doxorubicinol in the InterOrgan tissue chip (Fig. 6) show that that the model matched closely the experimental data, correctly predicting doxorubicin metabolism into doxorubicinol by liver tissue, and its diffusion into tissue chambers and the reservoir over time. The peak drug concentration in the heart tissue chamber was the lowest, most likely due to some absorption of the drug by the elastomer pillars supporting the tissue. Also, for the first timepoint, the calculated doxorubicin concentration in the reservoir did not fit well the experimental data, presumably due to the model assumption of perfect mixing. The concentration profile of doxorubicin and doxorubicinol in the Mixed tissue chip (Supplementary Fig. 9) reached a peak faster than in the InterOrgan tissue chip, presumably due to the less resistance to drug transport in the absence of endothelial barrier.
Figure 6 |. Experimental data and PK model of doxorubicin treatment in the InterOrgan tissue chip.

a-b, Doxorubicin (a) and doxorubicinol (b) levels, measured over time by UPLC-MSMS within all tissue chambers and in the reservoir (red bar), compared with prediction of the computational PK model (blue line). Data are mean ± SD.
The concentration-time profiles for the Mixed tissue chip were consistently above those for the InterOrgan tissue chip (Extended Data Fig. 10). This result is in line with the fewer barriers to drug diffusion between the fluidic channels and the tissue chambers in the Mixed tissue chip has. The higher concentration of the metabolite doxorubicinol in the liver chamber is also expected as the primary drug doxorubicin is metabolized by liver.
An in vitro PD model of doxorubicin
We then studied if the InterOrgan tissue chip was able to model doxorubicin PD, by exposing all tissue chip configurations to the same concentration of doxorubicin (30 μM) over 72 hours, as in PK studies. Liver tissues showed decreased albumin production and stable urea production (Fig. 7A&B), as seen clinically34. Cardiac excitability decreased and cardiac troponin I release increased, both of which are clinical measures of cardiac cell damage (Fig. 7C&D). Bone tissues displayed decreased bone sialoprotein levels only in the InterOrgan tissue chip (Fig. 7E), with stable TRAP responses (Fig. 7F) and decreased cellularity (Fig. 7G), suggesting that osteoblasts are more sensitive to doxorubicin than osteoclasts, as observed in pre-clinical studies35. As expected, the endothelium showed decreased resistance in response to doxorubicin (Fig. 7H). Decreased sensitivity of skin tissues in the InterOrgan 4-tissue tissue chip, where the peak concentrations of doxorubicin are lowered by the endothelial barrier function, is more physiological than in the Mixed tissue chip.
Figure 7 |. PD model of doxorubicin toxicity in the multi-organ tissue chip.

a-b, Liver specific measurements of albumin (a) and urea (b) secretion after 72 hours (n=3–5 biological replicates). c-d, Cardiac specific measurements of cardiac troponin secretion (c) and cardiac contractility (d) after 72 hours (n=3–6 biological replicates). e-g, Bone specific measurements of secreted bone sialoprotein (e), TRAP activity (f), and immunohistochemical images (g) after 72 hours (n=3–6 biological replicates). Scale bar, 100 μm. h, Vascular transendothelial electrical resistance (TEER) values as a measure of barrier integrity after 72 hours (n=3–5 biological replicates). i-k, Volcano plots detailing significant miRNA fold changes after doxorubicin treatment between the InterOrgan and Mixed tissue chips for cardiac tissues cultured in the presence of the other 3 tissues (i), in isolation as single tissues (j), or perfusate from the 4-tissue InterOrgan and Mixed conditions (k). l-m, Principal component analysis of miRNA fold changes (l) and tissue chip specific depiction of miRNA fold changes (m) after doxorubicin exposure for clinically relevant miRNAs (shaded region on graph) identified in doxorubicin induced cardiomyopathy within pediatric patients16. Data is shown as mean ± SD and statistics determined by two-way ANOVA.
Identification of miRNA biomarkers of doxorubicin cardiotoxicity
Current monitoring of cardiotoxicity in patients receiving doxorubicin includes measurements of left ventricular ejection fraction (LVEF) and cardiac troponin serum levels. Recently, miR-1 has been identified as a superior biomarker for predicting patients who will develop doxorubicin induced cardiotoxicity36. We also detected increased cardiac troponin I levels after doxorubicin treatment and changes in miRNAs including miRNA1 (Fig. 7C, I–M).
Differential miRNA expression was measured in the heart tissues and vascular flow by the GeneChip™ miRNA 4.0 Array following doxorubicin treatment to predict doxorubicin induced cardiotoxicity and subsequent cardiomyopathy for heart tissues cultured in the presence of the other tissues (4-tissue) or in isolation (1-tissue) (Fig. 1D), as shown in Fig. 7I–M and Supplementary Fig. 10–12)14,16,19. Only the InterOrgan tissue chip showed differential upregulation of miR-1 after doxorubicin treatment (p=0.0028). The InterOrgan configuration showed the highest fold change (FC) of miR-1 (InterOrgan vs. Mixed FC=38.84, p =0.0015, Fig. 7I), followed by the 1-tissue tissue chip (InterOrgan vs. Mixed FC=18.84, p=0.0245, Fig. 7J). Evaluation of the perfusate from the InterOrgan and Mixed conditions also showed upregulated miR-1 only in the InterOrgan tissue chip (4-tissue InterOrgan vs. Mixed Perfusate FC=4.69, p =0.0048, Fig. 7K).
Identification of miRNA biomarkers of doxorubicin cardiomyopathy
We were able to detect early miRNA biomarkers of doxorubicin cardiomyopathy suggested by a recent clinical study identifying 17 miRNAs differentially expressed in pediatric cancer patients who developed left ventricle failure following treatment with doxorubicin (Fig. 7L&M)16. Following Principal Component Analysis (PCA) dimensionality reduction, the statistically significant similarity of the clinical data cluster to the InterOrgan cluster (p=0.0021), but not to the Mixed model cluster (p=0.11), (Fig. 7L) suggests that linking of matured tissues by vascular flow provides a more physiological context than tissues cultured in common medium. When miRNAs were prioritized based on their statistical significance for subsequent cardiomyopathy in the clinical study, 85.7% of the miRNAs in the InterOrgan model showed similar fold changes versus only 28.6% of the miRNAs in the Mixed 4-tissue model, and 71.4% for both the InterOrgan and Mixed 1-tissue models (Fig. 7M).
To further probe the capabilities of the InterOrgan tissue chip, we determined the differential activities of miRNAs that were identified in pediatric16 and adult19 clinical studies following doxorubicin treatment, by measuring the Normalized Enrichment Scores (NES) of their targets in underexpressed genes computed by GSEA analysis26. Individual miRNA targets were assessed using miRDB database37.
Differential activity of previously validated pediatric miRNAs was highly significant in the InterOrgan tissue chip, consistent with clinical results for 10 out of 17 miRNAs17 (58.8%) versus 5 out of 17 miRNAs (29.4%) for the Mixed model (Supplementary Fig. 10A–C)16. For Isolated tissues, the presence of vascular endothelium increased specificity, with 12 vs 9 out of the 17 miRNAs, respectively, being consistent (Supplementary Fig. 11A–C)16.
miRNA expression was also assessed for the vascular flow, to mimic the clinical serum measurements. The InterOrgan perfusate showed miRNA activity consistent with clinical results for 8 out of the 17 (47.1%) pediatric miRNA biomarkers, compared to the Mixed model showing consistent activity for only 4 out of 17 miRNAs (23.5%) (Supplementary Fig. 12)16. Overall, the InterOrgan tissue chip outperformed the Mixed tissue chip, as evidenced by stronger clustering with clinical data (Fig. 9M), and the enrichment scores and fold changes more consistent with those seen clinically (Fig. 9I-K, Supplementary Fig. 11&12).
A set of miRNAs that were related to doxorubicin cardiotoxicity in adult clinical study were evaluated similarly in the multi-tissue tissue chips (Supplementary Fig. 10D&E) and the Isolated tissues (Supplementary Fig. 11D&E)19. We observed highly significant activity of miR-1273a, reported as a central regulator of pathways related to doxorubicin-induced heart failure in adult patients, in the InterOrgan tissue chip (FC = −45.23, NES = −43.24, p = 3.3e-19) (Supplementary Fig. 10D&E and 11D&E)19.
Overall, the InterOrgan tissue chip presented significant activation or inactivation (by GSEA) consistent with clinical data for 10 of 17 miRNAs (Supplementary Fig. 10). The Isolated heart tissue with endothelial barrier presented a similar consistent response (Supplementary Fig. 11). Critically, while all 17 miRNAs showed differential activity in the Mixed 4-tissue tissue chip, only five were in agreement with published results (29.4%), while 12 showed differential activities opposite to clinical observation (Supplementary Fig. 10).
These data suggest significant effects inter-organ communication, endothelial-mediated partitioning, and metabolic processing of the drug on treatment responses to doxorubicin.
SUMMARY AND LIMITATIONS OF THE STUDY
The InterOrgan tissue chip maintained the matured phenotypes of distinctly different human tissues (heart, liver, bone, skin) linked by vascular flow, over 4 weeks of culture. Endothelial barrier provided each tissue to be cultured in with its own optimized environment, while enabling their communication by cytokines, circulating cells and exosomes. These conditions allowed recapitulation of clinical PK/PD profiles of doxorubicin and miRNA biomarkers of cardiotoxicity. We believe that further studies of the InterOrgan tissue chip may lead to patient-specific models of systemic pathologies for testing new therapies and early biomarkers of drug toxicity.
However, this tissue chip also has limitations. Several important organs that can influence drug PK/PD (kidney, gut, fat) are not included, while their functions are important for studies of drugs acting systemically. Future work should also continue validating the model with different routes of administration, dosing schemes, metabolism, and elimination for both doxorubicin and other drugs. For example, skin tissue in the tissue chip allows PK/PD studies of topical drugs and environmental compounds.
We observed lower concentrations of doxorubicin in the heart chamber, presumably due to its absorption by the polydimethylsiloxane (PDMS) pillars. In future studies, such unwanted drug absorption could be reduced by coating or chemically modifying the PDMS surface. Because the focus was on predicting doxorubicin induced cardiomyopathy, we chose a single high dose clinically shown to be the main causative factor associated with doxorubicin induced cardiomyopathy. However, evaluating the effects of lower doses delivered over long periods of time and different drug formulations is of high importance for future studies.
Furthermore, the InterOrgan tissue chip would greatly benefit from multiplexing and standardization to better control multi-tissue homeostasis, drug delivery and sampling, and obtain more robust quantitative readouts. To increase translation, future work should adopt a more scalable version of the tissue chip on a multi-well plate footprint, to allow automated handling and compatibility with existing high-content imaging systems.
An interesting observation was that the endothelial barrier, one of the defining components of the tissue chip, maintained its permeability in heart, skin, and bone compartments and decreased it in the liver compartment. This change is in line with the high permeability of liver endothelium, but also indicates that the initial endothelium has plasticity that should be further investigated.
Vascular barriers enabled recapitulation of clinically relevant PK/PD profiles, as only the InterOrgan tissue chip showed clinically relevant biomarkers of doxorubicin cardiotoxicity (miR-1)14 and cardiomyopathy16. Linking tissues by vascular flow markedly increased the specificity of cardiac miRNA biomarker changes aligning with those seen clinically16, with the InterOrgan 4-tissue tissue chip showing a specificity of 81.8% versus 36.4% for the Mixed tissue chip. The need for connecting multiple tissues to reproduce clinical predictions was further supported by the most differentially expressed miRNA in the InterOrgan tissue chip, hsa-miR-1273a19.
This study suggests that the InterOrgan tissue chip can serve as a patient-specific model for developmental testing of new therapeutic regimens and biomarkers of drug toxicity, based on its ability to maintain the biological fidelity of each tissue while also allowing their communication. Medium separation is particularly important for tissues derived from hiPSC, that maintain some developmental plasticity and are not fully matured. The endothelial barrier between the tissues and vascular flow promotes physiological cell and tissue responses, due to the paracrine signaling, selective transport of drugs and secreted factors, and immune cell extravasation. Interestingly, the role of endothelial barrier was more pronounced as the biological complexity increased in the four-tissue tissue chip as compared to the single tissue tissue chip. However, there is much more to be learned in continued studies of human InterOrgan tissue chips.
