

Abstract
Red blood cell (RBC) shape and deformability are supported by a planar network of short actin filament (F-actin) nodes (∼37 nm length, 15–18 subunits) interconnected by long spectrin strands at the inner surface of the plasma membrane. Spectrin-F-actin network structure underlies quantitative modeling of forces controlling RBC shape, membrane curvature, and deformation, yet the nanoscale organization and dynamics of the F-actin nodes in situ are not well understood. We examined F-actin distribution and dynamics in RBCs using fluorescent-phalloidin labeling of F-actin imaged by multiple microscopy modalities. Total internal reflection fluorescence and Zeiss Airyscan confocal microscopy demonstrate that F-actin is concentrated in multiple brightly stained F-actin foci ∼200–300 nm apart interspersed with dimmer F-actin staining regions. Single molecule stochastic optical reconstruction microscopy imaging of Alexa 647-phalloidin-labeled F-actin and computational analysis also indicates an irregular, nonrandom distribution of F-actin nodes. Treatment of RBCs with latrunculin A and cytochalasin D indicates that F-actin foci distribution depends on actin polymerization, while live cell imaging reveals dynamic local motions of F-actin foci, with lateral movements, appearance and disappearance. Regulation of F-actin node distribution and dynamics via actin assembly/disassembly pathways and/or via local extension and retraction of spectrin strands may provide a new mechanism to control spectrin-F-actin network connectivity, RBC shape, and membrane deformability.
INTRODUCTION
Plasma membranes of metazoan cells are supported by a thin planar network of short, actin filament (F-actin) nodes (∼37 nm length; 15–18 subunits) cross-linked by long, flexible spectrin strands forming a membrane skeleton that provides mechanical resilience and creates micron-scale domains of transmembrane proteins such as ion channels and adhesion receptors (Bennett and Baines, 2001; Bennett and Lorenzo, 2013). The spectrin-F-actin network was first discovered in mammalian red blood cells (RBCs), which rely on the membrane skeleton for their biconcave disk shape, remarkable deformability, and stability during their transits through circulation (Chien, 1987; Mohandas and Evans, 1994; Mohandas and Gallagher, 2008). The network provides multisite attachments for RBC transmembrane proteins, including the anion channel (band 3), glycophorin A (GPA), and the glucose transporter (Glut4), restricting their lateral mobility in the membrane (Mohandas and Gallagher, 2008; Kusumi et al., 2012b; Alenghat and Golan, 2013; Machnicka et al., 2014). Tethering of membrane proteins to the network also facilitates formation of complexes with kinases and/or metabolic enzymes, promoting regulatory signaling and metabolic pathways (Machnicka et al., 2014; Lux, 2016). Genetic defects in spectrin-F-actin network components result in abnormal RBC shapes and deformability in human hemolytic anemias (Mohandas and Evans, 1994; Mohandas and Gallagher, 2008; Fowler, 2013; Gallagher, 2013) and affect adaptations of RBCs to invasion by Plasmodium sp. in malaria (Dhermy et al., 2007; Zuccala and Baum, 2011; Gokhin and Fowler, 2016).
Our current molecular understanding of spectrin-F-actin network organization is based principally on negative staining electron microscopy (EM) of spread RBC membrane skeletons in which (α1β1)2–spectrin tetramers are extended to their full ∼200 nm length (Fowler, 2013; Gokhin and Fowler, 2016; Lux, 2016). These images reveal a periodic 2D quasihexagonal lattice of short ∼37 nm F-actin nodes connected by ∼200 nm-long (α1β1)2–spectrin tetramers, with 5–7 spectrins attached to each F-actin node (Byers and Branton, 1985; Shen et al., 1986; Liu et al., 1987). However, with a native membrane surface area of 135 μm2 and ∼30,000–40,000 F-actin nodes in the membrane skeleton of each RBC (Fowler, 2013; Lux, 2016), the spread network of extended (α1β1)2–spectrin tetramers would encompass a surface area ∼9-fold greater than the cell membrane, which is not the case. Thus in the native RBC membrane, the 2D lattice must be condensed with spectrin tetramers ∼1/3 their extended length, such that F-actin nodes are expected to be ∼70 nm apart on average. This is supported by superresolution fluorescence microscopy showing an ∼80 nm periodic spacing of F-actin nodes and associated proteins, although frequent irregularities or holes in the lattice were also observed (Pan et al., 2018). A presumption of uniform periodicity in network dimensions in resting RBCs forms the basis for many quantitative models of forces accounting for a biconcave shape (Discher et al., 1998; Li et al., 2005, 2007; Fedosov et al., 2010; Peng et al., 2013; Li and Lykotrafitis, 2014; Chen and Boyle, 2017; Fai et al., 2017), with condensation of spectrin strands providing the potential for network extensibility in membrane deformations under flow and mechanical perturbations (McGough and Josephs, 1990; Ursitti et al., 1991; Terada et al., 1996; Nans et al., 2011; Brown et al., 2015). Interestingly, a recent modeling study by Feng and colleagues (Feng et al., 2020) incorporates observed wide variations in spectrin strand length from 46 ± 15 nm (Nans et al., 2011), i.e., nonperiodic network dimensions, to create a constitutive model of the membrane that provides a better explanation of the observed physical behavior of the RBC membrane during deformations.
These models are predicated on the idea that the short F-actin nodes form stable linkages for the spectrin strands in the network and do not polymerize or depolymerize. This assumption is challenged by observations that RBCs contain cytosolic actin monomers (G-actin) at concentrations sufficient to allow barbed-end assembly, comprising about 5% of total RBC actin (Gokhin et al., 2015). Moreover, a subset of the membrane skeleton F-actin can assemble and disassemble based on decreased or increased cytosolic G-actin after treatment of RBCs with jasplakinolide (Jasp) or latrunculin A (LatA), respectively. Additionally, fluorescence recovery after photobleaching (FRAP) of F-actin in live RBCs labeled with a fluorescent Jasp derivative [SiR-actin (Lukinavicius et al., 2013)] indicated a ∼30% mobile fraction with a slow recovery (∼2.5 min) (Gokhin et al., 2015). Perturbation of the G/F-actin ratio by actin drugs affects RBC membrane tension and leads to increased cell deformability in microfluidic channels (Gokhin et al., 2015; Gokhin and Fowler, 2016). However, it is unknown whether or how actin assembly/disassembly dynamics may influence nanoscale F-actin node distribution and network organization in the membrane skeleton. RBCs are an ideal cellular system to investigate this question because RBCs have no internal organelles and F-actin is present exclusively as short filaments (nodes) in the membrane skeleton (Fowler, 2013; Gokhin and Fowler, 2016), thus removing confounding contributions from cytoplasmic populations of other F-actin.
Here we examined directly the nanoscale distribution and dynamics of F-actin in intact RBCs using fluorescent-phalloidin labeling of cells imaged by total internal reflection fluorescence (TIRF) microscopy (Mattheyses et al., 2010), Zeiss AiryScan confocal microscopy (Huff, 2015), and stochastic optical reconstruction microscopy (STORM) (Xu et al., 2013; Pan et al., 2018). Our results demonstrate that F-actin appears in an irregular patchy distribution with brightly staining F-actin foci about 200–300 nm apart, likely corresponding to clusters of F-actin nodes. The F-actin foci are sensitive to LatA-induced F-actin disassembly and undergo nanoscale local motions in live cells over a timescale of seconds, suggesting transient formation and dissolution of clusters of F-actin nodes. This implies that nanoscale lateral organization of the spectrin-F-actin network depends on local motions of F-actin nodes constrained by spectrin strand lengths and influenced by dynamic actin assembly and disassembly, which in turn may alter spectrin attachment sites at nodes and affect network connectivity. These data will necessitate reconsideration of current molecular, structural, and mechanical models of RBC shape and membrane deformations, which are predicated on the idea that stable F-actin nodes and associated membrane proteins are uniformly distributed across the membrane.
RESULTS
F-actin is nonuniformly distributed across the membrane
To evaluate the organization of the F-actin network in RBCs in situ, we imaged F-actin by fluorescent-phalloidin labeling of fixed and permeabilized human RBCs under conditions that preserve cell integrity (Smith et al., 2018). Epifluorescence images focused on the edge of a biconcave cell double-labeled with rhodamine-phalloidin (rho-phalloidin) and an antibody to the membrane marker GPA reveal uniformly distributed, bright rim staining corresponding to the membrane at the edge of the biconcave disk (Figure 1A). A smaller fuzzy circle near the center of the cell corresponds to the tangentially oriented membrane at the edge of the dimple. Due to the superimposition of the out-of-focus light in epifluorescence microscopy, it is impossible to obtain more detailed information regarding the lateral distribution of F-actin in the plane of the membrane.
