

Abstract
Transcriptional factor EB (TFEB) is a master regulator of genes required for autophagy and lysosomal function. The nuclear localization of TFEB is blocked by the mechanistic target of rapamycin complex 1 (mTORC1)-dependent phosphorylation of TFEB at multiple sites including Ser-211. Here we show that inhibition of PIKfyve, which produces phosphatidylinositol 3,5-bisphosphate on endosomes and lysosomes, causes a loss of Ser-211 phosphorylation and concomitant nuclear localization of TFEB. We found that while mTORC1 activity toward S6K1, as well as other major mTORC1 substrates, is not impaired, PIKfyve inhibition specifically impedes the interaction of TFEB with mTORC1. This suggests that mTORC1 activity on TFEB is selectively inhibited due to loss of mTORC1 access to TFEB. In addition, we found that TFEB activation during inhibition of PIKfyve relies on the ability of protein phosphatase 2A (PP2A) but not calcineurin/PPP3 to dephosphorylate TFEB Ser-211. Thus when PIKfyve is inhibited, PP2A is dominant over mTORC1 for control of TFEB phosphorylation at Ser-S211. Together these findings suggest that mTORC1 and PP2A have opposing roles on TFEB via phosphorylation and dephosphorylation of Ser-211, respectively, and further that PIKfyve inhibits TFEB activity by facilitating mTORC1-dependent phosphorylation of TFEB.
INTRODUCTION
Lysosomes are acidic organelles, which are crucial for the degradation of many different proteins and lipids. The degradative products, including amino acids, are then released and reused for cell survival (Chen et al., 2017; Inpanathan and Botelho, 2019; Oyarzún et al., 2019). In addition, lysosomes serve as storage sites for several ions including calcium (Ca2+). Ca2+ release from lysosomes occurs in response to starvation through the transient receptor potential mucolipin 1 channel (Medina et al., 2015; Wang et al., 2015b; Cao et al., 2017) and the two-pore segment channels 1 and 2 (Ogunbayo et al., 2018). This local Ca2+ elevation causes the activation of enzymes such as protein phosphatases, which participate in the maintenance of cellular homeostasis (Medina et al., 2015; Martina et al., 2016; Zhang et al., 2016; Zhitomirsky et al., 2018). Moreover, lysosomes serve as a platform for several intracellular signaling pathways including the mechanistic target of rapamycin (mTOR) complex 1 (mTORC1).
In response to amino acid stimulation, the Rag small GTPases, which exist as heterodimers, become active when the complex contains GTP-bound RagA or RagB with GDP-bound RagC or RagD. The active heterodimers recruit cytoplasmic mTORC1 to the surface of the lysosomal membrane (Sekiguchi et al., 2001; Gao and Kaiser, 2006; Sancak et al., 2008, 2010; Bar-Peled et al., 2012) where mTORC1 is activated by another small GTPase, Rheb (Saucedo et al., 2003; Stocker et al., 2003). Active mTORC1 phosphorylates its downstream targets including p70S6K (S6K1), 4E-BP1, unc-51-like kinase 1 (ULK1), and transcriptional factor EB (TFEB) (Martina et al., 2012; Roczniak-Ferguson et al., 2012; Settembre et al., 2012; Lim and Zoncu, 2016; Saxton and Sabatini, 2017; Yao et al., 2017; Lawrence and Zoncu, 2019). Interestingly, a recent study demonstrated that TFEB phosphorylation by mTORC1 on the lysososme does not require Rheb (Napolitano et al., 2020).
TFEB and the related protein TFE3 have been recently identified as critical transcriptional factors that regulate multiple genes involved in lysosomal biogenesis, autophagy, and additional stress response pathways (Sardiello et al., 2009; Settembre et al., 2011, 2012; Martina et al., 2012, 2014; Roczniak-Ferguson et al., 2012; Raben and Puertollano, 2016). Under nutrient-rich conditions, TFEB binding to Rag GTPases results in a specific substrate-dependent recruitment mechanism and results in mTORC1-dependent phosphorylation of TFEB (Napolitano et al., 2020). mTORC1 phosphorylates TFEB at multiple sites including Ser-142 and Ser-211 (Roczniak-Ferguson et al., 2012; Settembre et al., 2012). TFEB phosphorylation at Ser-211 promotes TFEB binding to 14-3-3 proteins (Settembre et al., 2012; Martina and Puertollano, 2013; Martina et al., 2014), and this phosphorylation likely masks a nuclear localization signal,and results in the retention of TFEB in the cytoplasm (Roczniak-Ferguson et al., 2012). In contrast, phosphorylation at Ser-142 induces the nuclear export of TFEB (Li et al., 2018; Napolitano et al., 2018). In the absence of nutrients, TFEB is dephosphorylated and translocates to the nucleus, where it enhances the transcription of target genes. TFEB dephosphorylation occurs during starvation-induced inactivation of mTORC1 and/or activation of protein phosphatase calcineurin/PPP3 (Medina et al., 2015; Zhang et al., 2016).
Phosphoinositide lipids (PIs) are essential for multiple intracellular events including signal transduction, remodeling of the actin cytoskeleton, and membrane transport (Sasaki et al., 2009; Balla, 2013; Ebner et al., 2019). These lipids are localized on the cytoplasmic leaflet of most organelles as well as the plasma membrane. There are seven PIs in mammalian cells. Generation of these PIs occurs via phosphorylation of phosphatidylinositol and polyphosphorylated phosphatidylinositol species via the addition of phosphate to the 3, 4, and 5 positions of the inositol ring.
Phosphatidylinositol 3,5-bisphosphate [PI(3,5)P2], which is among the least abundant PI, has critical roles in membrane traffic, ion homeostasis in lysosomes, as well as the regulation of the function and size of late endosomes/lysosomes (Ho et al., 2012; Shisheva, 2012; Jin et al., 2016; Hasegawa et al., 2017). PI(3,5)P2 is generated from phosphatidylinositol 3-phosphate (PI3P) by a phosphoinositide 5-kinase PIKfyve (Fab1 in yeast) which forms a complex with the regulators Vac14 and Fig4 to control the kinase activity. Indeed, suppression of PIKfyve/Fab1, or its conserved positive regulators, leads to loss of cellular PI(3,5)P2 levels in both mammalian cells (Chow et al., 2007; Zolov et al., 2012; Takasuga et al., 2013) and yeast (Duex et al., 2006a,b). In addition, in mammalian cells, PIKfvye is also responsible for most of the cellular pools of PI5P (Shisheva, 2012; Zolov et al., 2012). Importantly, multiple studies have shown that depletion or inhibition of PIKfyve expands the size and volume of late endosomes/lysosomes (Ikonomov et al., 2002; Duex et al., 2006b; Rutherford et al., 2006; Chow et al., 2007; Jefferies et al., 2008; Cai et al., 2013; Compton et al., 2016), which fits with several studies that show that PIKfyve regulates multiple lysosomal proteins. Moreover, inhibition of PIKfyve induces the nuclear translocation of TFEB (Choy et al., 2018); however, the molecular mechanisms underlying PIKfyve-dependent regulation of TFEB regulation were unclear.
Here we find that PIKfyve is a positive regulator of TFEB retention on the lysosome and prevents TFEB translocation to the nucleus via regulation of the mTORC1-dependent phosphorylation site on TFEB, Ser-211. Upon PIKfyve inhibition, TFEB is dephosphorylated and translocates into the nucleus. Surprisingly, we found that mTORC1 is not globally inhibited during PIKfyve inhibition; phosphorylation of each of several mTORC1 substrates tested, including S6K1, 4E-BP1, and ULK1, occurs during PIKfyve inhibition, which initially raised the possibility that the role of PIKfyve in TFEB phosphorylation was independent of mTORC1. We further found that during PIKfyve inhibition, the dephosphorylation of TFEB as well as its nuclear translocation is prevented by blocking protein phosphatase 2A (PP2A) activity either by knockdown or via the addition of okadaic acid, an inhibitor of PP2A. Conversely, activation of PP2A via expression of the catalytic subunit of PP2A, but not its inactive mutant, was sufficient to induce the dephosphorylation of TFEB. These findings show that PP2A is a regulator of the mTORC1 phosphorylation site, Ser-211. Importantly, we found that PIKfyve inhibition impedes the interaction of TFEB with mTORC1, which suggests that PIKfyve inhibition selectively inhibits mTORC1 activity on TFEB by interfering with mTORC1 access to TFEB. In this situation PP2A becomes dominant over mTORC1 for the control of TFEB phosphorylation status. Together these findings suggest that mTORC1 and PP2A have opposing roles on TFEB via phosphorylation and dephosphorylation of Ser-211, respectively. Furthermore, these findings reveal that PIKfyve inhibits TFEB activity by facilitating mTORC1-dependent phosphorylation of TFEB on Ser-211.