METHODS
1. Multi-tissue chip
Tissue Chip design:
The InterOrgan tissue chip is designed to support the culture and communication of multiple types of engineered tissues, each maintained within its own optimal medium. Tissue communication occurs via recirculating vascular medium across an endothelium that serves as a selectively permeable barrier. The tissue chip is sized to fit onto a standard glass microscope slide. It has a modular design, with four culture chambers that can each contain 1.5 mL of tissue specific medium, and a reservoir for the recirculating vascular medium. Two ports and a channel enable recirculation of vascular medium via a peristaltic pump. A glass slide at the bottom enables real time imaging. Each chamber has an insert with a porous nylon mesh serving as a substrate for the endothelial layer.
Tissue chip fabrication:
The tissue chip has four different components: (i) a body with the tissue chambers, media reservoir, and flow channel, (ii) two clamps, (iii) an O-ring, and (iv) a glass slide at the bottom. The body was fabricated from polysulfone (McMaster-Carr) using a 3-axis computer numerical control (CNC) milling machine (Haas OM2). The clamps and tubing transfer lid, a secondary spacer between the cell culture dish and the lid to pass tubing in and out without introducing gaps, were machined in the same manner from polycarbonate (McMaster-Carr). The nylon mesh inserts were made via overmolding using an injection molding machine (A.B. Plastic Injectors, AB-200) and polypropylene thermoplastic (Flint Hills Resources, P9M7R-056). An aluminum tool (McMaster-Carr, alloy 7075) was CNC machined for this process and porous nylon meshes with different pore sizes (Millipore) were laser cut into 11 mm circles using a 30 W CO2 laser (ULS VersaLaser 3.50). The meshes were clamped into a multi-cavity aluminum tool, and polypropylene was injected to form the structure of the mesh insert. An O-ring was installed around the structure to provide a seal (Viton, dash 011, 60 A durometer, McMaster-Carr).
The remaining components of the tissue chip are off-the-shelf: 100 mm cell culture dish, 1 × 25 × 75 mm glass slide, Pharmed BPT tubing (1.6 mm inner diameter, Cole-Parmer, EW-96880–03; and 2.29 mm inner diameter, Pharmed, Cole-Parmer, EW-95714–44), 3-way stopcock valve (Smiths Medical, ASD MX9311L), Luer elbow (Cole-Parmer, EW-45508–84), male and female polypropylene connectors (Cole-Parmer EW-45518–00 and EW-45508–00, respectively), O-rings (McMaster-Carr, cat. no. 1283N47), and a peristaltic pump (Cole-Parmer, EW-07557–00 and EW-07519–25). Connections between tubing components were made with appropriately sized barbed Luer connectors (polypropylene, Cole-Parmer, EW-50621–95). Post-fabrication, the tissue chip components were subjected to ultrasonic cleaning and autoclaving on a dry cycle.
Tissue chip assembly:
Tissue chip components were removed from sterile packaging in a biosafety cabinet. A standard glass microscope slide was placed onto the bottom surface of the polysulfone body using a silicone O-ring. Two polycarbonate clamps were press-fit around the slide, silicone O-ring, and tissue chip to provide a compression seal. Luer elbows were press-fit into Luer-taper ports at both ends of the tissue chip. A loop of Pharmed tubing was attached to a peristaltic section and a 3-way valve, to connect the Luer elbows at the inlet and outlet ports. The entire assembly was inserted into a 100 mm cell culture dish and a tubing transfer lid was placed on top. Tubing was installed into the slots of the lid, while a standard lid is used to protect the tissue chip from the above. With the tubing assembly completed, 12 mL of endothelial medium were infused via the Luer lock and 3-way valve to prime the flow loop and remove air. Once primed, the endothelialized mesh inserts were installed into each tissue chamber, and the assembled tissue chip was then moved into an incubator and connected to a peristaltic pump.
Estimation of small molecule absorption and diffusive transport.
To determine absorption of small molecules by the mesh inserts, fluorescein isothiocyanate (FITC)-labeled dextran (3 kDa, 10 μM) was added to a standard cell culture well with a mesh insert. Aliquots were taken over time and assayed for fluorescence on a spectrophotometer (Biotek). To determine diffusion through the mesh inserts, FITC-labeled dextran (3 kDa, 10 μM) was added to the bottom chamber of a mesh insert, without vascular flow. Aliquots were taken from the top and bottom of the mesh insets over time and assayed for fluorescence on a spectrophotometer (Biotek). To determine distribution of a small molecule within the tissue chip, FITC-dextran (3 kDa, 10 μM) was introduced in the reservoir and the tissue chips were connected to a peristaltic pump run at a flow rate of 3.3 ml min−1. Aliquots were taken from different locations in the tissue chip over time and assayed for fluorescence on a spectrophotometer (Biotek).
Culture media:
The tissue chip contained 1.5 mL of media in each tissue compartment (liver, heart, bone, and skin), and 12 mL of recirculating media. The composition of each medium is summarized in Supplementary Table 2.
2. Study design
Tissue chip validation over 4 weeks of culture:
To validate the tissue chip, we studied four different configurations: (1) InterOrgan system (n=12), where the multi-chamber tissue chips contain endothelialized mesh inserts to separate tissue-specific niches from vascular flow; (2) Mixed system (n=6), containing mesh inserts without endothelial barrier, allowing tissue and vascular culture media to mix rapidly, a condition equivalent to the use of common media for all tissues; and (3–4) Isolated system (n = 6), with or without endothelial barrier, with each tissue cultured separately in the same volume of tissue-specific medium (1.5 mL). Conditions (1) and (2) had a perfusate flow channel running on a peristaltic pump with a hydrodynamic shear at the mesh of 1.88 dynes cm−2. Monocytes (50,000 CD14+ cells) were added into the reservoir at the beginning of the experiment and after 14 days. Recirculating flow was maintained for four weeks. Every other day, 1 mL of medium was changed in each tissue chamber and in the vascular reservoir; medium samples were immediately frozen at −20 ºC for subsequent analysis. Similarly, 1 mL of media from Isolated tissues was taken and replenished every other day. At the end of the 4-week study, tissues were harvested and sectioned for proteomics (immediately flash frozen) and histology (immediately fixed in 4 % paraformaldehyde).
Pharmacodynamic (PD) and pharmacokinetic (PK) models of doxorubicin toxicity:
After validating the multi-tissue tissue chip as described above, we modeled doxorubicin’s PK/PD. We designed a multi-compartment computational model of the tissue chip for mathematical simulations of absorption, distribution, metabolism, and secretion of doxorubicin. We compared the functional and molecular responses of tissues within the InterOrgan tissue chip (doxorubicin exposed n = 6; negative controls n = 3), Mixed tissue chip (doxorubicin exposed n = 6; negative control n = 3), and Isolated setting (doxorubicin exposed n = 6; negative control n = 3, with and without endothelial barrier). All groups were exposed to the same cumulative dose of doxorubicin (30 μM, 17,400 ng mL−1). In the InterOrgan and Mixed groups, we administered the drug into the reservoir and followed up for the 72 hours without media change. Small media samples were collected from the reservoir and the tissue chambers at 1, 24, and 72 hours for Ultra-Performance Liquid Chromatography-Mass Spectrometry (UPLC-MS/MS) quantification of nonmetabolized drug and its main metabolite. At 72 hours, the supernatant was collected, function was assessed for all tissues, and the heart tissues were sampled for evaluation of cardiotoxicity by miRNA analysis.
3. Tissue formation and maturation
Heart:
Heart muscle was formed and matured as described previously11,38. Briefly, a ratio of 75% human induced pluripotent stem cells (hiPSC)-derived cardiomyocytes (CM) (WTC-11 cell line, obtained through a material transfer agreement from B. Conklin, Gladstone Institute) were combined with 25% normal human dermal fibroblasts (NHDF, Lonza) in 84 μL of 33.3 mg mL−1 human fibrinogen (Sigma-Aldrich, F3879), and crosslinked with 16 μL of 100 U mL−1 thrombin from human plasma (Sigma-Aldrich, T6884) to form a hydrogel between two flexible pillars. After 20 minutes of crosslinking at 37 °C in 5% CO2, cardiac medium was added (RPMI 1640 (Thermo Fisher Scientific, 11875–093), B-27 supplement (serum free, Thermo Fisher Scientific, 17504044), ascorbic acid (Sigma-Aldrich, A8960) and penicillin/streptomycin (Gibco by Life Technologies, 15070063) supplemented with 0.02 mg mL−1 of aprotinin (Sigma-Aldrich, A3428). After 1 week of compaction, heart tissues were transferred to the maturation chip where they were subjected to electromechanical conditioning at a frequency increasing from 2 Hz to 6 Hz (0.33 Hz per day, biphasic stimulation, 2 ms pulse duration, 4.5 V/cm field intensity).
Liver:
HiPSC-derived hepatocytes were purchased from Cellular Dynamics International (iCell Hepatocytes 2.0) and thawed at room temperature. An AggreWell plate with 400 μm microwells (STEMCELL Technologies, 34411) was prepared according to the manufacture’s protocol. Hepatocytes (10 million cells) were mixed with NHDF (Lonza, 10 million cells) in hepatocyte culture medium (HCM, Lonza, CC-3198). The dual cell suspension was then added to 20 wells (approximately 500,000 hepatocytes and 500,000 NHDF in each well) with 2 mL of HCM per well. After 48 hours of culture at 37 °C and 5% CO2, cell aggregates were harvested and encapsulated in fibrin hydrogel formed from fibrinogen (84% of volume) and thrombin (16% of volume). The cells in hydrogel were placed into 48-well tissue culture-treated plates (Corning), using 200 μL per well. The hydrogel was allowed to cross-link in a cell culture incubator for 20 minutes, after which 1 mL HCM supplemented with 0.22 mg mL−1 of aprotinin were added. Tissues were allowed to polymerase for at least 48 hours before being used in experiments.
Bone:
Bovine calf metacarpal joints were purchased in bulk and stored at −40 °C (Lampire Biological Laboratories, 19D24003). A band saw was used to cut approximately 4 cm tall trabecular bone sections from the distal end of metacarpal. A CNC milling machine was then used to generate bone cores with a cross section of 4 mm x 8 mm that were cut into 1 mm thick sections using an IsoMet low speed wafering saw. Each section (4 mm wide x 8 mm long x 1 mm thick), was decellularized using our previously established protocols39, to remove all cellular material while preserving the bone matrix composition and architecture. Bone scaffolds were processed in batch by following the following step-wise protocol on an orbital shaker: (i) phosphate buffered saline (PBS) with 0.1% ethylenediaminetetraacetic acid (EDTA) (w/v) for 1 hour at room temperature, (ii) 10 mM tris, 0.1% EDTA (w/v) in deionized (DI) water overnight at 4 °C, (iii) 10 mM Tris, 0.5% sodium dodecyl sulfate (w/v) in DI water for 24 hours at room temperature, and (iv) 100 U ml−1 DNase, 1 U ml−1 RNase, 10 mM Tris in DI water for 6 hours at 37 °C. The resulting bone matrix was lyophilized and weighed to ensure that each piece had an appropriate matrix density for cell seeding (12 – 18 mg per scaffold). For sterilization, bone scaffolds were subjected to 70% ethanol treatment overnight under ultraviolet light, and incubated in Dulbecco’s Modified Eagle Medium (DMEM) overnight.
