

SUMMARY
The integration of HIV-1 DNA into the host genome results in single-strand gaps and 2-nt overhangs at the ends of viral DNA, which must be repaired by cellular enzymes. The cellular factors responsible for the DNA damage repair in HIV-1 DNA integration have not yet been well defined. We report here that HIV-1 infection potently activates the Fanconi anemia (FA) DNA repair pathway, and the FA effector proteins FANCI-D2 bind to the C-terminal domain of HIV-1 integrase. Knockout of FANCI blocks productive viral DNA integration and inhibits the replication of HIV-1. Finally, we show that the knockout of DNA polymerases or flap nuclease downstream of FANCI-D2 reduces the levels of integrated HIV-1 DNA, suggesting these enzymes may be responsible for the repair of DNA damages induced by viral DNA integration. These experiments reveal that HIV-1 exploits the FA pathway for the stable integration of viral DNA into host genome.
Graphical Abstract
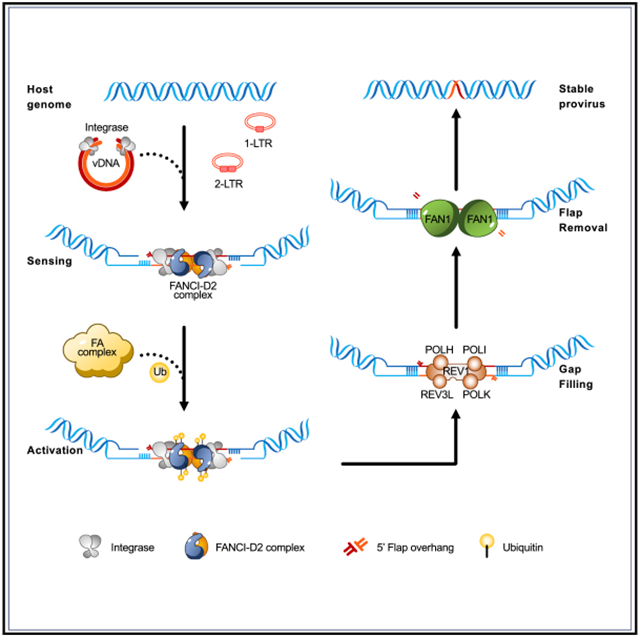
In brief
Fu et al. show that HIV-1 integrase interacts with FANCI-D2 complex, and HIV-1 DNA integration activates the Fanconi anemia (FA) pathway. They discover that the depletion of FA proteins and down-stream enzymes blocks viral DNA integration, suggesting that the FA pathway is exploited by HIV-1 to repair DNA damage induced by viral DNA integration.
INTRODUCTION
Mammalian cells are constantly challenged by DNA damage resulting from endogenous and exogenous sources. To maintain genome integrity, cells have developed multiple pathways to repair various types of DNA lesions (Chatterjee and Walker, 2017). The Fanconi anemia (FA) pathway repairs the DNA lesion of interstrand crosslinks (ICLs) that arise from exposure to ICL-inducing agents such as mitomycin C and aldehydes (Ceccaldi et al., 2016). The FA pathway is composed of at least 22 FA genes (FANCA–FANCW), and mutations of any one of these genes cause FA, a disorder characterized by hypersensitivity to DNA interstrand crosslinking agents, bone marrow failure, leukemia predisposition, and congenital abnormalities (Ceccaldi et al., 2016; Fang et al., 2020; Nalepa and Clapp, 2018). In the presence of ICLs, FANCM recognizes the stalled replication forks at the sites of ICLs and recruits the FA core complex, a ubiquitin E3 ligase composed of 10 proteins (FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, FAAP100, FAAP20, and FAAP24), to the DNA damage sites (Huang et al., 2010, 2013; Shakeel et al., 2019). The FA core complex subsequently catalyzes the monoubiquitination of two FA effector proteins, FANCD2 and FANCI (FANCI-D2) (Wang et al., 2021). The ubiquitylated FANCI-D2 is directed to the ICL region, where it orchestrates various steps to resolve the ICL damage (Ceccaldi et al., 2016). Specifically, the FANCI-D2 complex first recruits nucleases SLX4, FAN1, and endonucleases ERCC4 or MUS81 to coordinate the nucleolytic incisions, which unhook the ICL from one of the parental DNA strands. Next, the translesion synthesis polymerases (REV1 or other DNA polymerases) bypass the crosslinked nucleotide to extend the nascent DNA and restore one intact DNA duplex. Finally, the DNA double-strand break (DSB) in the other DNA duplex is repaired by homologous recombination for the completion of ICL repair. In addition to the repair of ICLs, the FA pathway is involved in other processes, including telomere length maintenance, cytokinesis, and herpesvirus DNA replication, to promote genome stability (Karttunen et al., 2014; Rodriguez and D’Andrea, 2017).
HIV-1 infection begins with the receptor-mediated fusion of cell and viral membranes with subsequent release of the viral core into the cytoplasm, followed by the reverse transcription of the RNA genome to form a linear double-stranded DNA (Suzuki and Craigie, 2007). A portion of this linear DNA gives rise to two circular DNA forms: homologous recombination between long terminal repeat (LTR) sequences at the ends of the linear DNA gives rise to 1-LTR circles, and ligation of the termini of the linear DNA by the non-homologous end-joining (NHEJ) DNA repair pathway gives rise to 2-LTR circles (Li et al., 2001). In a crucial step in the HIV-1 life cycle, the linear viral DNA molecules are inserted into the host genome by the viral integrase (IN) to form the integrated provirus. The integrated HIV-1 DNA is replicated along with cellular DNA during cycles of cell division, persists in the host cell, and is actively transcribed in permissive cells to give rise to progeny virus (Nakajima et al., 2001; Sakai et al., 1993).
The process of HIV-1 DNA integration consists of sequential enzymatic reactions catalyzed by viral IN and cellular DNA repair enzymes (Lesbats et al., 2016). The reaction catalyzed by IN occurs in two steps, referred to as 3′-processing and strand transfer. During 3′-processing, HIV-1 IN binds both ends of viral DNA and removes a dinucleotide (5′-GT-3′) at the 3′ end of each viral LTR DNA via hydrolysis of a phosphodiester bond, which liberates the 3′-hydroxyl groups of highly conserved 5′-CA-3′ dinucleotides (Chen et al., 1999; Hare et al., 2010). Following the nuclear entry, the HIV-1 IN cofactor LEDGF/p75 tethers viral DNA to the targeted host genome (Ciuffi et al., 2005; Ferris et al., 2010; Llano et al., 2006), and IN catalyzes the next reaction step, strand transfer. In strand transfer, the HIV-1 IN catalyzes a nucleophilic attack by the processed 3′-hydroxyls to join the 3′ viral DNA ends to 5′-O-phosphates of the target host DNA. Because the 5′ ends of viral DNA are not joined to the host DNA, the strand transfer results in hemi-integration intermediates, leaving a pair of single-strand gaps and 2-nt overhangs (5′ flap) at the ends of viral DNA. Formation of the stable integrated proviral DNA (fully integrated provirus) requires the repair of single-strand gaps by DNA polymerases, the removal of the 2-nt overhangs by 5′-flap nucleases, and finally a DNA ligase to join the 5′ viral DNA ends to the host DNA (Lesbats et al., 2016). Although enzymes capable of performing these repair steps in vitro have been identified, the enzymes functioning in vivo remain unknown (Faust and Triller, 2002; Rumbaugh et al., 1998; Yoder and Bushman, 2000).
Here, we discover that the FANCI-D2 complex interacts with HIV-1 IN and is required for the formation of stable integrated proviral DNA. Moreover, we show that the integration of HIV-1 DNA potently activates the FA pathway by stimulating the monoubiquitination of FANCI-D2. Finally, we identify the DNA polymerases (REV1, POLH, POLI, POLK, and REV3L) and the flap nuclease (FAN1) that acts downstream of the FANCI-D2 complex and are required for the generation of stable provirus. Our study has established the FA pathway as critical cellular machinery activated and used by HIV-1 to repair DNA damage for the formation of stable integrated proviral DNA in the HIV-1 life cycle.
RESULTS
HIV-1 IN interacts with FANCD2 and FANCI
The integration of HIV-1 DNA into the host genome is catalyzed by the viral enzyme IN (Figure 1A). In a search for host factors involved in HIV-1 DNA integration, we analyzed a mass spectrometry database of human proteins interacting with the HIV-1 IN (Jager et al., 2011) and discovered that peptides derived from the FANCI protein were identified in samples co-purifying with IN, suggesting that FANCI is an HIV-1 IN-interacting protein. Co-immunoprecipitation (coIP) experiments were used to validate the interaction of HIV-1 IN with FANCI. FLAG-tagged HIV-1 IN was stably expressed in 293A cells, and cell lysates were treated with benzonase for the removal of DNA and RNA. Immunoprecipitation of HIV-1 IN resulted in efficient coIP of endogenous FANCI, and also FANCD2, the other subunit of the FANCI-D2 complex (Figure 1B). The results indicated that FANCI did interact with HIV-1 IN. We examined the intracellular localization of HIV-1 IN, FANCD2, and FANCI in 293A cells stably expressing FLAG-tagged HIV-1 IN. HIV-1 IN, FANCD2, and FANCI were greatly localized in the nucleus, and both FANCD2 and FANCI colocalized with HIV-1 IN (Figures S1A and S1B). However, FANCI is not a binding factor for all retroviral INs, as the IN of Moloney leukemia virus (MLV) did not interact with FANCI (Figure S1C). Of note, cells did not express MLV IN at levels as high as HIV-1 IN because the CDS (coding sequence) of HIV-1 IN was codon optimized, but the MLV IN CDS was not.
Figure 1. HIV-1 IN interacts with FANCI and FANCD2.
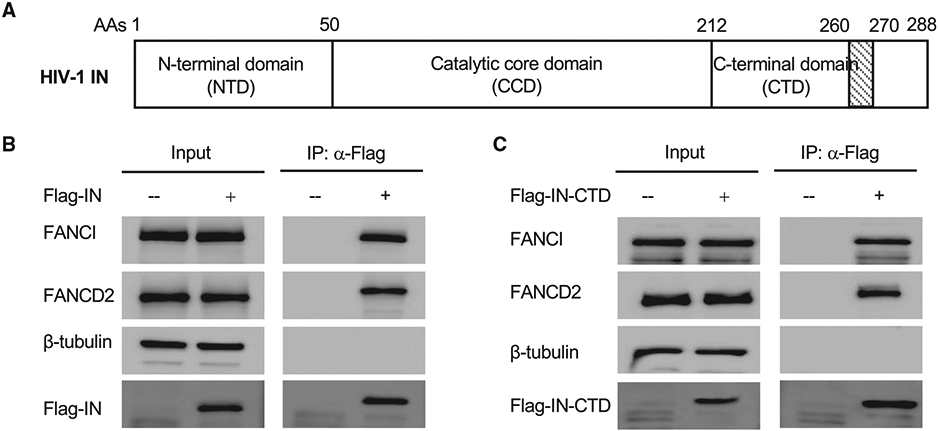
(A) Schematic domain organization of HIV-1 IN.