RESULTS
PIKfyve does not globally regulate mTORC1 activity
In mammalian cells, mTORC1 activity, as measured by phosphorylation of S6K1 and 4E-BP1, is not altered during PIKfyve inhibition (Wang et al., 2015b; Choy et al., 2018). To investigate this further, we inhibited PIKfyve with apilimod (Cai et al., 2013) or YM201636 (Jefferies et al., 2008) and tested the phosphorylation status of several additional mTORC1 substrates. Consistent with previous reports, we found that treatment with apilimod had no obvious impact on the phosphorylation of S6K1, 4E-BP1, and ULK1 (Figure 1, A and B). In addition, these substrates were phosphorylated in the presence of YM201636, another inhibitor of PIKfyve (Supplemental Figure S1A). Vps34 is a PI3-kinase which provides approximately two-thirds of the cellular PI3P pool including two-thirds of the pool utilized by PIKfyve (Devereaux et al., 2013; Ikonomov et al., 2015). Accordingly, inhibition of Vps34 provides another approach to lower PI(3,5)P2 and PI5P. Similar to PIKfyve inhibition, we found that inhibition of Vps34 with VPS34-IN1 (Bago et al., 2014) resulted in a less than 30% drop in the phosphorylation of S6K1 (Supplemental Figure S1A). Together these results suggest that in HeLa cells, PIKfyve does not regulate the activity of mTORC1. Similarly, in MEF cells inhibition of PIKfyve with apilimod or YM201636, or inhibition of Vps34 with VPS34-IN1, did not result in a loss of S6K1 phosphorylation (Supplemental Figure S1B). Moreover, in HEK293T cells, treatment with apilimod or YM201636 did not inhibit the phosphorylation S6K1 (Supplemental Figure S1C). Furthermore, it is unlikely that serum destabilizes apilimod. We did not observe differences in S6K1 phosphorylation when apilimod was added to media that includes amino acids and glucose but is serum free (Figure 1C). This indicates that factors in serum do not affect the stability of apilimod or contribute to the lack of effect of apilimod on mTORC1 activity.
FIGURE 1:

PIKfyve inhibition does not suppress mTORC1 activity. (A) HeLa cells treated with the indicated drugs (or DMSO vehicle control) were cultured for 2 h in the growth medium or HBSS for 2 h and then analyzed by immunoblot using the indicated antibodies. (B) Quantitation of phospho-S6K1 signal intensities from immunoblots in A, following normalization to the total S6K1 protein (mean ± SD; three independent experiments). **P < 0.01. (C) HeLa cells treated with apilimod were cultured for 2 h in the indicated medium (FBS[+], 10% FBS-DMEM; FBS[-], DMEM; HBSS) and then analyzed by immunoblot using anti-phospho-S6K1 and anti-S6K1 antibodies. Representative immunoblot from three independent experiments.
The PI3-kinase/Akt pathway is upstream of mTORC1 activation (Dienstmann et al., 2014). Therefore we tested whether Akt activation is altered by inhibition of PIKfyve. However, apilimod treatment did not change in the phosphorylation of Akt sites Thr-308 or Ser-473 (Figure 1A).
Inhibition of PIKfyve results in the dephosphorylation of TFEB Ser-211 and the translocation of TFEB to the nucleus
TFEB, a direct downstream target of mTORC1, is a critical transcription factor that regulates lysosomal and autophagic genes (Settembre et al., 2011; Raben and Puertollano, 2016). Phosphorylated TFEB is inactive and is localized in the cytoplasm, whereas when mTORC1 activity is suppressed either by starvation or by an mTOR inhibitor, e.g., Torin1, TFEB translocates to the nucleus and induces the expression of genes that are regulated by TFEB. Similarly, overexpression of phosphatases that act on TFEB such as calcineurin/PPP3 (Medina et al., 2015; Zhang et al., 2016) results in TFEB activation and translocation to the nucleus. As a first test of whether the phosphorylation status of TFEB is altered by PIKfyve inhibition, we performed immunoblot analysis using an anti-TFEB antibody. Phosphorylated TFEB migrates more slowly compared with dephosphorylated TFEB. In control cells grown in nutrient-rich media, TFEB is phosphorylated and is detected as an upper band; however, when mTORC1 is suppressed, via starvation or mTORC1 inhibition, TFEB is dephosphorylated and migrates faster (Figure 1A). Notably, on apilimod treatment only the lower band (dephosphorylated form) of TFEB is observed, despite the fact that other mTORC1 substrates, p-S6K1, p-4E-BP1, and p-ULK1, are observed. To test whether this band shift reflects the dephosphorylation of TFEB at a previously studied phosphorylation site, we used a commercially available anti-phospho-TFEB (Ser-211) antibody, which is directed against a known mTORC1 site. As expected, we detected phospho-TFEB (Ser-211) in nutrient-fed DMSO-treated cells; conversely, when mTORC1 was inhibited in cells starved in Hank’s balanced salt solution (HBSS) or treated with Torin1, phospho-TFEB (Ser-211) was not detected. Notably, phosphorylation at Ser-211 was also not present following apilimod treatment (Figure 1A). This indicates that phosphorylation at this site is dependent on PIKfyve activity.
Since dephosphorylated TFEB normally translocates into the nucleus, we also tested the localization of TFEB-GFP in HeLa cells treated with apilimod. Consistent with the loss of phosphorylation at TFEB Ser-211, apilimod treatment resulted in TFEB localization to the nucleus, whereas in DMSO-treated control cells, TFEB-GFP displayed a cytoplasmic pattern (Figure 2, A and B) (Wang et al., 2015b; Choy et al., 2018). Moreover, treatment with YM201636, another PIKfyve inhibitor, or VPS34-IN1, an inhibitor of VPS34, also resulted in the localization of TFEB to the nucleus (Supplemental Figure S2, A and B). We also found that TFE3, another MiT/TFE family’s transcriptional factor, showed strong nuclear localization in apilimod- or YM201636-treated cells (Supplemental Figure S3, A and B), which indicates that similar to TFEB, TFE3 localization to the cytoplasm also depends on PIKfyve activity.
FIGURE 2:

PIKfyve inhibition induces the dephosphorylation of TFEB and nuclear translocation of TFEB. (A) HeLa cells stably expressing TFEB-GFP treated with the indicated drugs were cultured for 1 h in the growth medium or HBSS for 1 h. Cells were fixed and stained with DAPI for nuclear staining and then analyzed by immunofluorescence microscopy. Bar, 10 µm. (B) Percentage of TFEB-GFP that is localized to the nucleus in A (mean ± SD; n > 100 cells from three independent experiments). **P < 0.01. (C) HeLa cells were transfected with siControl or siPIKfyve for 72 h and then analyzed by immunoblot using the indicated antibodies. (D) HeLa cells stably expressing TFEB-GFP were transfected with siControl or siPIKfyve for 72 h. Cells were fixed and stained with DAPI for nuclear staining and then analyzed by immunofluorescence microscopy. Bar, 10 µm. (E) Percentage of TFEB-GFP that is localized to the nucleus in D (mean ± SD; n > 50 cells from three independent experiments). **P < 0.01.
Since TFEB and TFE3 are critical transcriptional factors for lysosomal and autophagy genes expression (Settembre et al., 2011; Shen and Mizushima, 2014; Raben and Puertollano, 2016), we tested whether these genes are up-regulated by PIKfyve suppression. As expected, the transcripts of genes for autophagy and lysosomal function examined in this study are significantly increased by PIKfyve inhibitors compared with a DMSO control (Supplemental Figure S4).
