

Abstract
Spatially and temporally controlled delivery of biologicals, including gene vectors, represents an unmet need for regenerative medicine and gene therapy applications. Here we describe a method of reversible attachment of serotype 2 adeno-associated viral vectors (AAV2) to metal surfaces. This technique enables localized delivery of the vector to the target cell population in vitro and in vivo with the subsequent effective transduction of cells adjacent to the metal substrate. The underlying bioengineering approach employs coordination chemistry between the bisphosphonic groups of polyallylamine bisphosphonates and the metal atoms on the surface of metallic samples. Formation of a stable polybisphosphonate monolayer with plentiful allyl-derived amines allows further chemical modification to consecutively append thiol-modified protein G, an anti-AAV2 antibody, and AAV2 particles. Herein we present a detailed protocols for the metal substrate modification, for the visualization of the metal surface-immobilized vector using direct and indirect fluorescent AAV2 labeling and scanning electron microscopy, for quantification of the surface-immobilized vector load with RT-PCR, and for the localized vector transduction in vitro and in vivo.
Keywords: serotype 2 adeno-associated viral vectors, metal substrate, the metal surface-immobilized vector, AAV2 labeling
1. Introduction
Gene therapy is a forefront experimental approach for the correction of somatic mutations and overexpression or blockage of specific proteins affecting physiological functions. Despite the significant therapeutic potential, currently only a small number of gene therapies are used clinically 1, 2. One of the central problems hindering a broader use of gene therapies in clinical medicine is a lack of practical solutions for targeting gene vectors in a precise manner 3, 4. Failure of targeted delivery results in the untoward transduction of non-diseased tissue and increases toxicity and immunogenicity of gene therapy interventions 5. A concept of substrate-mediated gene delivery developed by Shea and colleagues 6–9 for non-viral gene vectors provides an opportunity for the spatially controlled gene therapy by implementing vector immobilization on the surfaces of biocompatible materials. Substrate-mediated gene delivery is especially advantageous in conjunction with medical implants that are in the extensive use in cardiology (stents, pacemakers), orthopedics (pins, rods, plates, joints), gynecology (intrauterine devices), urology (stents, artificial sphincters) and dental medicine (tooth implants). Theoretically, the surface of these devices can be modified prior to implantation in the human body to carry an array of gene vectors that would be released onto the implant/tissue interface and locally transduce the implant-contacting tissue to bestow biological properties facilitating tissue remodeling and integration of the implant. While originally substrate-mediated gene transfer techniques were developed to accommodate non-viral vectors, work from our laboratory 10–13 and others 14, 15 has expanded the scope of this approach to include adenoviral (Ad) 10–12, lentiviral (LV) 16 and adeno-associated viral (AAV) 13–15 vectors. Compared to non-viral counterparts, viral vectors possess much higher transduction efficiency, albeit at the expense of higher toxicity and immunogenicity, that can potentially be curtailed by the limited vector spread attainable with substrate-mediated delivery.
Our earlier work capitalized on the reversible attachment of reporter and therapeutic Ad vectors to the polyallylamine bisphosphonate-modified surfaces of metal stents used for relieving atherosclerotic plaque obstruction of blood flow in the coronary arteries. The chemical and biological engineering aspects of Ad vector immobilization via affinity adaptors 11 or through spontaneously hydrolysable cross-linkers 10, 12 were previously reported including in method-oriented publications 17. Due to relatively low toxicity and inflammatory activation, AAV vectors are currently the leading viral vector type in human clinical trials 18. Our recent efforts concentrated on engineering AAV delivery platforms consisting of AAV2 vector particles reversibly immobilized to the metal surface for use in gene delivery stents 13.
2. Materials
2.1. Immobilization of AAV2 vectors on a stainless steel substrate
Commercial sources for metal samples were: 316 L grade stainless steel foil – Goodfellow, Coraopolis, PA, cat #LS515178; stainless steel mesh disks – Electron Microscopy Sciences, Hatfield, PA, cat #E200-SS; 304-grade stainless steels multilink stents – Laserage Technology, Waukegan, IL, cat #LCP039669.
Isopropanol (cat #IX0235) and chloroform (cat #BDH1109) were from VWR, Radnor, PA. Orbital shaker incubator was from Sheldon Manufacturing, Cornelius, OR, model #1575R.
20-ml borosilicate glass scintillation vials are from Wheaton, Millville, NJ, cat #986546. A high-temperature oven was from Sheldon Manufacturing, Cornelius, OR, model #1350FM.
PABT was synthesized in the lab as published 10. To prepare a needed volume of PABT working solution, dissolve 0.5%−2% PABT (w/v) and 0.1%−0.4% potassium bicarbonate (w/v) (Sigma-Aldrich, St. Louis, MO, cat #237205) in double-distilled water (DDW).
To prepare 50 ml of 0.1M acetic buffer, dissolve 410 mg of anhydrous sodium acetate (Alfa Aesar, Tewksbury, MA, cat #A13184) in 49.7 ml of DDW and add 0.286 ml of glacial acetic acid (Fisher Scientific, Waltham, MA, cat #A38-212).
Tris(2-carboxyethyl)phosphine hydrochloride (TCEP·HCl) was from Sigma-Aldrich, St. Louis, MO, cat #C4706. To prepare a working solution of TCEP, dissolve 48 mg of TCEP in 4 ml of 0.1M acetic buffer.
PEI(PDT) was synthesized in the lab as published 10. To degas DDW, attach a 500-ml round bottom flask with a standard 24/40 connection (Fisher Scientific, Waltham, MA, cat #10-067G) to a lyophilizer (FreeZone, Labconco, Kansas City, MO, model #710201010) and turn the vacuum on. Boiling will start when the pressure falls below 12 mbars. Keep degassing water until boiling stops (typically, 1–2 minutes). Prepare a needed volume of working PEI(PDT) solution mix by mixing 1.7 volume parts of PEI(PDT) stock, 5.4 parts of degassed DDW, and 1 part of 0.4 M solution of sodium acetate (Alfa Aesar, Tewksbury, MA, cat #A13184) in degassed DDW.