(B) 293A cells were stably transduced with empty lentiviral vector (−) or lentiviral vector expressing FLAG-tagged full-length HIV-1 IN (+). Cell lysates were treated with benzonase. FLAG-tagged IN was immunoprecipitated by anti-FLAG antibody. The co-immunoprecipitated FANCI and FANCD2 were probed by western blot.
(C) 293A cells were stably transduced with empty lentiviral vector (−) or lentiviral vector expressing flag-tagged CTD of HIV-1 IN (+). Cell lysates were treated with benzonase. FLAG-tagged IN CTD was immunoprecipitated by anti-FLAG antibody. The co-immunoprecipitated FANCI and FANCD2 were probed by western blot.
See also Figure S1.
HIV-1 IN consists of three independent domains: an N-terminal domain (NTD), the catalytic core domain (CCD), and a C-terminal domain (CTD) (Figure 1A). The NTD enhances the tetramerization in the context of full-length IN and is required for integration activity (Hare et al., 2009). The CCD is responsible for the catalytic activities of the IN enzyme. The CTD is involved in DNA and RNA binding (Jenkins et al., 1997; Kessl et al., 2016). To localize the domain of IN required for FANCI binding, we tested a series of deletion mutants of IN for coIP with FANCI, compared with the empty vector control. Deletion of the CTD abolished the interaction between IN and FANCI, while IN with deletion of the NTD could still interact with FANCI (Figure S1C). The CCD alone did not interact with FANCI (Figure S1C). Immunoprecipitation of HIV-1 IN CTD resulted in efficient co-immunoprecipitation of endogenous FANCI and FANCD2 (Figure 1C). Deletion of the C-terminal 18 amino acids (271–288 aas) of IN did not affect its interaction with FANCI, but further deletion of 261–270 aas, which disrupted the structure of CTD, abolished the interaction of IN with FANCI (Figure S1D). All of these results indicated that HIV-1 IN uses its CTD to interact with FANCI and FANCD2.
FANCD2 and FANCI are required for productive HIV-1 DNA integration and expression of viral DNA
The FANCI-D2 complex plays a central role in the FA DNA repair pathway. To test the importance of FANCD2 and FANCI in HIV-1 infection, we generated FANCD2 knockout (KO) (Figure 2A) and FANCI knockout (Figure 2F) lines of 293A cells by CRISPR-Cas9 methods, and infected parental (wild type [WT]) and FANCD2- or FANCI-deficient (KO) cells with vesicular stomatitis virus G protein (VSV-G) pseudotyped HIV-Luc (luciferase) reporter virus. At 12, 24, and 48 h post-infection, cells were harvested and assayed for Luc expression. The KO of FANCD2 or FANCI decreased the expression of the Luc reporter gene by ~5-fold, while re-expression of FANCD2 or FANCI in KO cells restored the expression of luciferase (Figures 2B and 2G). The results suggest that HIV-1 infection is inhibited in FANCD2 or FANCI KO cells. The functions of FANCD2 and FANCI in HIV-1 infection were independent of DNA replication. KO of FANCD2 or FANCI inhibited HIV-1 infection in non-dividing cells in which the cell cycle was arrested by aphidicolin (Figure S2A).
Figure 2. FANCD2 and FANCI are required for HIV-1 DNA integration.
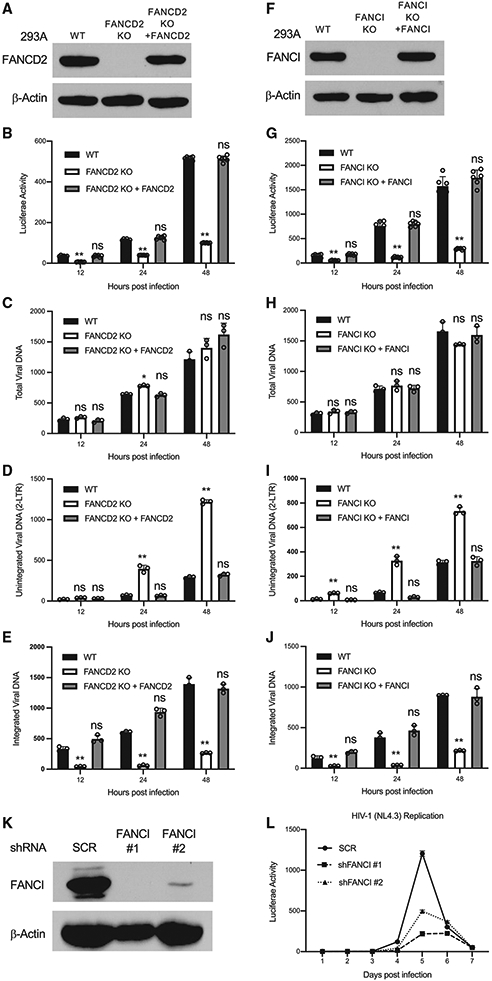
(A–J) FANCD2 (A–E) and FANCI (F–J) are required for HIV-1 DNA integration. Indicated cells were infected with VSV-G pseudotyped HIV-Luc virus. The expression of indicated proteins was determined by western blot (A and F). Luciferase activities (B and C), total viral DNA (C and H), unintegrated viral DNA (D and I), and integrated viral DNA (E and J) were measured at indicated time points post-infection. Data are represented as means ± SDs from 3 independent experiments (n = 3). ns p > 0.05; *p < 0.05; **p < 0.01.
(K and L) Knockdown of FANCI inhibits the replication of HIV-1. MT-4 cells stably expressing control shRNA (SCR) or shRNAs targeting FANCI were infected with HIV-1(NL4.3) at 1 ng CA/106 cells. The expression of FANCI was determined by western blot (K). Viral spreading was monitored by assay for the infectivity of supernatants in TZM-bl cells engineered with the HIV-1 Tat-responsive luciferase reporter gene (L). Data presented are means ± SEMs from 2 technical replicates and are representative of 2 independent experiments.
See also Figure S2.
To determine which step of HIV-1 infection is inhibited in FANCD2- and FANCI-KO cells, we monitored the levels of total viral DNA, unintegrated viral DNA, and integrated viral DNA at different time points post-virus infection by PCR. The KO of FANCD2 or FANCI had no detectable effect on the levels of total viral DNA (Figures 2C and 2H), suggesting that steps of HIV-1 infection before reverse transcription are not affected in FANCD2- or FANCI-KO cells. However, KO of FANCD2 or FANCI markedly increased the levels of unintegrated viral DNA (Figures 2D and 2I) and decreased the levels of integrated viral DNA (Figures 2E and 2J). Of note, the method we used to assay for integrated viral DNA, Alu-Gag PCR (Liszewski et al., 2009), scores hemi-integration intermediates or fully integrated proviruses, whether or not the single-strand gaps at the viral DNA ends are repaired. The decrease in integrated viral DNA is not explained by a selective outgrowth of uninfected cells during cell culture, because the DNA harvest and PCR readouts are performed as early as 12 h post-infection, before even a single round of cell division; nor is it explained by selective loss of infected cells, because there is no indication that cell death was observed under the microscope in this time frame. The results suggest that the FANCI-D2 complex plays a significant role in the integration of HIV-1 DNA into the host genome. However, it seems that FANCD2 and FANCI are not involved in the integration of viral DNA from all retroviruses, as KO of FANCI had no effect on the infection of an MLV reporter virus (Figure S2B), which is consistent with the observation that FANCI did not interact with MLV IN (Figure S1C).
To further confirm the importance of FANCI in HIV-1 integration, we assessed the impact of FANCI on the kinetics of replication by WT HIV-1. We generated MT-4 T cell lines stably expressing a control short hairpin RNA (shRNA) (SCR) or shRNAs targeting FANCI (Figure 2K) and infected these lines with replication-competent HIV-1 (strain NL4.3), and the production of progeny virus was monitored at different time points post-infection. Depletion of FANCI by shRNA markedly inhibited the replication of HIV-1 in MT-4 cells (Figure 2L).
HIV-1 infection activates the FA pathway
Upon sensing of a DNA damage, the cellular FA pathway is activated to repair the DNA damage, and the activation of the FA pathway can be monitored by the monoubiquitination of FANCD2 and FANCI (Sims et al., 2007). To investigate the impact of HIV-1 infection on the cellular FA pathway, we infected 293A cells with increasing amounts of VSV-G pseudotyped HIV-mCherry reporter virus and monitored the monoubiquitination of FANCD2 (Ub-FANCD2). Levels of Ub-FANCD2 were low in mock infected cells, and increased levels were easily detected in cells infected with HIV-1 at MOI 5 and 25 (Figure 3A). Another hallmark of FA pathway activation is the accumulation of FA proteins and other DNA damage response proteins into discrete nuclear foci (Smogorzewska et al., 2007). We observed that HIV-1 infection enhanced the formation of FANCD2 foci, γH2AX foci, and RPA1 foci in infected cells (Figures S3A and S3B). γH2AX associates with retroviral DNA at integration sites (Daniel et al., 2004b). The FANCD2 foci colocalized with γH2AX foci (Figure S3C), suggesting that FANCD2 was recruited to the integration sites to coordinate post-integration DNA repair. Importantly, HIV-1 DNA integration was required to activate the FA pathway, since the treatment of cells with the IN inhibitor raltegravir, which blocks the insertion of HIV-1 DNA into the host genome, abolished the monoubiquitination of FANCD2 induced by HIV-1 infection (Figure 3A), but had no effect on the monoubiquitination of FANCD2 induced by mitomycin C (MMC) treatment (Figure S3D). It has been reported that ataxia-telangiectasia mutated and Rad3-related (ATR) kinase-mediated phosphorylation promotes the monoubiquitination of FANCD2 (Tan et al., 2020). However, we observed that ATR was not required for FA activation induced by HIV-1 infection, because treatment of cells with the ATR inhibitor VE-821 had no effect on the monoubiquitination of FANCD2 in HIV-1-infected cells (Figure S3E). This result is consistent with a previous study showing that ATR is dispensable for HIV-1 integration (Ariumi et al., 2005).
Figure 3. HIV-1 infection activates the FA pathway.
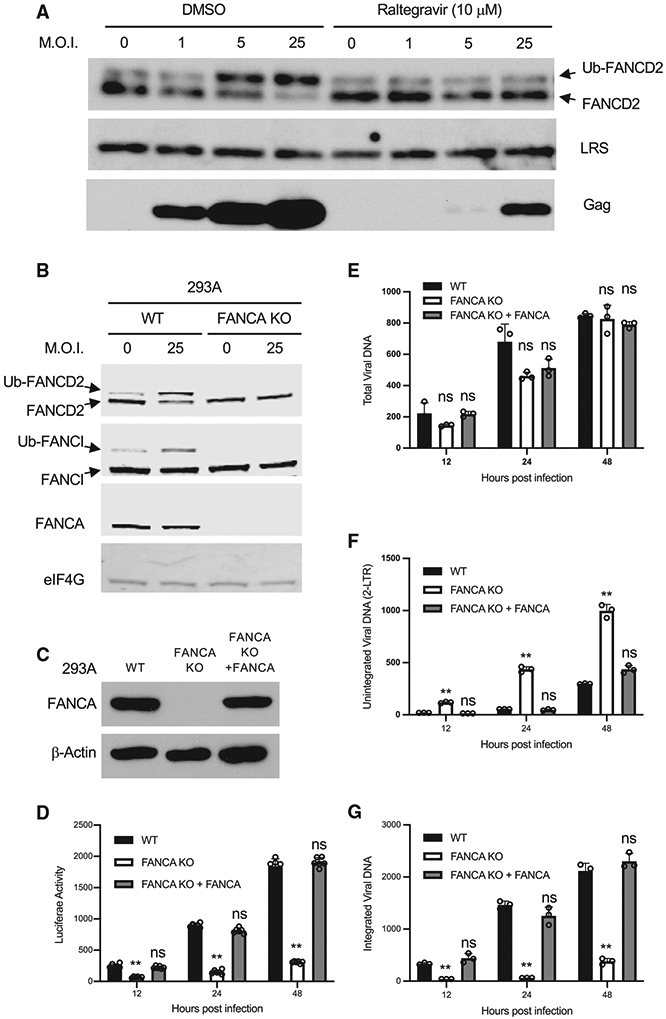
(A) Cells were infected with VSV-G pseudotyped HIV-mCherry reporter virus at indicated MOI and treated with DMSO or IN inhibitor raltegravir. A total of 24 h post-infection, FANCD2, LRS, and Gag in the infected cells were probed by western blot.