To eliminate possible off-target effects of PIKfyve inhibitors on TFEB phosphorylation, we also performed PIKfyve knockdown experiments. Similar to treatment with apilimod, PIKfyve knockdown showed the loss of TFEB phosphorylation (Figure 2C) as well as the nuclear translocation of TFEB (Figure 2, D and E). Conversely, to determine the impact of elevated PIKfyve activity, we utilized a hyperactive mutant of PIKfyve (PIKfyve-KYA) which elevates the levels of PI(3,5)P2 and PI5P (McCartney et al., 2014) and tested the localization of TFEB in cells expressing PIKfyve-wild type (WT) or PIKfyve-KYA under starved conditions. Importantly, the expression of PIKfyve-KYA, but not PIKfyve-WT or vector control, suppressed the starvation-induced nuclear localization of TFEB-GFP (Supplemental Figure S5, A and B). Thus increasing PI(3,5)P2 and/or PI5P levels suppresses the nuclear localization of TFEB. Together, these results suggest that cellular PI(3,5)P2 and/or PI5P levels play a role in maintaining TFEB/TFE3 localization in the cytoplasm.
PP2A plays a role in the dephosphorylation of TFEB
The phosphatase, calcineurin/PPP3 dephosphorylates TFEB during starvation (Medina et al., 2015). Therefore we tested whether calcineurin/PPP3 activity plays a role in the phosphorylation of TFEB Ser-211 during PIKfyve inhibition. We cotreated cells with FK-506 (Tacrolimus), a potent calcineurin/PPP3 inhibitor, and apilimod. However, apilimod-dependent TFEB dephosphorylation, as well as its nuclear translocation, was not suppressed by cotreatment with FK-506 even at a high dose (Supplemental Figure S6, A–C). These data indicate that dephosphorylation of TFEB during PIKfyve inhibition does not depend on calcineurin/PPP3.
In addition to calcineurin, PP2A was recently reported to participate in the dephosphorylation of TFEB in response to oxidative stress (Martina and Puertollano, 2018). As the first test of a more general role for PP2A in dephosphorylation of TFEB, we simultaneously treated cells with apilimod and okadaic acid, an inhibitor of PP2A activity. Notably, treatment with okadaic acid restored the apilimod-dependent loss of TFEB phosphorylation in a dose-dependent manner (Figure 3, A and B). Similar results were obtained in cells simultaneously treated with okadaic acid and YM201636, another PIKfyve inhibitor (Supplemental Figure S7, A and B). Importantly, consistent with the okadaic acid-dependent restoration of the phosphorylation of TFEB (Ser-211), the cytoplasmic localization of TFEB was also rescued during cotreatment of cells with apilimod and okadaic acid, while treatment with apilimod alone resulted in TFEB-GFP localization to the nucleus (Figure 3, C and D). These data raised the possibility that PP2A activity is negatively regulated by PIKfyve, and thus when PIKfvye is inhibited, there is enhanced dephosphorylation of TFEB by PP2A.
FIGURE 3:

Treatment with okadaic acid restores dephosphorylation and nuclear translocation of TFEB induced by PIKfyve inhibition. (A) HeLa cells treated with 1 µM apilimod (or DMSO vehicle control) and okadaic acid at the indicated concentration were cultured for 1 h in the growth medium and then analyzed by immunoblot using the indicated antibodies. (B) Quantitation of phospho-TFEB signal intensities from immunoblots in A, following normalization to the total TFEB protein (mean ± SD; three independent experiments). (C) HeLa cells stably expressing TFEB-GFP treated with 1 µM apilimod and 400 nM okadaic acid were cultured for 1 h in the growth medium. Cells were fixed and then analyzed by immunofluorescence microscopy. Bar, 10 µm. (D) Percentage of TFEB-GFP that is localized to the nucleus in C (mean ± SD; n > 50 cells from three independent experiments). **P < 0.01.
Global PP2A activity does not require PIKfyve
PP2A dephosphorylates several targets including the extracellular signal-regulated kinase (ERK)1/2, glycogen synthase kinase 3 (GSK3) β, c-Myc, and eukaryotic translation initiation factor 4E (elF4E) (Yeh et al., 2004; Letourneux et al., 2006; Jeon et al., 2010; Li et al., 2010). Therefore we tested whether inhibition of PIKfyve enhances the dephosphorylation of any of these substrates. We found that apilimod treatment showed little effect on the dephosphorylation of both ERK1/2 and GSK3β. However, apilimod accelerated the dephosphorylation of eIF4E, a substrate of MNK kinases (Figure 4, A and B) (Ueda et al., 2004). Importantly, all of the PP2A substrates tested showed enhanced phosphorylation in the presence of okadaic acid, which indicates that in the cell line used, HeLa cells, these substrates are all targets of PP2A. These observations suggest that PIKfyve does not directly inhibit all forms of PP2A activity but leaves open the possibility that PIKfyve may play a role in regulating PP2A activity on some of its substrates.
FIGURE 4:

PP2A knockdown suppresses PIKfyve-inhibition-dependent dephosphorylation and nuclear translocation of TFEB. (A) HeLa cells treated with 1 µM apilimod (or DMSO vehicle control) and okadaic acid at the indicated concentration were cultured for 1 h in the growth medium and then analyzed by immunoblot using the indicated antibodies. (B) Quantitation of phospho-elF4E signal intensities from immunoblots in A, following normalization to the total elF4E protein (mean ± SD; three independent experiments). **P < 0.01. (C) HeLa cells transfected with siControl, siPP2A-Cα and siPP2A-Cβ were treated with 1 µM apilimod (DMSO as a control) for 2 h and then analyzed by immunoblot using the indicated antibodies. (D) Quantitation of phospho-TFEB signal intensities from immunoblots in C, following normalization to the total TFEB protein (mean ± SD; three independent experiments). **P < 0.01. (E) HeLa cells transfected with siControl, siPP2A-Cα, and siPP2A-Cβ were treated with 1 µM apilimod (or DMSO vehicle control) for 2 h. Cells were fixed and then analyzed by immunofluorescence microscopy. Bar, 10 µm. (F) Percentage of TFEB-GFP that is localized to the nucleus in E (mean ± SD; n > 50 cells from three independent experiments). **P < 0.01.
Apilimod-induced dephosphorylation of TFEB is suppressed by PP2A knockdown
In addition to inhibiting PP2A, okadaic acid has many off-target effects. Thus we specifically knocked down PP2A. In cells transfected with siRNAs against both PP2A catalytic subunit α/β (PPP2CA and PPP2CB), we detected TFEB phosphorylation at Ser-211 even when PIKfyve was inhibited (Figure 4, C and D). Importantly, apilimod-dependent translocation of TFEB to the nucleus was suppressed by the knockdown of both catalytic subunits of PP2A (Figure 4, E and F). Moreover, we confirmed that the ectopic expression of mouse PPP2CA (resistant to human siRNA) could decrease the phosphorylation of TFEB in PPP2CA-depleted cells (Supplemental Figure S8, A and B), indicating that the increase in TFEB phosphorylation by the PPP2CA knockdown is not due to off-target effects of the siRNA. Although we could not detect ectopic mouse PPP2CA with the anti-PPP2CA antibody specific for human PPP2CA, ectopic mouse PPP2CA expression was detected using an anti-FLAG antibody (Supplemental Figure S8A). These results suggest that PP2A activity contributes to the dephosphorylation of TFEB during PIKfyve inhibition.
We next tested whether PP2A activity is sufficient to dephosphorylate TFEB. Previous studies showed that overexpression of ectopic PPP2CA, a catalytic subunit of PP2A, results in the dephosphorylation of some of its targets (Li et al., 2010). Thus we transfected HeLa cells with a plasmid that overexpresses FLAG-tagged PPP2CA WT or catalytic-inactive mutant (D85N) (Sun et al., 2010; Reynhout et al., 2019) and tested the impact on TFEB phosphorylation. Notably, overexpression of PPP2CA WT, but not D85N, resulted in dephosphorylation of TFEB (Figure 5, A and B), which indicates that PP2A activity plays a role in the phosphorylation status of TFEB. Together, our data strongly suggest that inhibition of PIKfyve results in the dephosphorylation of TFEB which can be suppressed by inactivation of PP2A.