Prepare a working solution of protein G-thiol by diluting 1 volume part of the stock solution (Protein Mods, Waunakee, WI, cat #PGT) with 4 part of PBS (Gibco, Gaithersburg, MD, cat #10010-023) to 0.1 mg/ml. Degas PBS as indicated in 2.1.7 prior to mixing with protein G-thiol.
Anti-AAV2 antibody (clone A20) was from GeneTex (Irvine, CA, cat #44496).
AAV2 vectors were from the University of Pennsylvania Vector Core. They are currently available from Addgene (Watertown, MA).
2.2. Fluorescent labeling of AAV2 and substrate immobilization of fluorescent AAV2 particles
Fisherbrand 7-ml scintillation vials were from Fisher Scientific (Waltham, MA, cat #03-337-26).
Mono-reactive Cy3-NHS ester was from GE Healthcare (Chicago, IL, cat #13101). To prepare 50 ml of 0.1 M carbonate/bicarbonate buffer (CBB) with pH 9.3, co-dissolve 53 mg of sodium carbonate (Fisher Scientific, Waltham, MA, cat #S263-500) and 378 mg of sodium bicarbonate (Fisher Scientific, Waltham, MA, cat #S233-500) in 50 ml of DDW. GENIE® SI-0236 Vortex-Genie 2 Mixer (Scientific Industries, Bohemia, NY, cat# SI-0236) was used to mix viral suspension with the dye.
Slide-A-Lyzer dialysis cassettes, 0.5–3 ml capacity, 20 kDa MWCO were from ThermoFisher Scientific (Waltham, MA), cat #66005.
Bovine serum albumin, fraction V (BSA) was from VWR, Randor, PA, cat #97061-420.
Flat bottom 96-well plates were from Corning Life Sciences (Durham, NC, cat #353072).
Photomicrographs were taking using a fluorescence microscopy (Nikon Instruments, Melville, NY, model Eclipse TE300) equipped with a digital camera (Leica Microsystems, Buffalo Grove, IL, model DMC 450).
2.3. Immunofluorescence staining of AAV2 immobilized on the surface of metal substrate
10% neutral buffered formalin was from VWR (Radnor, PA, cat #10790-714).
Goat medium was from Rockland Immunochemicals (Limerick, PA, cat #D104-00-0050).
Secondary Cy3.5-labeled goat anti-mouse antibody was from Rockland Immunochemicals (Limerick, PA, cat #610-112-121).
2.4. Scanning electron microscopy of AAV2-laden stainless steel surfaces
Sodium cacodylate buffer (0.2 M, pH 7.4) and 2% glutaraldehyde in sodium cacodylate buffer were obtained from Electron Microscopy Sciences (Hatboro, PA, cat# 11652 and 16536-15, respectively).
Ethanol was from Fisher Scientific (Waltham, MA, cat #A405P-4). Hexamethyldisilazane (HMDS) was from Electron Microscopy Sciences (Hatboro, PA, cat#16700).
Imaging was conducted using a scanning electron microscope (model Quanta250; FEI, Hillsboro, OR).
2.5. Quantification of vector load with RT-PCR
24-well plates were from Genesee Scientific (Morrisville, NC, cat #25–107).
Nucleases-free PCR tubes were from Molecular BioProducts, Toronto, ON, Canada, cat #3412A).
QIAamp DNA micro kit was from Qiagen (Germantown, MD, cat #56304).
Rat IL-4 primers (Fwd: 5′-ACA GGA GAA GGG ACG CCA T-3′ and Rvs: 5′-GAA-GCC-CTA-CAG-ACG-AGC-TCA-3′) were from Integrated DNA technologies, Coralville, IA.
Real time PCR system was from Applied Biosystems (Wilmington, DE, model 7500 fast). Data was analyzed with 7500 Software (v2.3).
2.6. Cell transduction with mesh disk-immobilized AAV2-eGFP particles
HEK-293 cells were from ATCC (Manassas, VA, cat #CRL-1573).
Dumont forceps from Fine Science Tools (Foster City, CA, cat #11254-20) were used to manipulate the mesh disks.
Cell culture incubator was from Forma Scientific (Marietta, OH, model 3110).
Fluorimeter was from Molecular Devices (San Jose, CA, model Gemini EM). Fluorimetric data was analyzed with SoftMax Pro 6.5.1 software.
2.7. Preparation and deployment of AAV2-eluting stents in the rat carotid model
Angioplasty balloon catheters were from NuMed (Hopkinton, NY, cat #CE-926-01).
Sprague-Dawley male rats (400–430 g) were from Taconic Biosciences (Rensselaer, NY, cat #SD-M).
Ketamine, meloxicam and normal saline were from Dechra Veterinary products (Overland Park, KS). Xylazine was from Bayer (Leverkusen, Germany). Cefazolin and heparin were from Lambert Vet Supply (Fairbury, NE, cat #9075 and 15086).
24G venous catheters were from Becton-Dickinson (Franklin Lakes, NJ, cat #381412).
Polyethylene terephthalate heat shrink tubing was from Nordson Medical (Salem, NH, cat #103-0311).
A balloon inflation device was from Merit Medical Systems (South Jordan, UT, cat #IN-4130)
Biodegradable 4.0 Vicryl suture was from Ethicon (Bridgewater NJ, cat # J214H). Skin staples and a stapler were from Fine Science Tools (Foster City, CA, cat #12040-02 and 12018-12, respectively).
2.8. Bioluminescence imaging of arterial gene expression
A rodent anesthesia machine (model RAS-4) was integrated with an IVIS imager (model Spectrum). Both devices were from PerkinElmer (Waltham, MA). Isoflurane was from Baxter (Deerfield, IL, cat #1001936040).
D-Luciferin (potassium salt) was from Gold Biotechnology (St. Louis, MO, cat #LUCK-100). Pluronic F-127 was from Sigma-Aldrich (St. Louis, MO, cat #P2443).
200-μl pipette tips were from USA Scientific (Ocala, FL, cat #1111-0816).
Nose cones for anesthesia were from Harvard Apparatus (Holliston, MA, cat #59-8255).
A Living Image software was from PerkinElmer (Waltham, MA, cat #128113).
Sterile saline solution was from Baxter (Deerfield, IL, cat #FE1323), and sterile gauzes were from Dukal (Ronkonkoma, NY, cat #1212).
3. Methods
3.1. Immobilization of AAV2 vectors on a stainless steel substrate (Fig. 1)
Fig. 1.