(B) The activation of the FA pathway by HIV-1 is dependent on FANCA. Parent 293A cells (WT) and FANCA KO cell lines were infected with VSV-G pseudotyped HIV-mCherry virus at indicated MOI, and the expression of indicated proteins were determined by western blot.
(C–G) Parental 293A cells (wild-type [WT]), the FANCA KO cell line (KO), and FANCA KO 293A cells reconstituted with WT FANCA (KO + FANCA) were infected with VSV-G pseudotyped HIV-Luc virus. The expression of indicated proteins were determined by western blot (C). Luciferase activities (D), total viral DNA (E), unintegrated viral DNA (F), and integrated viral DNA (G) were measured at indicated times post-infection. Data are represented as means ± SDs from 3 independent experiments (n = 3). nsp > 0.05; *p < 0.05; **p < 0.01.
See also Figure S3.
In the FA pathway, the FA core complex, a ubiquitin E3 ligase composed of 10 FA proteins, mediates the monoubiquitination of FANCD2 and FANCI and activates the FA pathway (Wang et al., 2021). FANCA is a subunit of the FA core complex and is required for the ubiquitin E3 ligase activity. To test the function of the FA core complex in HIV-1-induced activation of FA pathway, we generated a FANCA knockout 293A cell line and infected these cells with VSV-G pseudotyped HIV-mCherry reporter virus at an MOI of 25. HIV-1 infection of the parental cells (WT) induced the monoubiquitination of both FANCD2 and FANCI, but infection of the FANCA-KO cells did not (Figure 3B). We also assessed the activation of the FA pathway by HIV-1 in a patient-derived, FANCA-deficient cell line. Ub-FANCD2 or Ub-FANCI could not be detected after infection of FANCA-deficient cells, while complementation of FANCA-deficient cells with WT FANCA restored the monoubiquitination of FANCD2 and FANCI upon HIV-1 infection (Figure S3F). All of these results indicate that the FA core complex is the ubiquitin E3 ligase that mediates the monoubiquitination of FANCD2 and FANCI induced by HIV-1 DNA integration.
To investigate the function of FANCA in HIV-1 DNA integration, we infected FANCA-KO cells with VSV-G pseudotyped HIV-Luc reporter virus and monitored viral DNA levels by qPCR. The KO of FANCA had no effect on the production of total viral DNA, but the levels of integrated viral DNA and the expression of Luc reporter decreased, with an accumulation of unintegrated HIV-1 DNA in FANCA-KO cells (Figures 3C–3G). Re-expression of WT FANCA in FANCA-KO cells restored the expression of Luc and the levels of integrated HIV-1 DNA (Figures 3D and 3G). Consistent with the notion that the FA pathway is not involved in the viral DNA integration of MLV, knockout of FANCA had no effect on the MLV infection (Figure S3G).
Host REV1 protein is required for the integration of HIV-1 DNA
HIV-1 DNA integration results in single-strand gaps and 2-nt overhangs (5′ flap) at the ends of the viral DNA, which are repaired by cellular enzymes (Lesbats et al., 2016). The activation of FA pathway by HIV-1 and the interaction between HIV-1 IN and FANCI-D2 suggest that the single-strand gaps and 5′ flap overhangs that result from HIV-1 DNA integration may be repaired by cellular enzymes downstream of FANCI-D2 in the FA pathway. The single-strand gaps generated during HIV-1 DNA integration need to be repaired by DNA polymerases. In the FA pathway, the activated FANCI-D2 complex recruits downstream FANC proteins and cellular enzymes to repair DNA damage (Rodriguez and D’Andrea, 2017). REV1 is a scaffold protein that recruits DNA polymerases involved in translesion synthesis (TLS) of damaged DNA (Ceccaldi et al., 2016). REV1 is regulated by the FA core complex (Kim et al., 2012) and associates with Ub-FANCD2 via the C-terminal ubiquitin-binding motif (UBM) (Yang et al., 2015).
To test whether REV1 is required for the integration of HIV-1 DNA, we generated REV1-KO lines of 293A cells (Figure 4A). The KO efficiency of REV1 was confirmed by western blot (Figure 4A). Parental (WT) and REV1-KO cells were infected with VSV-G pseudotyped HIV-Luc reporter virus as before, and infection was monitored by assessing viral DNAs by qPCR and by determining Luc activity. KO of REV1 had no effect on the production of total viral DNA, but the levels of integrated viral DNA and the expression of Luc reporter decreased, with an accumulation of unintegrated HIV-1 DNA in REV1-KO cells (Figures 4B-4E). The association of REV1 with FANCD2 is indispensable for the function of REV1, since re-expression of WT REV1, but not a mutant REV1 lacking the UBM (ΔCTD), restored the expression of Luc and the integration of viral DNA that were lost in the REV1-KO line (Figures 4A, 4B, and 4E). KO of REV1 in FANCD2 KO cells did not further inhibit HIV-1 infection (Figures S4A and S4B), suggesting that REV1 and FANCD2 worked in the same pathway in regulating HIV-1 DNA integration. Consistent with the notion that the FA pathway is not involved in the viral DNA integration of MLV, KO of REV1 had no effect on the MLV infection (Figure S4C).
Figure 4. REV1 is required for HIV-1 DNA integration.
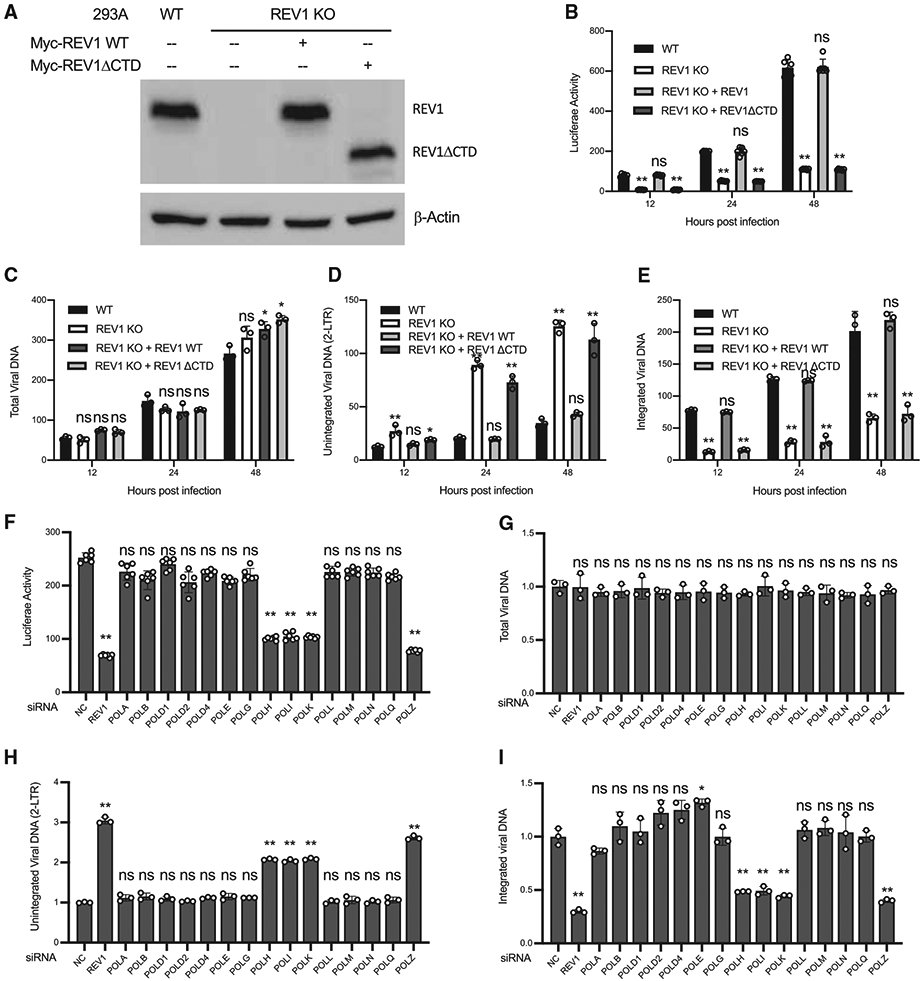
(A–E) Parental 293A cells (WT), the REV1 KO 293A cells (KO), and REV1 KO 293A cells reconstituted with indicated variants of REV1 were infected with VSV-G pseudotyped HIV-Luc virus. The expression of indicated proteins was determined by western blot (A). Luciferase activities (B), total viral DNA (C), unintegrated viral DNA (D), and integrated viral DNA (E) were measured at indicated times post-infection.
(F–I) REV1 and DNA polymerases POLH, POLI, POLK, and POLZ are required for HIV-1 DNA integration. 293A cells were transfected with indicated siRNA and then infected with VSV-G pseudotyped HIV-Luc virus. Luciferase activities (F), total viral DNA (G), unintegrated viral DNA (H), and integrated viral DNA (I) were measured at indicated times post-infection. NC, non-targeting control siRNA. Data are represented as means ± SDs from 3 independent experiments (n = 3). ns p > 0.05; *p < 0.05; **p < 0.01.
See also Figure S4.
We next sought to determine which DNA polymerases are responsible for the repair of the single-strand gaps in HIV-1 DNA integration. We used small interfering RNAs (siRNAs) to deplete 293A cells of each of 15 individual DNA polymerases followed by infection with VSV-G pseudotyped HIV-Luc reporter virus (Figures 4F and S4D). We found that knockdown of POLH, POLI, POLK, or REV3L markedly decreased the expression of Luc reporter (Figure 4F). Knockdown of any of these DNA polymerases had no effect on the production of viral DNA (Figure 4G). However, in the cells with depletion of any of the POLH, POLI, POLK, or REV3L polymerases, but not other DNA polymerases, unintegrated HIV-1 DNA accumulated (Figure 4H) and the integration of HIV-1 DNA was inhibited (Figure 4I). Notably, POLH, POLI, POLK, and REV3L, but not other DNA polymerases, have been shown to interact with REV1 (Murakumo et al., 2001; Ohashi et al., 2004; Sharma et al., 2012). Thus, the data suggest that Rev1 recruits DNA polymerases POLH, POLI, POLK, and REV3L to repair the single-strand gaps in HIV-1 DNA integration.