FIGURE 5:
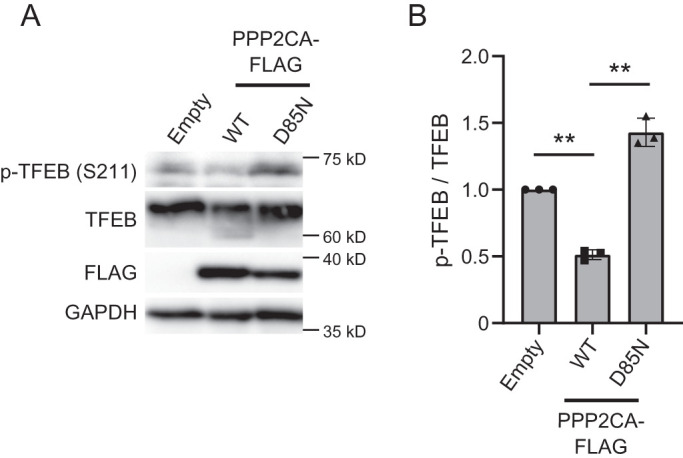
Overexpression PPP2AC results in the dephosphorylation of TFEB. (A) HeLa cells transfected with a control vector or plasmid, which overexpresses PPP2AC (WT, D85N)-FLAG, were incubated for 1 d in the growth medium and then analyzed by immunoblot using the indicated antibodies. (B) Quantitation of phospho-TFEB signal intensities from immunoblots in A and normalized to total TFEB protein (mean ± SD; three independent experiments). **P < 0.01.
Constitutively active Rag GTPase prevents TFEB dephosphorylation during PIKfyve inhibition
A recent study showed that TFEB phosphorylation requires the activity of Rag GTPases but not Rheb (Napolitano et al., 2020). Therefore we tested if the activity of Rag GTPase is involved in PIKfyve-dependent TFEB phosphorylation. We utilized a constitutively active Rag GTPase mutant pair (RagA QL and RagC SN) whose expression maintains the phosphorylation of mTORC1 substrates such as S6K1 even during amino acid starvation (Kim et al., 2008; Yoshida et al., 2011). Consistent with previous studies, the phosphorylation level of S6K1 was maintained in HBSS-treated cells expressing RagA QL and RagC SN (Figure 6A). Although apilimod or HBSS treatment significantly reduced the phosphorylation of TFEB in control cells, the expression of RagA QL and RagC SN rendered TFEB phosphorylation resistant to apilimod- or starvation-induced dephosphorylation (Figure 6, A and B). These data suggest that the presence of active Rag GTPase is essential for the phosphorylation of TFEB, and that PIKfyve acts upstream of Rag GTPases to maintain levels of TFEB phosphorylation.
FIGURE 6:
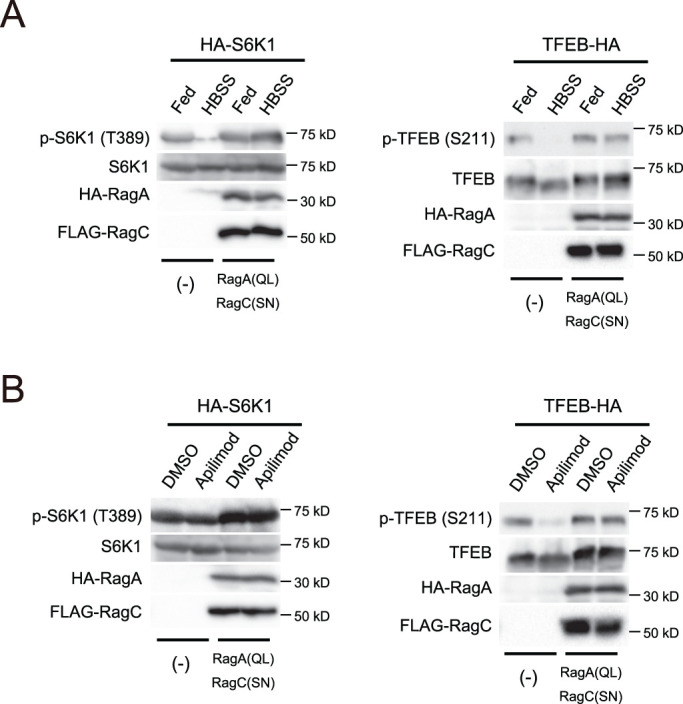
Constitutive active mutants of Rag GTPases suppress the dephosphorylation of TFEB by PIKfyve inhibition. (A) HEK293T cells expressing HA-S6K1 or TFEB-HA with/without HA-RagA (QL) and FLAG-RagC (SN) were cultured for 1 h in the growth medium or HBSS for 1 h and then analyzed by immunoblot using the indicated antibodies. Representative immunoblot from three independent experiments. (B) HEK293T cells expressing HA-S6K1 or TFEB-HA with/without HA-RagA (QL) and FLAG-RagC (SN) were treated with 1 µM apilimod (or DMSO vehicle control) and then analyzed by immunoblot using the indicated antibodies. Representative immunoblot from three independent experiments.
PIKfyve suppression impedes the interaction of TFEB with mTOR
That phosphorylation of TFEB was not impaired in apilimod-treated cells expressing active Rag GTPase (Figure 6B) suggested that PIKfyve may play a key role in mTORC1-dependent phosphorylation of TFEB but not in other mTORC1 substrates. Under nutrient-rich conditions, mTORC1 binds directly to TFEB (Roczniak-Ferguson et al., 2012). Thus we tested whether PIKfyve activity is required for this association. Similar to previous studies, an interaction between mTOR and TFEB was observed in control DMSO-treated cells. Notably, this interaction was impaired in cells treated with apilimod (Figure 7, A and B). However, the binding of mTOR with Raptor was not affected by apilimod, suggesting that the mTORC1 complex is stable. We also tested whether apilimod treatment affects TFEB binding to PP2A. We found that TFEB interacts with endogenous PP2A (PPP2CA and PPP2CB); importantly, the interaction was not affected by apilimod treatment (Figure 7, C and D). These results strongly suggest that apilimod treatment suppresses mTORC1 binding to TFEB but not to PP2A.
FIGURE 7:
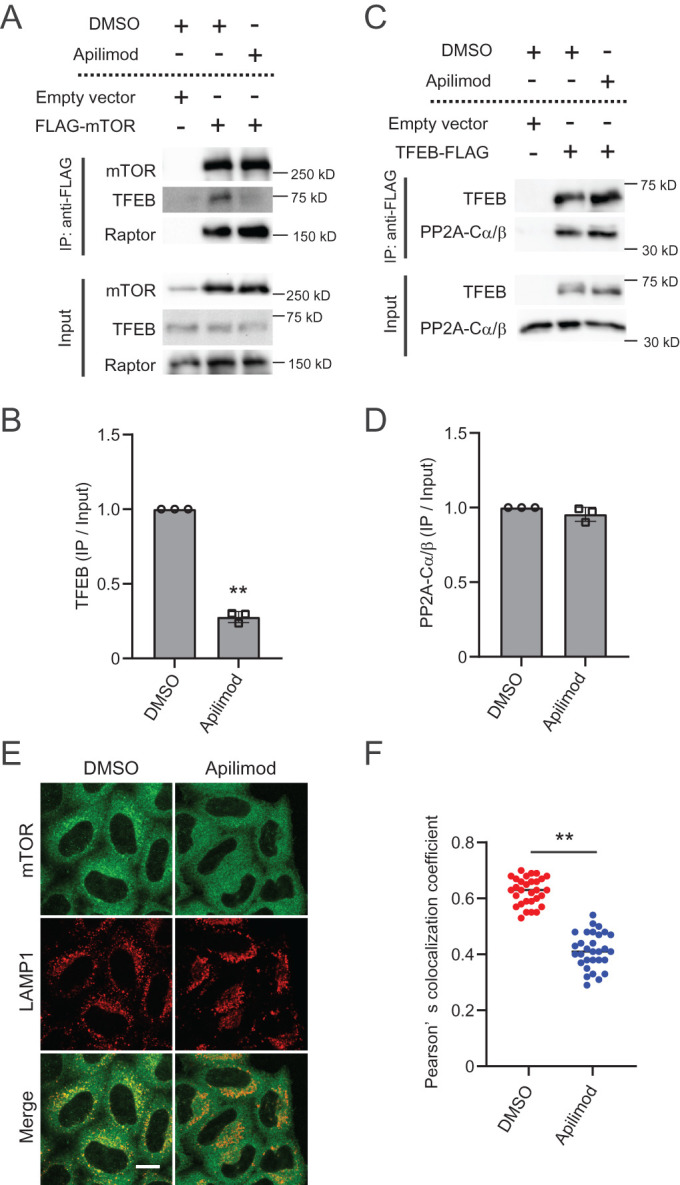
The interaction of TFEB with mTOR, but not with PP2A, is disrupted by PIKfyve suppression. (A) Lysates from HEK293T cells transfected with FLAG-mTOR were immunoprecipitated with anti-FLAG antibodies and then analyzed by immunoblot using the indicated antibodies. (B) Quantitation of immunoprecipitated TFEB signal intensities from immunoblots in A and normalized to input TFEB protein (mean ± SD; three independent experiments). **P < 0.01. (C) Lysates from HEK293T cells transfected with TFEB-FLAG were immunoprecipitated with anti-FLAG antibodies and then analyzed by immunoblot using the indicated antibodies. (D) Quantitation of immunoprecipitated PP2A-Cα/β signal intensities from immunoblots in C and normalized to input PP2A-Cα/β protein (mean ± SD; three independent experiments). **P < 0.01. (E) HeLa cells were treated with 1 µM apilimod (or DMSO vehicle control) for 2 h. Cells were fixed and stained with the indicated antibodies and then analyzed by immunofluorescence microscopy. Bar, 10 µm. (F) Quantitation of colocalization ratio between mTOR and LAMP1 in E (mean ± SD; n > 40 cells from three independent experiments). **P < 0.01.