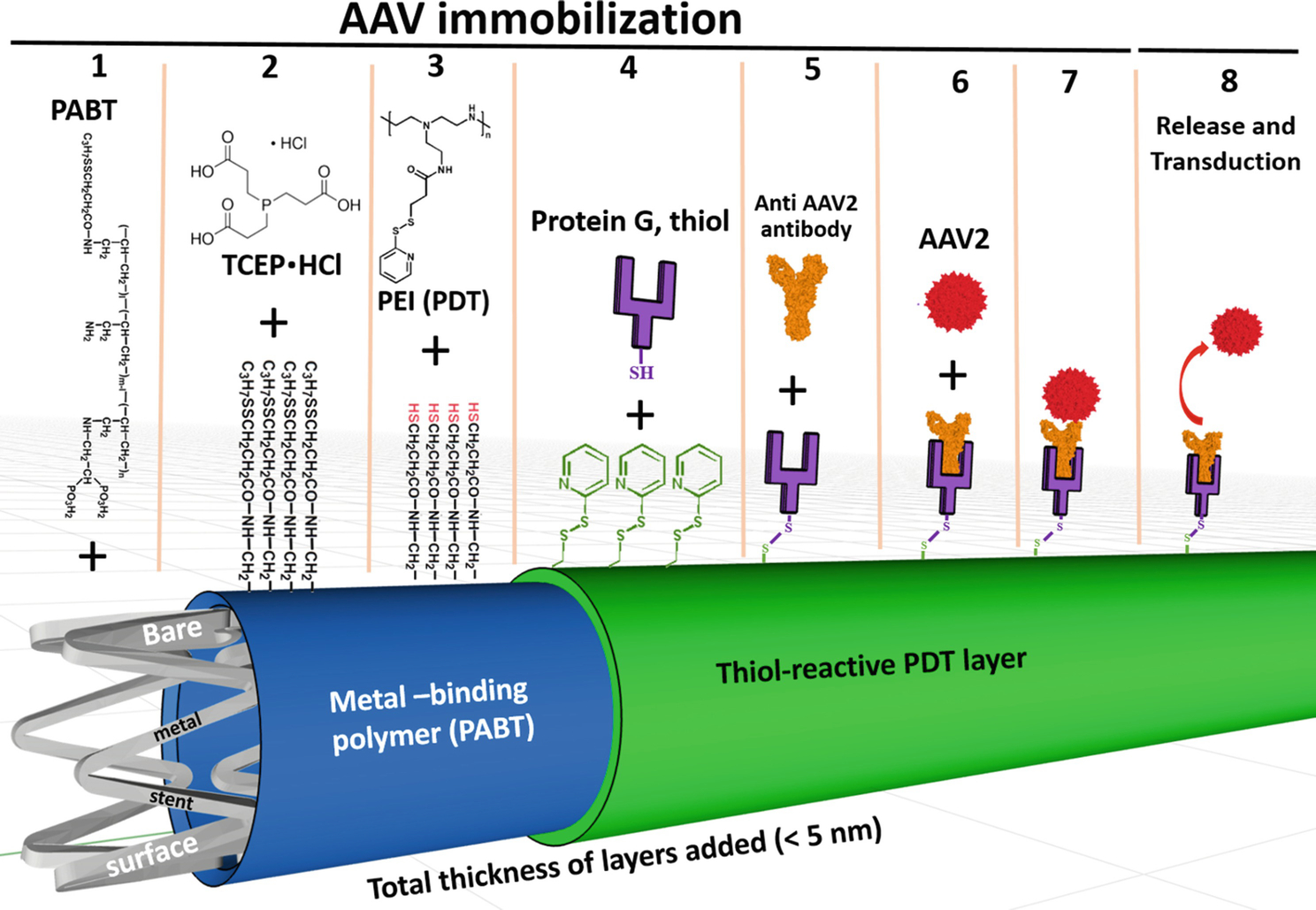
Schematic representation of reversible immobilization of AAV2 vectors on the bare metal surface through PABT/PEI(PDT)/protein G/anti-AAV2 antibody tethering.
Abbreviations: PABT-polyallylamine bisphosphonate with latent thiol groups; TCEP·HCl - tris(2-carboxyethyl)phosphine hydrochloride; PEI(PDT) polyethyleneimine with installed pyridyldithio groups; AAV2 – adeno-associated viral vector of serotype 2.
Cut square samples of stainless steel foil (10 mm × 10 mm). Use stainless steel mesh disks and stainless steel stents as provided. These will be referred to collectively as samples or specimens.
Wash the samples in 10 ml of isopropanol at 60° C with shaking (200 rpm) twice for 5 min each. Wash the samples in 10 ml of chloroform at 60° C with shaking (200 rpm) for 5 min (Note 1).
Relocate the samples in a new borosilicate glass vial and heat in an oven at 220° C for 20 min (Note 2). Remove the vial from the oven and let it cool for 10 min.
Incubate the samples in 1–4 ml of 0.5%−2% PABT at 72° C with shaking for 1–4 hours. The samples need to be completely covered with PABT solution (Note 3).
Aspirate PABT solution. Rinse with DDW three times.
Incubate the samples in 1–4 ml of TCEP working solution at room temperature (RT) with shaking (200 rpm) for 10 min.
Rinse the samples 5 times with degassed DDW. Relocate the samples into a new vial. Rinse additional 5 times (Note 4).
Incubate the samples in 0.1–2% working solution of PEI(PDT) at 37° C under argon atmosphere with shaking (200 rpm) for 1 hour.
Aspirate PEI(PDT) solution, rinse the samples 5 times with degassed DDW. Relocate the samples into a new vial, rinse twice with degassed PBS.
Incubate the samples in 1–4 ml of protein G-thiol thiol working solution at 37° C with shaking (200 rpm) in argon atmosphere for 1 hour.
Aspirate protein G-thiol solution and rinse 5 times in PBS. Relocate the samples into a new vial, rinse 5 times in PBS (Note 5).
Incubate in 1–4 ml of working solution of anti-AAV2 antibody (clone A20) at 37° C with shaking (100 rpm) for 1 hour.
Aspirate antibody solution and rinse 5 times in PBS. Relocate the samples into a new vial, rinse 5 times in PBS (Note 6).