FIGURE 1:

TIRF microscopy reveals bright F-actin foci at the RBC membrane. (A) Differential interference contrast (DIC) and epifluorescence microscopy images of a biconcave RBC stained with Alexa 488-anti GPA antibodies (488-GPA) (green) and Rhodamine (Rho)-phalloidin (red); merge on far right. Bar, 3 μm. (B) TIRF images of a flattened RBC stained with Alexa 488-GPA (green) and Rho-phalloidin (red); merge on far right. Bar, 1 μm. GPA staining is relatively smooth and continuous, compared with uneven F-actin staining. (C) Coefficients of variation for GPA and F-actin staining intensities, determined from images as in B. p < 0.0001. N = 19 cells. (D) Left, unprocessed TIRF image of a flattened RBC stained with Rho-phalloidin. Right, higher magnification deconvolved image of portion of cell in red box in left panel. Blue dots, centroids of F-actin foci determined in Nikon Elements. Bars, 1 μm. Quantification of (E) F-actin foci density (spots/μm3) and (F) nearest-neighbor distance (nm) from high magnification deconvolved images of RBCs stained with Rho- or Alexa 488-phalloidin. No significant differences are observed for RBCs stained with different phalloidins or fixed under the different conditions indicated. Each point represents data from an individual cell. (G) DIC or TIRF microscopy images of a biconcave or flattened RBC stained with Alexa 488-phalloidin (488-phalloidin). Bar, 4 μm. (H) Quantification of F-actin foci density (spots/μm3) and (F) nearest-neighbor distance (nm) from high magnification deconvolved images of biconcave or flattened RBCs stained with Alexa 488-phalloidin. Each point represents data from an individual cell. No significant differences are observed between biconcave and flattened RBCs.
Therefore, we turned to TIRF microscopy, which selectively illuminates fluorophores within 100–200 nm from the coverslip at or near the cell membrane (Mattheyses et al., 2010). Since the membrane surface area visible in TIRF images of biconcave-shaped RBCs is limited, we first examined fixed cells that had flattened during cytospinning onto the coverslip (113 × g). Comparison of F-actin with GPA staining in these flattened cells reveals an irregular, patchy appearance of F-actin staining contrasting with the smooth, continuous GPA staining across the membrane (Figure 1B). The irregular pattern for F-actin staining is also evident from the significantly greater coefficient of variation for F-actin pixel intensities as compared with GPA for each cell (Figure 1C). Following deconvolution, at higher magnification, the uneven F-actin staining appears as spots or foci of varying brightness, whose centroids can be identified using the General Analysis tools in Nikon Elements image analysis software (Figure 1D). Quantification reveals a density of ∼4–5 spots/μm2 and an average nearest-neighbor distance of ∼300–350 nm (Figure 1, E and F).
To rule out the possibility that fixation conditions or loss of biconcave RBC shape may induce artifactual rearrangements of F-actin, we compared several fixation conditions and biconcave versus flattened cells. The irregular F-actin staining pattern, spot density, and nearest-neighbor distances are similar for RBCs fixed with 4% paraformaldehyde (PFA) overnight or for 4 h, or with 4% PFA plus 0.01% glutaraldehyde for 30 min (Figure 1, E and F). We also compared rho-phalloidin to Alexa 488-phalloidin labeling and observed a similar irregular F-actin staining pattern for both probes (Figure 1, E and F). While the spot density tended to be slightly less and nearest-neighbor distances correspondingly slightly greater for rho-phalloidin than Alexa 488-phalloidin, this did not achieve statistical significance (Figure 1, E and F). These small differences may be because Alexa 488-phalloidin staining of RBC F-actin is brighter than rho-phalloidin, enabling better detection of more closely spaced, dimmer fluorescence spots. The irregular F-actin staining pattern, spot density, and nearest-neighbor distances are similar for fixed RBCs that remained biconcave or were flattened mechanically during deposition on the coverslip by cytospinning (Figure 1, G, H, and I). These experiments demonstrate that a patchy F-actin distribution (F-actin foci) along the membrane is a robust feature of RBCs.
F-actin foci are exclusively associated with the membrane
To explore F-actin distribution on the membrane at higher resolution and to rule out potential contributions from F-actin in the cytoplasm, we used sensitive Zeiss AiryScan confocal fluorescence microscopy to image GPA and F-actin staining in fixed biconcave RBCs. This method has an XY resolution of ∼140 nm and an XZ resolution of ∼300 nm (Huff, 2015), somewhat better than the maximum XY resolution of TIRF microscopy (∼200 nm) (Mattheyses et al., 2010). Maximum intensity projections of Z stacks of RBCs stained with Alexa 488–conjugated anti-GPA revealed smooth and continuous GPA staining along the membrane (Figure 2A). By contrast, in separate cells, the Alexa 488-phalloidin staining for F-actin was clearly discontinuous and patchy (Figure 2A). We also double-labeled cells with Alexa 488-anti-GPA and rho-phalloidin, or with Alexa 488-phalloidin and rhodamine-wheat germ agglutinin (WGA), which labels GPA, the major sialylated membrane protein (Figure 2B). In RBCs with an orthogonal orientation that had attached to the coverslip at their rims, single XY optical sections through the middle of the cells demonstrated that the GPA (or WGA) and F-actin staining were exclusively associated with the membrane and displayed a similar intensity at the rim as compared with the dimple (Figure 2B). Due to the oversampling of Z slices and the Z stretch, it is not possible to evaluate local F-actin distribution at the membrane from these optical sections through the middle of the cell.
FIGURE 2:

Superresolution Zeiss AiryScan confocal fluorescence microscopy reveals bright F-actin foci at the RBC membrane. (A) Maximum intensity projections of an AiryScan confocal Z stack of RBCs stained with Alexa 488-antibodies to GPA (488-GPA) or with Alexa 488-phalloidin for F-actin. GPA staining is smooth and continuous, while F-actin staining is irregular and patchy. Bar, 4 μm. (B) Single XY optical sections from Z stacks of RBCs stained with Alexa 488-GPA and Rho-phalloidin (left vertical panels), or with Alexa 488-phalloidin and rhodamine-WGA (right vertical panels). Merges at bottom. Bar, 1 μm. Images of the membrane in cross-section at the rim and dimple regions of cells attached end-on to the coverslip show that F-actin staining is exclusively associated with the membrane, colocalizing with GPA or WGA, with similar intensity at the dimple and rim. (C) Single XY or XZ optical sections from Z stacks of RBCs attached parallel to the coverslip, stained with Alexa 488-phalloidin. Left, XY optical section immediately adjacent to the coverslip, revealing irregular and patchy F-actin staining along the membrane (Slice 1). Middle, XY optical section through the middle of the cell revealing bright uneven F-actin staining along the membrane at the rim. Bar, 1 μm. Right, XZ optical section from the middle of the same cell, oriented orthogonally to the XY section in Slice 2. Membrane curvature and oversampling of fluorescence in optical sections leads to apparently continuous F-actin staining along the rim in the XY image in Slice 2. This precludes accurate evaluation of F-actin distribution in cross-sectional views of the RBC membrane in either XY or XZ views. Right graph, coefficient of variation for Alexa 488-phalloidin staining at the membrane is significantly higher than for rhodamine-WGA staining of GPA. Each point represents data from an XY optical section at Slice 1 of an individual cell. p < 0.0014. N = 14. (D) Left image, single XY optical section from a confocal Z stack of RBC stained with Alexa 488-phalloidin. Right image, higher magnification image of portion of cell in red box in left panel. Blue dots, centroids of F-actin foci determined in Nikon Elements. Bars, 1 μm. Right graphs, Quantification of F-actin foci density (spots/μm2) and nearest-neighbor distance (nm) from high magnification single optical sections from images as shown on left. Each point represents data from an individual cell. N = 16 cells.
Therefore, we examined single XY optical sections from the rim region of the RBC in the plane of the membrane at the coverslip, which again revealed an irregular pattern of Alexa 488-phalloidin F-actin staining intensity across the RBC membrane (Figure 2C, Slice 1). Similar to TIRF images, the coefficient of variation of pixel intensities was significantly greater for the F-actin than for the WGA (Figure 2C, right panel). A patchy distribution of F-actin is also detected in the dimple region in an optical section through the middle of the cell (Figure 2C, Slice 2), but due to the curvature at the edges of the dimple it is difficult to identify discrete foci for analysis (Figure 2C, XZ view). Quantitative analysis of F-actin distributions in the rim region by determination of the centroid positions for F-actin foci showed about ∼12 spots/μm2 with a nearest-neighbor distance of ∼200 nm (Figure 2D). In AiryScan images, the F-actin spot density is about two to three times greater, and nearest-neighbor distances are about half of those determined from TIRF images at the rim region (Figure 1, G, H, and I). This can be ascribed to the increased XY resolution of the AiryScan versus TIRF (∼140 nm vs. ∼200 nm), as well as to a greater sensitivity of AiryScan fluorescence image detection compared with TIRF, likely allowing discrimination of more closely spaced, dimmer spots. In summary, the irregular F-actin staining distribution across the RBC membrane observed with both imaging modalities indicates that a large proportion of the RBC F-actin in the spectrin-F-actin network is present in small regions, or foci, exclusively associated with the RBC membrane.