Flap nuclease FAN1 is required for the integration of HIV-1 DNA
Following the repair of the single-strand gap, removal of the 2-nt flap sequence is the next step in the formation of stable integrated proviral DNA. FAN1 (FANCD2- and FANCI-associated nuclease 1) is a nuclease downstream of the FANCI-D2 complex in the FA pathway (Kratz et al., 2010; Liu et al., 2010; MacKay et al., 2010; Smogorzewska et al., 2010). FAN1 interacts with monoubiquitinated FANCD2 and FANCI via its ubiquitin-binding zinc finger (UBZ) domain, and the nuclease (NUC) domain of FAN1 is responsible for the 5′ flap nuclease activity (Figure 5A). To analyze whether FAN1 plays a role in the integration of HIV-1 DNA, we generated FAN1 KO lines of 293A cells (Figure 5B). Parental (WT) and FAN1-KO cells were infected with VSV-G pseudotyped HIV-Luc reporter virus as before, and infection was monitored by assessing viral DNAs by qPCR and by determining Luc activity. As seen with the KO of FANCD2 and FANCI, the KO of FAN1 had no effect on the production of total viral DNA, but the levels of integrated viral DNA and the expression of Luc reporter decreased, with an accumulation of unintegrated HIV-1 DNA in FAN1-KO cells (Figures 5C-5F). KO of FAN1 in FANCD2-KO cells did not further inhibit HIV-1 infection (Figures S5A and S5B), suggesting that FAN1 and FANCD2 worked in the same pathway in regulating HIV-1 DNA integration. Consistent with the notion that the FA pathway is not involved in the viral DNA integration of MLV, KO of FAN1 had no effect on the MLV infection (Figure S5C).
Figure 5. Flap nuclease FAN1 is required for HIV-1 DNA integration.
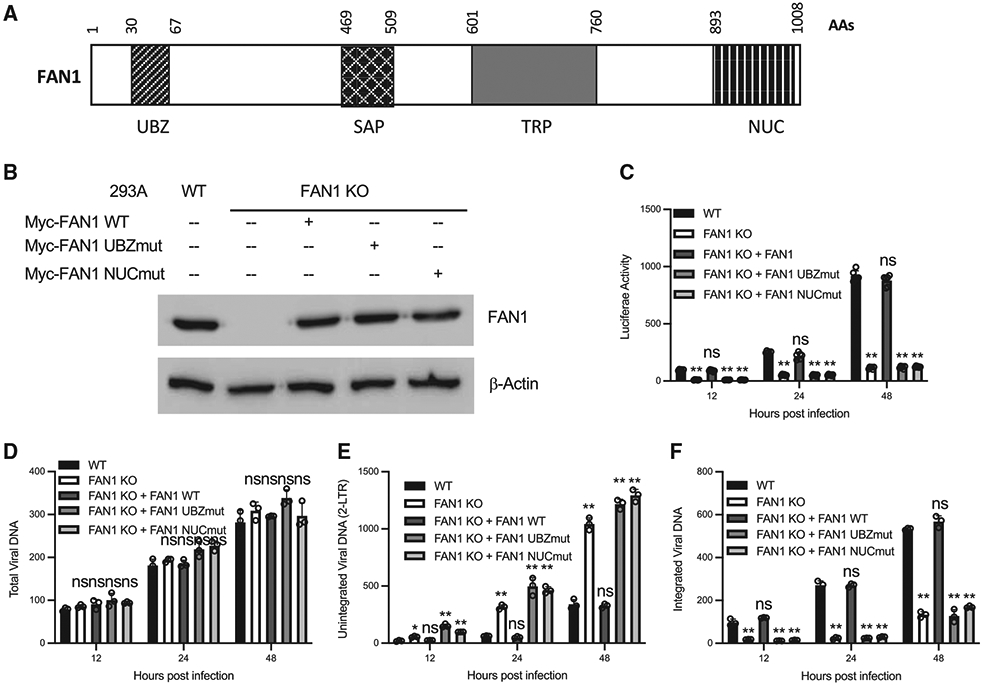
(A) Schematic domain organization of FAN1. UBZ, ubiquitin-binding zinc finger; SAP, SAF-A/B, Acinus, and PIAS; TPR, tetratricopeptide repeat; NUC, nuclease domain.
(B–F) Parental 293A cells (WT), the FAN1-KO 293A cells (KO), and the FAN1-KO 293A cells reconstituted with indicated variants of FAN1 were infected with VSV-G pseudotyped HIV-Luc virus. The expression of indicated proteins were determined by western blot (B). Luciferase activities (C), total viral DNA (D), unintegrated viral DNA (E), and integrated viral DNA (F) were measured at indicated times post-infection. Data are represented as means ± SDs from 3 independent experiments (n = 3). ns p > 0.05; *p < 0.05; **p < 0.01.
See also Figure S5.
To probe the domains of FAN1 required for HIV-1 DNA integration, we re-expressed WT FAN1 or mutants deficient in either the UBZ domain (UBZmut) or the NUC domain (NUCmut) in the FAN1-KO cell line. Re-expression of WT FAN1, but not the mutant forms, restored the expression of Luc and the integration of viral DNA that were lost in the FAN1-KO line (Figures 5C and 5F). Thus, both the UBZ domain and the NUC domain of FAN1 were required for the integration of HIV-1 DNA. All of these results suggested that the FANCD2 and FANCI complex attracted FAN1 to remove the 5′ flap overhang sequence generated in viral DNA integration.
The DNA repair nuclease SNM1A (Buzon et al., 2018) and other nucleases (SLX1 and XPF-ERCC1) associated with scaffold protein SLX4 (Kim et al., 2011) are also involved in the FA pathway to repair the DNA ICL. However, these nucleases are not required for the integration of HIV-1 DNA, since KO of SNM1A (Figures S5D and S5E) or SLX4 (Figures S5F and S5G) had no effect on HIV-1 infection. We also tested other DNA repair factors, including Ku70 and FEN1. KO of Ku70 or FEN1 inhibited the infection of HIV-1 (Figures S5H-S5J). The relationship between different DNA repair factors (Ku70, FEN1, FA proteins) needs to be explored in future studies.
DISCUSSION
The integration of viral DNA into the host genome, a defining step for the replication of all retroviruses, results in single-strand gaps and 2-nt overhangs at the 5′ ends of the viral DNA, which must be repaired by cellular DNA repair pathway to form stable integrated proviral DNA. Here, we show that HIV-1 infection activates the FA pathway and the activated FA pathway is required for the stable formation of integrated HIV-1 DNA. However, the FA pathway is not required for the integration of viral DNA from another retrovirus, MLV, since KO of key FA proteins (FANCI and FANCA) had no effect on MLV infection (Figures S2A and S3G). The FA effector protein FANCI interacts with the IN of HIV-1, but not MLV (Figure S1C), suggesting that the specificity of the HIV-1 IN for FANCI binding is responsible for the selective usage of FA pathway in HIV-1 integration. The interactions of FANCI with INs from other retroviruses and the breadth of the usage of FA pathway in retroviral DNA integration need to be explored in the future. Both retroviral (MLV) and lentiviral (HIV-1) vectors are used in gene therapy for FA (Rio et al., 2018). Because FA deficiency did not affect the infection of MLV, the retroviral vector may have advantages of gene transduction efficiency in FA patients.
In the presence of ICLs, stalled replication forks at ICLs are sensed by FANCM to activate the FA pathway (Huang et al., 2010). How the FA pathway senses DNA damage in HIV-1 DNA integration remains unclear. One possible mechanism is that DNA replication forks, which collapse at the single-strand gaps flanking viral DNA, activate the FA pathway to repair DNA damage in HIV-1 DNA integration. Another possibility is that the flap DNA structure at the ends of viral DNA are recognized directly by FANCM, since FANCM preferentially binds to ssDNA and flap DNA over double-stranded DNA (dsDNA) (Ciccia et al., 2007).
We used the Alu-Gag PCR method (Liszewski et al., 2009) to assess the levels of integrated viral DNA. Using this assay, we detected a significant decrease in the levels of integrated viral DNA after KO of many components of the FA pathway. One limitation of this method is that it does not discriminate between the hemi-integration intermediates (with gaps unrepaired) and the stably integrated viral DNAs (with gaps repaired), since PCR amplification can occur across the joint in either case. The decreased levels of integrated viral DNA in FA-deficient cells could result from several distinct mechanisms.
One possibility is that the step of first strand transfer is affected. Although we think it is unlikely that the FA pathway acts directly on the process of strand transfer, the activated FA pathway could increase the available levels of integration-competent linear viral DNA by blocking the formation of circularized viral DNA. Upon the entry of HIV-1 DNA into the nucleus, the viral DNA can either be circularized by the NHEJ DNA repair pathway to form unintegrated 2-LTR circular DNA or it can be inserted into the host genome by IN to form the stably integrated proviral DNA. The activated FA pathway is known to suppress the NHEJ (Adamo et al., 2010; Pace et al., 2010). Thus, it is conceivable that the activation of the FA pathway and the recruitment of the FANCI-D2 complex by IN to the viral DNA would inhibit the formation of 2-LTR circular DNA and enhance the insertion of viral DNA into the host genome. KO of FA genes and the stimulation of NHEJ would result in higher levels of 2-LTR circular viral DNA and reduce the insertion of the linear DNA into the host genome. A similar mechanism has been reported for the function of the FA pathway in the replication of herpesvirus, where FA activation counteracts NHEJ to antagonize viral DNA circularization and repairs gaps in linear viral DNA to support productive viral growth (Karttunen et al., 2014).
A second possibility is that the viral DNA may be rapidly excised from the host genome if the gaps are not repaired. In theory, the strand transfer reaction is reversible, being isoenergetic and not requiring any high-energy donor such as ATP (Chiu and Davies, 2004). HIV-1 IN bears disintegration activity in vitro to cleave branched DNAs that mimic hemi-integration intermediates (Chow and Brown, 1994; Chow et al., 1992), but during strand transfer in vivo, the reconfiguration of the active site followed by the dissociation of metal ion B is thought to prevent the disintegration activity of IN (Hare et al., 2012; Maertens et al., 2010). However, if the gaps at the viral DNA ends are not repaired and persist, it is conceivable that cells excise the viral DNA, either by IN or by other cellular DNA damage repair mechanisms.
A third possibility is that the unrepaired gaps are triggering cell death and thus the loss of viral DNA signal. It has been reported that infection of ligase-minus or other DNA damage mutant cells causes the loss of provirus signal via the loss of viable cells by apoptosis (Daniel et al., 2004a). However, this is unlikely to be the case in our experiments because we did not see obvious cell death in FA-deficient cells upon HIV-1 infection (MOI at ~0.5), and our readouts of integration occur very soon after infection. This possibility also is not consistent with our observation that the levels of unintegrated HIV-1 DNA increased rather than decreased after infection in FA-deficient cells.