Incubate the samples in 1–4 ml of working suspension of AAV2 at 28° C with shaking for 1 hour.
Aspirate AAV2 suspension and rinse 5 times in sterile PBS. Use immediately or store immersed in sterile PBS at 4° C for up to 2 hours.
3.2. Fluorescent labeling of AAV2 and substrate immobilization of fluorescent AAV2 particles (Fig. 2)
Fig. 2.

Representative photomicrographs of stainless steel mesh disks coated with Cy3-labeled AAV2 vector immobilized on the surface of the PABT/PEI(PDT)/protein G-treated meshes in the presence (A) and without (B) capturing anti-AAV2 antibody. Rhodamine filter set.
Thaw one 100 μl aliquot of AAV2 encoding any transgene (5×1012 VG/ml). Transfer AAV2 suspension in a 7-ml glass bottle and dilute the AAV2 prep with 100 μl PBS.
Dissolve the content of one vial of monoreactive Cy3-LC-NHS (0.15 mg) in 1 ml of CBB by pipetting up and down several times.
Add immediately 200 μl of Cy3-LC-NHS solution to the virus prep. Briefly mix vigorously by vortexing. Incubate in darkness with shaking at 28° C for 1 hour.
Load the reaction mixture (0.4 ml) in a 0.1–0.5 ml dialysis cassette with a 20 kDa cut-off. Dialyze against 2 liter of PBS for 24 hours with 3 changes of buffer.
Evacuate very lightly colored pinkish dialyzate (~0.35 ml) from the cassette and dilute with 350 μl of 2% BSA/PBS.
In parallel, prepare 4 mesh disks with the surface-immobilized AAV2 as described in 3.1.1 – 3.1.13. Prepare 2 additional no capturing antibody control disks by omitting steps 3.1.12 – 3.1.13.
Place the A20-conjugated and control mesh disks that were not reacted with anti-AAV2 antibody individually in the wells of a 96-well plate.
Add 100 μl of the formulation comprising Cy3-labeled AAV2 in 1% BSA/PBS to each well. Protect the plate from light and incubate at RT with minimal shaking for 1 hour.
Wash the mesh disks with PBS (3 times, 5 minutes each).
Image the mesh disks using a fluorescent microscope with an inverted optic using a rhodamine filter set at 40–200× magnification and acquire the representative images.
3.3. Immunofluorescence staining of AAV2 immobilized on the surface of metal substrate (Fig. 3)
Fig. 3.
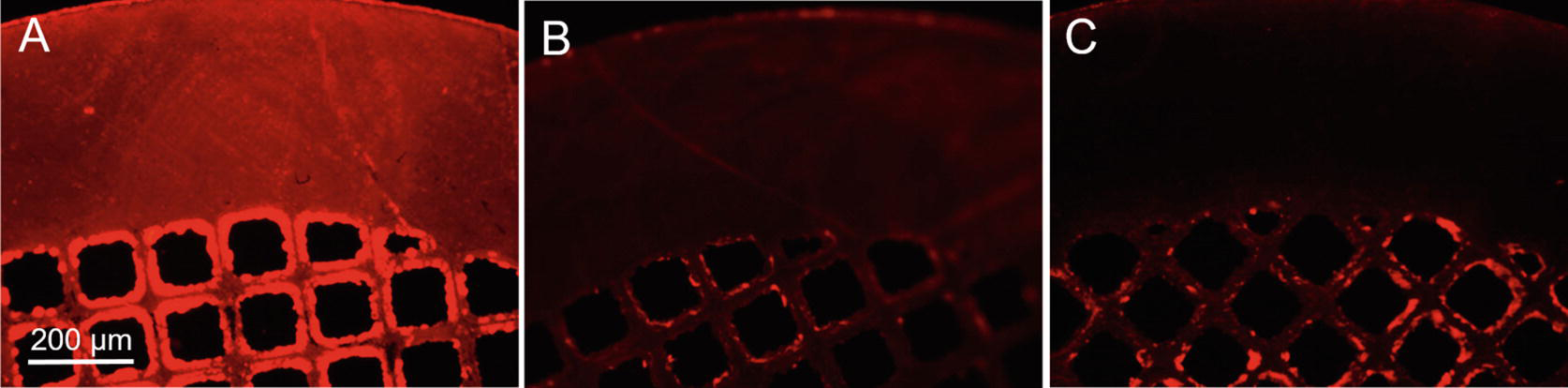
Representative photomicrographs of immunofluorescence detection of AAV2 vectors immobilized on the surface of stainless steel mesh disks. The disks were formulated with (A, C) or without (B) addition of capturing anti-AAV2 antibody. Immunofluorescence protocol included (A, B) or omitted (C) incubation with the detection anti-AAV2 antibody. Rhodamine filter set.
Prepare 4 mesh disks with the surface-immobilized AAV2 as described in 3.1.1 – 3.1.15. Prepare 2 additional disks with (no capturing antibody control) by omitting steps 3.1.12 – 3.1.13.
Relocate the disks in the individual wells of a 96-well plate. Fix the mesh disks with 10% neutral formalin at RT for 15 min. Rinse the wells with the mesh disks thrice with PBS.
Incubate in 10% goat serum/PBS at RT for 20 min, aspirate the blocking solution, do not wash.
Incubate 2 out of 4 properly conjugated mesh disks and 2 no capturing antibody controls with primary mouse monoclonal anti-AAV2 antibody (clone A20) diluted 1:100 in 1% BSA/PBS at RT for 1 hour.
Wash in PBS (3 times, 5 minutes each).
Incubate all 6 mesh disks (including those that were not treated with A20) with secondary Cy3.5-labeled goat anti-mouse IgG antibody diluted 1:200 in 1% BSA/PBS at RT for 45 minutes.
Wash in PBS (3 times, 5 minutes each).
Image the mesh disks using a fluorescent microscope with inverted optics equipped with rhodamine filter set at 40–200× magnification and acquire the representative images.
3.4. Scanning electron microscopy of AAV2-laden stainless steel surfaces (Fig. 4)
Fig. 4.