Bone-marrow derived mesenchymal stromal cells (MSC) (Lonza) or hiPSC-derived MSC were expanded and seeded into the bone matrix scaffolds using 4 × 105 cells per scaffold suspended in 40 μL of medium (DMEM supplemented with 10% (v/v) HyClone fetal bovine serum (FBS), 1% penicillin/streptomycin, and 1 ng mL−1 of basic fibroblast growth factor, bFGF), according to established protocols40. To differentiate hiPSC (WT-11 cell line) into MSC, the STEMdiff mesenchymal progenitor kit (STEMCELL technologies, 0529) was used according to the manufacturer protocol. The cells were allowed to attach for two hours, and supplemented with additional medium (DMEM supplemented with 10% (v/v) HyClone FBS, 1% penicillin/streptomycin, and 1 ng mL−1 bFGF) overnight. The following day, osteogenic differentiation was initiated by changing to osteogenic medium consisting of low glucose DMEM supplemented with 1 μm dexamethasone, 10 mm β-glycerophosphate, and 50 μm L-ascorbic acid-2-phosphate (all from Sigma-Aldrich). Each scaffold was incubated in 4 mL of osteogenic medium, with media changes three times a week, for three weeks, allowing for the MSC to differentiate into functional, maturing osteoblasts.
Primary monocytes were expanded, seeded into the bone scaffolds, and differentiated into functional, mature osteoclasts using our previously developed protocols21. Briefly, peripheral blood mononuclear cells (PBMC) were isolated from buffy coats of human blood (fully deidentified samples obtained from the New York Blood Center) by density gradient centrifugation with Ficoll-Paque PLUS (GE Healthcare, 17–1440-02). Following manufacturers protocol, immunomagnetic isolation of monocytes (Big Easy EasySep Magnet, Stem Cell Technologies, 180001) using negative selection (EasySep Human Monocyte Isolation Kit, Stem Cell Technologies, 19359) was performed. For the following two days, 8 × 106 monocytes were cultured on 25 cm2 ultralow attachment flasks (Corning, 3815) with 10 mL of maintenance medium (RPMI 1640, ATCC, 30–2001) supplemented with 10% heat-inactivated human serum (Corning, 35–060), 1% penicillin/streptomycin, and 20 ng mL−1 recombinant human macrophage colony-stimulating factor (M-CSF, PeproTech, 300–25) at 37 °C in a humidified incubator at 5% CO2. Human CD14+ monocytes were then seeded at a concentration of 4 × 105 cells per scaffold, using 40 μL of medium, allowed to attach for two hours at 37°C in a humidified incubator at 5% CO2, and subsequently differentiated for two weeks into osteoclasts in Minimum Essential Medium Eagle - alpha modification (α-MEM, Sigma, M4526) supplemented with 10% (v/v) heat-inactivated HyClone FBS, 1% penicillin/streptomycin, l-Glutamine (Gibco, 25030–081), 20 ng mL−1 recombinant human M-CSF (PeproTech, 300–25) and 40 ng mL−1 recombinant human sRANK Ligand (PeproTech, 310–01). Cytokines were replenished every three days. Cells were maintained at 37 °C in a humidified incubator at 5% CO2.
Skin:
Skin tissues were formed following a previously described protocol22. Briefly, 1.5 × 105 NHDF were embedded in 1 mL of 3 mg mL−1 type I collagen matrix (Millipore, 08–115), and the polymerized cell-containing hydrogel was incubated on a transwell mesh (BD Biosciences) for five to seven days in DMEM supplemented with 10% FBS. Then, 2.5 × 105 keratinocytes were seeded onto the matrix, and incubated in epidermalization medium containing a 3:1 mixture of DMEM and HAM’S F-12, 0.1 % FBS, 2 nM triiodothyronine (T3, Sigma, T5516), 5 ng ml−1 insulin (Sigma, I9278), 0.4 μg ml−1 hydrocortisone (Sigma, H0888), and 10 ng ml−1 epidermal growth factor (EGF, Millipore, 01–107) for an additional six days to ensure keratinocytes were confluent enough to cover the surface. The composite culture was raised to the air-liquid interface for seven days in a cornification medium with high calcium concentration (1.8 mM), without growth factors, to induce epidermal differentiation. By culturing skin tissues on top of a floating device inside the tissue chamber in the InterOrgan tissue chip, we were able to maintain the air-liquid interface over the entire duration of the experiment.
Endothelial barriers:
To optimize the hydrodynamic shear stress, we tested three different conditions: zero stress, 1.88, and 6.27 dynes cm−2. The barriers were prepared as described below and exposed to incrementally increasing shear stress: 0.5 dynes cm−2 for 12 hours, 1.0 dynes cm−2 for 24 hours, 1.88 dynes cm−2 for 24 hours, and 6.27 dynes cm−2 for 48 hours. Overall, we determined that a maximum shear stress rate of 1.88 dynes cm−2 was the most ideal, and subsequently used the following protocol for endothelial tissue formation within the studies described herein: 0.5 dynes cm−2 for 12 hours, 1.0 dynes cm−2 for 24 hours, 1.88 dynes cm−2 for 24 hours.
We also optimized the pore size of the nylon mesh by testing four different sizes (5, 10, 20, and 40 μm) and prepared the endothelial barrier in them as described below. Custom mesh inserts were fabricated as described above, autoclaved on a wet cycle, coated with fibronectin (1:100 from 10 ug mL−1 stock, Sigma-Aldrich, F4759) for 45 minutes and washed twice with PBS. Overall, we determined that 20 μm pore sizes were the largest pore size we could reliably use to establish confluent endothelial barriers, promoting cellular mediated communication and minimizing substrate area, and subsequently used these 20 μm pore size transwells for all studies described herein.
Human MSC were expanded in monolayers and dissociated with trypsin between passage five and eight, and were seeded using 150,000 cells in 50 μL volume to the top of each insert. The MSC suspension (50 μL) was left on each mesh insert for one hour to enable cell attachment. After one-hour, additional medium was added to each well (2 mL per well), fully immersing the cell coated meshes within the wells of the ultra-low attachment plate (Corning, 3473), and cultured at 37 °C with 5% CO2 for 24 hours.
Human umbilical venous endothelial cells (HUVEC) were expanded to passage number 5 to 8. To differentiate hiPSC (WTC-11 cell line) into endothelial cells (EC), the STEMdiff Endothelial differentiation kit (STEMCELL technologies, 08005) was used according to the manufacturer’s protocol. Within this study, HUVEC were used for all data except for Figure 3E–L where hiPSC-derived EC were used, as this study uses the WTC-11 hiPSC line to derive cells for all tissues to demonstrate the tissue chip’s utility in “patient-on-a-chip” models (i.e. using the same genetic background for all tissues). The bottom surface of each MSC inset was coated with 400,000 HUVEC and an additional 50,000 MSC. To this end, the MSC medium was removed, allowing for each insert to stay only slightly hydrated and flipped over to the bottom side. A 20 μL cell suspension of HUVEC/MSC was added twice, 15 minutes apart, to the bottom surface of each insert and incubated at 37 °C and 5% CO2 in between the two cell additions, allowing for incremental attachment of cells prior to addition of endothelial media (EGM-2, Lonza, CC-3162). Each insert was estimated to have a total of 400,000 HUVEC and 200,000 MSC, to mimic the dynamics between vascular populations, represented by the endothelium and perivascular supporting cells (pericytes) in blood vessels. After 48 hours, mesh inserts with adherent cells were placed into the tissue chips, and exposed to hydrodynamic shear stress of 0.5 dynes cm−2 for 12 hours, 1 dynes cm−2 for 24 hours, and 1.88 dynes cm−2 for 24 hours. Shear stress at the endothelial layer was modeled using SOLIDWORKS Flow Simulation software.
Immune cells:
To differentiate hiPSC (WTC-11 cell line) into monocytes, the STEMdiff Monocyte kit (STEMCELL technologies, 05320) was used according to the manufacturers protocol. Primary human CD14+ monocytes were isolated by using magnetic activated cell sorting (MACS) using CD14+ sorting beads (Miltenyi Biotec) from a human leukopak (New York Blood Center). The isolated cells were maintained in a buffer solution on ice for less than three hours prior to introduction into the tissue chip. Primary or hiPSC-derived CD14+ monocytes (50,000 cells) were introduced into the reservoir and circulated through the tissue chip for the duration of the experiment. After two weeks of the four weeks experiment, monocytes were replenished by introducing an additional 50,000 CD14+ cells into circulation through the reservoir.
CD63 Exosome Crosstalk:
HiPSC-derived CM were transfected with a pCT-CD63-GFP Exosome Cyto-Tracer (System Biosciences, CYT0120-VA-1). Due to decreased expression of transfected hiPSC after their differentiation into cardiomyocytes, cells were transfected at day 12 of the hiPSC-derived CM differentiation and directly used to make heart tissues. The CD63-GFP label was tracked using the IVIS Spectrum Optical Imaging System (PerkinElmer), in Columbia University’s Oncology Precision Therapeutics and Imaging Core. Engineered tissues were placed next to one another in the same field of view and compared directly with multiple imaging views (top, side) using an IVIS 200 Spectrum device. The IVIS Living Imaging software was used to convert the signal to the normalized Radiant Efficiency (Emission light [photons sec−1 cm−2 str−1]; Excitation light [μW cm−2]). Fluorescence was measured by selecting the same region of interest for each tissue and quantifying the sum of the Radiant Efficiency of all fluorescent pixels within the region of interest. Exported images showing the Radiant Efficiency as a heat map were generated within the IVIS Living Imaging software (PerkinElmer).
Cryoinjury studies.
Vascularized InterOrgan tissue chips were assembled as described above, and each tissue chip contained two heart tissues in two middle chambers, with the first and last chamber remaining empty. One of the two heart tissues was exposed to cryoinjury by dry ice for five seconds, while the other served as a control. Immune cells (200,000 CD14+ monocytes) were labeled with Vybrant™ DiD Cell-Labeling Solution (ThermoFisher) to enable tracking, introduced into the reservoir, and circulated in the tissue chip. The tissue chips were maintained for seven days without media change, and the heart tissues were imaged with an IVIS Spectrum Optical Imaging System (PerkinElmer), in the Columbia University’s Oncology Precision Therapeutics and Imaging Core. For imaging, the heart tissues were removed from the InterOrgan tissue chip to avoid autofluorescence. The healthy and injured tissues were aligned next to one another in the same field of view and compared directly in multiple imaging views (top, side) using the IVIS 200 Spectrum device. The IVIS Living Imaging software (PerkinElmer) was used to analyze the images by converting the signal to the normalized Radiant Efficiency (photons sec−1 cm−2 str−1]/Excitation light [μW cm−2]). The fluorescence was measured by selecting the same region of interest for each tissue and subsequently quantifying the sum of the Radiant Efficiency of all fluorescent pixels within the region of interest. Exported images showing the Radiant Efficiency as a heat map were generated within the IVIS Living Imaging software (PerkinElmer).
4. Supernatant and functional assays
Supernatant samples were collected from each chamber and reservoir, frozen immediately, and subsequently thawed and used for several assays as described below. Over four weeks of culture in the tissue chip, 1 mL of supernatant was sampled every two days, while for the doxorubicin experiments 100 to 250 μL samples were taken at different time points (1, 24, and 72 hours). Supernatant samples were stored at −20 °C for less than three months before use; once thawed, no supernatant sample was left at 4 °C for more than one week and thawed no more than two times.
Cytokine profiles:
CCL20 and CXCL12 readings were obtained from 50 μL of tissue supernatant taken after 28 days of culture using the Immune Monitoring 65-Plex Human ProcartaPlex™ Panel (Thermo Fisher Scientific, EPX650–10065-901), according to the manufacturer’s instructions. Samples were allowed to incubate overnight at 4 °C, run on a Luminex-200 and analyzed by Luminex software by comparing to the included standards.
Heart function assays:
Cardiac excitability, force, and beat rate were obtained using our previously established protocols11,38. Cardiac troponin I secretion was determined using the Human Cardiac Troponin I ELISA Kit (Abcam, ab200016) according to the manufacturer’s instructions.
Liver function assays:
To assess liver function, supernatant samples were analyzed for albumin and urea secretion using a Human ELISA kit (Bethyl, E88–129) and a Urea Nitrogen Test Kit (Fisher Scientific, SB-0580–250), respectively. Assays were performed according to the manufacturer’s instructions.