It is well defined that three sequential DNA repair activities (DNA polymerase, 5′ flap nuclease, and ligase) are required to complete the integration process-ligation of the 5′ viral DNA ends with host chromosomal DNA. Early studies synthesized DNAs containing the gap and 5′ flap structures present in retroviral integration intermediates, and used biochemical assays to test candidate cellular enzymes for their ability to repair these DNAs in vitro. DNA polymerase POLB, flap nuclease FEN1, and ligase I were shown to be sufficient to repair the synthesized DNA substrate in vitro (Yoder and Bushman, 2000). However, cell-based studies showed that POLB is dispensable for HIV-1 infection (Goetze et al., 2017). Here, we show that REV1-interacting polymerases POLH, POLI, POLK, and REV3L (Figure 4), and flap nuclease FAN1 (Figure 5) play substantial roles in HIV-1 infection, suggesting that these are in fact the enzymes that execute the DNA damage repair in viral DNA integration. As REV1 and FAN1 are downstream DNA repair enzymes of FA effector proteins FANCI-D2, all of these results support the notion that the FA pathway is exploited by HIV-1 for viral DNA integration. The finding suggest that the components of the FA pathway constitute potential new targets for the development of inhibitors of HIV-1 replication.
Limitations of the study
Although we showed that knockdown of FANCI inhibited HIV-1 replication in T cell line MT4, most of our mechanistic studies were performed in 293A cells, a cell line that is efficiently suitable for gene KOs and HIV-1 reporter virus infection. We cannot exclude the possibility that some FA proteins may not function in the same way in T cells as in 293A cells. The method we used to assay for integrated viral DNA, Alu-Gag PCR, scores hemi-integration intermediates (strand transfer) or fully integrated proviruses, whether or not the single-strand gaps at the viral DNA ends are repaired. Due to this technical limitation, we cannot fully dissect the functions of FA pathway in strand transfer or post-integration DNA repair.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yiping Zhu (yiping_zhu@urmc.rochester.edu).
Material availability
This study did not generate new unique reagents.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original dataset or code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and culture conditions
Mammalian cell lines 293A (female, Invitrogen, Cat# R70507), HEK293T (female, ATCC, Cat# CRL-11268), TZM-bl (female, NIH AIDS Reagent Program Cat#8129), GM6914 (male, Coriell), and their derivatives were maintained at 37°C and 5% CO2 in DMEM supplemented with 10% inactivated fetal bovine serum, 2 mM glutamine, penicillin, and streptomycin. MT-4 (male, NIH AIDS Reagent Program Cat#120) cells were cultured at 37°C and 5% CO2 in RPMI 1640 supplemented with 10% inactivated fetal bovine serum.
METHOD DETAILS
DNA construction
Replication-defective retroviral vectors pNCA-Luc (MLV vector expressing firefly luciferase, MLV-Luc) has been described before (PMID: 27866901). pNL4.3-Luc (HIV-1 vector expressing firefly luciferase, HIV-Luc) was obtained from the NIH AIDS Reagent Program (Cat# ARP-3418). pNL4.3-mCherry (HIV-mCherry) was constructed by replacing the luciferase CDS in pNL4.3-Luc with mCherry CDS. Plasmid pCMV-Intron expresses WT Gag and GagPol from NB-tropic MLV; pCMVdeltaR8.2 expresses HIV-1 Gag and GagPol; pMD2.G expresses the vesicular stomatitis virus (VSV) envelope glycoprotein.
pCMV-HF is a mammalian expression vector for the expression of proteins with HA and Flag at the N-terminus, and has been described previously (PMID: 21876179). Coding sequences for HIV-1 Integrase (human codon optimized; WT, mutations with deletions or truncations) and MLV Integrase were cloned into pCMV-HF. pLvx-EF1-IRES-Neo was constructed by replacing the CMV promoter in pLvx-IRES-Neo (Clontech, #632181) with the EF1 promoter. FANCA, FANCD2, and FANCI CDS were cloned into pLvx-EF1-IRES-Neo to produce pLvx-EF1-IRES-Neo-FANCA, pLvx-EF1-IRES-Neo-FANCD2, pLvx-EF1-IRES-Neo-FANCI. The coding sequences for FAN1(WT, UBZmut with C44A_C47A mutations, NUCmut with E975A_K977A mutations) and REV1 (WT: CDS 1-1249 AAs; ΔCTD: CDS 1-1070) were cloned into expression vector pCMV-Myc (Takara, Cat# 635689) to produce pCMV-Myc-FAN1, pCMV-Myc-FAN1-UBZmut, pCMV-Myc-FAN1-NUCmut, pCMV-Myc-REV1, and pCMV-Myc-REV1deltaCTD. The cDNAs for FANCA, FANCD2, FANCI, FAN1, and REV1 are sgRNA-resistant with silent-mutations of the PAM (NGG) sequences. The oligos for the cDNA cloning are listed in key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FANCI | Bethyl | A301-254A; RRID:AB_890616 |
| FANCA | Bethyl | A301-980A; RRID:AB_1547945 |
| FANCD2 | Abcam | ab108928; RRID:AB_10862535 |
| Myc tag | Cell Signaling Technology | 2278; RRID:AB_490778 |
| Flag tag | Sigma | F3165; RRID:AB_259529 |
| Flag | Cell signaling | 14793S; RRID:AB_2572291 |
| β-Actin | Sigma | A5316; RRID:AB_476743 |
| eIF4G | Santa Cruz | Sc-133155; RRID:AB_2095748 |
| HIV-1 p24 | Abcam | Ab9071; RRID:AB_306981 |
| β-Tubulin | ABclonal | A12289; RRID:AB_2861647 |
| Phospho-Histone H2AX-S139 Rabbit pAb | ABclonal | AP0099; RRID:AB_2771168 |
| Anti-gamma H2A.X (phospho S139) antibody [3F2] | Abcam | ab22551; RRID:AB_447150 |
| RPA1 | Proteintech | 67973-1-Ig |
| FITC-labeled Goat Anti-Rabbit IgG (H + L) | Beyotime | A0562 |
| Cy3-labeled Goat Anti-Rabbit IgG (H + L) | Beyotime | A0516 |
| Anti-Mouse IgG (whole molecule)–TRITC, antibody produced in goat | Sigma | T5393 |
| FAN1 Polyclonal Antibody | Proteintech | 17600-1-AP; RRID:AB_1059801 |
| REV1 Rabbit pAb | ABclonal | A8493; RRID:AB_2771985 |
| Anti-SLX4 | Abcam | ab169114 |
| SNM1A Rabbit pAb | ABclonal | A14312; RRID:AB_2761176 |
| Ku70 Rabbit pAb | ABclonal | A7330; RRID:AB_2767867 |
| FEN1 Polyclonal Antibody | Proteintech | 14768-1-AP; RRID:AB_2102780 |
| LRS antibody | Abcam | ab31534; RRID:AB_776011 |
| Bacterial and virus strains | ||
| Trans10 Chemically Competent Cell | Transgen Biotech | CD101-01 |
| HIV-1 (NL4.3) | NIH AIDS Reagent Program | 114 |
| Chemicals, peptides, and recombinant proteins | ||
| DNase I | Sigma | D5025 |
| Benzonase | Sigma | E1014 |
| Puromycin | Sigma | P9620 |
| RNase A | Thermo Fisher Scientific | AM2271 |
| Lipofectamine 2000 | Thermo Fisher Scientific | 11668019 |
| TRIzol Reagent | Thermo Fisher Scientific | 15596018 |
| ANTI-FLAG M2 Magnetic Beads | Sigma | M8823 |
| passive lysis buffer | Promega | E1941 |
| Aphidicolin | Abacm | ab142400 |
| Dimethyl sulfoxide (DMSO) ACS | AMRESCO | 0231-500ML |
| Propidium iodide | Sigma | P4170 |
| Paraformaldehyde,4% | Solarbio | P1110 |
| DAPI Staining Solution | Beyotime | C1005 |
| Critical commercial assays | ||
| Luciferase Assay System | Promega | E1501 |
| SuperReal PreMix Plus (SYBR Green) | Tiangen | FP205 |
| DNeasy Blood & Tissue Kit | Qiagen | 69506 |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | CRL-11268 |
| TZM-bl | NIH AIDS Reagent Program | 8129 |
| MT-4 | NIH AIDS Reagent Program | 120 |
| GM6914 | Coriell | GM6914 |
| 293A | Invitrogen | R70507 |
| Oligonucleotides | ||
| qGAPDH-F | This study | TCGGAGTCAACGGATTTG |
| qGAPDH-R | This study | GCATCGCCCCACTTGATT |
| qLate RT product-F | This study | TGTGTGCCCGTCTGTTGTG |
| qLate RT product-R | This study | GAGTCCTGCGTCGAGA |
| qInt-1F | This study | GCCTCCCAAAGTGCTGGGATTACAG |
| qInt-1R | This study | GTTCCTGCTATGTCACTTCC |
| qInt-2F | This study | TTAAGCCTCAATAAAGCTTGCC |
| qInt-2R | This study | GTTCGGGCGCCACTGCTAGA |
| q2-LTR-F | This study | CCCTCAGACCCTTTTAGTCAGTG |
| q2-LTR-R | This study | TGGTGTGTAGTTCTGCCAATCA |
| qMito-F | This study | ACCCACTCCCTCTTAGCCAATATT |
| qMito-R | This study | GTAGGGCTAGGCCCACCG |
| qFAN1-F | This study | GTGGCCCCAGGAAGAAGAAA |
| qFAN1-R | This study | AATCACTTTGGCCAGGGGTT |
| qREV1-F | This study | CTGGTCCCTGGCGAGGGCACTGG |
| qREV1-R | This study | TCTGCATAGCAGCATCTGATCG |