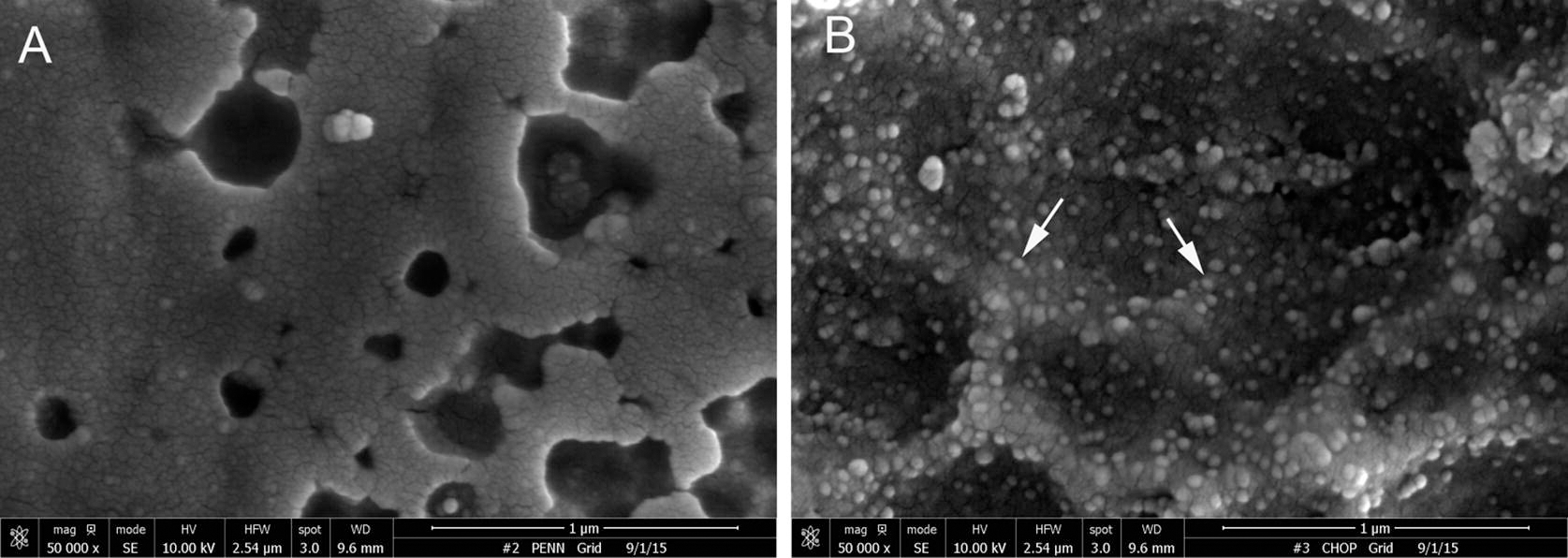
Representative scanning electron microscopy images of stainless steel foil surface modified according to the PABT/PEI(PDT)/protein G/anti-AAV2 antibody protocol AAV2 protocol and either omitted AAV2 incubation (A) or added AAV2 (B). Arrows point to the individual AAV2 particles (B). Original magnification is 80.000×.
Prepare one piece of stainless steel foil (3 mm × 3 mm) with the surface-immobilized AAV2 as described in 3.1.1 – 3.1.15. Prepare one piece of control bare metal surface by abrogating the surface modification protocol after the step 3.1.13.
Wash the samples in cacodylate buffer and fix in 2% glutaraldehyde in cacodylate buffer at RT overnight. Wash extensively with cacodylate buffer.
Dehydrate via serial aqueous ethanol solutions (50%, 70%, 95% and 100%; 10 min, 2 times each). Continue with 2 parts of 100% ethanol/1 part HMDS, 1 part of 100% ethanol/1 part HMDS and 1 part of ethanol/2 parts of HMDS for 15 min each, then with HMDS (15 min, 3 times). Leave in the last change of HMDS in the fume hood overnight to evaporate HMDS.
Mount foils on specimen stubs and sputter-coat samples with gold/palladium (80%/20%) alloy at 2.5 mV for 2 min to achieve ~10 nm thickness of the coating.
Image the control and AAV2-modified surfaces at 80.000× magnification using a scanning electron microscope and save representative images.
3.5. Quantification of vector load with RT-PCR
Prepare four 1 cm × 1 cm stainless steel foil samples with the surface-immobilized AAV2 as described in 3.1.1 – 3.1.15 using 4.5×1012 AAV2-IL4 particles in the step 3.1.14.
Either place the foils into the individual wells of a 24-well plate and store at −80° C for up to 2 weeks, or proceed immediately to step 3.
Cut each foil into 9 square pieces and fit them in a 200 μl PCR tube (Note 7). Add 180 μl of ALT buffer and 20 μl of proteinase K (both from QIAamp DNA micro kit). Add 10 μl of AAV2-IL4 at the titer of 1.5 × 1013/ml followed by 180 μl of ALT buffer and 20 μl of proteinase K. Incubate at 56° C for 2 hours.
Perform all subsequent steps of DNA isolation and purification in accordance to QIAamp DNA micro kit instructions. In a final step, elute DNA originated from each of 4 foil samples and a control sample of a known number of AAV2 particles in 50 μl of ultrapure water.
Serially dilute DNA eluted from control AAV2-IL4 lysate 10-fold to create calibration curve.
Run an RT-PCR reaction with viral DNA eluted from the foil specimens and calibration curve samples using Sybr Green chemistry and the following primers specific to rat IL-4 gene sequence: Fwd: 5′-ACA GGA GAA GGG ACG CCA T-3′ and Rvs: 5′-GAA-GCC-CTA-CAG-ACG-AGC-TCA-3′. Analyze viral DNA amplification data with 7500 Software (v2.3) to determine absolute number of AAV2 particles associated with the surface and immobilization density of AAV2 vector.
3.6. Cell transduction with mesh disk-immobilized AAV2-eGFP particles (Fig. 5)
Fig. 5.