Single molecule TIRF/STORM imaging demonstrates an irregular distribution of F-actin
To further analyze RBC nanoscale F-actin organization, we performed STORM of RBC membranes stained with saturating concentrations of Alexa 647-phalloidin (0.4 μM) according to Pan et al. (2018) and imaged by TIRF using a penetration depth suitable for imaging a single layer of the cell membrane (Xu et al., 2013; Pan et al., 2018). Using this approach, we obtained a map of Alexa 647-phalloidin localizations across the RBC surface (Figure 3, A and B) with a localization accuracy of ∼21 to 23 nm (Supplemental Figure S1A). To determine whether the individual localizations were due to the same dye molecules blinking multiple times, we calibrated the blinking properties under our experimental conditions using Alexa 647–conjugated IgG molecules adsorbed to poly-l-lysine-coated coverslips. Under our imaging conditions, the majority (>60%) of the IgG molecules show a single localization, ∼20% have two localizations, and ∼17% have three or more localizations (Supplemental Figure 1B). Since the commercial IgG-Alexa 647 conjugates have several (3–8) dye molecules per IgG molecule (average 5 per IgG for the lot used), the total blinks observed underestimate the number of Alexa 647-phalloidin molecules under our experimental conditions. Consequently, the localizations that we see for Alexa 647-phallloidin in RBCs most likely correspond to only 1 out of 5 phalloidin molecules blinking a single time for the duration of the acquisition. This may be due to our use of Vectashield as a mounting medium which quenches Alexa 647 dyes, leading to bleaching and resulting in reduced reblinking (Arsic et al., 2020).
FIGURE 3:

TIRF/STORM imaging and Ripley H function analysis reveals a nonrandom, clustered distribution of individual Alexa 647-phalloidin localizations at the RBC membrane. (A) STORM images of Alexa 647-phalloidin localizations in a RBC, acquired as in Methods. Red box shown at higher magnification in panel B. Bar, 1 μm. (B) High magnification image of boxed region in panel A. (C, D) Scatter plot of distribution of Alexa 647-phalloidin localizations in a RBC. The 4 × 4 μm2 red box in panel C is shown at higher magnification in panel D. Bar in D, 1 μm. (E) Normalized Ripley H test on STORM images of Alexa 647 phalloidin localizations as a function of radius r. Results from images (4 × 4 μm2) for 6 different cells were averaged. Error bars, SD. The dashed line indicates the expected value for the homogeneous Poisson process (known as complete spatial randomness). Data for RBCs indicates localization clustering with a peak at a ∼70 nm radius and extending up to a ∼600 nm radius, with the distribution becoming random at larger radii.
Visual inspection of STORM images revealed an irregular distribution of fluorescence localization (Figure 3, A and B). To quantitatively test whether this uneven distribution is random or clustered, we utilized the Ripley K test (Ripley, 1977), which quantifies clusters by comparing the spatial distribution of particles with a random point pattern as a function of distance in the given area of interest (Ripley, 1977). We performed the Ripley analysis over a 4 μm × 4 μm square region of each RBC (Figure 3, C and D) and plotted the normalized form of Ripley’s K function (Ripley’s H function, H(r) = L(r)-r) (Kiskowski et al., 2009) against intermolecular distance (r) from 0 to 1 μm to capture potential clustering at short and long length scales (Figure 3E). Consistent with our visual inspection, the Ripley H test suggested a high degree of clustering over the 0–600 nm range, while the distribution became random at length scales greater than ∼600 nm (Figure 3E). Further analysis of Ripley test results for individual RBCs shows that in each case the experimental data fall above randomly distributed data indicated by the simulation envelope (Supplemental Figure S2). The single peak at r ∼70 nm is roughly correlated with the radius of the predominant mean cluster size (Kiskowski et al., 2009; Caetano et al., 2015). Based on our localization accuracy of 21 to 23 nm (Supplemental Figure S1A), a cluster of localizations with a radius of ∼70 nm (i.e., diameter of ∼140 nm) may represent localizations from a group of several F-actin nodes, each labeled with multiple Alexa 647-phalloidin molecules. The long shoulder in the experimental data at higher radii up to r ∼600 nm is also greater than expected for a random distribution (Figure 3E; Supplemental Figure S2). This likely represents higher-order collections of individual F-actin nodes that may correspond to the irregular brightly stained F-actin foci observed in TIRF and Airyscan images of RBC membranes (Figures 1 and 2).
To further analyze whether localizations were clustered, we employed the density-based spatial clustering of applications with noise (DBSCAN) algorithm (Pedregosa et al., 2011; Schubert et al., 2017). In this analysis, localizations are considered part of a cluster if they have a number of neighbors equal to or greater than a minimum number for a specified radius, r (Supplemental Figure S3C). Based on the localization accuracy (Supplemental Figure S1A), we used a minimum cluster size of 2 and a radius of 20 nm as the minimum distance at which two localizations could be distinguished (Pageon et al., 2016) and compared the experimentally determined distributions with randomly generated synthetic data sets (Supplemental Figure S3, F and G). The experimental data showed a wide range of cluster sizes extending from 2–11 to >200 as compared with the random data where relatively few large clusters are observed, with none larger than 50 (Supplemental Figure S3F). In the experimental data, approximately 20% of localizations are associated with small clusters (2–10), while 80% are in larger clusters (≥11) (Supplemental Figure S3G). The significantly greater frequency of large clusters in experimental data (∼80% as compared with synthetic data (∼40%) indicates that the experimentally observed distribution of clusters is not random. The 20% of clusters with 2–10 localizations in the experimental data may be derived from single F-actin nodes, while the 80% of clusters with 11+ localizations may represent signals from groups of F-actin nodes. We conclude that both the Ripley and the DBSCAN tests indicate that the Alexa 647-phalloidin fluorescence localizations are not randomly distributed and instead are present in clusters of varying sizes in the RBC membrane, in agreement with the irregular distribution of F-actin staining observed by TIRF and Airyscan microscopy.
F-actin foci are perturbed by actin-depolymerizing drugs
We showed previously that a subset of RBC actin could assemble and disassemble based on incorporation of exogenous rhodamine-actin into the membrane skeleton and altered G/F actin ratio after treatment with LatA or Jasp (Gokhin et al., 2015). To investigate whether the F-actin foci observed here depend on actin polymerization or depolymerization, we treated RBCs with varying concentrations of LatA, Jasp or cytochalasin D (CytoD) in a physiological buffer at 37°C, followed by fixation, rho-phalloidin staining, and imaging by TIRF microscopy. TIRF microscopy was used to assess F-actin foci distribution since the efficient image acquisition and analysis allow us to analyze cells from many experimental conditions. Three parameters were assessed: numbers of F-actin foci (spots/μm2), foci intensity (coefficient of variation), and nearest-neighbor distance (nm). The most striking changes were observed on LatA treatment where the number of foci increased ∼15%, their intensity decreased ∼20%, and nearest-neighbor distance decreased ∼10% (Figure 4, A, D, and G). This suggests that LatA sequestration of actin monomers led to disassembly of some F-actin nodes, resulting in loss of brighter F-actin foci and the appearance of more foci of lower intensity located closer together. By contrast, Jasp had little to no effect on these parameters (Figure 4, B, E, and H); however, since Jasp competes with rho-phalloidin for binding to F-actin, pretreatment of RBCs with Jasp may obscure any potential changes. CytoD treatment appeared to have effects opposite to that of LatA, resulting in a ∼10% greater nearest-neighbor distance, trending toward a decreased number of spots and increased spot intensity, although these two latter parameters did not achieve statistical significance (Figure 4, C, F, and I). The effects of LatA and CytoD on F-actin foci distribution indicate that their formation and/or maintenance may depend on actin polymerization and barbed-end assembly.
FIGURE 4:
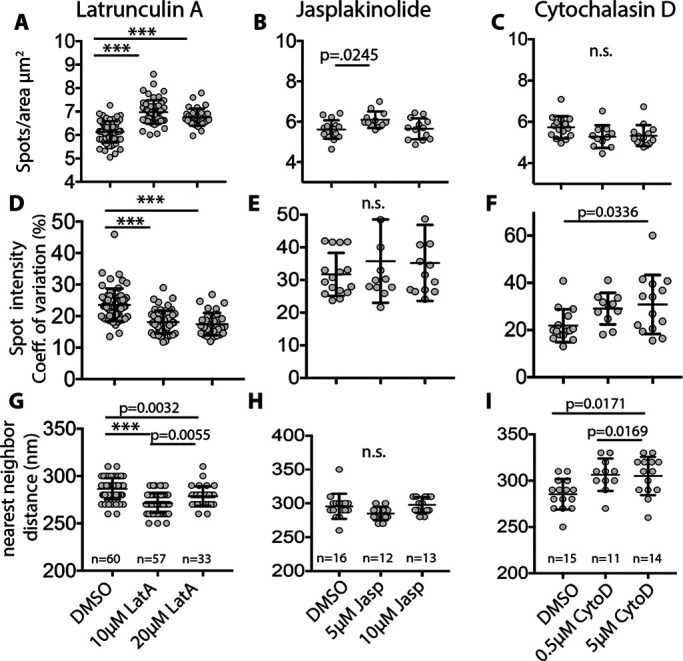
Treatment of RBCs with LatA and CytoD alters the distribution of F-actin foci in RBCs. RBCs were treated with (A, D, G) LatA, (B, E, H) Jasp, or (C, F, I) CytoD for 4 h at 37°C as in Methods. (A, B, C) Quantification of F-actin foci density (spots/μm2), (D, E, F) spot intensity (coefficient of variation, %), and (G, H, I) nearest-neighbor distance (nm), from high magnification deconvolved TIRF microscopy images of RBCs stained with Alexa 488-phalloidin. LatA treatment leads to significant increases in F-actin spots/μm2, and decreases in spot intensity and nearest-neighbor distances. CytoD leads to a significant increase in nearest-neighbor distances and a small but significant decrease in spot intensity, while Jasp appears to have no significant effect on these parameters. Each point represents data from an individual cell. N as indicated on graphs.