| qPOLA-F | This study | GGACCAACACATCTAGCCTGGA |
| qPOLA-R | This study | GGTCTGGTTTCAAAGCCATTGCC |
| qPOLB-F | This study | TGCAGAGTCCAGTGGTGACATG |
| qPOLB-R | This study | ATGAACCTTTTGTAACTGCTCCAC |
| qPOLD1-F | This study | ACTACACGGGAGCCACTGTCAT |
| qPOLD1-R | This study | GCGTGGTGTAACACAGGTTGTG |
| qPOLD2-F | This study | ACTGACCCGTTCATCTTCCCAG |
| qPOLD2-R | This study | CAACAGCACTGTCTGGTCCTCA |
| qPOLD4-F | This study | CTGCTGAGGCAGTTTGACCTGG |
| qPOLD4-R | This study | TCTTCAGCACCTGCCACACCTC |
| qPOLE-F | This study | ACGCTGGAAGAGGTGTATGGCT |
| qPOLE-R | This study | GGAACGGTTCTCAGAGATGAGC |
| qPOLG-F | This study | AGATGGAGAACTTGCGAGCTGC |
| qPOLG-R | This study | CACGTCGTTGTAAGGTCCATTGC |
| qPOLH-F | This study | GTGCCAGTTACCAGCTCAGA |
| qPOLH-R | This study | AGGTAATGAGGGCTTGGATG |
| qPOLI-F | This study | CTACTTCACGCTCTGGCAAGCA |
| qPOLI-R | This study | GTGGTATCTAGTGGAGACTCCC |
| qPOLK-F | This study | CCAGACATCACAACCATTCC |
| qPOLK-R | This study | TCAAGGCTTCCAGACTGATG |
| qPOLL-F | This study | CCATAAGCCTGTCACCTCGTAC |
| qPOLL-R | This study | GCTCTCACTGATATGGTCCAGC |
| qPOLM-F | This study | TGTGAGGAGGTGGAGAGAGTTC |
| qPOLM-R | This study | TCGGAGGTCATCTAAGGTTCGC |
| qPOLN-F | This study | CGAGCAATAACCAGCTTCGAGAG |
| qPOLN-R | This study | GGATGAAGGTCTCGCAGAGCAT |
| qPOLQ-F | This study | CTTGTGGCATCTCCTTGGAGCA |
| qPOLQ-R | This study | AATCCCTTGGCTGGTCTCCATC |
| qREV3L-F | This study | GTCTGAGACTATTTACCAGGAACC |
| qREV3L-R | This study | CCTTCCAAGGAAAAGTCTCCCTC |
| siNC | This study | UAAGGCUAUGAAGAGAUAC |
| siPOLA | This study | GCACGCAAUAAAGACAAGA |
| siPOLB | This study | GCAGCAUCUGUUAUAGCAA |
| siPOLD1 | This study | CCCUCAAGGUACAAACAUU |
| siPOLD2 | This study | CCAUUGAUGGAGUCAGAUU |
| siPOLD4 | This study | CCUCAGGACAGAAGCGAGA |
| siPOLE | This study | CGGAAGCAGAUUUAAGGUG |
| siPOLG | This study | GGUGCACAGACUUUAUGUA |
| siPOLH | This study | GCUCGUGCAUUUGGAGUCA |
| siPOLI | This study | GGAAAUUAUGAUGUGAUGA |
| siPOLK | This study | CCAAUAGACAAGCUGUGAU |
| siPOLL | This study | GGGAGAAGAAGCAGAAGAG |
| siPOLM | This study | GCGACACAUGUUGUGAUGG |
| siPOLN | This study | GCACCCAAUUCAGAUUACU |
| siPOLQ | This study | GGAAUGCCAUUUUCAAUUA |
| siREV3L | This study | AUGAGUAUGGAUCAUAUAC |
| siREV1 | This study | AUCGGUGGAAUCGGUUUGG |
| FANCA sgRNA-1 | This study | AGACGCATACTGACCACTCG |
| FANCA sgRNA-2 | This study | CCTCCACTCACAAGATCGTG |
| FANCA sgRNA-3 | This study | GACACACAGAACCTTCCGAG |
| FANCD2 sgRNA-1 | This study | AGTTGACTGACAATGAGTCG |
| FANCD2 sgRNA-2 | This study | TCATAGAGGCCAGCTAAACA |
| FANCD2 sgRNA-3 | This study | TTGACAATAGGTGTTTGACC |
| FANCI sgRNA-1 | This study | GTATCCAGTTGGTGGAATCG |
| FANCI sgRNA-2 | This study | AAATGAAACCAAGTTCTACG |
| FANCI sgRNA-3 | This study | TATGACTGTATTCTTATACC |
| FAN1 sgRNA-1 | This study | TGCCGGTTTAAGTCATATCT |
| FAN1 sgRNA-2 | This study | CTGATTGATAAGCTTCTACG |
| FAN1 sgRNA-3 | This study | GACTTCGTTCAAGTGGATCC |
| REV1 sgRNA-1 | This study | GCGAGCTGAAAATGATGGCT |
| REV1 sgRNA-2 | This study | AATTCGACCAGAATGGATTG |
| REV1 sgRNA-3 | This study | AAACTAATGATGTTGCATGG |
| SNM1A sgRNA-1 | This study | ACGAAGACCATCTTTCCTCA |
| SNM1A sgRNA-2 | This study | GAAGTGCCCCTTGGAAATGC |
| SNM1A sgRNA-3 | This study | AGGTGTCTGCCCTATCAATG |
| SLX4 sgRNA-1 | This study | ACGGAGGTTTCTTCTCTGCA |
| SLX4 sgRNA-2 | This study | CTGCGCTAGCTTTTTCCAAA |
| SLX4 sgRNA-3 | This study | CGGTTCACTTGCTTGCCATT |
| Ku70 sgRNA-1 | This study | CGAGGGCGATGAAGAAGCAG |
| Ku70 sgRNA-2 | This study | TTCCAAGACATGATGGGCCA |
| Ku70 sgRNA-3 | This study | TGATCGAGATCTCTTGGCTG |
| FEN1 sgRNA-1 | This study | GCCGTTCTCCATCATGCGAA |
| FEN1 sgRNA-2 | This study | AAGCCGCCACAGCTCAAGTC |
| FEN1 sgRNA-3 | This study | CTTGCCATCAAAGACATACA |
| pCDH-CMV-MCS-EF1-Puro-IN-F | This study | GGATTCCTGGACGGCATTGACAAGGC |
| pCDH-CMV-MCS-EF1-Puro-IN-R | This study | TTAGTCCTCATCTTGACGAGAGGC |
| pCMV-HF-INΔCTD-F | This study | ATGCAGAAGCAGATCACCAAGATCC |
| pCMV-HF-INΔCTD-R | This study | TTAGTCCTCATCTTGACGAGAGGC |
| pCMV-Myc-FAN1-F | This study | ATGATGTCAGAAGGGAAACCT |
| pCMV-Myc-FAN1-R | This study | TTAGCTAAGGCTTTGGCTCTTAGC |
| pCMV-Myc-FAN1 UBZ MUT C44A_C47A | This study | CCACCTGCTAAACTTGCCGCACCCGTTGCAAGTAAAATGGTGCCTAGATATG |
| pCMV-Myc-FAN1 NUC MUT E975A K977A | This study | CACTTTAAGCTGGTGGCAGTTGCAGGCCCCAATGATCGTC |
| pCMV-Myc-REV1-F | This study | ATGAGGCGAGGTGGATGGAGG |
| pCMV-Myc-REV1-R | This study | TTATGTAACTTTTAATGTGCTTC |
| pCMV-Myc-REV1 ΔCTD-R | This study | ATTCTTTGGCACAGATGCGCTGGCTCA |
| pCMV-Myc-FAN1-NGG-MUT-F-1 | This study | AGTAAAATGGTGCCAAGATATGACTTAA |
| pCMV-Myc-FAN1-NGG-MUT-R-1 | This study | AGGTGCCGGTTTAAGTCATATCTTGGCACC |
| pCMV-Myc-FAN1-NGG-MUT-F-2 | This study | TCCTGACAAAAAAAGGCCACGTAG |
| pCMV-Myc-FAN1-NGG-MUT-R-2 | This study | ATAAGCTTCTACGTGGCCTTTTTTTG |
| pCMV-Myc-FAN1-NGG-MUT-F-3 | This study | GTTCAAGTGGATCCTGGGCAGGTT |
| pCMV-Myc-FAN1-NGG-MUT-R-3 | This study | TTTATTAAGCCAACCTGCCCAGGATC |
| pCMV-Myc-REV1-NGG-MUT-F-1 | This study | GAAAATGATGGCTGCGAAACA |
| pCMV-Myc-REV1-NGG-MUT-R-1 | This study | CCCACCCCATGTTTCTCAGCCAT |
| pCMV-Myc-REV1-NGG-MUT-F-2 | This study | TTCGACCAGAATGGATTGTAGAAAGC |
| pCMV-Myc-REV1-NGG-MUT-R-2 | This study | CCAGCTTTGATGCTTTCTACAATC |
| pCMV-Myc-REV1-NGG-MUT-F-3 | This study | AACTAATGATGTTGCATGGCGGTCAATA |
| pCMV-Myc-REV1-NGG-MUT-R-3 | This study | GAATAATATACATGGTATTGACCGCCAT |
| pLvx-EF1-IRES-Neo-FANCD2-NGG-MUT-F | This study | GTGATGAAATCAACATACCACGACT |
| pLvx-EF1-IRES-Neo-FANCD2-NGG-MUT-R | This study | ACTGACAATGAGTCGTGGTATGTTG |
| pLvx-EF1-IRES-Neo-FANCA-NGG-MUT-F | This study | GTTGCCTCTAGCGTCGGACAGATCT |
| pLvx-EF1-IRES-Neo-FANCA-NGG-MUT-R | This study | AGCCGTGCAGATCTGTCCGACGCTAG |
| pLvx-EF1-IRES-Neo-FANCI-NGG-MUT-F | This study | TATACACTTGCTGTATCCAGTTGGTG |
| pLvx-EF1-IRES-Neo-FANCI-NGG-MUT-R | This study | TTCCACCAACTGGATACAGCAAGTG |
| Recombinant DNA | ||
| pNCA-Luc | Addgene | 67793 |
| pNL4-3 | NIH AIDS Reagent Program | 114 |
| pNL4.3-Luc | NIH AIDS Reagent Program | 3418 |
| pMLV-luc | This study | N/A |
| lentiCRISPR v2 | Addgene | 52961 |
| pCMVdeltaR8.2 | Addgene | 12263 |
| pMD2.G | Addgene | 12259 |
| pCMV-HF | N/A | PMID: 21876179 |
| pCMV-Myc | TAKARA | 635689 |
| pLvx-IRES-Neo | TAKARA | 632181 |
| pLvx-EF1-IRES-Neo | This study | N/A |
| pCMV-Intron | This study | N/A |
| pNL4.3-mCherrey | This study | N/A |
| pCMV-HF-IN | This study | N/A |
| pCDH-CMV-MCS-EF1-Puro-IN | This study | N/A |
| pCMV-HF-INΔCTD | This study | N/A |
| pLvx-EF1-IRES-Neo-FANCA | This study | N/A |
| pLvx-EF1-IRES-Neo-FANCD2 | This study | N/A |
| pLvx-EF1-IRES-Neo-FANCI | This study | N/A |
| pCMV-Myc-FAN1 | This study | N/A |
| pCMV-Myc-REV1 | This study | N/A |
| pCMV-Myc-FAN1UBZmut | This study | N/A |
| pCMV-Myc-FAN1NUCmut | This study | N/A |
| pCMV-Myc-REV1ΔCTD | This study | N/A |
| Software and algorithms | ||
| GraphPad Prism 8 | GraphPad software | N/A |
| FlowJo V10 | FLOWJO software | N/A |
| Image J | Image J | N/A |
Plasmids expressing non-targeting control shRNA (SCR; target sequence GCAAGCTGACCCTGAAG) was purchased from Dharmacon (Cat# ID: RHS6848). Plasmids expressing shRNAs targeting FANCI (#1-TRCN0000242773-target sequence CATGTG GAAGGCACCATTATT; #2-TRCN0000242774-target sequence ATGTAAGCTCGGAGCTAATAT) were purchased from Sigma.
DNA transfection, virus package, and infection
All the DNA transfections were performed using lipofectamine 2000 (Invitrogen) following the manufacturer’s protocol.
To package HIV-1 based VSV-G pseudotyped reporter viruses, viral vectors (pNL4.3-Luc or pNL4.3-mCherry) together with pMD2.G, were transfected into HEK293T cells. To package MLV vector based VSV-G pseudotyped viruses, viral vectors (pNCA-Luc) together with pCMV-Intron and pMD2.G were transfected into HEK293T cells.
To package lentiviral vector based VSV-G pseudotyped viruses, viral vectors (expressing shRNA or cDNA) together with pCMVdel-taR8.2 (expressing HIV-1 Gag and Gag-Pol) and pMD2.G were transfected into HEK293T cells.