Bone function assays:
To assess the bone’s ability to remodel its matrix, supernatant samples were analyzed for tartrate-resistant acid phosphatase (TRAP) activity (Kamiya Biomedical Company, KT-008) and bone sialoprotein (Mybiosource, MBS261861) after 28 days of culture. These assays were performed according to the manufacturer’s instructions. Micro-computed tomography (μCT) was performed within the OPTIC core using the Quantum FX μCT imaging system (PerkinElmer). Bones were places within 48 well plates and 4 wells were imaged at a time.
Skin function assays:
To assess the transepithelial/transendothelial electrical resistance (TEER) of skin tissues, we used a tissue resistance measurement chamber (EndOhm, World Precision Instruments). Each chamber contained a pair of concentric electrodes providing accurate resistance measurements, according to the manufacturer’s protocol. We recorded two to three technical replicates per tissue and calculated the tissue resistance by multiplying the measured resistance (Ohm) with the effective tissue area (cm2).
Endothelial barriers:
To assess the TEER of endothelial barriers, we used the tissue resistance measurement chamber (EndOhm, World Precision Instruments) described above. We recorded two to three technical replicates per barrier, and calculated the barrier resistance from the measured resistance (Ohm) and the effective barrier area (cm2). To assess dextran diffusion through the barrier, two different sizes of FITC-dextran (Thermo Fisher Scientific, 3 kDa, D3307; kDa, D1864) were added to the reservoir of tissue chips containing endothelial barriers that were prepared as described above. Chips were connected to the peristaltic pump and medium was sampled from each chamber (at 0, 1, 2, 6, 12, 24, and 48 hours) to measure the fluorescent signal by a spectrophotometer (BioTek, Synergy HTX). According to the previously reported method41, permeability was calculated based on the equation below:
, where Ct is the concentration in tissue chambers, Co is the initial concentration, V is the volume of the media within the vascular chamber, A is the area of the membrane with cells, and Δt is the assay time.
5. End-point assays
Tissue preparation:
Tissue samples were bisected for proteomic and histologic analyses. One half of the sample was snap frozen using liquid nitrogen and stored at −80 °C for less than one month before being analyzed for proteomics. The second half of each tissue sample was fixed for 24 hours in paraformaldehyde (PFA), washed in PBS, and submitted to the Herbert Irving Comprehensive Cancer Center (HICCC) Molecular Pathology Lab at Columbia University for paraffin embedding and sectioning.
Immunostaining:
Heart, bone, liver, and skin constructs were fixed in 4% PFA for 24 hours, embedded in paraffin, and sectioned for histological and immunofluorescence examination at 5 μm. All tissues were processed for hematoxylin & eosin (H&E), trichrome, and bone tissues were processed for picrosirius red staining by the HICCC Molecular Pathology Lab at Columbia University. Paraffin-embedded tissue blanks were hydrated, processed for antigen-retrieval using a 10 mM sodium citrate buffer for 20 min in heat, and permeabilized with 0.25% (v/v) Triton-X for 20 minutes. Samples were then blocked for two hours with 10% FBS and stained as follows. Heart samples were incubated with a primary antibody for actinin alpha 2 (1:100, Invitrogen, 701914) overnight at 4 °C. Liver samples were incubated with primary antibodies for albumin (1:100, ThermoFisher, A80–229F) and cytochrome P450 enzyme CYP3A4 (1:50, Millipore, AB1254) overnight at 4 °C. Bone samples were stained using the TRAP staining kit (Kamiya Biomedical Company, KT-008) according to the manufacturer’s directions. Bone samples were also incubated with an antibody for bone sialoprotein II (1:500, Millipore Sigma, AB1854) and osteocalcin (1:500, Millipore Sigma, AB10911). Skin samples were incubated with primary antibodies for keratin 14 (1:100, Biolegend, PRB-155P) and vimentin (1:100, Santa Cruz Biotechnology, sc-6260) overnight at 4 °C. After washing with PBS, samples were incubated with fluorophore-conjugated secondary antibodies (1:200, Invitrogen) for two hours at room temperature. Slides were covered with cover-slips using mounting medium containing 4’,6-diamidino-2phenylindole (DAPI) (Prolong Mountant with NucBlue, Invitrogen, P36981) and examined using either a Zeiss LSM 5 Exciter confocal laser scanning microscope or Nikon Ti Eclipse inverted confocal microscope.
Endothelial layers were fixed in PFA at 37 °C for two minutes. After aspirating fixation solution, samples were washed delicately using PBS supplemented with 1 mM CaCl2 and 0.5 mM MgCl2. Samples were subsequently permeabilized using 0.5% Triton X-100 at 37 °C for ten minutes. Once washed using supplemented PBS, endothelial barriers were stored at 4 °C for less than three weeks prior to staining. For staining, a dilution of 1:250 for VE-cadherin (Sino Biological, 10433-MM01, or Abcam, ab33168), and F-actin (Thermo Fisher Scientific, Alexa Fluor 647 phalloidin) in 2% BSA was added to each sample at 4 °C overnight. Samples were subsequently washed with supplemented PBS three times for five minutes each. For secondary staining, 1:400 488 Goat anti-mouse IgG in 2% BSA, 1:400 Phalloidin, and 1:1000 DAPI were added to each endothelial barrier sample. Samples were kept in the dark on a shaker overnight and washed three times for five minutes each the next day prior to imaging.
RNA sequencing:
Engineered heart tissues were flash frozen in RNAlater (ThermoFisher, AM7021) and sent to GENEWIZ for Standard RNA-seq with polyA selection using an Illumina HiSeq, 2×150bp configuration, single index, per lane, and subsequent analysis, as described below. Sequence reads were trimmed to remove possible adapter sequences and nucleotides of poor quality using Trimmomatic v.0.36. The trimmed reads were mapped to the Homo sapiens GRCh38 reference genome available on ENSEMBL using the STAR aligner v.2.5.2b (a splice aligner that detects splice junctions and incorporates them to help align the entire read sequences) to generate BAM files. Unique gene hit counts were calculated by using featureCounts from the Subread package v.1.5.2. Only unique reads that fell within exon regions were counted. The gene hit counts table was used for downstream differential expression analysis.
Using DESeq2, gene expressions were compared between the control tissues (engineered heart tissues cultured for 4 weeks without electromechanical stimulation) and matured tissues (engineered heart tissues cultured for 4 weeks with electromechanical stimulation at the frequency increasing from 2Hz to 6Hz by 0.33Hz per day11). The Wald test was used to generate p-values and log2 fold changes. Genes with p < 0.05 and absolute log2 fold change >1 were selected as differentially expressed genes for each comparison. The differentially expressed genes bi-clustering heat map was generated to visualize the expression profile of the top 30 genes sorted by their adjusted p-values, and identify co-regulated genes across the treatment conditions.
The Volcano plot was generated to include only the most statistically significant differentially expressed genes and show the global transcriptional change across the groups. All genes were plotted with each data point representing a gene. The log2 fold change of each gene (x-axis) was plotted against the log10 of its pvalue (y-axis). Genes with p < 0.05 and a log2 fold change > 1, indicated by red dots, represented upregulated genes. Genes with p < 0.05 and a log2 fold change < 1, indicated by blue dots, represented downregulated genes.
Gene Set Enrichment Analysis:
The differentially expressed genes obtained from RNA sequencing of matured heart tissues were ranked from highest to lowest expression and uploaded to GSEA software26,42. Enrichment plots were directly exported from the program.
Ingenuity Pathway Analysis:
Data were analyzed through the use of IPA Networks (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuitypathway-analysis), and the results were exported directly and used without modifications.
Flow Cytometry:
Prior to analysis by flow cytometry, the cells were labeled with the following antibodies or dyes: FITC anti-human CD45 Antibody (1:100, Biolegend, 304006), Brilliant Violet 605 anti-human CD14 (1:100, Biolegend, 301834), Brilliant Violet 421 anti-human ITGAM (1:100, Biolegend, 301324), PE anti-human CD68 Antibody (1:100, Biolegend, 333807), DRAQ5 (1:100, Biolegend, 424101), BUV395 Mouse Anti-Human ITGAX (CD11b) (1:100, BD Biosciences, 563787). Cells were measured using a FACSAria flow cytometer (BD Biosciences) and data were analyzed using FlowJo (Tree Star).
Quantitative Proteomics:
Proteomics sample preparation and tandem mass tag (TMT) labeling were performed as described earlier43 with minor modifications. Briefly, frozen tissues were lysed by bead-beating in 8 M urea, 1% SDS, 200 mM EPPS (pH 8.5) and protease inhibitor. Samples were reduced with 5 mM TCEP and alkylated with 10 mM iodoacetamide (IAA) that was quenched with 10 mM DTT. A total of 50 μg of protein was chloroform-methanol precipitated. Protein pellets were reconstituted in 200 mM EEPS (pH 8.5) and protein concentration was determined using a Pierce™ BCA Assay Kit (Thermo Fisher Scientific, 23225). Total protein from each sample (2 – 25 μg) was digested overnight at room temperature with Lys-C protease at a 50:1 protein-to-protease ratio while shaking. Trypsin was added at a 100:1 protein-to protease ratio, and the samples were incubated for 6 hours at 37 °C. Digested peptides were quantified using a Nanodrop at 280 nm and 2 to 25 μg of peptide from each sample were labeled with 200 μg TMT reagent using 10-plex TMT kit. TMT labels were checked by pooling 100 ng of each sample and were mixed at 1:1 across all channels using normalization factor samples. Bulk samples were fractionated using Pierce™ High pH Reversed-Phase Peptide Fractionation Kit (Thermo Fisher Scientific, 84868) and each fraction was dried down in a speed-vac. Dried peptides were dissolved in 10 μl of 3% acetonitrile/0.1% formic acid and injected using SPS-MS3.
Liquid Chromatography-Mass Spec/Mass Spec tissue proteomics:
Fractioned peptides were separated using Thermo Scientific™ UltiMate™ 3000 RSLCnano system and Thermo Scientific EASY Spray™ source with Thermo Scientific™ Acclaim™ PepMap™100 2 cm x 75 μm trap column and Thermo Scientific™ EASY-Spray™ PepMap™ RSLC C18. 50 cm x 75 μm ID column with a 5–30% acetonitrile gradient in 0.1% formic acid over 127 minutes at a flow rate of 250 nL/min. After each gradient, the column was washed with 90 % buffer B for 5 minutes and re-equilibrated with 98% buffer A (0.1% formic acid, 100% high-performance liquid chromatography (HPLC)-grade water) for 40 minutes. For BPRP-separated proteome fractions, the full MS spectra were acquired in the Orbitrap at a resolution of 120,000. The 10 most intense MS1 ions were selected for MS2 analysis. The isolation width was set at 0.7 Da and isolated precursors were fragmented by CID at a normalized collision energy (NCE) of 35 % and analyzed in the ion trap using “turbo” scan speed. Following acquisition of each MS2 spectrum, a synchronous precursor selection (SPS) MS3 scan was collected on the top 10 most intense ions in the MS2 spectrum. SPS-MS3 precursors were fragmented by higher energy collision-induced dissociation (HCD) at an NCE of 65% and analyzed using the Orbitrap.
Proteomic analysis:
Raw mass spectrometric data were analyzed using Proteome Discoverer 2.2 to perform database search and TMT reporter ions quantification. TMT tags on lysine residues and peptide N termini (+229.163 Da) and the carbamidomethylation of cysteine residues (+57.021 Da) were set as static modifications, while the oxidation of methionine residues (+15.995 Da) and deamidation (+0.984) on asparagine and glutamine were set as a variable modification43. Data were searched against UniProt Human database with peptide-spectrum match (PSM) and protein-level FDR at 1% FDR. The signal-to-noise (S/N) measurements of each protein were normalized so that the sum of the signal for all proteins in each channel was constant, to account for equal protein loading. Protein identification and quantification were imported into Perseus44 for multiple-sample tests for statistical analysis (FDR < 0.05 or FDR < 0.01) to identify proteins demonstrating statistically significant changes in abundance.