F-actin foci undergo local nanoscale motions in living cells
Next, we wondered whether F-actin foci are present in live RBCs and whether they are stable features of the spectrin-F-actin network over time. To address this, we imaged live RBCs labeled with a cell-permeable, infrared-fluorescent Jasp conjugate, referred to as SiR-actin (SiR-actin) (Lukinavicius et al., 2013; Gokhin et al., 2015). We used TIRF microscopy to visualize F-actin foci at the RBC rim closest to the coverslip (Figure 5, A and B) and a Leica-SP8 Hyvolution system to visualize F-actin foci in the dimple region of biconcave RBCs (Figure 5C). Both the rim and the dimple regions of live RBCs show a patchy distribution of SiR-actin fluorescence intensity, similar to that observed in the rim region at the membrane of fixed RBCs imaged by TIRF microscopy (Figures 1 and 5D). Visual inspection of successive time-lapse images of live RBCs collected for intervals of 2 s revealed the appearance and disappearance of some foci with others persisting in the same location for several frames (Figure 5, A–C). Some of the individual F-actin foci also display local lateral movements, or appear and disappear, as indicated by the red and green colors present in merges of sequential time-lapse images pseudocolored in red and green. The F-actin foci do not appear to undergo long-range directed translocation or flow. In controls with fixed RBCs, F-actin foci did not change in position or appearance during time-lapse imaging, as shown by the yellow color in merges of sequential time-lapse images pseudocolored in red and green (Figure 5D). We could not further extend these qualitative observations since we were unable to image the SiR-actin in live RBCs for shorter time intervals or for more than a total of 10 s due to extensive quenching by hemoglobin and photobleaching. Nevertheless, these observations in living cells indicate that the F-actin foci undergo dynamic local motions in situ.
FIGURE 5:

Time-lapse imaging of F-actin foci in live RBCs reveals dynamic local movements. (A) DIC and epifluorescence images of a SiR-actin-labeled biconcave RBC before collection of sequential TIRF microscopy images of the same RBC for 2 s intervals, over 10 s. Bar, 4 μm. (B) Higher magnification sequential images of a portion of the same RBC for 2 s intervals, over 10 s (Red box in left panel denotes region shown; Bar, 4 μm). Red arrows, foci which appear and disappear or display local motions over time. Bar, 1 μm. (C) Images of a SiR-actin-labeled biconcave RBC acquired by Leica-SP8 Hyvolution microscopy (see Methods). Far left panel, XY optical section near the middle of the cell, revealing F-actin foci along the membrane at the rim, and en face on the membrane of the dimple region (red box). Right panels, higher magnification sequential images of the dimple region collected for 2.8 s intervals, over 10 s. Red arrows, foci which appear and disappear or display local motions over time. The dim fluorescence of the SiR-actin in the RBCs as well as photobleaching precluded collecting more images for shorter time intervals, or over a longer time period. Bars, 1 μm. (D) TIRF images of a fixed RBC labeled with Alexa 488-phalloidin collected for 2 s intervals, over 10 s (red box in left panel denotes region shown in time-lapse). Bars, 1 μm. (A–D) Bottom rows in each series show sequential images that are pseudo-colored in green and red to illustrate presence or absence of colocalization of spots in sequential time-lapse images. Red and green colors indicate dynamic changes in F-actin foci patterns in live RBCs, while the yellow indicates no changes in foci positions in the fixed RBC.
DISCUSSION
We report here that the F-actin nodes are irregularly distributed in the RBC membrane skeleton, forming clusters that are visualized as brightly stained foci by fluorescent phalloidins using TIRF and Airyscan confocal microscopy. Quantitative analysis reveals ∼12 foci/μm2, ∼200 nm apart, across the entire cell, based on superresolution Airyscan confocal microscopy. With a RBC surface area of ∼135 μm2, this comes to ∼1620 foci/cell, which could contain up to ∼18–20 F-actin nodes each, if all the F-actin nodes were in foci, based on the ∼30,000–40,000 F-actin nodes/cell (Fowler, 2013). However, some F-actin nodes are certainly present in the dimmer staining regions between the brightly stained foci, so this is likely an overestimate. A nonuniform distribution of F-actin nodes is not unique to human RBCs, as brightly stained F-actin foci are also observed in mouse RBCs imaged by TIRF microscopy (Sui et al., 2017).
Notably, in live RBCs, the F-actin foci undergo dynamic local motions, forming and disappearing over a timescale of seconds, based on TIRF and STED microscopy of SiR-actin-labeled live RBCs. Moreover, pretreatment of RBCs with LatA to sequester G-actin and depolymerize F-actin, followed by fixation and imaging, results in reduced foci intensity but greater density. This suggests that LatA treatment leads to depolymerization of F-actin nodes within foci and implies that F-actin foci formation depends on actin polymerization. Pretreatment with CytoD had a smaller but opposite effect, suggesting that barbed-end assembly may play a role in foci formation. However, it is unlikely that F-actin node assembly/disassembly alone can explain F-actin foci formation and dynamics since biochemical approaches indicate that only a small portion (<5%) of the F-actin in the RBC membrane skeleton is susceptible to LatA-induced depolymerization (Gokhin et al., 2015). We therefore propose that individual F-actin nodes undergo local motions in live RBCs, forming transient clusters of varying sizes, contributing to the appearance and disappearance of the brightly stained F-actin foci observed by TIRF and Airyscan microscopy (Figure 6A). In this scenario, the spectrin strands connecting F-actin nodes would not be uniform lengths; closely spaced nodes would be connected by spectrin strands with shorter end-to-end lengths and more distant nodes by spectrin strands with longer end-to-end lengths. Thus F-actin foci dynamics in live RBCs would be enabled by transient variations in spectrin strand lengths, leading to coalescence and dissolution of foci, respectively. Indeed, EM and atomic force microscopy of native, unspread membrane skeletons reveals a dense meshwork with a wide range of spectrin strand lengths, from ∼20 to ∼80 nm (Ursitti et al., 1991; Terada et al., 1996; Takeuchi et al., 1998; Swihart et al., 2001). A more recent cryo-EM study of Triton-extracted membrane skeletons also identified highly convoluted spectrin strands with a wide range of lengths, 46 ± 15 nm, connected to short F-actins, consistent with nonuniform network organization (Nans et al., 2011).
FIGURE 6:

Models to explain irregular patterns of F-actin node distribution in RBCs. (A) Schematic of a spectrin-F-actin network with irregularly distributed, uniform-length F-actin nodes. Clusters of closely spaced F-actin nodes in foci (green shading) are connected by shorter, condensed spectrin strands, while sparsely distributed nodes are connected by longer, extended spectrin strands (unshaded regions). (B) Schematic of a spectrin-F-actin network with relatively uniformly distributed, but variable-length F-actin nodes, with clusters of longer filaments (green shaded regions) interspersed with clusters of shorter filaments (no shading), resulting in regions of higher and lower density of F-actin labeling by phalloidins. These models are not mutually exclusive.
In a second model (Figure 6B), the addition of actin subunits and elongation of some F-actin nodes, along with the loss of subunits and shortening of others, could result in more or less fluorescent-phalloidin binding, leading to regions of brighter and dimmer F-actin staining, i.e., formation of F-actin foci. In this scenario, dynamic fluctuations of foci position and intensity could be due to cooperative actin subunit association and dissociation, resulting in more or less F-actin staining (bright foci or dim interfoci regions, respectively). While negative staining EM studies of highly expanded membrane skeleton preparations have demonstrated a relatively uniform F-actin length distribution [37 ± 3 nm (Byers and Branton, 1985); 33 ± 5 nm (Shen et al., 1986)], these preparations depended on low ionic strength incubation steps that could have led to actin subunit dissociation, as well as mechanical shear forces that may have affected F-actin lengths (Kuhlman and Fowler, 1997). It may be significant that a cryo-EM study of the RBC membrane skeleton (Nans et al., 2011) identified structures resembling F-actin nodes ranging in length from 17 to 37 nm (27 nm ± 7 nm). Certainly these two models (Figure 6, A and B) are not mutually exclusive and spectrin strand condensation and expansion may operate together with cooperative actin assembly/disassembly to result in lateral motions of F-actin nodes to form and dissociate from foci. Such dynamic nanoscale lateral motions of F-actin nodes, along with subunit polymerization and depolymerization, could account for the ∼30% mobile fraction of F-actin (t½ ∼150 s) that we had observed previously by FRAP analysis of SiR-actin-labeled live RBCs (Gokhin et al., 2015; Gokhin and Fowler, 2016). Additional studies tracking foci in live RBCs with actin perturbing drugs will be required to establish the relative contributions of F-actin node assembly/disassembly versus their local lateral motions to F-actin foci dynamics.