For all the virus packaging, 40 h after transfection, supernatants were collected and filtered through a 45 μm membrane to produce virus preparations.
The HIV-mCherry virus was further concentrated by ultracentrifuge. The supernatant medium from cells (9 ml) was layered above 1 ml of 25% sucrose in the TEN buffer [10mM Tris-Cl (pH 8.0), 0.1M NaCl, 1mM EDTA (pH 8.0)]. Samples were centrifuged at 100,000 × g (~ 28,000 rpm) for 2 h at 4°C (SW55 rotor, Beckman). The virus-like particle pellets were resuspended in 100 μl of DMEM and stored in −80°C.
Unless otherwise indicated, viruses were 3-fold diluted with cell culture medium containing 20 mM HEPES (pH 7.5) and 4 μg/ml polybrene. Cell culture medium was changed 3 h post infection.
Luciferase assay
Luciferase activity was assayed 40 h after infection, using the Luciferase Assay System (Promega).
Reverse transcription
To measure the mRNA levels, total RNA was extracted using TRIzol (Invitrogen, Cat#15596026) reagent. One microgram of RNA was digested with DNase I at 37°C for 30 min and used as the template for Reverse transcription. Reverse transcription was performed using High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific).
Quantitative PCR (qPCR)
Quantitative PCR was performed in the ABI 7500 Fast Real-Time PCR System using SuperReal Premix Plus with SYBR green I (Tiagen, Cat# FP205). The PCR cycle program was: 1) 50°C 2 min, 1 cycle; 2) 95°C 10 min, 1 cycle; 3) 95°C 15 s -> 60°C 30 s -> 72°C 30 s, 40 cycles; 4) 72°C 10 min, 1 cycle. The primers used for real time PCR are listed in key resources table.
CRISPR-mediated gene knockout
sgRNAs (3 sgRNAs per gene, sequences are listed in key resources table) were cloned into lentiCRISPR v2. For each gene, a mixture of three plasmids encoding sgRNAs were transfected into 293A cells using lipofectamine 2000 for two days. The transfected cells were selected in 1 μg/ml puromycin for two weeks. The resulting gene knockout cell line is a mixed population of cells stably expressing sgRNAs targeting gene of interest. All gene knockouts were confirmed by Western blot using specific antibodies. To rescue the expression of a gene in the KO cells, KO cells were transfected with plasmids expressing the corresponding gene with silent-mutations of the all the three PAM (NGG) sequences.
Quantitation of viral DNA
To measure the levels of viral DNA, the reporter viruses for infection were pretreated with DNase I at 37°C for 30 min to prevent possible contamination from the plasmids used for transfection. Total DNA from cells infected with viruses was extracted using the DNeasy Blood & Tissue kit (Qiagen).
Total viral DNA (late RT product) and unintegrated viral DNA (2-LTR circular DNA) were quantified by qPCR, using mitochondrial DNA as an internal control. Sequences of the PCR primers were as follows: primers for the late RT product (5′-TGTGTGCCCGTCTGTTGTG-3’, 5′-GAGTCCTGCGTCGAGA-3′); primers for 2-LTR circular DNA (5′-CCCTCAGACCCTTTTAGTCAGTG-3’, 5′-TGGTGTGTAGTTCTGCCAATCA-3′); and primers for mitochondrial DNA (5′-ACCCACTCCCTCTTAGCCAATATT-3’, 5′-GTAGGGCTAGGCCCACCG-3′).
The integrated viral DNA was quantitated by nested PCR. The first round of PCR was performed with primers int-1F (5′-GCCTCCCAAAGTGCTGGGATTACAG-3’) and int-1R (5′-GTTCCTGCTATGTCACTTCC-3′), which bind to the Alu and HIV-1 gag DNA sequences, respectively, to amplify the Alu-HIV DNA. The PCR conditions for the first round were as follows: 15 cycles of 95°C for 10 s, 63°C for 10 s, and 72°C for 2 min 50 s. The PCR products were used as templates for the second round of qPCR, using primers int-2F (5′-TTAAGCCTCAATAAAGCTTGCC-3’) and int-2R (5′-GTTCGGGCGCCACTGCTAGA-3′).
siRNA transfection
Target sequences for siRNAs are listed in key resources table. For siRNA transfection, 105 cells were seeded in 6-well plates. 24 h later, siRNAs were transfected into cells by Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. After another 24 h, the same siRNA transfection was performed for the second time. 6 h after the second transfection, the siRNA transfected cells were infected with virus for further experiments.
Co-immunoprecipitation and immunoblotting
For co-immunoprecipitation, cell lysate (lysis buffer: CelLytic M, Sigma C2978) was clarified by centrifugation at 4°C for 15 min at 12000 rpm. For nuclease treatment, cell lysates were treated with Benzonase (250 U/ml supplemented with 10 mM MgCl2). ANTI-FLAG M2 Magnetic Beads (Sigma Cat# M8823) were incubated with 800 μl cell lysate at 4°C for 4 h. The beads were then washed 3 times with TBST. The bound proteins on the beads were eluted with 40 μl 1X SDS sample buffer and subjected to SDS-PAGE and Western blot analysis.
For immunoblotting, cells were lysed in the passive lysis buffer (Promega, Cat# E1941) for 10 min. The lysate was clarified by centrifugation at 4°C for 15 min at 12000 rpm. The samples were heated at 95°C in SDS sample buffer and resolved by SDS-PAGE electrophoresis, transferred to a PVDF membrane and probed with specific antibodies by Western blot.
HIV-1 replication assay
HIV-1 viruses were produced by transfecting HEK293T cells with pNL4.3 using lipofectamine 2000 (Invitrogen) following the manufacturer’s protocol. For HIV-1 replication in MT-4 cells, MT-4 cells stably expressing control shRNA (SCR) or shRNAs targeting FANCI were transduced with HIV-1 virus (0.1 ng p24/106 cells) by spin infection (3500 rpm at room temperature for 2 h) and then cultured at 37°C. 3 h post infection, cells were washed twice with PBS and then cultured in 1 ml medium in 24-well plate. Every day post infection, culture supernatants (50 μl) were taken out, and half of cells and medium (500 μl) were transferred to a new well containing 500 μl fresh medium. The Trypan Blue dye (Invitrogen, Cat# 15250061) exclusion test was used to determine the number of viable cells. To monitor the spreading of HIV-1 virus, 30 ul of supernatants were used to infect TZM-bl cells and luciferase activities were measured 24 h post infection.
Fluorescent staining and confocal microscopy
For Fluorescent staining assays, the indicated cells were sub-cultured onto coverslips overnight and then cells were infected with VSV-G pseudotyped HIV-1 reporter viruses. At indicated hours post infection, cells were fixed with 4% paraformaldehyde, washed with PBS, permeabilized with 0.2% Triton X-100. After blocking in staining buffer (10% serum and 0.1% Tween 20 in PBS), samples were incubated with the indicated antibody, washed with PBS and incubated with Fluorescent secondary antibody. Subsequently, the samples were washed with PBS, and mounted with DPAI Staining Solution (Beyotime). The subcellular localizations of the stained proteins were photographed using a Laser Confocal Microscope (Zeiss LSM700).
Image quantifications were performed using plugins in Image J. We counted and averaged foci (fluorescence intensity >= 200, size >= 75 pixel^2) numbers in ~ 10 cells at each time point from three representative images.
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless otherwise indicated, all data presented are mean ± SD from three independent experiments. p values are from paired two-sided t-tests.
Supplementary Material
Highlights.
HIV-1 infection activates the Fanconi anemia (FA) DNA repair pathway
HIV-1 integrase interacts with FANCI-D2 complex
Depletion of FA proteins and downstream enzymes inhibits HIV-1 DNA integration
ACKNOWLEDGMENTS
This work was supported by NCI grant R01 CA 30488 to S.P.G., the Chinese Academy of Sciences (XDB29010206) to G.G., the National Natural Science Foundation of China (81921005) to G.G., and a University of Rochester start-up fund to Y.Z.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110840.