Notably, the levels of mTOR on lysosomes were reduced in cells treated with apilimod under amino acid-replete conditions (Figure 7, E and F). This suggests that a small pool of mTORC1 at lysosomes is sufficient to phosphorylate S6K1, but more lysosomal mTORC1 may be necessary for TFEB phosphorylation. Our observations suggest that upon inhibition of PIKfyve activity, localization of mTORC1 to lysosomes is impaired and results in a loss of interaction between mTORC1 and TFEB. Thus dephosphorylation of TFEB by PP2A activity becomes dominant, and dephosphorylated TFEB translocates to the nucleus (Figure 8). Taken together, PI(3,5)P2 and/or PI5P on late endosomes and lysosomes play a crucial role in regulating TFEB activity by facilitating mTORC1 access to TFEB.
FIGURE 8:

A model for TFEB regulation by mTORC1 and PP2A. Under normal fed conditions, lysosomal localized mTORC1 phosphorylates TFEB, leading to cytoplasmic retention of TFEB. In PIKfyve-suppressed cells, the interaction between TFEB and mTORC1 is impaired, but TFEB can bind to PP2A, which promotes the dephosphorylation of TFEB, leading to the translocation of TFEB to the nucleus. See Results and Discussion for details.
DISCUSSION
In this study, we present mechanistic insight into how PIKfyve inhibition causes TFEB to translocate into the nucleus. We found that during PIKfyve inhibition, TFEB translocation to the nucleus correlates with dephosphorylation of Ser-211, a well-characterized mTORC1 phosphorylation site. Moreover, we found that PIKfyve activity is required for mTORC1 phosphorylation of TFEB on Ser-211. In addition, our studies suggest that PIKfyve acts upstream of the Rag GTPases. When PIKfyve inhibition disrupts mTORC1 interaction with TFEB, PP2A activity on TFEB is not impaired, which results in the dephosphorylation of TFEB and its translocation to the nucleus.
The precise molecular mechanisms by which PIKfyve is required for the lysosomal localization of mTORC1 or the interaction between TFEB and mTORC1 and whether PIKfyve has a role in the activation of Rag small GTPases warrant future investigation. Previously identified Rag GTPases regulators, including the KICSTOR-GATOR1 complex, the FLCN-FNIP complex, and SLC38A9, are constitutively or temporally localized on the lysosomal membrane (Petit et al., 2013; Rebsamen et al., 2015; Wang et al., 2015a; Wolfson et al., 2017; Lawrence et al., 2019). It is tempting to speculate that PIKfyve-dependent generation of lysosomal PI(3,5)P2 or/and PI5P might play a key role in the localization or/and the activity of these Rag GTPase regulators. While blockade of PIKfyve activity effectively decreased lysosomal mTOR localization and TFEB phosphorylation and increased nuclear TFEB expression (Figures 1, 2, and 7E), it showed little effect on S6K1 phosphorylation, a major direct substrate of mTORC1.
While it is unclear how apilimod preferentially inhibits TFEB phosphorylation, it possible that a small amount of lysosomal mTORC1 localization during PIKfyve inhibition is sufficient for the phosphorylation of other mTORC1 substrates. Intriguingly, a recent study demonstrated that PI(3,5)P2 on lysosomes plays a critical role in the recruitment of the TSC complex, the GTPase-activating protein (GAP) for Rheb GTPase (Fitzian et al., 2021). Therefore when TSC1 recruitment is impaired by loss of PI(3,5)P2 due to PIKfyve inhibition, the levels of Rheb activity are likely enhanced, which could lead to the activation of the limited amount of lysosomal mTORC1 that remains. This may be sufficient to phosphorylate other active Rheb-dependent mTORC1 substrates in cells lacking PIKfyve activity. In support of this hypothesis, mTORC1 phosphorylation of TFEB requires the amino acid-mediated activation of Rag GTPases but is insensitive to Rheb (Napolitano et al., 2020).
Together these previous observations and our data may explain why the inhibition of PIKfyve activity predominantly suppresses TFEB phosphorylation with little impact on other mTORC1 substrates.
Identification of PIKfyve as a key suppressor of TFEB activation raises questions about the role of PIKfyve in the regulation of TFEB-dependent transcriptome. Notably apilimod, the PIKfyve inhibitor used in these studies, was initially identified in a screen for compounds that lower expression of IL-12 (Cai et al., 2013). Importantly, apilimod-dependent inhibition of IL-12 expression is due to transcriptional up-regulation of the repressor ATF3 (Cai et al., 2014). However, plasma membrane- and lysosome-related steps upstream of IL-12 expression were not perturbed by apilimod. Thus there is a precedence for PIKfyve-dependent regulation of transcription. It is unclear whether ATF3 expression is regulated via TFEB. However, PIKfyve activity may also regulate other transcriptional regulators. Importantly, most studies of PIKfyve have focused on the roles of PIKfyve in the endomembrane system, and studies that provide a comprehensive view of transcripts and transcription factors that rely on PIKfyve have not been yet pursued. That PIKfyve is a negative regulator of TFEB, as well as a role for PIKfyve in the negative regulation of ATF3 expression, highlights the importance of pursuing future studies to determine genes that are either positively or negatively regulated by PIKfyve (Peña-Llopis et al., 2011; Roczniak-Ferguson et al., 2012).
MATERIALS AND METHODS
Request a protocol through Bio-protocol.
Reagents and antibodies
The following antibodies were used: rabbit anti-p70S6K (Cell Signaling Technologies; #2708), rabbit anti-p-p70S6K (Thr389) (Cell Signaling Technologies; #9205), rabbit anti-Akt (Cell Signaling Technologies; #9272), rabbit anti-p-Akt (Thr308) (Cell Signaling Technologies; #13038), rabbit anti-p-Akt (Ser473) (Cell Signaling Technologies; #4051), rabbit anti-ULK1 (Cell Signaling Technologies; #8054), rabbit anti-p-ULK1 (Ser757) (Cell Signaling Technologies; #6888), rabbit anti-TFEB (Cell Signaling Technologies; #4240), rabbit anti-p-TFEB (Ser211) (Cell Signaling Technologies; #37681), rabbit anti-4E-BP1 (Cell Signaling Technologies; #9644), rabbit anti-p-4E-BP1 (Thr37/46) (Cell Signaling Technologies; #2855), rabbit anti-mTOR (Cell Signaling Technologies; #2983), rabbit anti-Raptor (Cell Signaling Technologies; #2280), rabbit anti-HA (Cell Signaling Technologies; #3724), mouse anti-α-tubulin (Thermo; 236-10501), mouse anti-GAPDH (Thermo; AM4300), mouse anti-DYKDDDDK (WAKO; clone 1E6), mouse anti-LAMP1 (Development Studies Hybridoma Bank; H4A3), mouse anti-PIKfyve (Development Studies Hybridoma Bank; 3C9), rabbit anti-TFE3 (Sigma; HPA023881), mouse anti-PP2A-Cα/β (Santa Cruz; sc-80665), and rabbit anti-PPP2CA (Proteintech; 13482-1-AP). Apilimod was from MedChem Express. Okadaic acid was from Santa Cruz. Torin1, YM201636, VPS34-IN1, and FK-506 were from Cayman Chemical.
Plasmids
Human PPP2CA gene was amplified by PCR with cDNA derived from HEK293T cells using the following primers: forward (fw) 5′-GCGAATTCACCATGGACGAGAAGGTGTTCACCAAG-3′ and reverse (rv) 5′-GCGGATCCCGCAGGAAGTAGTCTGGGGTACGACGA-3′.