Benchmarking proteomic data for engineered tissues against adult tissues:
The identified proteins demonstrating statistically significant changes in abundance were compared to published adult tissue datasets from the Human Protein Atlas45, as follows. Because the methodology used to generate each dataset varies greatly, direct comparisons could not be made. Instead, within each tissue dataset, individual protein expression levels were exported into Excel (Microsoft) and subsequently tertiled into “Low”, “Medium”, and “High” expression levels, as is done in the Human Protein Atlas. Proteins that were not expressed were labelled as “Not detected” to avoid skewing comparisons with false low counts.
The percent of shared proteins was calculated by determining the number of proteins expressed (at any level) in the engineered tissue dataset versus the published adult tissue dataset, as a percentage of shared proteins (expressed in both tissues) within the total number of proteins45. To further determine the correlation of the shared protein expression levels between the engineered tissues and the corresponding published data45, the number of matching genes expressed as “Low”, “Medium”, or “High” in both tissue sets were calculated as a percentage over the total number of shared proteins expressed overall.
Heatmaps were generated using the tertiled data, by selecting a list of proteins according to two criteria: (i) the protein should be considered “tissue enriched” by the Human Protein Atlas, and (ii) the protein should be expressed in both datasets to enable comparisons. The lists of proteins for each heatmap were generated using only the proteins listed as highly expressed within each tissue per Human Protein Atlas. When the suggested protein was not expressed in one of the datasets, we continued down the list until completing data for 15 proteins per tissue. Comparisons between engineered and adult tissues were made according to tissue type, with engineered bone lacking a proper comparison. Bone is deemed a “rare” tissue by the Human Protein Atlas, and further literature searches did not yield more closely matching datasets, therefore the engineered bone protein data were compared to adult bone marrow protein data, the closest tissue.
Proteomic data GO, KEGG, differential expression and pathway analysis:
The identified proteins demonstrating statistically significant changes in abundance were subsequently used to perform gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses as follows. GO analysis of shared highly expressed proteins in all tissues within each culture condition (InterOrgan, Mixed, Isolated) was performed using ShinyGO v0.646. First, gene lists were assembled using Excel (Microsoft) by filtering for genes (as determined from the corresponding proteins in the description corresponding to each protein accession number) listed as highly expressed in the heart, liver, skin, and bone tissues, according to the previous methodology where each data set was tertiled within Excel (Microsoft). This list was uploaded to the ShinyGO v0.61 server with the following settings: “Human” as the “Best matching species”, “0.05” as the “P-value cutoff (FDR)”, and “30” as the “# of most significant terms to show”.
The resulting networks were directly exported from the site and used without editing. Tissue specific GO analysis was conducted using PANTHER (Protein Analysis Through Evolutionary Relationships) Classification System47 using lists in Excel (Microsoft), by filtering for genes (determined from the corresponding proteins in the description corresponding to each protein accession number) listed as highly expressed in the engineered tissue (e.g., heart) for each of the culture conditions (InterOrgan, Mixed, Isolated). The input genes were compared against all genes within the Homo sapiens reference list and a PANTHER Overrepresentation Test for GO cellular component complete was performed using a FISHER test with FDR correction. Further expression analysis for engineered tissues was performed by integrated Differential Expression and Pathway (iDEP) analysis, where the protein expression datasets generated for each engineered tissue were uploaded and pathway analysis was performed, using the Human reference dataset, using GSEA for GO Biological Process and KEGG pathways for the top 30 pathways with a significance cutoff (FDR) of 0.2 and a gene set size minimum between 15 and 200048.
6. Drug studies
Cardiac dose response studies:
Heart tissues were imaged at baseline and at each sequential dosage for 20 seconds at 100 frames per second under brightfield illumination using a Zyla 4.2 sCMOS camera (Andor) and NIS-Elements software (Nikon) or Pike F-032b (Allied Vision Technologies) camera. The resulting videos were analyzed for pixel movement in a custom MATLAB code described previously38. The resulting traces were used to determine the beat frequency and plot it as a function of increasing dosages to get an EC50 value.
Doxorubicin and doxorubicinol concentrations:
Concentrations of doxorubicin and its metabolic product doxorubicinol were measured in cell culture supernatants using Ultra-Performance Liquid Chromatography-Mass Spectrometry (UPLC-MS/MS) system at the Biomarkers Core Laboratory, Irving Institute for Clinical and Translational Research (Columbia University). Doxorubicin and doxorubicinol were assayed simultaneously in samples spiked with internal standard (daunorubicin) using Agilent 6410 LCMS/MS under positive ESI MRM mode (Transitions used: doxorubicin 544.2>398.1; doxorubicinol: 546.2>400.1). All compounds were quantitated by comparing the integrated peak areas of unknown against those of known amounts of purified standards.
7. miRNA analysis
miRNA characterization:
miRNA was isolated using miRNeasy Mini Kit from (Qiagen, 217004). Samples were shipped on dry ice to Advanced BioMedical Laboratories and assayed using the miRNA 4.0 Genechip Array (Thermo Fisher Scientific). Results were analyzed using the Transcriptome Analysis Console (TAC) 4.0 software (Thermo Fisher Scientific). Additional Ingenuity Pathway Analysis was performed using the methodology described above.
Gene set enrichment analysis:
To assess the activity of miRNAs, we performed one-tail gene set enrichment analysis (GSEA) of their targets, as reported in MultiMiR (release 3.11)49 and RBio-mirGS (update 0.2.12)50 using the R biocoductor packages. Specifically, we assessed whether miRNA targets were enriched in downregulated or upregulated genes, after doxorubicin treatment, indicating miRNA activity gain or loss, respectively. As a result, the Normalized Enrichment Score is negative when the miRNA activity increases and positive when its activity decreases. Thus, to avoid confusion, we inverted the NES sign, such that a positive or negative NES corresponds to a miRNA activity gain or loss, respectively. Since the analytical form of the null distribution for the NES statistic is not known, p-values were computed using an empirical null distribution, generated by random gene shuffling. GSEA was performed using fgsea (v1.13.5)51 package in R.
Sensitivity and Specificity calculations:
To assess the sensitivity and specificity of miRNA expression, we calculated whether or not the fold change demonstrated within the InterOrgan or Isolated group was consistent with what was seen clinically16,19. The results were tallied as true positive, false positive, false negative, or true negative. From these tallies, the specificity was calculated as described below.
8. Computational modeling
Models of each tissue chip compartment were developed using ordinary differential equations (ODE) based distributed multi-compartment reduced-order (MCRO) models, using CFD Research’s Computational Biology (CoBi) tools. These models were applied to simulate in vitro absorption, distribution, metabolism, and excretion (ADME) of doxorubicin.
General approach:
A compartmental mathematical model describing drug transport in various organ segments (fluidic channel, endothelial barrier, membrane, tissue, and tank) was derived using the general total fluid mass and species mass balance equations:
| (1) |
where is the velocity vector and the right-hand side term represents the sum of source inflows and outflows. In spite of a constant fluid density assumption, we retain the temporal derivative to enable treatment of time-depending volumes, as described below. The general species mass balance expressed for a concentration C is given as,
| (2) |
where D is the diffusion coefficient and SC is the generalized source term from chemical reactions. Integrating spatial terms for convection and diffusion into individual fluxes across the computational control volume boundaries and treating these as source terms in the ODE, the above mass and specie transport equations can be approximated as:
| (3) |
| (4) |
where V is the compartment volume, C is the specie concentration inside the compartment, Cin is the convective inlet concentration, Q is the volumetric flow rate, and J+, J- are the permeation (diffusive) fluxes across the boundaries of the computational control volume (organ device barriers). Because of the convection dominated flow conditions, streamwise diffusion has been neglected and only transverse fluxes (across barriers) have been retained. Note that the volume integration of the original partial differential equations resulted in a set of ODE.
The transverse flux between two adjacent sub compartments, F, and endothelial barrier, E, can be expressed as:
| (5) |
where S is the fluidic channel-endothelium barrier (F-E) interface area, is the drug concentration in the fluidic compartment, is the concentration in the endothelium barrier compartment, kp is the partition coefficient, and are the half-distances to the F-E interface, and is the effective permeability coefficient. Depending on the compound partition coefficient, kp, there may be a concentration discontinuity at the fluid-membrane interface. The model also accounts for drug partitioning into the tubing, tank and reservoir materials for cellular drug metabolism, and for the effects of intake/efflux transporters. Because each tank is subdivided into sub-compartments, Vi, in the vertical (tank axis) direction and due to no convective flow in the tank, the species mass balance equations in the tank account for vertical diffusion flux as:
| (6) |
and:
| (7) |
| (8) |
where A is the tank cross-section area, Ci- Ci and Ci+, are concentrations in the lower, current and upper sub-compartments, and δ- and δ+ are the distances between centers of adjacent sub compartments. For drug development applications, a set of two or more species conservation equations must be solved for both the primary compound and for its metabolite(s). These sets of equations are coupled via a drug enzymatic transformation reaction term in the cell layers, where a sink term of the primary compound becomes a source term in the metabolite conservation equation. Drug transport and conservation equations may include the compound metabolic clearance term calculated using the organ-specific intrinsic clearance, CL,int, which is typically expressed using the Michaelis–Menten kinetics model:
| (9) |
where represents the maximum reaction rate and the Michaelis constant Km is the compound concentration at which the reaction rate is half of 31. A detailed description of the mathematical models for each organ chamber is provided in the following sections.
Liver tissue chamber:
The compartmental mathematical model describing drug transport and metabolism can be derived using general mass balance equations with all flux terms integrated over the compartment boundaries. The resultant equations are expressed in the form of a set of ODE representing the species mass balance in each compartment. A schematic of the liver tissue chamber is shown in Fig. 6B. The model accounts for the primary drug (doxorubicin) and its main metabolite (doxorubicinol). In all equations below, doxorubicin is represented by the superscript 1, while doxorubicinol is represented by the superscript 2. The equations are primarily written for doxorubicin, but are also applicable for doxorubicinol, unless stated otherwise. The fluidic channel is separated into two different parts. One portion of the fluidic channel is exactly below the membrane and is called liver fluidic channel (LFC), while the other portion of the fluidic channel called liver inlet fluidic channel (LIFC) connects the LFC to the tubing. The mass balance equations for the LFC and the LIFC can be written as:
| (10) |
| (11) |
where Ci and Vi (i = LIFC, LFC, LBM, TB20) represent the species concentration and volume of individual compartments, and Ji-j describes diffusive flux between compartments i and j. The subscripts TL,20 and LBM represent the last discretized section of the tubing lumen and the bottom layer of the membrane. The flow rate is represented by Qf.
Endothelial barrier
The endothelial barrier consists of three layers: a nylon mesh and cells on both sides. The top and bottom layers are functionalized and account for drug partitioning. The mass balance equations for the three layers of the membrane can be written as:
| (12) |
| (13) |
| (14) |
where the subscript LMM represents the middle layer of the membrane, LTM represents the top layer of the membrane, and LBT represents the bottom layer of the tank.
The tank containing the liver tissue is separated into three layers. The liver tissue is situated in the bottom layer of the tank. The mass balance for the three layers of the tank can be written as:
| (15) |
| (16) |
| (17) |
where the subscript LMT represents the middle layer of the tank, LTT represents the top layer of the tank, and L represents the liver tissue.
It was experimentally observed that some amount of drug is absorbed by the tissue chip and the tank. The model accounts for this drug loss by including a clearance flow rate term (Qcl) in the bottom layer of the tank. The media is sampled from the middle layer of the tank.
The liver tissue is modeled to metabolize doxorubicin into its metabolite, doxorubicinol. The mass balance equation for the liver tissue can be written as:
| (18) |
| (19) |
where is the metabolic transformation rate of doxorubicin to doxorubicinol and SL,el is the compound elimination rate. The metabolic transformation rate is typically expressed using Michaelis Menten kinetics model, given as:
| (20) |
where is the maximum reaction rate and the Michaelis constant Km is the compound concentration at which the reaction rate is half of .