What might be the driving force for spectrin strand condensation and foci formation by F-actin nodes? An attractive mechanism is cross-linking of F-actin nodes by nonmuscle myosin IIA (NMIIA). NMIIA forms bipolar filaments in RBCs that associate with F-actin in the membrane skeleton via ATP-dependent interactions of their motor domains (Fowler et al., 1985; Smith et al., 2018). NMIIA motor activity promotes membrane tension and maintains RBC biconcave shape, controlling RBC deformability (Alimohamadi et al., 2020). Each NMIIA bipolar filament is ∼300–400 nm long with ∼30 motor domains at each end (Fowler et al., 1985; Smith et al., 2018; Pal et al., 2020). It is tempting to speculate that NMIIA contractility operating on F-actin nodes could transiently generate tension to gather nodes into foci and contribute to the formation and dynamics of foci in live RBCs. Our previous computational modeling of force distributions in biconcave-shaped RBCs, together with immunostaining for NMIIA, indicates that the forces exerted on the membrane are greater, and the NMIIA filaments are more concentrated at the dimple region as compared with the rim. This suggests that NMIIA-dependent dynamics of F-actin foci could be enhanced at the dimple as compared with the rim. Additional studies using 3D high-resolution imaging approaches with fluorescent probes for NMIIA and F-actin and investigation of F-actin foci dynamics in live RBCs treated with chemical inhibitors of NMIIA activity (Smith et al., 2018), or in RBCs with NMIIA mutations (Smith et al., 2019) will be necessary to further explore these ideas.
On the other hand, the appearance of bright foci due to variations in lengths of F-actin nodes may arise from some nodes being biochemically distinct from others with a different complement of associated F-actin-binding proteins, resulting in more or less stable and consequently longer or shorter F-actin nodes. Previous immunolocalization EM studies of dematin (an F-actin bundling protein), tropomodulin1 (the RBC pointed end capping protein), α,β adducin (the RBC barbed-end capping protein), and tropomyosin (an F-actin stabilizing protein) indicated that some but not all F-actin nodes were occupied by each of these F-actin-binding proteins (Derick et al., 1992; Ursitti and Fowler, 1994). Such differences in occupancy could lead to differences in relative rates of actin subunit assembly or disassembly, or in affinity for spectrin that could contribute to dynamic rearrangements of F-actin nodes and consequent foci formation (Gokhin et al., 2015; Gokhin and Fowler, 2016). Future studies will be required to colocalize dematin, α,β-adducin, tropomodulin1, and tropomyosin proteins with respect to the F-actin foci detected by TIRF and Airyscan imaging in RBCs.
Our STORM imaging of Alexa 647-phalloidin localization and Ripley H function and DBSCAN analysis of clustering in experimental and random synthetic data sets also support a nonuniform distribution of F-actin nodes in fixed RBCs. At first glance, these observations appear to be in conflict with a previous STORM analysis of F-actin node distribution, which identified a periodic hexagonal lattice containing ∼80 F-actin nodes/μm2 in native, unspread RBC membrane skeletons (Pan et al., 2018). However, this study also observed frequent irregularities or “holes” ∼200 nm in diameter in the periodic lattice which were depleted of F-actin nodes and their associated proteins. These holes may correspond to the dimmer staining regions we observe by TIRF and Airyscan microscopy. Moreover, the reported densities of ∼80 F-actin nodes/μm2 from Alexa 647-phalloidin staining, or ∼110 nodes/μm2 from antibody labeling of protein 4.1R at F-actin nodes (Pan et al., 2018), correspond to only ∼11,000 or ∼15,000 nodes, respectively, for a 135 μm2 RBC membrane surface area, considerably less than the ∼30,000–40,000 F-actin nodes (15–18 subunits long) per RBC, based on biochemical approaches (Fowler, 2013). A more recent proteomics study calculated ∼106 actin subunits/RBC, which could form >60,000 F-actin nodes, each 15 subunits long (Gautier et al., 2018). While this discrepancy was interpreted as partial labeling, it could also be explained by F-actin nodes that may not have been resolved by 3D STORM and/or did not follow a periodic pattern. Clearly the locally dynamic behavior of F-actin foci (i.e., transient clusters of F-actin nodes) observed here in live RBCs would be expected to transiently disrupt periodic polygonal patterning of the spectrin-actin membrane skeleton.
Studies of membrane protein mobility were pioneered in RBCs where mobility was shown to be restricted by direct anchorage of transmembrane proteins to the spectrin-F-actin network, or by trapping within “corrals” created by the extended spectrin tetramers of the lattice (Fowler and Branton, 1977; Fowler and Bennett, 1978; Golan and Veatch, 1980; Sheetz, 1983; Kodippili et al., 2012). Quantitative analyses of membrane protein mobility in RBCs have generally presumed a periodic network, with corral dimensions corresponding to a triangular lattice of extended, ∼200 nm-long spectrin tetramers linked to uniformly distributed F-actin nodes (Kodippili et al., 2020). Our data here indicate that mechanisms of RBC membrane protein mobility will require reevaluation based on dense nanoscale clusters of F-actin nodes interspersed with regions of sparsely distributed nodes. This implies considerable heterogeneity in corral dimensions that could influence membrane protein mobility and the frequency and prevalence of their interactions. Indeed, nanoscale clustering of membrane proteins has emerged as a prevalent feature of membrane protein dynamics and plasma membrane organization in other cells (Kusumi et al., 2012a,b; Garcia-Parajo et al., 2014; Nicolson, 2014; Bennett and Lorenzo, 2016).
The irregular distribution of F-actin nodes in native RBCs appears to contrast with neurons where superresolution fluorescence microscopy reveals a regular 1D periodic lattice with circumferential rings of F-actin connected by extended (α2β2)2–spectrin tetramers ∼190 nm long along axons and dendrites (Xu et al., 2013; Zhong et al., 2014; D’Este et al., 2015; Leterrier et al., 2015, 2021). However, the RBC proteins, dematin, tropomyosin3.1, and the actin capping proteins, tropomodulin1 and αβ-adducin are present in the axonal F-actin rings (Abouelezz et al., 2020; Zhou et al., 2020). This suggests that the F-actin rings could consist of many short filaments (F-actin nodes) distributed in an elongated circumferentially arranged “cluster” around the axon or dendrite.
Nanoscale F-actin foci are also likely to be characteristic of spectrin-F-actin networks in nonerythroid cells. A recent study from the Gauthier lab shows that large micron-sized spectrin-rich domains on the plasma membrane are located between F-actin-rich stress fibers in mouse embryonic fibroblasts (Ghisleni et al., 2020). While the large spectrin-rich domains are F-actin poor with respect to the stress fibers, close inspection of images in this study reveals uneven F-actin staining in the spectrin-rich regions visualized by TIRF microscopy, resembling the irregular F-actin distribution we observe at the RBC plasma membrane. Inasmuch as spectrin-rich domains restrict clathrin-mediated endocytosis (Jenkins et al., 2015; Ghisleni et al., 2020), further investigation of the nanoscale distribution and dynamics of spectrin and F-actin in these plasma membrane regions and how it may preclude or convert to one favoring endocytosis will be a fascinating area of future investigation.
In conclusion, we expect that the relatively simple RBC model system will continue to provide an opportunity to scrutinize the molecular and structural basis for membrane protein organization and dynamics and the role of spectrin-F-actin networks in determination of membrane functional properties in metazoan cells.
MATERIALS AND METHODS
Fluorescence staining of RBCs for TIRF and Airyscan imaging
Whole blood was collected from healthy human donors into EDTA tubes (BD Diagnostics) by the Normal Blood Donor Service at the Scripps Research Institute (La Jolla, CA). For phalloidin staining, 20 μl of whole blood was added to 1 ml of 4% PFA (Electron Microscopy Sciences) in Dulbecco’s phosphate-buffered saline (DPBS, Life Technologies), mixed, and incubated overnight at room temperature, unless otherwise indicated. In some experiments, cells were fixed in 4% PFA for 4 h at room temperature, or in a mixture of 4% PFA and 0.01% glutaraldehyde (Electron Microscopy Sciences) in DPBS for 30 min at room temperature, followed by washing and incubation with 0.1% sodium borohydride in DPBS for 20 min at room temperature. Fixed RBCs were washed three times in DPBS by centrifuging for 5 min at 1000 × g, permeabilized in DPBS + 0.3% TX-100 for 10 min, and then blocked in 4% bovine serum albumin (BSA), 1% normal goat serum in DPBS (blocking buffer, BB), and kept in BB at 4°C for up to 1 wk; 20 μl of fixed, permeabilized, and blocked RBCs plus 180 μl of BB were then incubated with fluorescent-phalloidin and fluorescent–conjugated GPA or fluorescent–conjugated WGA for 1–2 h at room temperature, followed by washing three times in BB as above. Fluorescent phalloidins were rho-phalloidin (Life Technologies R415) or Alexa 488-phalloidin (Life Technologies) at a final concentration of 130 nM, for TIRF and Airyscan imaging. Fluorescent-GPA was Alexa 488-GPA (BD Biosciences GA-R2 1:100) and Alexa 555-WGA (Molecular Probes 2 µg/ml). Stained cells were cytospun (Thermo Scientific Cytospin 4 1000 rpm for 3 min) onto 1.5-thick coverslips and mounted with ProLong Gold mounting medium (Molecular Probes #P36934) onto slides prior to imaging. For live cell microscopy, cells in 20 μl of whole blood from a normal donor were washed twice in 1.3 ml of DPBS/0.05% BSA. Washed cells were diluted 1:9 and stained with 50 nM SiR-actin (Spirochrome/Cytoskeleton) for 30 min (Lukinavicius et al., 2013). Stained cells were settled on Mat-tek dishes (MatTek, Ashland, MA) with a 1.5-size coverslip bottom for imaging.