The product was introduced into pcDNA3.1 hygro(+) with C-terminal FLAG-tag (Thermo Fisher Scientific). PPP2CA inactive mutant D85N was cloned into the same plasmid.
Mouse PPP2CA gene was amplified by PCR with cDNA derived from MEFs using the following primers: fw 5′-GCGGATCCACCATGGACGAGAAGTTGTTCACCAAGG-3′ and rv 5′-GCGCGGCCGCCAGGAAGTAGTCTGGGGTACGACGA-3′ and cloned into pcDNA3.1 hygro(+) with C-terminal FLAG-tag.
Human RagA and RagC genes were amplified by PCR with cDNA derived from HEK293T cells using the following primers: RagA fw 5′-GCAGATCTATGCCAAATACAGCCATGAACAAAAAG-3′, RagA rv 5′-GCGTCGACTCAACGCATAAGGAGACTGTGCTTGGG-3′, RagC fw 5′-GCAGATCTATGTCCCTGCAGTACGGGGCGGAGG-3′, and RagC rv 5′-GCGTCGACCTAGATGGCGTTTCGTGGCGTGCCA-3′ and cloned into pCMV5-HA or pFLAG-CMV2 plasmid. RagA (Q66L) and RagC (S75N) were also cloned into the same plasmids.
Human TFEB gene was amplified by PCR with cDNA derived from HEK293T cells using the following primers: fw 5′-GCGGATCCACCATGGCGTCACGCATAGGGTTGCG-3′ and rv 5′-GCGATATCCAGCACATCGCCCTCCTCCATGC-3′ and cloned into pcDNA3.1 hygro(+) with C-terminal FLAG-tag.
pcDNA3.1-6xHA-PIKfyve (WT, KYA) used in this study was described previously (Giridharan et al., 2022).
pRK7-HA-S6K1 and pcDNA3-FLAG-mTOR were kindly provided by John Blenis (Cornell University) and Jie Chen (University of Illinois at Urbana-Champaign), respectively.
Cell culture and transfection
HeLa cells, HeLa cells stably expressing TFEB-GFP, Atg7 WT MEFs, and HEK293T cells were grown in DMEM supplemented with 10% fetal bovine serum (FBS), 5 U/ml penicillin, and 50 U/ml streptomycin in a 5% CO2 incubator at 37°C. Transfection was performed using Lipofectamine 2000 (Thermo Fisher Scientific) or PEI Max (Polysciences). Experiments were carried out 24 h after transfection.
RNA interference
For knockdown of human PP2A-Cα and PP2A-Cβ, siRNAs were purchased from Santa Cruz (sc-43509 and sc-39175), and for knockdown of human PIKfyve, siRNA was purchased from Thermo Fisher Scientific (Silencer Select siRNA #4392420); 10 nM siRNAs were transfected into HeLa cells with Lipofectamine RNAiMAX (Thermo Fisher Scientific), and the expression levels were assessed after 48 or 72 h by immunoblots.
Immunoprecipitation
Cells transfected with the indicated plasmids were washed twice with phosphate-buffered saline (PBS) and lysed with lysis buffer A (25 mM HEPES, pH 7.4, 0.5% Triton X-100, 5 mM EDTA, 150 mM NaCl) for immunoprecipitation of TFEB-FLAG or lysis buffer B (40 mM HEPES, pH 7.4, 0.3% CHAPS, 1 mM EDTA, 150 mM NaCl, 10 mM β-glycerophosphate) for immunoprecipitation of FLAG-mTOR, supplemented with the protease inhibitor cocktail (Nacalai tesque). The cell lysates were then incubated with ANTI-FLAG M2-agarose (Sigma-Aldrich) for 4-8 h at 4°C with rotation. Complexes were washed three times in lysis buffer. Immunoprecipitated proteins were analyzed by SDS–PAGE and immunoblotting.
Western blotting
Samples were subjected to SDS–PAGE and transferred to nitrocellulose membranes (Fisher scientific) or polyvinylidene difluoride membranes (Millipore). The transferred membranes were blocked with PBS-T (0.1% Tween 20 and PBS) containing 2% skim milk for 30 min and were then incubated overnight at 4°C with primary antibodies. Membranes were washed three times for 5 min with TBS-T and incubated for 1 h at room temperature (RT) with HRP–conjugated secondary antibodies (Jackson ImmunoResearch). The protein bands were visualized by enhanced chemiluminescence using an x-ray film or ChemiDoc (Bio-Rad).
Immunofluorescence microscopy
Cells were fixed with 4% PFA in PBS for 10 min at RT and then were permeabilized with 0.1% saponin or 0.05% TritonX-100 in PBS for 10 min at RT, washed twice with PBS, and blocked with PBS containing 0.1% gelatin for 30 min at RT or overnight at 4°C. Cells were incubated with primary antibodies in PBS containing 0.1% gelatin for 60 min. After three washes with PBS, cells were incubated with the appropriate secondary antibodies in PBS containing 0.1% gelatin for 50 min. After a brief wash with PBS, coverslips were mounted onto slides using ProLong Gold antifade reagent with DAPI (Thermo Fisher Scientific) and observed under a Leica SP5 Inverted 2-Photon FLIM Confocal equipped with 63×/NA 1.40 oil immersion objective lens (Leica).
RNA extraction and quantitative RT-PCR
Total RNA was extracted from cells using Sepasol-RNA I Super G (Nacalai tesque) according to the manufacture’s protocol. Total RNA was rv transcribed using Transcriptor First Strand cDNA Synthesis Kit (Roche). Quantitative real-time PCR reactions were performed using an EvaGreen reagent (Biotium) and LightCycler 96 Systems (Roche). The primer sequences used in this method are:
Human Beclin1: fw 5′-AGCTGCCGTTATACTGTTCTG-3′ and rv 5′-ACTGCCTCCTGTGTCTTCAATCTT-3′
Human LC3B: fw 5′-AGCAGCATCCAACCAAAATC-3′ and rv 5′-CTGTGTCCGTTCACCAACAG-3′
Human Atg5: fw 5′-TGGATTTCGTTATATCCCCTTTAG-3′ and rv 5′-CCTAGTGTGTGCAACTGTCCA-3′
Human ATP6V1H: fw 5′-GGAAGTGTCAGATGATCCCCA-3′ and rv 5′-CCGTTTGCCTCGTGGATAAT-3′
Human CTSA: fw 5′-CAGGCTTTGGTCTTCTCTCCA-3′ and rv 5′-TCACGCATTCCAGGTCTTTG-3′
Human CTSB: fw 5′-AGTGGAGAATGGCACACCCTA-3′ and rv 5′-AAGAAGCCATTGTCACCCCA-3′
Human LAMP1: fw 5′-ACGTTACAGCGTCCAGCTCAT-3′ and rv 5′-TCTTTGGAGCTCGCATTGG-3′
Human MCOLN1: fw 5′-TTGCTCTCTGCCAGCGGTACTA-3′ and rv 5′-GCAGTCAGTAACCACCATCGGA-3′
Human β-actin: fw 5′-CACCATTGGCAATGAGCGGTTC-3′ and rv 5′-AGGTCTTTGCGGATGTCCACGT-3′.
Differences between samples were calculated using the ∆∆Ct method.
Statistical analysis
Statistically significant differences were determined using the Student’s t test or one-way ANOVA with post-hoc test. Differences were considered significant if P < 0.05.
Supplementary Material
Acknowledgments
We thank Haoxing Xu (University of Michigan, Ann Arbor, MI) for providing HeLa cells stably expressing TFEB-GFP; Masaaki Komatsu (Juntendo University, Tokyo, Japan) for providing Atg7 WT MEFs; Naoko Sakamoto for technical assistance.
This study was supported in part by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant No. JP19K22381 (to J.H.), the National Institutes of Health Grant R01- NS099340 (to L.S.W.), the ONO Medical Research Foundation (to J.H.), and the Takeda Science Foundation (to J.H.)
Abbreviations used:
- elF4E
eukaryotic translation initiation factor 4E
- ERK
extracellular signal-regulated kinase
- FBS
fetal bovine serum
- fw
forward
- GAP
GTPase-activating protein
- GSK3
glycogen synthase kinase 3
- HBSS
Hank’s balanced salt solution
- mTOR
mechanistic target of rapamycin
- mTORC1
mechanistic target of rapamycin complex 1
- PBS
phosphate-buffered saline
- PI
phosphoinositide lipid
- PI(3,5)P 2
phosphatidylinositol 3,5-bisphosphate
- PI3P
phosphatidylinositol 3-phosphate
- PP2A
protein phosphatase 2A
- rv
reverse
- RT
room temperature
- TFEB
transcriptional factor EB
- ULK1
unc-51-like kinase 1
- WT
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E21-06-0309) on January 12, 2022.