The flux terms in the above compartmental mass balance equations are expressed using permeability limiting equations. The diffusive flux terms can be written as:
| (21) |
| (22) |
| (23) |
| (24) |
| (25) |
| (26) |
| (27) |
where A is the interfacial surface area, P is the permeability, fu is the unbound fraction of the drug, and kp is the partition coefficient (based on the LogP value of the drug). The subscripts represent the associated compartments. The permeabilities are calculated as:
| (28) |
| (29) |
| (30) |
| (31) |
| (32) |
| (33) |
| (34) |
where δ is the effective thickness of a compartment and D is the diffusion coefficient of the drug in the compartment. The parameters used in the model are given in Supplementary Table 3. All variables used in the computational model and their meaning are described in Supplementary Table 4.
Heart tissue chamber:
A compartmental schematic of the heart tissue chamber is shown in Fig. 6C. The fluidic channels associated with the heart tissue chamber are named heart inlet fluidic channel (HIFC) and heart fluidic channel (HFC). HFC is situated exactly below the heart tissue chamber’s membrane. The top and bottom functionalized layers of the membrane are represented by the acronyms HTM and HBM. The middle layer of the membrane is represented as HMM. The top, middle and bottom layers of the tank are named as HTT, HMT, and HBT, respectively. The heart tissue (represented as H) is situated in the middle layer of the tank. The two polydimethylsiloxane (PDMS) pillars supporting the heart tissue are modeled as a single compartment that interacts with the second and third layers of the tank. The mass balance, diffusive flux and permeability calculation equations for the fluidic channels and the membrane layers of the heart tissue chamber are the same as those of the liver tissue chamber. Convective fluid flow brings in drug from the LFC compartment to the HIFC compartment. The main modeling differences in the heart tissue chamber are the position of the heart tissue, inclusion of the PDMS pillars and the absence of metabolism and elimination. Unlike the liver tissue, the heart tissue interacts with the second layer of the tank. The mass balance equations of the bottom, middle and top layers of the tank can be written as:
| (35) |
| (36) |
| (37) |
where the subscript PD represents the PDMS compartment. The diffusive flux exchange between the two layers of the tank and the PDMS pillar is given as:
| (38) |
| (39) |
The permeabilities and are calculated in the similar manner as given in equation (34). As there is no metabolic transformation or elimination in the heart tissue, the mass balance is given as:
| (40) |
Bone tissue chamber:
A schematic of the bone tissue chamber is shown in Fig. 6D. The fluidic channels associated with the bone tissue chamber are named as bone inlet fluidic channel (BIFC) and bone fluidic channel (BFC). BFC is situated exactly below the bone tissue chamber’s membrane. The top and bottom functionalized layers of the membrane are represented by the acronyms BTM and BBM. The middle layer of the membrane is represented as BMM. The top, middle and bottom layers of the tank are named as BTT, BMT and BBT, respectively. The bone tissue (represented as B) is situated in the first layer of the tank and on top of the BTM, and is treated as a porous media comprised of fluid, osteoblast cells (OB) and osteoclast cells (OC). The drug is assumed to bind to the receptors on the cells. The fluidic channel is modeled for the convective fluid flow that brings in the drug from the HFC to the BIFC. The mass balance, diffusive flux and permeability equations for the BIFC, BFC, BMM, BBM, BMT and BTT compartments are similar to that of the liver tissue chamber. As the bone tissue is situated directly above the top layer of the membrane, the diffusive flux is modeled for the membrane-bone interface using the following equations for the top layer of the membrane, bottom layer of the tank and bone tissue:
| (41) |
| (42) |
| (43) |
where and are nonlinear saturable bindings on the cell receptors and can be written as:
| (44) |
| (45) |
where k1f and k1r are the forward and reverse binding rate constants, and and are the maximum density of the binding sites (receptors).
The mass balance for the receptors can be written as:
| (46) |
| (47) |
Though the current model has the capability to include binding of the drug to the receptors, it is not included for the validation. The and terms are assumed to be zero. The diffusive flux between the top layer of the membrane and the bone tissue (), and between the bone tissue and the bottom layer of the tank () can be written as:
| (48) |
| (49) |
where is the surface area of the bone tissue in contact with the bottom layer of the tank (excludes bottom surface area of the bone tissue). The permeabilities are calculated in a similar manner as described in equation (32), as there will be no drug partitioning when moving from the functionalized top layer of the membrane to the bone tissue.
Skin tissue chamber:
The skin tissue chamber is modeled such that the skin tissue (represented as S) rests in the top layer of the tank to maintain an air-liquid interface. The rest of the compartments are modeled in the same way as for the liver tissue chamber. The fluidic channels associated with the skin tissue chamber are named as skin inlet fluidic channel (SIFC) and skin fluidic channel (SFC). The skin tissue chamber has another fluidic channel section named as skin outlet fluidic channel (SOFC) that connects the SFC to the reservoir. SFC is situated exactly below the skin tissue chamber’s membrane. The top and bottom functionalized layers of the membrane are represented by the acronyms STM and SBM. The middle layer of the membrane is represented as SMM. The top, middle and bottom layers of the tank are STT, SMT and SBT, respectively.
A compartmental schematic of the skin tissue chamber is shown in Fig. 6E. The mass balance, diffusive flux and permeability equations for the SIFC, SFC, SBM, SMM, STM and SMT compartments are similar to that of the liver tissue chamber. The mass balance for the SBT compartment is as described in equation (35). The fluidic channel is modeled such that the convective fluid flow brings in the drug from the BFC to the SIFC. The mass balance equation for the outlet fluidic channel compartment can be written as:
| (50) |
The mass balance equation for the top layer of the tank and the skin tissue can be written as:
| (51) |
| (52) |
The interfacial surface area used to calculate the diffusive flux excludes the top surface area of the skin tissue as it is in contact with the air and not the media.
Reservoir and tubing system:
The compartmental mathematical model describing the drug transport in the reservoir and tubing is also expressed in the form of a set of ODE representing the species mass balance in each compartment. The reservoir compartment is assumed to be well mixed and the tubing is discretized into twenty individual sections. The model accounts for the outer PharMed BPT shell around the tubing lumen. A compartmental schematic of the reservoir and tubing model is shown in Fig. 6F. The reservoir (RES) is connected to the SOFC compartment from the skin tissue chamber and the first discretized cell of the tubing lumen (TL, 1). The mass balance for the reservoir compartment can be written as:
| (53) |
The drug transport in the tubing lumen is majorly due to convective fluid flow. The model also accounts for drug absorption by the outer shell of the tubing. The mass balance equation for the first discretized section of the tubing lumen and the first discretized section of the BPT shell (TB, 1) can be written as:
| (54) |
| (55) |
where subscripts TB,2 and TL,2 represent the second BPT shell cell and the second lumen cell, respectively. The mass balance equations for the lumen and the BPT shell cells can be written as:
| (56) |
| (57) |
where TL is tubing lumen and TB is BPT shell. The subscript “i” corresponds to the respective discretized cell number and can have a value between 2 and 19 (i = 2 – 19). The mass balance equation for the last discretized cell of the lumen and BPT shell can be written as:
| (58) |
| (59) |
The diffusive flux between adjacent lumen discretized cells can be expressed as:
| (60) |
| (61) |
where ATL is the available interfacial area between two lumen cells and PTL is the drug permeability for the lumen.
The diffusive flux between adjacent BPT shell discretized cells can be expressed as:
| (62) |
| (63) |
where ATB is the available interfacial area between two BPT shell cells and PTB is the drug permeability for the BPT shell.
The diffusive flux between the lumen cell and the BPT shell cell can be expressed as
| (64) |
where ATLB is the available interfacial area between the lumen cell and the BPT shell cell, and PTLB is the drug permeability for the lumen-BPT shell interface.
The drug permeability for the lumen and for the BPT shell can be calculated as described in equation (32). The drug permeability for the lumen-BPT shell interface can be calculated as described in equation (28).
Non-endothelialized model:
The non-endothelialized model (Mixed condition) is based on the primary model of the entire tissue chip. The equations representing the species mass balance, diffusive flux and permeabilities for the reservoir, tubing, inlet fluidic channels, second and third layer of the tank (including first layer of the tank for the bone tissue chamber), and the tissue compartments (except for the bone tissue) are expressed as previously described for the entire tissue chip model. In this model, the nylon mesh separating the tank from the fluidic channel consists of a single layer. The top and bottom functionalized layers of the membrane are removed and thus the membrane does not account for the drug partitioning. In the liver, heart, and skin tissue chambers, the nylon mesh is directly in contact with the first layer of the tank, therefore the changed equations are similar for all of them.
A generalized compartmental schematic is shown in Supplemental Figure 12D. The compartments for which the equations are the same as the primary model, are not included in the schematic. For this model, the bottom layer of the tank, membrane and the fluidic channel below the membrane are represented by the acronyms BT, MM and FC, respectively. The mass balance equations for these compartments can be written as:
| (65) |
| (66) |
| (67) |
where the subscript IFC represents inlet fluidic channel, MT represents the middle layer of the tank and L represents the liver tissue. The flux JBT-L is only applicable for the liver tissue chamber and is not included for the heart tissue chamber and the skin tissue chamber.
The flux terms in the above compartmental mass balance equations can be written as:
| (68) |
| (69) |
The diffusive fluxes JBT-MT and JBT-L are same as described previously in equations (25) and (27), respectively. The permeabilities are calculated as:
| (70) |
| (71) |
In the bone tissue chamber, the membrane is in contact with the tissue and thus the equations slightly vary than for the other tissue chambers. The compartmental schematic for the non-endothelialized model of the bone tissue chamber is shown in Supplemental Figure 12E. The compartments for which the equations are the same as the primary model, are not included in the schematic. For this model, the bone tissue, membrane and the fluidic channel below the membrane are represented by the acronyms B, BMM and BFC, respectively (similar to the primary model). The mass balance equations for these compartments can be written as,
| (72) |
| (73) |
| (74) |
where the subscripts BIFC and BBT represent the inlet fluidic channel and bottom tank layer of the bone tissue chamber, respectively.
The flux terms in the above compartmental mass balance equations can be written as:
| (75) |
| (76) |
The diffusive flux JBBT-B is same as described previously in equations (49). The permeabilities are calculated as:
| (77) |
| (78) |
Mass and volume conservation:
To ensure proper model performance, we tested if the total mass and volume in the system was conserved during a simulation. We simulated a constant drug concentration of 1 g cm−3 in the reservoir, assumed no sampling or drug metabolism in this simulation, and assess the mass and volume in the system over more than 60 hours.
Parametric Study:
In the current InterOrgan and Mixed models, the tissue chambers are discretized into three layers. Chamber discretization is typically used to capture transport delays due to local concentration gradients. In the current models, tissue chamber discretization was optimized using a single tissue chamber test case (without tissue construct), using a similar perfusion media flow rate of 1.5 mL min−1. The test case model for doxorubicin included the following compartments: inlet fluidic channel, fluidic channel, membrane, and tissue chamber (Supplementary Figure 5F). Three test cases with varying spatial subdivision were performed, where the tissue chamber was discretized with one, three, and five layers. In the main text we have used the time step Δt = 10 seconds for the simulation period of 72 hours. To analyze the effect of time step, we performed simulations with reduced time steps Δt of 1.0 seconds and 0.01seconds for the total simulation time of 4 hours. Drug concentrations time profiles were monitored at the bottom and top of the chamber.
For Δt = 1.0 seconds, the three spatial resolution test cases showed similar overlapping concentration-time profiles. For Δt = 0.01 seconds, a concentration gradient was observed in the five-layer case for the first seven seconds; however, all concentration-time profiles overlapped for the remainder of the simulation time. This demonstrates that the original time step of Δt =10 seconds and three-layer spatial discretization are adequate for the primary simulations. Our initial approach of discretizing the tissue chamber into three cells allows us to position the individual tissues according to the experimental setup.