Drug treatment of RBCs
Freshly drawn human blood (see above) was centrifuged for 10 min at 1000 × g, serum and buffy coat were aspirated off, and RBCs were resuspended in 20 vol of HEPES-buffered saline (HBS) optimized for human RBCs (145 mM NaCl, 5 mM KCl, 1 mM MgCl2, 10 mM glucose, 2 mM adenosine, 10 mM HEPES, pH 7.4). RBCs were washed 3× by centrifugation at 1000 × g, aspiration of the supernatant, and resuspension in 20 vol of HBS. CytoD (C8273), LatA (L5163), and Jasp (J4580) were purchased from Sigma-Aldrich, and stock solutions were prepared in DMSO and stored at –20°C until use. Intact, washed RBCs were treated with DMSO or actin perturbing drugs at 5% hematocrit in a shaking water bath at 37°C for 4 h. Treated cells were fixed in 4% PFA in PBS overnight at 4°C, permeabilized in DPBS + 0.3% TX-100 for 10 min, and then blocked in 4% BSA, 1% normal goat serum in DPBS overnight at 4°C. Permeabilized and blocked RBCs were then incubated with 130 nm Alexa 488-phalloidin (Life Technologies) for 1–2 h at room temperature, followed by washing three times in DPBS. Stained cells were cytospun onto coverlips and mounted with ProLong Gold mounting medium (Invitrogen) prior to TIRF imaging.
Fluorescence microscopy
Fixed fluorescently stained RBCs were imaged using a Nikon Ti inverted microscope with a 100× Apochromat oil objective (NA 1.49) either by epifluorescence microscopy using vertical illumination with 488 and 561 laser lines and an ORCA-Flash 4.0 V2 Digital CMOS camera (Hamamatsu) or by TIRF illumination with the same laser lines and DIC microscopy. Images were acquired using NIS-Elements 5.0 software (Figures 1, 3–5). Fixed RBCs were also imaged using a sensitive Zeiss LSM 880 Airyscan laser scanning confocal microscope with a 63× or 100× 1.46 NA oil Plan Apo objective (Figure 2). Z stacks were acquired at a digital zoom of 1.7 and a Z-step size of 0.168 μm (Figure 2). Note that standard confocal laser scanning microscopy was unable to image fluorescent-phalloidin staining in RBCs due to the dim signal as a consequence of quenching from the abundant hemoglobin (Smith et al., 2018), relatively low sensitivity of standard confocal microscopy, and photobleaching during confocal image acquisition.
Time-lapse images of live SiR-actin-labeled RBCs were acquired using a Nikon Ti inverted microscope by TIRF illumination as above. As a control, time-lapse images of fixed RBCs (4% PFA, overnight) labeled with Alexa 488-phalloidin were acquired using an ANDOR Dragonfly system by TIRF illumination with a 100× objective (N.A. 1.4). Live cell images of SiR-actin-labeled RBCs were also acquired using a Leica TCS SP8 STED 3× microscope equipped with a HC PL APO CS2 93×/1.30 glycerin objective lens. A 3D time series was collected with a detector window set to collect fluorescence emission at 655–751 nm. A white light laser was used to excite the fluors. STED images were collected using depletion lasers of 592, 660, and 775 nm; HyD detectors; and time gating of fluorescence emission between 0.3 and 6.5 ns. Bidirectional scanning at a frequency of 8000 Hz, with a scan resolution of 512 × 512 pixels and a line averaging of 5. Z stacks, were collected with a step size of 0.333 µm every 2.8 s. Postacquisition, the images were deconvolved using SVI Huygens Essential software.
Image analysis for TIRF and Zeiss AiryScan images
TIRF images were deconvolved using the 2D deconvolution module in Nikon Elements, NIS-Elements 5.0 software. The General Analysis tool in Elements was used to pinpoint centroids in the segmented deconvolved images and to calculate the nearest-neighbor distances between the spots. AiryScan confocal stacks of overnight-fixed RBCs stained with GPA or WGA for membrane and phalloidin for F-actin were processed using Zen 2.3 software and analyzed using Volocity 6.3.0 software. The outer 4–6 Z slices of the Airyscan stacks, corresponding to the outer 300–500 nm of each cell, were cropped in Nikon Elements. Extended focus projections of these stacks were exported into Tiff images and spot identification and density (spot/μm2) measurements were made using the Nikon Elements software as for TIRF images. For Figure 2C, pixel intensity measurements of the WGA and phalloidin stains were performed using similar thresholding parameters in the find object option in the measurement module of Volocity 6.3.0. Coefficients of variation were calculated from the SD and mean intensity values for each stain.
STORM imaging of F-actin
STORM imaging was performed as in Xu et al. (2013) and Pan et al. (2018), with minor modifications. Briefly, whole blood was obtained as above and washed in DPBS (Life Technologies [2.67 mM KCl, 1.47 mM KH2PO4, 137.93 mM NaCl, 8.06 mM Na2HPO4-7H2O]) containing 10 mM glucose and 5 mg/ml BSA. Cells were washed twice in this buffer by centrifugation at 1200 rpm for 5 min in a Beckman Avanti centrifuge swinging bucket rotor and resuspended at 3.0 × 106 cells in PBS with 10 mM glucose and 5 mg/ml BSA. RBCs were allowed to settle onto poly-l-lysine-coated coverslips (squeaky clean coverslips coated with a 0.1% (wt/vol) solution of poly-l-lysine [Sigma-Aldrich P8920] for 1–2 h room temperature, then dried and kept at 4°C until ready for use) for 10 min at room temperature (1.5 × 106 cells per coverslip). Attached cells were permeabilized in 0.0015% saponin in cytoskeleton buffer (CB; 10 mM MES, pH 6.1, 150 mM NaCl, 5 mM EGTA, 5 mM glucose, and 5 mM MgCl2) (Xu et al., 2013; Pan et al., 2018) for 4 min at room temperature, fixed in either 2% glutaraldehyde or 4% PFA (Electron Microscopy Sciences) in CB for 20 min room temperature, and reduced with freshly prepared 0.1% sodium borohydride in PBS for 20 min at room temperature. Cells were stained with 0.4 µM Alexa 647-phalloidin (Life Technologies) for 40 min in 3% BSA in PBS, washed in 3% BSA in DPBS, mounted with ∼20 μl Vectashield (Vector Laboratories Cat. No. H-1000), and sealed with nail polish.
To calibrate the blinking behavior of Alexa 647 under the experimental conditions used for this study, we used Alexa 647–conjugated Donkey anti-goat IgG (Invitrogen A31573, Batch 2181018, containing 5 molecules of dye per IgG molecule). The IgG molecules were attached to poly-L-lysine-coated coverslips at a density of ∼100 IgG molecules/μm2, coverslips were mounted with Vectashield as above, and the IgG molecules were imaged under the same conditions as the Alexa 647-phalloidin stained RBC membrane skeletons.
STORM imaging was performed on a Nikon Ti inverted microscope with a 100× Apochromat oil objective (NA 1.49) using TIRF illumination with the 647 nm laser line and an ORCA-Flash 4.0 V2 Digital CMOS camera (Hamamatsu) (Figure 3); 100,000 images, 10 ms each, were acquired using the STORM acquisition module in NIS-Elements 5.0 software. STORM images were processed for peak height of 175 with drift correction and analyzed using the DBSCAN function embedded in Nikon Elements 5.0 to visualize clustering of Alexa 647-phalloidin localizations (Supplemental Figure S3, D and E). Data were exported to Excel for statistical analysis and graph plotting in Prism 8.2 software, or for Ripley and DBSCAN analysis as indicated below.
Ripley clustering analysis
We used the open source library for the Ripley test analysis in MATLAB with parameters Klim, nSteps, and Nsimul (https:github.com/aludnam/MATLAB/tree/master/PatternAnalysis). Klim denotes the range of the radial distance, nSteps indicates the number of steps for the position of the K function estimation, and Nsimul represents the number of simulations for the envelope computation. In this study, we set 0 < Klim < 1 μm, nSteps = 100, and Nsimul = 100. We performed the Ripley analysis over a 4 μm × 4 μm square region of each RBC sample, and we modified the open source algorithm to plot Ripley’s H function (H(r) = L(r)-r) as a function of radial distance r.