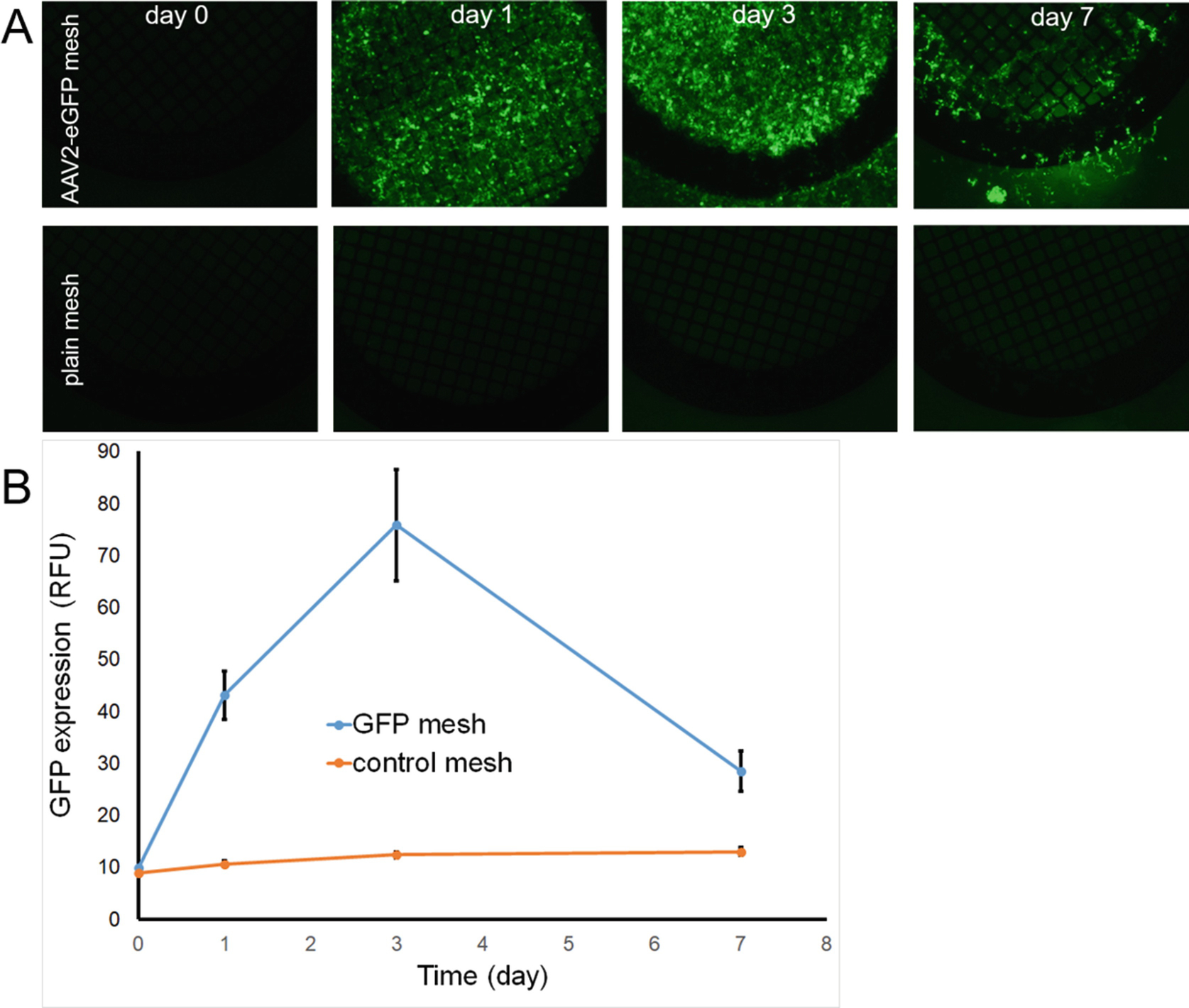
Time course of eGFP expression in HEK293 cells after transduction with mesh disks formulated with AAV2-eGFP immobilized according to the PABT/PEI(PDT)/protein G/anti-AAV2 antibody tethering protocol. Serial fluorescence microscopy (FITC filter set; 40 × original magnification) of HEK293 cells transduced with AAV2-eGFP meshes (A, upper panel) versus control meshes (A, lower panel) at the time of mesh disk placement, and 1, 3 and 7 days post-transduction supplemented by fluorimetry of the respective wells (B).
Grow HEK-293 cells to 75–85% confluence in a 96-well plate.
Formulate mesh disks with the surface-immobilized AAV2 as per 3.1.1–3.1.15 using 2×1011 AAV2-eGFP particles in the step 3.1.14. Formulate additional mesh disks according to the steps 3.1.1–3.1.3 to serve as the plain mesh controls.
Place the mesh disks individually on top of the cell monolayer (Note 8).
Place the 96-well plate back in the cell culture incubator for 24 hours.
After 24 hours, assay the plate using fluorimetry with 485 nm excitation and 538 nm emission wavelengths.
Inspect the wells with mesh disks using a fluorescence microscope with an inverted optic using a FITC filter set at 40–100× magnification. Acquire digital images at the same setting (magnification, acquisition time, etc) for the wells treated with the control and AAV2-GFP functionalized-mesh disks.
Return the plate into the cell culture incubator. Repeat steps 3.5.1–3.5.6 at 2, 3 and 7 days after the placement of the disks into the wells.
3.7. Preparation and deployment of AAV2-eluting stents in the rat carotid model
Formulate gene delivery stents as described in 3.1.1–3.1.15. Use 5×1011 AAV2-Luc particles in the step 3.1.14.
Mount the stents onto angioplasty balloon catheters and manually crimp the stents onto balloons. Keep the mounted stents immersed in sterile PBS until use.
Anesthetize male Sprague-Dawley rats (380–430 g) with intraperitoneal administration of ketamine (100 mg/kg) and xylazine (5 mg/kg). When full anesthesia is achieved as judged by the lack of reaction to toe pinch, shave and aseptically prep the neck and upper chest of the animal and drape it with a sterile fabric.
Administer analgesic (meloxicam; 0.5 mg/kg), saline solution (10 ml/kg) and wide-spectrum antibiotic, cefazolin (20 mg/kg) through the subcutaneous or intramuscular injection.
Wipe the tail with 70% alcohol and catheterize the lateral tail vein with a 24G catheter. Administer heparin (200 IU/kg) through the catheter and flush with 0.2 ml of saline solution.
After verifying the depth of anesthesia with toe pinch, perform a midline incision through the skin and neck fascia. Use blunt and sharp dissection to isolate the left external carotid artery. Apply a permanent ligature onto the distal segment of the external carotid artery. Apply a temporary ligature to the internal carotid artery at the most proximal position adjacent to the carotid bifurcation.