REFERENCES
- Adamo A, Collis SJ, Adelman CA, Silva N, Horejsi Z, Ward JD, Martinez-Perez E, Boulton SJ, and La Volpe A (2010). Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol. Cell 39, 25–35. 10.1016/j.molcel.2010.06.026. [DOI] [PubMed] [Google Scholar]
- Ariumi Y, Turelli P, Masutani M, and Trono D (2005). DNA damage sensors ATM, ATR, DNA-PKcs, and PARP-1 are dispensable for human immunodeficiency virus type 1 integration. J. Virol 79, 2973–2978. 10.1128/jvi.79.5.2973-2978.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzon B, Grainger R, Huang S, Rzadki C, and Junop MS (2018). Structure-specific endonuclease activity of SNM1A enables processing of a DNA interstrand crosslink. Nucleic Acids Res. 46, 9057–9066. 10.1093/nar/gky759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Sarangi P, and D’Andrea AD (2016). The Fanconi anaemia pathway: new players and new functions. Nat. Rev. Mol. Cell Biol 17, 337–349. 10.1038/nrm.2016.48. [DOI] [PubMed] [Google Scholar]
- Chatterjee N, and Walker GC (2017). Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen 58, 235–263. 10.1002/em.22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Wei SQ, and Engelman A (1999). Multiple integrase functions are required to form the native structure of the human immunodeficiency virus type I intasome. J. Biol. Chem 274, 17358–17364. 10.1074/jbc.274.24.17358. [DOI] [PubMed] [Google Scholar]
- Chiu TK, and Davies DR (2004). Structure and function of HIV-1 integrase. Curr. Top. Med. Chem 4, 965–977. 10.2174/1568026043388547. [DOI] [PubMed] [Google Scholar]
- Chow SA, and Brown PO (1994). Juxtaposition of two viral DNA ends in a bimolecular disintegration reaction mediated by multimers of human immunodeficiency virus type 1 or murine leukemia virus integrase. J. Virol 68, 7869–7878. 10.1128/jvi.68.12.7869-7878.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow SA, Vincent KA, Ellison V, and Brown PO (1992). Reversal of integration and DNA splicing mediated by integrase of human immunodeficiency virus. Science 255, 723–726. 10.1126/science.1738845. [DOI] [PubMed] [Google Scholar]
- Ciccia A, Ling C, Coulthard R, Yan Z, Xue Y, Meetei AR, Laghmani EH, Joenje H, McDonald N, de Winter JP, et al. (2007). Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Mol. Cell 25, 331–343. 10.1016/j.molcel.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Ciuffi A, Llano M, Poeschla E, Hoffmann C, Leipzig J, Shinn P, Ecker JR, and Bushman F (2005). A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med 11, 1287–1289. 10.1038/nm1329. [DOI] [PubMed] [Google Scholar]
- Daniel R, Greger JG, Katz RA, Taganov KD, Wu X, Kappes JC, and Skalka AM (2004a). Evidence that stable retroviral transduction and cell survival following DNA integration depend on components of the nonhomologous end joining repair pathway. J. Virol 78, 8573–8581. 10.1128/jvi.78.16.8573-8581.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel R, Ramcharan J, Rogakou E, Taganov KD, Greger JG, Bonner W, Nussenzweig A, Katz RA, and Skalka AM (2004b). Histone H2AX is phosphorylated at sites of retroviral DNA integration but is dispensable for postintegration repair. J. Biol. Chem 279, 45810–45814. 10.1074/jbc.m407886200. [DOI] [PubMed] [Google Scholar]
- Fang CB, Wu HT, Zhang ML, Liu J, and Zhang GJ (2020). Fanconi anemia pathway: mechanisms of breast cancer predisposition development and potential therapeutic targets. Front. Cell Dev. Biol 8, 160. 10.3389/fcell.2020.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust EA, and Triller H (2002). Stimulation of human flap endonuclease 1 by human immunodeficiency virus type 1 integrase: possible role for flap endonuclease 1 in 5’-end processing of human immunodeficiency virus type 1 integration intermediates. J. Biomed. Sci 9, 273–287. 10.1007/bf02256074. [DOI] [PubMed] [Google Scholar]
- Ferris AL, Wu X, Hughes CM, Stewart C, Smith SJ, Milne TA, Wang GG, Shun MC, Allis CD, Engelman A, and Hughes SH (2010). Lens epithelium-derived growth factor fusion proteins redirect HIV-1 DNA integration. Proc. Natl. Acad. Sci. U S A 107, 3135–3140. 10.1073/pnas.0914142107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetze RW, Kim DH, Schinazi RF, and Kim B (2017). A CRISPR/Cas9 approach reveals that the polymerase activity of DNA polymerase beta is dispensable for HIV-1 infection in dividing and nondividing cells. J. Biol. Chem 292, 14016–14025. 10.1074/jbc.m117.793661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S, Di Nunzio F, Labeja A, Wang J, Engelman A, and Cherepanov P (2009). Structural basis for functional tetramerization of lentiviral integrase. PLoS Pathog. 5, e1000515. 10.1371/journal.ppat.1000515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S, Gupta SS, Valkov E, Engelman A, and Cherepanov P (2010). Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 464, 232–236. 10.1038/nature08784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S, Maertens GN, and Cherepanov P (2012). 3’-processing and strand transfer catalysed by retroviral integrase in crystallo. EMBO J. 31, 3020–3028. 10.1038/emboj.2012.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Liu S, Bellani MA, Thazhathveetil AK, Ling C, de Winter JP, Wang Y, Wang W, and Seidman MM (2013). The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol. Cell 52, 434–446. 10.1016/j.molcel.2013.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Kim JM, Shiotani B, Yang K, Zou L, and D’Andrea AD (2010). The FANCM/FAAP24 complex is required for the DNA interstrand crosslink-induced checkpoint response. Mol. Cell 39, 259–268. 10.1016/j.molcel.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager S, Cimermancic P, Gulbahce N, Johnson JR, McGovern KE, Clarke SC, Shales M, Mercenne G, Pache L, Li K, et al. (2011). Global landscape of HIV-human protein complexes. Nature 481, 365–370. 10.1038/nature10719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins TM, Esposito D, Engelman A, and Craigie R (1997). Critical contacts between HIV-1 integrase and viral DNA identified by structure-based analysis and photo-crosslinking. EMBO J. 16, 6849–6859. 10.1093/emboj/16.22.6849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karttunen H, Savas JN, McKinney C, Chen YH, Yates JR 3rd, Hukkanen V, Huang TT, and Mohr I (2014). Co-opting the Fanconi anemia genomic stability pathway enables herpesvirus DNA synthesis and productive growth. Mol. Cell 55, 111–122. 10.1016/j.molcel.2014.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessl JJ, Kutluay SB, Townsend D, Rebensburg S, Slaughter A, Larue RC, Shkriabai N, Bakouche N, Fuchs JR, Bieniasz PD, and Kvaratskhelia M (2016). HIV-1 integrase binds the viral RNA genome and is essential during virion morphogenesis. Cell 166, 1257–1268.e12. 10.1016/j.cell.2016.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Yang K, Dejsuphong D, and D’Andrea AD (2012). Regulation of Rev1 by the Fanconi anemia core complex. Nat. Struct. Mol. Biol 19, 164–170. 10.1038/nsmb.2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Lach FP, Desetty R, Hanenberg H, Auerbach AD, and Smogorzewska A (2011). Mutations of the SLX4 gene in Fanconi anemia. Nat. Genet 43, 142–146. 10.1038/ng.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratz K, Schopf B, Kaden S, Sendoel A, Eberhard R, Lademann C, Cannavo E, Sartori AA, Hengartner MO, and Jiricny J (2010). Deficiency of FANCD2-associated nuclease KIAA1018/FAN1 sensitizes cells to interstrand crosslinking agents. Cell 142, 77–88. 10.1016/j.cell.2010.06.022. [DOI] [PubMed] [Google Scholar]
- Lesbats P, Engelman AN, and Cherepanov P (2016). Retroviral DNA integration. Chem. Rev 116, 12730–12757. 10.1021/acs.chemrev.6b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Olvera JM, Yoder KE, Mitchell RS, Butler SL, Lieber M, Martin SL, and Bushman FD (2001). Role of the non-homologous DNA end joining pathway in the early steps of retroviral infection. EMBO J. 20, 3272–3281. 10.1093/emboj/20.12.3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liszewski MK, Yu JJ, and O’Doherty U (2009). Detecting HIV-1 integration by repetitive-sampling Alu-gag PCR. Methods 47, 254–260. 10.1016/j.ymeth.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Ghosal G, Yuan J, Chen J, and Huang J (2010). FAN1 acts with FANCI-FANCD2 to promote DNA interstrand cross-link repair. Science 329, 693–696. 10.1126/science.1192656. [DOI] [PubMed] [Google Scholar]
- Llano M, Saenz DT, Meehan A, Wongthida P, Peretz M, Walker WH, Teo W, and Poeschla EM (2006). An essential role for LEDGF/p75 in HIV integration. Science 314, 461–464. 10.1126/science.1132319. [DOI] [PubMed] [Google Scholar]
- MacKay C, Declais AC, Lundin C, Agostinho A, Deans AJ, MacArtney TJ, Hofmann K, Gartner A, West SC, Helleday T, et al. (2010). Identification of KIAA1018/FAN1, a DNA repair nuclease recruited to DNA damage by monoubiquitinated FANCD2. Cell 142, 65–76. 10.1016/j.cell.2010.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maertens GN, Hare S, and Cherepanov P (2010). The mechanism of retroviral integration from X-ray structures of its key intermediates. Nature 468, 326–329. 10.1038/nature09517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakumo Y, Ogura Y, Ishii H, Numata S, Ichihara M, Croce CM, Fishel R, and Takahashi M (2001). Interactions in the error-prone postreplication repair proteins hREV1, hREV3, and hREV7. J. Biol. Chem 276, 35644–35651. 10.1074/jbc.m102051200. [DOI] [PubMed] [Google Scholar]
- Nakajima N, Lu R, and Engelman A (2001). Human immunodeficiency virus type 1 replication in the absence of integrase-mediated dna recombination: definition of permissive and nonpermissive T-cell lines. J. Virol 75, 7944–7955. 10.1128/jvi.75.17.7944-7955.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalepa G, and Clapp DW (2018). Fanconi anaemia and cancer: an intricate relationship. Nat. Rev. Cancer 18, 168–185. 10.1038/nrc.2017.116. [DOI] [PubMed] [Google Scholar]
- Ohashi E, Murakumo Y, Kanjo N, Akagi J.i., Masutani C, Hanaoka F, and Ohmori H (2004). Interaction of hREV1 with three human Y-family DNA polymerases. Genes Cells 9, 523–531. 10.1111/j.1356-9597.2004.00747.x. [DOI] [PubMed] [Google Scholar]
- Pace P, Mosedale G, Hodskinson MR, Rosado IV, Sivasubramaniam M, and Patel KJ (2010). Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science 329, 219–223. 10.1126/sci-ence.1192277. [DOI] [PubMed] [Google Scholar]
- Rio P, Navarro S, and Bueren JA (2018). Advances in gene therapy for Fanconi anemia. Hum. Gene Ther 29, 1114–1123. 10.1089/hum.2018.124. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, and D’Andrea A (2017). Fanconi anemia pathway. Curr. Biol 27, R986–R988. 10.1016/j.cub.2017.07.043. [DOI] [PubMed] [Google Scholar]
- Rumbaugh JA, Fuentes GM, and Bambara RA (1998). Processing of an HIV replication intermediate by the human DNA replication enzyme FEN1. J. Biol. Chem 273, 28740–28745. 10.1074/jbc.273.44.28740. [DOI] [PubMed] [Google Scholar]
- Sakai H, Kawamura M, Sakuragi J, Sakuragi S, Shibata R, Ishimoto A, Ono N, Ueda S, and Adachi A (1993). Integration is essential for efficient gene expression of human immunodeficiency virus type 1. J. Virol 67, 1169–1174. 10.1128/jvi.67.3.1169-1174.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakeel S, Rajendra E, Alcon P, O’Reilly F, Chorev DS, Maslen S, Degliesposti G, Russo CJ, He S, Hill CH, et al. (2019). Structure of the Fanconi anaemia monoubiquitin ligase complex. Nature 575, 234–237. 10.1038/s41586-019-1703-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Hicks JK, Chute CL, Brennan JR, Ahn JY, Glover TW, and Canman CE (2012). REV1 and polymerase zeta facilitate homologous recombination repair. Nucleic Acids Res. 40, 682–691. 10.1093/nar/gkr769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims AE, Spiteri E, Sims RJ 3rd, Arita AG, Lach FP, Landers T, Wurm M, Freund M, Neveling K, Hanenberg H, et al. (2007). FANCI is a second monoubiquitinated member of the Fanconi anemia pathway. Nat. Struct. Mol. Biol 14, 564–567. 10.1038/nsmb1252. [DOI] [PubMed] [Google Scholar]
- Smogorzewska A, Desetty R, Saito TT, Schlabach M, Lach FP, Sowa ME, Clark AB, Kunkel TA, Harper JW, Colaiacovo MP, and Elledge SJ (2010). A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol. Cell 39, 36–47. 10.1016/j.molcel.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER 3rd, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D’Andrea AD, and Elledge SJ (2007). Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 129, 289–301. 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, and Craigie R (2007). The road to chromatin - nuclear entry of retroviruses. Nat. Rev. Microbiol 5, 187–196. 10.1038/nrmicro1579. [DOI] [PubMed] [Google Scholar]
- Tan W, van Twest S, Murphy VJ, and Deans AJ (2020). ATR-mediated FANCI phosphorylation regulates both ubiquitination and deubiquitination of FANCD2. Front. Cell Dev. Biol 8, 2. 10.3389/fcell.2020.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Wang R, Peralta C, Yaseen A, and Pavletich NP (2021). Structure of the FA core ubiquitin ligase closing the ID clamp on DNA. Nat. Struct. Mol. Biol 28, 300–309. 10.1038/s41594-021-00568-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Liu Z, Wang F, Temviriyanukul P, Ma X, Tu Y, Lv L, Lin YF, Huang M, Zhang T, et al. (2015). FANCD2 and REV1 cooperate in the protection of nascent DNA strands in response to replication stress. Nucleic Acids Res. 43, 8325–8339. 10.1093/nar/gkv737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder KE, and Bushman FD (2000). Repair of gaps in retroviral DNA integration intermediates. J. Virol 74, 11191–11200. 10.1128/jvi.74.23.11191-11200.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original dataset or code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.