DBSCAN clustering analysis
We used the DBSCAN algorithm (Schubert et al., 2017) implemented in the Python library scikit-learn (Pedregosa et al., 2011) for the clustering tasks with parameters epsilon (eps) and min_samples (Supplemental Figure S3, F–I), where eps denotes the maximum distance between two samples to be considered as the neighborhood of each other and min_samples indicates the number of samples in a neighborhood for a point to be considered as a core point, including itself. In this study, we did the clustering analysis for eps = 20 nm and ran the algorithm for min_samples = 2. After clustering the data, the algorithm gathers statistics on the results of the clusters, including the number of molecules per cluster size and the number of outliers, which are the molecules that are too far from any other molecules. To generate the random data sets, we determined the total number of localizations per cell in each experimental STORM data set and then distributed the same number of random points uniformly within the half of the RBC surface area (∼65 µm2) for each of six cells (example of one cell shown) (Supplemental Figure S3, F–I).
Statistical analysis
Data are presented in dot plots as mean ± SD (SD). Differences between means were detected using unpaired t tests. When more than one comparison was made, means were compared using one-way ANOVAs followed by Tukey’s multiple comparisons test. Statistical significance was defined as p < 0.05. Statistical analysis was performed using GraphPad Prism 7.03 software. Box and whisker plots show median values (horizontal center line), third and first quartiles (top and bottom of boxes), and the minimum and maximum of the data (whiskers), as well as the mean (+ sign).
Supplementary Material
Acknowledgments
We gratefully acknowledge Scott Henderson and the Scripps Microscopy Core for assistance with Leica SP8 Hyvolution microscopy of live RBCs, Megan Coffin and Jeff Caplan at the University of Delaware for assistance with TIRF imaging of fixed RBCs, and Bob Tian for helpful discussions about STORM analysis. This work was supported by National Institutes of Health/National Heart, Lung and Blood Institute Grant R01-HL083464 (to V.M.F.) and National Institutes of Health/National Institute of General Medicine Grant R01-GM132106 (to P.R.). H.A. gratefully acknowledges partial support from a Siebel Scholarship.
Abbreviations used:
- BB
blocking buffer
- BSA
bovine serum albumin
- CB
cytoskeleton buffer
- CytoD
cytochalasin D
- DBSCAN
density-based spatial clustering of applications with noise
- DPBS
Dulbecco’s phosphate-buffered saline
- EM
electron microscopy
- eps
epsilon
- FRAP
fluorescence recovery after photobleaching
- Glut4
glucose transporter
- GPA
glycophorin A
- HBS
HEPES-buffered saline
- Jasp
jasplakinolide
- LatA
latrunculin A
- NMIIA
nonmuscle myosin IIA
- PFA
paraformaldehyde
- RBC
red blood cell
- rho-phalloidin
rhodamine-phalloidin
- STORM
stochastic optical reconstruction microscopy
- TIRF
total internal reflection fluorescence.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E21-03-0107) on January 12, 2022.
REFERENCES
- Abouelezz A, Stefen H, Segerstrale M, Micinski D, Minkeviciene R, Lahti L, Hardeman EC, Gunning PW, Hoogenraad CC, Taira T, et al. (2020). Tropomyosin Tpm3.1 is required to maintain the structure and function of the axon initial segment. iScience 23, 101053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alenghat FJ, Golan DE (2013). Membrane protein dynamics and functional implications in mammalian cells. Curr Top Membr 72, 89–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alimohamadi H, Smith AS, Nowak RB, Fowler VM, Rangamani P (2020). Non-uniform distribution of myosin-mediated forces governs red blood cell membrane curvature through tension modulation. PLoS Comput Biol 16, e1007890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsic A, Stajkovic N, Spiegel R, Nikic-Spiegel I (2020). Effect of Vectashield-induced fluorescence quenching on conventional and super-resolution microscopy. Sci Rep 10, 6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett V, Baines AJ (2001). Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol Rev 81, 1353–1392. [DOI] [PubMed] [Google Scholar]
- Bennett V, Lorenzo DN (2013). Spectrin- and ankyrin-based membrane domains and the evolution of vertebrates. Curr Top Membr 72, 1–37. [DOI] [PubMed] [Google Scholar]
- Bennett V, Lorenzo DN (2016). An adaptable spectrin/ankyrin-based mechanism for long-range organization of plasma membranes in vertebrate tissues. Curr Top Membr 77, 143–184. [DOI] [PubMed] [Google Scholar]
- Brown JW, Bullitt E, Sriswasdi S, Harper S, Speicher DW, McKnight CJ (2015). The physiological molecular shape of spectrin: a compact supercoil resembling a Chinese finger trap. PLoS Comput Biol 11, e1004302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers TJ, Branton D (1985). Visualization of the protein associations in the erythrocyte membrane skeleton. Proc Natl Acad Sci USA 82, 6153–6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caetano FA, Dirk BS, Tam JH, Cavanagh PC, Goiko M, Ferguson SS, Pasternak SH, Dikeakos JD, de Bruyn JR, Heit B (2015). MIiSR: Molecular interactions in super-resolution imaging enables the analysis of protein interactions, dynamics and formation of multi-protein structures. PLoS Comput Biol 11, e1004634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Boyle FJ (2017). An enhanced spring-particle model for red blood cell structural mechanics: application to the stomatocyte-discocyte-echinocyte transformation. J Biomech Eng 139, 121009. [DOI] [PubMed] [Google Scholar]
- Chien S (1987). Red cell deformability and its relevance to blood flow. Annu Rev Physiol 49, 177–192. [DOI] [PubMed] [Google Scholar]
- D’Este E, Kamin D, Gottfert F, El-Hady A, Hell SW (2015). STED nanoscopy reveals the ubiquity of subcortical cytoskeleton periodicity in living neurons. Cell Rep 10, 1246–1251. [DOI] [PubMed] [Google Scholar]
- Derick LH, Liu SC, Chishti AH, Palek J (1992). Protein immunolocalization in the spread erythrocyte membrane skeleton. Eur J Cell Biol 57, 317–320. [PubMed] [Google Scholar]
- Dhermy D, Schrevel J, Lecomte MC (2007). Spectrin-based skeleton in red blood cells and malaria. Curr Opin Hematol 14, 198–202. [DOI] [PubMed] [Google Scholar]
- Discher DE, Boal DH, Boey SK (1998). Simulations of the erythrocyte cytoskeleton at large deformation. II. Micropipette aspiration. Biophys J 75, 1584–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fai TG, Leo-Macias A, Stokes DL, Peskin CS (2017). Image-based model of the spectrin cytoskeleton for red blood cell simulation. PLoS Comput Biol 13, e1005790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedosov DA, Caswell B, Karniadakis GE (2010). A multiscale red blood cell model with accurate mechanics, rheology, and dynamics. Biophys J 98, 2215–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Waugh RE, Peng Z (2020). Constitutive model of erythrocyte membranes with distributions of spectrin orientations and lengths. Biophys J 119, 2190–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler V, Branton D. (1977). Lateral mobility of human erythrocyte integral membrane proteins. Nature 268, 23–26. [DOI] [PubMed] [Google Scholar]
- Fowler VM (2013). The human erythrocyte plasma membrane: a Rosetta Stone for decoding membrane-cytoskeleton structure. Curr Top Membr 72, 39–88. [DOI] [PubMed] [Google Scholar]
- Fowler VM, Bennett V (1978). Association of spectrin with its membrane attachment site restricts lateral mobility of human erythrocyte integral membrane proteins.. J Supramol Struct 8, 215–221. [Google Scholar]
- Fowler VM, Davis JQ, Bennett V (1985). Human erythrocyte myosin: identification and purification. J Cell Biol 100, 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher PG (2013). Abnormalities of the erythrocyte membrane. Pediatr Clin North Am 60, 1349–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Parajo MF, Cambi A, Torreno-Pina JA, Thompson N, Jacobson K (2014). Nanoclustering as a dominant feature of plasma membrane organization. J Cell Sci 127, 4995–5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier EF, Leduc M, Cochet S, Bailly K, Lacombe C, Mohandas N, Guillonneau F, El Nemer W, Mayeux P (2018). Absolute proteome quantification of highly purified populations of circulating reticulocytes and mature erythrocytes. Blood Adv 2, 2646–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghisleni A, Galli C, Monzo P, Ascione F, Fardin MA, Scita G, Li Q, Maiuri P, Gauthier NC (2020). Complementary mesoscale dynamics of spectrin and acto-myosin shape membrane territories during mechanoresponse. Nat Commun 11, 5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhin DS, Fowler VM (2016). Feisty filaments: actin dynamics in the red blood cell membrane skeleton. Curr Opin Hematol 23, 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhin DS, Nowak RB, Khoory JA, de la Piedra A, Ghiran IC, Fowler VM (2015). Dynamic actin filaments control the mechanical behavior of the human red blood cell membrane. Mol Biol Cell 26, 1699–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golan DE, Veatch W (1980). Lateral mobility of band 3 in the human erythrocyte membrane studied by fluorescence photobleaching recovery: evidence for control by cytoskeletal interactions. Proc Natl Acad Sci USA 77, 2537–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff J (2015). The airyscan detector from ZEISS: Confocal imagingwith improved signal-to-noise ratio and super-resolution. Nature Methods 12. [Google Scholar]