Make a 2-mm arteriotomy incision in the mid segment of the left external carotid artery located between two ligatures.
Using fine forceps to elevate the proximal lip of arteriotomy, insert the tip of a 2-French Fogarty catheter into the external carotid artery and advance it to the proximal segment of the common carotid artery.
Inflate the balloon of the Fogarty catheter with 50 μl of saline and pass the catheter 3 times from the aortic arch to the carotid bifurcation in order to denude the endothelium and inflict mechanical trauma to the media.
Slide a 3–4 cm segment of PET (polyethylene terephthalate) tubing (1.06 mm OD, 0.91 mm ID) over the Fogarty catheter and into the common carotid artery. Withdraw the deflated Fogarty catheter.
Insert the angioplasty catheter with the balloon-mounted stent hrough the PET tubing and advance it into the mid-section of the common carotid artery. Avoid rubbing the stent against the tube or vessel wall.
Deploy the stent at 10–14 atm for 30 sec using an Inflation Device. Deflate the balloon and withdraw the angioplasty catheter.
Ligate the external carotid artery immediately proximal to the arteriotomy site, release the temporary ligature on the internal carotid artery and check for resumption of blood flow through the common carotid artery.
Wash the surgical field with sterile saline solution and dab with a sterile gauze.
Repair the operative wound in layers with running 4.0 Vicryl suture and staple the skin. Recover the animal on a warming pad until recumbent and return to the cage (Note 9).
3.8. Bioluminescence imaging of arterial gene expression (Fig. 6)
Fig. 6.
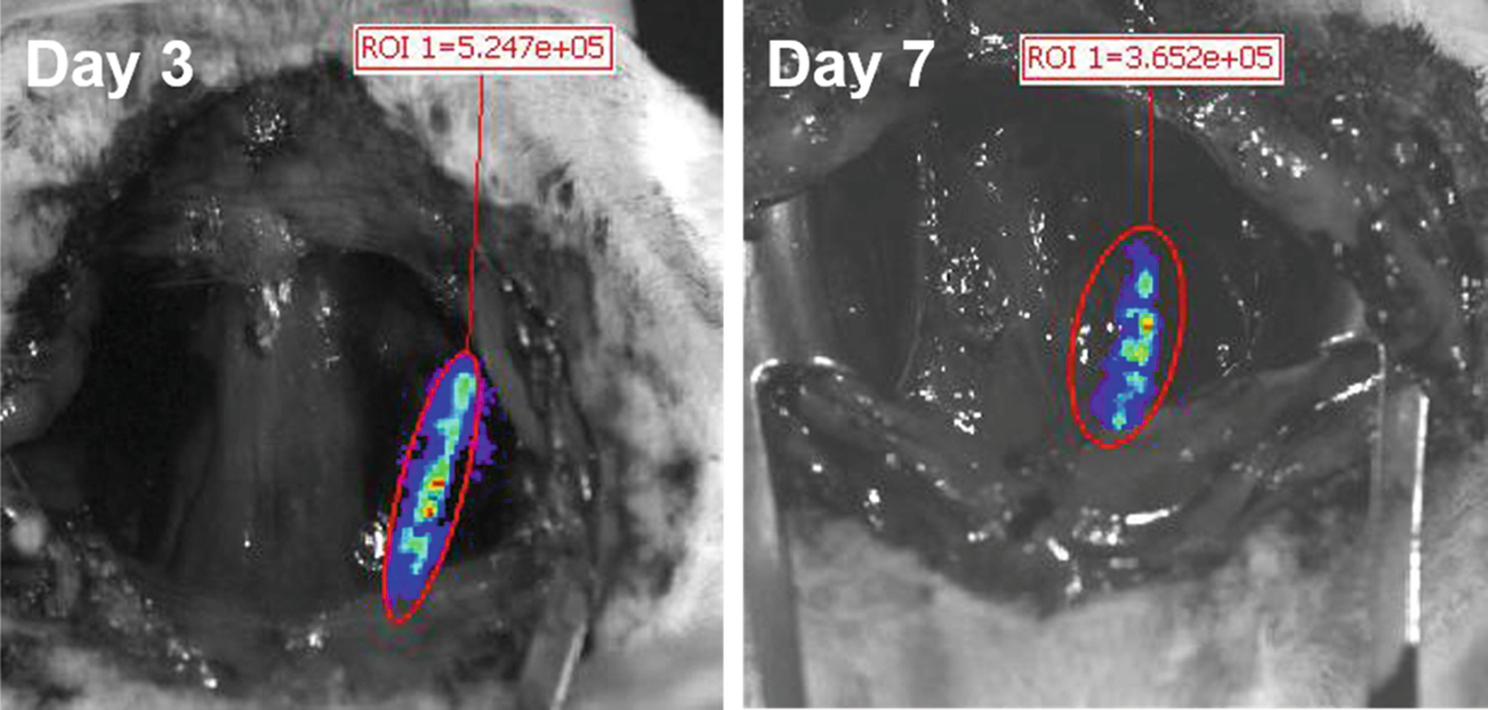
Serial bioluminescence imaging (days 3 and 7) of a rat carotid artery with a deployed endovascular stent formulated with AAV2-Luc vectors immobilized according to the PABT/PEI(PDT)/protein G/anti-AAV2 antibody tethering protocol.
At time points ranging between 2 days – 3 months after deployment of AAV2-eluting stent in the carotid artery, anesthesize the rat using isoflurane inhalation (2–4% isoflurane in oxygen; 2–3 liters/min flow rate).
Applying aseptic procedure, remove the surgical staples and reopen the operative wound. Using blunt dissection of the tissues, obtain the access to the stented segment of the left common carotid artery.
Prepare a mixture of 50 mg/ml Luciferin in PBS and 25% Pluronic F-127 in PBS (1:4 v/v) and store it on ice (Note 10).
Using a cooled pipette tip, apply 200 μl of a chilled Luciferin/Pluronic mixture directly to the exposed segment of the common carotid artery. Wait until gel solidifies.
Place the rat in the supine position inside the IVIS-Spectrum apparatus and adjust the animal’s position in relation to the field of view using the light markers.