9. Statistics
All measurements reported were taken from distinct samples. Data were analyzed in Excel (Microsoft) and graphed in Prism (GraphPad). Data are shown as mean ± standard deviation, for a given number of biological replicates. Significant differences were defined by p < 0.05 for all statistical methods, unless otherwise noted. Differences between the experimental groups were analyzed by unpaired, two-tailed Student’s t-test, or one-way or multi-way ANOVA. Post hoc pairwise analysis was done using Tukey’s HSD test.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Source data for all graphs in the main and Extended Data Figures are available in Supplementary Information. Any additional source data are available upon request.
Code availability
The study used a combination of commercial and open-source software packages (NIS-Elements from Nikon, Excel (Microsoft), Prism (GraphPad), MATLAB (MathWorks), CoBI (CFD Research), FlowJo (BD Biosciences), SOLIDWORKS Flow Simulation (SolidWorks), IVIS Living Image (PerkinElmer), GSEA (UC San Diego and Broad Institute), Transcriptome Analysis Console (Thermo Fisher Scientific). that are specified in the Methods section. The custom-designed software that will be available per request.
Extended Data
Extended Data Figure 1 |. Platform configurability and modularity.

a, Photograph of the assembled platform. b, Top view of the platform including each compartment chamber and reservoir for circulating media. c, Bottom view of the platform. d, Configurability of the platform can be established through connecting reactors in series for scaling of engineered organs. e, Alternative platform design for single-organ culture with perfusion and vascular barrier. f, Alternative platform design for dual-organ culture with perfusion and vascular barrier. g, Platform with tubing attached to a peristaltic pump during integrated culture. h, Computational fluidic model of shear stress during perfusion with the vascular barrier, at a flow rate of 1.3 mL/min to yield a shear stress of 1.88 dyn/cm2 at the vascular barrier interface.
Extended Data Figure 2 |. Platform design details for integration of engineered tissues.

a, Images and dimensions of the engineered transwell insert and its location within the platform. b, Top view of the platform reservoir to detail routing of fluid from the channel into the reservoir and subsequent pump driven routing into the tubing via the elbow connector. c, Schematic side view of the platform with measurements. d, Schematic images detailing fluidic routing of vascular media via the designed channel entry and exit ports. e, Platform details.
Extended Data Figure 3 |. Characterization of tissue specific maturation.
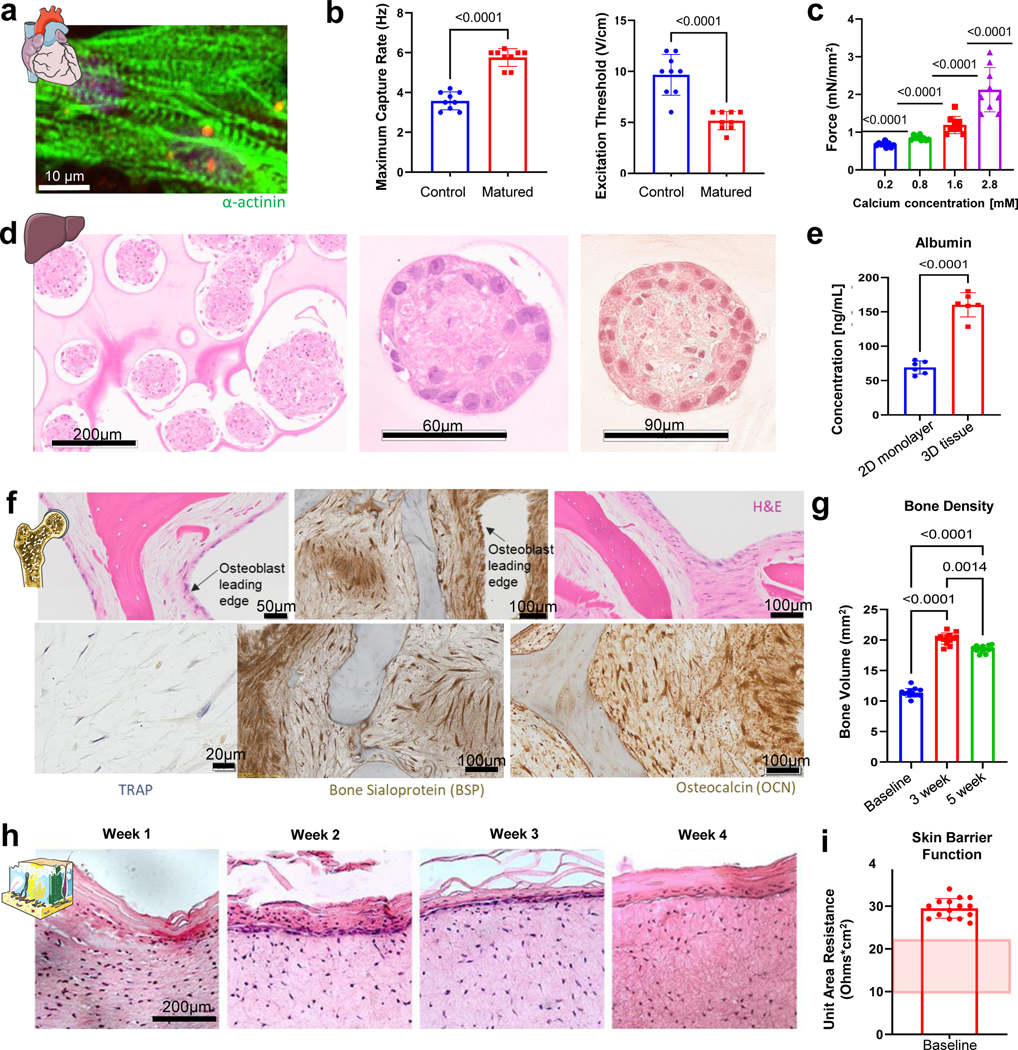
a-c, Electromechanically matured cardiac tissues show aligned alpha actinin expression (a) functional improvements in maximum capture rate and excitation threshold (b) (n=8–9 biological replicates), and increased force responses when exposed to increasing calcium concentrations (c) (n=9 biological replicates). d-e, Liver tissues are matured via co-culture of cells in 3D aggregated as detailed by immunohistochemical staining and increased albumin secretion (e) (n=6 biological replicates). f, Immunohistochemical staining of engineered bone slices stain positive for osteocalcin, TRAP and bone sialoprotein. g, micro-computed tomography imaging of bone scaffolds over time details bone remodeling during the osteoblastic and osteolytic phases of induced bone maturation (n=13 biological replicates). h, Immunohistochemical staining of engineered skin slices detail the formation of the epidermis and dermis over 4 weeks of maturation. i, TEER values detail the barrier function of the engineered skin, with reported values detailed by the red shaded region7 (n=15 biological replicates). Data is shown as mean ± SD and statistics determined by unpaired t-test (b, c, e) or one-way ANOVA (g, i).
Extended Data Figure 4 |. Establishment of mature, selectively permeable vascular barrier.

a-b, Immunofluorescence imaging of vascular barriers and (b)FITC-Dextran transport through vascular barriers cultured on various transwell pore sizes (n=7–10 biological replicates). c-d, (c) Immunofluorescence imaging of vascular barriers and (d) FITC-Dextran transport through vascular barriers cultured under different shear stress conditions (n=7–8 biological replicates). e, Barrier function demonstrated by tracking tagged dextran molecules of different size (n=3 biological replicates). Data is shown as mean ± SD and statistics determined by two-way or mixed ANOVA.
Extended Data Figure 5 |. Immune cell isolation, maturation, and characterization.

a, Initial cell population was made up of >98% CD14+, ITGAM+ monocytes. b, Brightfield image detailing monocyte adherence to barrier surface within platform. c, Flow cytometry characterization of monocyte viability after 3 days of culture (n=3–4 biological replicates). d, Monocyte specification and differentiation over two weeks of culture (n=3 biological replicates). Data is shown as mean ± SD and statistics determined by two-way ANOVA.
Extended Data Figure 6 |. Immune function over four-week culture.

a-b, Heatmap of all measured cytokines (a) and individual cytokine expression for select cytokines (b) over 28 days of culture in the different platform configurations (InterOrgan n=5 biological replicates, Mixed and Isolated n=3 biological replicates). Data is shown as mean ± SD and statistics determined by one-way ANOVA.
Extended Data Figure 7 |. Proteomic breakdown of bone, liver, and skin tissues studied over four weeks in (i) InterOrgan multi-tissue platform, (ii) in the Mixed media approach, and (iii) in Isolation.
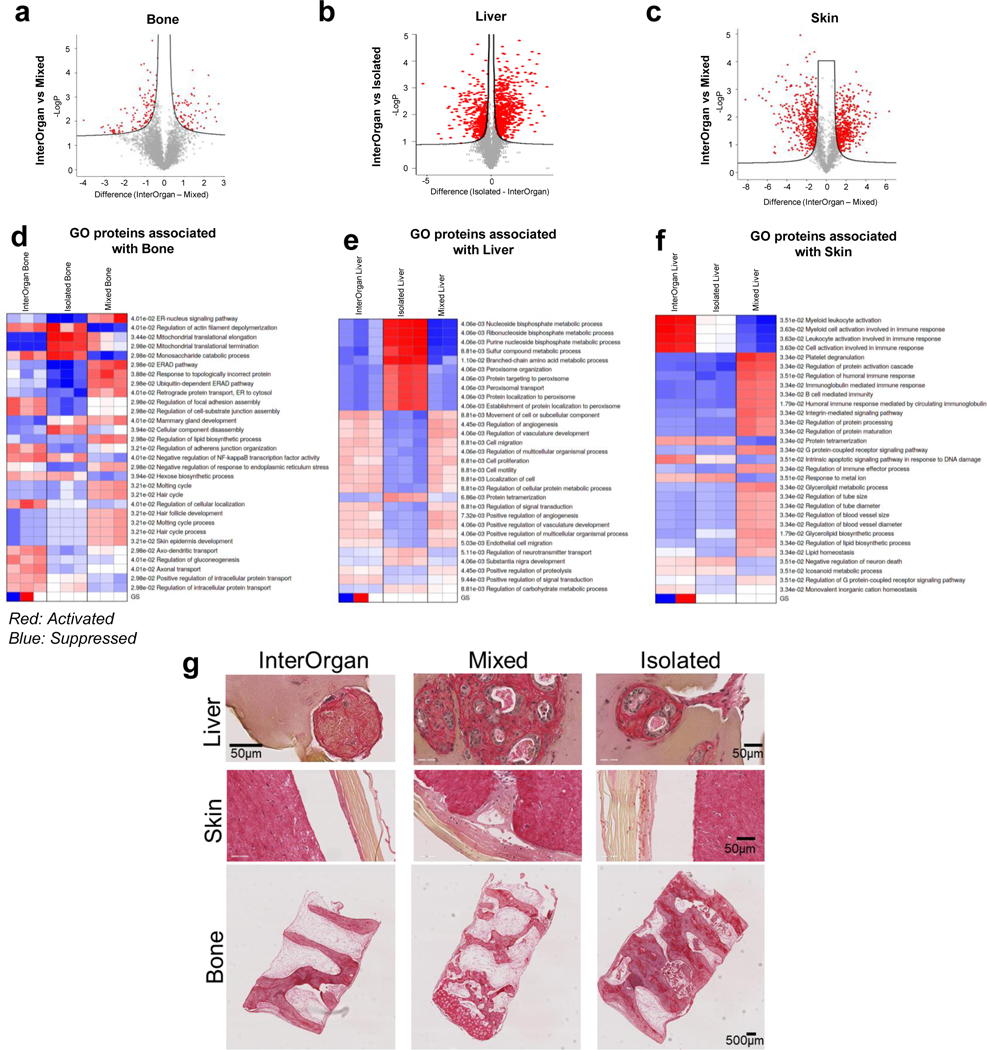
a-c, Proteomic breakdown of engineered bone (6,000+ unique proteins; a), liver (2,000+ proteins; b), and skin (2,000+ proteins; c) studied over 28 days in the (i) integrated InterOrgan multi-tissue platform, (ii) platform with mixed media, and (iii) tissues cultured in isolation. Comparison of integrated versus mixed conditions via differential protein abundances is represented by Volcano plots. d-f, PGSEA plots of the top 30 GO Biological Process pathways, with red indicating activated pathways and blue indicating suppressed pathways for bone (d), liver (e), and skin (f). g, Immunohistochemical staining for picrosirius red details collagen within liver, skin and bone tissues after 28 days.