- Jenkins PM, He M, Bennett V (2015). Dynamic spectrin/ankyrin-G microdomains promote lateral membrane assembly by opposing endocytosis. Sci Adv 1, e1500301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiskowski MA, Hancock JF, Kenworthy AK (2009). On the use of Ripley’s K-function and its derivatives to analyze domain size. Biophys J 97, 1095–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodippili GC, Giger K, Putt KS, Low PS (2020). DARC, Glycophorin A, Band 3, and GLUT1 Diffusion in Erythrocytes: Insights into Membrane Complexes. Biophys J 119, 1749–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodippili GC, Spector J, Hale J, Giger K, Hughes MR, McNagny KM, Birkenmeier C, Peters L, Ritchie K, Low PS (2012). Analysis of the mobilities of band 3 populations associated with ankyrin protein and junctional complexes in intact murine erythrocytes. J Biol Chem 287, 4129–4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman PA, Fowler VM (1997). Purification and characterization of an alpha 1 beta 2 isoform of CapZ from human erythrocytes: cytosolic location and inability to bind to Mg2+ ghosts suggest that erythrocyte actin filaments are capped by adducin. Biochemistry 36, 13461–13472. [DOI] [PubMed] [Google Scholar]
- Kusumi A, Fujiwara TK, Chadda R, Xie M, Tsunoyama TA, Kalay Z, Kasai RS, Suzuki KG (2012a). Dynamic organizing principles of the plasma membrane that regulate signal transduction: commemorating the fortieth anniversary of Singer and Nicolson’s fluid-mosaic model. Annu Rev Cell Dev Biol 28, 215–250. [DOI] [PubMed] [Google Scholar]
- Kusumi A, Fujiwara TK, Morone N, Yoshida KJ, Chadda R, Xie M, Kasai RS, Suzuki KG (2012b). Membrane mechanisms for signal transduction: the coupling of the meso-scale raft domains to membrane-skeleton-induced compartments and dynamic protein complexes. Semin Cell Dev Biol 23, 126–144. [DOI] [PubMed] [Google Scholar]
- Leterrier C (2021). Putting the axonal periodic scaffold in order. Curr Opin Neurobiol 69, 33–40. [DOI] [PubMed] [Google Scholar]
- Leterrier C, Potier J, Caillol G, Debarnot C, Rueda Boroni F, Dargent B (2015). Nanoscale architecture of the axon initial segment reveals an organized and robust scaffold. Cell Rep 13, 2781–2793. [DOI] [PubMed] [Google Scholar]
- Li H, Lykotrafitis G (2014). Erythrocyte membrane model with explicit description of the lipid bilayer and the spectrin network. Biophys J 107, 642–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Dao M, Lim CT, Suresh S (2005). Spectrin-level modeling of the cytoskeleton and optical tweezers stretching of the erythrocyte. Biophys J 88, 3707–3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Lykotrafitis G, Dao M, Suresh S (2007). Cytoskeletal dynamics of human erythrocyte. Proc Natl Acad Sci USA 104, 4937–4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SC, Derick LH, Palek J (1987). Visualization of the hexagonal lattice in the erythrocyte membrane skeleton. J Cell Biol 104, 527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukinavicius G, Umezawa K, Olivier N, Honigmann A, Yang G, Plass T, Mueller V, Reymond L, Correa IR Jr, Luo ZG, et al. (2013). A near-infrared fluorophore for live-cell super-resolution microscopy of cellular proteins. Nat Chem 5, 132–139. [DOI] [PubMed] [Google Scholar]
- Lux SE (2016). Anatomy of the red cell membrane skeleton: unanswered questions. Blood 127, 187–199. [DOI] [PubMed] [Google Scholar]
- Machnicka B, Czogalla A, Hryniewicz-Jankowska A, Boguslawska DM, Grochowalska R, Heger E, Sikorski AF (2014). Spectrins: a structural platform for stabilization and activation of membrane channels, receptors and transporters. Biochim Biophys Acta 1838, 620–634. [DOI] [PubMed] [Google Scholar]
- Mattheyses AL, Simon SM, Rappoport JZ (2010). Imaging with total internal reflection fluorescence microscopy for the cell biologist. J Cell Sci 123, 3621–3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGough AM, Josephs R. (1990). On the structure of erythrocyte spectrin in partially expanded membrane skeletons. Proc Natl Acad Sci USA 87, 5208–5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohandas N, Evans E (1994). Mechanical properties of the red cell membrane in relation to molecular structure and genetic defects. Annu Rev Biophys Biomol Struct 23, 787–818. [DOI] [PubMed] [Google Scholar]
- Mohandas N, Gallagher PG (2008). Red cell membrane: past, present, and future. Blood 112, 3939–3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nans A, Mohandas N, Stokes DL (2011). Native ultrastructure of the red cell cytoskeleton by cryo-electron tomography. Biophys J 101, 2341–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolson GL (2014). The Fluid-Mosaic Model of Membrane Structure: still relevant to understanding the structure, function and dynamics of biological membranes after more than 40 years. Biochim Biophys Acta 1838, 1451–1466. [DOI] [PubMed] [Google Scholar]
- Pageon SV, Nicovich PR, Mollazade M, Tabarin T, Gaus K (2016). Clus-DoC: a combined cluster detection and colocalization analysis for single-molecule localization microscopy data. Mol Biol Cell 27, 3627–3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal K, Nowak R, Billington N, Liu R, Ghosh A, Sellers JR, Fowler VM (2020). Megakaryocyte migration defects due to nonmuscle myosin IIA mutations underlie thrombocytopenia in MYH9-related disease. Blood 135, 1887–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan L, Yan R, Li W, Xu K (2018). Super-resolution microscopy reveals the native ultrastructure of the erythrocyte cytoskeleton. Cell Rep 22, 1151–1158. [DOI] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. (2011). Scikit-learn: Machine learning in python. J Mach Learn Res 12, 2825–2830. [Google Scholar]
- Peng Z, Li X, Pivkin IV, Dao M, Karniadakis GE, Suresh S (2013). Lipid bilayer and cytoskeletal interactions in a red blood cell. Proc Natl Acad Sci USA 110, 13356–13361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripley BD (1977). Modeling spatial patterns. J Roy Stat Soc B Met 39, 172–212. [Google Scholar]
- Schubert E, Sander J, Ester M, Kriegel H, Xu X (2017). DBSCAN revisited, revisited: Why and how you should (still) use DBSCAN. ACM Trans Database Syst 42.3, 1–21. [Google Scholar]
- Sheetz MP (1983). Membrane skeletal dynamics: role in modulation of red cell deformability, mobility of transmembrane proteins, and shape. Semin Hematol 20, 175–188. [PubMed] [Google Scholar]
- Shen BW, Josephs R, Steck TL (1986). Ultrastructure of the intact skeleton of the human erythrocyte membrane. J Cell Biol 102, 997–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AS, Nowak RB, Zhou S, Giannetto M, Gokhin DS, Papoin J, Ghiran IC, Blanc L, Wan J, Fowler VM (2018). Myosin IIA interacts with the spectrin-actin membrane skeleton to control red blood cell membrane curvature and deformability. Proc Natl Acad Sci USA 10.1073/pnas.1718285115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AS, Pal K, Nowak RB, Demenko A, Zaninetti C, Da Costa L, Favier R, Pecci A, Fowler VM (2019). MYH9-related disease mutations cause abnormal red blood cell morphology through increased myosin-actin binding at the membrane. Am J Hematol 94, 667–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui Z, Gokhin DS, Nowak RB, Guo X, An X, Fowler VM (2017). Stabilization of F-actin by tropomyosin isoforms regulates the morphology and mechanical behavior of red blood cells. Mol Biol Cell 28, 2531–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swihart AH, Mikrut JM, Ketterson JB, Macdonald RC (2001). Atomic force microscopy of the erythrocyte membrane skeleton. J Microsc 204, 212–225. [DOI] [PubMed] [Google Scholar]
- Takeuchi M, Miyamoto H, Sako Y, Komizu H, Kusumi A (1998). Structure of the erythrocyte membrane skeleton as observed by atomic force microscopy. Biophys J 74, 2171–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terada N, Fujii Y, Ohno S (1996). Three-dimensional ultrastructure of in situ membrane skeletons in human erythrocytes by quick-freezing and deep-etching method. Histol Histopathol 11, 787–800. [PubMed] [Google Scholar]
- Ursitti JA, Fowler VM (1994). Immunolocalization of tropomodulin, tropomyosin and actin in spread human erythrocyte skeletons. J Cell Sci 107 (Pt 6), 1633–1639. [DOI] [PubMed] [Google Scholar]
- Ursitti JA, Pumplin DW, Wade JB, Bloch RJ (1991). Ultrastructure of the human erythrocyte cytoskeleton and its attachment to the membrane. Cell Motil Cytoskel 19, 227–243. [DOI] [PubMed] [Google Scholar]
- Xu K, Zhong G, Zhuang X (2013). Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science 339, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong G, He J, Zhou R, Lorenzo D, Babcock HP, Bennett V, Zhuang X (2014). Developmental mechanism of the periodic membrane skeleton in axons. Elife 3, e04581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Han B, Nowak R, Lu Y, Heller E, Xia C, Chishti AH, Fowler VM, Zhuang X (2020). Proteomic and functional analyses of the periodic membrane skeleton in neurons. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccala ES, Baum J (2011). Cytoskeletal and membrane remodelling during malaria parasite invasion of the human erythrocyte. Br J Haematol 154, 680–689. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.