Maintain isoflurane anesthesia with a nose cone while the animal inside the imaging chamber.
Engage the Living Image software (version 4.2 or higher) on the computer interfaced with the IVIS-Spectrum. From the dropdown menu choose the position “B” (6.6 cm camera to object distance) and the “medium” binnng factor in the acquisition control panel window. Type “2.5 cm” in the subject height box and 1 min in the acquisition time box.
Two minutes after application of the Luciferin/Pluronic gel, start image acquisition by clicking the “Aquire” button on the screen (Note 11).
After acquiring the image, remove the animal from the imager, wash off Pluronic gel with sterile saline and dry the wound with sterile gauze.
Close the wound in layers with 4.0 Vicryl suture.
Monitor the animal until completely recovered and return it to its cage.
Repeat imaging at later time points to study the kinetic of transgene expression in the artery treated with AAV2-Luc-eluting stent.
4. Notes
Non-clinical grade steel materials often contain surface impurities such as lubricants used in the manufacture of drawn metal sheets and tubes. Removal of these impurities is essential for the proper interaction of PABT with the surface.
Heating promotes surface oxidation, thus increasing PABT capacity to create coordination bonds with metal surface.
Avoid using scintillation bottles that have aluminum foil layered cap. PABT will react with the metal coating of the cap.
Even low concentrations of residual TCEP will interfere with PEI(PDT) conjugation to the surface thiols. Therefore, multiple washings are required. However, the washing step needs to be performed quickly to minimize oxidation of thiols with the ambient air.
Extensive washing is required since the remnants of unwashed protein G will compete with the surface-immobilized protein G for the antibody binding.
Extensive washing is required since the remnants of unwashed anti-AAV2 antibody will compete with the surface-immobilized anti-AAV2 antibody for the vector binding.
Work carefully. Avoid mechanically dislodging immobilized AAV2 vector from the surface. Use fine forceps to hold the foils.
Use fine forceps to transfer the mesh disks from the vial to the wells. Avoid bending the disks. Bending impedes close alignment of the disk to the cell monolayer, thus reducing cell transduction.
While no signs of pain or discomfort are typically exhibited by the post-operative animals beyond the first 12 hours after the procedure, consider extension of meloxicam therapy (0.5 mg/kg, SC daily for 72 hours), if necessary.
This formulation presents a viscous solution at 4°C and immediately turns to gel upon contact with tissue at 37°C.
Image acquisition time can vary from 1 to 6 min depending on the anticipated signal strength.
6. Acknowledgements
The authors are willing to acknowledge the following funding sources: 1) R01HL137762 (NIH-NHBLI) and 2) Erin Fund (The Children’s Hospital of Philadelphia). Ms. Susan Kerns provided an excellent administrative support for the project.
7. References
- 1.Shahryari A, Saghaeian Jazi M, Mohammadi S et al. Development and Clinical Translation of Approved Gene Therapy Products for Genetic Disorders. Frontiers in genetics 2019;10:868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wei W, Luo J. Thoughts on Chemistry, Manufacturing, and Control of Cell Therapy Products for Clinical Application. Human gene therapy 2019;30:119–126. [DOI] [PubMed] [Google Scholar]
- 3.Humbert O, Davis L, Maizels N. Targeted gene therapies: tools, applications, optimization. Critical reviews in biochemistry and molecular biology 2012;47:264–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waehler R, Russell SJ, Curiel DT. Engineering targeted viral vectors for gene therapy. Nature reviews Genetics 2007;8:573–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hacein-Bey-Abina S, Von Kalle C, Schmidt M et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science (New York, NY) 2003;302:415–419. [DOI] [PubMed] [Google Scholar]
- 6.Bengali Z, Rea JC, Gibly RF et al. Efficacy of immobilized polyplexes and lipoplexes for substrate-mediated gene delivery. Biotechnology and bioengineering 2009;102:1679–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bengali Z, Shea LD. Gene Delivery by Immobilization to Cell-Adhesive Substrates. MRS bulletin 2005;30:659–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jang JH, Bengali Z, Houchin TL et al. Surface adsorption of DNA to tissue engineering scaffolds for efficient gene delivery. Journal of biomedical materials research Part A 2006;77:50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pannier AK, Anderson BC, Shea LD. Substrate-mediated delivery from self-assembled monolayers: effect of surface ionization, hydrophilicity, and patterning. Acta biomaterialia 2005;1:511–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fishbein I, Alferiev I, Bakay M et al. Local delivery of gene vectors from bare-metal stents by use of a biodegradable synthetic complex inhibits in-stent restenosis in rat carotid arteries. Circulation 2008;117:2096–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fishbein I, Alferiev IS, Nyanguile O et al. Bisphosphonate-mediated gene vector delivery from the metal surfaces of stents. Proceedings of the National Academy of Sciences of the United States of America 2006;103:159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fishbein I, Forbes SP, Chorny M et al. Adenoviral vector tethering to metal surfaces via hydrolyzable cross-linkers for the modulation of vector release and transduction. Biomaterials 2013;34:6938–6948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fishbein I, Guerrero DT, Alferiev IS et al. Stent-based delivery of adeno-associated viral vectors with sustained vascular transduction and iNOS-mediated inhibition of in-stent restenosis. Gene therapy 2017;24:717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee EJ, Robinson TM, Tabor JJ et al. Reverse transduction can improve efficiency of AAV vectors in transduction-resistant cells. Biotechnology and bioengineering 2018;115:3042–3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McConnell KI, Gomez EJ, Suh J. The identity of the cell adhesive protein substrate affects the efficiency of adeno-associated virus reverse transduction. Acta biomaterialia 2012;8:4073–4079. [DOI] [PubMed] [Google Scholar]
- 16.Brunger JM, Huynh NP, Guenther CM et al. Scaffold-mediated lentiviral transduction for functional tissue engineering of cartilage. Proceedings of the National Academy of Sciences of the United States of America 2014;111:E798–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fishbein I, Forbes SP, Adamo RF et al. Vascular gene transfer from metallic stent surfaces using adenoviral vectors tethered through hydrolysable cross-linkers. Journal of visualized experiments : JoVE 2014:e51653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang D, Tai PWL, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nature reviews Drug discovery 2019;18:358–378. [DOI] [PMC free article] [PubMed] [Google Scholar]