Extended Data Figure 8 |. Proteomic breakdown of engineered cardiac tissues studied over four weeks in (i) InterOrgan multi-tissue platform, (ii) in the Mixed media approach, and (iii) in Isolation.

a, excitation threshold and b, maximum capture rate of cardiac tissues for each experimental condition (i-iii) (InterOrgan and Mixed n=6 biological replicates, Isolated n=3 biological replicates). c, PCA clustering of each experimental condition (i-iii). d, Comparison of integrated versus mixed conditions via differential protein abundances. e, Proteins important to cardiac tissue function, structure, energetics, and calcium handling. f, PGSEA pathway analysis showing the top 30 GO Biological Process pathways related to disease and function in integrated vs. mixed conditions, with red indicating activated and blue indicating suppressed pathways.
Extended Data Figure 9 |. Development of a multi-compartment computational model of the multi-tissue platform.

a, Schematic of the entire mechanistic multi-compartment model. All tissue tanks have a similar configuration that is composed of a cylindrical tank divided in 3 sub-compartments (TT, MT, and BT), an endothelial membrane with 3 layers (TM, MM, and BM), a fluidic inflow segment (IFC), and a fluidic perfusion segment (FC). All the individual compartments were represented by species mass balance equations, calculated using drug flux (J) and volumetric medium flow (Qf). b, Schematic of the liver tissue chamber. c, Schematic of the heart tissue chamber. d, Schematic of the bone tissue chamber. e, Schematic of the skin tissue chamber. f, Schematic of the reservoir and tubing.
Extended Data Figure 10 |. Comparison of the InterOrgan and Mixed computational PK models.

a-b, Doxorubicin (a) and doxorubicinol (b) levels over time within all tissue chambers and in the reservoir predicted by the computational PK model for the InterOrgan (blue line) and the Mixed platform (red bat).
Supplementary Material
Acknowledgements
The authors gratefully acknowledge funding of their research by NIH (UG3 EB025765, P41 EB002520 and R01 CA249799 to G.V.-N.; R35 CA197745, S10 OD012351, and S10 OD021764 to A.C; UL1 TR001873 to the Irving Institute for Clinical and Translational Research; P30 CA013696 to the Confocal and Specialized Microscopy Shared Resource), NSF (Engineering Resource Center NSF16478 to C.S.C. and G.V.-N., Graduate Research Fellowship DGE1644869 to D.N.T.), and F.C.T. (PD/BD/105819/2014 to D.T.). We thank Pamela L. Graney, Ece Öztürk, Maria C. Samaritano, N. Valerio Dorrello, and Barry Fine for helpful discussions. We also thank Renu Nandakumar and Changhong Qiao (Biomarkers Core Laboratory at the Irving Institute for Clinical Translational Research), Michael Kissner (Columbia Stem Cell Initiative Flow Cytometry Core), Christopher Damoci (Oncology Precision Therapeutics and Imaging Core Shared Resource), Molecular Pathology, Confocal and Specialized Microscopy, and Oncology Precision Therapeutics and Imaging Core Shared Resources at the Columbia University Herbert Irving Comprehensive Cancer Center for technical support.
Competing interests
G.V.-N., K.R-B., and Y.Z. are co-founders of TARA Biosystems, a company that has licensed some of the cardiac tissue engineering methodologies, and hold equity in the company. G.V.-N. is also serving on the Board of Directors at TARA Biosystems and is being compensated for this role. A.C. is founder, equity holder, and consultant of DarwinHealth, Inc., a company that has licensed some of the algorithms used in this manuscript from Columbia University. Columbia University is also an equity holder in DarwinHealth, Inc.
Footnotes
Additional information [please do not modify this section]
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41551-01X-XXXX-X.
References
- 1.Di L.et al. A perspective on the prediction of drug pharmacokinetics and disposition in drug research and development. Drug Metab. Dispos 41, 1975–1993 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Mak IW, Evaniew N.& Ghert M.Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Transl. Res 6, 114–118 (2014). [PMC free article] [PubMed] [Google Scholar]
- 3.Tuntland T.et al. Implementation of pharmacokinetic and pharmacodynamic strategies in early research phases of drug discovery and development at Novartis Institute of Biomedical Research. Front. Pharmacol 5, 174 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pound P.& Ritskes-Hoitinga M.Is it possible to overcome issues of external validity in preclinical animal research? Why most animal models are bound to fail. J. Transl. Med 16, 304 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huh D.et al. Reconstituting organ-level lung functions on a chip. Science 328, 1662–1668 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wikswo JP The relevance and potential roles of microphysiological systems in biology and medicine. Exp. Biol. Med. Maywood NJ 239, 1061–1072 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Polacheck WJ et al. A non-canonical Notch complex regulates adherens junctions and vascular barrier function. Nature 552, 258–262 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shinha K, Nihei W, Ono T, Nakazato R.& Kimura H.A pharmacokinetic-pharmacodynamic model based on multi-organ-on-a-chip for drug-drug interaction studies. Biomicrofluidics 14, 044108 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trapecar M.et al. Gut-liver physiomimetics reveal paradoxical modulation of IBD-related inflammation by short-chain fatty acids. Cell Syst. 10, 223–239.e9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Low LA, Mummery C, Berridge BR, Austin CP & Tagle DA Organs-on-chips: Into the next decade. Nat. Rev. Drug Discov 20, 345–361 (2021). [DOI] [PubMed] [Google Scholar]
- 11.Ronaldson-Bouchard K.et al. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 556, 239–243 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Novak R.et al. Robotic fluidic coupling and interrogation of multiple vascularized organ chips. Nat. Biomed. Eng 4, 407–420 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ronaldson-Bouchard K.& Vunjak-Novakovic G.Organs-on-a-chip: A fast track for engineered human tissues in drug development. Cell Stem Cell 22, 310–324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leger KJ et al. Circulating microRNAs: Potential markers of cardiotoxicity in children and young adults treated With anthracycline chemotherapy. J. Am. Heart Assoc 6, e004653 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McAleer CW et al. Multi-organ system for the evaluation of efficacy and off-target toxicity of anticancer therapeutics. Sci. Transl. Med 11, eaav1386 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Oatmen KE et al. Identification of a novel microRNA profile in pediatric patients with cancer treated with anthracycline chemotherapy. Am. J. Physiol. Heart Circ. Physiol 315, H1443–H1452 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Tacar O, Sriamornsak P.& Dass CR Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol 65, 157–170 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Thorn CF et al. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharmacogenet. Genomics 21, 440–446 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yadi W.et al. Bioinformatic analysis of peripheral blood miRNA of breast cancer patients in relation with anthracycline cardiotoxicity. BMC Cardiovasc. Disord 20, 43 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schepers A, Li C, Chhabra A, Seney BT & Bhatia S.Engineering a perfusable 3D human liver platform from iPS cells. Lab. Chip 16, 2644–2653 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Villasante A.et al. Tissue-engineered model of human osteolytic bone tumor. Tissue Eng. Part C Methods 23, 98–107 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Itoh M.et al. Generation of 3D skin equivalents fully reconstituted from human induced pluripotent stem cells (iPSCs). PloS One 8, e77673 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neil JE, Brown MB & Williams AC Human skin explant model for the investigation of topical therapeutics. Sci. Rep 10, 21192 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davies PF Flow-mediated endothelial mechanotransduction. Physiol. Rev 75, 519–560 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirschi KK & D’Amore PA Pericytes in the microvasculature. Cardiovasc. Res 32, 687–698 (1996). [PubMed] [Google Scholar]
- 26.Subramanian A.et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeng H, Wang J, Clouse H, Lagrutta A.& Sannajust F.Resolving the reversed rate effect of calcium channel blockers on human-induced pluripotent stem cell-derived cardiomyocytes and the impact on in vitro cardiac safety evaluation. Toxicol. Sci. Off. J. Soc. Toxicol 167, 573–580 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Uhlén M.et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Prantil-Baun R.et al. Physiologically based pharmacokinetic and pharmacodynamic analysis enabled by microfluidically linked organs-on-chips. Annu. Rev. Pharmacol. Toxicol 58, 37–64 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Herland A.et al. Quantitative prediction of human pharmacokinetic responses to drugs via fluidically coupled vascularized organ chips. Nat. Biomed. Eng 4, 421–436 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Przekwas A.& Somayaji MR Chapter 10 - Computational pharmacokinetic modeling of organ-on-chip devices and microphysiological systems. in Organ-on-a-chip (eds. Hoeng J, Bovard D.& Peitsch MC) 311–361 (Academic Press, 2020). doi: 10.1016/B978-0-12-817202-5.00011-5. [DOI] [Google Scholar]
- 32.Somayaji MR, Das D.& Przekwas A.Computational approaches for modeling and analysis of human-on-chip systems for drug testing and characterization. Drug Discov. Today 21, 1859–1862 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Chramiec A.et al. Integrated human organ-on-a-chip model for predictive studies of anti-tumor drug efficacy and cardiac safety. Lab. Chip 20, 4357–4372 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y.et al. Taurine zinc solid dispersions attenuate doxorubicin-induced hepatotoxicity and cardiotoxicity in rats. Toxicol. Appl. Pharmacol 289, 1–11 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Fan C.et al. Combination breast cancer chemotherapy with doxorubicin and cyclophosphamide damages bone and bone marrow in a female rat model. Breast Cancer Res. Treat 165, 41–51 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Rigaud VO-C et al. Circulating miR-1 as a potential biomarker of doxorubicin-induced cardiotoxicity in breast cancer patients. Oncotarget 8, 6994–7002 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Y.& Wang X.miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 48, D127–D131 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ronaldson-Bouchard K.et al. Engineering of human cardiac muscle electromechanically matured to an adult-like phenotype. Nat. Protoc 14, 2781–2817 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhumiratana S.et al. Tissue-engineered autologous grafts for facial bone reconstruction. Sci. Transl. Med 8, 343ra83 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marcos-Campos I.et al. Bone scaffold architecture modulates the development of mineralized bone matrix by human embryonic stem cells. Biomaterials 33, 8329–8342 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas A.et al. Characterization of vascular permeability using a biomimetic microfluidic blood vessel model. Biomicrofluidics 11, 024102 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mootha VK et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34, 267–273 (2003). [DOI] [PubMed] [Google Scholar]
- 43.Navarrete-Perea J, Yu Q, Gygi SP & Paulo JA Streamlined Tandem Mass Tag (SL-TMT) protocol: An efficient strategy for quantitative (phospho)proteome profiling using tandem mass tag-synchronous precursor selection-MS3. J. Proteome Res 17, 2226–2236 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tyanova S.et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Wang D.et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol 15, e8503 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ge SX, Jung D.& Yao R.ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinforma. Oxf. Engl 36, 2628–2629 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mi H, Muruganujan A, Ebert D, Huang X.& Thomas PD PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47, D419–D426 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ge SX, Son EW & Yao R.iDEP: An integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinformatics 19, 534 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ru Y.et al. The multiMiR R package and database: Integration of microRNA-target interactions along with their disease and drug associations. Nucleic Acids Res. 42, e133 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang J.& Storey KB RBiomirGS: An all-in-one miRNA gene set analysis solution featuring target mRNA mapping and expression profile integration. PeerJ 6, e4262 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Korotkevich G.et al. Fast gene set enrichment analysis. bioRxiv 060012 (2021) doi: 10.1101/060012. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data for all graphs in the main and Extended Data Figures are available in Supplementary Information. Any additional source data are available upon request.