REFERENCES
- Bago R, Malik N, Munson MJ, Prescott AR, Davies P, Sommer E, Shpiro N, Ward R, Cross D, Ganley IG, et al. (2014). Characterization of VPS34-IN1, a selective inhibitor of Vps34, reveals that the phosphatidylinositol 3-phosphate-binding SGK3 protein kinase is a downstream target of class III phosphoinositide 3-kinase. Biochem J 463, 413–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balla T (2013). Phosphoinositides: Tiny lipids with giant impact on cell regulation. Physiol Rev 93, 1019–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM (2012). Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 150, 1196–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Xu Y, Cheung AK, Tomlinson RC, Alcázar-Román A, Murphy L, Billich A, Zhang B, Feng Y, Klumpp M, et al. (2013). PIKfyve, a class III PI Kinase, is the target of the small molecular IL-12/IL-23 inhibitor apilimod and a player in toll-like receptor signaling. Chem Biol 20, 912–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Xu Y, Kim Y-M, Loureiro J, Huang Q (2014). PIKfyve, a class III lipid kinase, is required for TLR-induced type I IFN production via modulation of ATF3. J Immunol 192, 3383–3389. [DOI] [PubMed] [Google Scholar]
- Cao Q, Yang Y, Zhong XZ, Dong XP (2017). The lysosomal Ca2+ release channel TRPML1 regulates lysosome size by activating calmodulin. J Biol Chem 292, 8424–8435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Wang K, Long A, Jia L, Zhang Y, Deng H, Li Y, Han J, Wang Y (2017). Fasting-induced hormonal regulation of lysosomal function. Cell Res 27, 748–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow CY, Zhang Y, Dowling JJ, Jin N, Adamska M, Shiga K, Szigeti K, Shy ME, Li J, Zhang X, et al. (2007). Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature 448, 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy CH, Saffi G, Gray MA, Wallace C, Dayam RM, Ou Z-YA, Lenk G, Puertollano R, Watkins SC, Botelho RJ (2018). Lysosome enlargement during inhibition of the lipid kinase PIKfyve proceeds through lysosome coalescence. J Cell Sci 131, jcs213587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton LM, Ikonomov OC, Sbrissa D, Garg P, Shisheva A (2016). Active vacuolar H+ atpase and functional cycle of rab5 are required for the vacuolation defect triggered by ptdins(3,5)p2 loss under PIKfyve or Vps34 deficiency. Am J Physiol - Cell Physiol 311, C366–C377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devereaux K, Dall’Armi C, Alcazar-Roman A, Ogasawara Y, Zhou X, Wang F, Yamamoto A, de Camilli P, Di Paolo G (2013). Regulation of mammalian autophagy by class II and III PI 3-Kinases through PI3P synthesis. PLoS One 8, 10–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienstmann R, Rodon J, Serra V, Tabernero J (2014). Picking the point of inhibition: A comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther 13, 1021–1031. [DOI] [PubMed] [Google Scholar]
- Duex JE, Nau JJ, Kauffman EJ, Weisman LS (2006a). Phosphoinositide 5-phosphatase Fig4p is required for both acute rise and subsequent fall in stress-induced phosphatidylinositol 3,5-bisphosphate levels. Eukaryot Cell 5, 723–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duex JE, Tang F, Weisman LS (2006b). The Vac14p-Fig4p complex acts independently of Vac7p and couples PI3,5P2 synthesis and turnover. J Cell Biol 172, 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebner M, Koch PA, Haucke V (2019). Phosphoinositides in the control of lysosome function and homeostasis. Biochem Soc Trans, BST20190158. [DOI] [PubMed] [Google Scholar]
- Fitzian K, Brückner A, Brohée L, Zech R, Antoni C, Kiontke S, Gasper R, Linard Matos AL, Beel S, Wilhelm S, et al. (2021). TSC1 binding to lysosomal PIPs is required for TSC complex translocation and mTORC1 regulation. Mol Cell 81, 2705-2721.e8. [DOI] [PubMed] [Google Scholar]
- Gao M, Kaiser CA (2006). A conserved GTPase-containing complex is required for intracellular sorting of the general amino-acid permease in yeast. Nat Cell Biol 8, 657–667. [DOI] [PubMed] [Google Scholar]
- Giridharan SSP, Luo G, Rivero-Rios P, Steinfeld N, Tronchere H, Singla A, Burstein E, Billadeau DD, Sutton MA, Weisman LS (2022). Lipid kinases VPS34 and PIKfyve coordinate a phosphoinositide cascade to regulate Retriever-mediated recycling on endosomes. eLife 11, e69709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa J, Strunk BS, Weisman LS (2017). PI5P and PI(3,5)P2: Minor, but essential phosphoinositides. Cell Struct Funct 60, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho CY, Alghamdi TA, Botelho RJ (2012). Phosphatidylinositol-3,5-bisphosphate: No longer the poor PIP 2. Traffic 13, 1–8. [DOI] [PubMed] [Google Scholar]
- Ikonomov OC, Sbrissa D, Mlak K, Kanzaki M, Pessin J, Shisheva A (2002). Functional dissection of lipid and protein kinase signals of PIKfyve reveals the role of PtdIns 3,5-P2 production for endomembrane integrity. J Biol Chem 277, 9206–9211. [DOI] [PubMed] [Google Scholar]
- Ikonomov OC, Sbrissa D, Venkatareddy M, Tisdale E, Garg P, Shisheva A (2015). Class III PI 3-kinase is the main source of PtdIns3P substrate and membrane recruitment signal for PIKfyve constitutive function in podocyte endomembrane homeostasis. Biochim Biophys Acta - Mol Cell Res 1853, 1240–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inpanathan S, Botelho RJ (2019). The lysosome signaling platform: Adapting with the times. Front Cell Dev Biol 7, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferies HB, Cooke FT, Jat P, Boucheron C, Koizumi T, Hayakawa M, Kaizawa H, Ohishi T, Workman P, Waterfield MD, et al. (2008). A selective PIKfyve inhibitor blocks PtdIns(3,5)P2 production and disrupts endomembrane transport and retroviral budding. EMBO Rep 9, 164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon KI, Jono H, Miller CL, Cai Y, Lim S, Liu X, Gao P, Abe JI, Li JD, Yan C (2010). Ca2+/calmodulin-stimulated PDE1 regulates the beta-catenin/TCF signaling through PP2A B56 gamma subunit in proliferating vascular smooth muscle cells. FEBS J 277, 5026–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin N, Lang MJ, Weisman LS (2016). Phosphatidylinositol 3,5-bisphosphate: Regulation of cellular events in space and time. Biochem Soc Trans 44, 177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL (2008). Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol 10, 935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence RE, Fromm SA, Fu Y, Yokom AL, Kim DJ, Thelen AM, Young LN, Lim CY, Samelson AJ, Hurley JH, et al. (2019). Structural mechanism of a Rag GTPase activation checkpoint by the lysosomal folliculin complex. Science (80-) 366, 971–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence RE, Zoncu R (2019). The lysosome as a cellular centre for signalling, metabolism and quality control. Nat Cell Biol 21, 133–142. [DOI] [PubMed] [Google Scholar]
- Letourneux C, Rocher G, Porteu F (2006). B56-containing PP2A dephosphorylate ERK and their activity is controlled by the early gene IEX-1 and ERK. EMBO J 25, 727–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Friedrichsen HJ, Andrews S, Picaud S, Volpon L, Ngeow K, Berridge G, Fischer R, Borden KLB, Filippakopoulos P, et al. (2018). A TFEB nuclear export signal integrates amino acid supply and glucose availability. Nat Commun 9, 2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Yue P, Deng X, Ueda T, Fukunaga R, Khuri FR, Sun SY (2010). Protein phosphatase 2A negatively regulates eukaryotic initiation factor 4E phosphorylation and eif4F assembly through direct dephosphorylation of mnk and eif4E. Neoplasia 12, 848–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim CY, Zoncu R (2016). The lysosome as a command-and-control center for cellular metabolism. J Cell Biol 214, 653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Chen Y, Gucek M, Puertollano R (2012). MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8, 903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Diab HI, Brady OA, Puertollano R (2016). TFEB and TFE 3 are novel components of the integrated stress response. EMBO J 35, 479–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Diab HI, Lishu L, Jeong-A L, Patange S, Raben N, Puertollano R (2014). The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci Signal 7, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Puertollano R (2013). Rag GTPases mediate amino acid-dependent recruitment of TFEB and MITF to lysosomes. J Cell Biol 200, 475–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Puertollano R (2018). Protein phosphatase 2A stimulates activation of TFEB and TFE3 transcription factors in response to oxidative stress. J Biol Chem 293, 12,525–12,534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCartney AJ, Zolov SN, Kauffman EJ, Zhang Y, Strunk BS, Weisman LS, Sutton MA (2014). Activity-dependent PI(3,5)P2 synthesis controls AMPA receptor trafficking during synaptic depression. Proc Natl Acad Sci USA 111, E4896–E4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, et al. (2015). Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol 17, 288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano G, Di Malta C, Esposito A, de Araujo MEG, Pece S, Bertalot G, Matarese M, Benedetti V, Zampelli A, Stasyk T, et al. (2020). A substrate-specific mTORC1 pathway underlies Birt–Hogg–Dubé syndrome. Nature 585, 597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano G, Esposito A, Choi H, Matarese M, Benedetti V, Di Malta C, Monfregola J, Medina DL, Lippincott-Schwartz J, Ballabio A (2018). mTOR-dependent phosphorylation controls TFEB nuclear export. Nat Commun 9, 3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogunbayo OA, Duan J, Xiong J, Wang Q, Feng X, Ma J, Zhu MX, Evans AM (2018). MTORC1 controls lysosomal Ca 2+ release through the two-pore channel TPC2. Sci Signal 11, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyarzún JE, Lagos J, Vázquez MC, Valls C, De la Fuente C, Yuseff MI, Alvarez AR, Zanlungo S (2019). Lysosome motility and distribution: Relevance in health and disease. Biochim Biophys Acta - Mol Basis Dis 1865, 1076–1087. [DOI] [PubMed] [Google Scholar]
- Peña-Llopis S, Vega-Rubin-De-Celis S, Schwartz JC, Wolff NC, Tran TAT, Zou L, Xie XJ, Corey DR, Brugarolas J (2011). Regulation of TFEB and V-ATPases by mTORC1. EMBO J 30, 3242–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit CS, Roczniak-Ferguson A, Ferguson SM (2013). Recruitment of folliculin to lysosomes supports the amino acid-dependent activation of rag gtpases. J Cell Biol 202, 1107–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben N, Puertollano R (2016). TFEB and TFE3: Linking Lysosomes to Cellular Adaptation to Stress. Annu Rev Cell Dev Biol 32, 255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebsamen M, Pochini L, Stasyk T, de Araújo ME, Galluccio M, Kandasamy RK, Snijder B, Fauster A, Rudashevskaya EL, Bruckner M, et al. (2015). SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 519, 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynhout S, Jansen S, Haesen D, van Belle S, de Munnik SA, Bongers EMHF, Schieving JH, Marcelis C, Amiel J, Rio M, et al. (2019). De novo mutations affecting the catalytic Cα subunit of PP2A, PPP2CA, cause syndromic intellectual disability resembling other PP2A-related neurodevelopmental disorders. Am J Hum Genet 104, 139–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM (2012). The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal 5, ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford AC, Traer C, Wassmer T, Pattni K, Bujny M V., Carlton JG, Stenmark H, Cullen PJ (2006). The mammalian phosphatidylinositol 3-phosphate 5-kinase (PIKfyve) regulates endosome-to-TGN retrograde transport. J Cell Sci 119, 3944–3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM (2010). Ragulator-rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM (2008). The rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science (80-) 320, 1496–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, et al. (2009). A gene network regulating lysosomal biogenesis and function. Science 325, 473–477. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Takasuga S, Sasaki J, Kofuji S, Eguchi S, Yamazaki M, Suzuki A (2009). Mammalian phosphoinositide kinases and phosphatases. Prog Lipid Res 48, 307–343. [DOI] [PubMed] [Google Scholar]
- Saucedo LJ, Gao X, Chiarelli DA, Li L, Pan D, Edgar BA (2003). Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat Cell Biol 5, 566–571. [DOI] [PubMed] [Google Scholar]
- Saxton RA, Sabatini DM (2017). mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi T, Hirose E, Nakashima N, Ii M, Nishimoto T (2001). Novel G proteins, Rag C and Rag D, interact with GTP-binding proteins, Rag A and Rag B. J Biol Chem 276, 7246–7257. [DOI] [PubMed] [Google Scholar]
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al. (2011). TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, et al. (2012). A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 31, 1095–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HM, Mizushima N (2014). At the end of the autophagic road: An emerging understanding of lysosomal functions in autophagy. Trends Biochem Sci 39, 61–71. [DOI] [PubMed] [Google Scholar]
- Shisheva A (2012). PIKfyve and its Lipid Products in Health and in Sickness. Curr Top Microbiol Immunol 362, 127–162. [DOI] [PubMed] [Google Scholar]
- Stocker H, Radimerski T, Schindelholz B, Wittwer F, Belawat P, Daram P, Breuer S, Thomas G, Hafen E (2003). Rheb is an essential regulator of S6K in controlling cell growth in Drosophila. Nat Cell Biol 5, 559–565. [DOI] [PubMed] [Google Scholar]
- Sun W, Wang H, Zhao X, Yu Y, Fan Y, Wang H, Wang X, Lu X, Zhang G, Fu S, et al. (2010). Protein phosphatase 2A acts as a mitogen-activated protein kinase kinase kinase 3 (MEKK3) phosphatase to inhibit lysophosphatidic acid-induced IκB kinase β/nuclear factor-κB activation. J Biol Chem 285, 21341–21348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasuga S, Horie Y, Sasaki J, Sun-Wada GH, Kawamura N, Iizuka R, Mizuno K, Eguchi S, Kofuji S, Kimura H, et al. (2013). Critical roles of type III phosphatidylinositol phosphate kinase in murine embryonic visceral endoderm and adult intestine. Proc Natl Acad Sci USA 110, 1726–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda T, Watanabe-Fukunaga R, Fukuyama H, Nagata S, Fukunaga R (2004). Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development. Mol Cell Biol 24, 6539–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Tsun ZY, Wolfson RL, Shen K, Wyant GA, Plovanich ME, Yuan ED, Jones TD, Chantranupong L, Comb W, et al. (2015a). Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science (80-) 347, 188–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Gao Q, Yang M, Zhang X, Yu L, Lawas M, Li X, Bryant-Genevier M, Southall NT, Marugan J, et al. (2015b). Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proc Natl Acad Sci 112, E1373–E1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfson RL, Chantranupong L, Wyant GA, Gu X, Orozco JM, Shen K, Condon KJ, Petri S, Kedir J, Scaria SM, et al. (2017). KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature 543, 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Jones E, Inoki K (2017). Lysosomal regulation of mTORC1 by amino acids in mammalian cells. Biomolecules 7, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, et al. (2004). A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol 6, 308–318. [DOI] [PubMed] [Google Scholar]
- Yoshida S, Hong S, Suzuki T, Nada S, Mannan AM, Wang J, Okada M, Guan KL, Inoki K (2011). Redox regulates mammalian target of rapamycin complex 1 (mTORC1) activity by modulating the TSC1/TSC2-Rheb GTPase pathway. J Biol Chem 286, 32651–32660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Cheng X, Yu L, Yang J, Calvo R, Patnaik S, Hu X, Gao Q, Yang M, Lawas M, et al. (2016). MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun 7, 12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhitomirsky B, Yunaev A, Kreiserman R, Kaplan A, Stark M, Assaraf YG (2018). Lysosomotropic drugs activate TFEB via lysosomal membrane fluidization and consequent inhibition of mTORC1 activity. Cell Death Dis 9, 1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolov SN, Bridges D, Zhang Y, Lee WW, Riehle E, Verma R, Lenk GM, Converso-Baran K, Weide T, Albin RL, et al. (2012). In vivo, Pikfyve generates PI(3,5)P2, which serves as both a signaling lipid and the major precursor for PI5P. Proc Natl Acad Sci USA 109, 17472–17477. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.